Contents
What is macrocephaly
Macrocephaly means increased head size or head circumference when the measured distance around the widest part of the skull is larger than expected for the child’s age and background, usually greater than the 98th percentile or 2 standard deviations above the age-related mean 1. The term relative macrocephaly can be used when the head size centile exceeds the centile for height, for example, head size at the 75th centile with height at the 5th centile for age and sex. Macrocephaly may be hereditary or the result of a central nervous system disorder (e.g., hydrocephalus, brain tumor), and imaging may be needed 2. Macrocephaly, the increased head circumference is linked to various events that can result in an increase of orbito-frontal head circumference for age, including anomalies of bone skull structures, subdural fluid collections, hydrocephalus, intracranial masses, and arteriovenous malformations 1. Macrocephaly has to be differentiated from megalencephaly, which is defined as increase in the size of the brain tissue 3. Megalencephaly is defined as an increased growth of cerebral structures related to dysfunctional anomalies during the various steps of brain development in the neuronal proliferation and/or migration phases or as a consequence of postnatal abnormal events that cause excessive cerebral growth.
Macrocephaly can result from enlargement of the skull bones or an increase in the volume of the intracranial structures like cerebrospinal fluid (CSF), hydrocephalus, cranial hyperostosis, blood, or enlarged brain (megalencephaly). Macrocephaly may be secondary to raised intracranial pressure or space-occupying lesions. Macrocephaly can also be a feature of various congenital syndromes and is then referred to as syndromic macrocephaly. These conditions may be the result of genetic disorders or disorders the child acquired before or after birth.
Evaluation should include a 3-generation family history, developmental and neurologic assessment, examination for limb asymmetry and cutaneous lesions, and brain MRI. Sometimes disproportionate macrocephaly is familial and not associated with other anomalies, complications, or developmental delays; this form is transmitted in an autosomal dominant pattern, so at least one parent has a large head circumference. The diagnoses to be considered include neurofibromatosis type 1, Fragile X syndrome, Sotos syndrome, and lysosomal storage disorders.
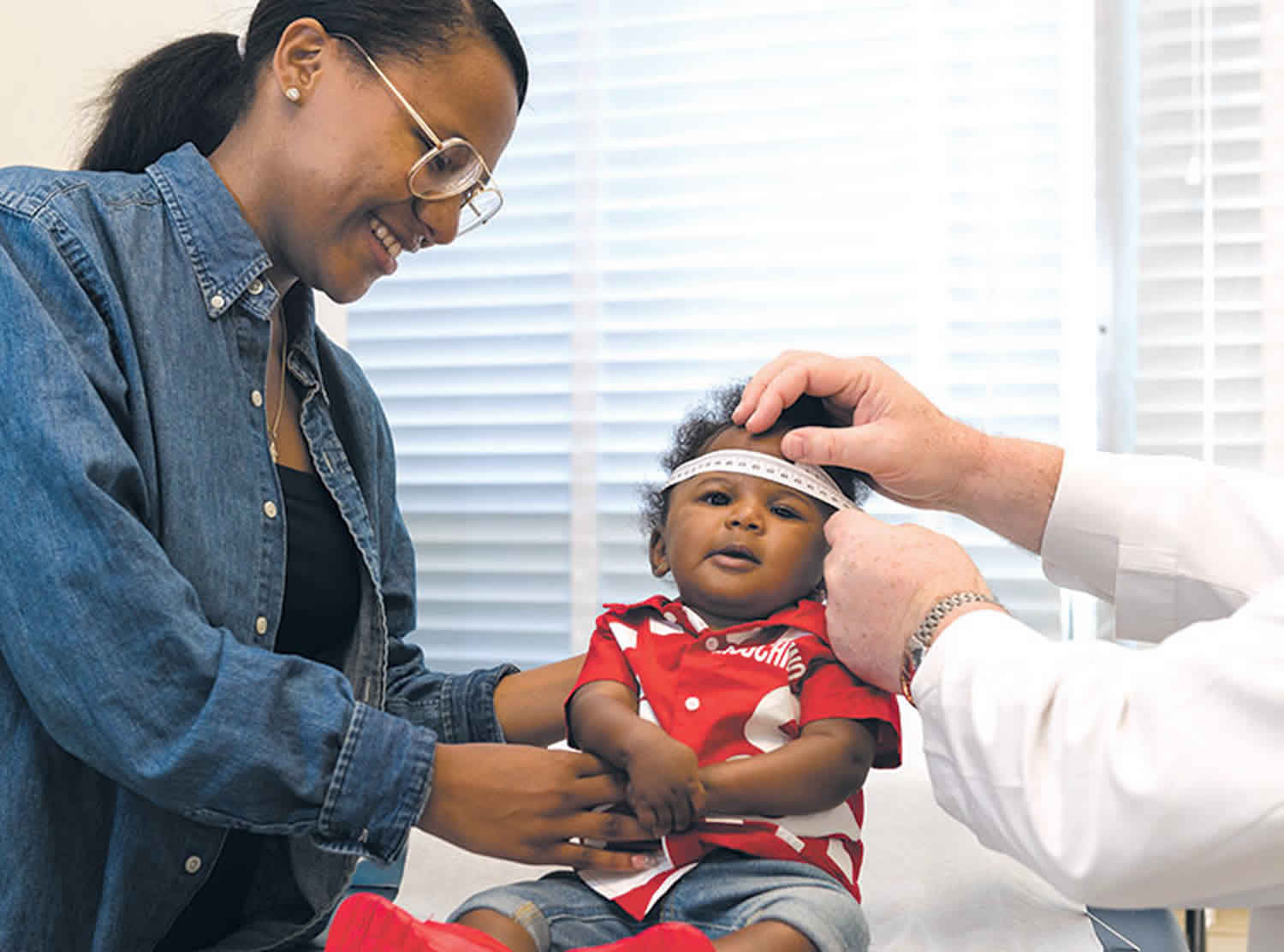
Head circumference is measured from just above the glabella (the most prominent point on the frontal bone above the root of the nose) to the most posterior prominent point of the occipital bone using a tape measure. Head circumference is measured to monitor head growth in infants and children. It is also known as occipitofrontal circumference and it denotes the size of the cranium. Macrocephaly can be the first manifestation of various congenital and acquired neurologic conditions or may be just a familial trait 4.
The measurement of the head circumference is a challenge for pediatricians, as it is not easy to carry out in young children. In some cases, a correct reading of the head measurement may be particularly unfeasible, as the child may be too much restless or the tape measure may be not placed properly and the patient’s hair may be too thick to give a correct result. A proper measure of the head circumference should be performed by putting the tape measure along the most prominent diameter of the occiput and the mid forehead; then, the results of the measurement must be checked with the head circumference growth charts, according to the age, gender, and height parameters 5. Correlation with the maternal and paternal head circumference is useful. The newborn brain is reported to weigh about 370 g and increases about 4-fold from infancy to childhood till reaching an adult’s weight of about 1500 g 6.
A newborn’s head is usually about 2 cm larger than the chest size. Between 6 months and 2 years, both measurements are about equal. After 2 years, the chest size becomes larger than the head.
Measurements over time that show an increased rate of head growth often provide more valuable information than a single measurement that is larger than expected.
Various gender-specific growth charts have been published, but the Centers for Disease Control (CDC) currently recommends that children in the United States between the ages of 0 and 2 years old are tracked with the World Health Organization (WHO) growth charts, and with the CDC growth charts after children turn 2 years old. Minor differences do exist between the two charts. Clinicians can easily tell the difference because charts from WHO data tend to stop at 24 months of age, whereas the charts from CDC data extend to 36 months of age 7.
Macrocephaly types
There are two types of macrocephaly:
- Disproportionate macrocephaly
- Proportionate macrocephaly
In disproportionate macrocephaly, the head is larger than appropriate for the child’s size; affected children are at risk of autism spectrum disorders, developmental disability, and seizures.
In proportionate macrocephaly, the head appears appropriately sized for the body (ie, the large head is associated with a large stature), and an overgrowth syndrome (e.g, growth hormone excess) should be considered.
Macrocephaly causes
Macrocephaly may be caused by any of the following:
- Benign familial macrocephaly (family tendency toward large head size)
- Canavan disease (condition that affects how the body breaks down and uses a protein called aspartic acid)
- Hydrocephalus (buildup of fluid inside the skull that leads to brain swelling)
- Bleeding inside the skull
- Disease in which the body is unable break down long chains of sugar molecules (Hurler or Morquio syndrome)
The causes of macrocephaly is diverse 8. The most common cause is benign familial macrocephaly characterized by enlargement of the subarachnoid spaces and accounts for almost 50% of cases.
Other causes are described below.
Enlargement of skull bones
- Hyperostosis cranii – associated with disorders such as osteogenesis imperfecta, achondroplasia, and osteopetrosis
- Secondary enlargement due to bone marrow expansion – as seen in thalassemia major
Increase in volume of cerebrospinal fluid
- Hydrocephalus
- Choroid plexus papilloma
- Benign familial macrocephaly
Megalencephaly
- Leukodystrophies – Canavan disease, Alexander disease, megalencephalic leukoencephalopathy with subcortical cysts
- Lysosomal storage disorders – Tay-sachs, mucopolysaccharoidosis, gangliosidosis
- Neurocutaneous disroders – Tuberous sclerosis, Sturge-weber syndrome, neurofibromatosis, Gorlin syndrome
- Autism spectrum disorder
- Other syndromes – Fragile X syndrome, Cowden syndrome, Sotos syndrome
Increased intracranial pressure (ICP)
- CNS infections
- Pseudotumor cerebri
- Subdural collections including hygromas
Mass lesions and increase in volume of blood
- Tumor
- Intraventricular hemorrhage, subdural hematoma, arteriovenous malformation
Benign familial macrocephaly
Benign familial macrocephaly is a primary macrocephaly and autosomal dominant 9 and multifactorial inheritances 10 had been proposed.
Asch and Myers 9 described 5 males in 2 generations of a family with occipitofrontal head circumferences greater than 2 standard deviation above the mean. All were neurologically and mentally normal. A maternal uncle of the first generation was said to have a large head. All were having a relatively long skull (dolichocephalic). By sonographic studies the ventricular system was enlarged in 3 of the 5. Similar families were reported by Platt and Nash 11 and by Day and Shutt 12.
Arbour et al. 10 measured head size in the parents and siblings of 23 patients with a head circumference more than 2 standard deviation above the mean and with no evidence of hydrocephalus or syndromic associations. In 12 of the 23, some degree of psychomotor impairment was present. It was found that head circumference of parents and sibs had a mean significantly greater than the population norm and a unimodal distribution. Probands with psychomotor impairment had bigger heads, and more had a history of birth difficulty than did unimpaired probands. They noted that macrocephaly in a parent or sibling of an unborn child may present a risk for birth injury to that child.
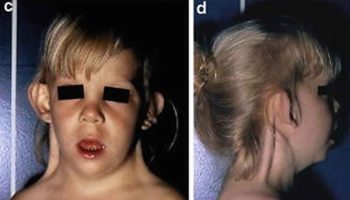
In the course of a clinical study of Sotos syndrome, Cole and Hughes 13 found that 6 of 79 probands who failed to fit that phenotype showed remarkable similarities to each other and to some of their first- and second-degree relatives. In addition to macrocephaly, clinical features included typical facies characterized by square outline with frontal bossing, ‘dished-out’ midface, biparietal narrowing, and long philtrum. Birth weight and length were normal or near normal with subsequent obesity. Cole and Hughes 13 were uncertain as to whether this represented a new entity or benign familial macrocephaly.
Diaz-Rodriguez et al. 14 reported a mother and son with benign familial macrocephaly who displayed the characteristic square facial appearance with frontal bossing and dished-out midfacies. There was also an unaffected sister.
Macrocephaly symptoms
Macrocephaly symptoms varies depending upon the underlying cause.
Increased pressure inside the head (increased intracranial pressure) often occurs with macrocephaly. Symptoms of this condition include:
- Eyes moving downward
- Irritability
- Vomiting
Macrocephaly diagnosis
Your health care provider usually finds macrocephaly or an increased head size in a baby during a routine well-baby exam.
A careful physical exam will be done. Other milestones for growth and development will be checked.
The following aspects must be assessed:
- Age at the onset of the disease
- Birth history – with information about the birth weight, length, and head size
- History of development and growth – with special attention paid to the chronology of attainment of milestones; and subsequently regression, if any. Time of closure of fontanels should also be established.
- History of other associated features (e.g., headache, vomiting, seizures, cognitive decline)
- History of fever, central nervous system infection, head injury, or intracranial hemorrhage
- Family history – metabolic disorders, neurocutaneous disorders, macrocephaly, and consanguineous marriages
In some cases, a single measurement is enough to confirm that there is a size increase that needs to be tested further. More often, repeated measurements of the head circumference over time are needed to confirm that the head circumference is increased and the problem is getting worse.
Diagnostic tests that may be ordered include:
- Head CT scan
- MRI of the head
Other tests that may prove helfpul for diagnosis include the following:
- Molecular genetic testing
- Metabolic tests
- Tests for other associations like electrocardiogram, echocardiography, ultrasound of the abdomen, and skeletal survey.
Macrocephaly treatment
The management is variable depending upon the cause of macrocephaly.
Benign familial macrocephaly does not necessitate treatment and children mostly remain asymptomatic. Periodic monitoring of head size is sufficient in these cases along with regular monitoring for physical growth and neurologic development.
Macrocephalic children with epilepsy require therapy with appropriate antiepileptic medication.
Surgical care
Neurosurgical intervention may be warranted in children with hydrocephalus and consequent symptoms of increased intracranial pressure. Various procedures may be considered for drainage of CSF in order to reduce the fluid volume including placement of a extraventricular drain or a ventriculoperitoneal (VP) shunt 15.
Another surgery reported in the literature is complete reconstruction of the skull in children with grossly deformed skull architecture. It has been tried in a case of macrocephaly due to hydrocephalus 16.
Long-Term Monitoring
It is recommended to measure and plot the head size monthly for six months to ascertain the growth curve of the child and to compare it to the normal curve. Physical and neurologic examination should be carried out regularly. Neuroimaging is not routinely recommended unless there is abnormal or rapid increase in head size or there is motor, cognitive, or growth retardation or loss of milestones.
- Pavone P, Praticò AD, Rizzo R, et al. A clinical review on megalencephaly: A large brain as a possible sign of cerebral impairment. Medicine (Baltimore). 2017;96(26):e6814. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5500017[↩][↩]
- Sniderman A. Abnormal head growth. Pediatr Rev. 2010;31(9):382–384.[↩]
- Pavone P, Praticò AD, Rizzo R, et al. A clinical review on megalencephaly: A large brain as a possible sign of cerebral impairment. Medicine (Baltimore). 2017;96(26):e6814. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5500017/[↩]
- Day RE, Schutt WH. Normal children with large heads–benign familial megalencephaly. Arch Dis Child. 1979 Jul. 54 (7):512-7[↩]
- Abnormal head growth. Sniderman A. Pediatr Rev. 2010 Sep; 31(9):382-4. https://pedsinreview.aappublications.org/content/31/9/382.long[↩]
- Blinkov SM, Glazer II. The Human Brain in Figures and Tables. A Quantitative Handbook. New York: Plenum Press; 1968.[↩]
- Clinical Growth Charts. https://www.cdc.gov/growthcharts/clinical_charts.htm[↩]
- Olney AH. Macrocephaly syndromes. Semin Pediatr Neurol. 2007 Sep. 14 (3):128-35.[↩]
- Asch, A. J., Myers, G. J. Benign familial macrocephaly: report of a family and review of the literature. Pediatrics 57: 535-539, 1976[↩][↩]
- Arbour, L., Watters, G. V., Hall, J. G., Fraser, F. C. Multifactorial inheritance of non-syndromic macrocephaly. Clin. Genet. 50: 57-62, 1996[↩][↩]
- Platt, M., Nash, A. Benign familial megalencephaly. (Abstract) Pediat. Res. 6: 426 only, 1972.[↩]
- Day, R. E., Shutt, W. H. Normal children with large heads–benign familial megalencephaly. Arch. Dis. Child. 54: 512-517, 1979[↩]
- Cole, T. R. P., Hughes, H. E. Autosomal dominant macrocephaly: benign familial macrocephaly or a new syndrome? Am. J. Med. Genet. 41: 115-124, 1991.[↩][↩]
- Diaz-Rodriguez, M., Becerra-Solano, L. E., Toscano-Flores, J. J., Banuelos-Robles, O., Duran-Gonzalez, J., Ramirez Duenas, M. L. Benign familial macrocephaly in a mother-son pair. Genet. Counsel. 21: 85-89, 2010[↩]
- Walker CT, Stone JJ, Jacobson M, Phillips V, Silberstein HJ. Indications for pediatric external ventricular drain placement and risk factors for conversion to a ventriculoperitoneal shunt. Pediatr Neurosurg. 2012. 48 (6):342-7.[↩]
- Mansour N, Sobel L, Lee M, Larumbe J, Stelnicki E. A new method for the treatment of macrocephaly caused by hydrocephalus. Cleft Palate Craniofac J. 2005 Jan. 42 (1):1-6.[↩]




{kind=link}