
Contents
What is infantile spasms
Infantile spasms often called West Syndrome, are a rare special type of seizure with both focal and generalized features. Infantile spasms onset is usually in the first year of life (over 90% of cases begin before 12 months of life), typically often begin between three and six months of age but some children may experience spasms as early as one month and appear as brief stiffening movements lasting one to two seconds each. The seizures often look like a sudden bending forward of the body with stiffening of the arms and legs lasting for 1-2 seconds; some children arch their backs as they extend their arms and legs. Infantile spasms can involve the whole body or only the head or a part of the face. Spasms typically repeat every few seconds in what is called a cluster, and the infant usually appears to recover or relax between each spasm. Clusters of spasms often occur after waking from sleep and often occur in multiple clusters and hundreds of seizures per day.
Infantile spasms is characterized by epileptic spasms, which consist of massive myoclonic jerks of the body, which can be extensor or flexor (or both) in nature. Infantile spasms often are accompanied by developmental problems and a characteristic interictal brain wave pattern on electroencephalography (EEG) testing called hypsarrhythmia. Most children, but not all, will have EEG readings of hypsarrhythmia. When all three features are present, the term “West syndrome” is commonly used. West syndrome is characterized by an electroclinical triad of (1) epileptic spasms, (2) hypsarrhythmia on EEG study, and (3) developmental stagnation or regression. West syndrome is considered to be catastrophic because of the frequent sequelae of global neurodevelopmental delay, significant intellectual disability, and medically refractory epilepsy.
Infantile spasms (West syndrome) usually stop by age five, but may be replaced by other seizure types. Many underlying disorders, such as birth injury, metabolic disorders, and genetic disorders can give rise to infantile spasms, making it important to identify the underlying cause. In some children, no cause can be found.
The incidence of infantile spasms is 2 to 3 per 10,000 live births 1, with a lifetime prevalence of 1.5 to 2 per 10,000 children 2. About 1,200 children in the US are diagnosed each year with infantile spasms. Infantile spasms is slightly more common in males, accounting for about 60% of cases, and a family history exists in 3% to 6% of cases 3.
Infantile spasms are so uncommon that most pediatricians will see only one or two infantile spasms cases during all the years of practice. Also infantile spasms often looks similar to common disorders such as a normal startle reflex, colic, or reflux.
It is very important to recognize that a child has infantile spasms as soon as it begins because 4:
- there are medications that may control the spasms
- the longer the spasms last before they are treated and controlled, the poorer the child may do developmentally
Unfortunately, children who develop infantile spasms are at great risk for developmental disability and autism, but some children will do well if they are treated early. Because the spells may be subtle, the diagnosis may be delayed for weeks or months
Infantile spasms was originally classified as a generalized epilepsy according to the 1989 International Epilepsy Classification 5. However, using the revised classification based on the 2001 International League Against Epilepsy report, this epileptic syndrome can be classified as: Level I (ictal phenomenology): clinical spasms, flexor, extensor, mixed or subtle; Level II (epilepsy seizure type): epileptic spasms; Level III (epilepsy syndrome): infantile spasm syndrome 6.
Determining the cause of infantile spasms is very important because it affects treatment and prognosis. After careful evaluation, the underlying cause can be identified in more than 70% of cases 4. There are dozens of disorders that are known to cause infantile spasms. When neurologists are able to identify the cause, it is labeled “symptomatic.” It is essential that an appropriate diagnostic evaluation be performed in every child. A diagnosis is important because it leads to specific treatment that may improve the long-term developmental outcome. In fact, some children with infantile spasms may ultimately lead normal lives, but only if they are diagnosed and treated correctly.
The goal of treatment for infantile spasms is for the seizures to stop and the EEG to improve (hypsarrhythmia should resolve). Standard first-line treatments for infantile spasms include several forms of hormonal therapy (including adrenocorticotrophic hormone [ACTH] or prednisone) or the anti-seizure medication vigabatrin (GABA aminotransferase inhibitor) 7. These treatments are highly effective but have serious side effects and should be administered in consulataton with a pediatric neurologist. Some children have spasms as the result of brain lesions, and surgical removal of these lesions may result in improvement. When standard treatments do not lead to improvement, other options such as the ketogenic diet and anti-seizure medications are considered. Regardless of the specific treatment chosen, it is critical to begin therapy as soon as possible. Ongoing infantile spasms (and hypsarrhythmia) have the potential to adversely impact all aspects of brain development.
To help make clear what infantile spasms look like, please watch these videos of a child having infantile spasms.
What do infantile spasms look like?
Infantile spasms were first described by the English physician, Dr. W.J. West in 1841 8. His description is as accurate today as it was then. It is a remarkable report because Dr. West was describing his own son and he was asking for help. The following is a quote from Dr. West’s report describing his son:
- “The child is now a year old; it was a remarkably fine, healthy child when born, and continued to thrive until he was four months old. It was at this time that I first observed slight bobbings of the head forward, which I then regarded as a trick, but were, in fact, the first indications of disease”
Like Dr. West’s son, many children with infantile spasms appear to be normal until the spasms begin. It seems that he was not concerned when he first saw the spasms because they did not appear to be serious. Just like Dr. West, parents today often tell us that they were not concerned at first. But, as the spasms become more obvious they realize that this is something serious. Dr. West then described what happened to his son over the next few months.
- “for these bobbings increased in frequency, and at length became so frequent and powerful, as to cause a complete heaving of the head forward toward his knees, and then immediately relaxing into the upright position: these bowings and relax things would be repeated alternately at intervals of a few seconds, and repeated from 10 to 20 or more times in each attack, which attack would not continue for more than two or 3 minutes; he sometimes has two, three, or more attacks in the day; they come on whether sitting or lying; just before they come on, he is all alive and in motion, making a strange noise, and then all of a sudden down goes his head and upwards his knees; he then appears frightened and screams out”
The spasms usually become more obvious as they did in this case. Sometimes they remain subtle, maybe just a brief head nod but nothing else. Dr. West‘s son had what’s called “flexor spasms” where the child’s head goes forward and the legs come up. The arms usually flare out from the body as the head and body come forward. Sometimes spasms occur with the head going back and the legs straightening out. These are called “extensor spasms.” Each spasm episode lasts just a second or two but they often come in clusters. Although the spasms may happen as single jerk event, clusters are more common and often occur on awakening in the morning or after a nap. By the time Dr. West wrote about his son, he had been having the spasms for several months and the effect on his development was obvious, as Dr. West wrote:
- “at one time he looked pale and exhausted, but lately he has regained his good looks, and independent of this affection, is a fine growing child, but he neither possesses the intellectual vivacity or the power of moving his limbs, a child his age; he never cries at the time of the attacks, or smiles or takes any notice, but looks placid and pitiful, yet his hearing and vision are good; he has no power of holding himself upright or using his limbs, and his head falls without support”
Unfortunately for Dr. West’s son, there were no effective treatments available in 1841 so he had the worst possible outcome. In addition to not developing mentally, he also seemed to be very weak and was not even able to control his head when he was seated.
Infantile spasms prognosis
The prognosis for children with infantile spasms is largely dependent on the underlying cause. The intellectual prognosis for children with infantile spasms is generally poor because many babies with infantile spasms have neurological impairment prior to the onset of spasms. Children who have rapid initiation of treatment, normal development prior to infantile spasms, and no identifiable cause may do well. Infantile spasms usually resolves by mid-childhood, but more than half of the children with infantile spasms will develop other types of seizures such as Lennox-Gastaut syndrome, an epileptic disorder of later childhood. In addition, children with infantile spasms are at a higher risk for autism. Shorter duration between the onset of infantile spasms and initiation of standard treatment appears to lead to an improved outcome; therefore early recognition of the seizures and early treatment are essential.
The spontaneous remission rate of infantile spasms in limited natural history studies is 30%. Although clinical spasms and the typical EEG pattern disappear by 3 to 4 years of age, up to 60% of children with infantile spasms go on to develop other types of seizures. The long-term prognosis for infantile spasms in terms of neurodevelopmental outcomes and development of other types of seizures remains dismal, although most studies reported better neurodevelopmental outcomes in idiopathic or cryptogenic cases.
The United Kingdom Infantile Spasms Study reported on the long-term outcomes of 77 patients with infantile spasms at 4 years and noted that whereas there was no significant difference between patients treated with either hormonal therapy or vigabatrin in outcomes for either neurodevelopmental delay or epilepsy, those patients with cryptogenic infantile spasms who received hormonal therapy had higher mean Vineland Adaptive Behavioral Scale scores 9. Also, a 3.9-point decrease in mean Vineland Adaptive Behavioral Scale score was observed for each increase in category of lead-time duration after controlling for the effects of treatment and etiology 9.
The 2012 updated guideline recommended a shorter lag time to treatment of infantile spasms with either hormonal therapy or vigabatrin to possibly improve long-term developmental outcomes 10.
These data emphasize the importance of prompt diagnosis and urgent treatment of children with infantile spasms to improve their long-term outcomes.
Dr. Riikonen 7 from Finland has followed 214 infantile spasms patients for 20–35 years. She has collected the best long-term follow-up studies of these patients. In her series, nearly 1/3 of the patients died during the follow-up period; many in the first 3 years of life. Many of the 24 patients who died by age 3 died of complications of therapy with ACTH. During the time the study was done, patients were treated with high-dose ACTH for extended periods of time. This caused immunosuppression , which greatly increased the risk of infection such as pneumonia. The current approach to treatment of using ACTH or prednisolone for much shorter periods of time is associated with a much lower risk of death. Of the 147 surviving patients, 25 (17%) had a favorable developmental outcome with an IQ of 85 or greater. Eleven others were somewhat lower with an IQ of 68–84. Thus, of the 214 patients diagnosed with infantile spasms, 31% died, 45% were developmentally disabled, but 24% had a reasonably favorable outcome. The outcome is dependent on two major factors. First and foremost is the underlying etiology or cause. Some etiologies will lead to death or mental retardation, whether or not the patient developed infantile spasms. However, children with cryptogenic infantile spasms or infantile spasms that due to treatable causes, such as focal cortical dysplasia, may have a normal or near normal developmental outcome if seizures are controlled. Thus, the goal of therapy is to achieve control as soon as possible, especially for children who may have the potential for normal intellectual development.
Infantile spasms symptoms
Clinically, the spasms appear in clusters and are characterized by brief, sudden contractions of the axial musculature. The clusters may occur several times daily, with up to 100 spasms per day. They appear to be temporally related to sleep, tending to occur as the infant falls asleep or awakens. Depending on the muscle groups involved, the spasms can be further subdivided into flexor, extensor, or mixed. The type of spasms may also be influenced by the position of the body at the time the spasms occur.
In flexor spasms, the infant appears to be in a self-hugging posture with sudden adduction of the arms. The abdominal muscles may be involved, with the infant bending at the waist; the term jackknife seizureis sometimes used for this manifestation. The combination of jackknife seizure plus adduction of the arms with or without neck flexion is called salaam seizures.
Extensor spasms look similar to an exaggerated Moro reflex. Mixed spasms are a combination of flexion of the neck, trunk, and arms, with leg extension. In some cases, the spasms may be subtle, manifesting as head nods and clusters of wide eye opening with eye deviation.
The cause of infantile spasms does not point to the clinical signs and symptoms, or appearance, of the spasms; however, the presence of asymmetry may indicate focal cortical pathology as a cause.
Developmental regression is almost always seen at the onset of infantile spasms, with decreased visual alertness often being the first symptom. The vast majority of children develop significant cognitive impairment when followed long-term. In many cases, the spasms evolve into other types of medically refractory epilepsy as the children grow older.
Infantile spasms causes
Infantile spasms may be classified into three groups: symptomatic, cryptogenic, and idiopathic. The term symptomatic is used to describe cases in which there are structural brain abnormalities or metabolic causes seen in a child with preexisting developmental delay. When there are no apparent causes identified in a child with developmental delay or some other neurologic impairment before the onset of spasms, the term cryptogenic infantile spasms is used. Idiopathic infantile spasms are those in which the child is developmentally normal, with a normal neurologic exam prior to onset of infantile spasms 11.
Cryptogenic cases constitute up to 15% of infantile spasms cases. The number of symptomatic cases has increased over the years due to advancement in neuroimaging technology, better metabolic testing, and the availability of genetic testing 11.
Symptomatic cases are further subdivided into prenatal, perinatal, and postnatal, depending on the timing of presumed causes. There are numerous disorders associated with symptomatic infantile spasms, but major categories include chromosomal abnormalities and genetic syndromes, disorders of cortical development, infections, metabolic conditions and vitamin deficiencies, trauma, vascular insults, and tumors. Tuberous sclerosis is a major symptomatic cause of infantile spasms, with up to 50% of patients with TS presenting with infantile spasms between 4 and 6 months of age. Several gene mutations that have been associated with infantile spasms include ARX, CDKL5, FOXG1, GRIN1, GRIN2A, MAGI2, MEF2C, SLC25A22, SPTAN1, and STXBP1. Children with these genetic disorders were formerly classified as cryptogenic or idiopathic. Classification of cases into these two categories will continue to dwindle as more and more genetic causes of infantile spasms are revealed by more sophisticated genetic screening. In fact, new insights into molecular genetic testing imply that all forms of infantile spasms may actually be symptomatic, and the International League Against Epilepsy has recently recommended replacing the symptomatic, cryptogenic, and idiopathic classification system of epilepsy syndromes. However, most studies on treatment and long-term outcomes of infantile spasms subdivide cases into either symptomatic or cryptogenic, and these studies do point to the cause as a contributing factor for long-term neurodevelopmental outcomes 11.
The cause of infantile spasms (West syndrome) is varied (see Figure 1 below), with the majority (~60%) being due to structural-metabolic causes while the rest are either of unknown cause or linked to genetic defects 12. The unknown cause group is the infants in which no structural-metabolic or genetic cause can be identified with the existing diagnostic tests. With the recent advances in methods for genetic diagnosis, an increasing number of associations of infantile spasms (West syndrome) with genetic defects has been made, which is now estimated to encompass the 12% of infants with infantile spasms (West syndrome) 13. The list of known genetic associations with infantile spasms can be found here (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4458451/bin/NIHMS689516-supplement-2.docx). The cause influences treatment response. A known structural-metabolic underlying cause diminishes significantly the chance of treatment response while the unknown cause group bears the best prognosis 7. On the other hand, knowing the cause may guide treatment selection, as is the case with tuberous sclerosis which is particularly responsive to vigabatrin 7.
Figure 1. Infantile spasms causes and diagnostic steps
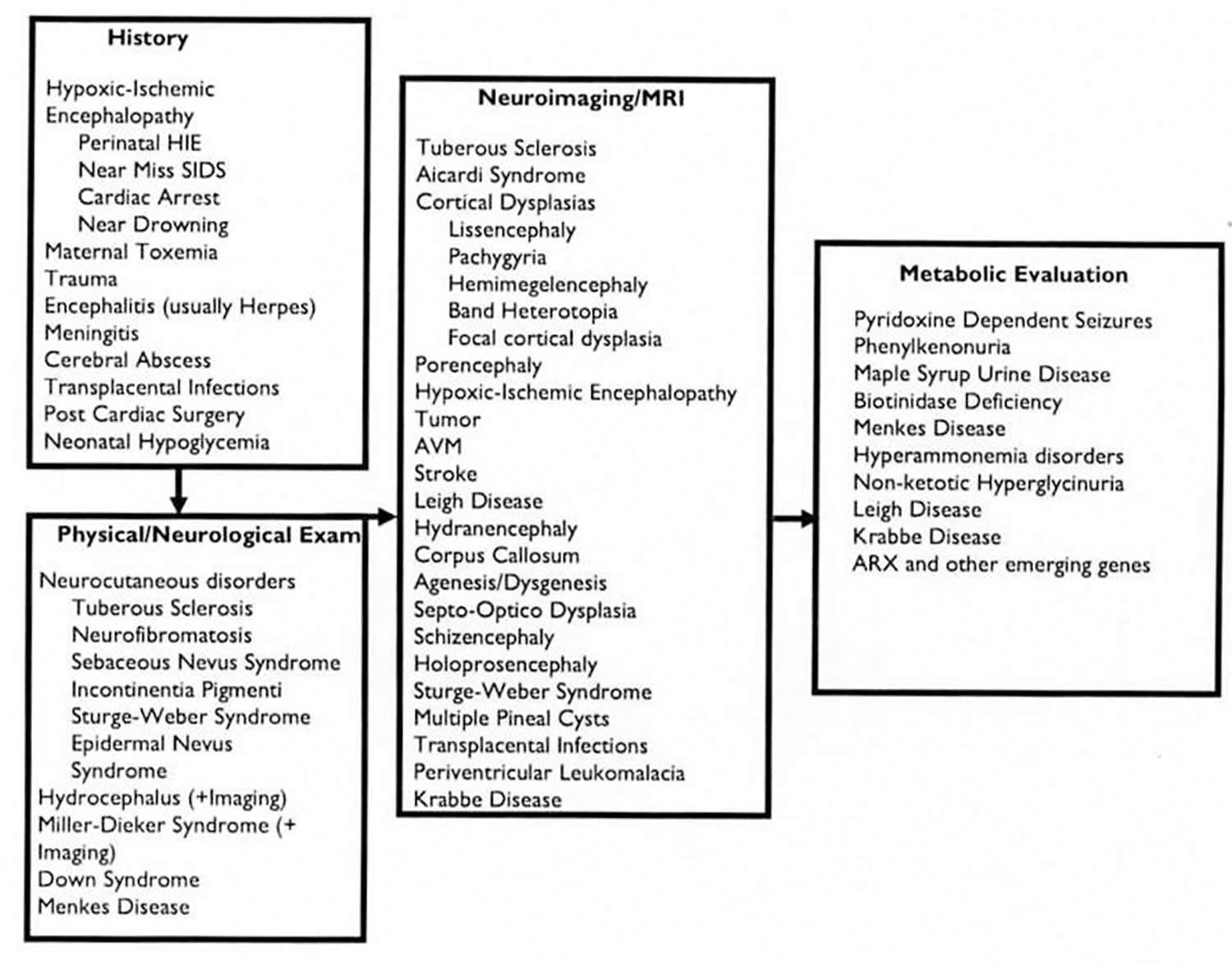
Infantile spasms diagnosis
Because of the critical importance of early treatment, children presenting with infantile spasms require prompt and comprehensive diagnostic evaluation. This includes a complete history, physical and neurologic examination, and an urgently obtained EEG. The latter is critical because the ultimate diagnosis of infantile spasms is made by the clinical history coupled with the EEG findings.
Doctors follow a standard evaluation process to try to identify the underlying cause of the infantile spasms. This process, as noted in the figure below, includes different steps a neurologist will perform to identify disorders that are associated with infantile spasms by:
- First, taking a history
- Second, a careful physical and neurologic examination
- Third, doing a magnetic resonance imaging (MRI) scan.
- Fourth, doing a electroencephalogram (EEG) test.
- Fifth, doing metabolic and genetic test. There are many metabolic and genetic disorders that have been associated with infantile spasms, but they are very rare.
History of the child
The first step is a careful history. Sometimes the underlying cause of infantile spasms is determined by taking a careful history of the patient. The history includes all information related to the pregnancy, labor and delivery as well as all of the events, including developmental milestones, which have occurred prior to the time of evaluation for the infantile spasms. A child may be developing normally, but then development stops, or even reverses. If that happens, it is an important clue. Some children may have seizures that occurred prior to the infantile spasms. Sometimes the seizures begin on one side of the body and that is also an important clue to understanding possible causes. Although there are many disorders associated with infantile spasms that may be identified by taking a careful history, two stand out as particularly important:
- A history of brain injury from lack of oxygen
- Brain injury from infection such as meningitis or encephalitis.
A child may be developing normally, but then development stops, or even reverses. If that happens, it is an important clue.
Physical and neurologic examination
The second step is a careful physical examination and neurologic examination of the child. Many patients may have been developmentally delayed prior to the onset of spasms. Several underlying disorders can be identified by careful examination, such as:
- Cerebral palsy: Some children may have signs of cerebral palsy. These symptoms would be a clue of a brain problem that occurred during development or at the time of birth, such as stroke causing brain damage.
- Down syndrome: If Down syndrome had not been identified prior to presentation with infantile spasms it will be apparent on the physical examination.
- Tuberous sclerosis: An examination of the skin, especially the characteristic of “ash leaf” spots, is evidence of tuberous sclerosis. Ash leaf spots are small areas of skin that appear to be white compared to the rest of the skin because they lacked normal pigment. Tuberous sclerosis is one of the more common associated disorders. Tuberous sclerosis also has very characteristic changes on the MRI brain scan that are diagnostic.
Several other disorders can be diagnosed by skin examination including neurofibromatosis and incontinentia pigmenti.
Neuroimaging
The third step is an MRI scan. Neuroimaging has been an important advancement in the last several decades in the diagnosis of the underlying etiologies or causes of infantile spasms. Both the MRI scan and computed tomography (CT) scan have been used to detect brain abnormalities in children with infantile spasms. However, the MRI scan is much more sensitive and more likely to discover an abnormality of the brain than the CT scan. A large list of neurologic abnormalities can be seen on MRI scan (including developmental brain abnormalities) and evidence of brain injury events (such as brain injury from lack of oxygen, trauma, brain tumor, or injury from past infection).
However, it is important to note that MRI performed in a child less than 2 years of age can still miss subtle cortical migrational anomalies due to immaturity of the white matter. Therefore, a child who has spasms in addition to other neurodevelopmental abnormalities, but a normal MRI scan in the first year of life, should have a repeat MRI at 2 years of age or older 15. Interictal positron emission tomography (PET) may detect areas of hypometabolism, which may correlate with cortical malformation. PET is not used in the routine diagnostic evaluation of infantile spasms; however, PET may be valuable in a child with spasms for whom the MRI is normal but a structural brain lesion is suggested by the asymmetry of spasms and a focally abnormal EEG pattern.
Once the doctor has completed a careful history, physical and neurologic examination, and performed an MRI scan, almost all of the underlying disorders that doctors know of which are associated with infantile spasms will have been identified.
Electroencephalogram (EEG)
Most children who have infantile spasms will have a very abnormal electroencephalogram (EEG) pattern called hypsarhythymia or modified hypsarhythmia. Hypsarhythmia is a very high-voltage, disorganized and asynchronous high-amplitude background with spikes and slow waves pattern of EEG. The hypsarrhythmia pattern is not present during the entire EEG in many cases. This EEG pattern can be seen in wakefulness and is enhanced during sleep. Therefore, an EEG study to rule out infantile spasms should ideally include both awake and sleep recording. A less chaotic pattern, called modified hypsarhythmia, actually may be more common than hypsarhythmia. Hypsarhythmia or modified hypsarhythmia is seen in about 2/3 of cases. Some children may have an EEG pattern that is less dramatic than hypsarhythmia or modified hypsarhythmia and in those cases, it is very important to see what the EEG does when the child has a cluster of spasms. At the time of a spasm, the EEG usually flattens out for a very brief time. This is called an “electrodecremental response” or burst suppression, which is a high-voltage sharp or slow-wave discharge followed by generalized voltage attenuation or flattening. In addition to hypsarrhythmia and burst suppression, there are several additional, less dramatic ictal EEG patterns associated with infantile spasms, including combinations of generalized sharp and slow-wave discharges and electrodecremental fast activity. Thus, the diagnosis of infantile spasms can be confirmed by observing either hypsarhythmia, modified hypsarhythmia or an electrodecremental response. Many patients will have all three.
- The diagnosis of infantile spasms can be confirmed by observing the patterns of hypsarhythmia, modified hypsarhythmia or an electrodecremental response on an EEG. The seizures’ appearance and EEG are so distinct that the clinical diagnosis of infantile spasms can be made with certainty in the most cases. Once it’s recognized, the diagnosis of infantile spasms is usually easy, but determining what caused the spasms may be difficult.
- It is important to emphasize that all of these abnormal EEG patterns represent a dynamic continuum that can occur at any point along the natural history of infantile spasms. Therefore, an EEG diagnosis of hypsarrhythmia is not required for diagnosing infantile spasms and initiating treatment. Any child with the clinical signs and symptoms of infantile spasms as described previously and who has any epileptiform activities on his or her EEG, be it hypsarrhythmia or some other paroxysmal abnormality, merits urgent treatment of the spasms. Conversely, if the child appears to be having infantile spasms by history but results from a waking and sleeping EEG are normal, the child does not have infantile spasms but rather could be experiencing a benign infantile myoclonus.
If the electroencephalogram (EEG) is normal, the diagnosis of infantile spasms should be reconsidered because there are benign disorders that may appear clinically similar to infantile spasms (for example, benign infantile myoclonus or benign familial infantile convulsions).
Metabolic and genetic studies
Finally, if the history, physical examination, neurologic examination, EEG and MRI scan do not reveal the underlying cause, then the genetic/metabolic evaluation may be considered. More than 50 genetic/metabolic diseases have been associated with infantile spasms. It is not always necessary to do the complete metabolic/genetic workup on every child. If the history or exam suggests the possibility of a metabolic or genetic disorder then the treating physician will undertake the evaluation. A rare cause of infantile spasms is a disorder called “pyridoxine (vitamin B6) dependent seizures.” It is sometimes useful to administer a trial of intravenous pyridoxine. If that is unsuccessful, blood and urine tests for metabolic or genetic diseases may be performed. A spinal tap (lumbar puncture) may be necessary since some disorders can be detected only in spinal fluid. If, at the conclusion of the history, EEG, MRI, and metabolic testing, no etiology has been identified, the cause of infantile spasms is then called “cryptogenic .” Cryptogenic means that the cause is hidden. It does not mean that there is not a cause; it just means that your doctors don’t know all the causes yet and don’t have a way to find them all.
More than 50 genetic/metabolic diseases have been associated with infantile spasms.
Blood and urine tests are performed for detecting a potential genetic, metabolic, or infectious etiology that can cause symptomatic infantile spasms. These tests also provide baseline results before initiation of treatment.
Blood tests should include but not be limited to complete blood and differential count; tests of glucose, electrolyte, pH, lactate, blood urea nitrogen, creatinine, aspartate aminotransferase, alanine transaminase, bilirubin, and alkaline phosphatase levels; test of thyroid function; and screens of serum amino acid, toxoplasma, and cytomegalovirus IgG and IgM or polymerase chain reaction (or both).
Urine tests should include urinalysis and amino acid and organic acid screens.
Electrocardiogram (ECG) and chest x-ray should be performed, especially when cardiac examination is abnormal. Consultations with ophthalmology, cardiology, and genetics can also be requested as needed.
Chromosomal studies with microarray and genetic tests for infantile epileptic encephalopathies may be indicated when there is a family history or when the above-listed blood, urine, and neuroimaging tests are negative.
Infantile spasms treatment
The goal of infantile spasm therapy is to achieve seizure control as soon as possible. This is very important because if the seizures cannot be controlled, the child is unlikely to develop typically. Unfortunately, even a 50%-90% reduction in the number of seizures does not provide for typical development. For example, if a child’s infantile spasms disorder was causing 100 spasms a day before treatment, but then only has one spasm per day after treatment, his therapeutic intervention is still considered unsuccessful. Unfortunately, for many patients, the underlying disorder does not allow for normal development even if the spasms are completely controlled (for example: brain damage from lack of oxygen or infection). Nevertheless, seizure control should still sought because it greatly improves the patient’s and the parents’ lives. For other patients, there may be an opportunity to greatly improve the developmental outcome. The patients with the best prognosis are those with a cryptogenic etiology and possibly some patients with tuberous sclerosis or a localized cortical dysplasia. In either case, the treatment that gives the child the best chance to achieve complete control of spasms is the treatment of choice. On rare occasions, the testing will reveal a metabolic or genetic disorder that has a specific therapy which would then be the treatment of choice.
Anticonvulsant medications
Medical treatment options are somewhat different for infantile spasms than for other seizure types. There are only two drugs that are approved by the FDA for the treatment of infantile spasms:
- Adrenocorticotropic hormone (ACTH)
- Vigabatrin
There is considerable variation in the management of infantile spasms, as evidenced by the US Consensus Report and a recent survey done on the current evaluation and treatment of infantile spasms among members of the Child Neurology Society 16. According to these sources, most neurologists use adrenocorticotrophic hormone (ACTH) as their preferred first-line treatment for infantile spasms not caused by tuberous sclerosis and vigabatrin as the first-line treatment of infantile spasms caused by tuberous sclerosis 17.
Medications that are used to treat older children or adults with epilepsy, such as phenobarbital, carbamazepine, or phenytoin, are rarely helpful. There is some evidence that one or two of the newer drugs or the ketogenic diet may be effective in some patients. So, even patients who fail treatment with ACTH and with vigabatrin may occasionally be controlled with other therapies. Therefore, it is very important to keep trying if the first treatments don’t seem to be successful.
First line medical therapies
Adrenocorticotropic Hormone (ACTH)
ACTH is the oldest of the approved medications for infantile spasms. It was the first drug that ever was shown to be successful in treating infantile spasms. In 1958, Sorel reported administering ACTH to seven patients, four of whom responded within a few days and only one of whom had no response at all. ACTH was used for more than 50 years before it was approved by the FDA in 2010 for the treatment of infantile spasms. One drawback from the use of ACTH is that it must be given by intramuscular injection once or twice each day. One or both parents are taught how to give the injections so that the shots can be given at home.
Questions regarding the optimal dose, formulation, and duration of ACTH treatment remain, although the 2012 updated guideline reported that low-dose ACTH is probably as effective as high-dose ACTH for short-term treatment of infantile spasms 10.
ACTH is a very powerful medication and has many of side effects. The majority of children will become very hungry and gain weight. Most become very irritable. More serious side effects include:
- High blood pressure (up to 37%)
- Irritability (37%-100%)
- Infection (14%)
- Cerebral atrophy (62%)
- Decreased glucose levels in the blood
- Stomach ulcer
- Growth retardation
- Heart problems
- Immunosuppression (difficulty fighting infection)
Higher dosage and longer duration of treatment correlate with higher incidence of side effects.
Different epilepsy centers show considerable variability in dosage (high vs low), formulation (natural ACTH in the United States vs synthetic ACTH in Canada, Japan, and Europe), and duration of therapy.
In one study, the risk of serious side effects with ACTH was 43%. ACTH is more likely to be effective at higher doses than lower doses. In addition, the more serious complications occur when the children have been on the medication for an extended period of time. Thus, by treating at high doses and by keeping the duration as short as possible, the child has the best chance for controlling the spasms and the risk of serious complications can be decreased. Children being treated with ACTH must see their doctors frequently for regular blood pressure measurements and blood tests to minimize the risks. The chance that ACTH will control the spasms at the highest doses may be as high as 80+%. Some children may have a return of the spasms when the medication has been tapered and stopped. Whether the medication should be repeated a second time is not clear. If ACTH treatment has not been successful within two weeks, it should be rapidly tapered and stopped and another medication should be tried.
Vigabatrin
In 1991, it was reported that a new medication, vigabatrin, showed remarkable efficacy with infantile spasms. Sixty-eight patients who had failed other therapies were treated with vigabatrin as add-on therapy, and 29 of them (43%) showed complete resolution of the spasms. It was also noted that 12 of 14 patients who had tuberous sclerosis responded with complete control. There is some evidence that vigabatrin also may improve developmental outcome in patients with tuberous sclerosis. It is important to understand how vigabatrin compares with ACTH. Vigevano et al. performed a study of children with newly diagnosed infantile spasms. The patients were given either ACTH or vigabatrin. Eleven of 23 vigabatrin patients responded (1 later relapsed) compared with 14 of 19 patients treated with ACTH (6 later relapsed). After relapses were taken into account, the long-term response rate was similar for both medications. In that study, ACTH was more effective for patients with perinatal hypoxic ischemic encephalopathy. Vigabatrin generally is well tolerated in young children. Most side effects are not serious. There are reports of decreased muscle tone, sleepiness, or difficulty sleeping. However, there is one side effect that is potentially more serious – visual field constriction (tunnel vision). Blindness has not been reported and central vision appears to be unaffected. The visual loss is usually very subtle; most people who had it were not aware that it had occurred until special testing was performed. It was hard for people to realize that they had a problem, so it took more than a decade to recognize that it occurs. There have been many reports showing that peripheral visual fields are constricted in 15 to 50% of adult patients. It is not known if visual field constriction occurs in very young children because there is no effective method of testing for it. However, given the tragic nature of infantile spasms, even if it is proven to occur in infants, visual field constriction may be an acceptable side effect to trade for seizure control and an improved opportunity for normal development. Despite the visual field issue, many pediatric epileptologists consider vigabatrin to be the drug of choice for children with infantile spasms that are due to tuberous sclerosis and for other conditions as well. Whether one chooses to treat initially with ACTH or vigabatrin is a decision made by the parents in consultation with their physician. There are circumstances where ACTH may be the best medication to choose and other circumstances where vigabatrin would be best. Most doctors recommend vigabatrin for patients who have infantile spasms associated with tuberous sclerosis.
Baseline visual field testing or, in the case of infants, electroretinography, should be done before initiation of treatment and regularly thereafter to monitor for retinal toxicity. The initial dose of vigabatrin is 50 mg/kg body weight which is rapidly titrated up to 150 mg/kg body weight divided twice a day. In order to minimize the risk of retinal toxicity, vigabatrin should be given for no longer than 6 months.
Prednisone
A possible alternative to the use of ACTH is an oral medication called prednisone or prednisolone. For many years it was not clear whether one was better than the other. Several studies showed the high-dose ACTH may be superior to what was then used as the dosage for prednisone. Some physicians are now using a much higher dose of prednisolone and finding that it is successful in treating many patients with infantile spasms. A recent study reported using 8 mg per kilogram of prednisolone for two weeks and if the medication was not successful , then trying ACTH. The main advantage of prednisolone is that it can be given orally whereas the ACTH requires daily injections.
Second line medical therapies
Pyridoxine
Pyridoxine (vitamin B6) dependency is a very rare cause of infantile spasms. A trial of 100-mg pyridoxine given intravenously should be administered if diagnosis remains in doubt after the history, examination, and MRI scan have been performed. An immediate normalization of the EEG suggests pyridoxine-dependent epilepsy. Continued oral administration of high doses of pyridoxine also may be effective for some patients who do not have pyridoxine-dependent seizures. In Japan, high-dose pyridoxine is considered the initial drug of choice by many pediatric neurologists, with reports that approximately 15% respond. While this response rate is lower than either ACTH or vigabatrin, pyridoxine is their first choice based on the safety profile. Side effects include loss of appetite, irritability, and vomiting—all of which are relatively common but modest compared with those associated with ACTH or vigabatrin. Pyridoxine has not found support outside of Japan and a few other epilepsy centers. But, given the low risk associated with its use, it seems reasonable to give patients a 1 to 2 week trial of 100 to 400-mg pyridoxine before starting other medications. If these “standard” medications fail, other therapies must be considered, including other antiepileptic drugs (valproate [valproic acid], topiramate, zonisamide) or vitamin B6 7.
Valproic Acid
Valproic acid probably has the best anecdotal or unscientific evidence of success in treatment, but there have been no prospective randomized studies of efficacy for infantile spasms. Doses range from 20 mg/kg/day to 100 mg/kg/day. Although none of the reported patients developed liver failure, it nevertheless is a risk in children less than 2 years of age and should be used with caution for children with infantile spasms. This is a difficult limitation to valporic acid’s use because almost all patients are less than 2 years of age. Thus, the risk/benefit ratio should be determined.
Clonazepam
One of the earliest non-steroid treatments for infantile spasms were with the benzodiazepines , including clonazepam and nitrazepam. Nitrazepam was never approved for use in the US, so clonazepam is the only one available that has evidence of efficacy. Clonazepam was first reported to be helpful in a few patients with infantile spasms in the 1960’s. It is rarely used today because there are so many better medications. Several of the new anticonvulsants have some evidence of efficacy.
Zonisamide
Zonisamide has shown some promise as an effective therapy for infantile spasms, but there have been no controlled or comparison trials to date. The Japanese experience suggests that zonisamide may be effective in about a 1/3 of patients. A recent report indicated that 5 of 25 patients with infantile spasms had a complete clinical and electrographic response (EEG pattern) to zonisamide within 1 to 2 weeks, with doses ranging from 8 to 32 mg/kg/day. Zonisamide is generally well tolerated. If the 30% or greater efficacy rates hold up in controlled studies, zonisamide could become a first-line therapy. However, most doctors are not finding the success rate to be that high.
Topiramate
In one study, topiramate was shown to be effective in 4 of 11 intractable infantile spasm patients in doses up to 25 mg/kg/day. Another study reported that topiramate reduced seizures in 43% of 14 infantile spasms patients, but 29% were made worse and none became seizure free.
Lamotrigine
Lamotrigine is another of the newer antiepileptic drugs with some anecdotal or scientific evidence of efficacy for infantile spasms, although there are no prospective controlled trials. One report of 3 patients who did not have a successful response to vigabatrin and ACTH treatment responded to lamotrigine after 1 dose. The usual dose of lamotrigine is 6 to 10 mg/kg/day. The major side effect is rash, which is dependent to some extent on how rapidly the dose is increased. The usual recommendation is to increase the dose slowly over 2 months to the minimum expected therapeutic dose. Given the severe nature of infantile spasms and the need to achieve control as soon as possible, taking 2 months to get to a therapeutic level obviously decreases the value of lamotrigine as a therapeutic option. However, if the lowest dose is effective, then lamotrigine becomes a drug to try when standard therapies have failed. Finally, there are three non-drug therapies that should be considered as options when other therapies have failed:
- Ketogenic diet
- High-dose intravenous immunoglobulin (IVIG)
- Surgery
Ketogenic Diet
The ketogenic diet is a decades-old therapy that is back in popularity. Two recent retrospective reports of 40 children with infantile spasms show diet may control spasms in 20 to 35% of patients who are intractable to other therapies 18, 19. In the past, there had been a question as to whether children less than 1 year of age could achieve and maintain ketosis. The recent reports indicate that young children can indeed achieve ketosis and may benefit from the diet. Most of the children tolerated the diet well, but there were adverse events, including renal stones, gastritis, hyperlipidemia, and gastroesophageal reflux.
High-dose intravenous immunoglobulin (IVIG)
High-dose intravenous immunoglobulin (IVIG) has been reported to be helpful in a variety of seizure disorders. One study reported that all six children in their study who had cryptogenic infantile spasms achieved complete remission, but only one of five symptomatic patients responded. Intravenous immunoglobulin doses ranged from 100 to 200 mg/kg/dose given every 2 to 3 weeks to 400 mg/kg/day for five consecutive days. Although there is little data, intravenous immunoglobulin could be considered a possible therapeutic option in patients whose other medical therapies have not been successful. However, the actual efficacy is unclear and the most appropriate dosing and duration have not been defined.
Surgery
The final nondrug therapy is removing an abnormal part of the brain (cortical resection). This option should be considered for patients who:
- Have failed other therapies including ACTH and Vigabatrin or both
- Have evidence of structural brain abnormalities in a defined area, such as developmental brain abnormalities, brain damage, or tuberous sclerosis.
Not many years ago, infantile spasms were considered to be a generalized seizure disorder, and thus surgery was not possible. It has become clear in the last several years that in spite of the generalized nature of the seizures, an area of cortical abnormality can often be discovered. Removal of the abnormality may lead to control of seizures and, possibly, improved developmental outcome. The majority of patients who have cortical resection have evidence of focal cortical abnormalities prior to surgical evaluation. For these patients, surgery should be considered early in the course rather than waiting for months or years. Selecting the appropriate candidates for surgery is usually more difficult in infantile spasms than for other types of epilepsy because of the generalized nature of the EEG abnormalities. The following should lead to a referral to a pediatric epilepsy surgery center for further evaluation:
- A careful review of the history (especially history of partial seizures that came first or accompanied infantile spasms)
- The presence of cortical disturbances on MRI scan
- Localized EEG abnormalities that suggest a localized cortical defect.
- Sidenvall R, Eeg-Olofsson O. Epidemiology of infantile spasms in Sweden. Epilepsia 1995;36:572-574.[↩]
- Trevathan E, Murphy CC, Yeargin-Allsopp M. The descriptive epidemiology of infantile spasms among Atlanta children. Epilepsia 1999;40:748-751.[↩]
- Riikonen R. Epidemiological data of West syndrome in Finland. Brain Dev 2001;23:539-541.[↩]
- Infantile Spasms. http://www.childneurologyfoundation.org/disorders/infantile-spasms[↩][↩]
- Commission on classification and terminology of the international league against epilepsy. Proposal for revised classification of epilepsies and epileptic syndromes. Epilepsia 1989;34:389-399.[↩]
- ILAE Commission Report. A proposed diagnostic scheme for people with epileptic seizures and with epilepsy: Report of the ILAE task force on classification and terminology. Epilepsia 2001;42:796-803.[↩]
- Recent advances in the pharmacotherapy of infantile spasms. Riikonen R. CNS Drugs. 2014 Apr; 28(4):279-90. https://www.ncbi.nlm.nih.gov/pubmed/24504827/[↩][↩][↩][↩][↩]
- West WJ. On a peculiar form of infantile convulsions. Lancet Neurol. 1841;1:724–725.[↩]
- Darke K, Edwards SW, Hancock E, et al. Developmental and epilepsy outcomes at age 4 years in the UKISS trial comparing hormonal treatments to vigabatrin for infantile spasms: a multi-centre randomized trial. Arch Dis Child 2010;95:382–386.[↩][↩]
- Go CY, Mackay MT, Weiss SK, et al. Evidence-based guideline update: medical treatment of infantile spasms. Report of the guideline development subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology 2012;78:1974-1980.[↩][↩]
- Osborne JP, Lux AL, Edwards SW, et al. The underlying etiology of infantile spasms (West syndrome): information from the United Kingdom Infantile Spasms Study (UKISS) on contemporary causes and their classification. Epilepsia 2010;51(10):2168-2174.[↩][↩][↩]
- Galanopoulou AS, Moshé SL. Pathogenesis and new candidate treatments for infantile spasms and early life epileptic encephalopathies: a view from preclinical studies. Neurobiology of disease. 2015;79:135-149. doi:10.1016/j.nbd.2015.04.015. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4458451/[↩]
- De novo mutations in synaptic transmission genes including DNM1 cause epileptic encephalopathies. EuroEPINOMICS-RES Consortium., Epilepsy Phenome/Genome Project., Epi4K Consortium. Am J Hum Genet. 2014 Oct 2; 95(4):360-70. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4185114/[↩]
- Infantile Spasms. http://www.childneurologyfoundation.org/disorders/infantile-spasms/[↩]
- Infantile Spasms. https://rarediseases.org/physician-guide/infantile-spasms/[↩]
- Pellock JM, Hrachovy R, Shinnar A, et al. Infantile spasms: a U.S. consensus report. Epilepsia 2010;51:2175-2189.[↩]
- Mytinger JR, Joshi S. The current evaluation and treatment of infantile spasms among members of the Child Neurology Society. J Child Neurol 2012;27:1289-1294.[↩]
- Ketogenic diet for infantile spasms refractory to first-line treatments: an open prospective study. Pires ME, Ilea A, Bourel E, Bellavoine V, Merdariu D, Berquin P, Auvin S. Epilepsy Res. 2013 Jul; 105(1-2):189-94. https://www.ncbi.nlm.nih.gov/pubmed/23357723/[↩]
- Infantile spasms treated with the ketogenic diet: prospective single-center experience in 104 consecutive infants. Hong AM, Turner Z, Hamdy RF, Kossoff EH. Epilepsia. 2010 Aug; 51(8):1403-7. https://www.ncbi.nlm.nih.gov/pubmed/20477843/[↩]





{kind=link}