Contents
- What is lipodystrophy
- Lipodystrophy symptoms
- Congenital generalized lipodystrophy
- Familial partial lipodystrophy
- What causes familial partial lipodystrophy
- Familial partial lipodystrophy signs and symptoms
- Familial partial lipodystrophy type 1 (Kobberling lipodystrophy)
- Familial partial lipodystrophy type 2 (Dunnigan lipodystrophy)
- Familial partial lipodystrophy type 3
- Familial partial lipodystrophy type 4
- Familial partial lipodystrophy type 5
- Familial partial lipodystrophy type 6 (Autosomal Recessive familial partial lipodystrophy)
- Familial partial lipodystrophy diagnosis
- Familial partial lipodystrophy treatment
- Acquired lipodystrophies
- Extremely rare genetic lipodystrophy syndromes
- Lipodystrophy treatment
What is lipodystrophy
Lipodystrophy is a group of rare disorders of diverse causes which are characterized by variable loss of body fat – either generalized (complete) or partial loss of adipose tissue 1. The loss of body fat may affect nearly the entire body (generalized), only certain body regions (partial) or small areas under the skin (localized) 1. Depending upon the severity and extent of body fat loss, patients may be predisposed to metabolic complications associated with insulin resistance 2. These metabolic complications include early onset of diabetes mellitus, hypertriglyceridemia (high triglycerides in blood) and hepatic steatosis (fatty liver) 3. In some patients, these metabolic complications are challenging to manage and can lead to complications including diabetic nephropathy and retinopathy, acute pancreatitis (from extreme hypertriglyceridemia and chylomicronemia), hepatic cirrhosis and premature cardiovascular disease. Other common clinical manifestations include polycystic ovarian syndrome (PCOS), acanthosis nigricans as a result of severe insulin resistance, and eruptive xanthomas due to extreme hypertriglyceridemia 2.
The loss of body fat can result from underlying genetic defects (genetic lipodystrophies including autosomal recessive or dominant subtypes) or from autoimmune mechanisms (acquired lipodystrophies including generalized or partial subtypes) or drugs (e.g. highly active antiretroviral therapy (HAART)-induced partial lipodystrophy in human immunodeficiency virus (HIV)-infected patients or localized lipodystrophies from insulin and other injected drugs) 3. The localized lipodystrophies and lipodystrophy in HIV-infected patients are the most prevalent subtype of lipodystrophies while the other genetic and acquired lipodystrophies are quite rare 2. Localized lipodystrophies do not predispose to metabolic complications as the loss of fat is trivial; however, other partial or generalized lipodystrophies cause variable predisposition to metabolic complications (Figure 1).
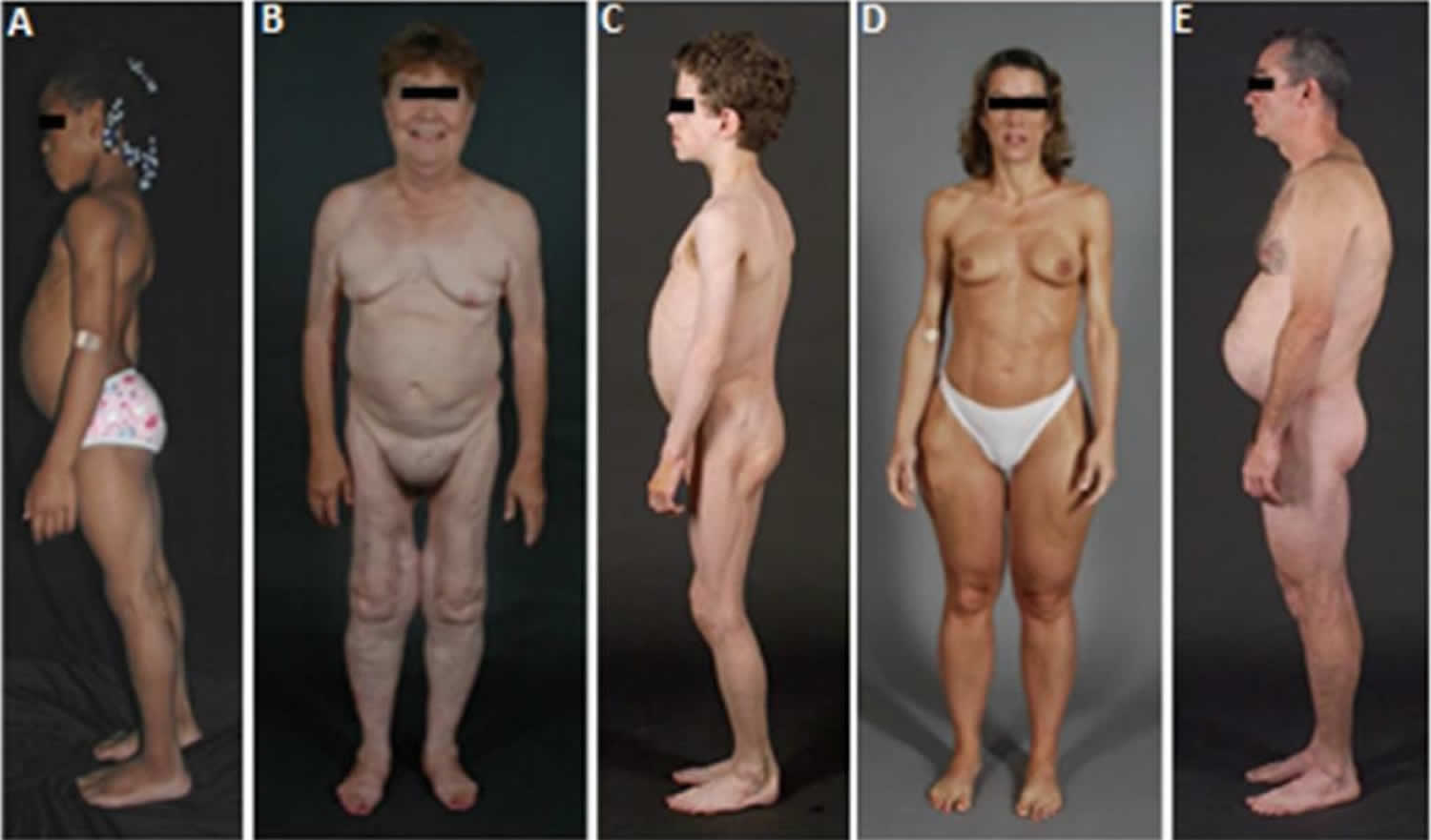
Figure 1. Lipodystrophy syndrome
Footnotes:
A) Lateral view of an 8-year-old African-American female with congenital generalized lipodystrophy (also known as Berardinelli-Seip congenital lipodystrophy), type 1 due to homozygous c.377insT (p.Leu126fs*146) mutation in AGPAT2. The patient had generalized loss of sc fat at birth and developed mild acanthosis nigricans in the axillae and neck later during childhood. She had umbilical prominence and acromegaloid features (enlarged mandible, hands and feet).
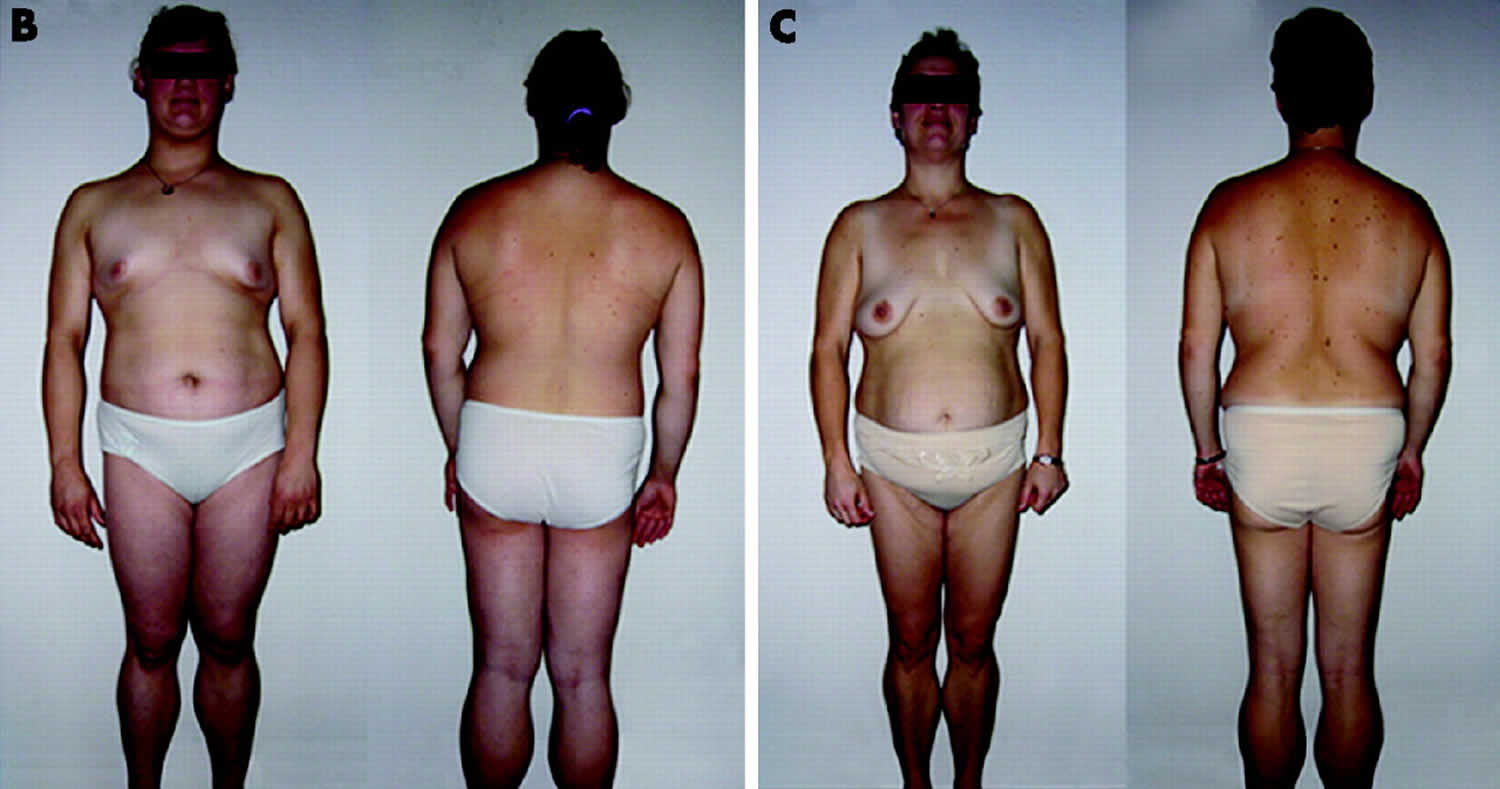
B) Anterior view of a 65-year-old Caucasian female with familial partial lipodystrophy of the Dunnigan variety due to heterozygous p.Arg482Gln mutation in LMNA. She noticed loss of sc fat from the limbs at the time of puberty and later lost sc fat from the anterior truncal region. The breasts were atrophic. She had increased sc fat deposits in the face, anterior neck, suprapubic and vulvar region, and medial parts of the knees.
C) Lateral view of an 8-year-old German boy with acquired generalized lipodystrophy. He started experiencing generalized loss of subcutaneous fat at age 3 with marked acanthosis nigricans in the neck, axillae and groin. He developed Crohn’s disease at age 11 requiring hemicolectomy at age 13.
D) Anterior view of a 39-year-old Caucasian female with acquired partial lipodystrophy (Barraquer-Simons syndrome). She noticed marked loss of sc fat from the face, neck, upper extremities, chest and abdomen at the age of 12 years but later developed increased sc fat deposition in the lower extremities.
E) Lateral view of a 39-year-old Caucasian male infected with human immunodeficiency (HIV) virus with protease inhibitor containing highly active antiretroviral therapy induced lipodystrophy. He had marked loss of sc fat from the face and limbs but had increased subcutaneous fat deposition in the neck region anteriorly and posteriorly showing buffalo hump. Abdomen was protuberant due to excess intra-abdominal fat. He had been on protease inhibitor containing antiretroviral therapy for more than 7 years.
[Source 4]The two main types of genetic lipodystrophies are congenital generalized lipodystrophy, an autosomal recessive syndrome (Table 2 and and 3) and familial partial lipodystrophy, mostly an autosomal dominant syndrome (Table 4). There are other extremely rare types which have been reported in approximately 30 patients or less (Table 8).
The major subtypes of lipodystrophy have been described in Table 1. However, it is important to note that given the heterogeneity of manifestations, variable patterns of fat loss and genetic bases that have yet to be identified, all lipodystrophy syndromes cannot be classified into these categories 5. Regardless of the etiology, patients with generalized lipodystrophy have extremely low serum levels of adipocytokines, such as leptin and adiponectin 6, whereas serum leptin and adiponectin levels in those with partial lipodystrophies can range from low to high. Marked hypoleptinemia may induce excessive appetite and can exacerbate metabolic complications of insulin resistance 3.
Table 1. General Classification of Major Lipodystrophy Subtypes
| Lipodystrophy Subtype | Main Characteristics |
|---|---|
| Congenital generalized lipodystrophy (CGL) | Presents with near total loss of body fat at birth or during infancy. Autosomal recessive inheritance. |
| Familial Partial lipodystrophy (FPL) | Presents with variable loss of sc fat from the upper and lower extremities and the truncal region at puberty or later. Autosomal dominant inheritance. |
| Acquired generalized lipodystrophy (AGL) | Characterized by gradual loss of sc fat from nearly all over the body. Associated with auto-immune diseases. |
| Acquired partial lipodystrophy (APL) | Characterized by gradual loss of fat from the upper body, including head, neck, upper extremities and truncal region during childhood. Associated with autoantibodies called complement 3 nephritic factor and in ~20% of patients with membranoproliferative glomerulonephritis. |
| HAART-induced lipodystrophy in HIV patients | Associated with therapy including HIV-protease inhibitors or nucleoside analogues. |
| Localized lipodystrophy | Usually due to insulin injections or other injectables such as steroids |
Abbreviations: HIV = human immunodeficiency virus; HAART = highly active antiretroviral therapy; sc = subcutaneous
Key points
- Lipodystrophy is a group of heterogeneous disorders characterized by varying degrees of body fat loss and predisposition to insulin resistance related metabolic complications.
- The two main subtypes of lipodystrophies are genetic and acquired lipodystrophies.
- Highly active antiretroviral therapy (HAART)-induced lipodystrophy in HIV-infected patients and drug-induced localized lipodystrophy are common subtypes followed by genetic and acquired autoimmune lipodystrophies.
- Common metabolic abnormalities and complications associated with lipodystrophies include insulin resistance and diabetes mellitus, hypertriglyceridemia and hepatic steatosis.
- Management options include diet and exercise, conventional anti-hyperglycemic agents and lipid-lowering therapy, and metreleptin therapy, which is the only drug approved specifically for generalized lipodystrophy.
Lipodystrophy symptoms
The extent of fat loss may determine the severity of metabolic complications. Some patients may have only cosmetic problems while others may also have severe metabolic complications.
Physical changes
The exact locations of fat tissue loss varies from person to person. For example, some people with lipodystrophy may have areas on their body that look very thin (face and arms), while other areas might appear large (hips or buttocks). Other people with lipodystrophy might have very little fat tissue on the lower areas of the body (legs and buttocks) and excess fat tissue on the upper areas of the body (abdomen, chin, and neck). Still others might have very little visible fat tissue anywhere on their bodies and may appear extremely muscular.
Internal changes
Because people with lipodystrophy are missing or have very low leptin, fat can be found in unusual places like the bloodstream, heart, kidneys, liver, and pancreas. Since fat is not meant to be in these places, it can lead to serious problems, such as insulin resistance, diabetes, high cholesterol, fatty liver disease, pancreatitis and heart disease.
Increase In Appetite
In congenital generalized lipodystrophy, acquired generalized lipodystrophy, and familial partial lipodystrophy, many patients have markedly increased appetites.
Congenital generalized lipodystrophy
Congenital generalized lipodystrophy (also called Berardinelli-Seip congenital lipodystrophy) is a rare condition characterized by an almost total lack of fatty (adipose) tissue in the body and a very muscular appearance. Adipose tissue is found in many parts of the body, including beneath the skin and surrounding the internal organs. It stores fat for energy and also provides cushioning. Congenital generalized lipodystrophy is part of a group of related disorders known as lipodystrophies, which are all characterized by a loss of adipose tissue. A shortage of adipose tissue leads to the storage of fat elsewhere in the body, such as in the liver and muscles, which causes serious health problems.
Congenital generalized lipodystrophy has an estimated prevalence of 1 in 10 million people worldwide. Between 300 and 500 people with the condition have been described in the medical literature. Although this condition has been reported in populations around the world, it appears to be more common in certain regions of Lebanon and Brazil.
The signs and symptoms of congenital generalized lipodystrophy are usually apparent from birth or early childhood. One of the most common features is insulin resistance, a condition in which the body’s tissues are unable to recognize insulin, a hormone that normally helps to regulate blood sugar levels. Insulin resistance may develop into a more serious disease called diabetes mellitus. Most affected individuals also have high levels of fats called triglycerides circulating in the bloodstream (hypertriglyceridemia), which can lead to the development of small yellow deposits of fat under the skin called eruptive xanthomas and inflammation of the pancreas (pancreatitis). Additionally, congenital generalized lipodystrophy causes an abnormal buildup of fats in the liver (hepatic steatosis), which can result in an enlarged liver (hepatomegaly) and liver failure. Some affected individuals develop a form of heart disease called hypertrophic cardiomyopathy, which can lead to heart failure and an abnormal heart rhythm (arrhythmia) that can cause sudden death.
People with congenital generalized lipodystrophy have a distinctive physical appearance. They appear very muscular because they have an almost complete absence of adipose tissue and an overgrowth of muscle tissue. A lack of adipose tissue under the skin also makes the veins appear prominent. Affected individuals tend to have a large chin, prominent bones above the eyes (orbital ridges), large hands and feet, and a prominent belly button (umbilicus). Affected females may have an enlarged clitoris (clitoromegaly), an increased amount of body hair (hirsutism), irregular menstrual periods, and multiple cysts on the ovaries, which may be related to hormonal changes. Many people with this disorder develop acanthosis nigricans, a skin condition related to high levels of insulin in the bloodstream. Acanthosis nigricans causes the skin in body folds and creases to become thick, dark, and velvety.
Researchers have described four types of congenital generalized lipodystrophy, which are distinguished by their genetic cause. The types also have some differences in their typical signs and symptoms. For example, in addition to the features described above, some people with congenital generalized lipodystrophy type 1 develop cysts in the long bones of the arms and legs after puberty. Type 2 can be associated with intellectual disability, which is usually mild to moderate. Type 3 appears to cause poor growth and short stature, along with other health problems. Type 4 is associated with muscle weakness, delayed development, joint abnormalities, a narrowing of the lower part of the stomach (pyloric stenosis), and severe arrhythmia that can lead to sudden death.
Patients with congenital generalized lipodystrophy can develop hyperphagia as a result of profound leptin deficiency in early childhood, and may have accelerated linear growth, advanced bone age and features suggestive of acromegaly such as enlarged hands, feet and jaw 6. Severe metabolic complications, along with hepatomegaly and splenomegaly, develop at an early age. Hyperinsulinemia leads to development of widespread acanthosis nigricans, followed by onset of diabetes mellitus during adolescence 7. Diabetes is generally ketosis-resistant. Some patients develop extreme hypertriglyceridemia especially after the onset of insulin-resistant diabetes mellitus and are prone to recurrent attacks of acute pancreatitis 7.
Fatty liver (hepatic steatosis) is common and severe, and can progress to steato-hepatitis, cirrhosis and liver failure 8. Female congenital generalized lipodystrophy patients have additional clinical features including hirsutism, clitoromegaly, irregular menstrual periods, polycystic ovaries, and/or infertility 9. There are four genetically distinct subtypes of congenital generalized lipodystrophy 10 and besides common clinical features listed above, each one has some peculiar clinical features (Tables 2).
Table 2. Subtypes of Congenital Generalized Lipodystrophy on the basis of genetic mutations
| Subtype | Gene | Molecular Basis | Prevalence |
|---|---|---|---|
| Congenital Generalized Lipodystrophy 1 | AGPAT2 | AGPAT enzymes play a key role in biosynthesis of triglycerides and phospholipids in various organs. AGPAT isoform 2 is highly expressed in the adipose tissue. | Most common subtype |
| Congenital Generalized Lipodystrophy 2 | BSCL2 | Seipin, encoded by BSCL2, plays a key role in fusion of small lipid droplets in the adipocytes and in adipocyte differentiation. | Second most common subtype |
| Congenital Generalized Lipodystrophy 3 | CAV1 | Caveolin 1, is an integral component of caveolae, which are present on adipocyte membranes. Caveolae translocate fatty acids and other lipids to lipid droplets. | Only one patient reported |
| Congenital Generalized Lipodystrophy 4 | PTRF | PTRF (also known as cavin-1) is involved in biogenesis of caveolae and regulates expression of caveolins 1 and 3. | About 20 patients reported |
Abbreviations: CGL = congenital generalized lipodystrophy; AGPAT2 = 1-acylglycerol-3-phosphate O-acyltransferase 2; BSCL2 = Berardinelli-Seip congenital lipodystrophy 2; CAV1 = caveolin 1; PTRF = polymerase I and transcript release factor.
Table 3. Unique clinical features in Congenital Generalized Lipodystrophy subtypes
| Affected feature | CGL type 1 (AGPAT2) | CGL type 2 (BSCL2) | CGL type 3 (CAV1) | CGL type 4 (PTRF) |
|---|---|---|---|---|
| Body fat loss | Only metabolically active adipose tissue is lost. Mechanical adipose tissue preserved. | Both metabolically active and mechanical adipose tissues are lost. | Absent metabolically active adipose tissue. Preserved mechanical and bone marrow adipose tissue. | Absent metabolically active adipose tissue. Preserved mechanical and bone marrow adipose tissue. |
| Cardiovascular complications | N/A | Cardiomyopathy | N/A | Cardiomyopathy, Catecholaminergic polymorphic ventricular tachycardia, prolonged QT, and sudden death. |
| Lytic bone lesions in long bones | Most frequent | Occasional | Not reported | Not reported |
| Gastrointestinal complications | N/A | N/A | Functional mega- esophagus | Congenital pyloric stenosis requiring surgery |
| Skeletal muscle | N/A | N/A | N/A | Congenital myopathy Developmental delay. Muscle weakness, Percussion-induced myotonia |
| Other features | N/A | Terato- zoospermia | Short stature, Hypocalcemia, Vitamin D resistance | Low bone density for age, distal metaphyseal deformation with joint stiffness, atlanto-axial instability. Late onset of lipodystrophy in infancy |
Abbreviations: CGL = congenital generalized lipodystrophy; N/A = not applicable.
[Source 11]Congenital generalized lipodystrophy signs and symptoms
Infants with all forms of congenital generalized lipodystrophy have a near total absence of body fat at birth or soon thereafter. They also have an extremely muscular appearance and may display prominent superficial veins. During early childhood, most children grow at an accelerated rate and have slightly enlarged hands, feet, and jaws (acromegaloid features). Infants and children have a markedly increased appetite and have been described as voracious eaters.
In individuals with congenital generalized lipodystrophy, fat deposits build up in areas of the body such as the muscles and liver. Consequently, affected individuals may develop abnormal enlargement of the muscles (muscular hypertrophy) or the liver (hepatomegaly). Some individuals may also have an abnormally enlarged spleen (splenomegaly). Hepatomegaly is often noticed during infancy. Fat accumulation in the liver (known as fatty liver or hepatic steatosis) may eventually cause scarring and damage to the liver (cirrhosis) and liver dysfunction. Ultimately, liver failure may develop in some patient, necessitating a liver transplant.
Individuals with congenital generalized lipodystrophy develop metabolic complications associated with insulin resistance. Some infants have a condition called acanthosis nigricans, a skin condition characterized by abnormally increased coloration (hyperpigmentation) and “velvety” thickening (hyperkeratosis) of the skin, particularly of skin fold regions, such as of the neck and groin and under the arms (axillae). Other complications of insulin resistance may occur at a young age (often between 15-20 years of age) including glucose intolerance, hypertriglyceridemia, and diabetes. These symptoms are often very difficult to control and diabetes is often severe. Some individuals may experience extreme hypertriglyceridemia and chylomicronemia a condition characterized by the accumulation of chylomicrons (lipoprotein particles carrying fat) in the plasma. In some cases, this can result in episodes of acute inflammation of the pancreas (pancreatitis). Acute pancreatitis can be associated with abdominal pain, chills, jaundice, weakness, sweating, vomiting, and weight loss.
Intellectual disability can occur in congenital generalized lipodystrophy, especially in cases caused by mutations of the BSCL2 gene (congenital generalized lipodystrophy type 2). However, the presence and/or severity of intellectual disability can vary dramatically from one person to another, even among members of the same family. Most cases have been associated with mild or moderate intellectual disability. Intellectual disability is not common in other forms of congenital generalized lipodystrophy.
After puberty, some women with congenital generalized lipodystrophy may develop polycystic ovary syndrome (PCOS). PCOS is characterized by an imbalance of female sex hormones. Affected women may also have too much androgen, a male hormone, in the body. PCOS can result in irregular menstrual periods or a lack of menstruation, oily skin that is prone to acne, multiple cysts on the ovaries, and mild hirsutism (a male pattern of hair growth). Hair may develop on the upper lip and chin. Most women with congenital generalized lipodystrophy are unable to conceive. However, in a few reported cases, affected women have had successful pregnancies. Affected men usually have normal reproductive capabilities. Recently, morphological defects in sperms were reported in a 20-year-old male with congenital generalized lipodystrophy, type 2.
Heart irregularities may occur in some cases, especially abnormal thickening of the muscular walls of the left lower chamber of the heart (hypertrophic cardiomyopathy). This condition can obstruct the flow of blood in and out of the heart. Some individuals may have no associated symptoms; others may develop shortness of breath upon exertion, fatigue, and excessive sweating. As affected individuals age, they may experience chest pain or discomfort, irregular heartbeats, dizziness or fainting usually upon heavy exertion, and, eventually, life-threatening complications such as congestive heart failure. Hypertrophic cardiomyopathy is most common is individuals with congenital generalized lipodystrophy types 2 and 4. It often develops in individuals around the age of 30, but has been reported in infants as well.
Additional findings have been reported in individuals with congenital generalized lipodystrophy including excessive sweating (hyperhidrosis). Some findings are more likely to be associated with a specific subtype of congenital generalized lipodystrophy such as the formation of bone cysts after puberty (more common in types 1 and 2), which can cause individuals to be prone to spontaneous fractures; bone marrow fat loss (more common in types 1and 2); and problems with vitamin D, reported in a patient with congenital generalized lipodystrophy type 3. Muscular dystrophy, a general term for disorders that cause muscle weakness and loss of muscle tissue, and pyloric stenosis (constriction of the opening of stomach into duodenum) is seen in individuals with congenital generalized lipodystrophy type 4. Irregular heartbeats (cardiac arrhythmias) and sudden death have also been associated with congenital generalized lipodystrophy type 4.
Individuals with congenital generalized lipodystrophy type 1 lack metabolically active fat, which is the fat plays a role in the storage and release of energy and is located in subcutaneous regions, intermuscular regions, the bone marrow and areas with the abdomen and chest (thoracic cavity), but mechanical fat is well preserved. Mechanical fat is the fat that supports and protects regions subjected to mechanical insults and is located in the palms, soles, eye sockets (orbits), scalp, and around the joints. Individuals with congenital generalized lipodystrophy type 2 are prone to having a more severe form of lipodystrophy because they also experience the loss of mechanical fat.
Congenital generalized lipodystrophy causes
Mutations in the AGPAT2, BSCL2, CAV1, and CAVIN1 genes cause congenital generalized lipodystrophy types 1 through 4, respectively. The proteins produced from these genes play important roles in the development and function of adipocytes, which are the fat-storing cells in adipose tissue. Mutations in any of these genes reduce or eliminate the function of their respective proteins, which impairs the development, structure, or function of adipocytes and makes the body unable to store and use fats properly. These abnormalities of adipose tissue disrupt hormones and affect many of the body’s organs, resulting in the varied signs and symptoms of congenital generalized lipodystrophy.
Some of the genes associated with congenital generalized lipodystrophy also play roles in other cells and tissues. For example, the protein produced from the BSCL2 gene is also present in the brain, although its function is unknown. A loss of this protein in the brain may help explain why congenital generalized lipodystrophy type 2 is sometimes associated with intellectual disability.
In some people with congenital generalized lipodystrophy, no mutations have been found in any of the genes listed above. Researchers are looking for additional genetic changes associated with this disorder.
Congenital generalized lipodystrophy is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition. Recessive genetic disorders occur when an individual inherits an abnormal gene for the same trait from each parent. If an individual receives one normal gene and one abnormal gene for the disease, the person will be a carrier for the disease, but usually will not show symptoms. The risk for two carrier parents to both pass the defective gene and, therefore, have an affected child is 25% with each pregnancy. The risk to have a child who is a carrier like the parents is 50% with each pregnancy. The chance for a child to receive normal genes from both parents and be genetically normal for that particular trait is 25%. The risk is the same for males and females.
The AGPAT2 gene contains instructions for creating (encoding) the enzyme AGPAT2, which is involved in the creation (synthesis) of triglycerides and fatty substances called phospholipids.
The BSCL2 gene encodes a protein known as seipin. Recent data suggest a role of seipin in fusion of lipid droplets and in fat cell differentiation.
The CAV1 gene encodes caveolin-1, which is expressed in caveolae, tiny structures on the surface of cells. Caveolae play a role in the formation of lipid droplets, most likely by transporting lipids and phospholipids from outside the cell to inside the cell. Lipid droplets are organelles, specialized subunits found within cells that have specific functions. One function of lipid droplets is the storage of lipids.
The PTRF gene encodes cavin, an essential protein factor in the creation (biogenesis) of caveolae.
Researchers believe that various genes and gene products associated with congenital generalized lipodystrophy are involved with the proper creation, function, and/or health of lipid droplets within adipocytes. Adipocytes are fat cells. Each adipocyte has a lipid droplet that accounts for approximately 90% of its cell volume. An adipocyte stores fats (triglycerides) within its lipid droplet. Mutations in the above mentioned genes ultimately lead to a loss of adipocytes and an inability to store fat. Consequently, fat is stored in other tissue of the body such as the liver and skeletal muscle causing symptoms such as liver disease and insulin resistance. The cause of other symptoms sometimes associated with congenital generalized lipodystrophy such as cardiomyopathy is unknown. More research is necessary to understand the exact, underlying mechanisms that ultimately cause congenital generalized lipodystrophy and its associated symptoms.
Congenital generalized lipodystrophy treatment
The treatment of congenital generalized lipodystrophy is directed toward the specific symptoms that are apparent in each individual. Treatment may require the coordinated efforts of a team of specialists. Pediatricians, surgeons, cardiologists, endocrinologists, nutritionists, and other healthcare professionals may need to systematically and comprehensively plan an affect child’s treatment.
Individuals with congenital generalized lipodystrophy and their families are encouraged to seek counseling after a diagnosis and before treatment because the diagnosis can cause anxiety, stress, and extreme psychological distress. Psychological support and counseling both professionally and through support groups is recommended for affected individuals and their families. Genetic counseling may be of benefit for affected individuals and their families as well.
Despite the lack of clinical trial evaluation, individuals with congenital generalized lipodystrophy are encouraged to follow a high carbohydrate, low-fat diet. Such a diet can improve chylomicronemia associated with acute pancreatitis. However, such diets may also raise very low density lipoprotein triglyceride concentration. It is important that children still consume sufficient energy for proper growth and development. Regular exercise and maintaining a healthy weight are also encouraged as a way to decrease the chances of developing diabetes.
Leptin is a hormone found in adipocytes. Severe lipodystrophy is sometimes associated with leptin deficiency. In 2014, the U.S. Food and Drug Administration approved Myalept (metreleptin for injection) as replacement therapy to treat the complications of leptin deficiency, in addition to diet, in patients with congenital generalized or acquired generalized lipodystrophy. Myalept is a recombinant analogue (laboratory-created form) of human leptin and is taken once a day by subcutaneous (under the skin) injection. It has been found to be beneficial for improving metabolic complications, such as diabetes and hypertriglyceridemia. The most common side effects are low blood sugar (hypoglycemia), headache, and decreased weight. Other side effects can be local reactions at injection site, possibility of developing leptin antibodies (which could result in severe infections or loss of treatment effectiveness) and rare cases of T- cell lymphoma. It is contraindicated in patients with general obesity not associated with congenital leptin deficiency and with prior severe hypersensitivity reactions to metreleptin or any of the product components. Myalept is available only through a restricted distribution program under a Risk Evaluation and Mitigation Strategy (REMS) which requires prescriber and pharmacy certification and special documentation. Detailed information about the medication for providers and patients can be found here: https://www.fda.gov/downloads/drugs/drugsafety/postmarketdrugsafetyinformationforpatientsandproviders/ucm388903.pdf
Individuals with extreme hypertriglyceridemia may be treated with fibric acid derivatives, statins, or n-3 polyunsaturated fatty acids from fish oils.
The characteristic loss of adipose tissue in individuals with congenital generalized lipodystrophy cannot be reversed. Consequently, cosmetic surgery may be beneficial in improving appearance. Individuals with severe facial lipodystrophy can undergo reconstructive facial surgery including fascial grafts from the thighs, free flaps from the anterolateral thigh, anterior abdomen, or temporalis muscle.
In some patients, liver disease can ultimately require a liver transplantation.
Additional therapies to treat individuals with congenital generalized lipodystrophy are symptomatic and supportive and follow regular, standard guidelines. Diabetes is treated with standard therapies. After the onset of diabetes, hyperglycemic drugs such as metformin and sulfonylureas may be recommended to treat hyperglycemia. Insulin can also be used to treat individuals with congenital generalized lipodystrophy and diabetes, although extremely high doses are often required. Although drug therapy is commonly used, there have been no clinical trials to establish the optimal use of drug therapy to treat the metabolic complications associated with congenital generalized lipodystrophy.
Special remedial education may be necessary for individuals with intellectual disability. Psychosocial support for the entire family is essential as well.
Familial partial lipodystrophy
Familial partial lipodystrophy is a rare condition characterized by an abnormal distribution of fatty (adipose) tissue 12. Adipose tissue is normally found in many parts of the body, including beneath the skin and surrounding the internal organs. It stores fat as a source of energy and also provides cushioning. In people with familial partial lipodystrophy, adipose tissue is lost from the arms, legs, and hips, giving these parts of the body a very muscular appearance. The fat that cannot be stored in the limbs builds up around the face and neck, and inside the abdomen. Excess fat in these areas gives individuals an appearance described as “cushingoid,” because it resembles the physical features associated with a hormonal disorder called Cushing disease. This abnormal fat distribution can begin anytime from childhood to adulthood.
Abnormal storage of fat in the body can lead to health problems in adulthood. Many people with familial partial lipodystrophy develop insulin resistance, a condition in which the body’s tissues cannot adequately respond to insulin, which is a hormone that normally helps to regulate blood sugar levels. Insulin resistance may worsen to become a more serious disease called diabetes mellitus. Some people with familial partial lipodystrophy develop acanthosis nigricans, a skin condition related to high levels of insulin in the bloodstream. Acanthosis nigricans causes the skin in body folds and creases to become thick, dark, and velvety.
Most people with familial partial lipodystrophy also have high levels of fats called triglycerides circulating in the bloodstream (high triglycerides or hypertriglyceridemia), which can lead to inflammation of the pancreas (pancreatitis). Familial partial lipodystrophy can also cause an abnormal buildup of fats in the liver (hepatic steatosis), which can result in an enlarged liver (hepatomegaly) and abnormal liver function. After puberty, some affected females develop multiple cysts on the ovaries, an increased amount of body hair (hirsutism), and an inability to conceive (infertility), which are likely related to hormonal changes.
Researchers have described at least six forms of familial partial lipodystrophy, which are distinguished by their genetic cause.
Table 4. Familial Partial Lipodystrophy subtypes
| Subtypes | Genetic Mutation | Prevalence |
|---|---|---|
| FPLD 1 (Kobberling-type) | Molecular basis unknown | Rare |
| FPLD2 (Dunnigan- type) | Missense mutations in LMNA | Most common subtype. More than 500 patients reported |
| FPLD3 | Heterozygous mutations in PPARG | Second most common subtype. About 30–50 patients reported. |
| FPLD4 | Heterozygous mutations in PLIN1 | Reported in three families |
| FPLD5 | Homozygous nonsense mutation in CIDEC (Autosomal recessive) | One patient reported |
| FPLD6 | Homozygous mutation in LIPE (Autosomal recessive) | Six patients reported |
| AKT2-linked lipodystrophy | Heterozygous mutation in AKT2 | Reported in one family |
Abbreviations: FPLD = familial partial lipodystrophy; LMNA = lamin A/C; PPARG = peroxisome proliferator-activated receptor gamma; PLIN1 = perilipin 1; CIDEC = cell death-inducing DFFA-like effector c; LIPE = hormone sensitive lipase; AKT2 = v-akt murine thymoma viral oncogene homolog 2.
Subdivisions of familial partial lipodystrophy:
- Familial partial lipodystrophy type 1 (Kobberling lipodystrophy)
- Familial partial lipodystrophy type 2 (Dunnigan lipodystrophy)
- Familial partial lipodystrophy type 3
- Familial partial lipodystrophy type 4
- Familial partial lipodystrophy type 5
- Familial partial lipodystrophy type 6 (Autosomal recessive familial partial lipodystrophy)
The most common form of Familial partial lipodystrophy is type 2, also called Dunnigan disease. In addition to the signs and symptoms described above, some people with this type of the disorder develop muscle weakness (myopathy), abnormalities of the heart muscle (cardiomyopathy), a form of heart disease called coronary artery disease, and problems with the electrical system that coordinates the heartbeat (the conduction system).
Familial partial lipodystrophy is a rare disease, affecting an estimated 1 in 1 million people overall. Type 2 is the most common form, with more than 500 cases reported in the medical literature. Women tend to be diagnosed with familial partial lipodystrophy more often than men, probably because a loss of fat from the hips and limbs is more easily recognized in women, and complications such as diabetes and hypertriglyceridemia (high triglycerides) occur more commonly in women.
What causes familial partial lipodystrophy
Familial partial lipodystrophy can be caused by mutations in several genes. Type 2 results from mutations in the LMNA gene. The other, less common forms of the disorder are caused by mutations in different genes.
LMNA and the other genes associated with familial partial lipodystrophy provide instructions for making proteins with a variety of functions, including important roles in fat storage. In particular, these proteins play important roles in the development and function of adipocytes, which are the fat-storing cells in adipose tissue. Mutations in any of the genes associated with familial partial lipodystrophy reduce or eliminate the function of their respective proteins, which impairs the development, structure, or function of adipocytes and makes the body unable to store and use fats properly. These abnormalities of adipose tissue alter hormone production and affect many of the body’s organs. However, it is unclear why the changes cause fat to be lost in some parts of the body and stored abnormally in others.
In some people with familial partial lipodystrophy, no gene mutations have been found. Researchers are looking for additional genetic changes that can cause this disorder.
Inheritance pattern
Most cases of familial partial lipodystrophy, including type 2, are inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder. In some cases, an affected person inherits the mutation from one affected parent. Other cases result from new mutations in the gene and occur in people with no history of the disorder in their family.
Rare cases of familial partial lipodystrophy appear to be inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
Familial partial lipodystrophy signs and symptoms
Familial partial lipodystrophy encompasses several subtypes differentiated by the underlying genetic mutation. The specific symptoms present, severity, and prognosis can vary greatly depending upon the specific type of familial partial lipodystrophy and the presence and extent of associated symptoms. The specific symptoms and severity can also vary among individuals with the same subtype and even among members of the same family. In addition, some subtypes of familial partial lipodystrophy have only been reported in a handful of individuals, which prevents physicians from developing an accurate picture of associated symptoms, severity, and prognosis. Therefore, it is important to note that affected individuals will not have all of the symptoms discussed below. Affected individuals should talk to their physician and medical team about their specific case, associated symptoms, and overall prognosis.
Common symptoms of familial partial lipodystrophy include selective, progressive loss of subcutaneous fat in the arms and legs and chest and trunk regions, abnormal accumulation of subcutaneous fat in other areas, and a variety of metabolic complications. Generally, women are more severely affected than men by the metabolic complications of familial partial lipodystrophy. Additional symptoms including those affecting the liver or heart may also occur.
Familial partial lipodystrophy type 1 (Kobberling lipodystrophy)
Kobberling lipodystrophy (familial partial lipodystrophy type 1) has only been reported in a handful of individuals. The symptoms are similar to those seen in familial partial lipodystrophy type 2, Dunnigan lipodystrophy. However, fat loss is generally confined to the arms and legs. Fat loss is usually more prominent on the lower (distal) portions of the arms and legs. Affected individuals have normal or slightly increased fat distribution on the face, neck, and trunk. In addition, some affected individuals may develop excess belly fat (central obesity). Metabolic abnormalities including insulin resistance, high blood pressure (hypertension), and severe hypertriglyceridemia have also been reported. This form of familial partial lipodystrophy has only been reported in women.
Familial partial lipodystrophy type 2 (Dunnigan lipodystrophy)
Familial partial lipodystrophy type 2 (Dunnigan lipodystrophy) is the most common form of familial partial lipodystrophy. Affected individuals usually have normal fat distribution during early childhood. However, around the time of puberty, fat in the arms and legs and trunk is gradually lost. In women, the loss of fat may be most striking in the buttocks and hips. At this time, fat may accumulate in other areas of the body including the face, causing a double chin, and the neck and upper back between the shoulder blades, causing a hump. Affected individuals may have a round face similar to individuals with Cushing’s syndrome. This characteristic distribution of fat and the overall muscular appearance makes the disorder more easily recognizable in women than men.
Insulin resistance is common and may be associated with a condition called acanthosis nigricans, a skin condition characterized by abnormally increased coloration (hyperpigmentation) and “velvety” thickening (hyperkeratosis) of the skin, particularly of skin fold regions, such as of the neck and groin and under the arms (axillae). An enlarged liver (hepatomegaly) is also common. Hepatomegaly is caused by the accumulation of fat in the liver (fatty liver or steatosis). Progressive accumulation of fat in the liver can cause scarring and damage to the liver (cirrhosis) and, eventually, liver dysfunction.
Other complications of insulin resistance may occur including glucose intolerance, hypertriglyceridemia, and diabetes. These symptoms are often very difficult to control and diabetes is often severe. Affected women are at a greater risk of developing diabetes than affected men and often experience more severe metabolic complications. Some individuals may experience extreme hypertriglyceridemia, resulting in episodes of acute inflammation of the pancreas (pancreatitis). Pancreatitis can be associated with abdominal pain, chills, jaundice, weakness, sweating, vomiting, and weight loss.
After puberty, some women with familial partial lipodystrophy may develop polycystic ovary syndrome (PCOS), a complex of symptoms that are not always present in every case. PCOS is often characterized by an imbalance of sex hormones as affected women may have too much androgen, a male hormone, in the body. PCOS can result in irregular menstrual periods or a lack of menstruation, oily skin that is prone to acne, cysts on the ovaries, failure of the ovary to release eggs, and mild hirsutism (a male pattern of hair growth). Hair may develop on the upper lip, chin and other parts of the body.
Individuals with familial partial lipodystrophy, Dunnigan variety are predisposed to coronary artery disease and other types of atherosclerotic vascular disease. In rare cases, in which individuals have a specific mutation of the lamin A/C (LMNA) gene, they are at an increased risk of developing disease of the heart muscles (cardiomyopathy), which can result in congestive heart failure and irregular heartbeats (cardiac arrhythmias) such as heart block or atrial fibrillation. Some individuals also develop muscular dystrophies (diseases of muscles causing loss of strength and joint contractures).
Familial partial lipodystrophy type 3
This form of familial partial lipodystrophy has only been reported in approximately 30 individuals. It is generally milder than the familial partial lipodystrophy type 2, Dunnigan variety. Consequently, it is believed that many cases may go undiagnosed. Fat loss is more prominent in the calves and forearms than in the upper arms and thighs. Diabetes, hypertriglyceridemia, hypertension, fatty liver, pancreatitis, and hirsutism have also been reported. Metabolic abnormalities are more prominent than the lipodystrophy in this form of the disorder.
Familial partial lipodystrophy type 4
This form of familial partial lipodystrophy has only been reported in a handful of individuals. Lipodystrophy is most prominent in the lower limbs and buttocks. Muscular hypertrophy may be prominent in the calves. Insulin resistance, severe hypertriglyceridemia, and diabetes were also reported.
Familial partial lipodystrophy type 5
This form of familial partial lipodystrophy has been reported in four members of one family who had hypertension, severe insulin resistance, and diabetes mellitus. Insulin resistance appears around the ages of 20 to 30. Lipodystrophy most prominently affects the arms and legs.
Familial partial lipodystrophy type 6 (Autosomal Recessive familial partial lipodystrophy)
This form of familial partial lipodystrophy has only been reported in one individual in the medical literature. The reported symptoms include partial lipodystrophy, severe insulin resistance, fatty liver, acanthosis nigricans, and diabetes.
Familial partial lipodystrophy diagnosis
A diagnosis of familial partial lipodystrophy is based upon identification of characteristic symptoms, a detailed patient history, and a thorough clinical evaluation. A diagnosis of familial partial lipodystrophy should be suspected in individuals who lose subcutaneous fat around puberty and gain a muscular appearance. Lipodystrophy, in general, should be suspected in individuals who are lean or “non-obese” and who present with early diabetes, severe hypertriglyceridemia, hepatic steatosis, hepatosplenomegaly, acanthosis nigricans and/or polycystic ovarian syndrome (PCOS).
Clinical Testing and Workup
Although the diagnosis of lipodystrophy is primarily clinical, a variety of tests can be used to aid in the diagnosis and/or rule out other conditions. A blood chemical profile may be conducted to assess the levels of glucose, lipids, liver enzymes, and uric acid.
The characteristic pattern of fat loss in the arms and legs and trunk, but fat gain in muscular fasciae can be noted on magnetic resonance imaging (MRI).
Molecular genetic testing can confirm a diagnosis of familial partial lipodystrophy in most cases. Molecular genetic testing can detect mutations in specific genes that cause familial partial lipodystrophy, but is only available on a clinical basis for only few genes such as LMNA.
Individuals with familial partial lipodystrophy may undergo tests to detect and/or evaluate the presence of potential complications including heart abnormalities. Holter monitoring, echocardiography, and a stress test are conducted for individuals suspected of have cardiomyopathy or coronary heart disease. A Holter monitor is a portable device that continually monitors the heart’s rhythms. An echocardiography uses reflected sound waves to create a picture of the heart. A stress test measures the heart’s ability to respond to external stress in a controlled environment.
Individuals with familial partial lipodystrophy may be evaluated to determine their leptin levels. Leptin is a hormone found in adipocytes. Some affected individuals have low levels of leptin.
Familial partial lipodystrophy treatment
The treatment of familial partial lipodystrophy is directed toward the specific symptoms that are apparent in each individual. Treatment may require the coordinated efforts of a team of specialists. Pediatricians, plastic surgeons, cardiologists, endocrinologists, nutritionists, and other healthcare professionals may need to systematically and comprehensively plan an affect child’s treatment.
Individuals with familial partial lipodystrophy and their families are encouraged to seek counseling after a diagnosis because the diagnosis can cause anxiety, stress, and extreme psychological distress. Psychological support and counseling both professionally and through support groups is recommended for affected individuals and their families. Genetic counseling may be of benefit for affected individuals and their families as well.
Despite the lack of clinical trial evaluation, individuals with familial partial lipodystrophy are encouraged to follow a high carbohydrate, low-fat diet. Such a diet can improve chylomicronemia associated with acute pancreatitis. Chylomicronemia is a condition characterized by the accumulation of fatty droplets called chylomicrons in the plasma. However, such diets may also raise very low density lipoprotein triglyceride concentration.
Because individuals with familial partial lipodystrophy have an increased risk of coronary heart disease, they should limit the intake of saturated and trans-unsaturated fats and dietary cholesterol. It is unknown whether such measures will be beneficial over the long term to reduce fatty liver or serum triglycerides levels, or whether they can improve glycemic control.
Regular exercise and maintaining a healthy weight are also encouraged as a way to decrease the chances of developing diabetes. In individuals with familial partial lipodystrophy, exercise and reducing energy intake can is also necessary to avoid excess fat deposition and accumulation in non-lipodystrophic areas such as the face, neck, and intra-abdominal region.
Individuals with extreme hypertriglyceridemia may be treated with fibric acid derivatives, statins, or omega-3 polyunsaturated fatty acids.
The characteristic loss of adipose tissue in individuals with familial partial lipodystrophy cannot be reversed. Consequently, cosmetic surgery may be beneficial in improving appearance and management metabolic complications. Procedures such as liposuction can be performed to remove excess, unwanted fat in areas where fat accumulates (e.g. chin).
In some cases, liver disease associated with familial partial lipodystrophy can ultimately require a liver transplantation.
Periodic cardiac examinations may be recommended for individuals with familial partial lipodystrophy to detect symptoms that can be potentially associated with the disorder including coronary heart disease and/or conduction defects. Affected individuals with heart abnormalities such as heart block or atrial fibrillation may require the use of a pacemaker. In some cases, a heart transplant may ultimately be necessary.
Additional therapies to treat individuals with familial partial lipodystrophy are symptomatic and supportive and follow regular, standard guidelines. Diabetes is treated with standard therapies. After the onset of diabetes, hyperglycemic drugs such as metformin and sulfonylureas may be recommended to treat hyperglycemia. Insulin can also be used to treat individuals with familial partial lipodystrophy and diabetes, although extremely high doses are often required. High blood pressure (anti-hypertensives) may also be recommended. Although drug therapy is commonly used, there have been no clinical trials to establish the optimal use of drug therapy to treat the metabolic complications in individuals with familial partial lipodystrophy.
Acquired lipodystrophies
HIV Lipodystrophy
Lipodystrophy is a condition characterized by abnormal fat distribution that can lead to either lipoatrophy (fat loss in the face, buttocks, arms, and legs) or lipohypertrophy (fat accumulation in specific areas of the body such as the neck, belly, upper torso, and breasts) 13. The term HIV-associated lipodystrophy refers to both abnormal central fat accumulation (lipohypertrophy) and localized loss of fat, most commonly in the mid portion of the face (lipoatrophy); however, some patients present only lipohypertrophy or lipoatrophy 14.
Fat buildup (also called lipohypertrophy) can occur:
- Around the organs in the belly (also called the abdomen).
- On the back of the neck between the shoulders (called a buffalo hump).
- In the breasts.
- Just under the skin. (The fatty bumps are called lipomas.)
Fat loss (also called lipoatrophy) tends to occur:
- In the arms and legs.
- In the buttocks.
- In the face.
While the exact mechanisms underlying HIV lipodystrophy are unknown, there are several hypotheses based on human and test tube studies that attempt to explain the unique pathophysiologic characteristics of this condition. Protease inhibitors and nucleoside reverse-transcriptase inhibitors, especially stavudine and zidovudine, have been suggested to inhibit both cytoplasmic retinoic acid–binding protein and low-density lipoprotein receptor–related protein 15. Although more research is needed to prove that there is a link between HIV medicines and lipodystrophy, some HIV medicines have been associated with the condition. While highly active antiretroviral therapy (HAART) is highly efficacious, the substantial negative impact on patient quality of life of the associated lipodystrophy reduces patient compliance. In a recent study of patients with HIV lipodystrophy, 90% of patients attributed the lipodystrophy to their HIV medications; 20% had thoughts of suicide secondary to the condition; and 20% stopped taking their HIV medications owing to concerns about development of the syndrome 16. Newer HIV medicines are less likely to cause lipodystrophy than HIV medicines developed in the past 17.
Table 5. The cause of Drug-induced Lipodystrophy in HIV-infected patients
| Type/Etiology | Pathogenesis and molecular basis |
|---|---|
| Protease inhibitor-induced | Protease inhibitors inhibit ZMPSTE24, which is important for the correct maturation and processing of prelamin A. Thus, protease inhibitors result in accumulation of toxic farnesylated prelamin A. May also cause dysregulation of transcription factors involved in adipogenesis. They may also inhibit glucose transporter 4 expression leading to insulin resistance. |
| Nucleoside reverse transcriptase inhibitor-induced | Nucleoside reverse transcriptase inhibitors (especially stavudine and zidovudine) inhibit mitochondrial polymerase-γ and subsequently cause mitochondrial toxicity. |
Abbreviations: PI = protease inhibitor; NRTI = nucleoside reverse transcriptase inhibitor; ZMPSTE24 = zinc mellatoproteinase STE24 = polymerase-γ, polymerase gamma.
In HIV-associated lipohypertrophy, the most typical areas for fat accumulation include the buffalo hump secondary to an enlarged dorsocervical fat pad, breast enlargement, and central truncal adiposity due to abdominal visceral fat accumulation (often referred to as “crix belly” or “protease paunch” for fat accumulation secondary to indinavir and other protease inhibitors) 20. Rarer presentations of lipodystrophy include multiple symmetric and asymmetric lipomatoses (symmetric lipomatoses extending from the breasts to the axillae have been reported) and suprapubic fat pads (pubic lipomas), which were reported in nearly 10% of patients with lipodystrophy 21. Involvement of the face is the most stigmatizing form of HIV-associated lipoatrophy, substantially reducing patient quality of life and inhibiting compliance with antiretroviral therapy 16. While a number of reports in the literature have described the dysmorphic features of the buffalo hump resulting from the enlargement of the dorsocervical fat pad, Figure 2 is a less-reported feature of the increase in neck circumference or “bullfrog neck” in patients with HIV lipodystrophy 22.
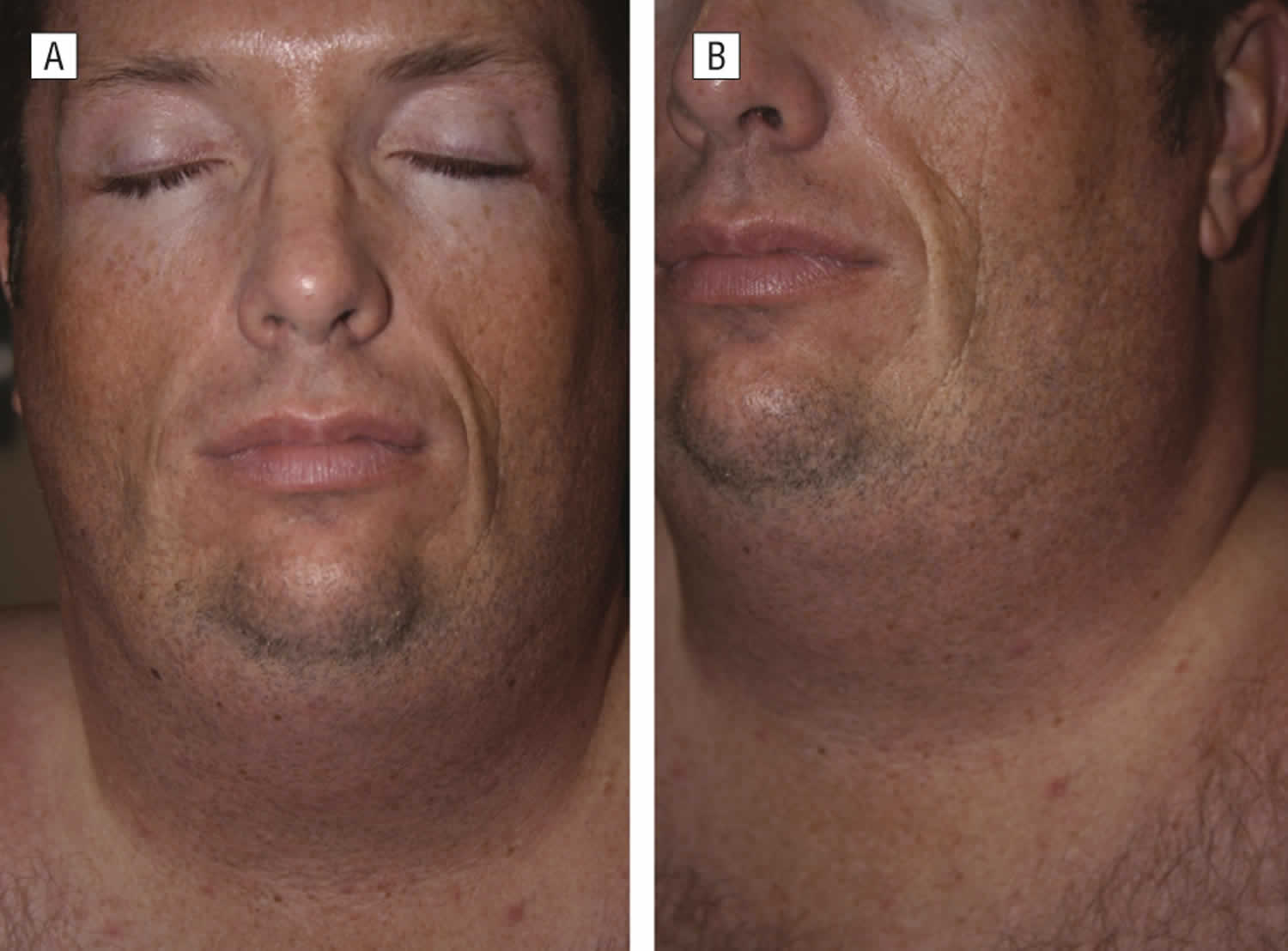
Figure 2. HIV lipodystrophy (38-year old white man with a 10-year history of HIV disease and HAART [highly active antiretroviral therapy])
Footnote:
(A) In frontal view, face and neck demonstrate facial lipoatrophy and circumferential neck lipohypertrophy.
(B) In lateral view, lower face and neck demonstrate facial lipoatrophy and circumferential neck lipohypertrophy.
[Source 13]Other risk factors for lipodystrophy in people with HIV include:
- Age: Older people are at higher risk.
- Race: Whites have the highest risk.
- Gender: Men are more likely to have fat loss in the arms and legs. Women are more likely to have buildup of breast and abdominal fat.
- Length and severity of HIV infection: The risk is higher with longer and more severe HIV infection.
If you are concerned about lipodystrophy, talk to your health care provider. They may recommend that you switch to a different HIV medicine.
How is lipodystrophy treated in people with HIV?
There are several ways to manage lipodystrophy. A healthy diet and daily exercise may help to build muscle and reduce fat buildup.
Liposuction (surgical removal of fat) is sometimes used to reduce a buffalo hump. Fat or a fat-like substance can be used as a filler for fat loss in the face. The filler is injected in the cheeks or around the eyes and mouth.
There are medicines that may help lessen the effects of lipodystrophy. Talk to your health care provider to discuss your treatment options.
Acquired Generalized Lipodystrophy
Acquired generalized lipodystrophy also known as Lawrence syndrome, is characterized by generalized loss of subcutaneous fat that occurs gradually in individuals who are born with a normal fat distribution. The fat loss typically begins in childhood or adolescence, but can rarely begin after 30 years of age 23. It can occur over a variable time period, ranging from a few weeks to months or years, and affects all sc areas of the body especially the face and extremities and may include the palms and soles. Orbital and bone marrow fat depots appear to be preserved, while intra-abdominal fat loss is variable. Acquired generalized lipodystrophy is more frequent in the females than males (3:1) 23. Acquired generalized lipodystrophy patients are predisposed to the same metabolic complications as other patients with lipodystrophies such as insulin resistance associated with diabetes mellitus and hypertriglyceridemia, with hypoleptinemia thought to be contributing to the pathogenesis. Usually these complications are quite severe in these patients. Most of the patients have associated autoimmune diseases, especially juvenile dermatomyositis, or panniculitis (pathologically infiltration of adipose tissue with inflammatory cells of various types resulting in loss of subcutaneous fat) (Table 6). In some patients, the underlying mechanism of fat loss is not clear (Idiopathic variety). Usually the metabolic complications are less severe in patients with panniculitis-associated acquired generalized lipodystrophy as compared to the other two subtypes.
Table 6. Classification of acquired generalized lipodystrophy
| Subtype | Prevalence | Clinical Features |
|---|---|---|
| Panniculitis- associated AGL | ~ 25% | Initial development of panniculitis (sc inflammatory nodules) followed by localized fat loss when these lesions heal. Ongoing panniculitis later on results in generalized loss of subcutaneous fat |
| Auto-immune AGL | ~ 25% | Gradual generalized fat loss associated with auto-immune diseases, especially juvenile dermatomyositis. Some patients have low levels of serum complement 4. |
| Idiopathic AGL | ~ 50% | Gradual generalized subcutaneous fat loss of unclear etiology. |
Abbreviations: AGL = acquired generalized lipodystrophy
Acquired Partial Lipodystrophy (Barraquer-Simons Syndrome)
Acquired partial lipodystrophy is characterized by gradual loss of subcutaneous fat from the upper body, i.e., the face, neck, upper extremities and upper trunk 24. Usually the lower abdomen, hips and lower extremities are spared and in fact, after puberty, especially female patients may accumulate excess fat there. Acquired partial lipodystrophy is more frequent in the females than males (4:1). It is frequently associated with autoimmune diseases. Most patients have a circulating auto-antibody called complement 3 nephritic factor, and have low circulating levels of serum complement 3 24. Approximately 20% of the patients develop membrano-proliferative glomerulonephritis and some of these patients develop end stage renal disease requiring renal transplantation. Rare patients have drusen on fundus examination. Metabolic complications are not seen as frequently as in other types of lipodystrophy 25.
Localized Lipodystrophies
Localized lipodystrophies are characterized by loss of fat from small areas, either single or multiple. Sometimes it can affect portions of the limbs or large contiguous areas on the trunk. Patients with localized lipodystrophies do not develop any metabolic abnormalities. There are several etiologies of localized lipodystrophies (Table 7) 26.
Table 7. Characteristics of different types of localized lipodystrophies
| Type | Etiology | Clinical Features |
|---|---|---|
| Drug-induced localized lipodystrophy | Insulin therapy (more common before purified/human insulin was available), steroids and antibiotics. High local production of TNF-α may cause dedifferentiation of adipocytes. Other mechanisms include presence of lipases, repeated trauma and/or auto- immune processes. | More common in patients with high titers of anti-insulin antibodies. May have deposition of IgA and C3 locally. Sometimes responds to local corticosteroids. |
| Pressure-induced localized lipodystrophy | Trauma and decreased perfusion caused by repeated pressure to the same area over a long period of time. | Fat atrophy localized to the area exposed to repeated pressure. This tends to improve when the pressure is avoided. |
| Panniculitis- associated localized lipodystrophy | Associated with serum ANA or anti dsDNA antibodies; may also have auto-immune diseases such as SLE. | Initial development of panniculitis (sc inflammatory nodules in several areas) followed by localized fat loss when these lesions heal. |
| Centrifugal lipodystrophy (lipodystrophia centrifugalis abdominalis infantalis) | Cause is unknown and most patients recover spontaneously with no intervention. | More common in Asians. Fat loss spreads in a centrifugal pattern from abdomen and groin area and is associated with peripheral panniculitis. It begins in infancy, stops spreading between the ages of 3 and 8 and then in most cases, resolves by itself. |
| Idiopathic localized lipodystrophy | Undetermined etiology. |
Abbreviations: TNF-α = tumor necrosis factor alpha; IgA = immunoglobulin A; C3 = complement 3; ANA = anti-nuclear antibodies; anti dsDNA Ab = anti-double stranded deoxyribonucleic acid antibodies; SLE = systemic lupus erythematous; AGL = acquired generalized lipodystrophy.
[Source 11]Extremely rare genetic lipodystrophy syndromes
Figure 8. Extremely rare genetic lipodystrophy syndromes
| Lipodystrophy Type | Gene | Molecular Basis | Clinical features |
|---|---|---|---|
| MAD type A | LMNA | Mutations may disrupt nuclear function resulting in premature cell death in many tissues. | Mandibular and clavicular hypoplasia, acro-osteolysis. Partial lipodystrophy affecting the extremities and trunk. |
| MAD type B | ZMPSTE24 | Mutations result in accumulation of farnesylated prelamin A that can disrupt nuclear function in several tissues. | Mandibular and clavicular hypoplasia, acro-osteolysis. More generalized loss of fat, premature renal failure, progeroid features. |
| JMP/CANDLE | PSMB8 | PSMB8 encodes subunit of immunoproteasomes that degrade abnormal/excess proteins in cells. | Joint contractures, muscle atrophy, microcytic anemia and panniculitis-induced lipodystrophy. Recurrent fevers, annular erythematous skin lesions, violaceous eyelid swelling, partial lipodystrophy. |
| SHORT syndrome | PIK3R1 | PIK3R1 plays a role in metabolic actions of insulin, mutations associated with insulin resistance. | Variable loss of subcutaneous fat, short stature, hyper-extensibility, ocular depression, teething delay. |
| MDP syndrome | POLD1 | Critical for DNA replication and repair. | Mandibular hypoplasia, deafness, and progeroid features. |
| Neonatal progeroid syndrome, type A | FBN1 | Fibrillin 1 | Generalized loss of body fat and muscle mass, and progeroid appearance at birth. Marfanoid habitus. |
| Neonatal progeroid syndrome, type B | CAV1 | Caveolin 1, present on adipocyte membranes, binds fatty acids and translocates them to lipid droplets. | Generalized loss of body fat and muscle mass, and progeroid appearance at birth. |
| Atypical Progeroid Syndrome | LMNA | Different heterozygous, mostly de novo mutations cause nuclear dysfunction. | Partial or generalized loss of subcutaneous fat, progeroid features. |
| Hutchinson- Gilford progeria | LMNA | Specific de novo mutations induce abnormal splicing and accumulation of truncated farnesylated prelamin A. | Generalized loss of subcutaneous fat, progeroid features. |
Abbreviations: MAD = mandibuloacral dysplasia; LMNA = lamin A/C; ZMPSTE24 = zinc metalloprotease STE24; CANDLE = chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature; PSMB8 = proteasome subunit beta 8; JMP = Joint contractures, Muscle atrophy, Microcytic anemia and Panniculitis-induced lipodystrophy; SHORT = Short stature, Hyper-extensibility or inguinal hernia, Ocular depression, Rieger anomaly and Teething delay; PIK3R1 = phosphoinositide-3-kinase regulatory subunit 1; MDP = Mandibular hypoplasia, Deafness, Progeroid features; POLD1 = polymerase (DNA) delta 1, catalytic subunit; CAV1 = caveolin 1
[Source 11]Lipodystrophy treatment
The treatment of lipodystrophy is focused on managing the metabolic abnormalities to prevent complications, and cosmetic appearance 1. Although there is no cure for lipodystrophy, morbidity and mortality can be improved through early intervention. Diet and exercise form an integral part of the treatment plan, although clinical trial data are not available.
A diet with a well-balanced macronutrient composition of about 50 – 60% carbohydrates, 20 – 30% fat and about 10 – 20% protein is appropriate for most patients 1. Over-feeding should be avoided, especially in infants and children (despite their lack of weight gain), as this can accelerate hepatic steatosis and worsen diabetes and hyperlipidemia. Energy restricted diets are more appropriate in adults, as children with growth and developmental needs may otherwise develop deficiencies.
Exercise, in the absence of contraindications, can help improve metabolic parameters, so patients should be encouraged to be physically active. Those who are predisposed to cardiomyopathy, such as patients with congenital generalized lipodystrophy, familial partial lipodystrophy type 2 (Dunnigan lipodystrophy), and progeroid syndromes should undergo a cardiac evaluation before engaging in an exercise program, and should avoid strenuous exercise. To avoid traumatic injuries, patients with severe hepatosplenomegaly and congenital generalized lipodystrophy patients with lytic lesions in the bones should avoid contact sports 1.
Strategies to reduce hypertriglyceridemia include medium chain triglyceride-based formulas in infants 27 and very low fat diets in older individuals. Any fat intake should be in the form of cis-mono-unsaturated fats and long chain omega-3 fatty acids. Patients who have developed acute pancreatitis secondary to hypertriglyceridemia, parental nutrition should be administered until they recover and they should subsequently be on an extremely low fat (total dietary fat less than 20 grams/day) diet. In patients who have not reached lipid-lowering goals after diet and lifestyle intervention, lipid-lowering drugs may be used 1.
Patients with insulin resistance and diabetes mellitus should be treated with conventional therapies, including both oral (metformin is the first-line drug) and insulin. Insulin therapy often provides the mainstay of treatment, and many patients require concentrated forms (such as U-500 regular insulin) because of severe insulin resistance. Whether thiazolidinediones are particularly efficacious in familial partial lipodystrophy patients with PPARG mutations (familial partial lipodystrophy type 3) remains unclear. Simple sugars should be avoided in favor of high-fiber complex carbohydrates consumed throughout the day in combination with protein and/or fat, to avoid blood glucose spikes. The treatment goals are similar to diabetic patients without lipodystrophy.
Hypertension, if uncontrolled, may be treated with angiotensin converting enzyme inhibitors (ACE inhibitors) or angiotensin receptor blockers (ARBs), as these medications also have favorable effects on proteinuria. No specific treatments have been shown to be particularly effective for hepatic steatosis or steato-hepatitis associated with lipodystrophy.
Generalized lipodystrophies are characterized by extremely low serum leptin levels 28, which led to research into recombinant human leptin (metreleptin) as a treatment option 29, and since then several long term studies have shown beneficial effects 30.
Metreleptin therapy has been shown to improve metabolic abnormalities in generalized lipodystrophy patients, including decreased serum triglyceride levels, increased insulin sensitivity and reduced hepatic steatosis 3. It is currently the only drug specifically approved for treatment of lipodystrophy 3. It is administered as a daily subcutaneous injection 31 and dose adjustments are made every 3 – 6 months based on metabolic parameters and weight change. The most common side effects include hypoglycemia and injection site reactions such as erythema and/or urticaria. The other side effects include development of neutralizing antibodies to metreleptin, and development of cutaneous T cell lymphomas especially in patients with acquired generalized lipodystrophy 32. The precise significance of neutralizing antibodies to leptin remains unclear at this time and some patients with acquired generalized lipodystrophy who have never received metreleptin therapy have also been reported to develop lymphomas. Because of paucity of data, approval of metreleptin for different types of lipodystrophy varies by country, depending on their regulatory boards.
Change in body shape caused by lipodystrophy can often lead to psychological distress, and sometimes even physical discomfort, such as from absent fat pads on the feet and buttocks. Patients should be referred to appropriate mental health providers for emotional distress. Plastic surgery may improve appearance in some people, though data are limited. Possible interventions include autologous fat transfer, dermal fillers or muscle grafts to treat facial lipoatrophy; surgical reduction or liposuction of areas with excessive fat; and breast implants for improved cosmetics in women.
- Hussain I, Garg A. LIPODYSTROPHY SYNDROMES. Dermatologic clinics. 2008;26(4):569-ix. doi:10.1016/j.det.2008.05.004. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4947059/[↩][↩][↩][↩][↩][↩][↩]
- Garg A. Clinical review#: Lipodystrophies: genetic and acquired body fat disorders. J Clin Endocrinol Metab. 2011;96(11):3313–3325.[↩][↩][↩]
- Brown RJ, Gorden P. Leptin Therapy in Patients with Lipodystrophy and Syndromic Insulin Resistance. In: Dagogo-Jack S, editor. Leptin: Regulation and Clinical Applications. Switzerland: Springer International Publishing; 2015.[↩][↩][↩][↩][↩]
- Garg A. Clinical review#: Lipodystrophies: genetic and acquired body fat disorders. J Clin Endocrinol Metab. 2011;96(11):3313–3325. https://academic.oup.com/jcem/article/96/11/3313/2834182[↩]
- Handelsman Y, Oral EA, Bloomgarden ZT, et al. The clinical approach to the detection of lipodystrophy – an AACE consensus statement. Endocr Pract. 2013;19(1):107–116 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4108221/[↩]
- Antuna-Puente B, Boutet E, Vigouroux C, et al. Higher adiponectin levels in patients with Berardinelli-Seip congenital lipodystrophy due to seipin as compared with 1-acylglycerol-3-phosphate-o-acyltransferase-2 deficiency. J Clin Endocrinol Metab. 2010;95(3):1463–1468.[↩][↩]
- Agarwal AK, Simha V, Oral EA, et al. Phenotypic and genetic heterogeneity in congenital generalized lipodystrophy. J Clin Endocrinol Metab. 2003;88(10):4840–4847.[↩][↩]
- Handelsman Y, Oral EA, Bloomgarden ZT, et al. The clinical approach to the detection of lipodystrophy – an AACE consensus statement. Endocr Pract. 2013;19(1):107–116.[↩]
- Garg A. Acquired and inherited lipodystrophies. N Engl J Med. 2004;350(12):1220–1234[↩]
- Hayashi YK, Matsuda C, Ogawa M, et al. Human PTRF mutations cause secondary deficiency of caveolins resulting in muscular dystrophy with generalized lipodystrophy. J Clin Invest. 2009;119(9):2623–2633[↩]
- Hussain I, Garg A. LIPODYSTROPHY SYNDROMES. Dermatologic clinics. 2008;26(4):569-ix. doi:10.1016/j.det.2008.05.004. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4947059[↩][↩][↩][↩][↩][↩]
- Familial partial lipodystrophy. https://ghr.nlm.nih.gov/condition/familial-partial-lipodystrophy[↩]
- Tierney EP, Hanke CW. “Bullfrog Neck,” a Unique Morphologic Trait in HIV LipodystrophyCase Series and Review of the Literature. Arch Dermatol. 2010;146(11):1279–1282. doi:10.1001/archdermatol.2010.341[↩][↩]
- Oh JHegele RA HIV-associated dyslipidaemia: pathogenesis and treatment. Lancet Infect Dis 2007;7 (12) 787- 796[↩]
- Mallon PW Pathogenesis of lipodystrophy and lipid abnormalities in patients taking antiretroviral therapy. AIDS Rev 2007;9 (1) 3- 15[↩]
- Huang JSLee DBecerra KSantos RBarber EMathews WC Body image in men with HIV. AIDS Patient Care STDS 2006;20 (10) 668- 677[↩][↩]
- HIV and Lipodystrophy. https://aidsinfo.nih.gov/understanding-hiv-aids/fact-sheets/22/61/hiv-and-lipodystrophy[↩]
- Hudon SE, Coffinier C, Michaelis S, Fong LG, Young SG, Hrycyna CA. HIV-protease inhibitors block the enzymatic activity of purified Ste24p. Biochem Biophys Res Commun. 2008;374(2):365–368.[↩]
- Lee H, Hanes J, Johnson KA. Toxicity of nucleoside analogues used to treat AIDS and the selectivity of the mitochondrial DNA polymerase. Biochemistry. 2003;42(50):14711–14719.[↩]
- De Luca AMurri RDamiano FAmmassari AAntinori A “Buffalo hump” in HIV-1 infection. Lancet 1998;352 (9124) 320[↩]
- Guaraldi GOrlando GSquillace N et al. Prevalence of and risk factors for pubic lipoma development in HIV-infected persons. J Acquir Immune Defic Syndr 2007;45 (1) 72- 76[↩]
- Vázquez E Banishing chipmunk cheeks and bullfrog neck. Treating these and other body changes from HIV drugs. Posit Aware 2006;17 (5) 20- 21[↩]
- Misra A, Garg A. Clinical features and metabolic derangements in acquired generalized lipodystrophy: case reports and review of the literature. Medicine (Baltimore) 2003;82(2):129–146.[↩][↩]
- Misra A, Peethambaram A, Garg A. Clinical features and metabolic and autoimmune derangements in acquired partial lipodystrophy: report of 35 cases and review of the literature. Medicine (Baltimore) 2004;83(1):18–34.[↩][↩]
- Chen D, Misra A, Garg A. Clinical review 153: Lipodystrophy in human immunodeficiency virus-infected patients. J Clin Endocrinol Metab. 2002;87(11):4845–4856.[↩]
- Garg A. Lipodystrophies. Am J Med. 2000;108(2):143–152.[↩]
- Wilson DE, Chan IF, Stevenson KB, Horton SC, Schipke C. Eucaloric substitution of medium chain triglycerides for dietary long chain fatty acids in acquired total lipodystrophy: effects on hyperlipoproteinemia and endogenous insulin resistance. J Clin Endocrinol Metab. 1983;57(3):517–523.[↩]
- Haque WA, Shimomura I, Matsuzawa Y, Garg A. Serum adiponectin and leptin levels in patients with lipodystrophies. J Clin Endocrinol Metab. 2002;87(5):2395.[↩]
- Oral EA, Simha V, Ruiz E, et al. Leptin-replacement therapy for lipodystrophy. N Engl J Med. 2002;346(8):570–578.[↩]
- Simha V, Subramanyam L, Szczepaniak L, et al. Comparison of efficacy and safety of leptin replacement therapy in moderately and severely hypoleptinemic patients with familial partial lipodystrophy of the Dunnigan variety. J Clin Endocrinol Metab. 2012;97(3):785–792.[↩]
- Rodriguez AJ, Mastronardi CA, Paz-Filho GJ. New advances in the treatment of generalized lipodystrophy: role of metreleptin. Ther Clin Risk Manag. 2015;11:1391–1400 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4577254/[↩]
- Brown RJ, Chan JL, Jaffe ES, et al. Lymphoma in acquired generalized lipodystrophy. Leuk Lymphoma. 2016;57(1):45–50. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4755279/[↩]




{kind=link}