Contents
- What is Langerhans cell histiocytosis
- Langerhans cell histiocytosis types
- Langerhans cell histiocytosis causes
- Langerhans cell histiocytosis pathology outlines
- Langerhans cell histiocytosis symptoms
- Langerhans cell histiocytosis skin
- Pulmonary Langerhans cell histiocytosis (PLCH)
- Langerhans cell histiocytosis of the central nervous system
- Langerhans cell histiocytosis of bones
- Langerhans cell histiocytosis of liver
- Langerhans cell histiocytosis of lymph nodes and thymus
- Langerhans cell histiocytosis of endocrine system
- Langerhans cell histiocytosis of eye
- Langerhans cell histiocytosis diagnosis
- Langerhans cell histiocytosis treatment
- Langerhans cell histiocytosis prognosis
- Langerhans cell histiocytosis life expectancy
What is Langerhans cell histiocytosis
Langerhans cell histiocytosis (LCH) previously referred to as Hand‐Schuller‐Christian disease, Letterer‐Siwe disease, eosinophilic granuloma or histiocytosis X, is a rare blood disorder in which abnormal accumulation of antigen-presenting dendritic cells called Langerhans cells (a type of white blood cell with CD1a, CD207 [Langerin] and and S100 expression) growing in certain tissues and organs including the bones, bone marrow, skin, lymph nodes, lungs and other areas and damage them 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19. Typically, Langerhans cell histiocytosis (LCH) disease causes granulomatous lesions in these organs 20, 21. According to Haupt and colleagues 22, the most commonly affected location is the skeleton, followed by the skin and the pituitary gland. Less-frequently impaired sites such as the bone marrow, liver, spleen, and lungs seem to coincide with a higher risk of severe progression of the disease 23.
The organs most commonly affected by Langerhans cell histiocytosis (LCH) are:
- Bone 77 percent. Bone involvement can cause pain, often in a very specific area where the disease is eroding the bone. The most commonly affected bones are the skull, jaw (particularly the lower jaw), ribs, pelvis, and vertebrae, but any bone can be affected.
- Skin 39 percent. Skin involvement often presents as single or multiple small red lumps, but the skin rash can also be flat, itchy, and flaky much like eczema or psoriasis. The scalp, ear canals, and vulva are common sites of skin involvement. Some patients develop mouth sores similar to canker sores, but often larger and flatter.
- Lymph nodes 19 percent. Lymph node involvement is usually found on radiology scans or by feeling painless, rubbery lumps in the neck, under the arms, or in the groin.
- Liver 16 percent. Liver involvement usually does not cause symptoms, but is detected by blood work or scans.
- Spleen 13 percent. Spleen involvement can be detected on physical exam or by a decrease in blood counts.
- Oral mucosa 13 percent
- Lung 10 percent. Lung involvement is particularly common in smokers and can cause a chronic cough or shortness of breath.
- Central nervous system (CNS) 6 percent. Central nervous system (brain and spinal cord) involvement most commonly affects the pituitary gland, a master gland that controls function of the thyroid, adrenal, and reproductive glands. A common manifestation of central nervous system (CNS) involvement of Langerhans cell histiocytosis is diabetes insipidus. Rarely, patients with CNS involvement can develop a condition called a neurodegenerative syndrome or LCH-associated abnormal CNS symptoms (LACS), which causes memory loss and problems with speech and balance.
The incidence of Langerhans cell histiocytosis is estimated at 2 to 20 per million, the majority of patients are children younger than 3 years (peak between one and three years of age) with a slight male predilection and the incidence in adults is approximately 1‐2 per million 8, 24, 25, 26. Langerhans cells, which help regulate the immune system, are normally found throughout the body, especially in the skin, lymph nodes, spleen, lungs, liver, and bone marrow. In Langerhans cell histiocytosis, excess immature Langerhans cells usually form tumors called granulomas. Many researchers now consider Langerhans cell histiocytosis to be a form of cancer, but this classification remains controversial.
The incidence of Langerhans cell histiocytosis (LCH) in children younger 15 years with a median age at diagnosis of 3 years is 2.6 to 8.9 cases per million per year 27, 28, 29, 30. Langerhans cell histiocytosis (LCH) incidence appears to be higher in Caucasians in northern European countries, and lower in Asia and Africa 31.
Langerhans cell histiocytosis (LCH) lesions contain varying proportions of the clonal pathological CD1a+, CD207+ dendritic cells (Langerhans cells) within an intense inflammatory infiltrate composed of macrophages, lymphocytes, eosinophils, multinucleated giant cells and, less commonly, neutrophils and plasma cells 32. Given the intense inflammatory infiltrate, it is not known whether Langerhans cell histiocytosis is a form of cancer or a cancer-like disease 33, 31, 19. Recent findings have led to the assumption that Langerhans cell histiocytosis (LCH) should be considered as an inflammatory neoplastic process comprising qualities of a tumor as well as immunogenic components, as it appears to be caused by somatic mutations in bone marrow progenitor cells 20, 21, 34.
Langerhans cell histiocytosis is now defined as an inflammatory myeloid neoplasm in the revised 2016 Histiocyte Society classification, and requires accurate diagnosis and appropriate treatment as necessary 31, 19.
Table 1. Clinical Classification of Langerhans cell histiocytosis (LCH)
| Clinical group | Description |
|---|---|
| Multisystem | Two or more systems involved |
| With risk organ involvement | Involvement of liver, spleen or bone marrow |
| Without risk organ involvement | Without involvement of liver, spleen or bone marrow |
| Single-system | Only 1 system involved |
| Single site | Skin, bone, lymph node, other (thyroid, thymus) |
| Multiple sites | Multifocal bone disease |
| Special site | Skull-base lesion with intracranial extension or vertebral lesion with intraspinal soft tissue extension |
| Pulmonary Langerhans cell histiocytosis (LCH) | Isolated lung disease |
| Central Nervous System (CNS) LCH | Tumorous lesions |
| Neurodegenerative disease | |
| LCH-associated abnormal CNS imaging (LACI) | |
| LCH-associated abnormal CNS symptoms (LACS) |
Langerhans cell histiocytosis is described as single-system disease or multisystem disease, depending on how many body systems are affected:
- Single-system Langerhans cell histiocytosis: Langerhans cell histiocytosis is found in one part of an organ or body system (unifocal) or in more than one part of that organ or body system (multifocal). Bone is the most common single place for Langerhans cell histiocytosis to be found.
- Multisystem Langerhans cell histiocytosis: Langerhans cell histiocytosis is found in two or more organs or body systems or may be found throughout the body. Multisystem Langerhans cell histiocytosis is less common than single-system Langerhans cell histiocytosis.
Langerhans cell histiocytosis may affect low-risk organs or high-risk organs:
- Low-risk organs include the skin, bone, lungs, lymph nodes, gastrointestinal tract, pituitary gland, thyroid gland, thymus, and central nervous system (CNS).
- High-risk organs include the liver, spleen, and bone marrow.
The cause and risk factors for developing Langerhans cell histiocytosis (LCH) is unknown and there has been debate as to whether Langerhans cell histiocytosis (LCH) is inflammatory, immune disorders, or cancer-like conditions 35, 36. Recently, through the use of genomics scientists have found that Langerhans cell histiocytosis (LCH) show gene changes (mutations) in early white blood cells. This leads to abnormal behavior in the cells. The abnormal cells then increase in various parts of body including the bones, skin, lungs, and other areas 37, 38, 39. Langerhans cell histiocytosis (LCH) has multiple cytokines involved, there is good survival in isolated lesions, and it can have spontaneous remissions. These characteristics support a reactive process. However, Langerhans cell histiocytosis (LCH) also can have organ infiltration. The widespread disease is associated with increased mortality, it typically responds to chemotherapy, and there has been at least one study that demonstrated an association with the BRAF‐V600E gene mutation. These characteristics support a neoplastic process. Langerhans cell histiocytosis is not known to be an inherited genetic disease.
Langerhans cell histiocytosis (LCH) includes a broad spectrum of clinical signs and symptoms in children and adults, ranging from self-healing lesions to life-threatening disseminated disease, which could result in improper treatment procedures, including surgical excision and irradiation 19, 7. The clinical manifestation of Langerhans cell histiocytosis varies from a single‐organ disease that could spontaneously go into remission, to a systemic and aggressive disease that can lead to death. Any organ can be involved, individually or in combination, but bone and skin have a higher frequency of involvement.
In approximately 80 percent of affected individuals, one or more granulomas develop in the bones, causing pain and swelling. The granulomas, which usually occur in the skull or the long bones of the arms or legs, may cause the bone to fracture.
Granulomas also frequently occur in the skin, appearing as blisters, reddish bumps, or rashes which can be mild to severe. The pituitary gland may also be affected; this gland is located at the base of the brain and produces hormones that control many important body functions. Without hormone supplementation, affected individuals may experience delayed or absent puberty or an inability to have children (infertility). In addition, pituitary gland damage may result in the production of excessive amounts of urine (diabetes insipidus) and dysfunction of another gland called the thyroid. Thyroid dysfunction can affect the rate of chemical reactions in the body (metabolism), body temperature, skin and hair texture, and behavior.
In 15 to 20 percent of cases, Langerhans cell histiocytosis affects the lungs, liver, or blood-forming (hematopoietic) system; damage to these organs and tissues may be life-threatening. Lung involvement, which appears as swelling of the small airways (bronchioles) and blood vessels of the lungs, results in stiffening of the lung tissue, breathing problems, and increased risk of infection. Hematopoietic involvement, which occurs when the Langerhans cells crowd out blood-forming cells in the bone marrow, leads to a general reduction in the number of blood cells (pancytopenia). Pancytopenia results in fatigue due to low numbers of red blood cells (anemia), frequent infections due to low numbers of white blood cells (neutropenia), and clotting problems due to low numbers of platelets (thrombocytopenia).
Other signs and symptoms that may occur in Langerhans cell histiocytosis, depending on which organs and tissues have Langerhans cell deposits, include swollen lymph nodes, abdominal pain, yellowing of the skin and whites of the eyes (jaundice), delayed puberty, protruding eyes, dizziness, irritability, and seizures. About 1 in 50 affected individuals experience deterioration of neurological function (neurodegeneration).
Diabetes insipidus is the most common initial sign of central nervous system (CNS) involvement in Langerhans cell histiocytosis 7. Isolated lung Langerhans cell histiocytosis, which is the most common manifestation in adult patients, is considered a specific type of Langerhans cell histiocytosis 7. Although pulmonary Langerhans cell histiocytosis cells have similar histopathologic characteristics, the majority of cell proliferation is polyclonal 40. Pulmonary Langerhans cell histiocytosis is strongly related to smoking; more than 90% of patients are smokers and quitting smoking could lead to remission without treatment 41, 42.
Most organs can be affected by Langerhans cell histiocytosis (LCH), but those more frequently affected in children are the bones (80% of cases), skin (33%), and the pituitary gland (25%), liver, spleen, hematopoietic system or lungs (15% each), lymph nodes (5%-10%), or the central nervous system (CNS) (2%-4% excluding the pituitary) 43, 22. In adults, lung involvement is more frequent than in children 19.
Langerhans cell histiocytosis is often diagnosed in childhood, usually between ages 2 and 3, but can appear at any age. Most individuals with adult-onset Langerhans cell histiocytosis are current or past smokers; in about two-thirds of adult-onset cases the disorder affects only the lungs. The diagnosis of Langerhans cell histiocytosis is based on clinical and radiological findings in combination with histopathological analyses identifying tissue infiltration by histiocytes with ultrastructural or immunophenotypic characteristics of Langerhans cells 19. It is recommended that biopsy confirmation of suspected LCH be performed in all cases, especially for patients requiring therapy 19.
Bone lesions are the most common manifestation of Langerhans cell histiocytosis, most often in flat bones such as the skull, mandible, ribs, pelvis, and spine 8. On imaging, bone lesions appear as osteolytic defects (“punched-out lesions”). On radiographs, calvarial lesions often demonstrate a double contour caused by asymmetrical involvement of the inner and outer tables, called the “hole within a hole” or “bevelled edge”-sign. MRI is superior in the evaluation of an associated soft-tissue mass or dural invasion. On MRI, osseous lesions most often demonstrate intermediate T1 and increased T2 signal intensity and avid contrast enhancement, as was the case in our patient. Involvement of the hypothalamic-pituitary axis is characterized by an absence of high signal intensity of the posterior pituitary on T1-weigthed imaging and a thickening and enhancement of the pituitary stalk. Both findings were also present in our case. In the absence of organ dysfunction, prognosis of children with either localized or multifocal Langerhans cell histiocytosis is excellent 44.
Treatment for Langerhans cell histiocytosis depends upon the individual patient; it may differ depending on the type and severity of the condition as well as what part(s) of the body are affected. In some cases, the disease will go away without any treatment at all. In other cases, depending on the extent of the disease, limited surgery and small doses of radiation therapy or chemotherapy may be needed. Treatment is planned after complete evaluation of the patient, with the goal of using as little treatment as possible to keep the disease under control 45.
No consensus exists for the best therapy for Langerhans cell histiocytosis, especially when multiple organs are involved. However, the Histiocyte Society has done many clinical trials to evaluate the effect of several treatments, which have resulted in recommendations by the Histiocyte Society.
Nine types of standard treatment are used to treat Langerhans cell histiocytosis:
- Chemotherapy. Chemotherapy is a cancer treatment that uses drugs to stop the growth of cancer cells, either by killing the cells or by stopping them from dividing. Chemotherapy that is taken by mouth or injected into a vein or muscle enters the bloodstream and can reach cancer cells throughout the body (systemic chemotherapy). Chemotherapy may also be applied to the skin in a cream or lotion (topical chemotherapy). Chemotherapy may be given by injection, by mouth, or applied to the skin to treat Langerhans cell histiocytosis (LCH).
- Surgery. Surgery may be used to remove Langerhans cell histiocytosis (LCH) lesions and a small amount of nearby healthy tissue. Curettage is a type of surgery that uses a curette (a sharp, spoon-shaped tool) to scrape LCH cells from bone. When there is severe liver or lung damage, the entire organ may be removed and replaced with a healthy liver or lung from a donor.
- Radiation therapy. Radiation therapy is a cancer treatment that uses high-energy x-rays or other types of radiation to kill cancer cells or keep them from growing. External radiation therapy uses a machine outside the body to send radiation toward the area of the body with cancer. Ultraviolet B (UVB) radiation therapy may be given using a special lamp that directs radiation toward Langerhans cell histiocytosis (LCH) skin lesions.
- Photodynamic therapy. Photodynamic therapy is a cancer treatment that uses a drug and a certain type of laser light to kill cancer cells. A drug that is not active until it is exposed to light is injected into a vein. The drug collects more in cancer cells than in normal cells. For Langerhans cell histiocytosis (LCH), laser light is aimed at the skin and the drug becomes active and kills the cancer cells. Photodynamic therapy causes little damage to healthy tissue. Patients who have photodynamic therapy should not spend too much time in the sun. In one type of photodynamic therapy, called psoralen and ultraviolet A (PUVA) therapy, the patient receives a drug called psoralen and then ultraviolet A radiation is directed to the skin.
- Immunotherapy. Immunotherapy is a treatment that uses the patient’s immune system to fight cancer. Substances made by the body or made in a laboratory are used to boost, direct, or restore the body’s natural defenses against cancer. Thalidomide is a type of immunotherapy used to treat Langerhans cell histiocytosis (LCH).
- Targeted therapy. Targeted therapy is a type of treatment that uses drugs or other substances to identify and attack specific cancer cells. There are different types of targeted therapy:
- BRAF inhibitors block proteins needed for cell growth and may kill cancer cells. The BRAF gene is found in a mutated (changed) form in some Langerhans cell histiocytosis (LCH) and blocking it may help keep LCH cells from growing.
- Vemurafenib and dabrafenib are BRAF inhibitors used to treat Langerhans cell histiocytosis (LCH).
- Trametinib is a BRAF inhibitor that is being studied in the treatment of certain childhood tumors for use alone or combined with dabrafenib.
- Monoclonal antibodies are immune system proteins made in the laboratory to treat many diseases, including cancer. As a cancer treatment, these antibodies can attach to a specific target on cancer cells or other cells that may help cancer cells grow. The antibodies are able to then kill the cancer cells, block their growth, or keep them from spreading. Monoclonal antibodies are given by infusion. They may be used alone or to carry drugs, toxins, or radioactive material directly to cancer cells.
- Rituximab is a monoclonal antibody used to treatLangerhans cell histiocytosis (LCH).
- BRAF inhibitors block proteins needed for cell growth and may kill cancer cells. The BRAF gene is found in a mutated (changed) form in some Langerhans cell histiocytosis (LCH) and blocking it may help keep LCH cells from growing.
- Other drug therapy. Other drugs used to treat eosinophilic granuloma (Langerhans cell histiocytosis) include the following:
- Steroid therapy, such as prednisone, is used to treat eosinophilic granuloma (Langerhans cell histiocytosis) lesions.
- Bisphosphonate therapy (such as pamidronate, zoledronate, or alendronate) is used to treat eosinophilic granuloma (Langerhans cell histiocytosis) lesions of the bone and to lessen bone pain.
- Anti-inflammatory drugs are drugs (such as pioglitazone and rofecoxib) that are commonly used to decrease fever, swelling, pain, and redness. Anti-inflammatory drugs and chemotherapy may be given together to treat adults with bone eosinophilic granuloma (Langerhans cell histiocytosis).
- Stem cell transplant. Chemotherapy is given to kill cancer cells. Healthy cells, including blood-forming cells, are destroyed by the eosinophilic granuloma (Langerhans cell histiocytosis) treatment. Stem cell transplant is a treatment to replace the blood-forming cells. Stem cells (immature blood cells) are removed from the blood or bone marrow of the patient or a donor and are frozen and stored. After the patient completes chemotherapy, the stored stem cells are thawed and given back to the patient through an infusion. These stem cells grow into (and restore) the body’s blood cells.
- Observation. Observation is closely monitoring a patient’s condition without giving any treatment until signs or symptoms appear or change.
- Clinical trials. A treatment clinical trial is a research study meant to help improve current treatments or obtain information on new treatments for patients with cancer. When clinical trials show that a new treatment is better than the standard treatment, the new treatment may become the standard treatment. Whenever possible, patients should take part in a clinical trial in order to receive new types of treatment for Langerhans cell histiocytosis (LCH). Some clinical trials are open only to patients who have not started treatment.
Generally, the choice of treatment is based on disease severity; patients with single lesions may respond well to local treatment, whereas patients with multisystem disease require systemic therapy 18. The International eosinophilic granuloma (Langerhans cell histiocytosis) Study of the Histiocyte Society proposes classifying Langerhans cell histiocytosis (LCH) cases by the number of systems involved and by the number of sites within that system (e.g., involving one or more bones, involving one or multiple lymph nodes). Although most of the trials are in children, the recommendations can also be used for adults.
Langerhans cell histiocytosis survival rates for patients without organ dysfunction is excellent, but mortality rates for patients with organ dysfunction may reach 20% 18. However, when treatment stops, new lesions may appear or old lesions may come back. This is called reactivation (recurrence) and may occur within 1 year after stopping treatment. Patients with multisystem disease are more likely to have a reactivation. Common sites of reactivation are bone, ears, or skin. Diabetes insipidus also may develop. Less common sites of reactivation include lymph nodes, bone marrow, spleen, liver, or lung. Some patients may have more than one reactivation. Langerhans cell histiocytosis disease reactivation rates remain above 30%, and standard second-line treatment is yet to be established 18. Treatment failure is associated with increased risks for death and long-term morbidity, including LCH-associated neurodegeneration. Early case series report promising clinical responses in patients with relapsed and refractory LCH treated with BRAF or MEK inhibitors, although potential for this strategy to achieve cure remains uncertain.
Figure 1. Langerhans cell histiocytosis diagnosis (LCH lesion histology)
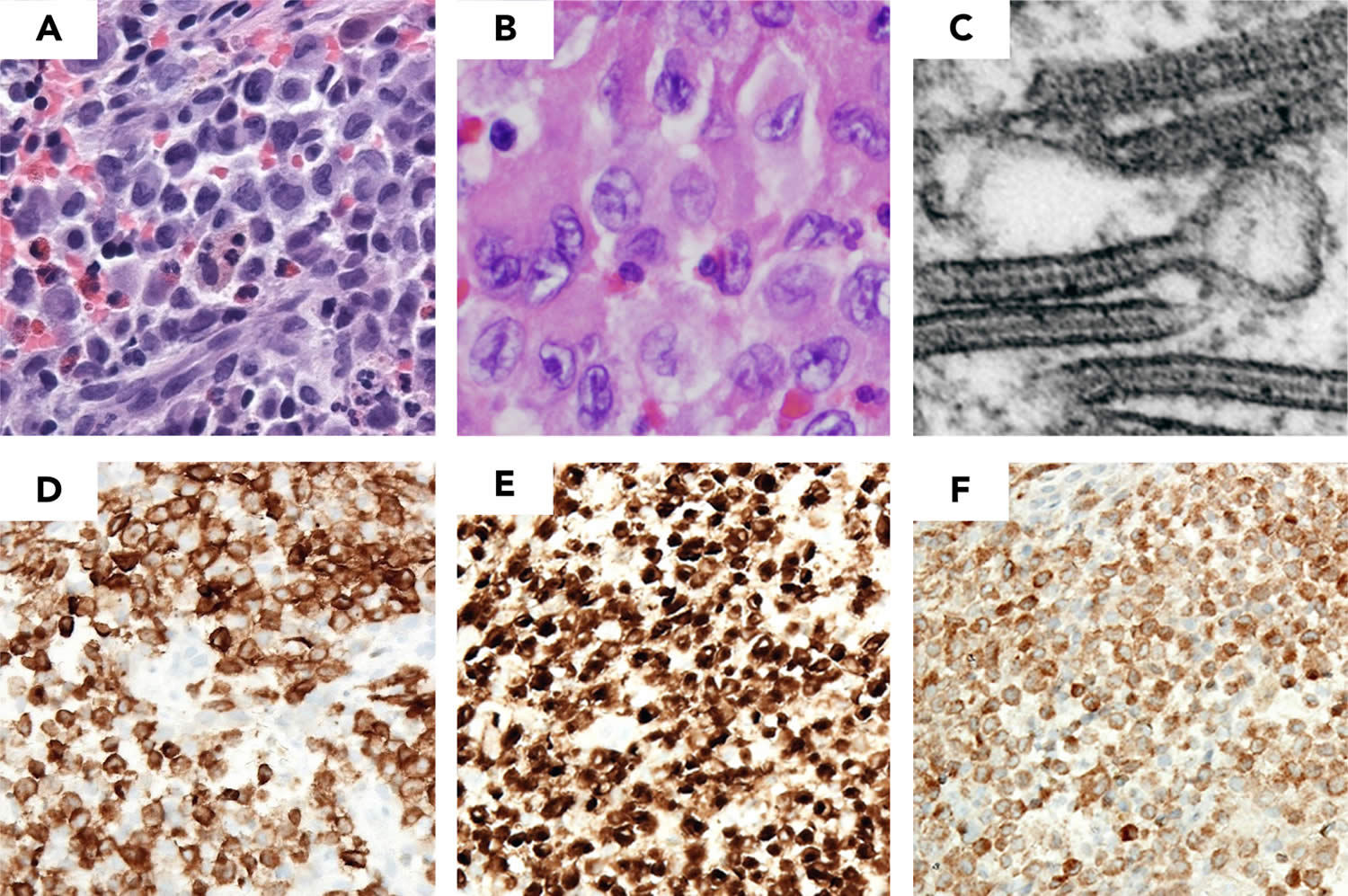
Footnotes: Images demonstrate typical histology of LCH lesion obtained from a bone biopsy with pathologic histiocytes and inflammatory infiltrate. (A-B) Hematoxylin and eosin stain demonstrates histiocytes with pale cytoplasm and reniform nuclei. (C) Birbeck granules on electron microscopy; immunohistochemistry strongly positive for (D) Langerin (CD207), (E) CD1a, and (F) S100a.
[Source 18 ]Figure 2. Histiocytosis
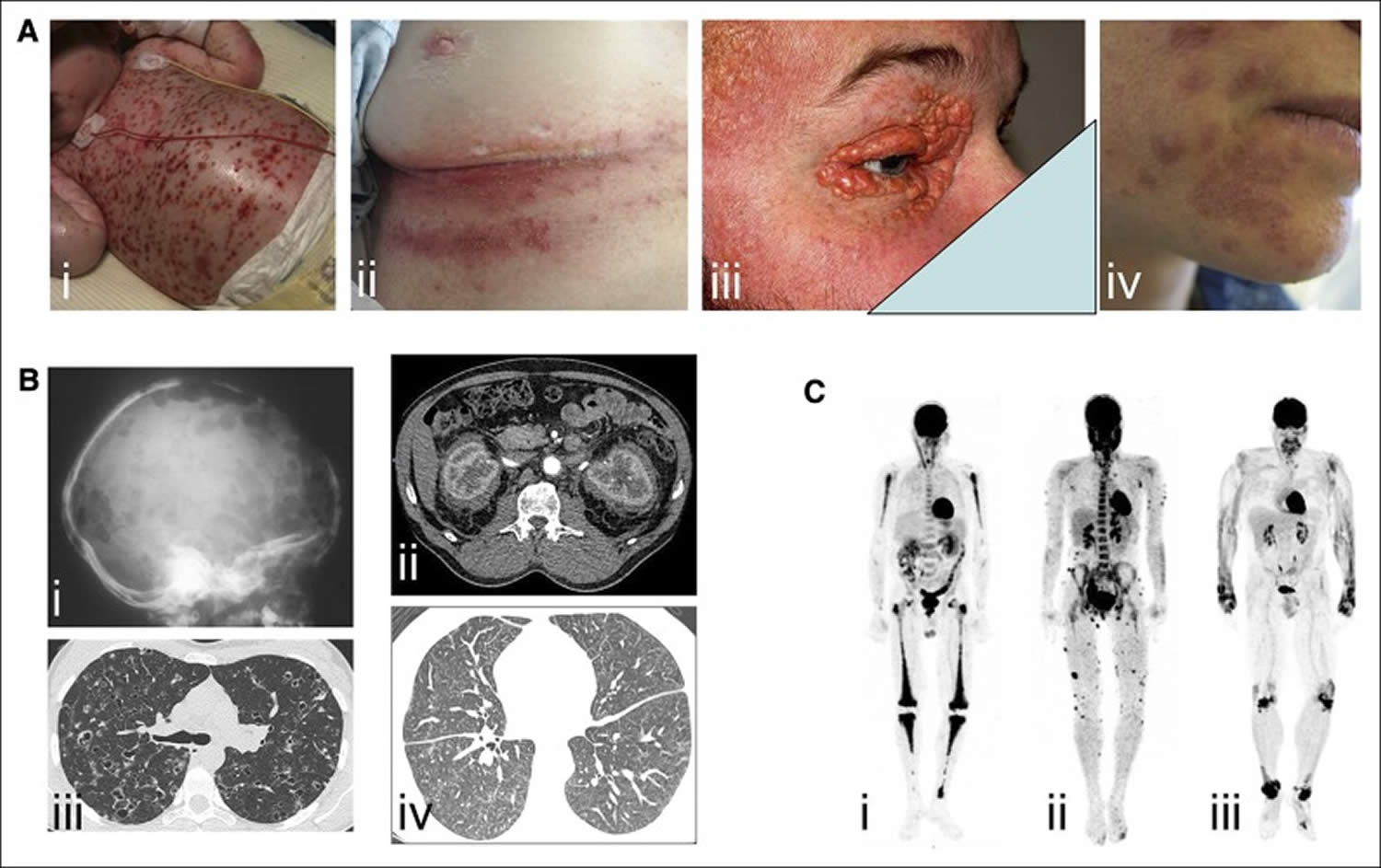
Footnotes: Examples of clinical involvement by histiocytoses. (A) Examples of cutaneous manifestations in (i) a child with multisystemic Langerhans cell histiocytosis (LCH), (ii) adult with intertrigo-like lesions, (iii) xanthelasma of Erdheim-Chester disease and (iv) skin manifestations of Rosai-Dorfman disease. (B) Radiographic imaging and CT scans of (i) lytic skull bone lesions and (ii) pulmonary nodules and cysts in Langerhans cell histiocytosis (LCH), (iii) CT scan revealing typical “hairy kidney” lesions and (iv) micronodular ground-glass opacities and thickening of interlobular pulmonary septa in Erdheim-Chester disease. (C) 18F-labeled fluorodeoxyglucose (PET) imaging revealing (i) bilateral and symmetric signal in femurs, tibiae, and humeri in Erdheim-Chester disease, (ii) cutaneous multiple lesions in Rosai-Dorfman disease, and (iii) signal over wrist, knees, and ankles of a patient with xanthoma disseminatum.
[Source 19 ]Figure 3. Langerhans cell histiocytosis
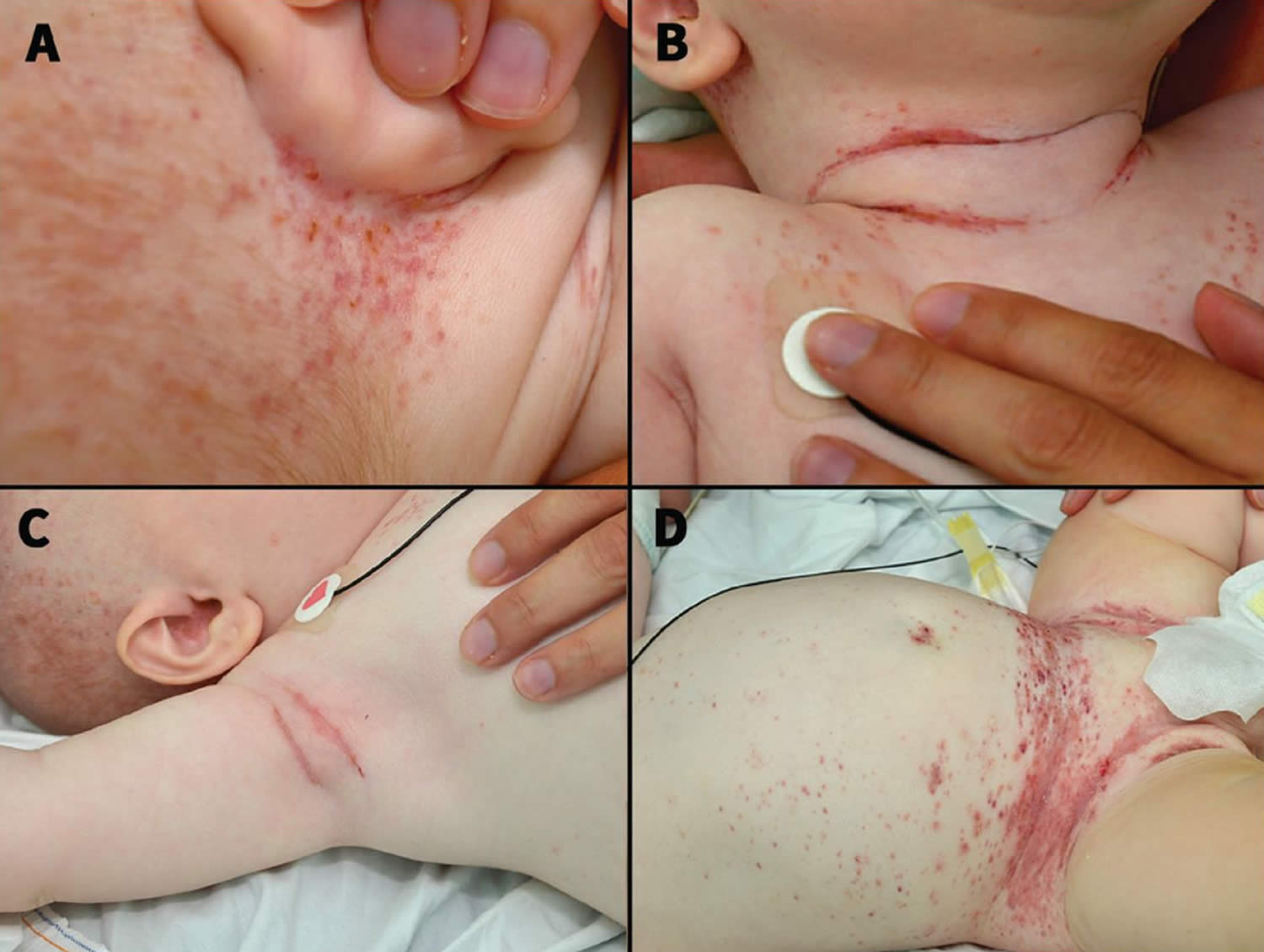
Footnotes: Photographs of a 5-month-old boy showing petechiae, confluent erythema, crusted papules and hyperemia in the skin creases involving the ears (A), neck (B), axilla (C) and groin (D). Vesicles and ulcers in the ear and groin creases, and areas of maceration in the neck, axilla and groin are seen. Kidney and liver function tests were normal. A lytic lesion on the patient’s left proximal femur was found on a skeletal survey. Skin and duodenal biopsies were positive for Langerhans cell histiocytosis, with involvement in multiple sites (dermatological, hematological, bone and gastrointestinal). The patient responded poorly to vinblastine and prednisolone; as he tested positive for the BRAF V600E mutation, he was started on dabrafenib, a BRAF kinase inhibitor, with clinical improvement.
[Source 46 ]Figure 4. Langerhans cell histiocytosis (LCH) skin lesions (self-healing Langerhans cell histiocytosis with BRAF-V600E mutation)
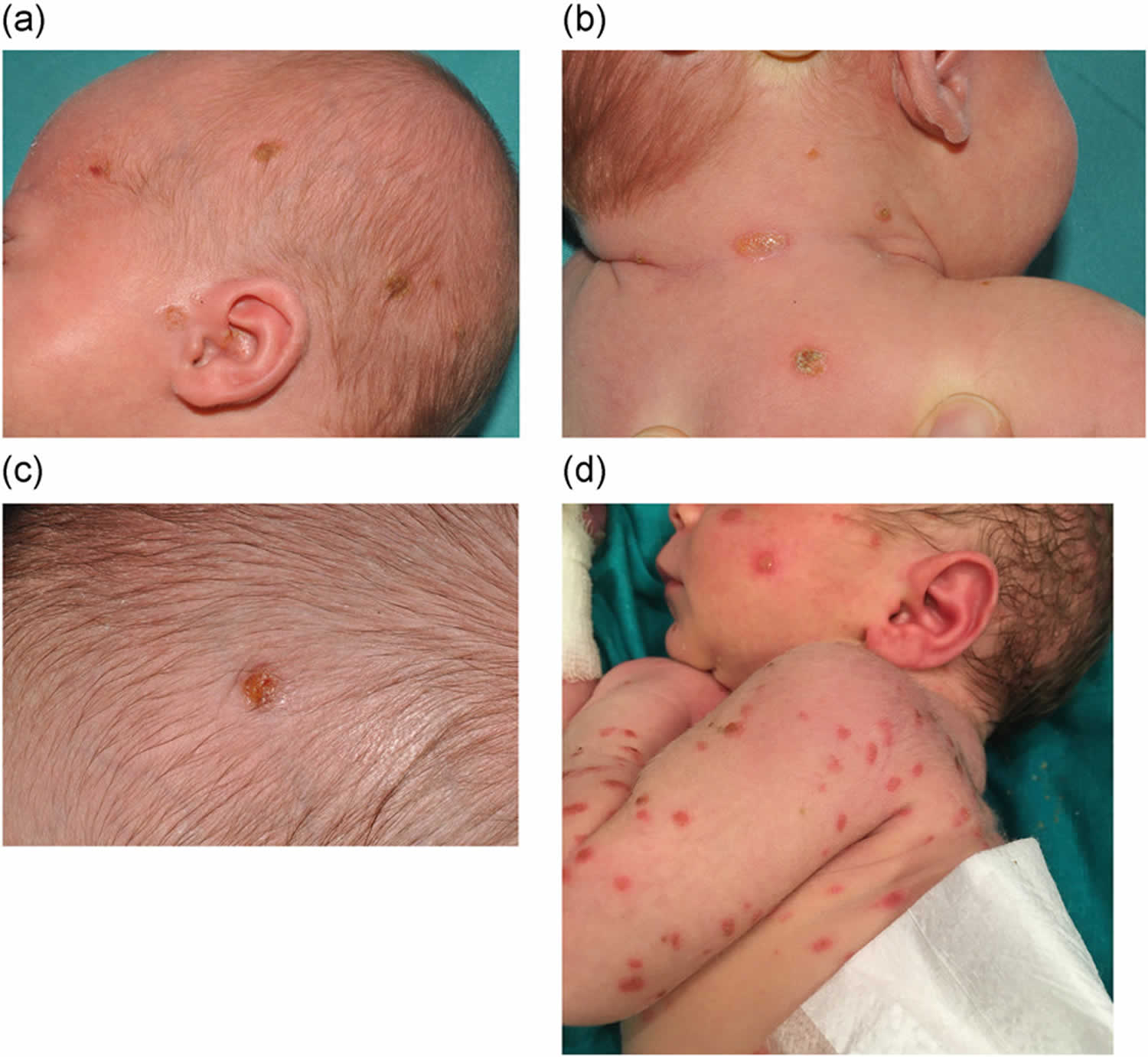
Footnotes: Self-healing Langerhans cell histiocytosis with BRAF-V600E mutation. A full-term female neonate presented multiple crusted vesicular lesions at birth. She was seen in our department at the age of 1 month, and examination revealed scattered, yellow-brown, crusted papules on her face, scalp, trunk and limbs. A skin biopsy revealed a dense infiltrate in the upper and mid dermis composed of mononuclear cells with indented nuclei. These cells were strong and diffusely positive for S100 protein, CD1a, langerin/CD207 and BRAF-V600E VE1. Ki-67 stain was noted in 80% of the nuclei. Lab tests, including TORCHs serologies, skeletal survey, chest X-ray and abdominal ultrasound were unremarkable. The cutaneous lesions started regressing at 2 months of age and had completely disappeared 4 months later, without any sign of systemic involvement after 1-year follow-up.
[Source 47 ]Figure 5. LCH skin lesions
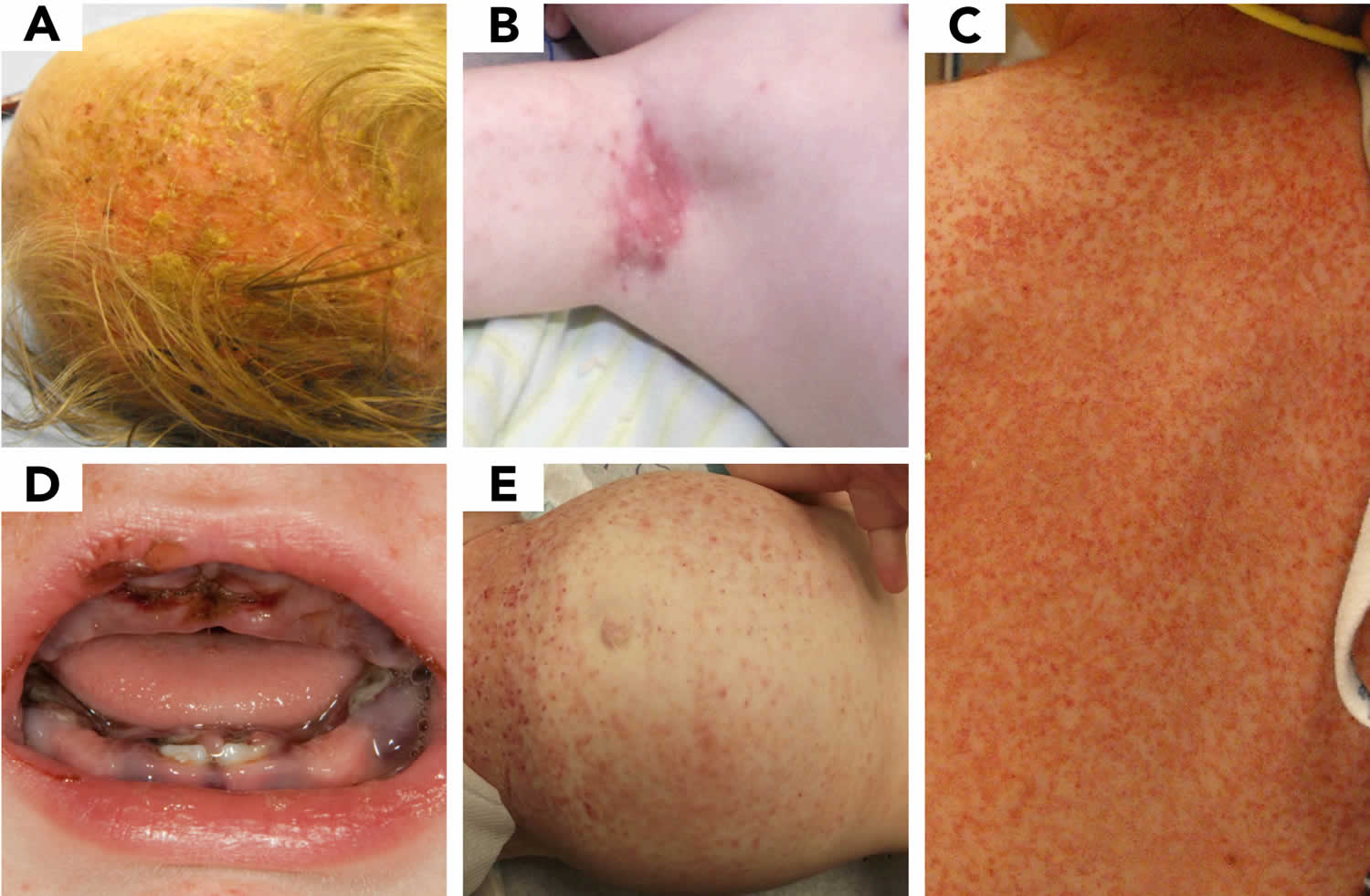
Footnote: A range of LCH skin disease is presented. (A) cradle cap, (B) eczema, (C) scarlet fever or scalded skin syndrome, (D) herpes gingivostomatitis, and (E) immune thrombocytopenic purpura.
[Source 18 ]Figure 6. LCH imaging
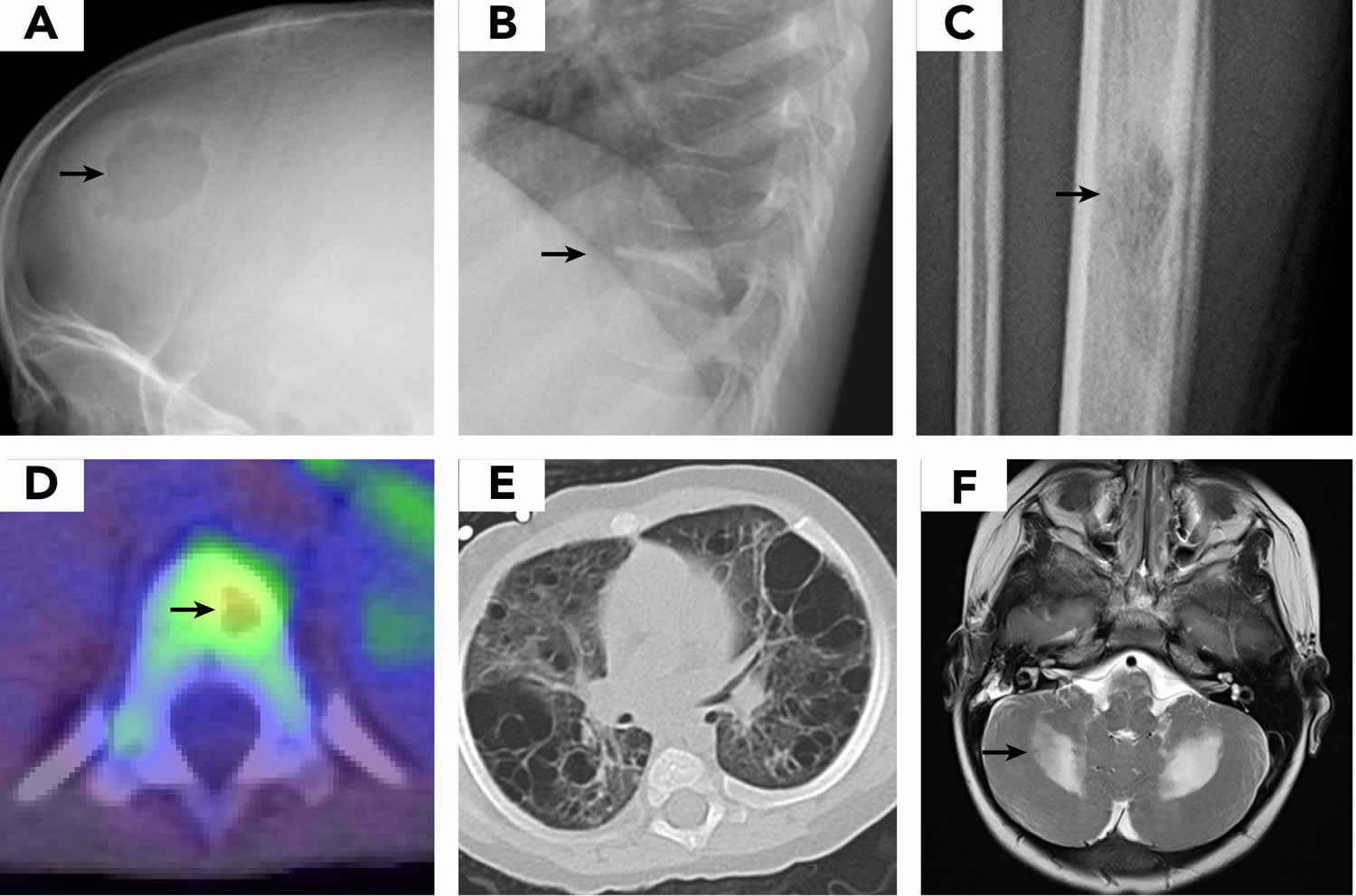
Footnotes: Images demonstrate typical presentation of LCH lesions including (A) single skull lesion on X-ray, (B) vertebra plana on X-ray, (C) femur lesion on X-ray, (D) vertebral lesion on positron emission tomography/computed tomography scan, and (E) cystic lung disease on computed tomography scan. (F) Brain magnetic resonance imaging (MRI) demonstrates T2 hyperintensity in cerebellum in LCH-associated neurodegeneration.
[Source 18 ]What is histiocytosis?
Histiocytosis is a general name for a group of disorders or “syndromes” that involve an abnormal increase in the number of specialized white blood cells that are called histiocytes 48. The term histiocyte refers to a bone marrow derived progenitor cell that differentiates, depending on the cytokine and growth factor milieu, into a dendritic antigen presenting cell (Langerhans cell, dermal dendrocyte) or a phagocytic cell (tissue macrophage) 49.
Recently, new knowledge about this family of diseases has led experts to develop a new classification. Five categories have been proposed 18, 19:
- L group — includes Langerhans cell histiocytosis (LCH), Erdheim-Chester disease, Indeterminate-cell histiocytosis and mixed Langerhans cell histiocytosis/Erdheim-Chester disease
- C group — includes non-Langerhans cell histiocytosis that involves the skin
- Cutaneous non-Langerhans cell histiocytosis
- Xanthomatous granuloma (XG) family: juvenile xanthogranuloma, adult xanthogranuloma, solitary reticulohistiocytoma, benign cephalic histiocytosis, generalized eruptive histiocytosis, progressive nodular histiocytosis
- Non-Xanthomatous granuloma (non-XG) family: Cutaneous Rosai-Dorfman disease, necrobiotic xanthogranuloma, other
- Cutaneous non-Langerhans cell histiocytosis with a major systemic component
- Xanthomatous granuloma (XG) family: xanthoma disseminatum
- Non-Xanthomatous granuloma (non-XG) family: multicentric reticulohistiocytosis
- Cutaneous non-Langerhans cell histiocytosis
- M group — includes Primary malignant histiocytoses and Secondary malignant histiocytoses
- R group — includes Rosai-Dorfman disease
- Familial Rosai-Dorfman disease
- Sporadic Rosai-Dorfman disease
- Classical Rosai-Dorfman disease
Extranodal Rosai-Dorfman disease - Rosai-Dorfman disease with neoplasia or immune disease
- Unclassified
- Classical Rosai-Dorfman disease
- H Group — includes hemophagocytic lymphohistiocytosis
- Primary hemophagocytic lymphohistiocytosis : Monogenic inherited conditions leading to hemophagocytic lymphohistiocytosis
- Secondary hemophagocytic lymphohistiocytosis (non-Mendelian hemophagocytic lymphohistiocytosis)
- Hemophagocytic lymphohistiocytosis of unknown/uncertain origin
Langerhans cell histiocytosis types
The first definitive case of Langerhans cell histiocytosis (LCH) was reported in 1893 by Alfred Hand 2. The patient was a 3‐year‐old boy with excessive urination (polyuria), bulging or protruding eyeballs (exophthalmos) and enlarged liver and spleen (hepatosplenomegaly); similar cases were reported by Henry Christian and Arthur Schuller 3, 4. The disease was named Hand‐Schuller‐Christian disease 7. Based on similar histopathological patterns, Hand‐Schuller‐Christian disease, Letterer‐Siwe disease, and eosinophilic granuloma were unified as histiocytosis X in 1953 and renamed Langerhans cell histiocytosis (LCH) in 1985 5. Depending on the number of lesions and systems involved, Langerhans cell histiocytosis (LCH) is composed of three categories 8:
- Eosinophilic granuloma is limited to one or a few bones with potential lung involvement.
- Hand-Schüller-Christian disease involves multiple bones, the reticuloendothelial system and the pituitary gland.
- Letterer-Siwe disease is disseminated disease with fulminant clinical course.
Eosinophilic granuloma (single-system disease)
Unifocal or multifocal single-system lesions (60 to 80% of Langerhans cell histiocytosis cases) occurs predominantly in older children and young adults, usually by age 30; incidence peaks between ages 5 and 10 years. Lesions most frequently involve bones, often with pain, the inability to bear weight, or both and with overlying tender (sometimes warm) swelling.
Congenital self-healing reticulohistiocytosis
Congenital self-healing reticulohistiocytosis previously called Hashimoto-Pritzker disease is a single-system disease with isolated skin lesions that occurs in neonates. Lesions generally resolve on their own or respond to topical treatment. Patients should be evaluated to exclude systemic disease.
Hand-Schüller-Christian disease (multisystem disease without risk organ involvement)
Hand‐Schuller‐Christian disease (15 to 40% of Langerhans cell histiocytosis cases) occurs in children aged 2 to 5 years and in some older children and adults. Classic findings in Hand‐Schuller‐Christian disease include involvement of the flat bones of the skull, ribs, pelvis, scapula, or a combination. Long bones and lumbosacral vertebrae are less frequently involved; the wrists, hands, knees, feet, and cervical vertebrae are rarely involved. In classic cases, patients have proptosis caused by orbital tumor mass. Rarely, vision loss or strabismus is caused by optic nerve or orbital muscle involvement. Tooth loss caused by apical and gingival infiltration is common in older patients.
Chronic otitis media and otitis externa due to involvement of the mastoid and petrous portions of the temporal bone with partial obstruction of the auditory canal are fairly common. Diabetes insipidus, the last component of the classic triad that includes flat bone involvement and proptosis, affects 5 to 50% of patients, with higher percentages in children who have systemic disease and involvement of the orbit and skull. Up to 40% of children with systemic disease have short stature. Hyperprolactinemia and hypogonadism can result from hypothalamic infiltration.
Letterer-Siwe disease (multisystem disease with risk organ involvement)
Letterer-Siwe disease (10% of Langerhans cell histiocytosis cases), a systemic disorder, is the most severe form of Langerhans cell histiocytosis. Typically, a child < 2 years presents with a scaly seborrheic, eczematoid, sometimes purpuric rash involving the scalp, ear canals, abdomen, and intertriginous areas of the neck and face. Denuded skin may facilitate microbial invasion, leading to sepsis. Frequently, there is ear drainage, lymphadenopathy, hepatosplenomegaly, and, in severe cases, hepatic dysfunction with hypoproteinemia and diminished synthesis of clotting factors. Anorexia, irritability, failure to thrive, and pulmonary manifestations (eg, cough, tachypnea, pneumothorax) may also occur. Significant anemia and sometimes neutropenia occur; thrombocytopenia is of grave prognostic significance. Parents frequently report precocious eruption of teeth, when in fact the gums are receding to expose immature dentition. Patients may appear abused or neglected.
Langerhans cell histiocytosis causes
The cause and risk factors for developing Langerhans cell histiocytosis (LCH) is unknown and there has been debate as to whether Langerhans cell histiocytosis (LCH) is inflammatory, immune disorders, or cancer-like conditions 35, 36. Recently, through the use of genomics scientists have found that Langerhans cell histiocytosis (LCH) show gene changes (mutations) in early white blood cells. This leads to abnormal behavior in the cells. The abnormal cells then increase in various parts of body including the bones, skin, lungs, and other areas 37, 38, 39. Langerhans cell histiocytosis (LCH) has multiple cytokines involved, there is good survival in isolated lesions, and it can have spontaneous remissions. These characteristics support a reactive process. However, Langerhans cell histiocytosis (LCH) also can have organ infiltration. The widespread disease is associated with increased mortality, it typically responds to chemotherapy, and there has been at least one study that demonstrated an association with the BRAF gene mutation. These characteristics support a neoplastic process. Langerhans cell histiocytosis is not known to be an inherited genetic disease.
Somatic mutations in the BRAF gene have been identified in the Langerhans cells of about 50% of individuals with Langerhans cell histiocytosis. Somatic gene mutations are acquired during a person’s lifetime and are present only in certain cells. These changes are not inherited. The BRAF gene provides instructions for making a protein that is normally switched on and off in response to signals that control cell growth and development. Somatic mutations cause the BRAF protein in affected cells to be continuously active and to transmit messages to the nucleus even in the absence of these chemical signals. The overactive protein may contribute to the development of Langerhans cell histiocytosis by allowing the Langerhans cells to grow and divide uncontrollably.
Changes in other genes have also been identified in the Langerhans cells of some individuals with Langerhans cell histiocytosis. Some researchers believe that additional factors, such as viral infections and environmental toxins, may also influence the development of this complex disorder.
Langerhans cell histiocytosis pathology outlines
The Langerhans histiocytosis cells in LCH lesions (LCH cells) are immature dendritic cells making up fewer than 10% of the cells present in the lesion 34, 50. The pathologic histiocytes or Langerhans cell histiocytosis (LCH) cells are classically large oval cells with abundant pink cytoplasm and a bean-shaped nucleus on hematoxylin and eosin stain. Langerhans histiocytosis cells (LCH cells) stain positively with antibodies to S100, CD1a, and/or anti-Langerin (CD207). Staining with CD1a or Langerin confirms the diagnosis of Langerhans cell histiocytosis, but care should be taken to correlate with clinical presentation in organs in which normal Langerhans cells occur 9. LCH cells, known for many years to be a clonal proliferation, have now been shown to likely derive from a myeloid precursor whose proliferation is uniformly associated with activation of the MAPK/ERK signaling pathway 51, 52.
Because LCH cells activate other immunologic cells, LCH lesions also contain other histiocytes, lymphocytes, macrophages, neutrophils, eosinophils, and fibroblasts, and they may contain multinucleated giant cells.
In the brain, the following three types of histopathologic findings have been described in Langerhans cell histiocytosis 53:
- Mass lesions in the meninges or choroid plexus with CD1a-positive LCH cells and predominantly CD8-positive lymphocytes.
- Mass lesions in connective tissue spaces with CD1a-positive LCH cells and predominantly CD8-positive lymphocytes that cause an inflammatory response and neuronal loss.
- Neurodegenerative lesions, consisting of cells staining for the mutant BRAF protein with CD14+, CD33+, and CD163+, identifying these as hematopoietic myeloid/monocytic cells. These are the pathologic Langerhans cells that have migrated into the brain and do not stain with CD1a or CD207 and have become microglia-like 54.
Normally, the Langerhans cell is a primary presenter of antigen to naive T lymphocytes. However, in Langerhans cell histiocytosis (LCH), the pathologic dendritic cell does not efficiently stimulate primary T-lymphocyte responses 55. Antibody staining for the dendritic cell markers, including CD80, CD86, and class 2 antigens, has been used to show that in Langerhans cell histiocytosis (LCH), the abnormal cells are immature dendritic cells that present antigen poorly and are proliferating at a low rate 56, 57, 55. Transforming growth factor-beta (TGF-beta) and interleukin (IL)-10 may be responsible for preventing LCH cell maturation in Langerhans cell histiocytosis (LCH) 56. The expansion of regulatory T cells in patients with LCH has been reported 57. The population of CD4-positive CD25(high) FoxP3(high) cells was reported to comprise 20% of T cells and appeared to be in contact with LCH cells in the lesions. These T cells were present in higher numbers in the peripheral blood of patients with LCH than in the peripheral blood of controls and returned to a normal level when patients were in remission 57.
BRAF, NRAS, and ARAF mutations
The theory for the genomic basis of Langerhans cell histiocytosis was advanced by a 2010 report of an activating mutation of the BRAF oncogene (V600E) that was detected in 35 of 61 cases (57%) 58. Multiple subsequent reports have confirmed the presence of BRAF V600E mutations in 50% or more of Langerhans cell histiocytosis cases in children 59, 60, 61. Other BRAF mutations that result in signal activation have been described 59, 62. ARAF mutations are infrequent in Langerhans cell histiocytosis but, when present, can also lead to RAS-MAPK pathway activation 63.
The RAS-MAPK signaling pathway (Figure 7) transmits signals from a cell surface receptor (e.g., a growth factor) through the RAS pathway (via one of the RAF proteins [A, B, or C]) to phosphorylate MEK and then the extracellular signal-regulated kinase (ERK), which leads to nuclear signals affecting cell cycle and transcription regulation. The V600E mutation of BRAF leads to continuous phosphorylation, and thus activation, of MEK and ERK without the need for an external signal. Activation of ERK occurs by phosphorylation, and phosphorylated ERK can be detected in virtually all Langerhans cell histiocytosis lesions 64, 58.
The presence of the BRAF V600E mutation in blood and bone marrow was studied in a series of 100 patients, 65% of whom tested positive for the BRAF V600E mutation by a sensitive quantitative polymerase chain reaction technique 61. Circulating cells with the BRAF V600E mutation could be detected in all high-risk patients and in a subset of low-risk multisystem patients. The presence of circulating cells with the mutation conferred a twofold increased risk of relapse. In a similar study that included 48 patients with BRAF V600E–mutated Langerhans cell histiocytosis, the BRAF V600E allele was detected in circulating cell-free DNA in 100% of patients with risk-organ–positive multisystem Langerhans cell histiocytosis, 42% of patients with risk-organ–negative Langerhans cell histiocytosis, and 14% of patients with single-system Langerhans cell histiocytosis 65.
The myeloid dendritic cell origin of Langerhans cell histiocytosis was confirmed by finding CD34-positive stem cells with the mutation in the bone marrow of high-risk patients. In those with low-risk disease, the mutation was found in more mature myeloid dendritic cells, suggesting that the stage of cell development at which the somatic mutation occurs is critical in defining the extent of disease in Langerhans cell histiocytosis. Langerhans cell histiocytosis is now generally considered to represent a myeloid neoplasm.
Figure 7. RAS-MAPK signaling pathway
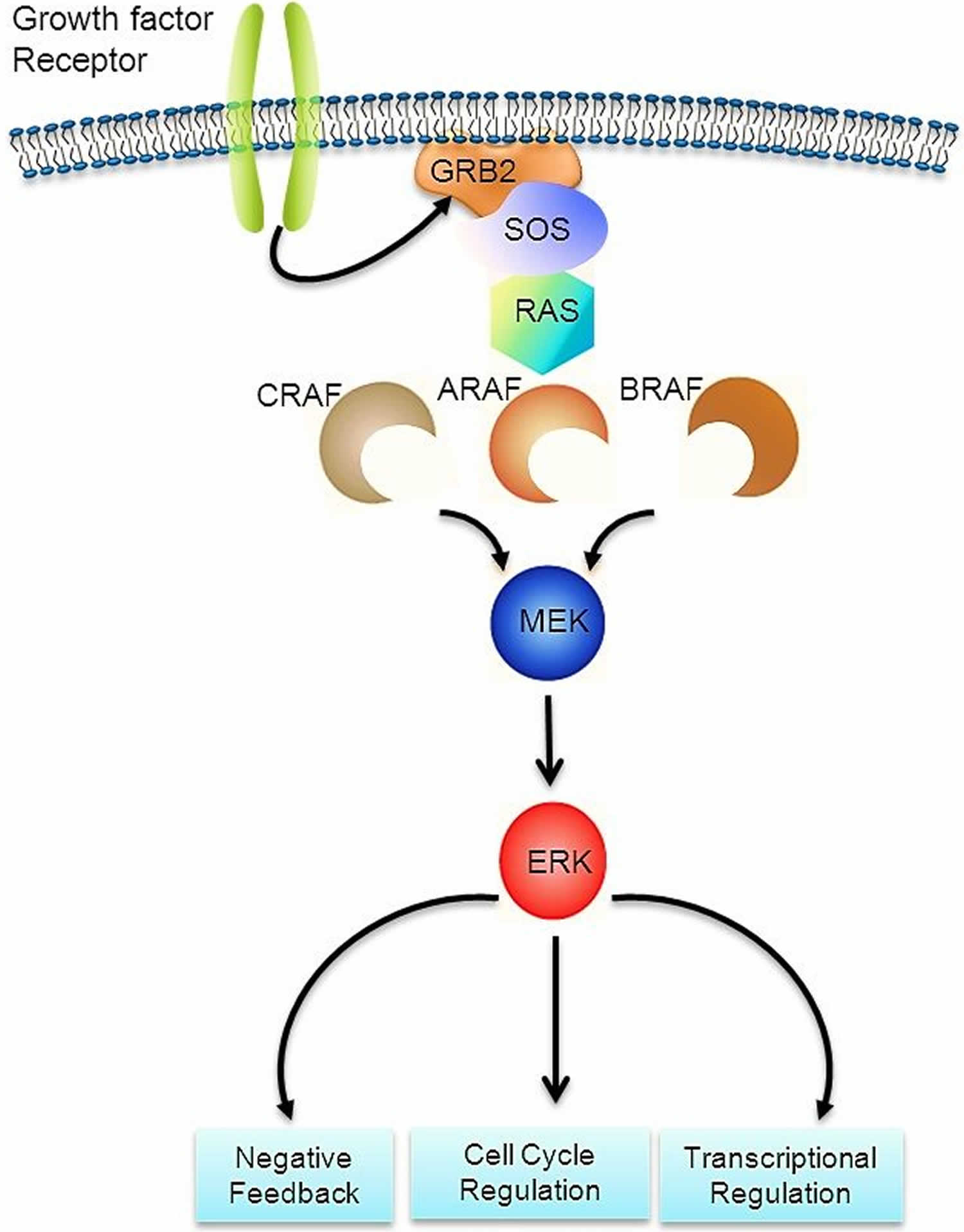
Other RAS-MAPK pathway alterations
Because RAS-MAPK pathway activation can be detected in all Langerhans cell histiocytosis cases, but not all cases have BRAF mutations, the presence of genomic alterations in other components of the pathway was suspected. The following genomic alterations were identified:
- MAP2K1 mutations. Whole-exome sequencing of BRAF-mutated versus BRAF–wild-type Langerhans cell histiocytosis biopsy tissue samples revealed that 7 of 21 BRAF–wild-type specimens had MAP2K1 mutations, while no BRAF-mutated specimens had MAP2K1 mutations 64. The mutations in MAP2K1 (which codes for MEK) were activating, as indicated by their induction of ERK phosphorylation 64. Another study showed MAP2K1 mutations exclusively in 11 of 22 BRAF–wild-type cases 66.
- In-frame BRAF deletions and FAM73A/BRAF fusions. In-frame BRAF deletions and in-frame FAM73A/BRAF fusions have occurred in the group of BRAF V600E and MAP2K1 mutation–negative cases 67.
Studies support the universal activation of ERK in Langerhans cell histiocytosis; ERK activation in most cases is explained by BRAF and MAP2K1 alterations 67, 64, 58. Altogether, these mutations in the MAP kinase pathway account for nearly 90% of the causes of the universal activation of ERK in Langerhans cell histiocytosis 67, 64, 58.
Cytokine analysis
Immunohistochemical staining has shown upregulation of many different cytokines/chemokines, both in LCH lesions and in the serum/plasma of patients with Langerhans cell histiocytosis 68, 69. In an analysis of gene expression in Langerhans cell histiocytosis by gene array techniques, 2,000 differentially expressed genes were identified. Of 65 genes previously reported to be associated with Langerhans cell histiocytosis, only 11 were found to be upregulated in the array results. The most highly upregulated gene in both CD207-positive and CD3-positive cells was SPP1 (encoding the osteopontin protein); other genes that activate and recruit T cells to sites of inflammation are also upregulated 70. The expression profile of the T cells was that of an activated regulatory T-cell phenotype with increased expression of FOXP3, CTLA4, and SPP1. These findings support a previous report on the expansion of regulatory T cells in Langerhans cell histiocytosis 70. There was pronounced expression of genes associated with early myeloid progenitors such as CD33 and CD44, which is consistent with an earlier report of elevated myeloid dendritic cells in the blood of patients with Langerhans cell histiocytosis 71. A model of Misguided Myeloid Dendritic Cell Precursors has been proposed, whereby myeloid dendritic cell precursors are recruited to sites of Langerhans cell histiocytosis by an unknown mechanism, and the dendritic cells, in turn, recruit lymphocytes by excretion of osteopontin, neuropilin-1, and vannin-1 70.
A study to evaluate possible biomarkers for central nervous system LCH examined 121 unique proteins in the cerebrospinal fluid (CSF) of 40 pediatric patients with Langerhans cell histiocytosis and compared them with controls, which included 29 patients with acute lymphoblastic leukemia, 25 patients with brain tumors, 28 patients with neurodegenerative diseases, and 9 patients with hemophagocytic lymphohistiocytosis. Only osteopontin proved to be significantly increased in the CSF of Langerhans cell histiocytosis patients with either neurodegeneration or mass lesions (pituitary), compared with all of the control groups. Analysis of osteopontin expression in these tissues confirmed an upregulation of the SPP1 gene 72.
Several investigators have published studies evaluating the level of various cytokines or growth factors in the blood of patients with Langerhans cell histiocytosis that have included many of the genes found not to be upregulated by the gene expression results discussed above 70. One explanation for elevated levels of these proteins is a systemic inflammatory response, with the cytokines/growth factors being produced by cells outside the LCH lesions. A second possible explanation is that macrophages in the LCH lesions produce the cytokines measured in the blood or are concentrated in lesions.
IL-1 beta and prostaglandin GE2 levels were measured in the saliva of patients with oral LCH lesions or multisystem high-risk patients with and without oral lesions; levels of both were higher in patients with active disease and decreased after successful therapy 73.
Langerhans cell histiocytosis symptoms
Langerhans cell histiocytosis symptoms and signs vary considerably depending on which organs are infiltrated.
Patients are divided into 2 groups based on organ involvement:
- Single system: Single system disease is unifocal or multifocal involvement of one of the following organs: bone, skin, lymph nodes, lungs, central nervous system, or other, rare locations (eg, thyroid, thymus). An example of single system disease is eosinophilic granuloma. Unifocal or multifocal eosinophilic granuloma (60 to 80% of Langerhans cell histiocytosis cases) occurs predominantly in older children and young adults, usually by age 30; incidence peaks between ages 5 and 10 years. Lesions most frequently involve bones, often with pain, the inability to bear weight, or both and with overlying tender (sometimes warm) swelling.
- Multisystem: Multisystem disease is disease in two or more organ systems. Risk organs may or not be affected. An example of multisystem disease without risk organ involvement is Hand-Schüller-Christian disease. An example of multisystem disease with risk organ involvement is Letterer-Siwe disease. In a large series of patients from the Mayo Clinic, 31% had multisystem eosinophilic granuloma (Langerhans cell histiocytosis) compared with 69% registered on the Histiocyte Society adult registry; this likely reflects referral bias 74. In the adult multisystem patients, the sites of disease included the following:
- Bone involvement can cause pain, often in a very specific area where the disease is eroding the bone. The most commonly affected bones are the skull, jaw (particularly the lower jaw), ribs, pelvis, and vertebrae, but any bone can be affected.
- Skin (50%). Skin involvement often presents as single or multiple small red lumps, but the skin rash can also be flat, itchy, and flaky much like eczema or psoriasis. The scalp, ear canals, and vulva are common sites of skin involvement. Some patients develop mouth sores similar to canker sores, but often larger and flatter.
- Mucocutaneous (40%).
- Diabetes insipidus (29.6%). In diabetes insipidus, your body is unable to make the hormone that allows the kidneys to reabsorb water. Conditions that cause the brain to make too little antidiuretic hormone (ADH) or vasopressin or disorders that block the effect of antidiuretic hormone (ADH) or vasopressin cause patients can have extreme thirst or make too much urine.
- Adults typically urinate an average of 1 to 3 quarts (about 1 to 3 liters) a day. People who have diabetes insipidus and who drink a lot of fluids may make as much as 20 quarts (about 19 liters) of urine a day. Symptoms of diabetes insipidus in adults include:
- Being very thirsty, often with a preference for cold water.
- Making large amounts of pale urine.
- Getting up to urinate and drink water often during the night.
- A baby or young child who has diabetes insipidus may have these symptoms:
- Large amounts of pale urine that result in heavy, wet diapers.
- Bed-wetting.
- Being very thirsty, with a preference for drinking water and cold liquids.
- Weight loss.
- Poor growth.
- Vomiting.
- Irritability.
- Fever.
- Constipation.
- Headache.
- Problems sleeping.
- Vision problems.
- Adults typically urinate an average of 1 to 3 quarts (about 1 to 3 liters) a day. People who have diabetes insipidus and who drink a lot of fluids may make as much as 20 quarts (about 19 liters) of urine a day. Symptoms of diabetes insipidus in adults include:
- Hepatosplenomegaly (16%). Liver involvement usually does not cause symptoms, but is detected by blood work or scans. Spleen involvement can be detected on physical exam or by a decrease in blood counts.
- Hypothyroidism (underactive thyroid) (6.6%).
- Hypothyroidism symptoms in adults may include:
- Tiredness.
- More sensitivity to cold.
- Constipation.
- Dry skin.
- Weight gain.
- Puffy face.
- Hoarse voice.
- Coarse hair and skin.
- Muscle weakness.
- Muscle aches, tenderness and stiffness.
- Menstrual cycles that are heavier than usual or irregular.
- Thinning hair.
- Slowed heart rate, also called bradycardia.
- Depression.
- Memory problems.
- Hypothyroidism in children and teens. In general, children and teens with hypothyroidism have symptoms similar to those in adults. But they also may have:
- Poor growth that leads to short stature.
- Delayed development of permanent teeth.
- Delayed puberty.
- Poor mental development.
- Hypothyroidism symptoms in adults may include:
- Lymphadenopathy or swollen lymph nodes (6%). Lymph node involvement is usually found on radiology scans or by feeling painless, rubbery lumps in the neck, under the arms, or in the groin.
- Lung involvement is particularly common in smokers and can cause a chronic cough or shortness of breath.
- Central nervous system (CNS) involvement most commonly affects the pituitary gland, a master gland that controls function of the thyroid, adrenal, and reproductive glands. A common manifestation of CNS is diabetes insipidus.
Involvement of the zygomatic, sphenoid, orbital, ethmoid, or temporal bones denotes a category of CNS (central nervous system) risk lesions that imparts a higher risk of neurodegenerative disease in the skull and front of the face.
Involvement of risk organs implies a worse prognosis. Risk organs include the liver, spleen, and organs of the hematopoietic system.
Patients with single system disease (unifocal, multifocal, and central nervous system (CNS) risk organs) and multisystem disease without risk organ involvement are considered low risk. Patients with multisystem disease and risk organ involvement are considered high risk.
Adult patients may have signs and symptoms of eosinophilic granuloma (Langerhans cell histiocytosis) for many months before receiving a definitive diagnosis and treatment. Eosinophilic granuloma in adults is often similar to that in children and appears to involve the same organs, although the incidence in an organ may be different. There is a predominance of lung disease in adults, usually occurring as single-system disease and closely associated with smoking and some unique biologic characteristics. Most adult isolated lung eosinophilic granuloma cases are polyclonal and possibly reactive, while fewer lung eosinophilic granuloma (Langerhans cell histiocytosis) cases are monoclonal 75.
A German registry with 121 registrants showed that 62% had single-organ involvement and 38% had multisystem involvement, while 34% of the total had lung involvement. The median age at diagnosis was 44 years ± 12.8 years. The most common organ involved was lung, followed by bone and skin. All organ systems found in childhood eosinophilic granuloma (Langerhans cell histiocytosis) were seen, including endocrine and central nervous system, liver, spleen, bone marrow, and gastrointestinal tract. The major difference is the much higher incidence of isolated pulmonary eosinophilic granuloma (Langerhans cell histiocytosis) in adults, particularly in young adults who smoke. Other differences appear to be the more frequent involvement of genital and oral mucosa. There may possibly be a difference in the distribution of bone lesions, but both groups suffer reactivations of bone lesions and progression to diabetes insipidus, although the exact incidence in adults is unknown 76.
Presenting symptoms from published studies are (in order of decreasing frequency):
- dyspnea or tachypnea,
- polydipsia and polyuria,
- bone pain,
- lymphadenopathy,
- weight loss,
- fever,
- gingival hypertrophy,
- ataxia, and
- memory problems.
The signs of eosinophilic granuloma (Langerhans cell histiocytosis) are:
- skin rash,
- scalp nodules,
- soft tissue swelling near bone lesions,
- lymphadenopathy,
- gingival hypertrophy, and
- hepatosplenomegaly.
Patients who present with isolated diabetes insipidus should be carefully observed for the onset of other symptoms or signs characteristic of eosinophilic granuloma (Langerhans cell histiocytosis). At least 80% of patients with diabetes insipidus had involvement of other organ systems, including bone (68%), skin (57%), lung (39%), and lymph nodes (18%) 77. However, isolated diabetes insipidus in adults is similar to that in pediatric patients, with progression from posterior to anterior pituitary/hypothalamus and to cerebellar involvement.
Langerhans cell histiocytosis skin
Langerhans cell histiocytosis (LCH) skin involvement is common, particularly in infants, where it presents as seborrheic eczema (seborrheic dermatitis), and in adults, where it may present as refractory eczema in an intertriginous area is where two skin areas may touch or rub together (e.g., the armpit, groin folds, gluteal cleft, skin folds of the breasts and between digits) and genital areas (Figure 3). Skin-only Langerhans cell histiocytosis occurs but it is less common in adults than in children. The prognosis for skin-only Langerhans cell histiocytosis is excellent, with an approximately 60% chance of regression with topical treatments and 100% probability of 5-year survival 78, 79, 80, 81, 82.
The lesions of congenital self-healing Langerhans cell histiocytosis are often present at or shortly after birth; can appear as eroded or ulcerated papules, pustules, or vesicles with hemorrhagic crusting; and may masquerade as diffuse neonatal hemangiomatosis or blueberry muffin rash (Figure 3) 81, 82. Close monitoring is required in infants, as reactivation or progression to multisystem involvement has been observed in up to 40% of cases 83, 84.
Thirty-seven percent of adults with Langerhans cell histiocytosis have skin involvement, usually as part of multisystem disease. The skin involvement is clinically similar to that seen in children and may take many forms 74. Skin involvement commonly presents as papules and intertrigo, with significant scaling and crusting, most commonly in the scalp, although mucosal involvement of the genitalia or oral cavity is also common 85, 86, 87. Infra-mammary and vulvar involvement may be seen in adult women with skin Langerhans cell histiocytosis.
Many patients have a papular rash with brown, red, or crusted areas ranging from the size of a pinhead to a dime. In the scalp, the rash is similar to that of seborrhea. Skin in the inguinal region, genitalia, or around the anus may have open ulcers that do not heal after antibacterial or antifungal therapy. The lesions are usually asymptomatic but may be pruritic or painful. In the mouth, swollen gums or ulcers along the cheeks, roof of the mouth, or tongue may be signs of eosinophilic granuloma.
In infants, signs or symptoms of Langerhans cell histiocytosis that affects the skin may include:
- Flaking of the scalp that may look like “cradle cap.”
- Flaking in the creases of the body, such as the inner elbow or perineum.
- Raised skin rash with brown or purple areas that occur anywhere on the body.
In children and adults, signs or symptoms of Langerhans cell histiocytosis that affects the skin and nails may include:
- Flaking of the scalp that may look like dandruff.
- Raised skin rash with red, brown, or crusted areas that may be itchy or painful. The rash can occur in the groin area or on the abdomen, back, or chest.
- Bumps or ulcers on the scalp.
- Ulcers behind the ears, under the breasts, or in the groin area.
- Fingernails that fall off or have discolored grooves that run across the nail.
Signs or symptoms of Langerhans cell histiocytosis that affects the mouth may include:
- Swollen gums.
- Sores on the roof of the mouth, inside the cheeks, or on the tongue or lips.
- Teeth that become uneven or fall out.
Diagnosis of Langerhans cell histiocytosis is usually made by skin biopsy performed for persistent skin lesions 74.
Pulmonary Langerhans cell histiocytosis (PLCH)
In children, lung involvement usually occurs in the context of multisystem disease, where it has been reported to occur in up to 35% of patients 88. Radiographic findings are typical for the presence of a reticulonodular pattern with bullae formation (Figure 6). In the absence of other risk organ involvement, pulmonary Langerhans cell histiocytosis (PLCH) is not a predictor of adverse outcome 89, 90, 91. Isolated pulmonary Langerhans cell histiocytosis is a rare presentation that is almost exclusive of adults with a smoking habit 92.
Isolated pulmonary Langerhans cell histiocytosis (PLCH) is primarily a disease of young adult smokers, with more than 90% of patients endorsing a smoking history 41, 93, 94. Pulmonary Langerhans cell histiocytosis (PLCH) presents with respiratory symptoms, mainly cough and shortness of breath on exertion, in approximately two-thirds of the cases. Less frequently, patients may present with spontaneous pneumothorax or with asymptomatic lesions on routine chest X-ray 95. Extrapulmonary organ involvement occurs in 10% to 15% of patients 93. High-resolution CT shows a pattern of bilateral reticulonodular and cystic changes, with apical and midlung predominance, sparing the bases and costophrenic angles 95. Transbronchial lung biopsies may be diagnostic of pulmonary Langerhans cell histiocytosis (PLCH) in expert centers; however, because of the focal nature of the disease, the diagnostic yield varies between 15% and 40%, and a thoracoscopic lung biopsy is usually recommended 95.
Signs or symptoms of pulmonary Langerhans cell histiocytosis may include:
- Collapsed lung. This condition can cause chest pain or tightness, trouble breathing, feeling tired, and a bluish color to the skin.
- Trouble breathing, especially in adults who smoke.
- Dry cough.
- Chest pain.
Pulmonary Langerhans cell histiocytosis (PLCH) can be diagnosed by bronchoscopy in about 50% of adult patients, as defined by characteristic CD1a immunostaining cells of at least 5% of cells observed 96. High-resolution lung computed tomography (CT) shows characteristic changes with cysts and nodules, more prevalent at the mid and upper zones. These changes have been characterized as pathognomonic for Pulmonary Langerhans cell histiocytosis 97.
Pathology of pulmonary Langerhans cell histiocytosis (PLCH) shows nodular lesions with the typical histology. In late disease, nodules are replaced by advanced bullous and cystic lesions, often in association with hyperinflation and honeycombing 98. BRAFV600E and MAP2K1 mutations have been reported at similar frequency as in extrapulmonary LCH, although lower mutation rates are identified in the more fibrotic lesions 99, 100.
The Langerhans cells in adult lung lesions were shown to be mature dendritic cells expressing high levels of the accessory molecules CD80 and CD86, unlike Langerhans cells found in other lung disorders 101. Pulmonary Langerhans cell histiocytosis in adults has been considered a primarily reactive process, rather than a clonal proliferation as seen in childhood Langerhans cell histiocytosis 75. However, ERK pathway mutations have been demonstrated in up to two-thirds of pulmonary Langerhans cell histiocytosis lesions in adults, suggesting a clonal process in a significant proportion of patients 102.
The course of pulmonary Langerhans cell histiocytosis in adults is variable and unpredictable 103.
Favorable prognostic factors for adult pulmonary Langerhans cell histiocytosis include the following:
- Minimal symptoms. Adults with pulmonary Langerhans cell histiocytosis who have minimal symptoms have a good prognosis, although some have steady deterioration over many years 104.
- Smoking cessation or treatment. Fifty-nine percent of patients do well with either spontaneous remission with cessation of smoking, or with some form of therapy 104. However, one study reported that smoking cessation did not increase the longevity of adults with pulmonary Langerhans cell histiocytosis, apparently because the tempo of disease is so variable 105.
- Lung transplantation. Patients receiving lung transplantation for treatment of pulmonary Langerhans cell histiocytosis have a 77% survival rate at 1 year and a 54% survival rate at 10 years, with a 20% chance of Langerhans cell histiocytosis recurrence 106.
Unfavorable prognostic factors for adult pulmonary Langerhans cell histiocytosis include the following:
- Altered pulmonary function. Lower forced expiratory volume/forced vital capacity (FEV1/FVC) ratio and higher residual volume/total lung capacity (RV/TLC) ratio are adverse prognostic variables 105. About 10% to 20% of patients have early severe progression to respiratory failure, severe pulmonary hypertension, and cor pulmonale. Adults who have progression with diffuse bullae formation, multiple pneumothoraces, and fibrosis have a poor prognosis 107.
- Age. Age older than 26 years is an adverse prognostic variable 105.
The remaining patients have a variable course, with stable disease in some patients and relapses and progression of respiratory dysfunction in others, some after many years 108. A natural history study of 58 Langerhans cell histiocytosis patients with pulmonary involvement found that 38% of patients had deterioration of lung function after 2 years 109. The most significant adverse prognostic variables were positive smoking statuses and low PaO2 levels at the time of inclusion.
The following results may be noted on diagnostic tests:
- Pulmonary function testing. The most frequent pulmonary function abnormality finding in patients with pulmonary Langerhans cell histiocytosis is a reduced carbon monoxide diffusing capacity in 70% to 90% of cases 105.
- CT scan. A high-resolution CT scan, which reveals a reticulonodular pattern classically with cysts and nodules, usually in the upper lobes and sparing the costophrenic angle, is characteristic of Langerhans cell histiocytosis 110. The presence of cystic abnormalities on high-resolution CT scans appears to be a poor predictor of which patients will have progressive disease 111.
- Biopsy. Despite the typical CT findings, most pulmonologists agree that a lung biopsy is needed to confirm the diagnosis. A study that correlated lung CT findings and lung biopsy results in 27 patients with pulmonary Langerhans cell histiocytosis observed that thin-walled and bizarre cysts had active LCs and eosinophils 112.
Langerhans cell histiocytosis of the central nervous system
Central nervous system (CNS) involvement in Langerhans cell histiocytosis (LCH-CNS) represents a spectrum of diseases ranging from active infiltration by Langerhans cell histiocytosis to long-term effects. Its prevalence has been noted to range from 3.4% to 57% 113. LCH-CNS can be divided in focal mass lesions and lesions associated with progressive neurodegeneration 113, 114.
Mass lesions tylically present in meninges, choroid plexus, and brain parenchyma. Characteristic neuroimaging findings include hypothalamic-pituitary involvement, often with diabetes insipidus, infundibular thickening, and absent bright spot in posterior pituitary; enlargement and enhancement of the pineal gland; thickening and enhancement of choroid plexus; or intraparenchymal masses.61 Among patients with anterior pituitary dysfunction, the most common deficiency is in antidiuretic hormone, followed by growth hormone (which occurs in up to 50% of patients with diabetes insipidus), gonadotropin, and thyrotropin 115, 116. Anterior pituitary dysfunction is more common in childhood-onset patients and in those with multisystem disease 115, 117. Diabetes insipidus, the hallmark of this dysfunction, has been reported to occur in up to 24% of patients with Langerhans cell histiocytosis, but in half of patients with multisystem disease 117, 118; in one third of cases, the diabetes insipidus precedes or is concurrent with the diagnosis of Langerhans cell histiocytosis, and in the remaining two-thirds of the cases, it is diagnosed later. 117, 88, 119. With the use of more comprehensive risk-adapted management of Langerhans cell histiocytosis, the incidence of endocrinopathies has decreased to 10% to 15% in recent large cohort studies 120, 121.
Signs or symptoms of Langerhans cell histiocytosis that affects the central nervous system (brain and spinal cord) may include:
- Loss of balance, uncoordinated body movements, and trouble walking.
- Trouble speaking.
- Trouble seeing.
- Headaches.
- Changes in behavior or personality.
- Memory problems.
These signs and symptoms may be caused by lesions in the central nervous system (CNS) or by neurodegenerative Langerhans cell histiocytosis (LCH-ND). Neurodegenerative Langerhans cell histiocytosis (LCH-ND) is characterized by progressive radiologic and clinical abnormalities. As recently reviewed by Yeh et al 113, two separate clinical forms are identified: LCH-associated abnormal CNS imaging (LACI), which includes asymptomatic patients with radiologic findings, and LCH-associated abnormal CNS symptoms (LACS), which describes patients with abnormal cognitive and psychological findings. LACI and LACS are associated with increased T2-weighted MRI signal in the dentate nucleus of the cerebellum, basal ganglia, and pons (Figure 6).
LCH-associated abnormal CNS symptoms (LACS) is a neurodegenerative syndrome of variable severity and course. The incidence of long-term neurodegeneration has been estimated to be between 1.9% and 11% and it seems to be higher in patients with multisystem disease, diabetes insipidus, history of involvement of bones of the skull base and orbit or BRAFV600E-mutated LCH 117, 122, 118, 123. Of particular therapeutic relevance are the skull-based lesions (CNS-risk lesions), as this risk association has been considered an indication for the use of systemic therapy, rather than local control measures only.
The appearance of clinical and radiographic signs of neurodegenerative Langerhans cell histiocytosis (LCH-ND) can occur with the initial LCH diagnosis, although it commonly occurs years later 113, 114, 124. Symptoms may initially include tremors, abnormal reflexes, gait disturbance, motor spasticity, ataxia, dysarthria, dysphagia, behavioral changes, learning disorder, or psychiatric problems. Some patients develop a progressive cerebellar syndrome, with spastic tetraparesis, pseudobulbar palsy, and cognitive deterioration 113, 114. Magnetic resonance imaging (MRI) shows a characteristic infratentorial predilection, with symmetric abnormalities of the dentate nuclei and of the white matter of the cerebellum and pons (Figure 6). Outside the infratentorial compartment, abnormalities of the basal ganglia, optic nerves, and tracts; dilatation of the Virchow-Robin spaces; or diffuse abnormalities of the hemispheric white matter consistent with leukoencephalopathy are also common 113, 114. Serial imaging and neurocognitive evaluations are recommended when the disease is suspected 113, 125.
Whether central nervous system (brain and spinal cord) involvement with degeneration represents active disease or a radiologic scar remains undefined. Until recently, the only histologic study of LCH-associated abnormal CNS symptoms (LACS) reported absence of CD1a+ histiocytes, an inflammatory collection of CD8+ lymphocytes with neuronal and axonal degeneration, and extensive myelin loss, supporting the view of a late consequence of an inflammatory phenomenon 126. However, a recent study supports hematopoietic origin of myeloid cells that share precursors with LCH lesion CD207+ cells 54. Clinical and radiological responses to BRAF inhibitors further support this view 127.
Langerhans cell histiocytosis of bones
The skull, including the skull base, is very commonly involved; typical locations include the bones of the orbit or the temporal bone (typically the mastoid). Involvement of the vertebral bodies is also common, and the presence of a vertebra plana is frequent. Pain and tumor formation in a localized area of bone is a very common presentation of Langerhans cell histiocytosis.
The relative frequency of bone involvement in adults differs from that in children; the frequency of mandible involvement is 30% in adults and 7% in children, and the frequency of skull involvement is 21% in adults and 40% in children 76. The frequency of vertebrae (13%), pelvis (13%), extremities (17%), and rib (6%) lesions in adults are similar to those found in children 76.
Signs or symptoms of Langerhans cell histiocytosis that affects the bone may include:
- Swelling or a lump over a bone, such as the skull, jawbone, ribs, pelvis, spine, thigh bone, upper arm bone, elbow, eye socket, or bones around the ear.
- Pain where there is swelling or a lump over a bone.
Children with eosinophilic granuloma (Langerhans cell histiocytosis) lesions in bones around the ears or eyes have a high risk of diabetes insipidus and other central nervous system diseases.
Signs or symptoms of Langerhans cell histiocytosis that affects the bone marrow may include:
- Easy bruising or bleeding.
- Fever.
- Frequent infections.
Radio-isotope imaging is recommended to assess the number of bone lesions; fluorodeoxyglucose–positron emission tomography (PET) scans can be useful in defining the extent of the disease and the response to therapy 128, 129.
Langerhans cell histiocytosis of liver
Liver involvement was reported in 27% of adult patients with Langerhans cell histiocytosis and multiorgan disease 130. Hepatomegaly (48%) and liver enzyme abnormalities (61%) were present. CT and ultrasound imaging abnormalities are often found.
Signs or symptoms of Langerhans cell histiocytosis that affects the liver or spleen may include:
- Swelling in the abdomen caused by a buildup of extra fluid.
- Trouble breathing.
- Yellowing of the skin and whites of the eyes (jaundice).
- Itching.
- Easy bruising or bleeding.
- Feeling very tired.
- Diarrhea.
- Bloody stools.
The early histopathologic stage of liver Langerhans cell histiocytosis includes infiltration of CD1a-positive cells and periductal fibrosis with inflammatory infiltrates with or without steatosis. The late stage is biliary tree sclerosis; treatment with ursodeoxycholic acid is suggested 130.
Langerhans cell histiocytosis of lymph nodes and thymus
Signs or symptoms of Langerhans cell histiocytosis that affects the lymph nodes or thymus may include:
- Swollen lymph nodes.
- Cough, trouble breathing, or fast breathing.
- Superior vena cava syndrome. This can cause coughing, trouble breathing, and swelling of the face, neck, and upper arms.
Langerhans cell histiocytosis of endocrine system
Signs or symptoms of Langerhans cell histiocytosis that affects the pituitary gland may include:
- Diabetes insipidus. This can cause a strong thirst and frequent urination.
- Slow growth.
- Early or late puberty.
- Being very overweight.
Signs or symptoms of Langerhans cell histiocytosis that affects the thyroid may include:
- Swollen thyroid gland.
- Hypothyroidism. This can cause tiredness, lack of energy, being sensitive to cold, constipation, dry skin, thinning hair, memory problems, trouble concentrating, and depression. In infants, this can also cause a loss of appetite and choking on food. In children and adolescents, this can cause behavior problems, weight gain, slow growth, and late puberty.
- Trouble breathing.
Langerhans cell histiocytosis of eye
Signs or symptoms of Langerhans cell histiocytosis that affects the eye may include:
- Vision problems or blindness.
Langerhans cell histiocytosis diagnosis
In addition to asking about your health history and doing a physical exam, your doctor may perform the following tests and procedures to diagnose Langerhans cell histiocytosis:
- Complete blood count (CBC) with differential: A procedure in which a sample of blood is drawn and checked for the following:
- The amount of hemoglobin (the protein that carries oxygen) in the red blood cells.
- The portion of the blood sample made up of red blood cells.
- The number and type of white blood cells.
- The number of red blood cells and platelets.
- Blood chemistry studies: A procedure in which a blood sample is checked to measure the amounts of certain substances released into the body by organs and tissues in the body. An unusual (higher or lower than normal) amount of a substance can be a sign of disease.
- Liver function test: A blood test to measure the blood levels of certain substances released by the liver. A high or low level of these substances can be a sign of disease in the liver.
- BRAF gene testing: A laboratory test in which a sample of blood or tissue is tested for certain mutations in the BRAF gene.
- Urinalysis: A test to check the color of urine and its contents, such as sugar, protein, red blood cells, and white blood cells.
- Water deprivation test: A test to check how much urine is made and whether it becomes concentrated when little or no water is given. This test is used to diagnose diabetes insipidus, which may be caused by eosinophilic granuloma (Langerhans cell histiocytosis).
- Bone marrow aspiration and biopsy: The removal of bone marrow and a small piece of bone by inserting a hollow needle into the hipbone. A pathologist views the bone marrow and bone under a microscope to look for signs of eosinophilic granuloma (Langerhans cell histiocytosis). The following test may be done on the tissue that was removed:
- Immunohistochemistry: A laboratory test that uses antibodies to check for certain antigens (markers) in a sample of a patient’s tissue. The antibodies are usually linked to an enzyme or a fluorescent dye. After the antibodies bind to a specific antigen in the tissue sample, the enzyme or dye is activated, and the antigen can then be seen under a microscope. This type of test is used to help diagnose cancer and to help tell one type of cancer from another type of cancer.
Tissue biopsy for histological diagnosis is necessary to confirm the diagnosis 35. CT-guided biopsy for eosinophilic granuloma has been effective for histological diagnosis, with low morbidity and a diagnostic accuracy of 70–100% 131. Although anecdotally excellent results with biopsy alone have been previously reported for patients with eosinophilic granulomas 132, biopsy should not be considered as a strategy for treatment of these patients but rather as a step to confirm diagnosis 131.
Diagnosis of Langerhans cell histiocytosis (LCH) requires a clonal neoplastic proliferation with expression of CD1a, CD207 (Langerin), and S100 (Figure 1) 18, 35. LCH cells are generally large, round to oval in shape, with a coffee-bean nuclear grove, and without the branching that characterizes inflammatory CD1a+ dendritic cells. On electron microscopy, pentalaminar cytoplasmic rod-shaped inclusions also known as Birbeck granules can be identified, although electron microscopy is no longer required for diagnosis in the presence of CD207+ staining. Because LCH cells activate and recruit other immunologic cells, microscopic examination shows an inflammatory pattern consisting of eosinophils, neutrophils, lymphocytes, and macrophages in addition to the Langerhans cells; this appearance is what is described as eosinophilic granuloma 133.
When the diagnosis is confirmed, workup for systemic involvement should include a skeletal survey, abdominal ultrasound, complete blood count (with bone marrow biopsy if indication of bone marrow involvement), and evaluation for diabetes insipidus 53, 134, 113.
CT scanning or MRI
Both CT scan and MRI are invaluable for assessing the hypothalamic-pituitary area. Fluorodeoxyglucose (FDG) PET scanning also is being used to assess patients with LCH. The technique is far more sensitive than bone scan for early detection of disease in the spleen, lymph nodes, and lung. In fact, FDG PET scan is now also being routinely used to monitor disease during treatment.
Other Tests
- Pulmonary function testing may help identify asymptomatic patients with lung involvement
- Small bowel series is recommended in patients with failure to thrive, diarrhea, and malabsorption
- Neurological and visual testing
- Auditory testing
- Recent studies suggest that CSF fluid can be used to detect biomarkers like glial fibrillary acidic protein to evaluate the onset of disease and response to therapy.
- Skin biopsy can establish the diagnosis 135
Langerhans cell histiocytosis treatment
Langerhans cell histiocytosis treatment varies greatly depending on the involved organs. If the disease is isolated, observation alone may be appropriate. Surgical removal of an isolated area is also a treatment option. Isolated skin lesions may resolve on their own, especially if they present in infancy (self-healing Langerhans cell histiocytosis), or may be treated with topical steroids, oral methotrexate, or thalidomide. Chemotherapy and radiation may be used for more systemic involved cases. There are several different chemotherapy protocols that have been used; currently, protocols are using prednisone and vinblastine, though other regimen options include vincristine, cytosine arabinoside, prednisone, cladribine, or pamidronate 136.
Late complications are common; therefore, follow up with oncology is crucial. Complications include diabetes insipidus, growth failure, delayed puberty, tooth loss, mandibular bone loss, hearing loss, secondary cancers, neurologic/cerebellar effects, liver disease, and pulmonary fibrosis.
Bone treatment
Various treatment options have been reported for Langerhans cell histiocytosis of bone, including observation and immobilization, indomethacin administration, methylprednisolone injections, radiofrequency ablation, local excision and curettage with or without bone grafting, chemotherapy and irradiation; results have been reported as satisfactory with a recurrence rate of less than 20 % 137. In general, the treatment of typical solitary lesions in asymptomatic patients is conservative 138. In patients with mild neurological deficits from solitary eosinophilic granulomas of the spine, immobilization and radiation therapy has been reported 139. Low-dose radiation therapy is advocated by some authors to be effective in the healing of lytic lesions and limiting disease progression 140; others argue that radiation therapy may damage endochondral growth plates and limit bone healing and reconstitution 141, or lead to secondary radiation-induced morbidity such as post-radiation sarcomas and myelitis 142. Although no clear correlation between the degree of vertebral collapse and the degree of neurological symptoms has been observed 143, in patients with severe pain and restriction of range of motion, and/or persistent spinal subluxation and neurological symptoms, surgical treatment is required 144. Chemotherapy is not recommended for solitary eosinophilic granuloma, and should be reserved for systemic involvement 145, or as initial therapy in children with solitary lesions in locations that preclude safe and complete surgical resection 146.
Since eosinophilic granuloma in children is known to resolve spontaneously with time, observation alone or biopsy alone to confirm the diagnosis have also been recommended as a treatment strategy 132. A previous study reported spontaneous resolution without recurrence of the lesions in six skeletally immature patients that had biopsy followed by observation alone (open biopsy in three and percutaneous in three), suggesting the intriguing possibility that surgery may result in a higher rate of recurrence than less aggressive procedures 132. However, patients, especially children with symptomatic bone lesions should not be left alone to let the disease take its natural course without a histological diagnosis. Moreover, although solitary eosinophilic granuloma is considered a benign lesion, without treatment, the time required for resolution is unpredictable and can be associated with significant morbidity secondary to unremitting pain, restricted activity, growth disturbance, or pathological fracture 147. Therefore, some experts recommend that these patients should undergo biopsy for histological diagnosis, and treatment is then considered 131.
Lung treatment
It is difficult to judge the effectiveness of various treatments for pulmonary Langerhans cell histiocytosis because patients can recover spontaneously or have stable disease without treatment.
Treatment options for adult patients with pulmonary Langerhans cell histiocytosis include the following:
- Smoking cessation. Smoking cessation is mandatory because of the apparent causal effect of smoking in pulmonary eosinophilic granuloma (Langerhans cell histiocytosis) 148. Most adult patients with eosinophilic granuloma (Langerhans cell histiocytosis) have gradual disease progression with continued smoking. The disease may regress or progress with the cessation of smoking 149. A study of 27 patients with pulmonary eosinophilic granuloma (Langerhans cell histiocytosis) observed that 52% of patients improved after a mean follow-up period of 14 months; most patients improved with smoking cessation, and some patients improved with steroid treatment. Four patients (15%) had stable disease at a mean follow-up of 26 months, and nine patients (33%) demonstrated disease progression during the mean follow-up of 22 months 150.
- Steroid therapy. It is not known whether steroid therapy is efficacious in the treatment of adult pulmonary eosinophilic granuloma (Langerhans cell histiocytosis) because reported case series did not control for smoking cessation 148.
- Chemotherapy. Some patients have been reported to respond to cladribine therapy 151.
- Lung transplantation. Lung transplantation may be necessary for adults with extensive pulmonary destruction from eosinophilic granuloma (Langerhans cell histiocytosis) 152. This multicenter study reported 54% survival at 10 years posttransplant, with 20% of patients having recurrent eosinophilic granuloma (Langerhans cell histiocytosis) that did not impact survival; longer follow-up of these patients is needed 152. Another study confirmed an approximate 50% survival at 10 years and improved hemodynamic changes associated with pulmonary arterial hypertension, but did not alter pulmonary function testing or incidence of pulmonary edema 104.
The best strategy for follow-up of pulmonary eosinophilic granuloma includes physical examination, chest radiographs, lung function tests, and high-resolution computed tomography (CT) scans 75.
Skin treatment
Treatment options for adult patients with single-system skin disease include the following:
- Surgical excision. Localized lesions can be treated by surgical excision, but as with bone, mutilating surgery, including hemivulvectomy, should be avoided unless the disease is refractory to all available therapy.
- Topical therapy. Topical therapies are described in greater detail in the childhood isolated skin involvement section of this summary and include the following:
- Systemic therapy. Systemic therapy for severe skin eosinophilic granuloma includes oral methotrexate, hydroxyurea, oral thalidomide, oral interferon-alpha, or combinations of interferon and thalidomide 108. Interferon and thalidomide are also used to treat chronic adult skin eosinophilic granuloma 109. Recurrences may occur after treatment is stopped but may respond to re-treatment. Oral isotretinoin has induced remission in some refractory cases of skin eosinophilic granuloma in adults 153. Chemotherapy is generally used for skin eosinophilic granuloma associated with multisystem disease in adults.
Chemotherapy for the treatment of other single-system disease and multisystem disease
Evidence (chemotherapy for the treatment of other single-system disease [not mentioned above] and multisystem disease):
- A single-center, retrospective review of 58 adult patients with eosinophilic granuloma (Langerhans cell histiocytosis) reported on the efficacy and toxicities of treatment with vinblastine/prednisone, cladribine, and cytarabine 110.
- Patients treated with vinblastine/prednisone had the worst outcome, with 84% not responding within 6 weeks or relapsing within a year.
- The no-response/relapse rate was 59% for cladribine and 21% for cytarabine.
- Grade 3 or 4 neurologic toxic effects occurred in 75% of patients treated with vinblastine.
- Grade 3 or 4 neutropenia occurred in 37% of patients treated with cladribine and in 20% of patients receiving cytarabine.
- A report on the treatment of adult patients with either vindesine and prednisone or cyclophosphamide, etoposide, vindesine, and prednisone showed that more than 70% of patients relapsed with either regimen 111.
- Etoposide has been used with some success in single-system and multisystem eosinophilic granuloma (Langerhans cell histiocytosis).
- Minimal toxicity was reported with the use of prolonged oral etoposide in adults with skin eosinophilic granuloma (Langerhans cell histiocytosis), while 3-day courses of intravenous etoposide (100 mg/m²/day) induced complete remission in a small number of patients with resistant single-system and multisystem disease 112.
- Another study at the same center found that azathioprine was the most successful drug for localized disease in adults, with the addition of etoposide for refractory and multisystem disease 154.
- For patients who do not respond to front-line therapy with etoposide, cladribine is effective for adults with skin, bone, lymph node, and probably pulmonary and central nervous system (CNS) disease 155.
- The first study that used cladribine to treat refractory and recurrent skin eosinophilic granuloma (Langerhans cell histiocytosis) disease reported on three patients (aged 33, 51, and 57 years) who received two to four courses of cladribine at 0.7 mg/kg intravenously over 2 hours/day for 5 days 156.
- In a series of five adults (one untreated and four with refractory eosinophilic granuloma (Langerhans cell histiocytosis) treated with cladribine at the same dose noted directly above), three patients achieved a complete remission and two patients achieved a partial remission 155.
- An adult lymphoma treatment regimen of methotrexate, doxorubicin, cyclophosphamide, vincristine, prednisone and bleomycin (MACOP-B) was used in three patients with multisystem eosinophilic granuloma (Langerhans cell histiocytosis) and four with single-system multifocal bone eosinophilic granuloma (Langerhans cell histiocytosis) from 1995 to 2007. Total duration of therapy was 12 weeks 157.
- Response was seen in all patients, two with partial response and five with complete response.
- Three recurrences were seen after therapy was stopped.
- Despite the small number of patients and the retrospective nature of the study, MACOP-B may be useful as salvage therapy in adult patients with eosinophilic granuloma (Langerhans cell histiocytosis) and deserves further study 158.
- Neurodegenerative CNS (central nervous system) disease. A case report suggests some benefit to treating neurodegenerative CNS Langerhans cell histiocytosis disease with infliximab, a tumor necrosis factor (TNF)-alpha inhibitor 159. However, the TNF inhibitors infliximab and etanercept have limited ability to cross the blood-brain barrier. Thalidomide, which also has anti-TNF activity, has been effective in adults with skin and bone eosinophilic granuloma (Langerhans cell histiocytosis) 160. One study reported an improvement in ataxia in a patient with eosinophilic granuloma (Langerhans cell histiocytosis) who was treated with vemurafenib 161.
- Pituitary Langerhans cell histiocytosis. A report of stereotactic radiosurgery for the treatment of pituitary eosinophilic granuloma (Langerhans cell histiocytosis) in adults showed efficacy in reducing the masses 162. However, radiation therapy is not considered the standard of care for children with pituitary involvement. Systemic chemotherapy with cytarabine and cladribine have been the preferred treatments 163.
Targeted therapies for the treatment of single-system and multisystem disease
Early reports on the use of targeted therapies for Langerhans cell histiocytosis patients with low-risk or high-risk Langerhans cell histiocytosis sites include the following:
- Tyrosine kinase inhibitors. Imatinib mesylate was effective in the treatment of four adult patients with eosinophilic granuloma (Langerhans cell histiocytosis) who had skin, lung, bone, and/or CNS involvement 164. Another adult patient with eosinophilic granuloma (Langerhans cell histiocytosis) did not respond to imatinib mesylate 165.
- MAP2K/ERK pathway inhibitors. The finding that most patients with eosinophilic granuloma (Langerhans cell histiocytosis) have BRAF and other RAS pathway mutations led to several reports of good responses to vemurafenib, a BRAF V600E inhibitor, in adult patients with eosinophilic granuloma (Langerhans cell histiocytosis), Erdheim-Chester (ECD) disease, or mixed ECD/eosinophilic granuloma (Langerhans cell histiocytosis), as well as in severe cutaneous eosinophilic granuloma (Langerhans cell histiocytosis) 166. Of four patients with eosinophilic granuloma (Langerhans cell histiocytosis) who were treated with vemurafenib on the VE-BASKET (NCT01524978) trial, one patient had a complete response and three patients had partial responses 161. Early results of targeted inhibitor therapy are encouraging, but many questions remain, particularly the optimal duration of therapy and the reactivation rate after therapy is discontinued. A BRAF inhibitor in combination with a MEK inhibitor have been shown to be effective in patients with melanoma who have BRAF mutations (with reduced toxicity), and this combination may be effective in patients with eosinophilic granuloma (Langerhans cell histiocytosis) 167. A number of clinical trials of BRAF and other RAS pathway inhibitors in adults and children with eosinophilic granuloma (Langerhans cell histiocytosis) are ongoing.
Follow-up tests
Langerhans cell histiocytosis patients should be monitored for many years because of the risk of reactivation (recurrence). Some of the tests that were done to diagnose Langerhans cell histiocytosis may be repeated. This is to see how well the treatment is working and if there are any new lesions. These tests may include:
- Physical exam.
- Neurological exam.
- Ultrasound exam.
- MRI.
- CT scan.
- PET scan.
Other tests that may be needed include:
- Brain stem auditory evoked response (BAER) test: A test that measures the brain’s response to clicking sounds or certain tones to detect some types of hearing loss.
- Pulmonary function test (PFT): A test to see how well the lungs are working. It measures how much air the lungs can hold and how quickly air moves into and out of the lungs. It also measures how much oxygen is used and how much carbon dioxide is given off during breathing. This is also called a lung function test.
- Chest x-ray: An x-ray of the organs and bones inside the chest. An x-ray is a type of energy beam that can go through the body and onto film, making a picture of areas inside the body.
The results of these tests can show if your condition has changed or if the cancer has recurred (come back). These tests are sometimes called follow-up tests or check-ups. Decisions about whether to continue, change, or stop treatment may be based on the results of these tests.
Follow-up of children with Langerhans cell histiocytosis
Because of the risk of reactivation (which ranges from 10% in single-system unifocal bone lesions to close to 50% in low-risk and high-risk multisystem Langerhans cell histiocytosis) and the risk of permanent long-term effects, Langerhans cell histiocytosis patients need to be monitored for many years.
Patients with diabetes insipidus and/or skull lesions in the orbit, mastoid, or temporal bones appear to be at higher risk of Langerhans cell histiocytosis CNS involvement and Langerhans cell histiocytosis CNS neurodegenerative syndrome. These patients should have MRI scans with gadolinium contrast at the time of Langerhans cell histiocytosis diagnosis and every 1 to 2 years thereafter for 10 years to detect evidence of CNS disease 168. The Histiocyte Society CNS Langerhans cell histiocytosis Committee does not recommend any treatment for radiologic CNS Langerhans cell histiocytosis of the neurodegenerative type if there is no associated clinical neurodegeneration and the MRI findings remain stable. However, careful neurological examinations and appropriate imaging with MRI are suggested at regular intervals 113.
Auditory brain stem response tests should be done at regular intervals to define the onset of clinical CNS Langerhans cell histiocytosis as early as possible, as this may affect response to therapy 169. When clinical signs are present, intervention may be indicated in patients with radiologic evidence of Langerhans cell histiocytosis-associated changes in the cerebellum. Available studies of different forms of therapy for CNS neurodegeneration suggest that the neurodegenerative changes may be stabilized or improved, but only if therapy is started early 169. Careful follow-up of patients at risk is critical.
For children with Langerhans cell histiocytosis in the lung, pulmonary function testing and chest CT scans are sensitive methods for detecting disease progression 170.
A 16-year follow-up study of patients from one institution suggested that children with Langerhans cell histiocytosis have an increased risk of developing adult smoker’s lung Langerhans cell histiocytosis compared with normal young adults who smoke. Ongoing re-education regarding this risk should be part of the routine follow-up of children with Langerhans cell histiocytosis at any site 170.
In summary, many patients with multisystem disease will experience long-term complications caused by their underlying disease and/or treatment. Endocrine and CNS complications are the most common. These long-term complications significantly affect health related quality of life in many of these patients 171.
Langerhans cell histiocytosis prognosis
The prognosis (chance of recovery) for people with Langerhans cell histiocytosis depends on whether the initial presentation was a “low-risk disease” (isolated to the skin, lymph nodes, or pituitary gland) or “high-risk disease” (involving the spleen, liver, bone marrow, lung, or skeleton). In general, patients who are young and those in which the disease is present in many parts of the body and organ dysfunction tend to have a poorer prognosis. Newborns who present only with skin lesions tend to do well. Therefore, the age at presentation is only important when multiple organs are affected.
The prognosis and treatment options depend on the following:
- Which organs or body systems are affected by eosinophilic granuloma (Langerhans cell histiocytosis).
- How many organs or body systems the eosinophilic granuloma (Langerhans cell histiocytosis) affects.
- Whether eosinophilic granuloma (Langerhans cell histiocytosis) is found in the liver, spleen, bone marrow, or certain bones in the skull.
- How quickly eosinophilic granuloma (Langerhans cell histiocytosis) responds to initial treatment.
- Whether there are certain mutations in the BRAF gene.
- Whether eosinophilic granuloma (Langerhans cell histiocytosis) has just been diagnosed or has come back (recurred).
High-risk patients like multisystem involvement, skeletally mature patients, more than one bone involvement, and skull-base bone involvement (sphenoid, ethmoid, orbital, and temporal bones) have a high risk for recurrence, complications, and subsequent worse prognosis. Operative treatment offers more prompt pain relief when compared to non-operative treatment 172. Over 10% of patients die of this disease, including a few cases with reactivations and long term morbidity 173.
The presence of hematopoietic dysfunction in the form of cytopenias is a poor prognostic sign. It occurs in the context of multisystem involvement, usually in very young children. Its pathophysiology is multifactorial, including direct involvement of the bone marrow as well as peripheral destruction resulting from hypersplenism from Langerhans cell infiltrates in the spleen 174.
Liver involvement also carries a very poor prognosis. Patients present with hypoalbuminemia, hepatomegaly, or conjugated hyperbilirubinemia. A well-described but rare complication in young children and in adults is the development of sclerosing cholangitis and hepatic fibrosis, which commonly evolve to end-stage liver failure 175, 176, 177.
In infants up to 1 year of age, Langerhans cell histiocytosis may go away without treatment. Children with Langerhans cell histiocytosis in high-risk organs and the gastrointestinal tract have a greater risk of not responding to treatment than patients with high-risk Langerhans cell histiocytosis and no disease in the gastrointestinal tract. High-risk Langerhans cell histiocytosis is usually seen in children younger than 2 years. Mortality may be low for isolated lesions (< 5%) and as high as 50% for the more widespread disease 178. Those with disseminated disease will die within 24-48 months 178. Those with localized disease do have a better prognosis but the quality of life is reduced.
Individuals who have liver, spleen, lung, or bone marrow involvement usually have a worse prognosis. In a study looking at patients from several centers, it was shown that the best prognostic indicator was the patient’s response to chemotherapy during the first six weeks of therapy. Therefore, it has been recommended by some that individuals who do not respond positively within the first six weeks of treatment should be treated more aggressively.
The prognosis for pulmonary eosinophilic granuloma varies and is related to smoking cessation. Those who continue to smoke experience disease progression, but those who quit disease stabilizes or regresses. Extreme age, large cysts, and honeycombing radiological, multiorgan involvement, and prolonged corticosteroid therapy have a worse prognosis.
Eosinophilic granuloma (Langerhans cell histiocytosis) in the skin, bones, lymph nodes or pituitary gland usually gets better with treatment and is called “low-risk.” Some patients have involvement in the spleen, liver and bone marrow. This is called “high-risk disease” and may be more difficult to treat. Some patients may develop long-term side effects such as diabetes insipidus, stunted growth, loss of teeth, bone defects, hearing loss, or neurologic problems; while other patients remain without side effects. In a few cases, the disease can be life-threatening.
Patients with eosinophilic granuloma (Langerhans cell histiocytosis) should usually have long term follow-up care to detect late complications of the disease or treatment. These may include problems of skeletal deformity or function, liver or lung problems, hormone abnormalities, dental issues or neurological and neurocognitive dysfunction.
Langerhans cell histiocytosis in adults
The natural history of adult Langerhans cell histiocytosis (LCH), with the exception of pulmonary Langerhans cell histiocytosis, is unknown 53. It is unclear whether there are significant differences from childhood Langerhans cell histiocytosis, although it appears that multisystem high-risk Langerhans cell histiocytosis in adults is less aggressive than childhood high-risk disease 53. The risk of reactivations is unknown but may be higher than in children with Langerhans cell histiocytosis. A reactivation rate of 62.5% has been reported in adults, compared with 36.8% in children 179. Sixty-four percent of adults with diabetes insipidus monitored for an average of 6 years developed other endocrine problems 180, 181.
Adult patients with Langerhans cell histiocytosis have higher rates of cancers than do age-matched patients without Langerhans cell histiocytosis by ratios of 2 to 4, depending on patient age 182. A review of 132 patients with Langerhans cell histiocytosis from a single institution found 31 patients with other cancers before their Langerhans cell histiocytosis diagnosis, 11 patients with concurrent malignancies, and 11 patients with other malignancies after their Langerhans cell histiocytosis diagnosis. Solid tumors comprised 74% of the malignancies, lymphomas comprised 17% of the cases, and hematologic malignancies comprised 9% of the cases. Seventy-one percent of the patients were smokers 182. These results are in contrast to an earlier study that was based on a literature review and institutional surveys that reported a higher incidence of lymphomas concurrent with the diagnosis of Langerhans cell histiocytosis 183.
Langerhans cell histiocytosis life expectancy
More than 50% of children under the age of 2 with disseminated Langerhans cell histiocytosis will die of the disease, but those with localized disease may have a prolonged life-span 35. For those who have been treated and do not have more lesions at 12 to 24 months, a full recovery can be expected 35. When there is lung involvement, the prognosis is not good. However, patients with isolated skin involvement and a solitary lymph node involvement tend to have a good prognosis 35.
Unfortunately, nearly 50% of patients with Langerhans cell histiocytosis are prone to a variety of complications which include the following 35:
- Musculoskeletal disability
- Skin scarring
- Diabetes insipidus
- Hearing impairment
- Neuropsychiatric problems like depression, anxiety, and intellectual impairment
- Pulmonary impairment
- Secondary malignancies like lymphoblastic leukemia and sodi tumors
- Growth retardation
- Liver cirrhosis.
Langerhans cell histiocytosis survival rate in children
Langerhans cell histiocytosis survival rate in children is closely linked to the extent of disease at presentation when high-risk organs (liver, spleen, and/or bone marrow) are involved, as well as the response to initial treatment 53. Many studies have confirmed the high mortality rate (35%) in patients with high-risk multisystem disease who do not respond well to therapy in the first 6 weeks 90. Because of treatment advances, including early implementation of additional therapy for poor responders, the outcome for children with Langerhans cell histiocytosis involving high-risk organs has improved 184, 177. Data from HISTSOC-Langerhans cell histiocytosis-III (NCT00276757) showed an overall survival (OS) rate of 84% for patients treated for 12 months with systemic chemotherapy 121.
For many years, lungs were thought to be high-risk organs, but isolated lung involvement in children with Langerhans cell histiocytosis is no longer considered to pose a significant risk of death 90.
Patients with single-system disease and low-risk multisystem disease do not usually die of Langerhans cell histiocytosis, but recurrent disease may result in considerable morbidity and significant late effects 117. Overall, recurrences have been found in 10% of patients with single-system unifocal disease, 25% of patients with single-system multifocal bone Langerhans cell histiocytosis, and 50% of patients with low-risk multisystem disease and those with high-risk multisystem disease who achieve nonactive disease status with chemotherapy. HISTSOC-Langerhans cell histiocytosis-III data showed a significant difference in reactivation rate for low-risk organ patients randomly assigned to receive 6 months of treatment (54%) versus 12 months of treatment (37%) 121. Similarly, the nonrandomized high-risk group of patients who were all treated for 12 months had a reactivation rate of 30%, compared with more than 50% in previous studies in which patients were treated with the same therapy for 6 months 121.
Most good-responding, high-risk patients who have a reactivation (30%) will do so in low-risk organs such as bone and will have the same risk of late effects as the low-risk multisystem patients 121. The major current treatment challenge is to reduce this overall 20% to 30% incidence of reactivations and the significant risk of serious permanent consequences in this group of patients.
Apart from disease extent, prognostic factors for children with Langerhans cell histiocytosis include the following:
- Age at diagnosis. Although age younger than 2 years was once thought to portend a worse prognosis, data from the HISTSOC-Langerhans cell histiocytosis-II study showed that patients aged 2 years or younger without high-risk organ involvement had the same response to therapy as did older patients 177. In contrast, the overall survival was poorer in neonates with risk-organ involvement compared with infants and children with the same extent of disease when patients were treated for only 6 months 177.
- Response to treatment. Response to therapy at 6 to 12 weeks has been shown to be a more important prognostic factor than age 185. The overall response to therapy is influenced by the duration and intensity of treatment 177, 184.
- Site of involvement.
- BRAF mutation. A study of 173 patients with the BRAF V600E mutation and 142 without the mutation revealed that the mutation occurred in 88% of patients with high-risk disease, 69% of patients with multisystem low-risk Langerhans cell histiocytosis, and 44% of patients with single-system low-risk Langerhans cell histiocytosis 186. The mutation was also found in 75% of patients with the neurodegenerative syndrome and 73% of patients with pituitary involvement. Resistance to initial treatment and relapse were higher in patients with the mutation 186.
An earlier study of 100 patients did not find these clinical correlations with the BRAF V600E mutation 187.
Late disease and treatment complications of childhood Langerhans cell histiocytosis
The reported frequency of long-term complications of Langerhans cell histiocytosis has ranged from 20% to 70%. Children with low-risk organ involvement (skin, bones, lymph nodes, or pituitary gland) have an approximately 20% chance of developing long-term sequelae 117, 119, 188. Patients with multisystem involvement have an approximately 70% incidence of long-term complications 117, 189, 116, 190. This wide variation in frequency results from case definition, sample size, therapy used, method of data collection, and follow-up duration.
Quality-of-life studies have reported the following:
- In one quality-of-life study of long-term survivors of skeletal Langerhans cell histiocytosis, the quality-of-life scores were not significantly different from those of healthy control children and adults 191. In addition, the quality-of-life scores were very similar between those with and without permanent complications.
- In another study of 40 patients who were carefully screened for late effects, adverse quality-of-life scores were found in more than 50% of patients 171. Seventy-five percent of patients had detectable long-term sequelae; hypothalamic/pituitary dysfunction (50%), cognitive dysfunction (20%), and cerebellar involvement (17.5%) were the most common side effects.
The late effects of Langerhans cell histiocytosis may occur in the following body systems:
- Endocrine. Patients with diabetes insipidus are at risk of panhypopituitarism and should be monitored carefully for adequacy of growth and development. In a retrospective review of 141 patients with Langerhans cell histiocytosis and diabetes insipidus, 43% developed growth hormone (GH) deficiency 189, 116, 190. The 5-year risk of growth hormone deficiency among children with Langerhans cell histiocytosis and diabetes insipidus was 35%, and the 10-year risk was 54%. There was no increased reactivation of Langerhans cell histiocytosis in patients who received growth hormone compared with those who did not 116. Growth and development problems are more frequent because of the young age at presentation and the more toxic effects of long-term prednisone therapy in the very young child.
- Hearing loss. Hearing loss has been found in 38% of children who were treated for Langerhans cell histiocytosis 189. 70% of patients with Langerhans cell histiocytosis in this study had ear involvement, which included aural discharge, mastoid swelling, and hearing loss. Of those with CT or MRI abnormalities in the mastoid, 59% had hearing loss 192.
- Neurological. Neurological symptoms secondary to vertebral compression of cervical lesions have been reported in 3 of 26 patients with Langerhans cell histiocytosis and spinal lesions 189. CNS Langerhans cell histiocytosis occurs most often in children with Langerhans cell histiocytosis of the pituitary or CNS-risk skull bones (mastoid, orbit, or temporal bone). Significant cognitive defects and MRI abnormalities may develop in some long-term survivors with CNS-risk skull lesions 193. Some patients have markedly abnormal cerebellar function and behavior abnormalities, while others have subtle deficits in short-term memory and brain stem–evoked potentials 194.
- Skeletal. Orthopedic problems from lesions of the spine, femur, tibia, or humerus may be seen in 20% of patients. These problems include vertebral collapse or instability of the spine that may lead to scoliosis and facial or limb asymmetry.
- Respiratory. Diffuse pulmonary disease may result in poor lung function with higher risk of infections and decreased exercise tolerance. These patients should be monitored with pulmonary function testing, including the diffusing capacity of carbon monoxide and ratio of residual volume to total lung capacity 170.
- Digestive. Liver disease may lead to sclerosing cholangitis, which rarely responds to any treatment other than liver transplant 176. Dental problems characterized by loss of teeth have been significant for some patients, usually related to overly aggressive dental surgery 195.
- Subsequent neoplasms. Bone marrow failure secondary to Langerhans cell histiocytosis or from therapy is rare and is associated with a higher risk of malignancy. Patients with Langerhans cell histiocytosis have a higher-than-normal risk of developing secondary cancers 183, 196.
Leukemia (usually acute myeloid leukemia) occurs after treatment, as does lymphoblastic lymphoma 53. Concurrent Langerhans cell histiocytosis and malignancy has been reported in a few patients, and some patients had their malignancy first, followed by development of Langerhans cell histiocytosis. Three patients with T-cell acute lymphoblastic leukemia (ALL) and aggressive Langerhans cell histiocytosis were reported and, as with all histiocytic disorders associated with or following lymphoblastic malignancies, the same genetic changes were found in both diseases, suggesting a shared clonal origin 197, 198, 199. One study reported two cases in which clonality with the same T-cell receptor gamma genotype was found 198. The authors of this study emphasized the plasticity of lymphocytes developing into Langerhans cells. The second study described one patient with Langerhans cell histiocytosis after T-cell ALL who had the same T-cell receptor gene rearrangements and activating mutations of the NOTCH1 gene 199.
An association between solid tumors and Langerhans cell histiocytosis has also been reported. Solid tumors associated with Langerhans cell histiocytosis include retinoblastoma, brain tumors, hepatocellular carcinoma, and Ewing sarcoma 53.
- Langerhans P. Über die Nerven der menschlichen Haut. Arch Abl B Pathol. 1868;44:325‐337.[↩]
- Hand AR. Polyuria and tuberculosis. Arch Pediatr. 1893;10:673‐675.[↩][↩]
- Schuller A. Uber eigenartige Schadeldefekte im Jugendalter. Fortschr Roentgenstr. 1915;23:12‐18.[↩][↩]
- Christian HA. Defects in membraneous bones, exophthalmos and diabetes insipidus: an unusual syndrome of dyspituitarism. Med Clin N Am. 1919;3:849‐871.[↩][↩]
- Histiocytosis syndromes in children. Writing Group of the Histiocyte Society. Lancet. 1987 Jan 24;1(8526):208-9.[↩][↩]
- Langerhans Cell Histiocytosis—Health Professional Version. https://www.cancer.gov/types/langerhans/hp[↩]
- Kobayashi M, Tojo A. Langerhans cell histiocytosis in adults: Advances in pathophysiology and treatment. Cancer Sci. 2018 Dec;109(12):3707-3713. doi: 10.1111/cas.13817[↩][↩][↩][↩][↩]
- Leenknegt B, Herregods N, Lemmerling M. Craniofacial and Intracranial Langerhans Cell Histiocytosis. J Belg Soc Radiol. 2020 Oct 23;104(1):57. doi: 10.5334/jbsr.2245[↩][↩][↩][↩]
- Chikwava K, Jaffe R. Langerin (CD207) staining in normal pediatric tissues, reactive lymph nodes, and childhood histiocytic disorders. Pediatr Dev Pathol. 2004 Nov-Dec;7(6):607-14. doi: 10.1007/s10024-004-3027-z[↩][↩]
- Huang WD, Yang XH, Wu ZP, Huang Q, Xiao JR, Yang MS, Zhou ZH, Yan WJ, Song DW, Liu TL, Jia NY. Langerhans cell histiocytosis of spine: a comparative study of clinical, imaging features, and diagnosis in children, adolescents, and adults. Spine J. 2013 Sep;13(9):1108-17. doi: 10.1016/j.spinee.2013.03.013[↩]
- Gao XM, Li J, Cao XX. Signaling pathways, microenvironment, and targeted treatments in Langerhans cell histiocytosis. Cell Commun Signal. 2022 Dec 19;20(1):195. doi: 10.1186/s12964-022-00917-0[↩]
- Le Louet S, Barkaoui MA, Miron J, Galambrun C, Aladjidi N, Chastagner P, Kebaili K, Armari-Alla C, Lambilliotte A, Lejeune J, Moshous D, Della Valle V, Sileo C, Ducou Le Pointe H, Chateil JF, Renolleau S, Piloquet JE, Portefaix A, Epaud R, Chiron R, Bugnet E, Lorillon G, Tazi A, Emile JF, Donadieu J, Héritier S. Childhood Langerhans cell histiocytosis with severe lung involvement: a nationwide cohort study. Orphanet J Rare Dis. 2020 Sep 9;15(1):241. doi: 10.1186/s13023-020-01495-5[↩]
- Kitticharoenjit P, Supakul N, Rujkijyanont P, Traivaree C, Photia A, Monsereenusorn C. Clinical characteristics and outcomes of Langerhans cell histiocytosis at a single institution in Thailand: a 20-year retrospective study. Asian Biomed (Res Rev News). 2021 Aug 20;15(4):171-181. doi: 10.2478/abm-2021-0022[↩]
- Kemps PG, Zondag TCE, Arnardóttir HB, Solleveld-Westerink N, Borst J, Steenwijk EC, van Egmond D, Swennenhuis JF, Stelloo E, Trambusti I, Verdijk RM, van Noesel CJM, Cleven AHG, Scheijde-Vermeulen MA, Koudijs MJ, Krsková L, Hawkins C, Egeler RM, Brok J, von Bahr Greenwood T, Svojgr K, Beishuizen A, van Laar JAM, Pötschger U, Hutter C, Sieni E, Minkov M, Abla O, van Wezel T, van den Bos C, van Halteren AGS. Clinicogenomic associations in childhood Langerhans cell histiocytosis: an international cohort study. Blood Adv. 2023 Feb 28;7(4):664-679. doi: 10.1182/bloodadvances.2022007947[↩]
- Angelini A, Mavrogenis AF, Rimondi E, Rossi G, Ruggieri P. Current concepts for the diagnosis and management of eosinophilic granuloma of bone. J Orthop Traumatol. 2017 Jun;18(2):83-90. doi: 10.1007/s10195-016-0434-7[↩]
- Zhao SS, Yan LF, Feng XL, Du P, Chen BY, Dong WT, Gao Y, He JB, Cui GB, Wang W. Incidence and radiological pattern of eosinophilic granuloma: a retrospective study in a Chinese tertiary hospital. J Orthop Surg Res. 2019 May 9;14(1):123. doi: 10.1186/s13018-019-1158-1[↩]
- Yamamoto T, Miyazaki T, Kurashima Y, Ohata K, Okawa M, Tanaka S, Uenishi T, Miyaji K, Fukumoto N. Solitary Hepatic Eosinophilic Granuloma Accompanied by Eosinophilia Without Parasitosis: Report of a Case. Int Surg. 2015 Jun;100(6):1011-7. doi: 10.9738/INTSURG-D-14-00126.1[↩]
- Rodriguez-Galindo C, Allen CE. Langerhans cell histiocytosis. Blood. 2020 Apr 16;135(16):1319-1331. https://doi.org/10.1182/blood.2019000934[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Emile JF, Abla O, Fraitag S, Horne A, Haroche J, Donadieu J, Requena-Caballero L, Jordan MB, Abdel-Wahab O, Allen CE, Charlotte F, Diamond EL, Egeler RM, Fischer A, Herrera JG, Henter JI, Janku F, Merad M, Picarsic J, Rodriguez-Galindo C, Rollins BJ, Tazi A, Vassallo R, Weiss LM; Histiocyte Society. Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood. 2016 Jun 2;127(22):2672-81. doi: 10.1182/blood-2016-01-690636[↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Delprat C, Aricò M. Blood spotlight on Langerhans cell histiocytosis. Blood. 2014 Aug 7;124(6):867-72. doi: 10.1182/blood-2014-02-556407[↩][↩]
- Berres ML, Merad M, Allen CE. Progress in understanding the pathogenesis of Langerhans cell histiocytosis: back to Histiocytosis X? Br J Haematol. 2015 Apr;169(1):3-13. doi: 10.1111/bjh.13247[↩][↩]
- Haupt R, Minkov M, Astigarraga I, Schäfer E, Nanduri V, Jubran R, Egeler RM, Janka G, Micic D, Rodriguez-Galindo C, Van Gool S, Visser J, Weitzman S, Donadieu J; Euro Histio Network. Langerhans cell histiocytosis (LCH): guidelines for diagnosis, clinical work-up, and treatment for patients till the age of 18 years. Pediatr Blood Cancer. 2013 Feb;60(2):175-84. doi: 10.1002/pbc.24367[↩][↩]
- Tran G, Huynh TN, Paller AS. Langerhans cell histiocytosis: A neoplastic disorder driven by Ras-ERK pathway mutations. J Am Acad Dermatol. 2018 Mar;78(3):579-590.e4. doi: 10.1016/j.jaad.2017.09.022[↩]
- Carstensen H, Ornvold K. The epidemiology of LCH in children in Denmark. Med Pediatr Oncol. 1993;21:387‐388.[↩]
- Baumgartner I, von Hochstetter A, Baumert B, Luetolf U, Follath F. Langerhans’-cell histiocytosis in adults. Med Pediatr Oncol. 1997 Jan;28(1):9-14. doi: 10.1002/(sici)1096-911x(199701)28:1<9::aid-mpo3>3.0.co;2-p[↩]
- Goyal G, Shah MV, Hook CC, Wolanskyj AP, Call TG, Rech KL, Go RS. Adult disseminated Langerhans cell histiocytosis: incidence, racial disparities and long-term outcomes. Br J Haematol. 2018 Aug;182(4):579-581. doi: 10.1111/bjh.14818[↩]
- Alston RD, Tatevossian RG, McNally RJ, Kelsey A, Birch JM, Eden TO. Incidence and survival of childhood Langerhans cell histiocytosis in Northwest England from 1954 to 1998. Pediatr Blood Cancer. 2007 May;48(5):555-60. doi: 10.1002/pbc.20884[↩]
- Guyot-Goubin A, Donadieu J, Barkaoui M, Bellec S, Thomas C, Clavel J. Descriptive epidemiology of childhood Langerhans cell histiocytosis in France, 2000-2004. Pediatr Blood Cancer. 2008 Jul;51(1):71-5. doi: 10.1002/pbc.21498[↩]
- Salotti JA, Nanduri V, Pearce MS, Parker L, Lynn R, Windebank KP. Incidence and clinical features of Langerhans cell histiocytosis in the UK and Ireland. Arch Dis Child. 2009 May;94(5):376-80. doi: 10.1136/adc.2008.144527[↩]
- Stålemark H, Laurencikas E, Karis J, Gavhed D, Fadeel B, Henter JI. Incidence of Langerhans cell histiocytosis in children: a population-based study. Pediatr Blood Cancer. 2008 Jul;51(1):76-81. doi: 10.1002/pbc.21504[↩]
- Swerdlow SH, Campo E, Harris NL, et al. WHO classification of tumours of haematopoietic and lymphoid tissues In: Weiss LM, Jaffe R, Facchetti F, eds. WHO classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: IARC; 2017:470‐472.[↩][↩][↩]
- McClain KL, Bigenwald C, Collin M, Haroche J, Marsh RA, Merad M, Picarsic J, Ribeiro KB, Allen CE. Histiocytic disorders. Nat Rev Dis Primers. 2021 Oct 7;7(1):73. doi: 10.1038/s41572-021-00307-9[↩]
- Arceci RJ, Brenner MK, Pritchard J. Controversies and new approaches to treatment of Langerhans cell histiocytosis. Hematol Oncol Clin North Am. 1998 Apr;12(2):339-57. doi: 10.1016/s0889-8588(05)70514-1[↩]
- Berres ML, Allen CE, Merad M. Pathological consequence of misguided dendritic cell differentiation in histiocytic diseases. Adv Immunol. 2013;120:127-61. doi: 10.1016/B978-0-12-417028-5.00005-3[↩][↩]
- Tillotson CV, Anjum F, Patel BC. Langerhans Cell Histiocytosis. [Updated 2023 Jul 17]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK430885[↩][↩][↩][↩][↩][↩][↩][↩]
- Bhatia S, Nesbit ME Jr, Egeler RM, Buckley JD, Mertens A, Robison LL. Epidemiologic study of Langerhans cell histiocytosis in children. J Pediatr. 1997 May;130(5):774-84. doi: 10.1016/s0022-3476(97)80021-2[↩][↩]
- Panaite L, Shadman M. Concurrent diagnosis of classical Hodgkin lymphoma and Langerhans cell histiocytosis. Blood. 2019 May 9;133(19):2109. doi: 10.1182/blood-2019-01-891937[↩][↩]
- Kambouchner M, Emile JF, Copin MC, Coulomb-Lherminé A, Sabourin JC, Della Valle V, Sileo C, Ducou Le Pointe H, Bégueret H, Galmiche L, Lambilliotte A, Paraf F, Piche M, Piguet C, Rullier A, Secq V, Serre I, Bernaudin JF, Donadieu J. Childhood pulmonary Langerhans cell histiocytosis: a comprehensive clinical-histopathological and BRAFV600E mutation study from the French national cohort. Hum Pathol. 2019 Jul;89:51-61. doi: 10.1016/j.humpath.2019.04.005[↩][↩]
- Narayanasamy Rajavelu T, Abimannane A, Chinnaiah Govindhareddy DK, Kayal S, Kar R. Langerhans’ Cell Histiocytosis Masquerading as Caroli’s Disease. J Pediatr Hematol Oncol. 2020 Oct;42(7):e620-e622. doi: 10.1097/MPH.0000000000001495[↩][↩]
- Yousem SA, Colby TV, Chen YY, Chen WG, Weiss LM. Pulmonary Langerhans’ cell histiocytosis: molecular analysis of clonality. Am J Surg Pathol. 2001 May;25(5):630-6. doi: 10.1097/00000478-200105000-00010[↩]
- Tazi A, de Margerie C, Naccache JM, Fry S, Dominique S, Jouneau S, Lorillon G, Bugnet E, Chiron R, Wallaert B, Valeyre D, Chevret S. The natural history of adult pulmonary Langerhans cell histiocytosis: a prospective multicentre study. Orphanet J Rare Dis. 2015 Mar 14;10:30. doi: 10.1186/s13023-015-0249-2[↩][↩]
- Vassallo R, Ryu JH, Schroeder DR, Decker PA, Limper AH. Clinical outcomes of pulmonary Langerhans’-cell histiocytosis in adults. N Engl J Med. 2002 Feb 14;346(7):484-90. doi: 10.1056/NEJMoa012087[↩]
- Laurencikas E, Gavhed D, Stålemark H, van’t Hooft I, Prayer D, Grois N, Henter JI. Incidence and pattern of radiological central nervous system Langerhans cell histiocytosis in children: a population based study. Pediatr Blood Cancer. 2011 Feb;56(2):250-7. doi: 10.1002/pbc.22791[↩]
- D’Ambrosio N, Soohoo S, Warshall C, Johnson A, Karimi S. Craniofacial and intracranial manifestations of langerhans cell histiocytosis: Report of findings in 100 patients. AJR Am J Roentgenol. 2008; 191(2): 589–97. DOI: 10.2214/AJR.07.3573[↩]
- LCH in Children. https://www.histio.org/lchinchildren[↩]
- Chanchlani N, Parke SC, Hart JW. Langerhans cell histiocytosis in a 5-month-old baby. CMAJ. 2021 Jan 4;193(1):E23. doi: 10.1503/cmaj.201151[↩]
- Ramírez-Lluch, M, Colmenero, I, Suárez, J, Carrión-Marrero, S, Rodríguez-Fuertes, F, Mateos-Mayo, A, et al. Three cases of congenital self-healing Langerhans cell histiocytosis with BRAF-V600E mutation. JEADV Clin Pract. 2022; 1: 53–57. https://doi.org/10.1002/jvc2.14[↩]
- Histiocytosis. https://medlineplus.gov/ency/article/000068.htm[↩]
- 15 – Histiocytic Proliferative Disorders. Dermatopathology Foundations in Diagnostic Pathology 2010, Pages 619-630. https://doi.org/10.1016/B978-0-443-06654-2.00015-9[↩]
- Badalian-Very G, Vergilio JA, Fleming M, Rollins BJ. Pathogenesis of Langerhans cell histiocytosis. Annu Rev Pathol. 2013 Jan 24;8:1-20. doi: 10.1146/annurev-pathol-020712-163959[↩]
- Willman CL, Busque L, Griffith BB, Favara BE, McClain KL, Duncan MH, Gilliland DG. Langerhans’-cell histiocytosis (histiocytosis X)–a clonal proliferative disease. N Engl J Med. 1994 Jul 21;331(3):154-60. doi: 10.1056/NEJM199407213310303[↩]
- Yu RC, Chu C, Buluwela L, Chu AC. Clonal proliferation of Langerhans cells in Langerhans cell histiocytosis. Lancet. 1994 Mar 26;343(8900):767-8. doi: 10.1016/s0140-6736(94)91842-2[↩]
- PDQ Pediatric Treatment Editorial Board. Langerhans Cell Histiocytosis Treatment (PDQ®): Health Professional Version. 2023 Apr 19. In: PDQ Cancer Information Summaries [Internet]. Bethesda (MD): National Cancer Institute (US); 2002-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK65799[↩][↩][↩][↩][↩][↩][↩]
- McClain KL, Picarsic J, Chakraborty R, et al. CNS Langerhans cell histiocytosis: Common hematopoietic origin for LCH-associated neurodegeneration and mass lesions. Cancer. 2018 Jun 15;124(12):2607-2620. doi: 10.1002/cncr.31348[↩][↩]
- Yu RC, Morris JF, Pritchard J, Chu TC. Defective alloantigen-presenting capacity of ‘Langerhans cell histiocytosis cells’. Arch Dis Child. 1992 Nov;67(11):1370-2. doi: 10.1136/adc.67.11.1370[↩][↩]
- Geissmann F, Lepelletier Y, Fraitag S, Valladeau J, Bodemer C, Debré M, Leborgne M, Saeland S, Brousse N. Differentiation of Langerhans cells in Langerhans cell histiocytosis. Blood. 2001 Mar 1;97(5):1241-8. doi: 10.1182/blood.v97.5.1241[↩][↩]
- Senechal B, Elain G, Jeziorski E, Grondin V, Patey-Mariaud de Serre N, Jaubert F, Beldjord K, Lellouch A, Glorion C, Zerah M, Mary P, Barkaoui M, Emile JF, Boccon-Gibod L, Josset P, Debré M, Fischer A, Donadieu J, Geissmann F. Expansion of regulatory T cells in patients with Langerhans cell histiocytosis. PLoS Med. 2007 Aug;4(8):e253. doi: 10.1371/journal.pmed.0040253[↩][↩][↩]
- Badalian-Very G, Vergilio JA, Degar BA, MacConaill LE, Brandner B, Calicchio ML, Kuo FC, Ligon AH, Stevenson KE, Kehoe SM, Garraway LA, Hahn WC, Meyerson M, Fleming MD, Rollins BJ. Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood. 2010 Sep 16;116(11):1919-23. doi: 10.1182/blood-2010-04-279083[↩][↩][↩][↩]
- Satoh T, Smith A, Sarde A, Lu HC, Mian S, Trouillet C, Mufti G, Emile JF, Fraternali F, Donadieu J, Geissmann F. B-RAF mutant alleles associated with Langerhans cell histiocytosis, a granulomatous pediatric disease. PLoS One. 2012;7(4):e33891. doi: 10.1371/journal.pone.0033891. Epub 2012 Apr 10. Erratum in: PLoS One. 2012;7(6). doi:10.1371/annotation/74a67f4e-a536-4b3f-a350-9a4c1e6bebbd. Mian, Sophie [corrected to Mian, Syed].[↩][↩]
- Sahm F, Capper D, Preusser M, Meyer J, Stenzinger A, Lasitschka F, Berghoff AS, Habel A, Schneider M, Kulozik A, Anagnostopoulos I, Müllauer L, Mechtersheimer G, von Deimling A. BRAFV600E mutant protein is expressed in cells of variable maturation in Langerhans cell histiocytosis. Blood. 2012 Sep 20;120(12):e28-34. doi: 10.1182/blood-2012-06-429597[↩]
- Berres ML, Lim KP, Peters T, Price J, Takizawa H, Salmon H, Idoyaga J, Ruzo A, Lupo PJ, Hicks MJ, Shih A, Simko SJ, Abhyankar H, Chakraborty R, Leboeuf M, Beltrão M, Lira SA, Heym KM, Bigley V, Collin M, Manz MG, McClain K, Merad M, Allen CE. BRAF-V600E expression in precursor versus differentiated dendritic cells defines clinically distinct LCH risk groups. J Exp Med. 2014 Apr 7;211(4):669-83. doi: 10.1084/jem.20130977. Epub 2014 Mar 17. Erratum in: J Exp Med. 2015 Feb 9;212(2):281.[↩][↩]
- Héritier S, Hélias-Rodzewicz Z, Chakraborty R, Sengal AG, Bellanné-Chantelot C, Thomas C, Moreau A, Fraitag S, Allen CE, Donadieu J, Emile JF. New somatic BRAF splicing mutation in Langerhans cell histiocytosis. Mol Cancer. 2017 Jul 6;16(1):115. doi: 10.1186/s12943-017-0690-z[↩]
- Nelson DS, Quispel W, Badalian-Very G, van Halteren AG, van den Bos C, Bovée JV, Tian SY, Van Hummelen P, Ducar M, MacConaill LE, Egeler RM, Rollins BJ. Somatic activating ARAF mutations in Langerhans cell histiocytosis. Blood. 2014 May 15;123(20):3152-5. doi: 10.1182/blood-2013-06-511139[↩]
- Chakraborty R, Hampton OA, Shen X, et al. Mutually exclusive recurrent somatic mutations in MAP2K1 and BRAF support a central role for ERK activation in LCH pathogenesis. Blood. 2014 Nov 6;124(19):3007-15. doi: 10.1182/blood-2014-05-577825[↩][↩][↩][↩][↩][↩]
- Héritier S, Hélias-Rodzewicz Z, Lapillonne H, Terrones N, Garrigou S, Normand C, Barkaoui MA, Miron J, Plat G, Aladjidi N, Pagnier A, Deville A, Gillibert-Yvert M, Moshous D, Lefèvre-Utile A, Lutun A, Paillard C, Thomas C, Jeziorski E, Nizard P, Taly V, Emile JF, Donadieu J. Circulating cell-free BRAFV600E as a biomarker in children with Langerhans cell histiocytosis. Br J Haematol. 2017 Aug;178(3):457-467. doi: 10.1111/bjh.14695[↩]
- Brown NA, Furtado LV, Betz BL, Kiel MJ, Weigelin HC, Lim MS, Elenitoba-Johnson KS. High prevalence of somatic MAP2K1 mutations in BRAF V600E-negative Langerhans cell histiocytosis. Blood. 2014 Sep 4;124(10):1655-8. doi: 10.1182/blood-2014-05-577361[↩]
- Chakraborty R, Burke TM, Hampton OA, et al. Alternative genetic mechanisms of BRAF activation in Langerhans cell histiocytosis. Blood. 2016 Nov 24;128(21):2533-2537. doi: 10.1182/blood-2016-08-733790[↩][↩][↩]
- Fleming MD, Pinkus JL, Fournier MV, Alexander SW, Tam C, Loda M, Sallan SE, Nichols KE, Carpentieri DF, Pinkus GS, Rollins BJ. Coincident expression of the chemokine receptors CCR6 and CCR7 by pathologic Langerhans cells in Langerhans cell histiocytosis. Blood. 2003 Apr 1;101(7):2473-5. doi: 10.1182/blood.V101.7.2473. Erratum in: Blood. 2003 Nov 15;102(10):3477.[↩]
- Annels NE, Da Costa CE, Prins FA, Willemze A, Hogendoorn PC, Egeler RM. Aberrant chemokine receptor expression and chemokine production by Langerhans cells underlies the pathogenesis of Langerhans cell histiocytosis. J Exp Med. 2003 May 19;197(10):1385-90. doi: 10.1084/jem.20030137[↩]
- Allen CE, Li L, Peters TL, Leung HC, Yu A, Man TK, Gurusiddappa S, Phillips MT, Hicks MJ, Gaikwad A, Merad M, McClain KL. Cell-specific gene expression in Langerhans cell histiocytosis lesions reveals a distinct profile compared with epidermal Langerhans cells. J Immunol. 2010 Apr 15;184(8):4557-67. doi: 10.4049/jimmunol.0902336[↩][↩][↩][↩]
- Rolland A, Guyon L, Gill M, Cai YH, Banchereau J, McClain K, Palucka AK. Increased blood myeloid dendritic cells and dendritic cell-poietins in Langerhans cell histiocytosis. J Immunol. 2005 Mar 1;174(5):3067-71. doi: 10.4049/jimmunol.174.5.3067[↩]
- McClain KL, Picarsic J, Chakraborty R, Zinn D, et al. CNS Langerhans cell histiocytosis: Common hematopoietic origin for LCH-associated neurodegeneration and mass lesions. Cancer. 2018 Jun 15;124(12):2607-2620. doi: 10.1002/cncr.31348[↩]
- Preliasco VF, Benchuya C, Pavan V, de la Cal C, Ganzinelli S, Sterin-Borda L. IL-1 beta and PGE2 levels are increased in the saliva of children with Langerhans cell histiocytosis. J Oral Pathol Med. 2008 Oct;37(9):522-7. doi: 10.1111/j.1600-0714.2008.00675.x[↩]
- Arzoo K, Sadeghi S, Pullarkat V: Pamidronate for bone pain from osteolytic lesions in Langerhans’-cell histiocytosis. N Engl J Med 345 (3): 225, 2001.[↩][↩][↩]
- Abbritti M, Mazzei MA, Bargagli E, et al.: Utility of spiral CAT scan in the follow-up of patients with pulmonary Langerhans cell histiocytosis. Eur J Radiol 81 (8): 1907-12, 2012.[↩][↩][↩]
- Kim HJ, Lee KS, Johkoh T, et al.: Pulmonary Langerhans cell histiocytosis in adults: high-resolution CT-pathology comparisons and evolutional changes at CT. Eur Radiol 21 (7): 1406-15, 2011.[↩][↩][↩]
- Kriz J, Eich HT, Bruns F, et al.: Radiotherapy in langerhans cell histiocytosis – a rare indication in a rare disease. Radiat Oncol 8: 233, 2013.[↩]
- Dennin MH, Roman CJ, Stein SL. Congenital Langerhans cell histiocytosis presenting in a 27-week-gestation neonate. Pediatr Dermatol. 2018 Mar;35(2):e140-e141. doi: 10.1111/pde.13416[↩]
- Gothwal S, Gupta AK, Choudhary R. Congenital Self Healing Langerhans Cell Histiocytosis. Indian J Pediatr. 2018 Apr;85(4):316-317. doi: 10.1007/s12098-017-2469-z[↩]
- Yu J, Rubin AI, Castelo-Soccio L, Perman MJ. Congenital Self-Healing Langerhans Cell Histiocytosis. J Pediatr. 2017 May;184:232-232.e1. doi: 10.1016/j.jpeds.2017.01.040[↩]
- Kalpana S, Lakshmi V. Systemic Congenital Langerhans Cell Histiocytosis Masquerading as Diffuse Neonatal Hemangiomatosis. Indian Pediatr. 2018 Jul 15;55(7):613.[↩][↩]
- Schmitt AR, Wetter DA, Camilleri MJ, Khan SP, Tollefson MM. Langerhans cell histiocytosis presenting as a blueberry muffin rash. Lancet. 2017 Jul 8;390(10090):155. doi: 10.1016/S0140-6736(17)30564-0[↩][↩]
- Lau L, Krafchik B, Trebo MM, Weitzman S. Cutaneous Langerhans cell histiocytosis in children under one year. Pediatr Blood Cancer. 2006 Jan;46(1):66-71. doi: 10.1002/pbc.20479[↩]
- Abla O, Egeler RM, Weitzman S. Langerhans cell histiocytosis: Current concepts and treatments. Cancer Treat Rev. 2010 Jun;36(4):354-9. doi: 10.1016/j.ctrv.2010.02.012[↩]
- Crickx E, Bouaziz JD, Lorillon G, de Menthon M, Cordoliani F, Bugnet E, Bagot M, Rybojad M, Mourah S, Tazi A. Clinical Spectrum, Quality of Life, BRAF Mutation Status and Treatment of Skin Involvement in Adult Langerhans Cell Histiocytosis. Acta Derm Venereol. 2017 Jul 6;97(7):838-842. doi: 10.2340/00015555-2674[↩]
- Ruiz Beguerie J, Fernández J, Stringa MF, Anaya J. Vulvar Langerhans cell histiocytosis and thalidomide: an effective treatment option. Int J Dermatol. 2017 Mar;56(3):324-326. doi: 10.1111/ijd.13377[↩]
- Jiang W, Li L, He YM, Yang KX. Langerhans cell histiocytosis of the female genital tract: a literature review with additional three case studies in China. Arch Gynecol Obstet. 2012 Jan;285(1):99-103. doi: 10.1007/s00404-011-2113-5[↩]
- Gao YJ, Su M, Tang JY, Pan C, Chen J. Treatment Outcome of Children With Multisystem Langerhans Cell Histiocytosis: The Experience of a Single Children’s Hospital in Shanghai, China. J Pediatr Hematol Oncol. 2018 Jan;40(1):e9-e12. doi: 10.1097/MPH.0000000000001016[↩][↩]
- Braier J, Latella A, Balancini B, Castaños C, Rosso D, Chantada G, Ripoli M, Goldberg J. Outcome in children with pulmonary Langerhans cell Histiocytosis. Pediatr Blood Cancer. 2004 Dec;43(7):765-9. doi: 10.1002/pbc.20112[↩]
- Ronceray L, Pötschger U, Janka G, Gadner H, Minkov M; German Society for Pediatric Hematology and Oncology, Langerhans Cell Histiocytosis Study Group. Pulmonary involvement in pediatric-onset multisystem Langerhans cell histiocytosis: effect on course and outcome. J Pediatr. 2012 Jul;161(1):129-33.e1-3. doi: 10.1016/j.jpeds.2011.12.035[↩][↩][↩]
- Morimoto A, Shioda Y, Imamura T, Kudo K, Kawaguchi H, Sakashita K, Yasui M, Koga Y, Kobayashi R, Ishii E, Fujimoto J, Horibe K, Bessho F, Tsunematsu Y, Imashuku S. Intensified and prolonged therapy comprising cytarabine, vincristine and prednisolone improves outcome in patients with multisystem Langerhans cell histiocytosis: results of the Japan Langerhans Cell Histiocytosis Study Group-02 Protocol Study. Int J Hematol. 2016 Jul;104(1):99-109. doi: 10.1007/s12185-016-1993-3[↩]
- Tazi A. Adult pulmonary Langerhans’ cell histiocytosis. Eur Respir J. 2006 Jun;27(6):1272-85. doi: 10.1183/09031936.06.00024004[↩]
- Tazi A, de Margerie-Mellon C, Vercellino L, Naccache JM, Fry S, Dominique S, Jouneau S, Lorillon G, Bugnet E, Chiron R, Wallaert B, Valeyre D, Chevret S. Extrathoracic investigation in adult patients with isolated pulmonary langerhans cell histiocytosis. Orphanet J Rare Dis. 2016 Feb 2;11:11. doi: 10.1186/s13023-016-0387-1[↩][↩]
- DeMartino E, Go RS, Vassallo R. Langerhans Cell Histiocytosis and Other Histiocytic Diseases of the Lung. Clin Chest Med. 2016 Sep;37(3):421-30. doi: 10.1016/j.ccm.2016.04.005[↩]
- Lorillon G, Tazi A. How I manage pulmonary Langerhans cell histiocytosis. Eur Respir Rev. 2017 Sep 6;26(145):170070. doi: 10.1183/16000617.0070-2017[↩][↩][↩]
- O’Kane D, Jenkinson H, Carson J: Langerhans cell histiocytosis associated with breast carcinoma successfully treated with topical imiquimod. Clin Exp Dermatol 34 (8): e829-32, 2009.[↩][↩]
- Morimoto A, Shioda Y, Imamura T, et al.: Nationwide survey of bisphosphonate therapy for children with reactivated Langerhans cell histiocytosis in Japan. Pediatr Blood Cancer 56 (1): 110-5, 2011.[↩]
- Roden AC, Yi ES. Pulmonary Langerhans Cell Histiocytosis: An Update From the Pathologists’ Perspective. Arch Pathol Lab Med. 2016 Mar;140(3):230-40. doi: 10.5858/arpa.2015-0246-RA[↩]
- Mourah S, How-Kit A, Meignin V, Gossot D, Lorillon G, Bugnet E, Mauger F, Lebbe C, Chevret S, Tost J, Tazi A. Recurrent NRAS mutations in pulmonary Langerhans cell histiocytosis. Eur Respir J. 2016 Jun;47(6):1785-96. doi: 10.1183/13993003.01677-2015[↩]
- Kamionek M, Ahmadi Moghaddam P, Sakhdari A, Kovach AE, Welch M, Meng X, Dresser K, Tomaszewicz K, Cosar EF, Mark EJ, Fraire AE, Hutchinson L. Mutually exclusive extracellular signal-regulated kinase pathway mutations are present in different stages of multi-focal pulmonary Langerhans cell histiocytosis supporting clonal nature of the disease. Histopathology. 2016 Sep;69(3):499-509. doi: 10.1111/his.12955[↩]
- Reichle A, Vogt T, Kunz-Schughart L, et al.: Anti-inflammatory and angiostatic therapy in chemorefractory multisystem Langerhans’ cell histiocytosis of adults. Br J Haematol 128 (5): 730-2, 2005.[↩]
- Taverna JA, Stefanato CM, Wax FD, et al.: Adult cutaneous Langerhans cell histiocytosis responsive to topical imiquimod. J Am Acad Dermatol 54 (5): 911-3, 2006.[↩]
- Brown RE: Bisphosphonates as antialveolar macrophage therapy in pulmonary langerhans cell histiocytosis? Med Pediatr Oncol 36 (6): 641-3, 2001.[↩]
- Le Pavec J, Lorillon G, Jaïs X, et al.: Pulmonary Langerhans cell histiocytosis-associated pulmonary hypertension: clinical characteristics and impact of pulmonary arterial hypertension therapies. Chest 142 (5): 1150-1157, 2012.[↩][↩][↩]
- Rieker J, Hengge U, Ruzicka T, et al.: [Multifocal facial eosinophilic granuloma: successful treatment with topical tacrolimus]. Hautarzt 57 (4): 324-6, 2006.[↩][↩][↩][↩]
- Vogel CA, Aughenbaugh W, Sharata H: Excimer laser as adjuvant therapy for adult cutaneous Langerhans cell histiocytosis. Arch Dermatol 144 (10): 1287-90, 2008.[↩][↩]
- McClain KL, Kozinetz CA: A phase II trial using thalidomide for Langerhans cell histiocytosis. Pediatr Blood Cancer 48 (1): 44-9, 2007.[↩]
- Zinn DJ, Grimes AB, Lin H, et al.: Hydroxyurea: a new old therapy for Langerhans cell histiocytosis. Blood 128 (20): 2462-2465, 2016.[↩][↩]
- Chang SE, Koh GJ, Choi JH, et al.: Widespread skin-limited adult Langerhans cell histiocytosis: long-term follow-up with good response to interferon alpha. Clin Exp Dermatol 27 (2): 135-7, 2002.[↩][↩]
- Cantu MA, Lupo PJ, Bilgi M, et al.: Optimal therapy for adults with Langerhans cell histiocytosis bone lesions. PLoS One 7 (8): e43257, 2012.[↩][↩]
- Duan MH, Han X, Li J, et al.: Comparison of vindesine and prednisone and cyclophosphamide, etoposide, vindesine, and prednisone as first-line treatment for adult Langerhans cell histiocytosis: A single-center retrospective study. Leuk Res 42: 43-6, 2016.[↩][↩]
- Tsele E, Thomas DM, Chu AC: Treatment of adult Langerhans cell histiocytosis with etoposide. J Am Acad Dermatol 27 (1): 61-4, 1992.[↩][↩]
- Yeh EA, Greenberg J, Abla O, Longoni G, Diamond E, Hermiston M, Tran B, Rodriguez-Galindo C, Allen CE, McClain KL; North American Consortium for Histiocytosis. Evaluation and treatment of Langerhans cell histiocytosis patients with central nervous system abnormalities: Current views and new vistas. Pediatr Blood Cancer. 2018 Jan;65(1). doi: 10.1002/pbc.26784[↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Grois N, Fahrner B, Arceci RJ, Henter JI, McClain K, Lassmann H, Nanduri V, Prosch H, Prayer D; Histiocyte Society CNS LCH Study Group. Central nervous system disease in Langerhans cell histiocytosis. J Pediatr. 2010 Jun;156(6):873-881.e1. doi: 10.1016/j.jpeds.2010.03.001[↩][↩][↩][↩]
- Sagna Y, Courtillot C, Drabo JY, Tazi A, Donadieu J, Idbaih A, Cohen F, Amoura Z, Haroche J, Touraine P. Endocrine manifestations in a cohort of 63 adulthood and childhood onset patients with Langerhans cell histiocytosis. Eur J Endocrinol. 2019 Sep;181(3):275-285. doi: 10.1530/EJE-19-0177[↩][↩]
- Donadieu J, Rolon MA, Pion I, Thomas C, Doz F, Barkaoui M, Robert A, Deville A, Mazingue F, David M, Brauner R, Cabrol S, Garel C, Polak M; French LCH Study Group. Incidence of growth hormone deficiency in pediatric-onset Langerhans cell histiocytosis: efficacy and safety of growth hormone treatment. J Clin Endocrinol Metab. 2004 Feb;89(2):604-9. doi: 10.1210/jc.2003-030907[↩][↩][↩][↩]
- Haupt R, Nanduri V, Calevo MG, Bernstrand C, Braier JL, Broadbent V, Rey G, McClain KL, Janka-Schaub G, Egeler RM. Permanent consequences in Langerhans cell histiocytosis patients: a pilot study from the Histiocyte Society-Late Effects Study Group. Pediatr Blood Cancer. 2004 May;42(5):438-44. doi: 10.1002/pbc.20021[↩][↩][↩][↩][↩][↩][↩]
- Donadieu J, Rolon MA, Thomas C, Brugieres L, Plantaz D, Emile JF, Frappaz D, David M, Brauner R, Genereau T, Debray D, Cabrol S, Barthez MA, Hoang-Xuan K, Polak M; French LCH Study Group. Endocrine involvement in pediatric-onset Langerhans’ cell histiocytosis: a population-based study. J Pediatr. 2004 Mar;144(3):344-50. doi: 10.1016/j.jpeds.2003.12.030[↩][↩]
- Chow TW, Leung WK, Cheng FWT, Kumta SM, Chu WCW, Lee V, Shing MMK, Li CK. Late outcomes in children with Langerhans cell histiocytosis. Arch Dis Child. 2017 Sep;102(9):830-835. doi: 10.1136/archdischild-2016-312185[↩][↩]
- Rigaud C, Barkaoui MA, Thomas C, Bertrand Y, Lambilliotte A, Miron J, Aladjidi N, Plat G, Jeziorski E, Galambrun C, Mansuy L, Lutz P, Deville A, Armari-Alla C, Reguerre Y, Fraitag S, Coulomb A, Gandemer V, Leboulanger N, Moshous D, Hoang-Xuan K, Tazi A, Heritier S, Emile JF, Donadieu J. Langerhans cell histiocytosis: therapeutic strategy and outcome in a 30-year nationwide cohort of 1478 patients under 18 years of age. Br J Haematol. 2016 Sep;174(6):887-98. doi: 10.1111/bjh.14140[↩]
- Gadner H, Minkov M, Grois N, Pötschger U, Thiem E, Aricò M, Astigarraga I, Braier J, Donadieu J, Henter JI, Janka-Schaub G, McClain KL, Weitzman S, Windebank K, Ladisch S; Histiocyte Society. Therapy prolongation improves outcome in multisystem Langerhans cell histiocytosis. Blood. 2013 Jun 20;121(25):5006-14. doi: 10.1182/blood-2012-09-455774[↩][↩][↩][↩][↩]
- Héritier S, Barkaoui MA, Miron J, Thomas C, Moshous D, Lambilliotte A, Mazingue F, Kebaili K, Jeziorski E, Plat G, Aladjidi N, Pacquement H, Galambrun C, Brugières L, Leverger G, Mansuy L, Paillard C, Deville A, Pagnier A, Lutun A, Gillibert-Yvert M, Stephan JL, Cohen-Aubart F, Haroche J, Pellier I, Millot F, Gandemer V, Martin-Duverneuil N, Taly V, Hélias-Rodzewicz Z, Emile JF, Hoang-Xuan K, Idbaih A, Donadieu J. Incidence and risk factors for clinical neurodegenerative Langerhans cell histiocytosis: a longitudinal cohort study. Br J Haematol. 2018 Nov;183(4):608-617. doi: 10.1111/bjh.15577[↩]
- Grois N, Pötschger U, Prosch H, Minkov M, Arico M, Braier J, Henter JI, Janka-Schaub G, Ladisch S, Ritter J, Steiner M, Unger E, Gadner H; DALHX- and LCH I and II Study Committee. Risk factors for diabetes insipidus in langerhans cell histiocytosis. Pediatr Blood Cancer. 2006 Feb;46(2):228-33. doi: 10.1002/pbc.20425[↩]
- Garg D, Pedapati R, Nakra T, Singh RK, Prabhakar A, Dash D, Bhatia R, Tripathi M. Langerhans cell histiocytosis presenting as a rapidly evolving frontotemporal syndrome. Neurol Sci. 2019 May;40(5):1055-1058. doi: 10.1007/s10072-019-3709-y[↩]
- Sieni E, Barba C, Mortilla M, Savelli S, Grisotto L, Di Giacomo G, Romano K, Fonda C, Biggeri A, Guerrini R, Aricò M. Early Diagnosis and Monitoring of Neurodegenerative Langerhans Cell Histiocytosis. PLoS One. 2015 Jul 15;10(7):e0131635. doi: 10.1371/journal.pone.0131635[↩]
- Grois N, Prayer D, Prosch H, Lassmann H; CNS LCH Co-operative Group. Neuropathology of CNS disease in Langerhans cell histiocytosis. Brain. 2005 Apr;128(Pt 4):829-38. doi: 10.1093/brain/awh403[↩]
- Eckstein OS, Visser J, Rodriguez-Galindo C, Allen CE; NACHO-LIBRE Study Group. Clinical responses and persistent BRAF V600E+ blood cells in children with LCH treated with MAPK pathway inhibition. Blood. 2019 Apr 11;133(15):1691-1694. doi: 10.1182/blood-2018-10-878363[↩]
- Kaste SC, Rodriguez-Galindo C, McCarville ME, Shulkin BL. PET-CT in pediatric Langerhans cell histiocytosis. Pediatr Radiol. 2007 Jul;37(7):615-22. doi: 10.1007/s00247-007-0467-4[↩]
- Phillips M, Allen C, Gerson P, McClain K. Comparison of FDG-PET scans to conventional radiography and bone scans in management of Langerhans cell histiocytosis. Pediatr Blood Cancer. 2009 Jan;52(1):97-101. doi: 10.1002/pbc.21782[↩]
- Chu T: Langerhans cell histiocytosis. Australas J Dermatol 42 (4): 237-42, 2001.[↩][↩]
- Mavrogenis AF, Abati CN, Bosco G, Ruggieri P. Intralesional methylprednisolone for painful solitary eosinophilic granuloma of the appendicular skeleton in children. J Pediatr Orthop. 2012;32(4):416–422. doi: 10.1097/BPO.0b013e3182561153[↩][↩][↩]
- Plasschaert F, Craig C, Bell R, Cole WG, Wunder JS, Alman BA. Eosinophilic granuloma. A different behaviour in children than in adults. J Bone Joint Surg Br. 2002;84(6):870–872. doi: 10.1302/0301-620X.84B6.12585[↩][↩][↩]
- Picarsic J, Jaffe R. Nosology and Pathology of Langerhans Cell Histiocytosis. Hematol Oncol Clin North Am. 2015 Oct;29(5):799-823. doi: 10.1016/j.hoc.2015.06.001[↩]
- Abla O, Jacobsen E, Picarsic J, Krenova Z, Jaffe R, Emile JF, Durham BH, Braier J, Charlotte F, Donadieu J, Cohen-Aubart F, Rodriguez-Galindo C, Allen C, Whitlock JA, Weitzman S, McClain KL, Haroche J, Diamond EL. Consensus recommendations for the diagnosis and clinical management of Rosai-Dorfman-Destombes disease. Blood. 2018 Jun 28;131(26):2877-2890. doi: 10.1182/blood-2018-03-839753[↩]
- Vielgut I, Liegl-Atzwanger B, Bratschitsch G, Leithner A, Radl R. Langerhans-cell histiocytosis of the cervical spine in an adult patient: Case report and review of the literature. J Orthop. 2017 Jun;14(2):264-267[↩]
- DiCaprio MR, Roberts TT. Diagnosis and management of langerhans cell histiocytosis. J Am Acad Orthop Surg. 2014 Oct;22(10):643-52. doi: 10.5435/JAAOS-22-10-643[↩]
- Brown CW, Jarvis JG, Letts M, Carpenter B. Treatment and outcome of vertebral Langerhans cell histiocytosis at the Children’s Hospital of Eastern Ontario. Can J Surg. 2005;48(3):230–236.[↩]
- Metellus P, Gana R, Fuentes S, Eusebio A, Adetchessi A, Dufour H, Grisoli F. Spinal Langerhans’ cell histiocytosis in a young adult: case report and therapeutic considerations. Br J Neurosurg. 2007;21(2):228–230. doi: 10.1080/02688690701268701[↩]
- Osenbach RK, Youngblood LA, Menezes AH. Atlanto-axial instability secondary to solitary eosinophilic granuloma of C2 in a 12-year-old girl. J Spinal Disord. 1990;3:408–412[↩]
- Raab P, Hohmann F, Kuhl J, Krauspe R. Vertebral remodeling in eosinophilic granuloma of the spine: a long-term follow-up. Spine. 1998;23:1351–1354. doi: 10.1097/00007632-199806150-00011[↩]
- Green NE, Robertson WW, Jr, Kilroy AW. Eosinophilic granuloma of the spine with associated neural deficit: report of three cases. J Bone Joint Surg Am. 1980;62:1198–1202. doi: 10.2106/00004623-198062070-00024[↩]
- Greenlee JD, Fenoy AJ, Donovan KA, Menezes AH. Eosinophilic granuloma in the pediatric spine. Pediatr Neurosurg. 2007;43(4):285–292. doi: 10.1159/000103308[↩]
- Yeom JS, Lee CK, Shin HY, Lee CS, Han CS, Chang H. Langerhans’ cell histiocytosis of the spine. Analysis of twenty-three cases. Spine. 1999;24:1740–1749. doi: 10.1097/00007632-199908150-00016[↩]
- Tanaka N, Fujimoto Y, Okuda T, Nakanishi K, Sumida T, Manabe H, Ochi M. Langerhans cell histiocytosis of the atlas. A report of three cases. J Bone Joint Surg Am. 2005;87(10):2313–2317. doi: 10.2106/00004623-200510000-00023[↩]
- Bertram C, Madert J, Eggers C. Eosinophilic granuloma of the cervical spine. Spine. 2002;27:1408–1413. doi: 10.1097/00007632-200207010-00007[↩]
- Levy EI, Scarrow A, Hamilton RC, Wollman MR, Fitz C, Pollack IF. Medical management of eosinophilic granuloma of the cervical spine. Pediatr Neurosurg. 1999;31(3):159–162. doi: 10.1159/000028853[↩]
- Corby RR, Stacy GS, Peabody TD, Dixon LB. Radiofrequency ablation of solitary eosinophilic granuloma of bone. AJR Am J Roentgenol. 2008;190(6):1492–1494. doi: 10.2214/AJR.07.3415[↩]
- Tazi A: Adult pulmonary Langerhans’ cell histiocytosis. Eur Respir J 27 (6): 1272-85, 2006.[↩][↩]
- Mogulkoc N, Veral A, Bishop PW, et al.: Pulmonary Langerhans’ cell histiocytosis: radiologic resolution following smoking cessation. Chest 115 (5): 1452-5, 1999.[↩]
- Kim HJ, Lee KS, Johkoh T, et al.: Pulmonary Langerhans cell histiocytosis in adults: high-resolution CT-pathology comparisons and evolutional changes at CT. Eur Radiol 21 (7): 1406-15, 2011[↩]
- Lorillon G, Tazi A: How I manage pulmonary Langerhans cell histiocytosis. Eur Respir Rev 26 (145), 2017.[↩]
- Dauriat G, Mal H, Thabut G, et al.: Lung transplantation for pulmonary langerhans’ cell histiocytosis: a multicenter analysis. Transplantation 81 (5): 746-50, 2006.[↩][↩]
- Tsambaos D, Georgiou S, Kapranos N, et al.: Langerhans’ cell histiocytosis: complete remission after oral isotretinoin therapy. Acta Derm Venereol 75 (1): 62-4, 1995.[↩]
- Chu T: Langerhans cell histiocytosis. Australas J Dermatol 42 (4): 237-42, 2001[↩]
- Pardanani A, Phyliky RL, Li CY, et al.: 2-Chlorodeoxyadenosine therapy for disseminated Langerhans cell histiocytosis. Mayo Clin Proc 78 (3): 301-6, 2003.[↩][↩]
- Saven A, Foon KA, Piro LD: 2-Chlorodeoxyadenosine-induced complete remissions in Langerhans-cell histiocytosis. Ann Intern Med 121 (6): 430-2, 1994.[↩]
- Derenzini E, Fina MP, Stefoni V, et al.: MACOP-B regimen in the treatment of adult Langerhans cell histiocytosis: experience on seven patients. Ann Oncol 21 (6): 1173-8, 2010.[↩]
- Gadner H: Treatment of adult-onset Langerhans cell histiocytosis–is it different from the pediatric approach? Ann Oncol 21 (6): 1141-2, 2010.[↩]
- Chohan G, Barnett Y, Gibson J, et al.: Langerhans cell histiocytosis with refractory central nervous system involvement responsive to infliximab. J Neurol Neurosurg Psychiatry 83 (5): 573-5, 2012.[↩]
- Sander CS, Kaatz M, Elsner P: Successful treatment of cutaneous langerhans cell histiocytosis with thalidomide. Dermatology 208 (2): 149-52, 2004.[↩]
- Diamond EL, Subbiah V, Lockhart AC, et al.: Vemurafenib for BRAF V600-Mutant Erdheim-Chester Disease and Langerhans Cell Histiocytosis: Analysis of Data From the Histology-Independent, Phase 2, Open-label VE-BASKET Study. JAMA Oncol 4 (3): 384-388, 2018.[↩][↩]
- Hong WC, Murovic JA, Gibbs I, et al.: Pituitary stalk Langerhans cell histiocytosis treated with CyberKnife radiosurgery. Clin Neurol Neurosurg 115 (5): 573-7, 2013.[↩]
- Dhall G, Finlay JL, Dunkel IJ, et al.: Analysis of outcome for patients with mass lesions of the central nervous system due to Langerhans cell histiocytosis treated with 2-chlorodeoxyadenosine. Pediatr Blood Cancer 50 (1): 72-9, 2008.[↩]
- Janku F, Amin HM, Yang D, et al.: Response of histiocytoses to imatinib mesylate: fire to ashes. J Clin Oncol 28 (31): e633-6, 2010.[↩]
- Wagner C, Mohme H, Krömer-Olbrisch T, et al.: Langerhans cell histiocytosis: treatment failure with imatinib. Arch Dermatol 145 (8): 949-50, 2009.[↩]
- Charles J, Beani JC, Fiandrino G, et al.: Major response to vemurafenib in patient with severe cutaneous Langerhans cell histiocytosis harboring BRAF V600E mutation. J Am Acad Dermatol 71 (3): e97-9, 2014.[↩]
- Haroche J, Cohen-Aubart F, Emile JF, et al.: Dramatic efficacy of vemurafenib in both multisystemic and refractory Erdheim-Chester disease and Langerhans cell histiocytosis harboring the BRAF V600E mutation. Blood 121 (9): 1495-500, 2013.[↩]
- Wnorowski M, Prosch H, Prayer D, Janssen G, Gadner H, Grois N. Pattern and course of neurodegeneration in Langerhans cell histiocytosis. J Pediatr. 2008 Jul;153(1):127-32. doi: 10.1016/j.jpeds.2007.12.042[↩]
- Allen CE, Flores R, Rauch R, Dauser R, Murray JC, Puccetti D, Hsu DA, Sondel P, Hetherington M, Goldman S, McClain KL. Neurodegenerative central nervous system Langerhans cell histiocytosis and coincident hydrocephalus treated with vincristine/cytosine arabinoside. Pediatr Blood Cancer. 2010 Mar;54(3):416-23. doi: 10.1002/pbc.22326[↩][↩]
- Bernstrand C, Cederlund K, Henter JI. Pulmonary function testing and pulmonary Langerhans cell histiocytosis. Pediatr Blood Cancer. 2007 Sep;49(3):323-8. doi: 10.1002/pbc.20707[↩][↩][↩]
- Nanduri VR, Pritchard J, Levitt G, Glaser AW. Long term morbidity and health related quality of life after multi-system Langerhans cell histiocytosis. Eur J Cancer. 2006 Oct;42(15):2563-9. doi: 10.1016/j.ejca.2006.05.031[↩][↩]
- Zhou Z, Zhang H, Guo C, Yu H, Wang L, Guo Q. Management of eosinophilic granuloma in pediatric patients: surgical intervention and surgery combined with postoperative radiotherapy and/or chemotherapy. Childs Nerv Syst. 2017 Apr;33(4):583-593. doi: 10.1007/s00381-017-3363-8[↩]
- Allen CE, Ladisch S, McClain KL. How I treat Langerhans cell histiocytosis. Blood. 2015 Jul 2;126(1):26-35. doi: 10.1182/blood-2014-12-569301[↩]
- Galluzzo ML, Braier J, Rosenzweig SD, Garcia de Dávila MT, Rosso D. Bone marrow findings at diagnosis in patients with multisystem langerhans cell histiocytosis. Pediatr Dev Pathol. 2010 Mar-Apr;13(2):101-6. doi: 10.2350/09-05-0651-OA.1[↩]
- Hatemi I, Baysal B, Senturk H, Behzatoglu K, Bozkurt ER, Ozbay G. Adult Langerhans cell histiocytosis and sclerosing cholangitis: a case report and review of the literature. Hepatol Int. 2010 Aug 4;4(3):653-8. doi: 10.1007/s12072-010-9205-3[↩]
- Braier J, Ciocca M, Latella A, de Davila MG, Drajer M, Imventarza O. Cholestasis, sclerosing cholangitis, and liver transplantation in Langerhans cell Histiocytosis. Med Pediatr Oncol. 2002 Mar;38(3):178-82. doi: 10.1002/mpo.1306[↩][↩]
- Gadner H, Grois N, Pötschger U, Minkov M, Aricò M, Braier J, Broadbent V, Donadieu J, Henter JI, McCarter R, Ladisch S; Histiocyte Society. Improved outcome in multisystem Langerhans cell histiocytosis is associated with therapy intensification. Blood. 2008 Mar 1;111(5):2556-62. doi: 10.1182/blood-2007-08-106211[↩][↩][↩][↩][↩]
- Tillotson CV, Patel BC. Langerhans Cell Histiocytosis. [Updated 2019 Jun 3]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2019 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK430885[↩][↩]
- Maia RC, de Rezende LM, Robaina M, Apa A, Klumb CE. Langerhans cell histiocytosis: differences and similarities in long-term outcome of paediatric and adult patients at a single institutional centre. Hematology. 2015 Mar;20(2):83-92. doi: 10.1179/1607845414Y.0000000173[↩]
- Malpas JS, Norton AJ. Langerhans cell histiocytosis in the adult. Med Pediatr Oncol. 1996 Dec;27(6):540-6. doi: 10.1002/(SICI)1096-911X(199612)27:6<540::AID-MPO6>3.0.CO;2-L[↩]
- Di Iorgi N, Allegri AE, Napoli F, Calcagno A, Calandra E, Fratangeli N, Vannati M, Rossi A, Bagnasco F, Haupt R, Maghnie M. Central diabetes insipidus in children and young adults: etiological diagnosis and long-term outcome of idiopathic cases. J Clin Endocrinol Metab. 2014 Apr;99(4):1264-72. doi: 10.1210/jc.2013-3724[↩]
- Ma J, Laird JH, Chau KW, Chelius MR, Lok BH, Yahalom J. Langerhans cell histiocytosis in adults is associated with a high prevalence of hematologic and solid malignancies. Cancer Med. 2019 Jan;8(1):58-66. doi: 10.1002/cam4.1844[↩][↩]
- Egeler RM, Neglia JP, Puccetti DM, Brennan CA, Nesbit ME. Association of Langerhans cell histiocytosis with malignant neoplasms. Cancer. 1993 Feb 1;71(3):865-73. doi: 10.1002/1097-0142(19930201)71:3<865::aid-cncr2820710334>3.0.co;2-0[↩][↩]
- Gadner H, Grois N, Arico M, Broadbent V, Ceci A, Jakobson A, Komp D, Michaelis J, Nicholson S, Pötschger U, Pritchard J, Ladisch S; Histiocyte Society. A randomized trial of treatment for multisystem Langerhans’ cell histiocytosis. J Pediatr. 2001 May;138(5):728-34. doi: 10.1067/mpd.2001.111331. Erratum in: J Pediatr 2001 Jul;139(1):170.[↩][↩]
- Minkov M, Prosch H, Steiner M, Grois N, Pötschger U, Kaatsch P, Janka-Schaub G, Gadner H. Langerhans cell histiocytosis in neonates. Pediatr Blood Cancer. 2005 Nov;45(6):802-7. doi: 10.1002/pbc.20362[↩]
- Héritier S, Emile JF, Barkaoui MA, et al. BRAF Mutation Correlates With High-Risk Langerhans Cell Histiocytosis and Increased Resistance to First-Line Therapy. J Clin Oncol. 2016 Sep 1;34(25):3023-30. doi: 10.1200/JCO.2015.65.9508[↩][↩]
- Berres ML, Lim KP, Peters T, Price J, et al. BRAF-V600E expression in precursor versus differentiated dendritic cells defines clinically distinct LCH risk groups. J Exp Med. 2014 Apr 7;211(4):669-83. doi: 10.1084/jem.20130977. Epub 2014 Mar 17. Erratum in: J Exp Med. 2015 Feb 9;212(2):281.[↩]
- Sakamoto K, Morimoto A, Shioda Y, Imamura T, Imashuku S; Japan LCH Study Group (JLSG). Long-term complications in uniformly treated paediatric Langerhans histiocytosis patients disclosed by 12 years of follow-up of the JLSG-96/02 studies. Br J Haematol. 2021 Feb;192(3):615-620. doi: 10.1111/bjh.17243[↩]
- Willis B, Ablin A, Weinberg V, Zoger S, Wara WM, Matthay KK. Disease course and late sequelae of Langerhans’ cell histiocytosis: 25-year experience at the University of California, San Francisco. J Clin Oncol. 1996 Jul;14(7):2073-82. doi: 10.1200/JCO.1996.14.7.2073[↩][↩][↩][↩]
- Komp DM. Long-term sequelae of histiocytosis X. Am J Pediatr Hematol Oncol. 1981 Summer;3(2):163-8.[↩][↩]
- Lau LM, Stuurman K, Weitzman S. Skeletal Langerhans cell histiocytosis in children: permanent consequences and health-related quality of life in long-term survivors. Pediatr Blood Cancer. 2008 Mar;50(3):607-12. doi: 10.1002/pbc.21322[↩]
- Nanduri V, Tatevossian R, Sirimanna T. High incidence of hearing loss in long-term survivors of multisystem Langerhans cell histiocytosis. Pediatr Blood Cancer. 2010 Mar;54(3):449-53. doi: 10.1002/pbc.22186[↩]
- Nanduri VR, Lillywhite L, Chapman C, Parry L, Pritchard J, Vargha-Khadem F. Cognitive outcome of long-term survivors of multisystem langerhans cell histiocytosis: a single-institution, cross-sectional study. J Clin Oncol. 2003 Aug 1;21(15):2961-7. doi: 10.1200/JCO.2003.05.048[↩]
- Mittheisz E, Seidl R, Prayer D, Waldenmair M, Neophytou B, Pötschger U, Minkov M, Steiner M, Prosch H, Wnorowski M, Gadner H, Grois N. Central nervous system-related permanent consequences in patients with Langerhans cell histiocytosis. Pediatr Blood Cancer. 2007 Jan;48(1):50-6. doi: 10.1002/pbc.20760[↩]
- Guimarães LF, Dias PF, Janini ME, de Souza IP. Langerhans cell histiocytosis: impact on the permanent dentition after an 8-year follow-up. J Dent Child (Chic). 2008 Jan-Apr;75(1):64-8.[↩]
- Egeler RM, Neglia JP, Aricò M, Favara BE, Heitger A, Nesbit ME, Nicholson HS. The relation of Langerhans cell histiocytosis to acute leukemia, lymphomas, and other solid tumors. The LCH-Malignancy Study Group of the Histiocyte Society. Hematol Oncol Clin North Am. 1998 Apr;12(2):369-78. doi: 10.1016/s0889-8588(05)70516-5[↩]
- Castro EC, Blazquez C, Boyd J, Correa H, de Chadarevian JP, Felgar RE, Graf N, Levy N, Lowe EJ, Manning JT Jr, Proytcheva MA, Senger C, Shayan K, Sterba J, Werner A, Surti U, Jaffe R. Clinicopathologic features of histiocytic lesions following ALL, with a review of the literature. Pediatr Dev Pathol. 2010 May-Jun;13(3):225-37. doi: 10.2350/09-03-0622-OA.1[↩]
- Feldman AL, Berthold F, Arceci RJ, Abramowsky C, Shehata BM, Mann KP, Lauer SJ, Pritchard J, Raffeld M, Jaffe ES. Clonal relationship between precursor T-lymphoblastic leukaemia/lymphoma and Langerhans-cell histiocytosis. Lancet Oncol. 2005 Jun;6(6):435-7. doi: 10.1016/S1470-2045(05)70211-4[↩][↩]
- Rodig SJ, Payne EG, Degar BA, Rollins B, Feldman AL, Jaffe ES, Androkites A, Silverman LB, Longtine JA, Kutok JL, Fleming MD, Aster JC. Aggressive Langerhans cell histiocytosis following T-ALL: clonally related neoplasms with persistent expression of constitutively active NOTCH1. Am J Hematol. 2008 Feb;83(2):116-21. doi: 10.1002/ajh.21044[↩][↩]





{kind=link}