Contents
What is Kartagener syndrome
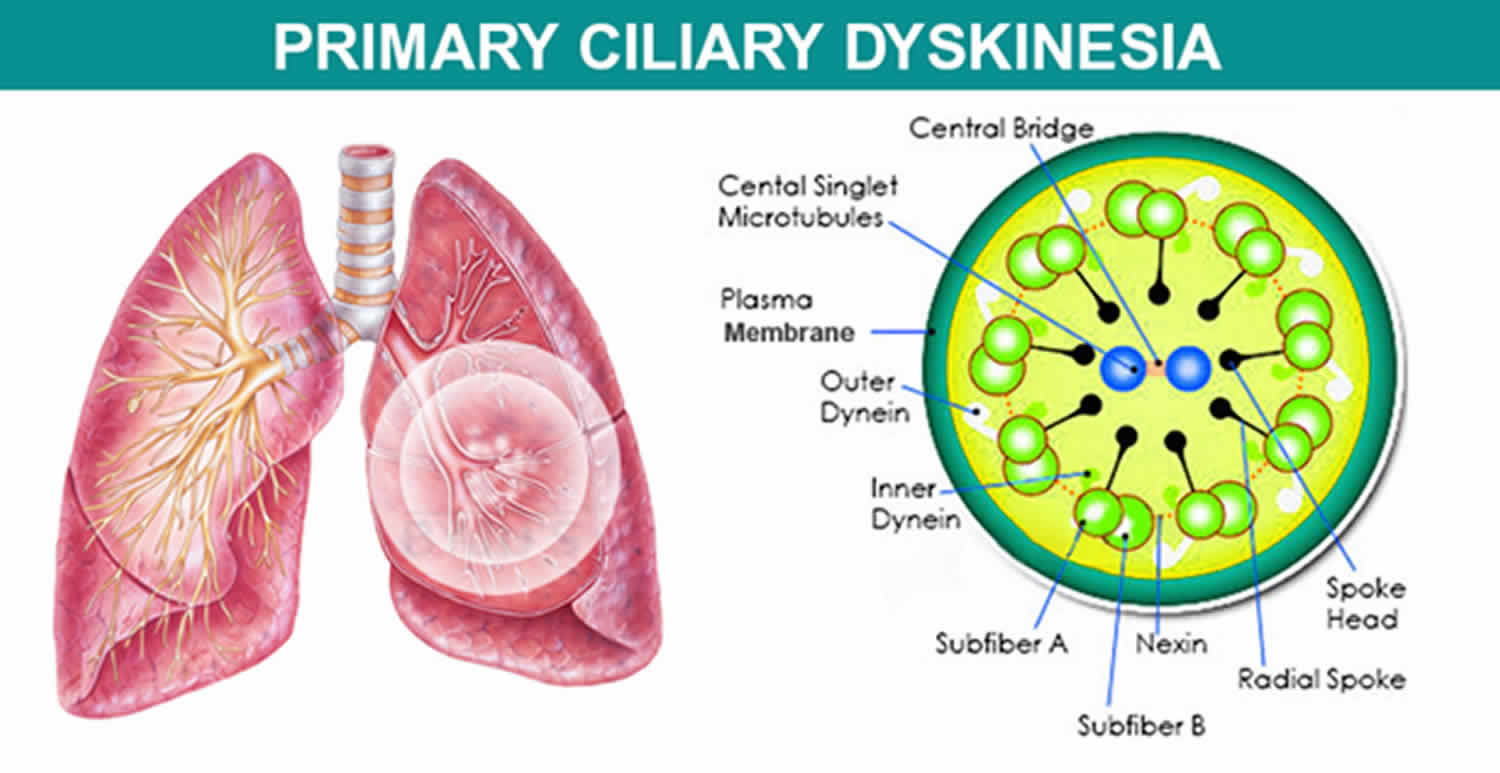
Kartagener syndrome is a type of primary ciliary dyskinesia that is characterized by triad of chronic sinusitis, bronchiectasis, and situs inversus totalis (mirror-image reversal of internal organs) 1. Kartagener syndrome signs and symptoms vary but may include neonatal respiratory distress; frequent lung, sinus and middle ear infections beginning in early childhood; and male and female infertility 2.
The signs and symptoms of Kartagener syndrome are caused by abnormal cilia and flagella. Cilia are microscopic, finger-like projections that stick out from the surface of cells. They are found in the linings of the airway, the reproductive system, and other organs and tissues. Flagella are tail-like structures, similar to cilia, that propel sperm cells forward.
In the respiratory tract, cilia move back and forth in a coordinated way to move mucus towards the throat. This movement of mucus helps to eliminate fluid, bacteria, and particles from the lungs. Most babies with Kartagener syndrome experience breathing problems at birth, which suggests that cilia play an important role in clearing fetal fluid from the lungs. Beginning in early childhood, affected individuals develop frequent respiratory tract infections. Without properly functioning cilia in the airway, bacteria remain in the respiratory tract and cause infection. People with Kartagener syndrome also have year-round nasal congestion and a chronic cough. Chronic respiratory tract infections can result in a condition called bronchiectasis, which damages the passages, called bronchi, leading from the windpipe to the lungs and can cause life-threatening breathing problems.
Some individuals with Kartagener syndrome have abnormally placed organs within their chest and abdomen. These abnormalities arise early in embryonic development when the differences between the left and right sides of the body are established. About 50 percent of people with Kartagener syndrome have a mirror-image reversal of their internal organs (situs inversus totalis). For example, in these individuals the heart is on the right side of the body instead of on the left. Situs inversus totalis does not cause any apparent health problems. When someone with primary ciliary dyskinesia has situs inversus totalis, they are often said to have Kartagener syndrome.
Approximately 12 percent of people with Kartagener syndrome have a condition known as heterotaxy syndrome or situs ambiguus, which is characterized by abnormalities of the heart, liver, intestines, or spleen. These organs may be structurally abnormal or improperly positioned. In addition, affected individuals may lack a spleen (asplenia) or have multiple spleens (polysplenia). Heterotaxy syndrome results from problems establishing the left and right sides of the body during embryonic development. The severity of heterotaxy varies widely among affected individuals.
Kartagener syndrome can also lead to infertility. Vigorous movements of the flagella are necessary to propel the sperm cells forward to the female egg cell. Because their sperm do not move properly, males with Kartagener syndrome are usually unable to father children. Infertility occurs in some affected females and is likely due to abnormal cilia in the fallopian tubes.
Another feature of primary ciliary dyskinesia is recurrent ear infections (otitis media), especially in young children. Otitis media can lead to permanent hearing loss if untreated. The ear infections are likely related to abnormal cilia within the inner ear.
Rarely, individuals with Kartagener syndrome have an accumulation of fluid in the brain (hydrocephalus), likely due to abnormal cilia in the brain.
Kartagener syndrome can be cause by changes (mutations) in many different genes that are inherited in an autosomal recessive manner. Although scientists have identified many of the genes associated with Kartagener syndrome, the genetic cause of some cases is unknown 3.
Reported prevalence of Kartagener syndrome is approximately 1:15,000 live births 4. Given difficulties in diagnosing primary ciliary dyskinesia, misdiagnosis is common, and therefore primary ciliary dyskinesia may be more prevalent than reported.
There is no cure for Kartagener syndrome. Treatment varies based on the signs and symptoms present in each person but may include airway clearance therapy and antibiotics 5.
Kartagener syndrome causes
Kartagener syndrome or primary ciliary dyskinesia can result from mutations in many different genes. There are currently 33 known genes associated with primary ciliary dyskinesia, with the majority following autosomal recessive inheritance 4. DNAI1 and DNAH5, which encode for components of the outer dynein arm complex in cilia, are the two most common genes associated with Kartagener syndrome. Two rare X-linked genes, RPGR and OFD1, have also been identified. These genes provide instructions for making proteins that form the inner structure of cilia and produce the force needed for cilia to bend. Coordinated back and forth movement of cilia is necessary for the normal functioning of many organs and tissues. The movement of cilia also helps establish the left-right axis (the imaginary line that separates the left and right sides of the body) during embryonic development.
Mutations in the genes that cause primary ciliary dyskinesia result in defective cilia that move abnormally or are unable to move (immotile). Because cilia have many important functions within the body, defects in these cell structures cause a variety of signs and symptoms.
Mutations in the DNAI1 and DNAH5 genes account for up to 30 percent of all cases of primary ciliary dyskinesia. Mutations in the other genes associated with this condition are found in only a small percentage of cases. In many people with primary ciliary dyskinesia, the cause of the disorder is unknown.
Kartagener syndrome inheritance pattern
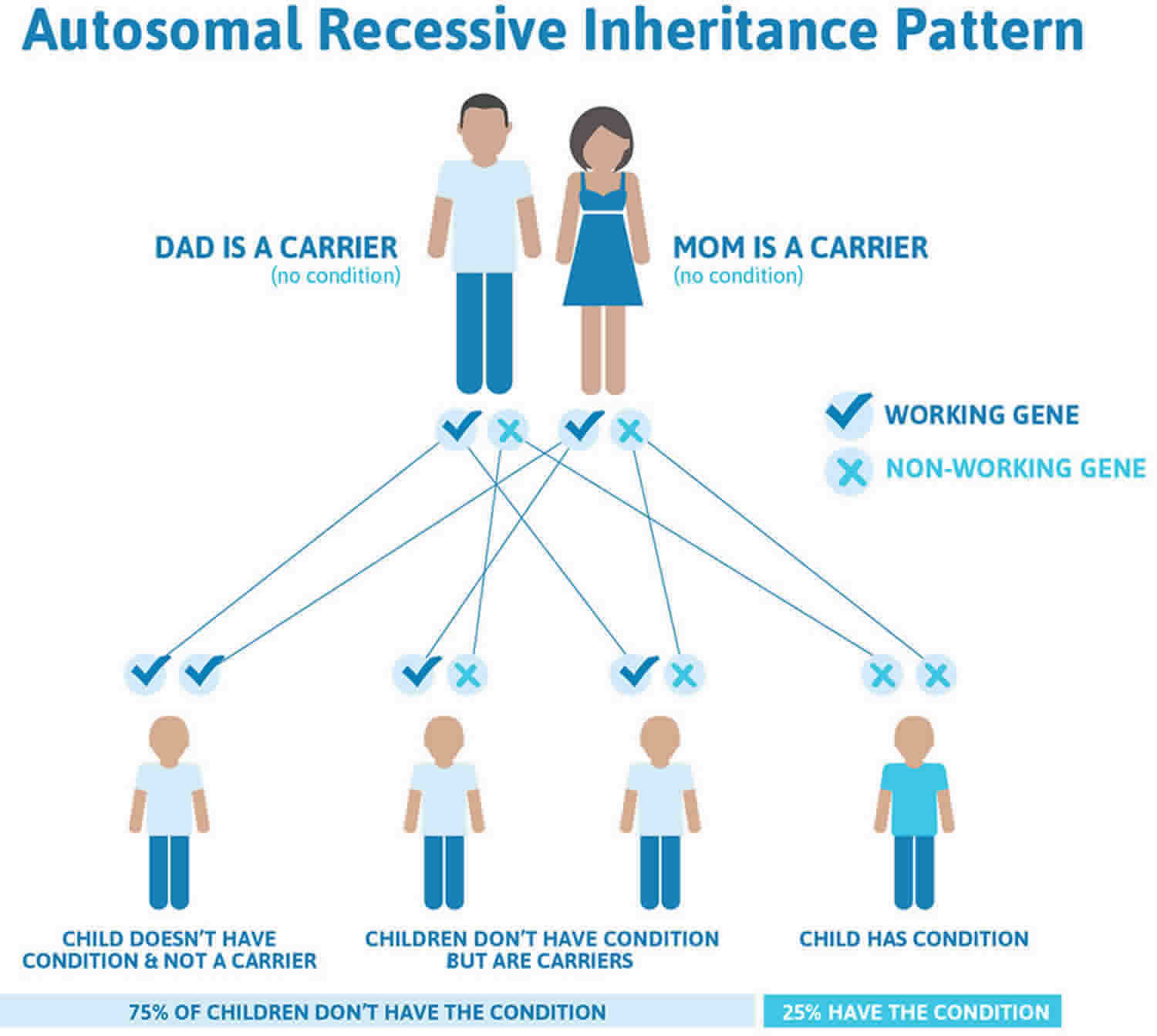
Kartagener syndrome is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
It is rare to see any history of autosomal recessive conditions within a family because if someone is a carrier for one of these conditions, they would have to have a child with someone who is also a carrier for the same condition. Autosomal recessive conditions are individually pretty rare, so the chance that you and your partner are carriers for the same recessive genetic condition are likely low. Even if both partners are a carrier for the same condition, there is only a 25% chance that they will both pass down the non-working copy of the gene to the baby, thus causing a genetic condition. This chance is the same with each pregnancy, no matter how many children they have with or without the condition.
- If both partners are carriers of the same abnormal gene, they may pass on either their normal gene or their abnormal gene to their child. This occurs randomly.
- Each child of parents who both carry the same abnormal gene therefore has a 25% (1 in 4) chance of inheriting a abnormal gene from both parents and being affected by the condition.
- This also means that there is a 75% ( 3 in 4) chance that a child will not be affected by the condition. This chance remains the same in every pregnancy and is the same for boys or girls.
- There is also a 50% (2 in 4) chance that the child will inherit just one copy of the abnormal gene from a parent. If this happens, then they will be healthy carriers like their parents.
- Lastly, there is a 25% (1 in 4) chance that the child will inherit both normal copies of the gene. In this case the child will not have the condition, and will not be a carrier.
These possible outcomes occur randomly. The chance remains the same in every pregnancy and is the same for boys and girls.
Figure 1 illustrates autosomal recessive inheritance. The example below shows what happens when both dad and mum is a carrier of the abnormal gene, there is only a 25% chance that they will both pass down the abnormal gene to the baby, thus causing a genetic condition
Key points to remember
- A person must inherit two copies of a abnormal gene, one from each parent, in order to be affected by the condition (25% chance). If a person inherits only one abnormal gene then they will be a carrier (50% chance). These outcomes occur randomly. They remain the same in every pregnancy and are the same for boys and girls.
- A abnormal gene cannot be corrected – it is present for life.
- A abnormal gene is not something that can be caught from other people. They can still be a blood donor, for example.
- People often feel guilty about a genetic condition which runs in the family. It is important to remember that it is no-one’s fault and no-one has done anything to cause it to happen.
Figure 1. Kartagener syndrome autosomal recessive inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Kartagener syndrome symptoms
The symptoms of Kartagener syndrome vary greatly in affected individuals. Symptoms often begin shortly after birth and can include coughing, gagging, choking and lung collapse (neonatal respiratory distress). Unlike other causes of respiratory distress in newborns, which occurs in the first few hours after birth, the respiratory distress with primary ciliary dyskinesia patients occurs 12 to 24 hours after birth in full term infants. Four main clinical features have been described in primary ciliary dyskinesia including unexplained neonatal respiratory distress in term infants, early onset year-round wet cough, early onset year-round nasal congestion, and laterality defects. If three or more of these features are met, the specificity for primary ciliary dyskinesia diagnosis is more than 96%. About half of patients with primary ciliary dyskinesia also have situs inversus totalis, including transposition of the right and left lung. Male infertility is common and occurs in nearly 100% of males. Females can present with reduced fertility or ectopic pregnancy due to abnormal fallopian tube transit of oocytes. Patients may also have chronic sinusitis and nasal congestion that does not change with seasons and does not resolve between viral infections. Persistent daily cough that begins in infancy is reported in nearly 100% of primary ciliary dyskinesia patients.
Affected individuals often experience chronic sinus, middle ear and lung infections as well as chronic coughing, excess mucus and hearing loss. The recurring respiratory infections can lead to an irreversible scarring and obstruction in the bronchi (bronchiectasis) and severe lung damage.
Cilia are also present in the ventricles of the brain and in the reproductive system so ciliary dysfunction can also affect other body systems. Affected men are often infertile because movement of sperm (motility) is abnormal. Kartagener syndrome may also be associated with infertility and ectopic pregnancy in females.
Movement of cilia may also be important in organ placement in the developing embryo. Approximately 50% of individuals with Kartagener syndrome have Kartagener syndrome in which the internal organs including the heart, liver, spleen and intestine are on the opposite side of the body (situs inversus totalis). Some individuals with Kartagener syndrome have a condition called heterotaxy (situs ambiguus) in which internal organs are abnormally positioned and have abnormal structure. Approximately, half of the Kartagener syndrome patients with heterotaxy have congenital heart defects that can be serious and life threatening.
Kartagener syndrome diagnosis
Primary ciliary dyskinesia is diagnosed definitively through examination of lung or sinus tissue obtained from a biopsy. Specific structural defects that are present in these tissues can be detected under an electron microscope. Early diagnosis is important in order to provide prophylactic treatment to prevent or decrease damage to the respiratory system from recurrent infections. Screening for levels of nasal nitric oxide (in patients over 5 years of age who can cooperate with palate closure maneuvers) is helpful to identify individuals who may have Kartagener syndrome and should proceed with a biopsy. Currently, mutations in 33 genes are known to be associated with Kartagener syndrome. These do not account for all cases of Kartagener syndrome and hence more Kartagener syndrome genes are yet to be identified. Kartagener syndrome clinical genetic testing is available for some of the 33 genes associated with Kartagener syndrome by the commercial laboratories and new genes are being added to their panels periodically.
Kartagener syndrome treatment
Kartagener syndrome is managed by a multidisciplinary team of healthcare workers. Kartagener syndrome patients should be managed in centers that specialize in primary ciliary dyskinesia and chronic pulmonary diseases.
Airway clearance therapy is used to keep the lung tissue healthy for as long as possible. This therapy may include routine washing and suctioning of the sinus cavities and ear canals. Antibiotics, bronchodilators, steroids and mucus thinners (mucolytics) are also used to treat Kartagener syndrome. Routine hearing evaluation is important for young children and speech therapy and hearing aids may appropriate for children with hearing loss and speech problems. Lung transplantation is an option for severe, advanced lung disease. Surgery may be indicated if heart defects are present.
Primary ciliary dyskinesia patients should be evaluated by a pulmonologist two to four times per year with spirometry and surveillance cultures of sputum. A chest radiograph should be completed at the time of diagnosis and during acute respiratory exacerbations. At least once after diagnosis, chest CT to evaluate for bronchiectasis should be considered. Additionally, children with primary ciliary dyskinesia should be evaluated by ear-nose-throat specialist one to two times per year and as needed in adults. An audiology assessment should be performed since there is a high incidence of recurrent otitis media with chronic middle ear effusions and associated complications of conductive hearing loss. Patients should be monitored for chronic rhinosinusitis and nasal polyps. Primary ciliary dyskinesia patients should receive daily chest physiotherapy, influenza vaccination, and pneumococcal vaccination as indicated. Antibiotics should be prescribed for acute respiratory exacerbations. In cases with recurrent respiratory infections, long-term oral or nebulized antibiotics can be considered. Gene editing has been proposed as a possible future treatment for primary ciliary dyskinesia. Ex vivo gene repair by site-specific recombination has been found to rescue ciliary beating, which could have tremendous application in primary ciliary dyskinesia patients.
- Tadesse A, Alemu H, Silamsaw M, Gebrewold Y. Kartagener’s syndrome: a case report. J Med Case Rep. 2018 Jan 10;12(1):5[↩]
- Zariwala MA, Knowles MR, Leigh MW. Primary Ciliary Dyskinesia. 2007 Jan 24 [Updated 2015 Sep 3]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2019. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1122[↩]
- Primary ciliary dyskinesia. https://ghr.nlm.nih.gov/condition/primary-ciliary-dyskinesia[↩]
- Stern BM, Sharma G. Ciliary Dysfunction (Kartagener Syndrome, Primary Ciliary Dyskinesia) [Updated 2019 Mar 1]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2019 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK448201[↩][↩]
- Primary Ciliary Dyskinesia (Kartagener Syndrome). https://emedicine.medscape.com/article/299299-overview[↩]




{kind=link}