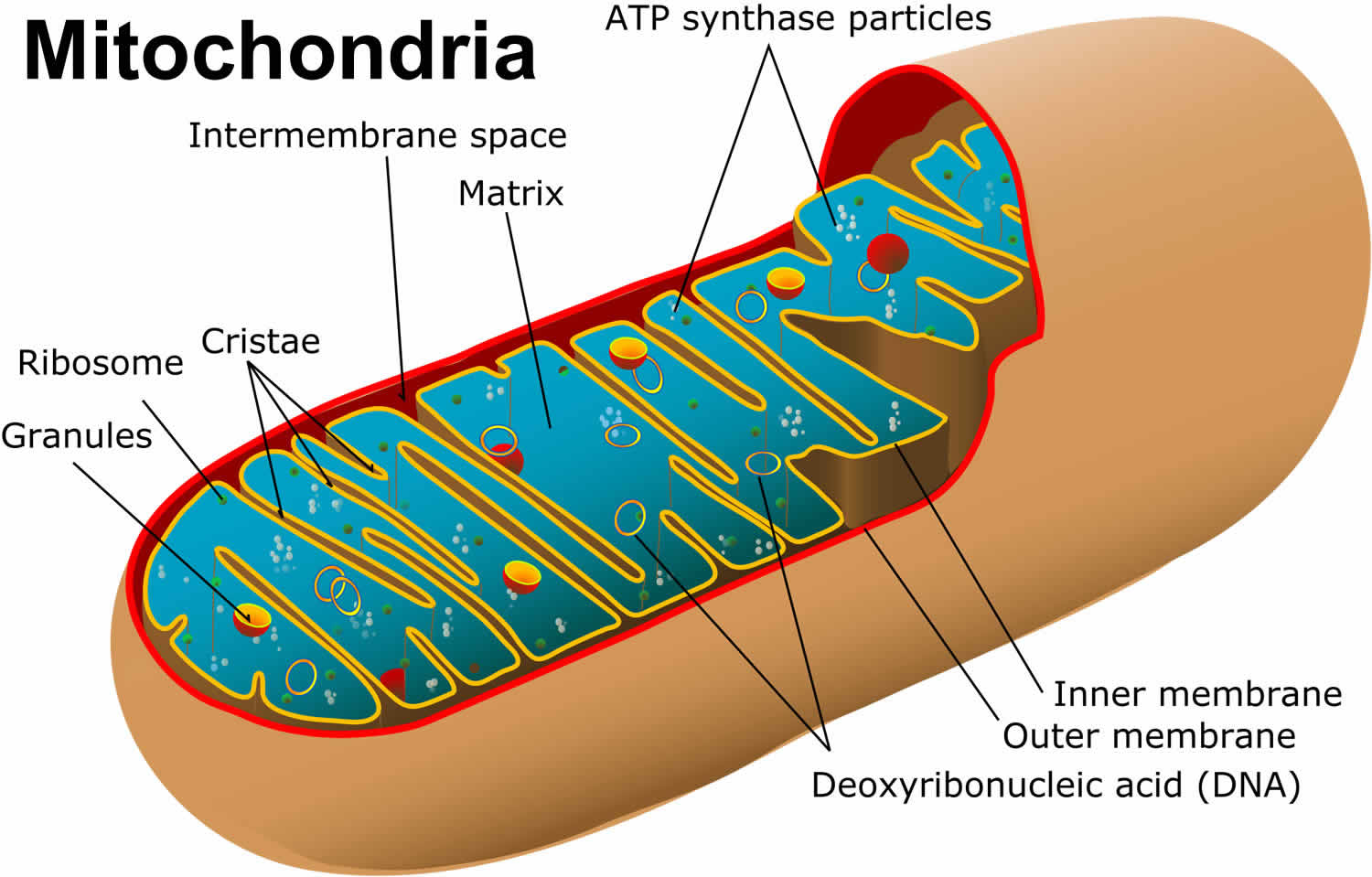
What is mitochondria
You have mitochondria present in every cell of your body except red blood cells. Mitochondria are membrane-bound cell organelles (mitochondrion, singular) that generate most of the chemical energy needed to power the cell’s biochemical reactions. The mitochondria in the cells throughout your body are responsible for creating more than 90% of the energy needed by your body to sustain life and support organ function. When mitochondria fail, less and less energy is generated within the cell. Cell injury and even cell death follow. If this process is repeated throughout the body, whole organ systems begin to fail – people get sick, and even die. Chemical energy produced by the mitochondria is stored in a small molecule called adenosine triphosphate (ATP). Mitochondria contain their own small chromosomes. Generally, mitochondria, and therefore mitochondrial DNA, are inherited only from the mother. Problems with mitochondria, the structures that produce energy for all cells, have been linked to the development of Parkinson’s disease.
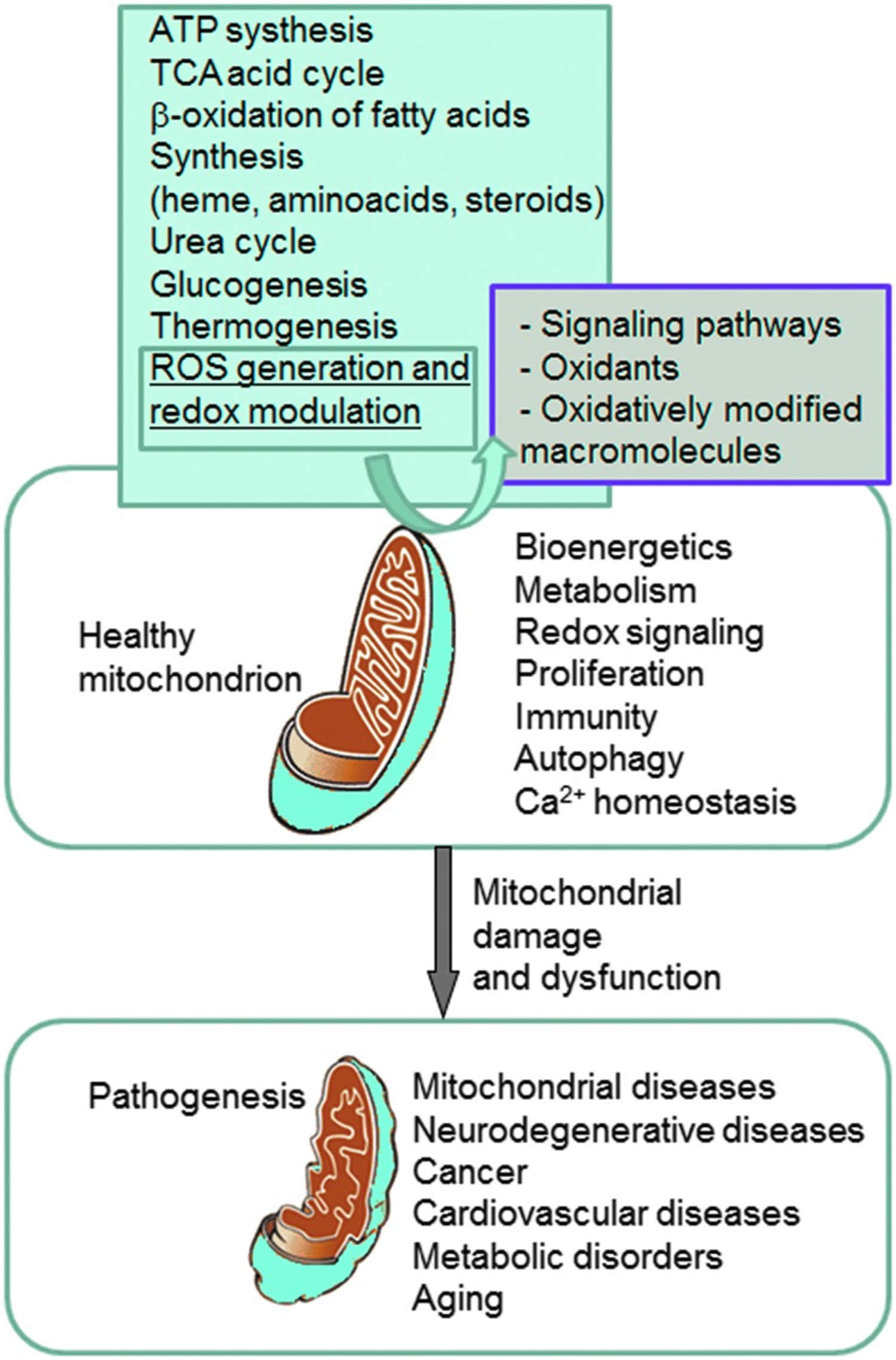
Mitochondria play a fundamental role in cell physiology; mitochondria organelles are involved in a variety of processes, including bioenergetics, various metabolic pathways, including crucial anabolic and catabolic reactions, such as ATP (adenosine triphosphate) synthesis, the tricarboxylic acid cycle (citric acid cycle or Kreb cycle), and biosynthetic processes, and govern fundamental cellular actions, including proliferation, immunity, and autophagy. Mitochondrial damage and malfunction have been related to the pathogenesis of a large number of human pathologies, such as mitochondrial diseases, neurodegenerative diseases, cancer, cardiovascular diseases, metabolic disorders, and aging. The participation of mitochondria in the redox equilibrium and redox signaling of the cell is also pivotal. Modification of the redox state and increased reactive oxygen species (ROS) production within mitochondria have major consequences for both mitochondrial and extramitochondrial processes and, ultimately, modulate fundamental cellular phenomena such as autophagy and apoptosis.
In people with mitochondrial disease, the parts of the body, such as the heart, brain, muscles and lungs, requiring the greatest amounts of energy are the most affected 1. Based upon recent epidemiological studies, mitochondrial disorders affect at least 1 in 8000 of the general population 2. Mitochondrial disease is difficult to diagnose, because it affects each individual differently. Symptoms can include seizures, strokes, severe developmental delays, inability to walk, talk, see, and digest food combined with a host of other complications. If three or more organ systems are involved, mitochondrial disease should be suspected.
Figure 1. Mitochondria cell
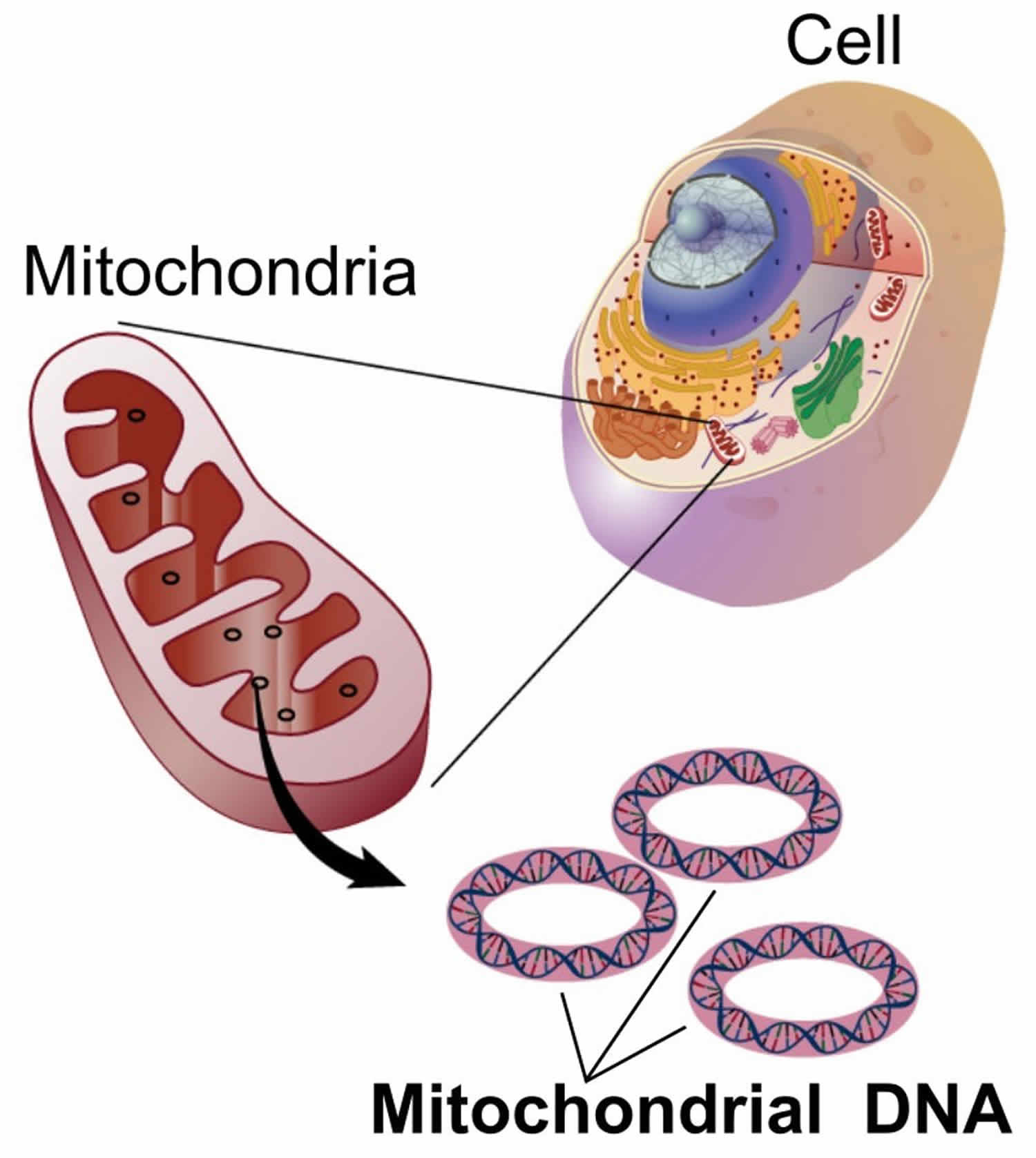
Mitochondrial diseases are the result of either inherited or spontaneous mutations in mitochondrial DNA (mtDNA) or nuclear DNA (nDNA) which lead to altered functions of the proteins or RNA molecules that normally reside in mitochondria. Problems with mitochondrial function, however, may only affect certain tissues as a result of factors occurring during development and growth that scientists do not yet understand. Even when tissue-specific isoforms of mitochondrial proteins are considered, it is difficult to explain the variable patterns of affected organ systems in the mitochondrial disease syndromes seen clinically.
Because mitochondria perform so many different functions in different tissues, there are literally hundreds of different mitochondrial diseases. Each disorder produces a spectrum of abnormalities that can be confusing to both patients and physicians in early stages of diagnosis. Because of the complex interplay between the hundreds of genes and cells that must cooperate to keep our metabolic machinery running smoothly, it is a hallmark of mitochondrial diseases that identical mtDNA mutations may not produce identical diseases. Genocopies are diseases that are caused by the same mutation but which may not look the same clinically.
The converse is also true: different mutations in mitochondrial DNA (mtDNA) and nuclear DNA (nDNA) can lead to the same diseases. In genetics, these are known as phenocopies. A good example is Leigh syndrome, which can be caused by about a dozen different gene defects. Leigh syndrome, originally a neuropathological description of the brain of one affected child, was described by Denis Leigh, the distinguished British physician, in 1951. It is characterized by bilaterally symmetrical MRI abnormalities in the brain stem, cerebellum, and basal ganglia, and often accompanied by elevated lactic acid levels in the blood or cerebrospinal fluid. Leigh syndrome may be caused by the NARP mutation, the MERRF mutation, complex I deficiency, cytochrome oxidase (COX) deficiency, pyruvate dehydrogenase (PDH) deficiency, and other unmapped DNA changes. Not all children with these DNA abnormalities will go on to develop Leigh syndrome, however.
Mitochondrial diseases are even more complex in adults because detectable changes in mitochondrial DNA (mtDNA) occur as we age and, conversely, the aging process itself may result from deteriorating mitochondrial function. There is a broad spectrum of metabolic, inherited and acquired disorders in adults in which abnormal mitochondrial function has been postulated or demonstrated.
Clinical features of mitochondrial diseases
Mitochondrial disorders principally affect tissues that are heavily dependent upon oxidative metabolism. These tissues include the central nervous system, peripheral nerves, eye, skeletal and cardiac muscle, and endocrine organs. Many individuals with mitochondrial diseases have a multi‐system disorder that often involves skeletal muscle and the central nervous system, but some individuals have a disorder that only affects one organ system 3. In general terms, the clinical features of mitochondrial disease can be divided into two groups: central neurological features (including encephalopathy, stroke‐like episodes, seizures, dementia and ataxia), and peripheral neurological features (including myopathy, ophthalmoplegia, and peripheral neuropathy). Some individuals have a mixture of central and peripheral features, whereas others have a pure central or peripheral phenotype.
Many individuals with mitochondrial disease have a clearly defined clinical phenotype (summarized in Table 1: Clinical syndromes associated with mitochondrial disease). Chronic progressive external ophthalmoplegia (CPEO), the Kearns‐Sayre syndrome (KSS) and Pearson syndrome are usually due to a deletion of mtDNA 4. Leber hereditary optic neuropathy (LHON), mitochondrial encephalomyopathy with lactic acidosis and stroke‐like episodes (MELAS), myoclonic epilepsy with ragged‐red fibres (MERRF), maternally inherited diabetes and deafness (MIDD), and neurogenic (or neuropathy) ataxia with retinitis pigmentosa (NARP) are usually due to point mutations of mtDNA 5. Unlike nuclear DNA, mtDNA is inherited down the maternal line, so these disorders either affect sporadic cases or they are passed from mother to child 6. Children presenting with a relapsing encephalopathy with prominent brain stem signs and lactic acidosis (Leigh syndrome) may have a mtDNA defect, or an underlying nuclear genetic defect causing a respiratory chain deficiency. These mutations can affect the genes that code for the subunits themselves (complexes I and II), genes important for the assembly of an intact respiratory chain (complexes III and IV), or genes involved in mitochondrial transcription or translation, and they are usually autosomal recessive 7. Mutations in the gene LPPRC cause a specific form of infantile COX deficiency found in the Saguenay‐Lac‐Saint‐Jean region of Canada 8.
Table 1. Clinical syndromes associated with mitochondrial disease
| Clinical syndrome | Primary features | Additional features |
| Chronic progressive external ophthalmoplegia (CPEO) | External ophthalmoplegia and bilateral ptosis | Mild proximal myopathy |
| Infantile myopathy and lactic acidosis (fatal and non‐fatal forms) | Hypotonia in the first year of life. Feeding and respiratory difficulties | Fatal form may be associated with a cardiomyopathy and/or the Toni‐Fanconi‐Debre syndrome |
| Kearns‐Sayre syndrome (KSS) | CPEO onset before age 20 with pigmentary retinopathy Plus one of the following: CSF protein greater than 1 g/l, cerebellar ataxia, heart block | Bilateral deafness Myopathy Dysphagia Diabetes mellitus and hypoparathyroidism Dementia |
| Leber hereditary optic neuropathy (LHON) | Subacute painless bilateral visual failure Males: females approx. 4:1 Median age of onset 24 years | Dystonia Cardiac pre‐excitation syndromes |
| Leigh syndrome (LS) | Subacute relapsing encephalopathy with cerebellar and brain‐stem signs presenting during infancy | Basal ganglia lucencies |
| Mitochondrial encephalomyopathy with lactic acidosis and stroke‐like episodes (MELAS) | Stroke‐like episodes before age 40 years Seizures and/or dementia Ragged‐red fibres and/or lactic acidosis | Diabetes mellitus Cardiomyopathy (hypertrophic leading to dilated) Bilateral deafness Pigmentary retinopathy Cerebellar ataxia |
| Myoclonic epilepsy with ragged‐red fibres (MERRF) | Myoclonus Seizures Cerebellar ataxia Myopathy | Dementia, optic atrophy Bilateral deafness Peripheral neuropathy Spasticity Multiple lipomata |
| Neurogenic weakness with ataxia and retinitis pigmentosa (NARP) | Late childhood or adult onset peripheral neuropathy with associated ataxia and pigmentary retinopathy | Basal ganglia lucencies Abnormal electroretinogram Sensorimotor neuropathy |
| Pearson Syndome | Sideroblastic anaemia of childhood Pancytopenia Exocrine pancreatic failure | Renal tubular defects |
A further group of mitochondrial disorders have recently been defined at the molecular level. These disorders result from a disorder of mtDNA maintenance. For some of these diseases the primary defect is an abnormality of the intra‐mitochondrial nucleoside pool. Most individuals with autosomal dominant PEO have a mutation in one of three genes: PEO1, ANT1 or POLG, which lead to the formation of multiple mtDNA deletions in muscle 10. Children presenting with mtDNA depletion syndrome may have mutations in the nuclear genes thymidine kinase 2 (TK2) in the myopathic form, or deoxyguanosine kinase (DGUOK) or POLG in the hepatic form 11. Secondary mtDNA multiple deletions are also a feature of mitochondrial neurogastrointestinal encephalomyopathy (MNGIE) which is also due to a disturbance of the intra‐mitochondrial nucleoside pool secondary to thymidine phosphorylase (TP) deficiency 12. A final important group are the disorders associated with coenzyme Q10 (ubiquinone) deficiency. This may present with childhood encephalopathy and seizures, recurrent rhabdomyolysis or ataxia with seizures. Case reports suggest that this disorder responds to Q10 replacement therapy 13.
A large proportion of individuals with mitochondrial disease do not have a clearly defined phenotype. There may be single or multi‐organ involvement including the heart, endocrine organs (particularly the pancreas) and the nervous system. Gastrointestinal complications are an under‐recognised but common feature of mitochondrial disorders. Mitochondrial disease should be considered in any patient presenting with an unexplained progressive multi‐system disorder with prominent neurological features 14.
Secondary mitochondrial disorders
Many genetic disorders are associated with abnormal mitochondrial function either as a secondary phenomenon, or because mitochondria play a crucial role in the pathophysiology of the disorder. To date these include three X‐linked conditions (Barth syndrome, sideroblastic anaemia with ataxia, deafness and dystonia) and a number of autosomal recessive (Friedreich ataxia, spastic paraparesis SPG4 and SPG13, Wilson disease) and autosomal dominant conditions (hereditary paragangliomas). Increasingly, common neurodegenerative disorders such as Parkinson’s disease and Alzheimer’s disease are beginning to enter this category as well. Disorders of mitochondrial fusion and fission also result in secondary mitochondrial dysfunction 15. Practically speaking, diseases caused by OPA1 mutation would likely have been covered in this review (autosomal dominant optic atrophy and progressive external ophthalmoplegia), on account of their highly similar clinical presentation with primary mitochondrial disorders. Other fusion/fission defects causing secondary mitochondrial dysfunction (hereditary motor and sensory neuropathy type 2a due to MFN2 mutation, and various other neurological diseases which involve fusion/fission defects in their pathogenesis) have not been included.
Clinical management of mitochondrial disease
There is currently no established treatment for mitochondrial disorders and the clinical management of individuals is largely supportive, but there have been a number of case reports and small trials describing the positive effects of a number of different drugs, vitamins and food supplements 9. Exercise therapy has also been shown to help with the muscle symptoms. A number of groups are developing treatments that act on the genetic or cellular level, but it is unlikely that these will be available for individuals in the near future 16.
A number of different pharmacological treatments and nutritional supplements have been used in individuals with mitochondrial disease, with varying reports of success. These include antioxidants (coenzyme Q10, idebenone, vitamin C, vitamin E and menadione), agents that specifically improve lactic acidosis (dichloroacetate and dimethylglycine, which is a component of pangamic acid (vitamin B15), agents that correct secondary biochemical deficiencies (carnitine, creatine), respiratory chain co‐factors (nicotinamide, thiamine, riboflavin, succinate, and coenzyme Q10), and hormones (growth hormone and corticosteroids) 17. Much of the evidence used to support specific treatments comes from single case reports, but there have been a number of small quasi‐randomised trials and open‐labelled case series. Improvements following dietary modification (for example, a ketogenic diet) and exercise therapy (for example, endurance training) have also been documented in individual cases, and open‐labelled trials 18. A recent 2012 Cochrane review 9 concluded that there is currently no clear evidence supporting the use of any of these agents in mitochondrial disorders. Further research is needed to establish the role of a wide range of therapeutic approaches. Furthermore, dichloracetate should not be used in adults because of the high incidence of potentially irreversible side‐effects.
Mitochondria function
Mitochondria contain enzymes involved in cellular metabolism, and are involved in many cell death pathways. It is therefore possible that mitochondria play a central role in many disease processes. Mitochondria also play a pivotal role in the final common pathway of aerobic metabolism ‐ oxidative phosphorylation (OXPHOS). Oxidative phosphorylation is carried out by the mitochondrial respiratory chain, which is a group of five multi‐subunit enzyme complexes situated on the inner mitochondrial membrane that generate adenosine triphosphate (ATP) from intermediary metabolites. Adenosine triphosphate is a high‐energy phosphate molecule that provides an energy source for all active cellular processes. The term ‘mitochondrial disorders’ usually refers to primary disorders of the mitochondrial respiratory chain.
The tricarboxylic acid (TCA) cycle also called the citric acid or Kreb cycle and electron transport chain are two important mitochondrial processes in cellular ATP (adenosine triphosphate) production. The overall pathway for tricarboxylic acid cycle (citric acid cycle or Kreb cycle) is as follows: catabolism of glucose in the cytosol produces two molecules of pyruvate, which pass through the mitochondrion’s double membrane and enter the tricarboxylic acid cycle (citric acid cycle or Kreb cycle) 19. When there are relatively high concentrations of ATP, pyruvate carboxylase is activated and shuttles pyruvate in the direction of gluconeogenesis. When energy demands and the concentration of ATP are relatively low, two pyruvate molecules pass through the pyruvate dehydrogenase (PDH) complex to produce two molecules of acetyl-coenzyme A (acetyl CoA), which enter the tricarboxylic acid cycle (citric acid cycle or Kreb cycle). Subsequently, each acetyl CoA produces three molecules of nicotinamide adenine dinuceotide (NADH) and two molecules of flavin adenine dinucleotide (FADH), for a total of six NADH and four FADH per one molecule of pyruvate 20. Additionally, acetyl CoA can be produced by oxidation of free fatty acids (FFA), which then requires the nutrient L-carnitine to shuttle the acetyl CoA into the mitochondria to enter the tricarboxylic acid cycle (citric acid cycle or Kreb cycle) 21. The electron transport chain consists of five enzymatic complexes (I–V) of integral membrane proteins. They are NADH-CoQ reductase (complex I), succinate-CoQ reductase (complex II), CoQ-cytochrome c reductase (complex III), cytochrome c oxidase (complex IV), and ATP synthase (complex V) 22. Electrons from NADH or FADH2 are transferred through a series of respiratory chain complexes to O2, which finally generates H2O. A proton gradient across the membrane is the driving force of F0F1-ATPase to produce ATP from adenosine diphosphate (ADP). ATP is transported to the cytosol by exchanging adenosine diphosphate (ADP) through an adenine nucleotide translocator and is important for various biological events 23. Additionally, mitochondria generate heat by a mechanism called the “proton leak.” Proton leak from the intermembrane space to the matrix reduces proton-motive force and generates heat instead of ATP. Much of the proton leak is a catalytic property of specific molecules termed uncoupling proteins (UCPs), such as UCP1, UCP2, and UCP3. UCPs play an important role in reducing the proton gradient and mitochondrial function by regulating both heat and reactive oxygen species (ROS) generation 23.
Figure 2. Citric acid cycle (tricarboxylic acid cycle or Kreb cycle)
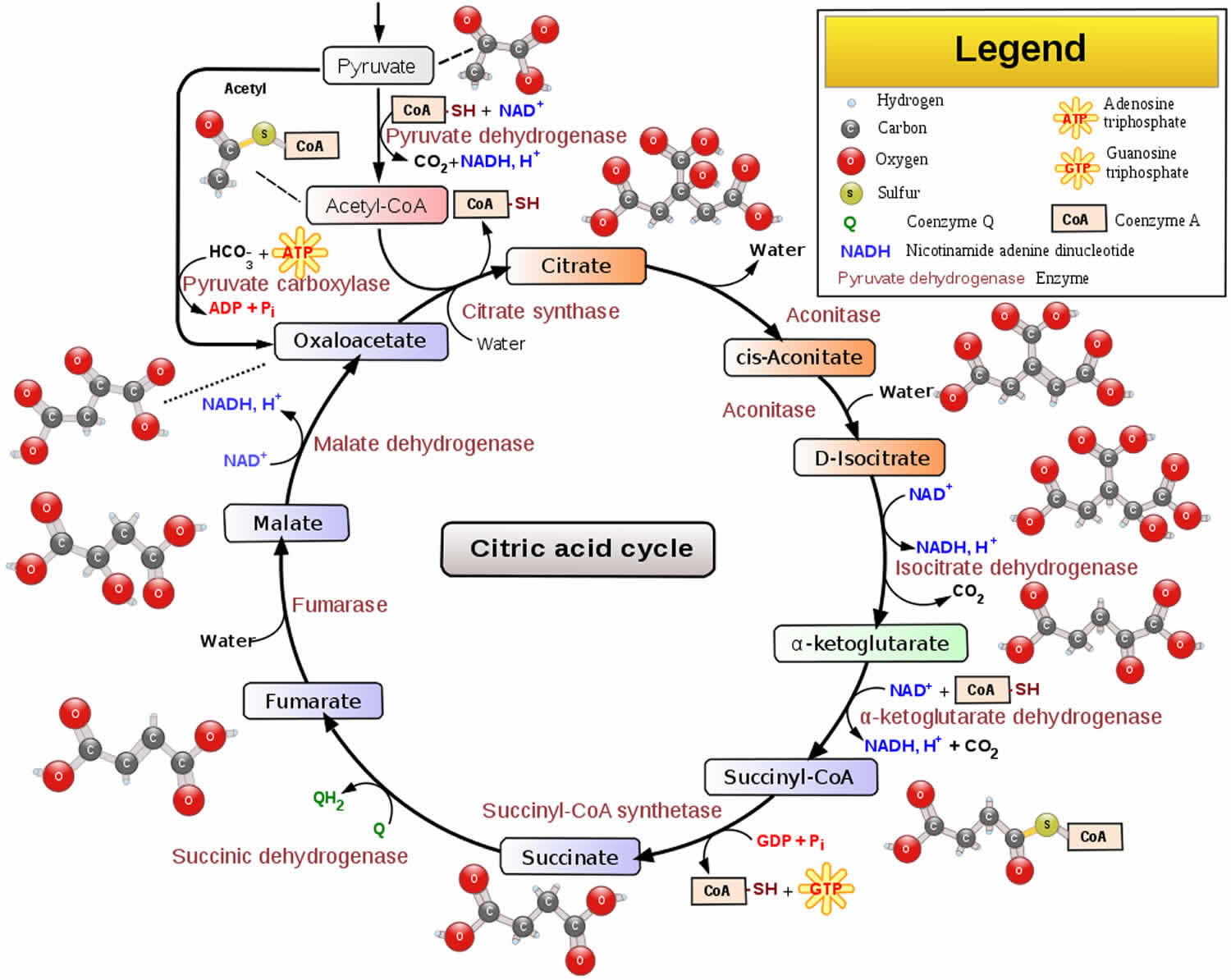 Footnotes: The citric acid cycle oxidizes two-carbon units, producing two molecules of CO2, one molecule of Guanosine-5′-triphosphate (GTP), and high-energy electrons in the form of NADH and FADH2.
Footnotes: The citric acid cycle oxidizes two-carbon units, producing two molecules of CO2, one molecule of Guanosine-5′-triphosphate (GTP), and high-energy electrons in the form of NADH and FADH2.The citric acid cycle is regulated primarily by the concentration of ATP and NADH. The key control points are the enzymes isocitrate dehydrogenase and α-ketoglutarate dehydrogenase.
Isocitrate dehydrogenase is allosterically stimulated by ADP, which enhances the enzyme’s affinity for substrates. The binding of isocitrate, NAD+, Mg2+, and ADP is mutually cooperative. In contrast, NADH inhibits iso-citrate dehydrogenase by directly displacing NAD+. ATP, too, is inhibitory. It is important to note that several steps in the cycle require NAD+ or FAD, which are abundant only when the energy charge is low.
A second control site in the citric acid cycle is α-ketoglutarate dehydrogenase. Some aspects of this enzyme’s control are like those of the pyruvate dehydrogenase complex, as might be expected from the homology of the two enzymes. α-Ketoglutarate dehydrogenase is inhibited by succinyl CoA and NADH, the products of the reaction that it catalyzes. In addition, α-ketoglutarate dehydrogenase is inhibited by a high energy charge. Thus, the rate of the cycle is reduced when the cell has a high level of ATP.
[Source 24]Oxygen consumed in mitochondrial is associated with production of reactive oxygen species (ROS) in most cells and tissues. Mitochondrial electron transport generates superoxide radical (O2•−) as an inevitable by-product in complex I and complex III 25. Biologically, reactive oxygen species (ROS) include O2•−, hydrogen peroxide (H2O2), and the hydroxyl radical (OH•). It has been estimated that about 0.2–2% of oxygen consumed are converted into O2•− by electron transport chain in human cells 26. However, the mitochondria of heart cells have high respiratory rates, and approximately 90% of basal cellular reactive oxygen species (ROS) are from mitochondria 27. Moreover, seven other sites were also suggested to generate O2•− in the mitochondria. They are cytochrome b five reductase, monoamine oxidase, dihydroorotate dehydrogenase, dehydrogenase of α-glycerophosphate, succinate dehydrogenase, aconitase, and α-ketoglutarate dehydrogenase complex 22. It should be recognized that reactive oxygen species (ROS) are derived from outside of mitochondria, such as oxygen radicals from peroxisomal β-oxidation of fatty acids, NAD(P)H oxidase, xanthine oxidase, arachidonic acid metabolism, microsomal P-450 enzymes, and prooxidant heme molecule 28. Several enzymatic means may protect against reactive oxygen species (ROS) in mitochondria. These include conversion of superoxide to H2O2 by superoxide dismutase (SOD), scavenging of H2O2 by catalase, glutathione peroxidase, and peroxiredoxin III 28. Ca2+ transport can cross the mitochon-drial inner membrane and is taken up by mitochondria both through a uniporter and a pulsed or rapid mode 29. The most studied Ca2+ influx mechanism is the uniporter, which transports Ca2+ down its electrochemical gradient 30. Several important functions have been proposed for these processes, including control of the metabolic rate for cellular energy production, modulation of cytosolic Ca2+ transients, and induction of apoptosis through release of cytochrome C 31.
The respiratory chain has a dual genetic basis 32. The vast majority of the respiratory chain subunits (more than 70) are the products of nuclear genes. These subunits are synthesized within the cytosol and are delivered into mitochondria by a peptide targeting sequence. By contrast, 13 essential respiratory chain subunits are synthesised within the mitochondrial matrix from small 16.5 kb circles of double‐stranded DNA called mitochondrial DNA (mtDNA) 33. MtDNA is different to nuclear DNA in a number of respects. First, there are many thousands of copies of mtDNA within each cell. MtDNA mutations may only affect a proportion of the mtDNA molecules, leading to a mixture of mutant and wild‐type mtDNA within the cell (heteroplasmy) 34. Single cell studies have shown that the proportion of mutant mtDNA must exceed a critical threshold level before the cell expresses a biochemical defect of the mitochondrial respiratory chain 35. This threshold varies from tissue to tissue, and partly explains the tissue‐selectivity seen in mitochondrial disorders 36. The percentage level of mutant mtDNA can also vary between and within individuals harbouring a pathogenic mtDNA defect, and this partly explains the clinical variability that is a hallmark of mtDNA disorders 37 in tissues of symptomatic relatives with MELAS: the role of mitotic segregation. Neurology 1993;43(8):1586‐90.)). In the last decade it has become clear that nuclear gene defects are a major cause of mitochondrial disease, affecting the assembly and structure of the respiratory chain protein complexes or other components of the respiratory chain, such as coenzyme Q10. Nuclear gene defects can also affect the maintenance of mtDNA, leading to secondary mitochondrial DNA mutations which accumulate during life; or they can affect the translation of the mitochondrial genome itself 38. Some of these disorders have a distinct clinical phenotype, but others cause an overlapping spectrum of phenotypes similar to primary disorders of mtDNA 39.
It takes about 3000 genes to make a mitochondrion. Mitochondrial DNA encodes just 37 of these genes; the remaining genes are encoded in the cell nucleus and the resultant proteins are transported to the mitochondria. Only about 3% of the genes necessary to make a mitochondrion (100 of the 3000) are allocated for making ATP (adenosine triphosphate). More than 95% (2900 of 3000) are involved with other functions tied to the specialized duties of the differentiated cell in which it resides. These duties change as you develop from embryo to adult, and your tissues grow, mature, and adapt to the postnatal environment. These other, non-ATP-related functions are intimately involved with most of the major metabolic pathways used by a cell to build, break down, and recycle its molecular building blocks. Cells cannot even make the RNA and DNA they need to grow and function without mitochondria. The building blocks of RNA and DNA are purines and pyrimidines. Mitochondria contain the rate-limiting enzymes for pyrimidine biosynthesis (dihydroorotate dehydrogenase) and heme synthesis (d-amino levulinic acid synthetase) required to make hemoglobin. In the liver, mitochondria are specialized to detoxify ammonia in the urea cycle. Mitochondria are also required for cholesterol metabolism, for estrogen and testosterone synthesis, for neurotransmitter metabolism, and for free radical production and detoxification. They do all this in addition to breaking down (oxidizing) the fat, protein, and carbohydrates you eat and drink.
Mitochondrial dysfunction, such as mitochondrial loss and overproduction of oxidants, has been suggested to be involved in the development of impaired insulin metabolic signaling/insulin resistance in various tissues of skeletal muscle, liver, fat, heart, and the kidney. Sedentary lifestyle, genetic factors, oxidative stress, maladaptive immunomodulation, and aging may adversely affect mitochondrial function, leading to impaired insulin metabolic signaling and progression of the cardiorenal metabolic syndrome (Figure 3). Physical activity plays an important role in the regulation of muscle mitochondrial function because aerobic exercise potently activates mitochondrial function. On the contrary, sedentary behavior is linked with reduced activity, with obesity and other metabolic disorders 40. Thus, it is likely that some mitochondrial defects in overweight or obese insulin-resistant subjects can be explained by low levels of physical activity. Mitochondrial proteins are encoded by both nuclear and mitochondrial genomes, and there is some evidence to suggest that mitochondria DNA (mtDNA) deletions or mutations in nuclear-encoded genes are linked with various abnormalities characterizing the cardiorenal metabolic syndrome. It has been hypothesized that the mitochondrial genome is more susceptible to various mutagenic stressors because mitochondrial genes are more proximal to the Reactive oxygen species (ROS) source 41. Previous research showed that a naturally occurring thymidine-to-cytidine mutation in the mitochondrial tRNAILE gene is associated with phenotypes of hypertension, hypercholesterolemia, and hypomagnesemia 23. A3243G, another mutation gene on mtDNA that encodes tRNA and causes impaired insulin secretion. In addition, patients with defects in acyl CoA dehydrogenase have phenotypes of cardiomyopathy, liver dysfunction, and neurological disorders 42. Furthermore, polymorphisms in the promoter of UCP2 are associated with decreased incidence of obesity, reduced insulin secretion, and a high prevalence of type 2 diabetes mellitus (T2DM) 23. Thus, genetic factors that are inherited through nuclear or mitochondrial genes may influence the pathogenesis of the cardiorenal metabolic syndrome and associated cardiovascular disease through functional impairment of mitochondria.
Figure 3. Mitochondria in health and disease
Reactive oxygen species (ROS) production occurs mainly at complex I and complex III in mitochondria 43. Under conditions of glucose and fatty acid overnutrition, nutrient overflow into cells prompts electrons transferring to oxygen without ATP production and further favors a state of increased Reactive oxygen species (ROS), which potentially leads to oxidative damage within mitochondria 44. Therefore, Reactive oxygen species (ROS) generated from mitochondria damages proteins, DNA, and lipid membrane components, which results in mitochondrial dysfunction. In aging, increasing mutations in mitochondrial DNA have been found 45. Old mitochondria have changes in morphology in addition to increased Reactive oxygen species (ROS) production and decreased ATP production. Abnormalities in aging include cumulative DNA damage, mitochondrial dysfunction, telomere loss, altered gene expression, and oxidative damages 46. In addition, mitochondrial biogenesis may be impaired by age-dependent accumulations of point mutations in human 46. Thus, increased Reactive oxygen species (ROS) production associated with aging also contribute to mitochondrial dysfunction in the cardiorenal metabolic syndrome.
Figure 4. Mechanisms of oxidative stress involving mitochondria
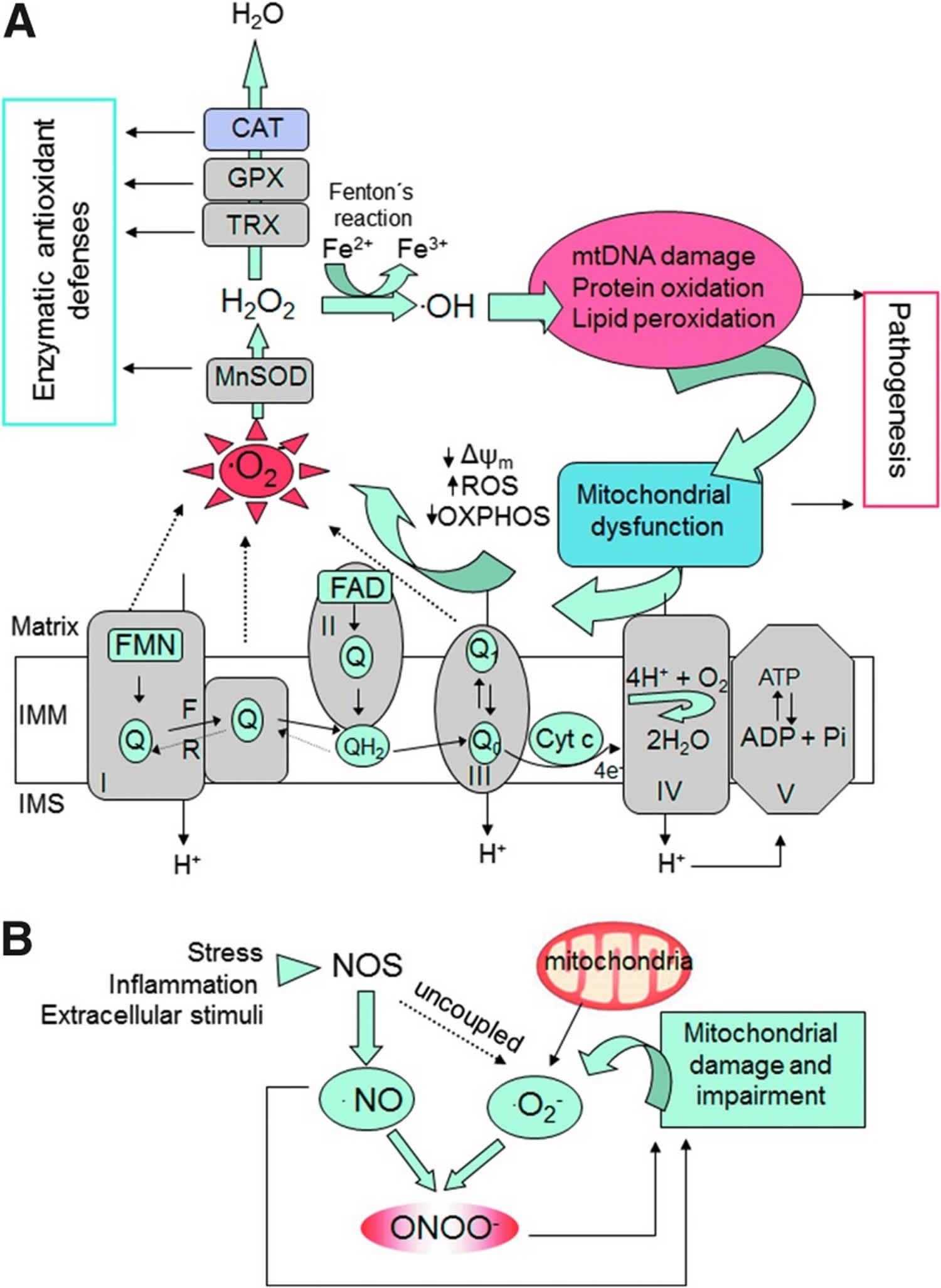
Footnotes: (A) The mitochondrial ETC reoxidizes reduced cofactors (NADH and FADH2) using molecular oxygen as the final electron acceptor, and the energy released in this process is captured in the form of ATP. Several sites of the ETC (CI and CIII and the reverse electron flow at Complex II) generate O2•−. This radical is further converted into H2O2 by mitochondrial SOD. Other antioxidant enzymes within mitochondria involve TRX and GPX, and in certain tissues (liver, cardiac muscle) also CAT. Through the Fenton reaction, H2O2 is converted into •OH, a molecule that produces oxidative cell injury through DNA damage, carboxylation of proteins, and lipid peroxidation. Damaged mitochondria are dysfunctional and further produce free radicals, thus generating a “vicious cycle.” (B) Mechanisms of nitrosative stress. •NO is produced by the activity of intracellular NOS. •NO can be combined with O2•− to produce ONOO−, a molecule that acts as a strong oxidant and can damage many cellular structures and alter their function. Reactive nitrogen species such as ONOO− contribute to further mitochondrial dysfunction. •NO, nitric oxide; •OH, hydroxyl radical; CAT, catalase; ETC, electron transport chain; F, forward; GPX, glutathione peroxidase; H2O2, hydrogen peroxide; IMM, inner mitochondrial membrane; IMS, intermembrane space; NOS, nitric oxide synthase; O2•−, superoxide anion; ONOO−, peroxynitrite; OXPHOS, oxidative phosphorylation; R, reverse; SOD, superoxide dismutase; TRX, thioredoxin; Δψm, mitochondrial membrane potential.
[Source 47]A recent study involving human embryos has discovered that the number of mitochondria is sharply reduced throughout development – from 1 million in a fertilized egg to only around 1,500 per cell in a 4-week-old embryo. Researchers also found that cells taken from older embryos had fewer mitochondrial mutations, meaning that cells with the most defects were somehow eliminated throughout embryonic development.
It is not yet clear how cells with the most mitochondrial mutations are selectively removed in human embryos. But because most of the harmful mutations were eliminated at the stage of embryonic development when cells start breathing more actively, scientists think that damaged mitochondria simply fail to produce enough energy for the cell to survive.
Many questions remain. For instance, why do cells with high levels of defective mitochondria sometimes escape these quality-control mechanisms, resulting in incurable disorders? Ultimately, greater understanding of these mechanisms should suggest better ways of estimating the risk of mitochondrial diseases, or even develop new interventions to prevent them completely.
Summary
Oxidative stress is implicated in a wide range of pathologies. Mitochondria generate most of the cell’s energy by oxidative phosporylation and are considered the main source of ROS in many cell types 47. Mitochondria-generated ROS are crucial signaling molecules and participants in many cellular adaptative mechanisms. However, when the redox balance is disrupted as a consequence of excessive ROS generation or insufficient scavenging, mitochondrial ROS (mtROS) become harmful. The continuous generation of ROS inside mitochondria and the specific molecular/morphological characteristics of these organelles versus other parts of the cell (both mtDNA and proteins are particularly susceptible to oxidative damage) make this cellular compartment highly vulnerable to the impact of ROS. Oxidative stress leads to mitochondrial impairment, both of which are features of the pathophysiology of a wide variety of diseases, including cardiometabolic diseases, neurodegenerative disorders, cancer, and other age-related diseases.
For decades now, numerous studies have focused on the potential of non-targeted antioxidant therapy for restoring normal physiology in oxidative stress conditions. In general, studies in cells or animal models have been successful, but clinical trials have led to disappointing and contradictory results 47.
Taking into account the importance of their role in human pathophysiology, mitochondria stand out as a key pharmacological target. Multiple compounds have been developed to treat mitochondrial dysfunction in different pathological situations with varying targets and mechanisms of action. The use of different mitochondria-targeting strategies, including lipophilic cations, cationic plastoquinone derivatives, liposomal carriers, peptide-based targeting, uncouplers, and other recently described compounds. The majority of these compounds have demonstrated their beneficial effects in different models of oxidative stress, and some of them have proved to be effective in clinical trials 47.
Moreover, multiple novel delivery tools have recently been developed and are currently under evaluation, which offers broad opportunities for mitochondrial modulation. Many of these approaches are promising according to the results obtained in vitro or in cell-free systems; however, additional in-depth studies, particularly in vivo, are lacking to ensure translation of findings to more clinically relevant settings. Specific animal disease models are necessary to address the ability of a given compound to prevent and/or treat oxidative stress-related dysfunction. Finally, human clinical trials under physiologically relevant conditions are crucial to assess the clinical relevance of these molecules.
- Schapira AH. Mitochondrial disease. Lancet 2006;368(9529):70‐82.[↩]
- Schaefer AM, McFarland R, Blakely EL, He L, Whittaker RG. Prevalence of mitochondrial DNA disease in adults. Annals of Neurology 2008;63(1):35‐9.[↩]
- DiMauro S, Schon EA. Mitochondrial DNA mutations in human disease. American Journal of Medical Genetics 2001;106(1):18‐26.[↩]
- Moraes CT, DiMauro S, Zeviani M, Lombes A, Shanske S, Miranda AF, et al. Mitochondrial DNA deletions in progressive external ophthalmoplegia and Kearns‐Sayre syndrome. New England Journal of Medicine 1989;320(20):1293‐9.[↩]
- Lamantea E, Tiranti V, Bordoni A, Toscano A, Bono F, Servidei S, et al. Mutations of mitochondrial DNA polymerase gammaA are a frequent cause of autosomal dominant or recessive progressive external ophthalmoplegia. Annals of Neurology 2002;52(2):211‐9.[↩]
- Chinnery PF, Howell N, Lightowlers RN, Turnbull DM. Genetic counseling and prenatal diagnosis for mtDNA disease. American Journal of Human Genetics 1998;63(6):1908‐11.[↩]
- Tucker EJ, Compton AG, Thorburn DR. Recent advances in the genetics of mitochondrial encephalomyopathies. Current Neurology and Neuroscience Reports 2010;10(4):277–85.[↩]
- Mootha VK, Lepage P, Miller K, Bunkenborg J, Reich M, Hjerrild M, et al. Identification of a gene causing human cytochrome c oxidase deficiency by integrative genomics. Proceedings of the National Academy of Sciences of the United States of America 2003;100(2):605‐10.[↩]
- Pfeffer G, Majamaa K, Turnbull DM, Thorburn D, Chinnery PF. Treatment for mitochondrial disorders. Cochrane Database of Systematic Reviews 2012, Issue 4. Art. No.: CD004426. DOI: 10.1002/14651858.CD004426.pub3[↩][↩][↩]
- Goethem G, Dermaut B, Lofgren A, Martin JJ, Broeckhoven C. Mutation of POLG is associated with progressive external ophthalmoplegia characterized by mtDNA deletions. Nature Genetics 2001;28(3):211‐2.[↩]
- Mandel H, Szargel R, Labay V, Elpeleg O, Saada A, Shalata A. The deoxyguanosine kinase gene is mutated in individuals with depleted hepatocerebral mitochondrial DNA. Nature Genetics 2001;29(3):337‐41.[↩]
- Nishino I, Spinazzola A, Hirano M. Thymidine phosphorylase gene mutations in MNGIE, a human mitochondrial disorder. Science 1999;283(5402):689‐92.[↩]
- Musumeci O, Naini A, Slonim AE, Skavin N, Hadjigeorgiou GL, Krawiecki N, et al. Familial cerebellar ataxia with muscle coenzyme Q10 deficiency. Neurology 2001;56(7):849‐55.[↩]
- Chinnery PF, Turnbull DM. Clinical features, investigation and management of patients with defects of mitochondrial DNA. Journal of Neurology, Neurosurgery and Psychiatry 1997;63(5):559‐63.[↩]
- Westermann B. Mitochondrial fusion and fission in cell life and death. Nature Reviews Molecular Cell Biology 2010;11(12):872‐4.[↩]
- Hassani A, Horvath R, Chinnery PF. Mitochondrial myopathies: developments in treatment. Current Opinion in Neurology 2010;23(5):459‐65.[↩]
- Chinnery PF, Turnbull DM. Epidemiology and treatment of mitochondrial disease. American Journal of Medical Genetics 2001;106(1):94‐101.[↩]
- Taivassalo T, Shoubridge EA, Chen J, Kennaway NG, Dimauro S, Arnold DL, et al. Aerobic conditioning in patients with mitochondrial myopathies: physiological, biochemical, and genetic effects. Annals of Neurology 2001;50(2):133‐41.[↩]
- Mandavia CH, Aroor AR, Demarco VG, Sowers JR. Molecular and metabolic mechanisms of cardiac dysfunction in diabetes. Life Sci. 2013;92:601–608[↩]
- Zhang Y, Sowers JR, Ren J. Pathophysiological insights into cardiovascular health in metabolic syndrome. Exp Diabetes Res. 2012;2012:320534[↩]
- Pieczenik SR, Neustadt J. Mitochondrial dysfunction and molecular pathways of disease. Exp Mol Pathol. 2007;83:84–92.[↩]
- Gao L, Laude K, Cai H. Mitochondrial pathophysiology, reactive oxygen species, and cardiovascular diseases. Vet Clin North Am Small Anim Pract. 2008;38:137–155.[↩][↩]
- Kim JA, Wei Y, Sowers JR. Role of mitochondrial dysfunction in insulin resistance. Circ Res. 2008;102:401–414.[↩][↩][↩][↩]
- By Narayanese, WikiUserPedia, YassineMrabet, TotoBaggins – http://biocyc.org/META/NEW-IMAGE?type=PATHWAY&object=TCA. Image adapted from :Image:Citric acid cycle noi.svg|(uploaded to Commons by wadester16), CC BY-SA 3.0, https://commons.wikimedia.org/w/index.php?curid=6217701[↩]
- Nicolson GL. Metabolic syndrome and mitochondrial function: molecular replacement and antioxidant supplements to prevent membrane peroxidation and restore mitochondrial function. J Cell Biochem. 2007;100:1352–1369.[↩]
- Whaley-Connell A, McCullough PA, Sowers JR. The role of oxidative stress in the metabolic syndrome. Rev Cardiovasc Med. 2011;12:21–29[↩]
- Demarco VG, Whaley-Connell AT, Sowers JR, Habibi J, Dellsperger KC. Contribution of oxidative stress to pulmonary arterial hypertension. World J Cardiol. 2010;2:316–324[↩]
- Sivitz WI, Yorek MA. Mitochondrial dysfunction in diabetes: from molecular mechanisms to functional significance and therapeutic opportunities. Antioxid Redox Signal. 2010;12:537–577.[↩][↩]
- Sowers JR, Bakris GL, Black HR, Giles TD. The cardiometabolic syndrome and calcium channel blocker combination drugs. J Cardiometab Syndr. 2007;2:207–212.[↩]
- Hayden MR, Tyagi SC, Kolb L, Sowers JR, Khanna R. Vascular ossification-calcification in metabolic syndrome, type 2 diabetes mellitus, chronic kidney disease, and calciphylaxis-calcific uremic arteriolopathy: the emerging role of sodium thiosulfate. Cardiovasc Diabetol. 2005;4:4.[↩]
- Gunter TE, Buntinas L, Sparagna G, Eliseev R, Gunter K. Mitochondrial calcium transport: mechanisms and functions. Cell Calcium. 2000;28:285–296.[↩]
- DiMauro S, Schon EA. Nuclear power and mitochondrial disease. Nature Genetics 1998;19(3):214‐5.[↩]
- Anderson S, Bankier AT, Barrell BG, Bruijn MH, Coulson AR, Drouin J, et al. Sequence and organization of the human mitochondrial genome. Nature 1981;290(5806):457‐65.[↩]
- Holt IJ, Harding AE, Morgan‐Hughes JA. Deletion of muscle mitochondrial DNA in patients with mitochondrial myopathies. Nature 1988;331(6158):717‐9.[↩]
- Schon EA, Bonilla E, DiMauro S. Mitochondrial DNA mutations and pathogenesis. Journal of Bioenergetics and Biomembranes 1997;29(2):131‐49.[↩]
- Wallace DC. Mitochondrial DNA mutations in diseases of energy metabolism. Journal of Bioenergetics & Biomembranes 1994;26(3):241‐50.[↩]
- Macmillan C, Lach B, Shoubridge EA. Variable distribution of mutant mitochondrial DNAs (tRNA(Leu[3243][↩]
- Spinazzola A, Zeviani M. Mitochondrial diseases: a cross‐talk between mitochondrial and nuclear genomes. Advances in Experimental Medicine and Biology 2009;652:69‐84.[↩]
- Zhu X, Peng X, Guan MX, Yan Q. Pathogenic mutations of nuclear genes associated with mitochondrial disorders. Acta Biochimica et Biophysica Sinica 2009;41(3):179‐87.[↩]
- Turner N, Heilbronn LK. Is mitochondrial dysfunction a cause of insulin resistance? Trends Endocrinol Metab. 2008;19:324–330.[↩]
- Sack MN. Type 2 diabetes, mitochondrial biology and the heart. J Mol Cell Cardiol. 2009;46:842–849.[↩]
- Sharef SW, Al-Senaidi K, Joshi SN. Successful treatment of cardiomyopathy due to very long-chain acyl-CoA dehydrogenase deficiency: first case report from Oman with literature review. Oman Med J. 2013;28:354–356.[↩]
- Whaley-Connell A, Sowers JR. Indices of obesity and cardiometabolic risk. Hypertension. 2011;58:991–993[↩]
- Liu J, Shen W, Zhao B, et al. Targeting mitochondrial biogenesis for preventing and treating insulin resistance in diabetes and obesity: hope from natural mitochondrial nutrients. Adv Drug Deliv Rev. 2009;61:1343–1352.[↩]
- de Cavanagh EM, Inserra F, Ferder L. Angiotensin II blockade: a strategy to slow ageing by protecting mitochondria? Cardiovasc Res. 2011;89:31–40[↩]
- Kim JA, Wei Y, Sowers JR. Role of mitochondrial dysfunction in insulin resistance. Circ Res. 2008;102:401–414[↩][↩]
- Apostolova N, Victor VM. Molecular Strategies for Targeting Antioxidants to Mitochondria: Therapeutic Implications. Antioxidants & Redox Signaling. 2015;22(8):686-729. doi:10.1089/ars.2014.5952. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4350006/[↩][↩][↩][↩]





{kind=link}