Contents
- What is muscle atrophy
- What causes muscle atrophy
- Spinal muscle atrophy
- Muscle atrophy symptoms
- Muscle atrophy diagnosis
- Muscle atrophy treatment
What is muscle atrophy
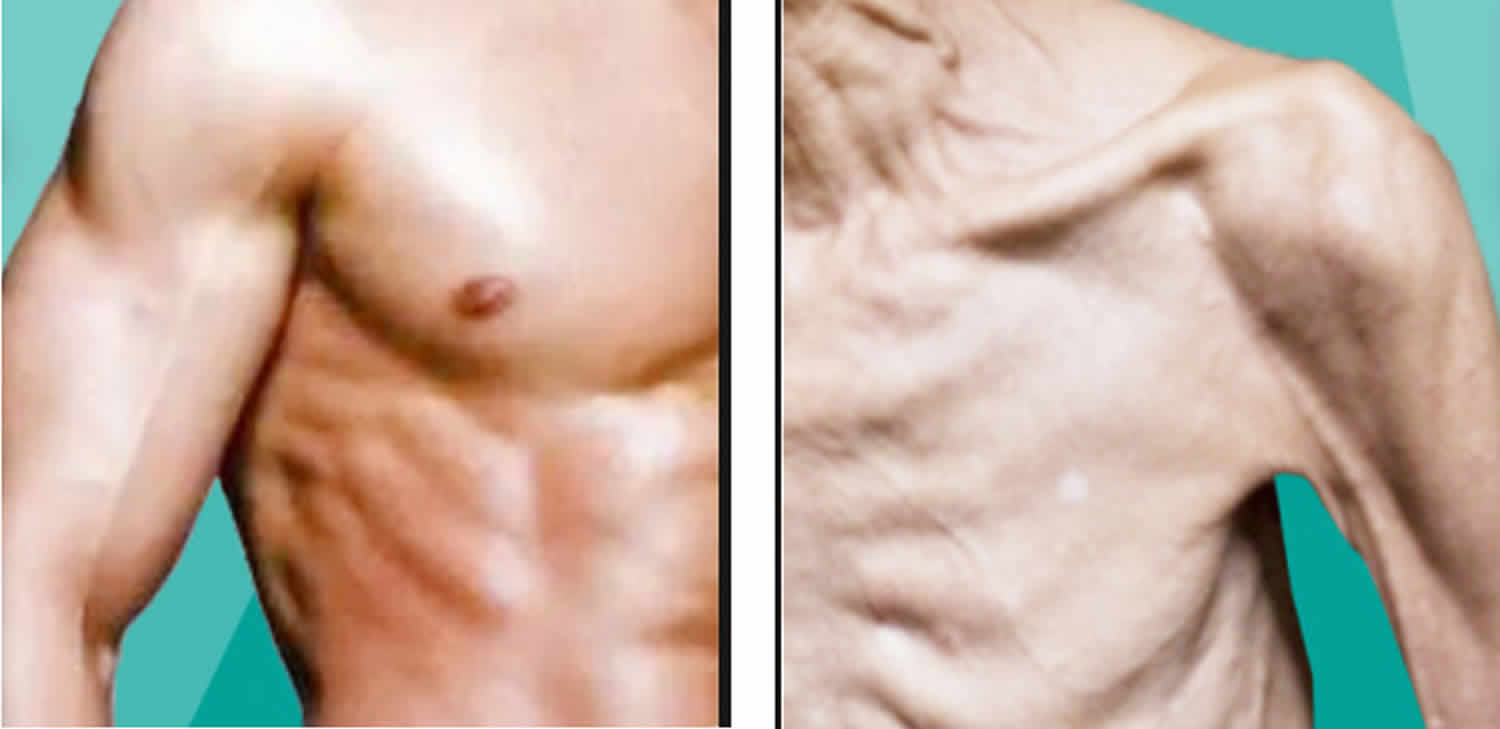
Muscle atrophy is the wasting or loss of muscle tissue that is used for movement (skeletal muscles). Although people can adapt to muscle atrophy, even minor muscle atrophy causes some loss of movement or strength.
Call your doctor for an appointment if you have unexplained or long-term muscle loss. You can often see muscle atrophy when you compare one hand, arm, or leg to the other.
Can muscle atrophy be reversed?
That depends on what is causing your muscle atrophy. Usually treating the underlying condition plus an exercise program may help treat muscle atrophy. Exercises may include ones done in a swimming pool to reduce the muscle workload, and other types of rehabilitation. Your health care provider can tell you more about this.
What causes muscle atrophy
There are three types of muscle atrophy: physiologic, pathologic, and neurogenic.
Table 1. Selected Causes of Primary Muscle Weakness
| Cause | Weakness | Age of onset/diagnosis | Systemic symptoms and findings | Laboratory abnormalities | Creatine kinase | Electromyogram | Muscle biopsy |
|---|---|---|---|---|---|---|---|
Drugs | |||||||
Alcohol | Proximal (may be distal) | Variable | Change in mental status; telangiectasia; peripheral neuropathy | Elevated transaminase and GGT levels; anemia; decreased vitamin B12 | Normal to elevated | Normal | Myopathic changes*; selected atrophy of type II muscle fibers |
Endocrine | |||||||
Adrenal insufficiency | Generalized | Variable | Hypotension; hypoglycemia; bronzing of the skin | Hyponatremia; hyperkalemia; ACTH assay; ACTH stimulation test | Normal | Myotonic discharges† | Diminished glycogen content |
Glucocorticoid excess | Proximal | Variable | Buffalo hump; striae; osteoporosis | Elevated urine-free cortisol, dexamethasone suppression, or corticotropin-releasing hormone stimulation tests | Normal | Myopathic MUAPs‡ | Selective atrophy of type II muscle fibers |
Parathyroid hormone (secondary hyperparathyroidism§) | Proximal, lower extremity more than upper extremity | Variable, older adult | Usually has associated comorbidities (cardiovascular disease, diabetes) | Hypocalcemia; uremia | Normal | Myopathic MUAPs‡ | Atrophy of type II muscle fibers; increased lipofuscin beneath cell membrane; calcium deposits in muscle |
Thyroid hormone (hyperthyroidism) | Proximal, bulbar | 40 to 49 years | Weight loss; tachycardia; increased perspiration; tremor | Elevated T4 and T3; TSH variable, depending on cause | Normal or elevated | Myopathic MUAPs‡ with or without fibrillation potentials∥ | Usually normal |
Thyroid hormone (hypothyroidism) | Proximal | 30 to 49 years | Menorrhagia; bradycardia; goiter; delayed relaxation of deep tendon reflexes | TSH | Elevated | With or without myopathic MUAPs‡ and fibrillation potentials∥ | Myopathic changes*; glycogen accumulation |
Inflammatory | |||||||
Dermatomyositis | Proximal | Variable, increased incidence with age | Gottron papules; heliotrope rash; calcinosis; interstitial lung disease; disordered GI motility | Elevated myoglobin; ANA positive; myositis autoantibodies may be present | Greater than 10 times normal elevations | Myopathic MUAPs‡ with fibrillation potentials∥ | Inflammatory infiltrate with myopathic changes* and replacement by adipose and collagen |
Inclusion body myositis | Distal, especially forearm and hand | At least 50 years (younger than 50 years: rare) | Dysphagia; extramuscular involvement not as common | Elevated myoglobin; positive ANA less common; myositis autoantibodies may be present | Elevated | Myopathic MUAPs‡ with fibrillation potentials∥ | Inflammatory infiltrate with vacuoles containing eosinophilic inclusions |
Polymyositis | Proximal | Variable, increased incidence with age | Interstitial lung disease, disordered GI motility; overlap with rheumatologic diseases more common | Elevated myoglobin; ANA positive; myositis autoantibodies may be present | Greater than 10 times normal elevations | Myopathic MUAPs‡ with fibrillation potentials∥ | Inflammatory infiltrate with myopathic changes* and replacement by adipose and collagen |
Rheumatologic | |||||||
Rheumatoid arthritis | Focal, periarticular, or diffuse | Adult | Symmetric joint inflammation (especially MCP, PIP joints); dry eyes and mouth | Elevated rheumatoid factor | Normal or elevated | No data | Atrophy of type II muscle fibers; may have overlap syndrome with polymyositis |
Systemic lupus erythematosus | Proximal | Adult | Malar rash; nephritis; arthritis | ANA, anti-DNA antibodies, depressed C3 and C4 | Normal to elevated | No data | Type II fiber atrophy; lymphocytic vasculitis; myositis |
Genetic | |||||||
Becker muscular dystrophy | Hip; proximal leg and arm | Late childhood to adulthood | Mental retardation; cardiomyopathy | None | Elevated | Myopathic MUAPs‡ with fibrillation potentials∥ | Myopathic changes*; decreased and patchy staining of dystrophin |
Limb-girdle muscular dystrophies** | Variable, usually proximal limb, pelvic, and shoulder girdle muscles | Variable | Variable, may have cardiac abnormalities | None | Variable, normal, or elevated | Myopathic MUAPs‡ +/– fibrillation potentials∥ | Myopathic changes*; may demonstrate absence of specific protein on immunohistochemical staining |
Myotonic dystrophy type 1 | Distal greater than proximal; foot drop; temporal and masseter wasting | Adolescence to adulthood | Conduction abnormalities; mental retardation; cataracts; insulin resistance | None | Normal to minimally elevated | Myopathic MUAPs‡; myotonic discharges∥ | Less necrosis and remodeling than in muscular dystrophies; atrophy of type I muscle fibers; ring fibers |
Metabolic | |||||||
Glycogen and lipid storage diseases; mitochondrial dis ease | Proximal | Variable | Variable; exercise intolerance and cardiomyopathy more common | Some glycogenoses associated with abnormal FIET†† | Variable, may increase with exercise | Normal or myopathic MUAPs‡ +/– fibrillation potentials∥ | Myopathic changes* with glycogen deposits, lipid deposits, or ragged red fibers (for glycogen, lipid, or mitochondrial disease, respectively) |
Abbreviations: GGT = gamma-glutamyltransferase; ACTH = adrenocorticotropin hormone; MUAPs = motor unit action potentials; T3 = triiodothyronine; T4 = thyroxine; TSH = thyroid-stimulating hormone; GI = gastrointestinal; ANA = antinuclear antibodies; MCP = metacarpophalangeal; PIP = proximal interphalangeal; EMG = electromyogram; FIET = forearm ischemic exercise testing.
Footnotes:
*—Myopathic changes are nonspecific and include atrophy, degeneration, and regeneration of muscle fibers.
†—Myotonic discharges are a type of prolonged burst of activity seen on insertion of the EMG needle.
‡—Myopathic MUAPs are shorter in duration, lower in amplitude, and polyphasic when compared to MUAPs from normal muscle.
∮—Secondary hyperparathyroidism usually caused by renal failure.
∥—Fibrillation potentials represent spontaneous activity on the part of single muscle fibers (not usually noted with nondiseased muscle).
¶—C3 and C4 are complement components.
**—Including facioscapulohumeral dystrophy.
††—FIET evaluates the rise of ammonia and lactate in the forearm during exertion.
[Source 1 ]Physiologic muscle atrophy
Physiologic muscle atrophy is caused by not using the muscles enough. This type of muscle atrophy can often be reversed with exercise and better nutrition. People who are most affected are those who:
- Have seated jobs, health problems that limit movement, or decreased activity levels
- Are bedridden
- Cannot move their limbs because of stroke or other brain disease
- Are in a place that lacks gravity, such as during space flights
Pathologic muscle atrophy
Pathologic muscle atrophy is seen with aging, starvation, and diseases such as Cushing disease (because of taking too much medicines called corticosteroids).
Other causes of muscle atrophy may include:
- Burns
- Long-term corticosteroid therapy
- Malnutrition
- Osteoarthritis
- Rheumatoid arthritis
Neurogenic muscle atrophy
Neurogenic muscle atrophy is the most severe type of muscle atrophy. Neurogenic muscle atrophy can be from an injury to, or disease of a nerve that connects to the muscle. Neurogenic muscle atrophy tends to occur more suddenly than physiologic muscle atrophy.
Examples of diseases affecting the nerves that control muscles:
- Amyotrophic lateral sclerosis (ALS, or Lou Gehrig disease)
- Damage to a single nerve, such as carpal tunnel syndrome
- Guillain-Barre syndrome
- Nerve damage caused by injury, diabetes, toxins, or alcohol
- Polio (poliomyelitis)
- Spinal cord injury
- Muscular dystrophy and other diseases of the muscle
- Spinal muscular atrophy
Spinal muscle atrophy
Spinal muscular atrophy is a serious genetic neuromuscular disease that makes your muscles weaker and causes problems with movement. Spinal muscular atrophy is characterized by muscle weakness and muscle atrophy. Spinal muscular atrophy gets worse over time, but there are treatments to help manage the symptoms.
Patients with spinal muscular atrophy generally start to show symptoms early in life, and the disease becomes more severe over time. spinal muscular atrophy is the leading genetic cause of death in infants and toddlers 2. The severity of spinal muscular atrophy varies; it ranges from very severe with symptoms starting to show in the first few months after birth, to mild with disease onset in adulthood.
Spinal muscular atrophy is caused by a loss of specialized nerve cells, called motor neurons that control muscle movement. The weakness tends to be more severe in the muscles that are close to the center of the body (proximal) compared to muscles away from the body’s center (distal). The muscle weakness usually worsens with age. There are many types of spinal muscular atrophy that are caused by changes in the same genes. The types differ in age of onset and severity of muscle weakness; however, there is overlap between the types. Other forms of spinal muscular atrophy and related motor neuron diseases, such as spinal muscular atrophy with progressive myoclonic epilepsy, spinal muscular atrophy with lower extremity predominance, X-linked infantile spinal muscular atrophy, and spinal muscular atrophy with respiratory distress type 1 are caused by mutations in other genes.
It’s not currently possible to cure spinal muscular atrophy, but research is ongoing to find possible new treatments.
Treatment and support is available to manage the symptoms and help people with spinal muscular atrophy have the best possible quality of life.
Treatment may involve:
- exercises and equipment to help with movement and breathing
- feeding tubes and diet advice
- braces or surgery to treat problems with the spine or joints
A range of healthcare professionals may be involved in your care, including specialist doctors, physiotherapists, occupational therapists, and speech and language therapists.
Spinal muscular atrophy types
There are several types of spinal muscular atrophy, which start at different ages. Some types cause more serious problems than others.
The main types of spinal muscular atrophy are 3:
- Type 0 spinal muscular atrophy – evident before birth and is the rarest and most severe form of spinal muscular atrophy. spinal muscular atrophy type 0 (congenital spinal muscular atrophy) presents with severe weakness and hypotonia present at birth. There may be a history of decreased in utero movements and joint contractures. Infants with spinal muscular atrophy type 0 have severe respiratory compromise and rarely survive past age six months 4.
- Type 1 spinal muscular atrophy (Werdnig-Hoffmann Disease) – develops in babies less than 6 months old and is the most severe type. Symptoms usually emerge within first six months of age. Patients typically exhibit limited movement, which makes even the simplest tasks (such as holding their head straight, eating, and swallowing) very difficult. The progressive weakening of the chest muscles could lead to respiratory infections, lung collapse and death. Many Type 1 patients die by the age of two 2.
- Type 2 spinal muscular atrophy (Dubowitz disease) – appears in babies who are 7-18 months old and is less severe than type 1. In type 2 spinal muscular atrophy, symptoms usually emerge in patients between six and eighteen months of age, and the progression of symptoms varies greatly. Type 2 spinal muscular atrophy patients are able to sit unassisted, but they do not walk independently. Due to the varied progression of symptoms, life expectancy ranges from early childhood to adulthood. The majority of type 2 spinal muscular atrophy patients do live into adulthood and many have normal life expectancies.
- Type 3 spinal muscular atrophy (Juvenile Spinal Muscular Atrophy) – develops after 18 months of age and is the least severe type affecting children. The onset of symptoms can appear at any time between eighteen months and early adulthood. People with spinal muscular atrophy type 3 spinal muscular atrophy often exhibit difficulty walking, have mild muscle weakness, and are still at risk for respiratory infections. These patients have a normal life expectancy.
- Type 4 spinal muscular atrophy (adult form of spinal muscular atrophy) – a less common form of spinal muscular atrophy that afflicts adults and is characterized by a slower progression of symptoms that particularly affect walking. Symptoms typically emerge after age thirty-five.
Babies with type 1 rarely survive beyond the first few years of life. Most children with type 2 survive into adulthood and can live long, fulfilling lives. Types 3 and 4 don’t usually affect life expectancy.
Spinal muscular atrophy type 0 is evident before birth and is the rarest and most severe form of the condition. Affected infants move less in the womb, and as a result they are often born with joint deformities (contractures). They have extremely weak muscle tone (hypotonia) at birth. Their respiratory muscles are very weak and they often do not survive past infancy due to respiratory failure. Some infants with spinal muscular atrophy type 0 also have heart defects that are present from birth (congenital) 5.
Spinal muscular atrophy type 1 (also called Werdnig-Hoffmann disease) is the most common form of the condition. It is a severe acute form of spinal muscular atrophy with muscle weakness evident at birth or within the first few months of life. Most affected children cannot control their head movements or sit unassisted. Children with this type may have swallowing problems that can lead to difficulty feeding and poor growth. They can also have breathing problems due to weakness of respiratory muscles and an abnormally bell-shaped chest that prevents the lungs from fully expanding. Most children with spinal muscular atrophy type 1 do not survive past early childhood due to respiratory failure 5.
Spinal muscular atrophy type 2 (also called Dubowitz disease) is an intermediate form of spinal muscular atrophy that is characterized by muscle weakness that develops in children between ages 6 and 12 months. Children with this type can sit without support, although they may need help getting to a seated position. However, as the muscle weakness worsens later in childhood, affected individuals may need support to sit. Individuals with spinal muscular atrophy type 2 cannot stand or walk unaided. They often have involuntary trembling (tremors) in their fingers, a spine that curves side-to-side (scoliosis), and respiratory muscle weakness that can be life-threatening. The life span of individuals with spinal muscular atrophy type 2 varies, but many people with this condition live into their twenties or thirties 5.
Spinal muscular atrophy type 3 (also called Kugelberg-Welander disease) is a mild form of spinal muscular atrophy that typically causes muscle weakness after early childhood. Individuals with this condition can stand and walk unaided, but over time, walking and climbing stairs may become increasingly difficult. Many affected individuals require wheelchair assistance later in life. People with spinal muscular atrophy type 3 typically have a normal life expectancy 5.
Spinal muscular atrophy type 4 is rare and often begins in early adulthood. Affected individuals usually experience mild to moderate muscle weakness, tremors, and mild breathing problems. People with spinal muscular atrophy type 4 have a normal life expectancy 5.
Spinal muscular atrophy affects 1 per 8,000 to 10,000 people worldwide. Spinal muscular atrophy type 1 is the most common type, accounting for about half of all cases. Types 2 and 3 are the next most common and types 0 and 4 are rare.
Other forms of spinal muscular atrophy include:
- Congenital spinal muscular atrophy with arthrogryposis (persistent contracture of joints with fixed abnormal posture of the limb) is a rare disorder. Manifestations include severe contractures, scoliosis, chest deformity, respiratory problems, unusually small jaws, and drooping of the upper eyelids.
- Kennedy’s disease, also known as progressive spinobulbar muscular atrophy, may first be recognized between 15 and 60 years of age. The onset of symptoms varies and includes weakness and atrophy of the facial, jaw, and tongue muscles, leading to problems with chewing, swallowing, and changes in speech. Early symptoms may include muscle pain and fatigue. Weakness in arm and leg muscles closest to the trunk of the body develops over time, with muscle atrophy and fasciculations. Individuals with Kennedy’s disease also develop sensory loss in the feet and hands. Nerve conduction studies confirm that nearly all individuals have a sensory neuropathy (pain from sensory nerve inflammation or degeneration). Affected individuals may have enlargement of the male breasts or develop noninsulin-dependent diabetes mellitus.
- Distal spinal muscular atrophy (DSMA) – a type of spinal muscular atrophy that mainly affects the hands, feet, lower arms, and lower legs.
- Spinal muscular atrophy with respiratory distress (SMARD) – a type of spinal muscular atrophy that’s usually diagnosed during a baby’s first year of life and can cause serious breathing problems.
- X-linked infantile spinal muscular atrophy (XL-SMA) is characterized by congenital hypotonia and areflexia and evidence of degeneration and loss of anterior horn cells (i.e., lower motor neurons) in the spinal cord and brain stem 6. Often congenital contractures and/or fractures are present. Intellect is normal. Life span is shortened because of progressive ventilatory insufficiency resulting from chest muscle involvement.
Spinal muscular atrophy causes
Mutations in the survival motor neuron-1 (SMN1) gene cause all types of spinal muscular atrophy described above. The number of copies of the SMN2 gene modifies the severity of the condition and helps determine which type develops.
The SMN1 and SMN2 genes both provide instructions for making a protein called the survival motor neuron (SMN) protein. Normally, most functional SMN protein is produced from the SMN1 gene, with a small amount produced from the SMN2 gene. Several different versions of the SMN protein are produced from the SMN2 gene, but only one version is functional; the other versions are smaller and quickly broken down. The SMN protein is one of a group of proteins called the SMN complex, which is important for the maintenance of motor neurons. Motor neurons transmit signals from the brain and spinal cord that tell skeletal muscles to tense (contract), which allows the body to move.
Most people with spinal muscular atrophy are missing a piece of the SMN1 gene, which impairs SMN protein production. A shortage of SMN protein leads to motor neuron death, and as a result, signals are not transmitted between the brain and muscles. Muscles cannot contract without receiving signals from the brain, so many skeletal muscles become weak and waste away, leading to the signs and symptoms of spinal muscular atrophy.
Typically, people have two copies of the SMN1 gene and one to two copies of the SMN2 gene in each cell. However, the number of copies of the SMN2 gene varies, with some people having up to eight copies. In people with spinal muscular atrophy, having multiple copies of the SMN2 gene is usually associated with less severe features of the condition that develop later in life. The SMN protein produced by the SMN2 genes can help make up for the protein deficiency caused by SMN1 gene mutations. People with spinal muscular atrophy type 0 typically have zero copies or one copy of the SMN2 gene in each cell, while those with type 1 generally have one or two copies, those with type 2 usually have three copies, those with type 3 have three or four copies, and those with type 4 have four or more copies. Other factors, many unknown, also contribute to the variable severity of spinal muscular atrophy.
Spinal muscular atrophy inheritance pattern
Spinal muscular atrophy is inherited in an autosomal recessive pattern, which means both copies of the SMN1 gene in each cell have mutations. In most cases, the parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition. In rare cases, a person with spinal muscular atrophy inherits an SMN1 gene mutation from one parent and acquires a new mutation in the other copy of the gene that occurs during the formation of reproductive cells (eggs or sperm) or in early embryonic development. In these cases, only one parent is a carrier of the SMN1 gene mutation.
Individuals who have more than the usual two copies of the SMN2 gene usually do not inherit the extra copies from a parent. They typically arise during a random error when making new copies of DNA (replication) in an egg or sperm cell or just after fertilization.
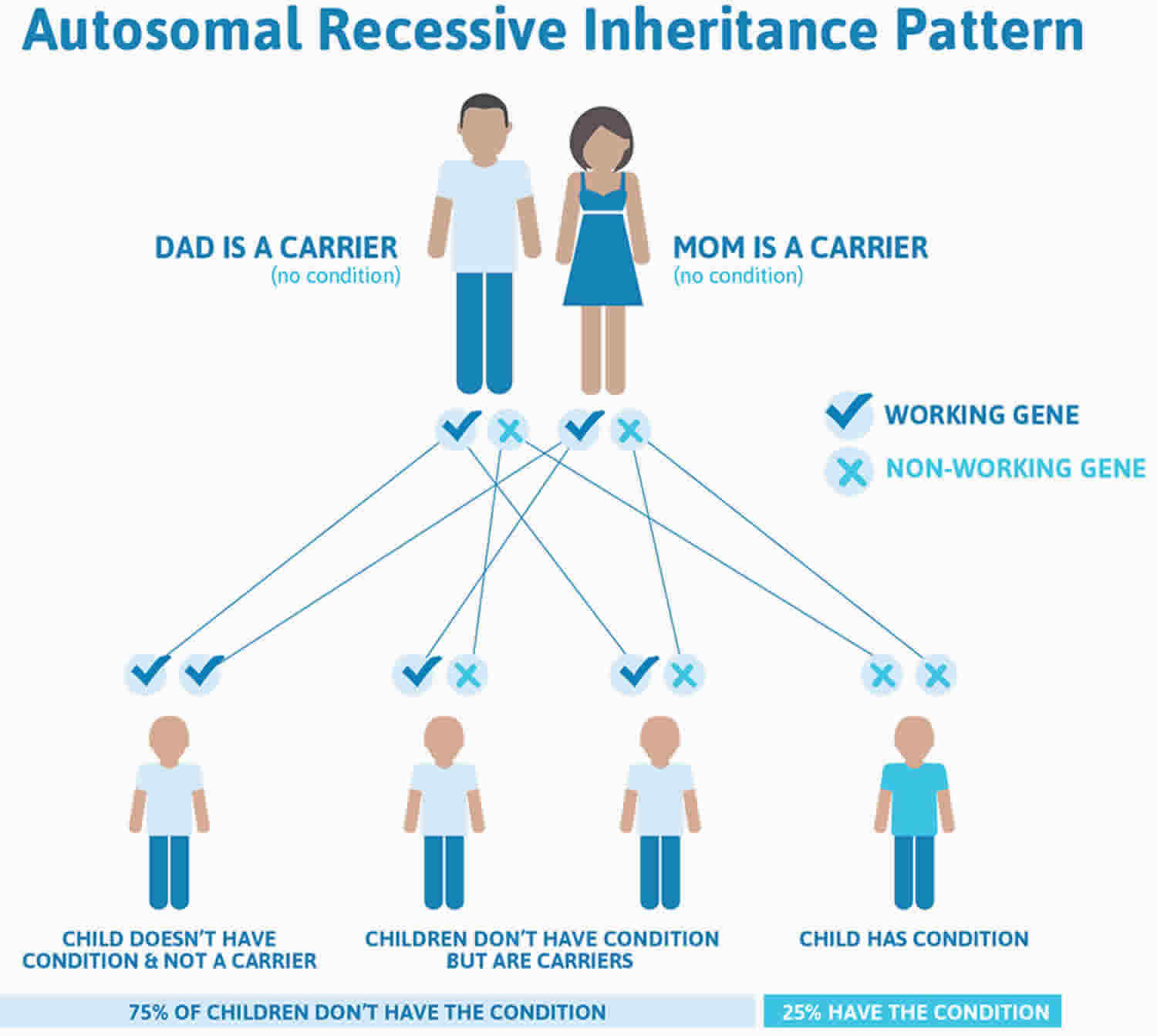
Autosomal recessive means two copies of the abnormal gene, one from each parent (one abnormal gene from mum and one abnormal gene from dad), is needed to cause the disorder or disease. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of spinal muscular atrophy. Approximately 1 out of 50 people is a carrier of spinal muscular atrophy. Autosomal recessive disorders are typically not seen in every generation of an affected family.
Autosomal recessive conditions are individually pretty rare, so the chance that you and your partner are carriers for the same recessive genetic condition are likely low. Even if both partners are a carrier for the same condition, there is only a 25% chance that they will both pass down the non-working copy of the gene to the baby, thus causing a genetic condition. This chance is the same with each pregnancy, no matter how many children they have with or without the condition.
- If both partners are carriers of the same abnormal gene, they may pass on either their normal gene or their abnormal gene to their child. This occurs randomly.
- Each child of parents who both carry the same abnormal gene therefore has a 25% (1 in 4) chance of inheriting a abnormal gene from both parents and being affected by the condition.
- This also means that there is a 75% ( 3 in 4) chance that a child will not be affected by the condition. This chance remains the same in every pregnancy and is the same for boys or girls.
- There is also a 50% (2 in 4) chance that the child will inherit just one copy of the abnormal gene from a parent. If this happens, then they will be healthy carriers like their parents.
- Lastly, there is a 25% (1 in 4) chance that the child will inherit both normal copies of the gene. In this case the child will not have the condition, and will not be a carrier.
These possible outcomes occur randomly. The chance remains the same in every pregnancy and is the same for boys and girls.
Figure 1 illustrates spinal muscular atrophy autosomal recessive inheritance. The example below shows what happens when both dad and mum is a carrier of the abnormal gene, there is only a 25% chance that they will both pass down the abnormal gene to the baby, thus causing a genetic condition.
Figure 1. Spinal muscular atrophy autosomal recessive pattern
Genetics counseling
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Spinal muscular atrophy symptoms
The symptoms of spinal muscular atrophy and when they first appear depend on the type of spinal muscular atrophy you have.
Spinal muscular atrophy should be suspected in individuals with the following:
- History of motor difficulties, especially with loss of skills
- Proximal muscle weakness
- Hypotonia
- Areflexia/hyporeflexia
- Tongue fasciculations
- Evidence of motor unit disease on physical examination
Typical spinal muscular atrophy symptoms include:
- floppy or weak arms and legs
- movement problems – such as difficulty sitting up, crawling or walking
- twitching or shaking muscles (tremors)
- bone and joint problems – such as an unusually curved spine (scoliosis)
- swallowing problems
- breathing difficulties
Spinal muscular atrophy doesn’t affect intelligence or cause learning disabilities.
Spinal muscular atrophy complications
Poor weight gain with growth failure, restrictive lung disease, scoliosis, joint contractures, and sleep difficulties are common complications of spinal muscular atrophy.
Nutrition
Wang et al 7 describe the nutritional difficulties experienced by children with spinal muscular atrophy 1. Bulbar dysfunction is universal in spinal muscular atrophy 1. The bulbar dysfunction eventually becomes a serious problem for persons with spinal muscular atrophy 2 and only very late in the course of disease for those with spinal muscular atrophy 3. Gastrointestinal dysmotility results in constipation, delayed gastric emptying, and potentially life-threatening gastroesophageal reflux with aspiration. Growth failure can be addressed with gastrostomy tube placement as needed.
Pulmonary
Individuals with spinal muscular atrophy, particularly type 1 and 2, show progressive respiratory decline. Respiratory failure is usually the cause of death in these subtypes. Decreased respiratory function leads to impaired cough with inadequate clearance of lower airway secretions, hypoventilation during sleep, and recurrent pneumonia.
Orthopedic
Scoliosis is a major problem in most persons with spinal muscular atrophy 2 and in half of those with spinal muscular atrophy 3. Before age ten years, approximately 50% of affected children (especially those who are nonambulatory) develop spinal curvatures of more than 50 degrees, which require surgery. Progressive scoliosis impairs lung function and in severe cases cardiac output. Progressive scoliosis is treated with surgery when appropriate. Joint contractures and hip dislocation are also common in spinal muscular atrophy.
Metabolic acidosis
An unexplained potential complication of spinal muscular atrophy is severe metabolic acidosis with dicarboxylic aciduria and low serum carnitine concentrations during periods of intercurrent illness or fasting 8. Whether these metabolic abnormalities are primary or secondary to the underlying defect in spinal muscular atrophy is unknown. Although the etiology of these metabolic derangements remains unknown, one recent report suggests that aberrant glucose metabolism may play a role 9. Prolonged fasting should be avoided in individuals with spinal muscular atrophy.
Spinal muscular atrophy diagnosis
Children with spinal muscular atrophy generally appear normal at birth, with symptoms developing as early as a few months after they are born. Symptoms of spinal muscular atrophy often overlap with other neuromuscular diseases, therefore diagnosis of spinal muscular atrophy can be challenging for a non-specialist. Please consult your doctor if you suspect any symptoms of the disease in your child.
A blood test is available that can indicate whether there are deletions or mutations of the SMN1 gene. This test identifies at least 95 percent of spinal muscular atrophy Types 1, 2, and 3. Other diagnostic tests may include electromyography (which records the electrical activity from the brain and/or spinal cord to a peripheral nerve root found in the arms and legs that controls muscles during contraction and at rest), nerve conduction velocity studies (which measure electrical energy by assessing the nerve’s ability to send a signal), muscle biopsy (used to diagnose neuromuscular disorders and may also reveal if a person is a carrier of a defective gene that could be passed on to children), and laboratory tests of blood, urine, and other substances.
Spinal muscular atrophy treatment
There is no cure for spinal muscular atrophy. Treatments help with symptoms and prevent complications. They may include machines to help with breathing, nutritional support, physical therapy, and medicines.
A team of different healthcare professionals will be involved in your or your child’s care. They’ll help come up with a care plan outlining the support and treatments you may need.
Medications
In December 2016 the U.S. Food and Drug Administration approved nusinersen (Spinraza ™) as the first drug approved to treat children and adults with spinal muscular atrophy. The drug is administered by intrathecal injection into the fluid surrounding the spinal cord. It is designed to increase production of the full-length SMN protein, which is critical for the maintenance of motor neurons.
Muscle relaxants such as baclofen, tizanidine, and the benzodiazepines may reduce spasticity. Botulinum toxin may be used to treat jaw spasms or drooling. Excessive saliva can be treated with amitriptyline, glycopyolate, and atropine or by botulinum injections into the salivary glands. Antidepressants may be helpful in treating depression.
Feeding and diet help
It’s important for people with spinal muscular atrophy, especially children, to get the right nutrients. This will help with healthy growth and development.
A dietitian can offer advice about feeding and diet.
If you or your child has difficulty feeding or swallowing, a feeding tube may sometimes be needed.
Several types of tube can be used, such as a tube attached directly to the stomach through the skin of the tummy (gastrostomy tube), or a tube passed through the nose and down the throat (nasogastric tube).
Breathing help
Respiratory problems are the primary causes of illness and the most common cause of death among children with spinal muscular atrophy Types 1 and 2. It is important for patients and families to learn and practice techniques that help maintain a clear airway, take measures to prevent respiratory problems, and minimize the impact of respiratory infection.
There are several treatments for the breathing problems that can affect people with spinal muscular atrophy.
These include:
- breathing exercises to strengthen the breathing muscles and make coughing easier
- a suction machine to help clear the throat if needed – this involves passing a thin, plastic tube to the back of the throat to suck away any mucus
- in more severe cases, a special machine that provides air through a mask or tube
You may be advised to have vaccinations against flu and pneumonia to reduce your risk of becoming seriously ill from these infections.
Assistive equipment
If you or your child have difficulty moving, an occupational therapist or physiotherapist can provide advice and support.
For example, they can advise you about things such as:
- mobility equipment – including walking frames and wheelchairs
- supports for the arms or legs (splints or braces)
- shoe inserts that make walking easier (orthotics)
Exercises and stretches
Exercises and stretches can help maintain strength and stop joints becoming stiff. Spinal muscular atrophy patients achieve their highest level of function when their torso, arm, leg, and neck muscles are properly maintained. A physical therapist can design an individualized physical therapy plan to support function, and help slow or prevent additional complications.
The amount of exercise you or your child can do will depend on your condition, but it’s best to try to stay as active as possible.
Treatments for spine problems
Some children with spinal muscular atrophy develop an unusually curved spine (scoliosis).
Treatments for this include:
- a specially made back brace to help support the back and encourage the spine to grow correctly
- spinal surgery – where the spine is straightened using metal hooks and rods, before being fused into place with pieces of bone
Research into new treatments
Research is being carried out into possible new treatments for spinal muscular atrophy.
You can ask your medical team about ongoing clinical trials into new treatments. You can also check the database of clinical trials for spinal muscular atrophy to see what research is happening at the moment here (https://www.clinicaltrials.gov/ct2/results?term=spinal+muscular+atrophy&Search=Search).
Previous research has suggested a new medicine called nusinersen (Spinraza) can help some people with spinal muscular atrophy.
It was recently approved in the US and by the European Medicines Agency, but will be assessed by the National Institute for Health and Care Excellence (NICE) before it can be made routinely available in England.
Spinal muscular atrophy life expectancy
The life expectancy for spinal muscular atrophy patients varies by type. Many patients with the severe form of spinal muscular atrophy (Type 1) die before the age of two. However, the life expectancy of Type 1 patients really depends on the severity of the disease at diagnosis, as well as treatment choices (which can include respiratory therapy, nutritional support, and physical therapy). Therefore, depending on the care provided and the severity of the disease, some Type 1 patients may live into adulthood.
Children with moderate to mild forms of spinal muscular atrophy (Types 2 and 3) live into adulthood and may have a normal life expectancy depending on the severity of respiratory, nutritional, and orthopedic symptoms.
Spinal muscular atrophy prognosis
Prognosis varies depending on the type of spinal muscular atrophy. Some forms of spinal muscular atrophy are fatal.
The course of Kennedy’s disease varies but is generally slowly progressive. Individuals tend to remain ambulatory until late in the disease. The life expectancy for individuals with Kennedy disease is usually normal.
People with spinal muscular atrophy may appear to be stable for long periods, but improvement should not be expected.
Muscle atrophy symptoms
Muscle atrophy symptoms are:
- muscle weakness
- wasting or loss of muscle tissue
Muscle atrophy diagnosis
Your doctor will perform a physical examination and ask about your medical history and symptoms, including:
- When did the muscle atrophy begin?
- Is it getting worse?
- What other symptoms do you have?
Your doctor will look at your arms and legs and measure muscle size. This may help determine which nerves are affected.
Tests that may be performed include:
- Blood tests
- CT scans
- Electromyography (EMG)
- MRI scans
- Muscle or nerve biopsy
- Nerve conduction studies
- X-rays
Muscle atrophy treatment
Muscle atrophy treatment may include physical therapy, ultrasound therapy and, in some cases, surgery to correct a contracture.
Muscle atrophy exercises
An exercise program may help treat muscle atrophy. Exercises may include ones done in a swimming pool to reduce the muscle workload, and other types of rehabilitation. Your health care provider can tell you more about this.
People who cannot actively move one or more joints can do exercises using braces or splints.
- Evaluation of the Patient with Muscle Weakness. Am Fam Physician. 2005 Apr 1;71(7):1327-1336. https://www.aafp.org/afp/2005/0401/p1327.html[↩]
- Spinal muscular atrophy FAQ. http://www.smafoundation.org/about-sma/faq/[↩][↩]
- Prior TW, Finanger E. Spinal Muscular Atrophy. 2000 Feb 24 [Updated 2016 Dec 22]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2019. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1352[↩]
- Dubowitz V. Very severe spinal muscular atrophy (SMA type 0): an expanding clinical phenotype. Eur J Paediatr Neurol. 1999;3:49–51.[↩]
- Spinal muscular atrophy. https://ghr.nlm.nih.gov/condition/spinal-muscular-atrophy[↩][↩][↩][↩][↩]
- Baumbach-Reardon L, Sacharow SJ, Ahearn ME. Spinal Muscular Atrophy, X-Linked Infantile. 2008 Oct 30 [Updated 2012 Sep 13]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2019. Available from: https://www.ncbi.nlm.nih.gov/books/NBK2594[↩]
- Wang CH, Finkel RS, Bertini ES, Schroth M, Simonds A, Wong B, Aloysius A, Morrison L, Main M, Crawford TO, Trela A; Participants of the International Conference on SMA Standard of Care. Consensus statement for standard of care in spinal muscular atrophy. American Society of Gene and Cell Therapy 19th Annual Meeting. 2007.[↩]
- Kelley RI, Sladky JT. Dicarboxylic aciduria in an infant with spinal muscular atrophy. Ann Neurol. 1986;20:734–6.[↩]
- Bowerman M, Swoboda KJ, Michalski JP, Wang GS, Reeks C, Beauvais A, Murphy K, Woulfe J, Screaton RA, Scott FW, Kothary R. Glucose metabolism and pancreatic defects in spinal muscular atrophy. Ann Neurol. 2012;72:256–68.[↩]




{kind=link}