
Contents
- What is MCAD deficiency
- What risks are posed to other members of an affected individual’s family?
- Where can I learn more about the risks posed to my family members and the importance of genetic testing?
- What happens to the fats when a baby with MCAD deficiency cannot break them down?
- Is it normal for a baby with MCAD deficiency to be hungry all the time?
- What is newborn screening?
- Are there factors which can cause a false positive test result for MCAD deficiency on newborn screen?
- What follow-up testing can be done to definitively determine if my child has MCAD deficiency?
- MCAD deficiency causes
- MCAD deficiency symptoms
- MCAD deficiency diagnosis
- MCAD deficiency treatment
- MCAD deficiency life expectancy
What is MCAD deficiency
MCAD deficiency is short for medium-chain acyl-coenzyme A dehydrogenase deficiency, which is an inherited metabolic disorder that prevents the body from converting certain fats to energy, particularly during periods without food (fasting). MCAD deficiency is a genetic disorder of mitochondrial fatty acid beta-oxidation. This means that fats in the body cannot efficiently be broken down and used for energy. The body relies on glucose (a sugar) for energy, and during times when the amount of glucose in the blood is too low, the body can break down fat stores and convert them to glucose. Enzymes called acyl-coenzyme A dehydrogenases are necessary for one of the steps in the biochemical pathway by which fat is broken down into glucose. Medium chain acyl-coenzyme A dehydrogenase (MCAD) is one of these enzymes. People with MCAD deficiency do not have enough of medium chain acyl-coenzyme A dehydrogenase enzyme needed to metabolize a group of fats called medium-chain fatty acids. Signs and symptoms usually begin by early childhood generally develop between 1 and 24 months of age, although they can sometimes first appear in adulthood and may include vomiting, lack of energy, and low blood sugar (hypoglycemia). Individuals with MCAD deficiency experience symptoms of metabolic crisis due to low blood sugar (hypoglycemia) after periods of prolonged fasting or in response to a common viral illness. These may include weakness, vomiting, and seizures. Rarely, coma or sudden death may occur. MCAD deficiency is inherited as autosomal recessive genetic condition.
MCAD deficiency is caused by mutations in the ACADM gene and inheritance is autosomal recessive 1. MCAD deficiency is usually diagnosed through newborn screening. An early diagnosis of this disorder is important in order to be able to prevent symptoms from occurring. MCAD deficiency is a known cause of sudden infant death syndrome (SIDS).
In the United States, the estimated incidence of MCAD deficiency is 1 in 17,000 people. MCAD deficiency is more common in people of northern European ancestry than in other ethnic groups.
A diagnosis of MCAD deficiency requires an evaluation of a person’s symptoms as well as the interpretation of several tests. Initial testing may include:
- Plasma acylcarnitine
- Urine organic acid
- Urine acylglycine
Further testing to confirm the diagnosis may include molecular genetic testing of the ACADM gene or biochemical genetic testing 2.
MCAD deficiency is included in many newborn screening programs, so a newborn with MCAD deficiency who does not yet exhibit symptoms may be diagnosed early. If a newborn screening result for MCAD deficiency is not in the normal range (“positive”), additional testing can then be ordered 3.
Treatment includes strict avoidance of fasting and avoidance of medium chain triglycerides in the diet 4. If not treated, people with MCAD deficiency are at risk of serious complications including sudden death 1.
What risks are posed to other members of an affected individual’s family?
The risk to other family members depends on the status of the individuals most closely related to the affected individual. At conception, the siblings of an affected individual have a 25% risk of being affected, a 50% risk of being asymptomatic carriers, and a 25% risk of being unaffected and not carriers. The risk could be 50% if one of the parents is also affected. Because asymptomatic parents and siblings may have MCAD deficiency, biochemical evaluation and/or molecular genetic testing should be offered to both parents and all siblings 2.
Where can I learn more about the risks posed to my family members and the importance of genetic testing?
Your family may benefit from consulting with a genetics professional who can help to determine who may be at risk for medium-chain acyl-coenzyme A dehydrogenase (MCAD) deficiency and who warrants further testing.
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
What happens to the fats when a baby with MCAD deficiency cannot break them down?
Medium chain acyl-CoA dehydrogenase deficiency is a fatty acid oxidation disorder in which people have problems breaking down fat into energy for the body. It is caused by an enzyme that does not work properly. MCAD deficiency occurs when an enzyme, called medium chain acyl-CoA dehydrogenase, is either missing or not working properly. This enzyme’s job is to break down certain fats in food into energy. It also breaks down fat already stored in the body. Energy from fat keeps us going whenever our bodies run low of their main source of energy, a type of sugar called glucose. Your body rely on fat when you don’t eat for a stretch of time – like when you miss a meal or when you sleep 5.
When the medium chain acyl-CoA dehydrogenase enzyme is missing or not working well, the body cannot use certain types of fat for energy, and must rely solely on glucose. Although glucose is a good source of energy, there is a limited amount available. Once the glucose has been used up, the body tries to use fat without success. This leads to low blood sugar, called hypoglycemia, and to the build up of harmful substances in the blood, which can lead to metabolic crises. Symptoms often happen after having nothing to eat for more than a few hours. Hypoglycemia can occur, with or without other symptoms of metabolic crisis, just by going too long without food. Hypoglycemia can cause a person to feel weak, shaky, or dizzy, and to have clammy, cold skin. If not treated, hypoglycemia can lead to coma and even death 5.
Is it normal for a baby with MCAD deficiency to be hungry all the time?
Infants and young children with MCAD deficiency need to eat frequently to prevent hypoglycemia or a metabolic crisis. In general, it is often suggested that these infants be fed every four to six hours. Some babies need to eat even more frequently than this. It is also important that infants be fed during the night. They may need to be woken up to eat if they do not wake up on their own. Your metabolic doctor and dietician will give you an appropriate feeding plan for your infant 5.
A low fat, high carbohydrate food plan is often recommended. Carbohydrates give the body many types of sugar that can be used as energy. For children needing this treatment, most food in the diet should be carbohydrates (bread, pasta, fruit, vegetables, etc.) and protein (lean meat and low-fat dairy foods). Your dietician can create a food plan with the correct type and amount of fat your child needs. Any diet changes should be made under the guidance of an experienced dietician 5.
What is newborn screening?
Newborn screening tests look for serious developmental, genetic, and metabolic disorders so that important action can be taken during the critical time before symptoms develop. Screening tests do not diagnose illnesses. They identify which babies need additional testing to confirm or rule out illnesses. Good screening tests have a low false-negative rate (if the test is normal, the child should be healthy), but may have a high false-positive rate (as many affected children as possible should test positive, even if this means many healthy children also test positive) 6.
An abnormal result on a newborn screen means that the child should have additional testing to confirm or rule out the condition. If a disorder is diagnosed on follow-up testing, appropriate treatment can be started right away, before symptoms appear 6.
Are there factors which can cause a false positive test result for MCAD deficiency on newborn screen?
The false positive rate for medium-chain acyl-coenzyme A dehydrogenase (MCAD) deficiency varies between screening programs because of differences in analysis of acylcarnitine (a type of fatty acid). Programs that screen for MCAD deficiency but not other fatty acid oxidation disorders often limit their analysis to octanoylcarnitine, the primary screening marker for MCAD deficiency. However, octanoylcarnitine is not specific for MCAD deficiency and is expected to be elevated in several other disorders including glutaric acidemia type 2. Consideration of other potential disorders in the differential diagnosis should minimize the false positive rate 2.
What follow-up testing can be done to definitively determine if my child has MCAD deficiency?
ACADM is the only gene known to be associated with medium-chain acyl-coenzyme A dehydrogenase (MCAD) deficiency. The biochemical diagnosis of MCAD deficiency can be confirmed by measurement of MCAD enzyme activity in fibroblasts or other tissues and/or by genetic testing to detect mutations in the ACADM gene 2.
For more information on genetic testing for MCAD deficiency, we would recommend you consult with a genetics professional.
MCAD deficiency causes
Mutations in the ACADM gene cause MCAD deficiency. This gene provides instructions for making an enzyme called medium-chain acyl-CoA dehydrogenase, which is required to break down (metabolize) a group of fats called medium-chain fatty acids. These fatty acids are found in foods and the body’s fat tissues. Fatty acids are a major source of energy for the heart and muscles. During periods of fasting, fatty acids are also an important energy source for the liver and other tissues.
Mutations in the ACADM gene lead to a shortage (deficiency) of the medium-chain acyl-coenzyme A dehydrogenase enzyme within cells. Without sufficient amounts of MCAD enzyme, medium-chain fatty acids are not metabolized properly. As a result, these fats are not converted to energy, which can lead to the characteristic signs and symptoms of this disorder such as lethargy and hypoglycemia. Medium-chain fatty acids or partially metabolized fatty acids may also build up in tissues and damage the liver and brain. This abnormal buildup causes the other signs and symptoms of MCAD deficiency.
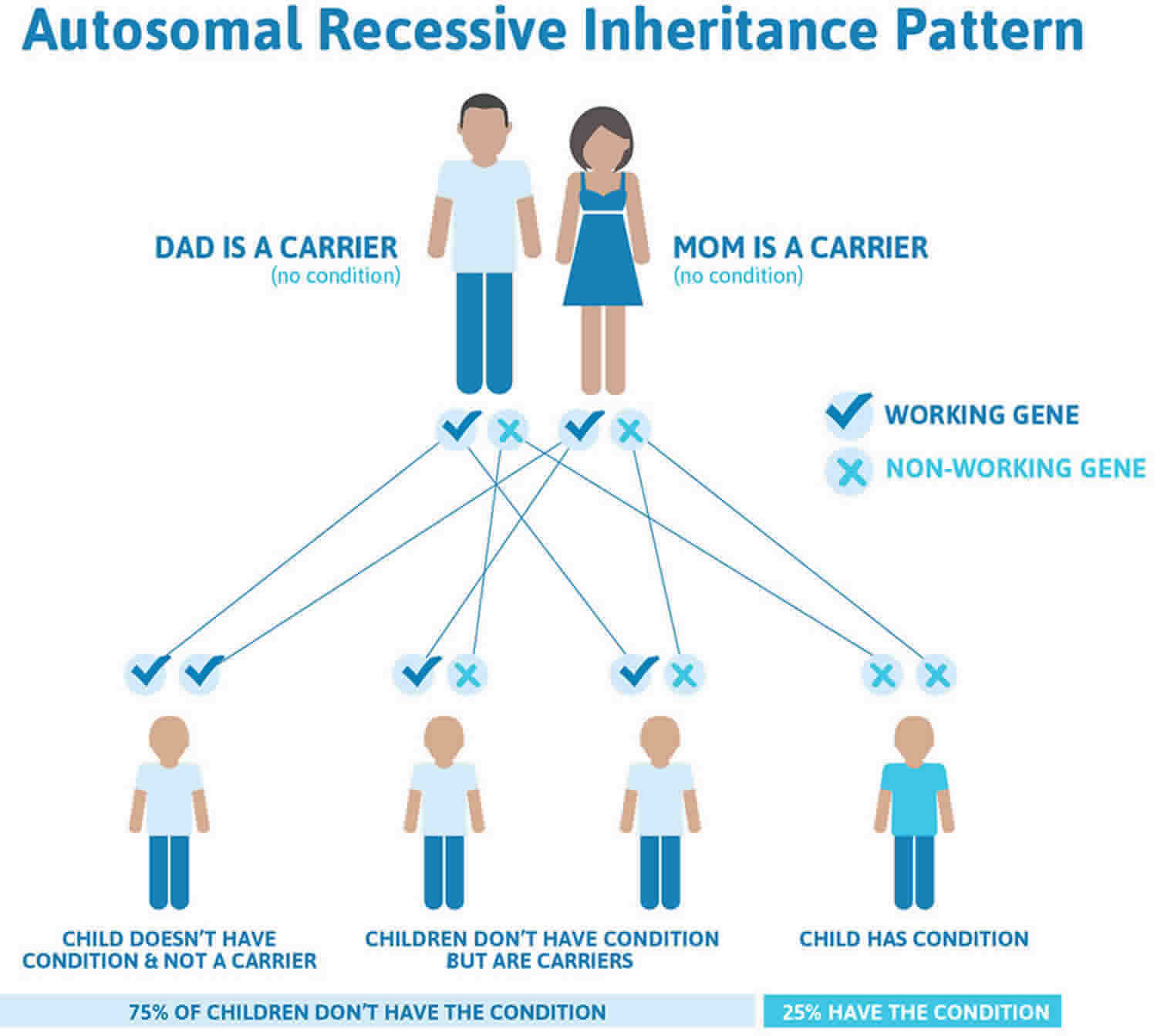
MCAD deficiency inheritance pattern
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
It is rare to see any history of autosomal recessive conditions within a family because if someone is a carrier for one of these conditions, they would have to have a child with someone who is also a carrier for the same condition. Autosomal recessive conditions are individually pretty rare, so the chance that you and your partner are carriers for the same recessive genetic condition are likely low. Even if both partners are a carrier for the same condition, there is only a 25% chance that they will both pass down the non-working copy of the gene to the baby, thus causing a genetic condition. This chance is the same with each pregnancy, no matter how many children they have with or without the condition.
- If both partners are carriers of the same abnormal gene, they may pass on either their normal gene or their abnormal gene to their child. This occurs randomly.
- Each child of parents who both carry the same abnormal gene therefore has a 25% (1 in 4) chance of inheriting a abnormal gene from both parents and being affected by the condition.
- This also means that there is a 75% ( 3 in 4) chance that a child will not be affected by the condition. This chance remains the same in every pregnancy and is the same for boys or girls.
- There is also a 50% (2 in 4) chance that the child will inherit just one copy of the abnormal gene from a parent. If this happens, then they will be healthy carriers like their parents.
- Lastly, there is a 25% (1 in 4) chance that the child will inherit both normal copies of the gene. In this case the child will not have the condition, and will not be a carrier.
These possible outcomes occur randomly. The chance remains the same in every pregnancy and is the same for boys and girls.
Figure 1 illustrates autosomal recessive inheritance. The example below shows what happens when both dad and mum is a carrier of the abnormal gene, there is only a 25% chance that they will both pass down the abnormal gene to the baby, thus causing a genetic condition.
Figure 1. MCAD deficiency autosomal recessive inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
MCAD deficiency symptoms
Symptoms of MCAD deficiency are characterized by metabolic crisis brought about by low blood sugar (hypoglycemia). Because infants are typically weaned from nighttime feedings sometime between 3 and 24 months of age, this is when the infant’s first experience with longer fasting would occur. Being previously healthy, a child with MCAD deficiency might suddenly experience severe symptoms at this point. A child could also have symptoms in response to a common and normally mild disease like a cold, since it can decrease appetite and increase the body’s metabolic requirements. Alternatively, an individual with a milder form of MCAD deficiency might first develop symptoms in adulthood, in response to an extreme metabolic stress such as surgery or severe illness. A person with MCAD deficiency who never entered a low blood sugar state would never experience the symptoms of the disease.
In the event of a hypoglycemic crisis, the affected individual might experience tiredness/weakness (lethargy), vomiting (emesis), seizures, coma, or sudden death (in 18% of first crises). Additional signs of the disease could include an enlarged liver (hepatomegaly), low blood sugar due to inefficient breakdown of fats (hypoketotic hypoglycemia), and elevated levels of certain substances in the blood or urine (e.g. acylglycines).
Secondary symptoms of MCAD deficiency that can develop after a person has experienced one or multiple metabolic crises are caused by damage to body tissues due to the hypoglycemic conditions during the events. These can include lasting muscle weakness and pain, as well as reduced tolerance to exercise. Affected individuals may acquire such brain disorders as an inability to understand or use language (aphasia) and attention deficit disorder due to damage to the brain. Women with MCAD deficiency may experience pregnancy complications such as HELLP syndrome (hemolysis, elevated liver enzymes, and low platelet count).
MCAD deficiency diagnosis
MCAD deficiency is usually diagnosed through newborn screening by a blood test. The test looks for the amount of chemicals known as acylcarnitines. High levels of a type of acylcarnitine called octanoylcarnitine are characteristic of MCAD deficiency, but this is not specific to this disorder. Moreover, if an infant develops the classic symptoms or dies after being otherwise healthy, a diagnosis of MCAD deficiency would be considered. This means that vomiting, lethargy, and seizures after a period of fasting or a common illness in a previously healthy individual would suggest MCAD deficiency. Additionally, low blood sugar (hypoketonic hypoglycemia) in response to fasting or illness would be a more measurable sign of the disorder. It is also prudent to consider family genetic history when making a diagnosis. Some individuals may never develop symptoms of MCAD deficiency or be diagnosed.
Clinical Testing/Workup
Various biochemical analyses can be used to further confirm the diagnosis of MCAD deficiency and to monitor the condition. Genetic testing to identify two copies of the pathogenic version (allele) of the ACADM gene may be done. It is possible to extract cells from the body, grow them in a culture dish, and run tests to directly assess the activity of MCAD or the cells’ ability to break down fat. Furthermore, urine analysis can be done to test for such metabolite markers as medium chain dicarboxylic acids or organic acids and acylglycines. Blood plasma tests to measure acylcarnitines like octanoylcarnitines (C8) may be done. In all cases, interpretation of these tests should take into consideration the symptomatic state of the person, as an asymptomatic individual may not show elevated levels of the various indicator molecules in the body. The results of the testing and confirmed diagnosis would provide valuable information for the genetic counseling of the individual’s family and any potential reproductive partner.
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
MCAD deficiency treatment
The focus of treatment for MCAD deficiency is the prevention of the symptoms. It is important to maintain blood sugar levels by avoiding fasting for long periods. Frequent feeding is encouraged, and it is helpful to consume sources of complex carbohydrates at bedtime in order to supply a steadier source of glucose overnight. Treatment requirements and recommendations will vary depending upon the severity of the deficiency—how much functional MCAD is present in the body.
It is advisable to consult a genetic metabolic specialist and undergo routine surveillance of the condition. Because of the higher feeding requirements and lower tolerance of exercise associated with MCAD deficiency, it is also helpful to talk to a dietician in order to ensure one is consuming a healthful balanced diet, although significant dietary modification is generally not required. In general, feeding infants with formulas high in medium chain fatty acids is inadvisable. In adults, excessive alcohol consumption can lead to a metabolic crisis. In any case, a metabolic crisis can be treated or reverted by consuming a glucose supplement or foods high in sugars. Glucose can be administered directly to the blood (IV) as well.
MCAD deficiency diet
Strict avoidance of fasting is the primary objective. Medium chain triglycerides should be avoided but no other special dietary restrictions are required. Guidelines are available for the safe interval time between feeds for infants and young children. The use of low dose carnitine supplementation in patients who develop a low blood carnitine status remains controversial. In symptomatic patients, simple carbohydrates are given by mouth (glucose tablets) or intravenously until blood glucose level is maintained at above 5 mmol/L. During intercurrent infections, an emergency regimen should be made available. Immediate medical attention is needed in case of decompensation. Artificial sweeteners should be avoided.
The United Mitochondrial Disease Foundation lists alcohol as a substance that people with mitochondrial disease should avoid, because alcohol has been known to hasten the progression of some mitochondrial disorders 7. In 2009, Lang 8 reviewed several cases of MCAD deficiency diagnosed in adulthood. Many of these cases had alcohol consumption as a precipitating factor for an episode of metabolic crisis. Alcohol inhibits the breakdown (oxidation) of fatty acids in the liver, which can lead to the toxic accumulation of fatty acids within cells. In the cases reviewed by Lang, the consumption of alcohol also led to vomiting, which may have caused symptoms due to a period of fasting.
MCAD deficiency life expectancy
The prognosis is excellent in diagnosed patients who avoid fasting and who are managed appropriately during an intercurrent illness/ metabolic crisis.
- Medium-chain acyl-CoA dehydrogenase deficiency. https://ghr.nlm.nih.gov/condition/medium-chain-acyl-coa-dehydrogenase-deficiency[↩][↩]
- Matern D, Rinaldo P. Medium-Chain Acyl-Coenzyme A Dehydrogenase Deficiency. 2000 Apr 20 [Updated 2015 Mar 5]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2019. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1424[↩][↩][↩][↩]
- MCAD deficiency. https://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=EN&Expert=42[↩]
- Medium chain acyl-CoA dehydrogenase deficiency. https://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=EN&Expert=42[↩]
- Fatty Acid Disorder. http://www.newbornscreening.info/Parents/fattyaciddisorders/MCADD.html[↩][↩][↩][↩]
- Newborn screening tests. https://medlineplus.gov/ency/article/007257.htm[↩][↩]
- Treatments & Therapies. https://www.umdf.org/what-is-mitochondrial-disease/treatments-therapies/[↩]
- Lang TF. Adult presentations of medium-chain acyl-CoA dehydrogenase deficiency (MCADD). Journal of Inherited Metabolic Disease. 2009; http://www.ncbi.nlm.nih.gov/pubmed/19821147[↩]





{kind=link}