
Contents
What is Morquio syndrome
Morquio syndrome also called mucopolysaccharidosis type IV (MPS IV), is a rare genetic metabolic condition in which the body is unable to break down long chains of sugar molecules called glycosaminoglycans (GAG) 1. Glycosaminoglycans (GAG) are long chains of sugar molecules used in the building of bones, cartilage, skin, tendons and many other tissues in the body. These sugar chains are submicroscopic and cannot be seen with the eye, but can be studied using special scientific instruments and analytical methods. Toxic levels of glycosaminoglycans (GAG) sugars accumulate in cell structures called lysosomes, leading to the various signs and symptoms associated with the condition. These glycosaminoglycans sugars accumulate in cells, blood, tendons and ligaments, causing damage over time. There is also evidence that GAG are bioactive. This means that their accumulation can cause activation of other chemical reactions in the body (i.e. they may trigger inflammation in joints). Babies may show little sign of the disease, but as more and more glycosaminoglycans (GAG) accumulates, symptoms start to appear. Sugar or foods normally eaten will not affect whether there is more or less buildup of glycosaminoglycans (GAG). Affected children generally develop features of Morquio syndrome (MPS IV) between the ages of 1 and 3. These signs and symptoms may include abnormalities of the skeleton, eyes, heart and respiratory system.
Affected children have a characteristic facial appearance that may include an enlarged head, broad mouth, prominent cheekbones, an unusually small nose, widely spaced and thinly enameled teeth, and widely separated eyes with subtle corneal clouding. The liver and spleen may be mildly enlarged. Children with Morquio syndrome (MPS IV) show marked growth retardation with short trunks and normal limbs from early in life. The elbows, wrists, hips, knees and other large joints are abnormally flexible, causing overall instability. Affected individuals exhibit a waddling gait with frequent falls. Early development and intelligence are typically normal, unlike other MPS storage disorders. High frequency hearing impairment is common.
Individuals with Morquio syndrome (MPS IV) are missing one of two specific enzymes which are essential in the breakdown of certain GAG called keratan sulfate (KS) and chondroitin-6-sulfate (CS).
There are two forms of Morquio syndrome (MPS IV):
- MPS IVA (MPS IV type A) is caused by changes (mutations) in the GALNS gene. MPS IVA is caused by a defect in the GALNS gene that instructs the body to make the enzyme N-acetyl- galactosamine-6-sulfate sulfatase (GALNS), which is also called galactosamine-6-sulfatase.
- MPS IVB (MPS IV type B) is caused by mutations in the GLB1 gene. MPS IVB is caused by a defect in the GLB1 gene that instructs the body to make the enzyme beta-galactosidase (GLB1). Because of this gene defect, cells either produce the enzymes in low amounts or not at all, and incompletely broken down glycosaminoglycans (GAG) remains stored inside cells in the body and begins to build up, causing progressive damage.
Both forms are inherited in an autosomal recessive manner. Treatment is based on the signs and symptoms present in each person 2.
The exact prevalence of Morquio syndrome (MPS IV) is unknown, although it is estimated to occur in 1 in 200,000 to 300,000 individuals. MPS IVA (95% of individuals affected by MPS IV) occurs more often than MPS IVB (5% of affected individuals).
What causes Morquio syndrome
Mutations in the GALNS and GLB1 genes cause Morquio syndrome (MPS IV). These genes provide instructions for producing enzymes involved in the breakdown of large sugar molecules called glycosaminoglycans (GAGs). GAGs were originally called mucopolysaccharides, which is where this condition gets its name. When Morquio syndrome (MPS IV) is caused by mutations in the GALNS gene it is called Morquio A syndrome (MPS IV type A or MPS IVA), and when it is caused by mutations in the GLB1 gene it is called Morquio B syndrome (MPS IV type B or MPS IVB). In general, the two types of Morquio syndrome (MPS IV) cannot be distinguished by their signs and symptoms.
Mutations in the GALNS and GLB1 genes reduce or completely eliminate the activity of the enzymes produced from these genes. Without these enzymes, GAGs accumulate within cells, specifically inside the lysosomes. Lysosomes are compartments in the cell that break down and recycle different types of molecules. Conditions such as Morquio syndrome (MPS IV) that cause molecules to build up inside the lysosomes are called lysosomal storage disorders. In Morquio syndrome (MPS IV), GAGs accumulate to toxic levels in many tissues and organs, particularly in the bones. The accumulation of GAGs causes the bone deformities in this disorder. Researchers believe that the buildup of GAGs may also cause the features of Morquio syndrome (MPS IV) by interfering with the functions of other proteins inside lysosomes and disrupting the movement of molecules inside the cell.
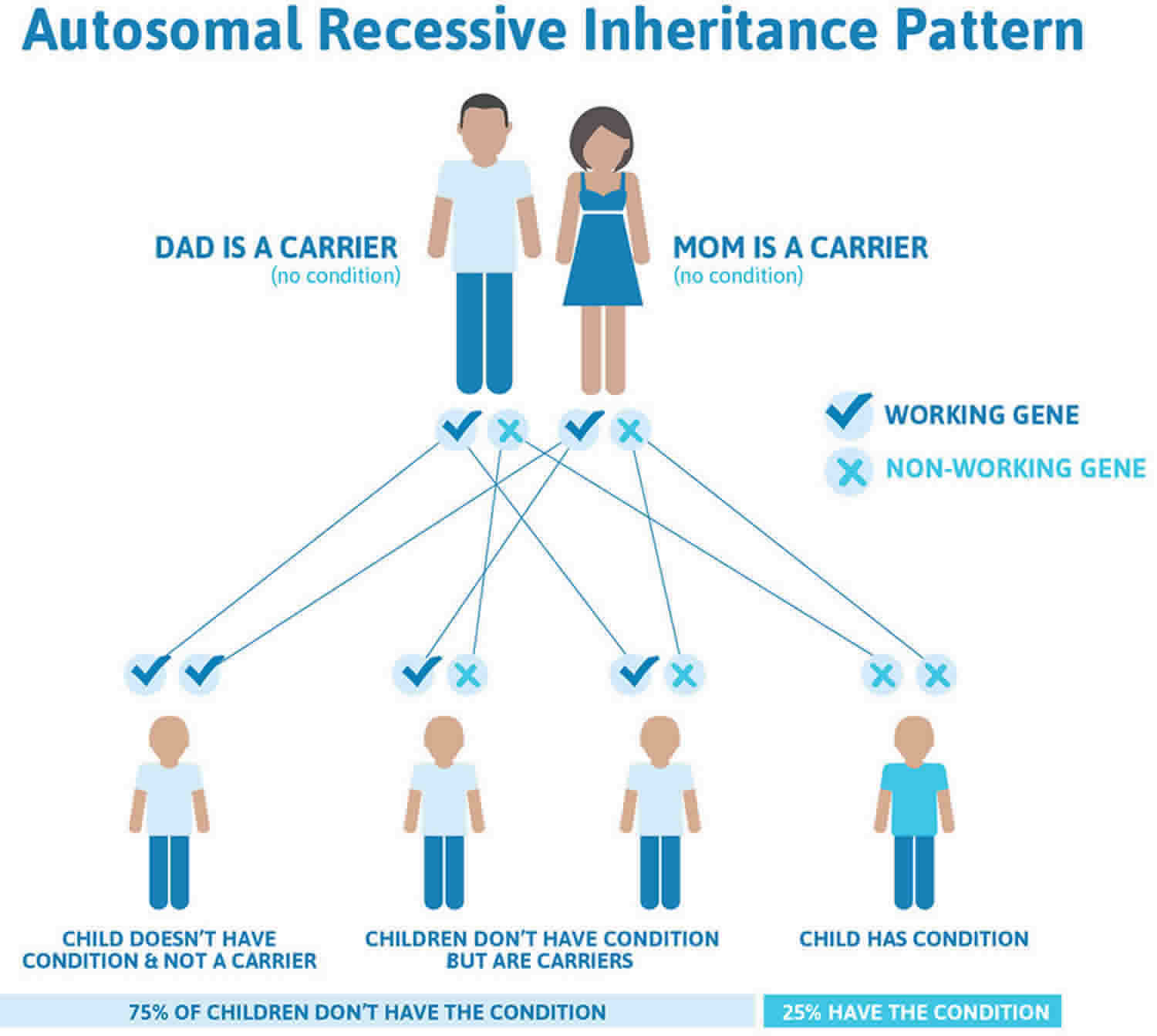
Morquio syndrome inheritance pattern
Morquio syndrome is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
It is rare to see any history of autosomal recessive conditions within a family because if someone is a carrier for one of these conditions, they would have to have a child with someone who is also a carrier for the same condition. Autosomal recessive conditions are individually pretty rare, so the chance that you and your partner are carriers for the same recessive genetic condition are likely low. Even if both partners are a carrier for the same condition, there is only a 25% chance that they will both pass down the non-working copy of the gene to the baby, thus causing a genetic condition. This chance is the same with each pregnancy, no matter how many children they have with or without the condition.
- If both partners are carriers of the same abnormal gene, they may pass on either their normal gene or their abnormal gene to their child. This occurs randomly.
- Each child of parents who both carry the same abnormal gene therefore has a 25% (1 in 4) chance of inheriting a abnormal gene from both parents and being affected by the condition.
- This also means that there is a 75% ( 3 in 4) chance that a child will not be affected by the condition. This chance remains the same in every pregnancy and is the same for boys or girls.
- There is also a 50% (2 in 4) chance that the child will inherit just one copy of the abnormal gene from a parent. If this happens, then they will be healthy carriers like their parents.
- Lastly, there is a 25% (1 in 4) chance that the child will inherit both normal copies of the gene. In this case the child will not have the condition, and will not be a carrier.
These possible outcomes occur randomly. The chance remains the same in every pregnancy and is the same for boys and girls.
Figure 1 illustrates autosomal recessive inheritance. The example below shows what happens when both dad and mum is a carrier of the abnormal gene, there is only a 25% chance that they will both pass down the abnormal gene to the baby, thus causing a genetic condition.
Figure 1. Morquio syndrome autosomal recessive inheritance pattern
Morquio syndrome symptoms
People affected by Morquio syndrome (mucopolysaccharidosis type IV or MPS IV) often develop signs and symptoms of the condition in early childhood between ages 1 and 3. Morquio syndrome is considered progressive; however, the rate at which symptoms worsen varies significantly among affected people. All people affected by Morquio syndrome develop skeletal problems such as scoliosis, knock-knees, short stature, pectus carinatum and variety of other abnormalities of the ribs, chest, spine, hips, and wrists 3. Another common feature of Morquio syndrome (MPS IV) is an underdeveloped odontoid process (a peg-like bone in the neck that helps stabilize the cervical vertebrae). This can misalign, compress and damage the spinal cord, leading to paralysis or even death 4.
Other signs and symptoms of Morquio syndrome include 4:
- Scoliosis or kyphosis
- Knock knees
- Heart and vision problems
- An enlarged liver (mild hepatomegaly)
- Short height
- Coarse facial features
- Hypermobile joints
- Corneal clouding and vision loss
- Heart valve abnormalities
- Respiratory complications, including airway obstruction, sleep apnea and restrictive lung disease
- Widely-spaced, discolored teeth with thin enamel
- Mild to moderate hearing loss
Skeletal X-rays typically show marked flattening of the vertebra. The long bones of the arms and legs are characteristically shorter and thicker than normal. The skull is large for the rest of the body. The connection between the first and second vertebrae in the neck is poorly developed and this abnormality can be life threatening. A trivial injury may cause the two vertebrae to slip on each other and compress the spinal cord. Surgery to stabilize the upper cervical spine, usually by spinal fusion, can be lifesaving but life expectancy is decreased somewhat despite surgery. The deformity of the chest causes a strain on the heart and lungs, which may eventually cause respiratory failure.
Figure 2. Morquio syndrome ulnar deviation of both wrists and joint enlargement in a male age 15 years with MPS IVA
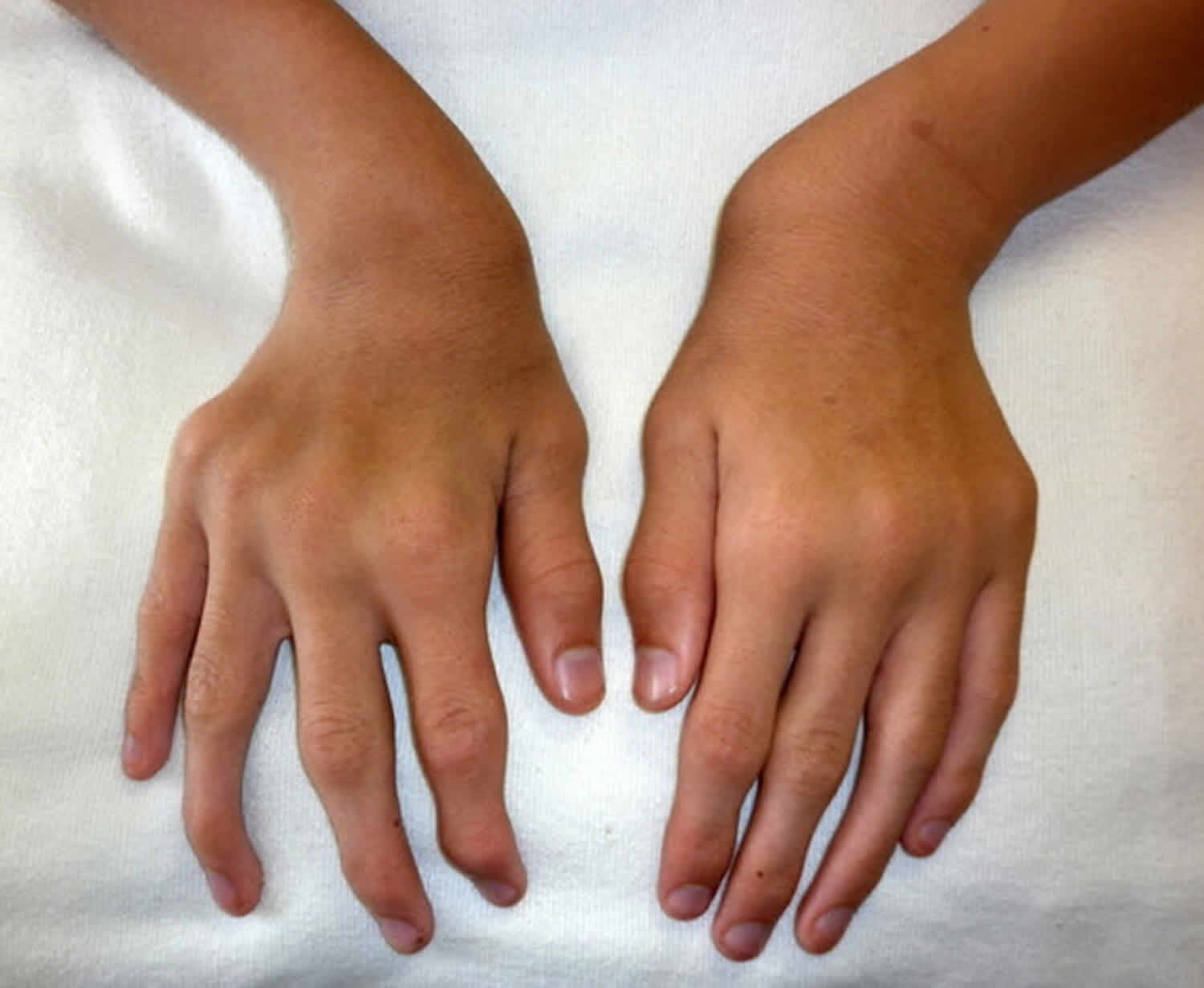
Figure 3. Morquio syndrome with shortened forearm and ulnar deviation of the wrist in a male age 15 years with MPS IVA
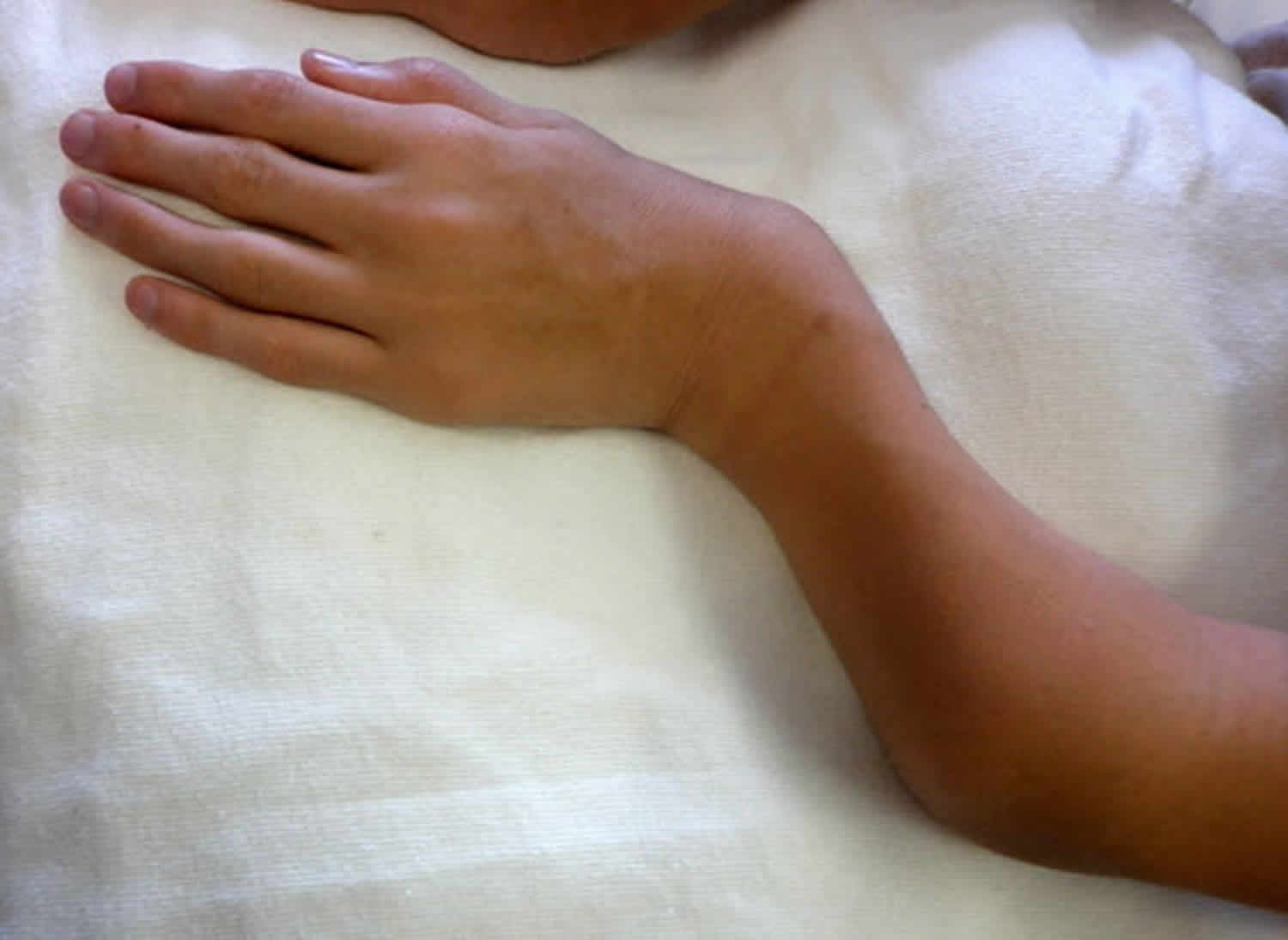
Figure 4. Morquio syndrome with pectus anomaly and short neck in a male age 15 years with MPS IVA
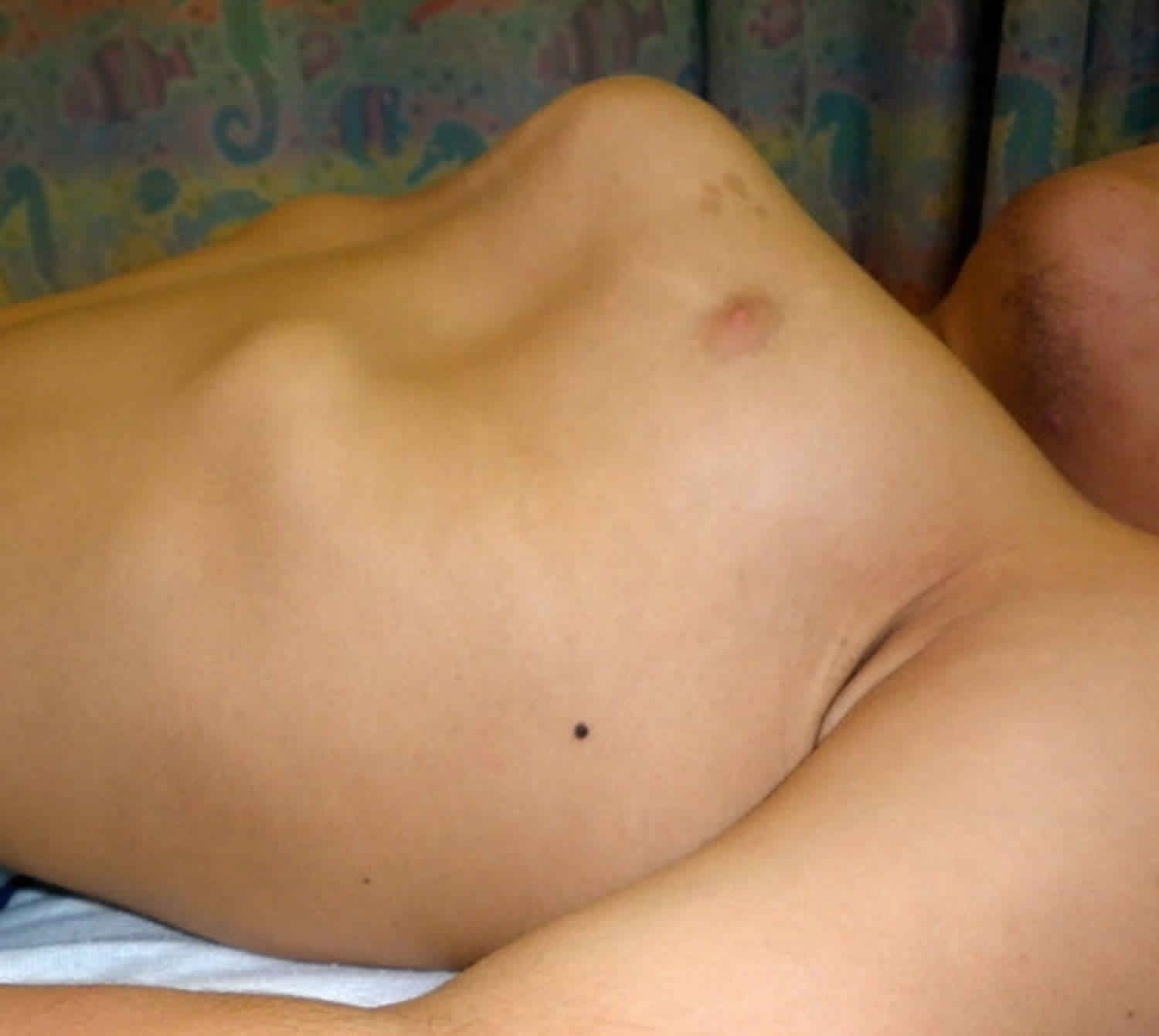
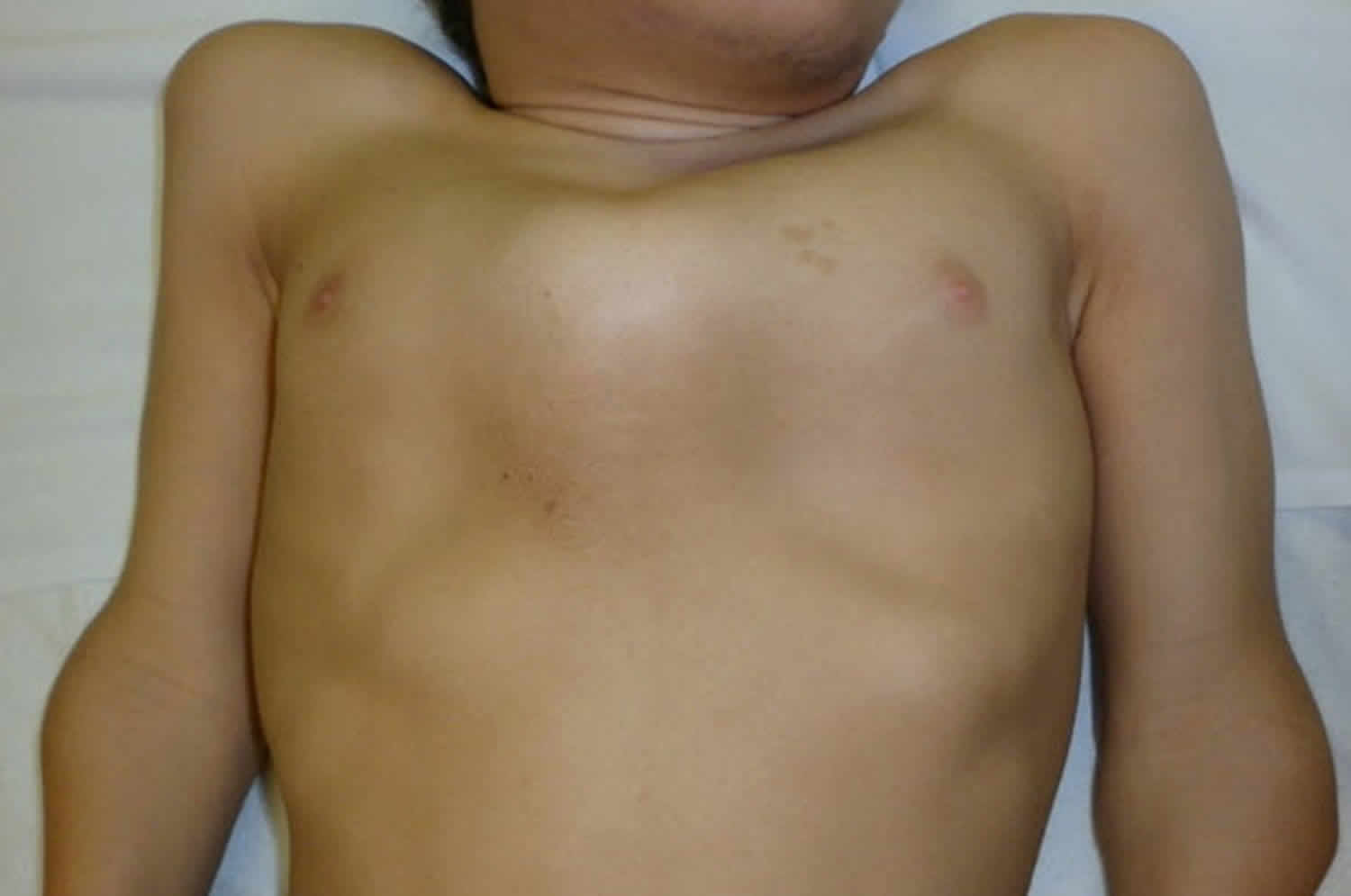
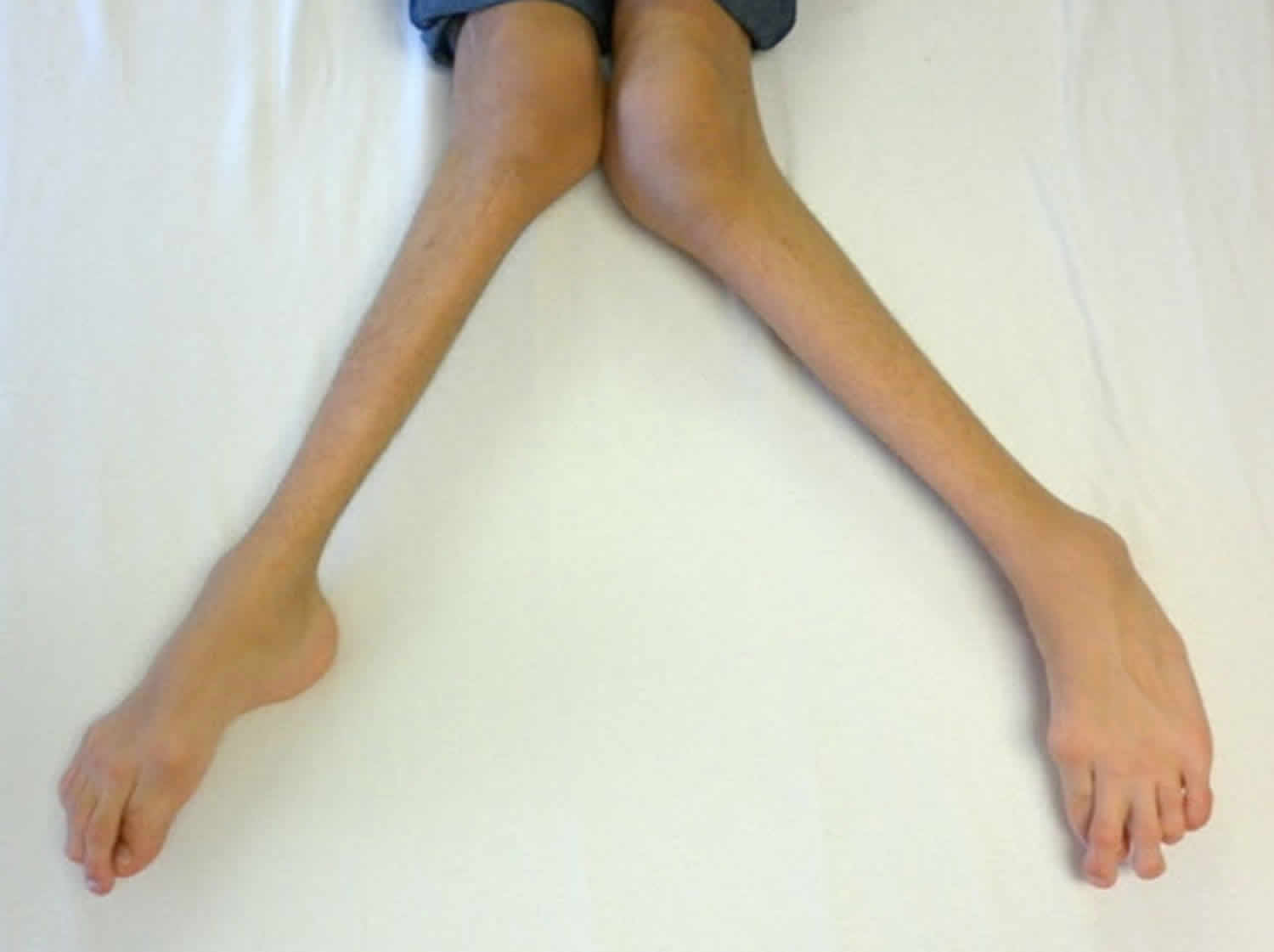
Figure 5. Morquio syndrome with severe genu valgum (knock-knee) in a male age 15 years with MPS IVA
Morquio syndrome diagnosis
Diagnosis of Morquio syndrome starts with a thorough medical history and physical exam.
Physical examination. In severe MPS IVA the following findings are usually observed between ages one and three years; in slowly progressive MPS IVA the following findings may not become evident until as late as the second decade of life:
- Marked disproportionate short stature with short trunk and normal limbs (arm span exceeds height)
- Ulnar deviation of the wrists (see Figures 2 and Figure 3)
- Pectus carinatum and flaring of the lower rib cage (Figure 4)
- Gibbus (short-segment structural thoracolumbar kyphosis resulting in sharp angulation of the back), kyphosis, and scoliosis
- Genu valgum (knock-knee) (Figure 5)
- Hypermobile joints
- Waddling gait with frequent falls
Your child’s doctor might order:
- Genetic testing
- X-ray images to produce images of your child’s bones
- MRI scans to produce images of organs and other structures
- Echocardiogram to examine the heart and its functioning
- Laboratory tests e.g., urine glycosaminoglycans (GAG) analysis. Excessive amounts of keratan sulfate will usually be present in the urine.
Diagnosis of MPS IVA is confirmed by low N-acetylgalactosamine-6-sulfate sulfatase (GALNS) enzyme activity in cultured blood or skin cells and/or molecular genetic testing to identify GALNS gene mutations.
MPS IVB diagnosis is confirmed by the finding of a beta-galactosidase deficiency in blood or skin cells and/or molecular genetic testing to identify GLB1 gene mutations.
Morquio syndrome treatment
The goals of managing Morquio syndrome (MPS IV) are to improve quality of life, to slow down the progression of the disease, and to prevent permanent tissue and organ damage. Currently there is no cure for Morquio syndrome (MPS IV); however, early intervention may help prevent irreversible damage. Treatment options for Morquio syndrome (MPS IV) include those aimed at disease management and supportive or palliative care (care that makes a person with a disease that cannot be cured more comfortable).
In 2014, the FDA approved a recombinant human N-acetylgalactosamine-6-sulfate sulfatase (GALNS) intravenous enzyme replacement therapy (elosulfase alfa, or Vimizim) for the treatment of MPS IVA (Morquio A syndrome). Vimizim is manufactured by BioMarin Pharmaceutical Inc. Vimizim (elosulfase alfa) is administered weekly via intravenous infusion.
There is no treatment for MPS IVB (Morquio B syndrome).
Other treatment of MPS IV is symptomatic and supportive. Surgery to decompress and fuse the bones of the upper neck to the base of the skull can prevent destabilization of the cervical vertebrae and potential damage to the spinal cord.
Management of affected individuals with MPS IV is best undertaken by multiple specialists, including: a physical therapist for physical rehabilitation, a psychiatrist for psychological support, educational professionals for learning optimization, and home care professionals for affected individuals with medical equipment dependence.
Surgeons may also play a crucial role in treating affected individuals. The placement of a bioprosthetic or prosthetic valve may be required for affected induvial with ventricular hypertrophy (overgrowth). Enlarged tonsils and adenoids may need to be removed in order to relieve upper-airway obstruction and sleep apnea. Additionally, ventilation tubes and hearing aids may be needed for individuals with hearing loss. Penetrating keratoplasty (corneal replacement) may be needed to treat corneal opacification (scarring or clouding of the cornea), which causes impaired vision.
Since children with MPS IVA are of normal intelligence, they usually attend regular classes, but they made need to sit close to the front of the classroom if they have difficulties hearing or seeing. They may also need to use a wheelchair around school grounds.
Genetic counseling is recommended for affected individuals and their families.
Morquio syndrome life expectancy
Disease severity varies significantly for individuals with Morquio syndrome (MPS IV), and it is not possible to predict the expected life span for a given individual. Those on the more slowly progressing end of the disease spectrum may have a reasonably normal lifespan. However, the availability of new and ever-improving treatments as well as other surgical procedures provides hope for better future outcomes for individuals affected by Morquio syndrome (MPS IV).
Morquio syndrome (MPS IV) has a highly variable phenotype. This means that some children may have many of the symptoms described below and may be severely affected while others may not experience all of the symptoms and have a milder presentation. There is currently no reliable way of telling from biochemical diagnostic tests how severe the disease will be. Detailed studies have shown that in individuals with attenuated, or slowly progressing, Morquio syndrome (MPS IV), a very small amount of active enzyme is working. This small amount of enzyme will digest some of the accumulating GAG, resulting in the disease being less severe than in an individual who has almost no enzyme activity.
DNA tests do not always correctly determine the severity of Morquio syndrome (MPS IV). Many different kinds of mutations(permanent changes) in the gene that produces the enzyme deficiency have been identified. The gene has been studied extensively to see if thereis any relationship betweenspecific genetic mutations and the symptoms of the disease. There are some common mutations of the gene that result in absolutely no enzyme being produced. If both copies of the defective gene inherited by an individual are of this kind, evidence suggests that the individual’s condition is likely to be at the severe end of the spectrum. Other common mutations of the gene cause very small amounts of defective enzyme to be produced, and still other mutations are not common at all and may only occur in a single known family. In these cases, it is virtually impossible to predict severity of disease using DNA analysis.
There is therefore no perfectly reliable way to determine the exact course of disease for individuals with Morquio syndrome (MPS IV). Even with the same small amount of enzyme activity, and even within the same family, there can be variations in severity that cannot be explained by the enzyme level or DNA mutation. It is important to remember that whatever name is given to your child’s condition, Morquio syndrome (MPS IV) is a spectrum with a variety of symptoms, and is extremely varied in its effects. This booklet addresses a wide range of possible symptomsthat individuals with Morquio syndrome (MPS IV) may encounter; however, parents should be aware that their child(ren) may not experience them all or to the degree described.
Early diagnosis of Morquio syndrome (MPS IV) is critical. The earlier Morquio syndrome (MPS IV) is diagnosed, the sooner potential treatment options can be explored and supportive care may be started to help you or your loved one, and potentially prevent some of the permanent damage that may be caused by the disease.
- Mucopolysaccharidosis type IV. https://rarediseases.info.nih.gov/diseases/12562/mucopolysaccharidosis-type-iv[↩]
- Mucopolysaccharidosis IV. https://rarediseases.org/rare-diseases/morquio-syndrome[↩]
- Di Cesare A, Di Cagno A, Moffa S, Teresa P, Luca I, Giombini A. A description of skeletal manifestation in adult case of morquio syndrome: radiographic and MRI appearance. Case Rep Med. 2012; 2012:324596[↩]
- Regier DS, Oetgen M, Tanpaiboon P. Mucopolysaccharidosis Type IVA. 2013 Jul 11 [Updated 2016 Mar 24]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2019. Available from: https://www.ncbi.nlm.nih.gov/books/NBK148668[↩][↩]



{kind=link}