
Contents
What is maple syrup urine disease
Maple syrup urine disease is a rare but serious inherited condition or genetic disorder characterized by deficiency of certain enzymes (branched-chain alpha-keto acid dehydrogenase complex) required to break down (metabolize) the three branched-chain amino acids (BCAAs) [Leucine, Isoleucine and Valine] in the body. The result of this metabolic failure is that all three branched-chain amino acids (BCAAs), along with their various byproducts, accumulate abnormally throughout the body. In the classic, severe form of maple syrup urine disease, the plasma concentrations of the BCAAs begin to rise within a few hours of birth. If untreated, symptoms begin to emerge, often within the first 24-48 hours of life. One of the characteristic symptoms of maple syrup urine disease is sweet-smelling urine, which gives the condition its name. Maple syrup urine disease is also characterized by poor feeding, vomiting, lack of energy (lethargy), abnormal movements, and delayed development. If untreated, maple syrup urine disease can lead to seizures, coma, and death.
Newborn Screening for maple syrup urine disease is performed throughout the US and in many other countries so that most such infants are detected through these programs. Where such screening is not available, infants with maple syrup urine disease usually present with the neurological signs in an advanced state. Early diagnosis and treatment stabilizes the infants and, if well performed, can largely mitigate against serious metabolic decompensations and long-term complications.
Maple syrup urine disease affects males and females in equal numbers. Maple syrup urine disease affects an estimated 1 in 185,000 infants worldwide 1. Due to a founder effect, the disorder occurs with greater frequency among individuals in the Old Order Mennonite population, with an estimated incidence of about 1 in 380 newborns 1. Maple syrup urine disease occurs in the Ashkenazi Jewish population with an incidence estimated at 1:26,000 live births.
Normally, your body breaks down protein foods such as meat and fish into amino acids. Any amino acids that are not needed are usually broken down and removed from the body. Babies with maple syrup urine disease are unable to break down branched-chain amino acids (BCAAs) – leucine, isoleucine and valine. Very high levels of these amino acids are harmful.
There are three or possibly four types of maple syrup urine disease: the classic type; intermediate type, intermittent type, and possibly a thiamine-responsive type 2. The various subtypes of maple syrup urine disease have different levels of residual enzyme activity, severity, and age of onset. All forms are inherited as autosomal recessive traits.
Maple syrup urine disease is often classified by its pattern of signs and symptoms. The most common and severe form of the disease is the classic type, which becomes apparent soon after birth. Variant forms of the disorder become apparent later in infancy or childhood and are typically milder, but they still lead to delayed development and other health problems if not treated.
The “non-specific” symptoms are those of increasing neurological dysfunction and include lethargy, irritability and poor feeding, followed soon by focal neurological signs such as abnormal movements and increasing spasticity, and shortly thereafter, by convulsions and deepening coma. If untreated, progressive brain damage is inevitable and death ensues usually within weeks or months. The finding that is unique to maple syrup urine disease is the emergence of a characteristic odor, reminiscent of maple syrup that can most readily be detected in the urine and earwax and may be smelled within a day or two of birth.
Maple syrup urine disease can be successfully managed through a specialized diet. However, even with treatment, both affected children and adults patients with maple syrup urine disease remain at high risk for developing episodes of acute illness (metabolic crises) often triggered by infection, injury, failure to eat (fasting) or even by psychological stress. During these episodes there is a rapid, sudden spike in amino acid levels necessitating immediate medical intervention.
Without treatment, maple syrup urine disease can lead to feeding difficulties, lethargy, seizures, urine and cerumen that smell like maple syrup, vomiting, coma, and death 3. Early diagnosis and treatment can optimize outcomes, although treated individuals have an increased rate of anxiety, depression, attention deficit hyperactivity disorder (ADHD), movement disorders, and small reductions in intelligence and global function 3.
Treatment consists of a protein-restrictive diet that limits the amount of branched-chain amino acids consumed, along with synthetic formula consisting of the other amino acids and various micronutrients 4. The goal of treatment is to maintain plasma leucine concentrations, with frequent monitoring, between 75 and 200 μmol/L for infants and children age five years and younger, and between 75 and 300 μmol/L for individuals older than five years of age 5. Even with dietary treatment, metabolic decompensation can occur during times of stress (e.g., infection) and must be treated promptly 3. Liver transplants may allow for relaxation of the diet, and can help prevent further brain damage but cannot reverse existing damage 6.
Other names for maple syrup urine disease:
- BCKD deficiency
- branched-chain alpha-keto acid dehydrogenase deficiency
- branched-chain ketoaciduria
- ketoacidemia
- MSUD
Maple syrup urine disease classifications
Four general classifications are used to identify the types of maple syrup urine disease: classic, intermediate, intermittent and thiamine-responsive. These terms refer to the amount and type of enzyme activity present in the affected child, which can vary considerably within each classification.
- Classic is the most common type of maple syrup urine disease. In classic maple syrup urine disease, little or no enzyme activity (usually less than 2% of normal) is present. Infants with classic maple syrup urine disease will show symptoms within the first several days of life. They generally have poor tolerance for the BCAAs, so protein must be severely restricted in their diet.
- Intermediate maple syrup urine disease is a variant of the classic type of the disease. Those with intermediate maple syrup urine disease have a higher level of enzyme activity (approximately 3-8% of normal). They can usually tolerate a greater amount of leucine. However, when ill or fasting, the child with intermediate maple syrup urine disease reacts just like a child with classic maple syrup urine disease. Management is similar for the intermediate and classic types of maple syrup urine disease.
- Intermittent maple syrup urine disease is a milder form of the disease because of the greater enzyme activity present (approximately 8-15% of normal). Often the child does not have symptoms until 12 to 24 months of age, usually in response to an illness or surge in protein intake. During episodes of illness or fasting, the BCAA levels elevate, the characteristic maple syrup (or burnt sugar) odor becomes evident, and the child can go into a metabolic crisis.
- Thiamine-responsive maple syrup urine disease is basically just what the name implies. Giving large doses of thiamine to the thiamine-responsive child will increase the enzyme activity which breaks down leucine, isoleucine and valine. In most cases only moderate protein restriction is needed for this more rare type of maple syrup urine disease.
At times the peculiar maple syrup smell in the urine or sweat can occur in older, healthy children or adults who are non-symptomatic. The reason for this is unknown. However, these persons should be checked for a milder form of maple syrup urine disease, especially if there are other symptoms of maple syrup urine disease.
Maple syrup urine disease life expectancy and prognosis
With strict dietary compliance and good medical care, children with maple syrup urine disease can, and do, lead relatively normal lives. Therapy must be started at the earliest possible age to achieve the best possible outcome. Careful control of the diet to insure the correct balance of the amino acid levels is the best strategy for the optimal function of persons with maple syrup urine disease. There always remains the risk of brain injury during extended times of elevated leucine levels.
Maple syrup urine disease symptoms
Symptoms of maple syrup urine disease usually appear within the first few days or weeks after birth. More general symptoms include:
- sweet-smelling urine and sweat
- poor feeding or loss of appetite
- weight loss
Babies with maple syrup urine disease may also have episodes known as a “metabolic crisis”, sometimes early in their life. Symptoms of a metabolic crisis include:
- lack of energy
- vomiting
- irritability
- breathing difficulties
It’s important to get medical help immediately if your baby develops symptoms of a metabolic crisis. Your doctor will give you advice to help you recognize the signs.
In some cases, a metabolic crisis may be triggered later in childhood by an infection or illness. The hospital will provide you with emergency treatment instructions to follow if your child is ill, which helps prevent these symptoms developing.
The symptoms and severity of maple syrup urine disease at onset varies greatly from patient to patient and largely relate to the amount of residual enzyme activity. Classic maple syrup urine disease is the most common and most severe form of maple syrup urine disease characterized by little to no enzyme activity.
Most infants with classic maple syrup urine disease show subtle emerging symptoms within 2-3 days; these include poor feeding at bottle or breast and increasing lethargy and irritability. As the decline continues, the infant further disengages and then starts to show increasing focal neurologic signs including athetoid [so called “fencing and “cycling”] movements together with increasing hypertonia, spasticity and opisthotonus progressing to convulsions and coma. There may be temporary episodes of extreme hypotonia. In the end, central neurologic function fails with respiratory failure and death. By the time that symptoms have emerged, a distinctive odor of maple syrup may be detected in cerumen, sweat, and urine. This is derived from one of the organic acids that accumulate along with the BCAAs as the disorder spirals out of control.
Once the disorder has been treated and stabilized, there remains a life long threat of recurrent metabolic decompensation. Even without any change in dietary intake, these episodes occur due to increased breakdown of protein resulting from a number of metabolic stresses. Infection, psychological stress, fasting, trauma, fasting or indeed any major change in dietary habits all cause a change in the metabolism of protein resulting in more of the BCAAs requiring to be metabolized. These episodes are characterized by emergence of the symptoms that are typical in an acute untreated case and due to elevated branched-chain amino acids (BCAAs) and metabolites. Every such episode has the potential to turn into a metabolic catastrophy and must be treated as vigorously as any episode in a newborn. Individuals with classic maple syrup urine disease may show a degree of intellectual limitation and may develop a variety of behavioral issues including attention deficient hyperactivity disorder (ADHD), impulsivity, anxiety and/or depression.
Additional complications with classic maple syrup urine disease include generalized loss of bone mass that may predispose individuals to fractures (osteoporosis), and inflammation of the pancreas (pancreatitis). Some individuals may develop a condition known as intracranial hypertension, in which increased blood pressure in the skull causes painful headaches that are sometimes associated with nausea and vomiting.
Intermediate maple syrup urine disease is characterized by greater levels of residual enzyme activity than is seen with classic maple syrup urine disease. The onset and symptoms of intermediate maple syrup urine disease may be neonatal, but the majority of children are diagnosed between the ages of five months and seven years. Affected children may experience seizures and neurological impairment and developmental delays of varying degrees. Some experience feeding problems, poor growth and the characteristic odor of maple syrup in their earwax, sweat, and urine shortly after birth. Other affected children may remain asymptomatic until later in life. Intermediate maple syrup urine disease patients are susceptible to the same neurologic conditions and extreme symptoms as those with classic maple syrup urine disease. Due to the indefinite distinction between the classic and intermediate forms of maple syrup urine disease, disease management principles are the same for both.
Intermittent maple syrup urine disease is characterized by normal growth and intellectual development and affected individuals often can tolerate normal levels of amino acids in their diet. Symptoms usually do not occur in this form until an affected child experiences stress, does not eat, or develops an infection. Symptoms may include lethargy, the characteristic odor of maple syrup in the earwax, sweat and urine, and ataxia. Affected children can develop metabolic crises that result in seizures, coma, brain damage, and, in rare cases, life-threatening neurological complications.
Thiamine-response maple syrup urine disease is a form of the disorder that responds to treatment with thiamine (vitamin B1). Thiamine helps the body convert carbohydrates into energy. The symptoms and clinical course of thiamine-responsive maple syrup urine disease resembles intermediate maple syrup urine disease. Symptoms are rarely present in the newborn period. Affected infants respond to large doses of thiamine, which boosts residual enzyme activity. No individuals with thiamine-responsive maple syrup urine disease have been treated solely with thiamine – most follow a combination of thiamine with a partially-restricted diet.
While the majority of patients fall into the categories above, several families with multiple affected members have been identified who do not fit the criteria for any of the above subtypes. These unique patients are deemed unclassified maple syrup urine disease.
It should be emphasized that in the presence of such apparently non-specific neurologic findings the diagnosis of maple syrup urine disease cannot be excluded by the absence of the maple syrup smell.
Maple syrup urine disease causes
Maple syrup urine disease is caused by changes (mutations) in three different genes: BCKDHA, BCKDHB and DBT encoding the E1α, E1β, and E2 subunits of the branched-chain α-ketoacid dehydrogenase enzyme complex 7. These three genes provide instructions for making proteins that work together as part of a complex. The protein complex is essential for breaking down the amino acids leucine, isoleucine, and valine, which are present in many kinds of food, particularly protein-rich foods such as milk, meat, and eggs. Mutations in these genes, BCKDHA, BCKDHB and DBT, result in absent or decreased activity of human branched-chain alpha-ketoacid dehydrogenase complex (BCKAD) enzymes. These enzymes are responsible for breaking down the branched chain amino acids leucine, isoleucine, and valine that are in protein-rich foods. The branched-chain amino acids (BCAAs) are the only amino acids that have a split main carbon chain. Accumulation of these amino acids and their toxic byproducts (ketoacids) results in the serious health problems associated with maple syrup urine disease. The toxicity of these amino acids seems to be restricted to the leucine; indeed, extra valine and isoleucine are often given during treatment. Accumulation of their respective ketoacids results in metabolic acidosis.
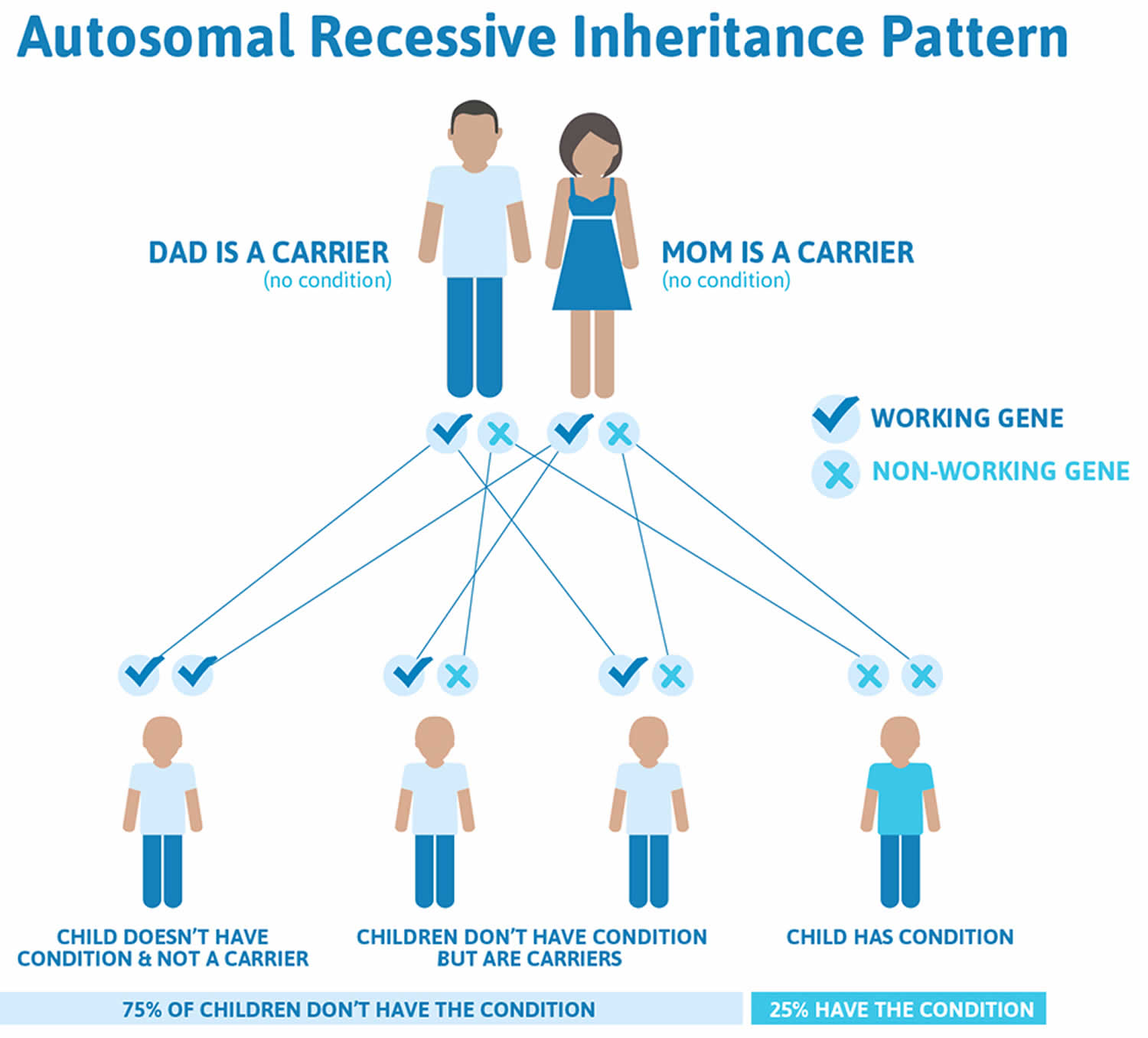
Maple syrup urine disease is inherited as an autosomal recessive genetic condition. Recessive genetic disorders occur when an individual inherits two copies of an altered gene for the same trait, one from each parent. If an individual inherits one normal gene and one gene for the disease, the person will be a carrier for the disease but usually will not show symptoms. The risk for two carrier parents to both pass the altered gene and have an affected child is 25% with each pregnancy. The risk to have a child who is a carrier like the parents is 50% with each pregnancy. The chance for a child to receive normal genes from both parents is 25%. The risk is the same for males and females.
Parents who are close relatives (consanguineous) have a higher chance than unrelated parents to both carry the same abnormal gene, which increases the risk to have children with a recessive genetic disorder.
If you’re a carrier of the affected genes and have a baby with a partner who’s also a carrier, your baby has:
- a 1 in 4 chance of developing the condition
- a 1 in 2 chance of being a carrier of maple syrup urine disease
- a 1 in 4 chance of receiving a pair of normal genes
Although it’s not possible to prevent maple syrup urine disease, it’s important to let your midwife and doctor know if you have a family history of the condition. Any further children you have can be tested for the condition as soon as possible and given appropriate treatment.
You may also wish to consider genetic counseling for support, information and advice about genetic conditions.
Figure 1. Maple syrup urine disease autosomal recessive inheritance pattern
Related Disorders
Symptoms of the following disorders can be similar to those of maple syrup urine disease. Comparisons may be useful for a differential diagnosis.
The urea cycle disorders are a group of rare disorders affecting the urea cycle, a series of biochemical processes in which nitrogen is converted into urea and removed from the body through the urine. The symptoms of all urea cycle disorders vary in severity and result from the excessive accumulation of ammonia in the blood and body tissues (hyperammonemia). Common symptoms include lack of appetite, vomiting, drowsiness, seizures, and/or coma. The liver may be abnormally enlarged (hepatomegaly). In some children, life-threatening complications may result. The urea cycle disorders include ornithine transcarbamylase (OTC) deficiency: carbamyl phosphate synthetase (CPS) deficiency; argininosuccinate synthetase deficiency (citrullinemia); argininosuccinate lyase (ASL) deficiency; arginase deficiency (argininemia); and N-acetylglutamate synthetase (NAGS) deficiency.
Propionic acidemia is a rare metabolic disorder caused by a deficiency of the enzyme propionyl CoA carboxylase, one of the enzymes necessary in the process of breaking down amino acids. Symptoms most commonly become apparent during the first weeks of life and may include hypotonia, poor feeding, vomiting, dehydration, and seizures. Without appropriate treatment, coma and potentially life-threatening complications may result. In rare cases, the condition may become apparent later during infancy and may be associated with less severe symptoms and findings. Propionic acidemia is inherited as an autosomal recessive genetic condition.
Methylmalonic acidemia (MMA) is an inborn error of metabolism in which people have trouble breaking down certain proteins and fats in food. The symptoms of MMA usually arise during early infancy, but can also begin in adulthood. Symptoms can include vomiting, dehydration, hypotonia, developmental delay, lethargy, enlarged liver and failure to thrive (inability to gain weight or reach developmental milestones). Over the course of the disease, patients can develop intellectual disability, chronic kidney disease, pancreatitis and feeding problems. There are multiple treatments for MMA: cobalamin and carnitine supplements are provided along with strict dietary control such as a low-protein diet. If supplements do not work effectively, doctors may recommend a diet free of certain amino acids; namely, methionine, threonine, isoleucine and valine. MMA is an autosomal recessive genetic disorder caused by mutations in five different genes: MMAA, MMAB, MMADHC, MCEE and MUT.
Glycine encephalopathy is an inborn error of metabolism characterized by the accumulation of large amounts of the amino acid glycine in blood and, particularly, the cerebrospinal fluid (CSF). The metabolic block occurs in the conversion of glycine into smaller molecules. There are four forms of this disorder: a relatively common neonatal form, an infantile form, a mild-episodic form, and a late-onset form. Common presenting symptoms of the disease include hypotonia, seizures, unexplained coma, and developmental delay in neonates and infants.
Maple syrup urine disease diagnosis
Many infants with maple syrup urine disease are identified through newborn screening programs. At around 5 days old, babies are offered newborn blood spot screening to check if they have maple syrup urine disease. This involves pricking your baby’s heel to collect drops of blood to test. Tandem mass spectrometry, an advanced newborn screening test that screens for more than 30 different disorders through one blood sample, has aided in the diagnosis of maple syrup urine disease. Infants with mild or intermittent forms of the disorder may have totally normal blood amino acids after birth and thus can be missed by newborn screening.
In places where testing for maple syrup urine disease is unavailable or where newborn screening fails to detect maple syrup urine disease, a diagnosis may be suspected based upon symptomatic findings (lethargy, failure to thrive, neurologic signs or, during a metabolic crisis, odor of maple syrup in earwax, sweat or urine). Tests to diagnose maple syrup urine disease may include urine analysis to detect high levels of keto acids (ketoaciduria) and blood analysis to detect abnormally high levels of amino acids.
An enzymatic diagnosis may be confirmed through analysis of white blood cells (lymphocytes) or cells taken from an affected individual’s skin. Early diagnosis, especially in suspected individuals, allows for management of asymptomatic infants before the onset of the usual clinical manifestations. Diagnosis through DNA testing is readily available and prenatal diagnosis is available.
Molecular genetic testing for mutations in the BCKDHA, BCKDHB and DBT genes is also available to confirm the diagnosis, and is necessary for purposes of carrier testing for at-risk relatives and prenatal diagnosis for at-risk pregnancies.
If maple syrup urine disease is diagnosed, treatment can be given straight away to reduce the risk of serious complications.
With early diagnosis and the correct treatment, the outcome can be greatly improved. However, treatment for maple syrup urine disease must be continued for life.
Without treatment, severe, life-threatening symptoms can develop, including seizures (fits) or falling into a coma. Some children with untreated maple syrup urine disease are also at risk of brain damage and developmental delay.
Maple syrup urine disease treatment
The treatment of classic, intermediate, intermittent, and thiamine-responsive maple syrup urine disease has two chief components: lifelong therapy to maintain acceptable amino acid levels in the body and immediate medical intervention for metabolic crises.
Maple syrup urine disease diet
Children diagnosed with maple syrup urine disease are first referred to a specialist metabolic dietitian and given a low-protein diet. This is tailored to reduce the amount of amino acids your baby receives, especially leucine, valine and isoleucine.
High-protein foods need to be limited, including:
- meat
- fish
- cheese
- eggs
- pulses
- nuts
Your dietitian will provide detailed advice and guidance, as your baby still needs some of these foods for healthy growth and development.
Individuals with maple syrup urine disease must remain on a protein-restrictive diet that limits the amount of branched-chain amino acids they take in. Protein-restriction must start as soon as possible after birth to promote proper growth and development. Artificially-made (synthetic) formulas are available that provide all the nutrients necessary for proper growth and development, but lack leucine, isoleucine and valine. It is particularly important to limit the amount of leucine in the diet. The three amino acids are added to the diet separately in small amounts so that affected individuals can grow and develop normally. The amount of leucine, isoleucine and valine that can be tolerated by a child varies based upon residual enzyme activity. Affected children must be regularly monitored to ensure that their amino acid levels remain within acceptable normal ranges.
Breastfeeding and baby milk also need to be monitored and measured, as advised by your dietitian. Regular baby milk contains the amino acids that need to be restricted, so a special formula is used instead. This contains all the vitamins, minerals and other amino acids your baby needs.
People with maple syrup urine disease need to follow a low-protein diet for the rest of their life to reduce the risk of a metabolic crisis. As your child gets older, they’ll eventually need to learn how to control their diet and will stay in contact with a dietitian for advice and monitoring.
Some physicians recommend a trial of thiamine therapy to determine whether an affected individual is thiamine-responsive. However, no individual with maple syrup urine disease has been treated solely with thiamine.
Even if affected individuals strictly follow a specialized diet, a risk of metabolic crisis still exists. Episodes of metabolic crisis require immediate medical intervention to lower the levels of branched-chain amino acids, especially leucine, in the blood plasma. Various techniques have been used to reduce plasma leucine levels including dialysis or a process in which plasma is removed from the body and passed through a filter before being returned to the body (hemofiltration).
The aim of aggressive therapy for metabolic crises is to try and reduce, and then reverse, the increased protein catabolism that is the root cause of such episodes. This means that ANY method to increase calories, to reduce protein catabolism [for energy needs] may be helpful. This includes a high glucose intake with intravenous glucose, if necessary, supplemented by a “glucose-insulin drip” since insulin is known to enhance endogenous protein synthesis. Intravenous fat is another important source of calories. In addition, it is essential to provide all the other amino acids in amounts sufficient to permit new protein synthesis. This is done by the judicious use of intra GI drips or more usually, parenteral nutrition IV using solutions that lack leucine. Many hospitals may use total parenteral nutrition solutions that lack branched-chain amino acid. In addition, insulin may be used to stimulate a metabolic process known as anabolism. During anabolism, various cellular components (including proteins) are combined (synthesized) to formed energy-rich compounds.
Other treatment is symptomatic and supportive. Early intervention is important in ensuring that children with maple syrup urine disease reach their highest potential.
Genetic counseling is recommended for affected individuals and their families.
Emergency treatment
If your baby develops an infection, such as a high temperature or cold, their risk of having a metabolic crisis increases. It’s possible to reduce the risk by changing to an emergency diet while they’re ill.
Your dietitian will provide detailed instructions, but the aim is to replace milk and foods containing protein with special high-sugar drinks and amino acid supplements.
If your baby can’t keep down their emergency feeds or has repeated diarrhea, contact the metabolic team at the hospital to let them know you’re heading straight to the accident and emergency department.
You should also be given a leaflet to bring with you in the event of an emergency in case the doctors have not seen maple syrup urine disease before.
Once in hospital, your baby can be monitored and treated with fluids given directly into a vein (intravenous fluids).
You should also take your baby to hospital if they develop the symptoms of a metabolic crisis, such as irritability, loss of energy or breathing difficulties.
Liver transplant
A liver transplant is sometimes an option to treat maple syrup urine disease. If a person with maple syrup urine disease receives a donated liver, they’ll no longer be at risk of a metabolic crisis and can have a normal diet. Liver transplantation has resulted in individuals who are symptom-free and able to eat protein-rich foods. The new liver supplies enough of the enzymes needed to breakdown the three amino acids that accumulate in maple syrup urine disease. However, availability of a donor liver and the high cost are hurdles to this procedure. For those that do undergo transplantation, success rates are very high. University of Pittsburgh Children’s Hospital and the Clinic for Special Children conducted a collaborative study involving 52 liver transplants between 2004 and 2013. Of these 52, 100% of the patients had disease-free survival and graft survival. More research is necessary to determine the long-term effects of liver transplantation on neurological development in individuals with maple syrup urine disease.
Furthermore, a liver transplant is a major procedure with its own risks. You will have to take medicine to suppress the immune system (immunosuppressant medication) for the rest of your life to stop your body rejecting the new liver.
It’s important to consider all the pros and cons before deciding whether or not to have a liver transplant. Your doctor will be able to discuss whether this is a suitable option.
- Maple syrup urine disease. https://ghr.nlm.nih.gov/condition/maple-syrup-urine-disease[↩][↩]
- Maple Syrup Urine Disease. https://rarediseases.org/rare-diseases/maple-syrup-urine-disease/[↩]
- K.A. Strauss, E.G. Puffenberger, D.H. Morton, Maple syrup urine disease, in: R. Pagon, M. Adam, T. Bird, et al. (Eds.), GeneReviews, University of Washington, Seattle, WA, 30 January 2006, pp. 1993–2013[↩][↩][↩]
- K.A. Strauss, B. Wardley, D. Robinson, C. Hendrickson, N.L. Rider, E.G. Puffenberger, D. Shellmer, A.B. Moser, D.H. Morton, Classical maple syrup urine disease and brain development: principles of management and formula design, Mol. Genet. Metab. 99 (4) (2010) 333–345.[↩]
- D.M. Frazier, C. Allgeier, C. Homer, B.J. Marriage, B. Ogata, F. Rohr, P.L. Splett, A. Stembridge, R.H. Singh, Nutrition management guideline for maple syrup urine disease: an evidence- and consensus-based approach, Mol. Genet. Metab. 112 (3) (2014) 210–217.[↩]
- V.M. Diaz, C. Camarena, Á. de la Vega, M. Martínez-Pardo, C. Díaz, M. López, F. Hernández, A. Andrés, P. Jara, Liver transplantation for classical maple syrup urine disease: long-term follow-up, J. Pediatr. Gastroenterol. Nutr. 59 (5) (2014) 636–639.[↩]
- P.R. Blackburn, J.M. Gass, F.P.E. Vairo, K.M. Farnham, H.K. Atwal, S. Macklin, E.W. Klee, P.S. Atwal, Maple syrup urine disease: mechanisms and management, Appl. Clin. Genet. 10 (2017) 10:57–66.[↩]





{kind=link}