
What is paraplegia
Paraplegia is loss of muscle function or severe weakness (where you can’t move parts of the body) or paralysis in both legs (but not the arms) and lower half of your body. Paralysis of both arms and legs (a loss of function below the neck) is quadriplegia or tetraplegia. Tetraplegia (quadriplegia) means that your arms, hands, trunk, legs and pelvic organs are all affected by your spinal cord injury. Paraplegia happens when something goes wrong with the way messages pass between your brain to your muscles e.g., damage to the spinal cord, various degenerations in the spinal cord and polyneuritis (an illness where sensory and motor nerves of the peripheral nervous system are affected e.g., Guillain-Barré syndrome or diabetic neuropathy (diabetes-related nerve damage)). Your spinal cord sends and receives signals between your brain and the rest of your body. A spinal cord injury often causes permanent changes in strength, feeling and other body functions below the site of the injury. The body parts that may be affected are the chest, stomach, hips, legs, and feet 1. Trauma to the thoracic nerves (spinal cord injury) from the T2 vertebra in the upper, middle, or lower back at the S5 can usually be called paraplegic 1. The spinal cord does not need to be completely severed to cause loss of function. Compression of the spinal cord or bruising or inflammation can also cause loss of function. Paralysis can be complete or partial. It can occur on one or both sides of your body. It can also occur in just one area, or it can be widespread.
The most common causes of spinal cord injuries in the United States are:
- Motor vehicle accidents. Auto and motorcycle accidents are the leading cause of spinal cord injuries, accounting for almost half of new spinal cord injuries each year.
- Falls. A spinal cord injury after age 65 is most often caused by a fall. Overall, falls cause more than 15 percent of spinal cord injuries.
- Acts of violence. Around 12 percent of spinal cord injuries result from violent encounters, often involving gunshot and knife wounds.
- Sports and recreation injuries. Athletic activities, such as impact sports and diving in shallow water, cause about 10 percent of spinal cord injuries.
- Alcohol. Alcohol use is a factor in about 1 out of every 4 spinal cord injuries.
- Diseases. Cancer, arthritis, osteoporosis and inflammation of the spinal cord also can cause spinal cord injuries.
One of the terms you will hear often in reference to your spinal cord injury is complete or incomplete.
- An incomplete injury means that the ability of the spinal cord to convey messages to or from the brain is not completely lost. Additionally, some sensation (even if it’s faint) and movement is possible below the level of injury.
- A complete injury is indicated by a total lack of sensory and motor function below the level of injury.
The absence of motor and sensory function below the injury area does not necessarily mean there are no remaining intact axons or nerves crossing the injury site, just that they are not functioning appropriately as a result of the trauma.
There are also two main ways that paralyzed muscles act in paraplegia:
- Flaccid paraplegia: This means that muscles don’t work at all and remain flaccid or limp.
- Spastic paraplegia: This is paraplegia that results in muscles that don’t work by themselves and contract uncontrollably.
The treatments for paraplegia depend on what caused the issue and where it is in your spinal cord. The injuries and conditions that can cause paraplegia can vary widely, and the potential to recover from them can also vary. Because of that, your doctor is the best person to explain the situation. He/she offer you guidance for your specific situation, including the possible treatments and the side effects or complications that can happen with those treatments.
Table 1. American Spinal Injury Association Impairment Scale
| Classification | Description |
|---|---|
| A | Complete: no motor or sensory function is preserved below the level of injury, including the sacral segments S4-S5 |
| B | Incomplete: sensory, but not motor, function is preserved below the neurologic level and some sensation in the sacral segments S4-S5 |
| C | Incomplete: motor function is preserved below the neurologic level, however, more than half of key muscles below the neurologic level have a muscle grade less than 3 (i.e., not strong enough to move against gravity) |
| D | Incomplete: motor function is preserved below the neurologic level, and at least half of key muscles below the neurologic level have a muscle grade of 3 or more (i.e., joints can be moved against gravity) |
| E | Normal: motor and sensory functions are normal |
What’s the difference between paraplegia and tetraplegia?
Paraplegia is paralysis that affects your legs. Tetraplegia also known as quadriplegia is paralysis that affects your arms and legs.
What’s the difference between paraplegia and hemiplegia?
Paraplegia is paralysis that affects your legs and sometimes the lower half of your body. Hemiplegia is paralysis that affects one side of your body, either left or right. It can involve your arm, leg or one side of your face, or a combination of these three.
Types of paraplegia
There are also two main ways that paraplegia can happen, complete and incomplete:
- Complete injury: This means a total loss of function, including the ability to feel sensation and move. Your body also can’t control automatic functions that rely on your spinal cord for relaying signals, such as controlling your bladder and bowels.
- Incomplete injury: This means a partial loss of function. You might still be able to feel or move body parts below the injury, but usually not as strongly as you could before the injury.
There are also two main ways that paralyzed muscles act in paraplegia:
- Flaccid paraplegia: This means that muscles don’t work at all and remain flaccid or limp.
- Spastic paraplegia: This is paraplegia that results in muscles that don’t work by themselves and contract uncontrollably.
People with paraplegia have different symptoms depending on how high or low the injury is in their spine. Paraplegia generally affects your legs, but may also affect your abdominal muscles, making it difficult to cough. It can also affect your chest muscles, making it difficult to take deep breaths.
Your spine has multiple sections. Each section has several vertebrae (singular is vertebra). Your vertebrae are interlocking bone segments that make up your backbone and protect your spinal cord.
The closer the spinal injury is to the skull, the more extensive is the impairment of the body’s ability to move and feel. If the lesion is low on the spine, say, in the sacral area, it is likely that there will be a lack of feeling and movement in the thighs and lower parts of the legs, the feet, most of the external genital organs, and the anal area. But the person will be able to breathe freely and move his head, neck, arms, and hands. By contrast, someone with a broken neck may be almost completely incapacitated, even to the extent of requiring breathing assistance 2.
Thoracic Nerves T1 – T5
- Corresponding nerves affect muscles, upper chest, mid-back and abdominal muscles.
- Arm and hand function is usually normal.
- Injuries usually affect the trunk and legs(also known as paraplegia).
- Most likely use a manual wheelchair
- Can learn to drive a modified car
- Can stand in a standing frame, while others may walk with braces
Thoracic Nerves T6 – T12
- Nerves affect muscles of the trunk (abdominal and back muscles) depending on the level of injury.
- Usually results in paraplegia
- Normal upper-body movement
- Fair to good ability to control and balance trunk while in the seated position
- Should be able to cough productively (if abdominal muscles are intact)
- Little or no voluntary control of bowel or bladder but can manage on their own with special equipment
- Most likely use a manual wheelchair
- Can learn to drive a modified car
- Some can stand in a standing frame, while others may walk with braces.
Lumbar Nerves L1 – L5
- Injuries generally result in some loss of function in the hips and legs.
- Little or no voluntary control of bowel or bladder, but can manage on their own with special equipment
- Depending on strength in the legs, may need a wheelchair and may also walk with braces
Sacral Nerves S1 – S5
- Injuries generally result in some loss of functioning the hips and legs.
- Little or no voluntary control of bowel or bladder, but can manage on their own with special equipment
- Most likely will be able to walk
Paraplegia causes
Spinal cord injuries may result from damage to the vertebrae, ligaments or disks of the spinal column or to the spinal cord itself.
A traumatic spinal cord injury may stem from a sudden, traumatic blow to your spine that fractures, dislocates, crushes or compresses one or more of your vertebrae. It also may result from a gunshot or knife wound that penetrates and cuts your spinal cord.
Additional damage usually occurs over days or weeks because of bleeding, swelling, inflammation and fluid accumulation in and around your spinal cord.
A nontraumatic spinal cord injury may be caused by arthritis, cancer, inflammation, infections or disk degeneration of the spine.
Vehicle crashes are currently the leading cause of spinal cord injury, closely followed by falls. Acts of violence (primarily gunshot wounds) and sports/recreation activities are also relatively common causes. About 78% of new spinal cord injury cases are male.
A recent estimate showed that the annual incidence of spinal cord injury is approximately 54 cases per one million people in the United States, or about 17,700 new spinal cord injury cases each year. New spinal cord injury cases do not include those who die at the location of the incident that caused the spinal cord injury.
The frequency of incomplete and complete paraplegia is virtually the same.
The number of people with spinal cord injury living in the United States is currently estimated to be approximately 288,000 persons, with a range from 247,000 to 358,000 persons. The average age at injury has increased from 29 years during the 1970s to 43 years currently.
Less than 1% of persons experienced complete neurological recovery by the time of hospital discharge.
Risk factors for paraplegia
Although a spinal cord injury is usually the result of an accident and can happen to anyone, certain factors may predispose you to a higher risk of sustaining a spinal cord injury, including:
- Being male. Spinal cord injuries affect a disproportionate amount of men. In fact, females account for only about 20 percent of traumatic spinal cord injuries in the United States.
- Being between the ages of 16 and 30. You’re most likely to suffer a traumatic spinal cord injury if you’re between the ages of 16 and 30.
- Being older than 65. Falls cause most injuries in older adults.
- Engaging in risky behavior. Diving into too-shallow water or playing sports without wearing the proper safety gear or taking proper precautions can lead to spinal cord injuries. Motor vehicle crashes are the leading cause of spinal cord injuries for people under 65.
- Having a bone or joint disorder. A relatively minor injury can cause a spinal cord injury if you have another disorder that affects your bones or joints, such as arthritis or osteoporosis.
Paraplegia prevention
Following this advice may reduce your risk of a spinal cord injury:
- Drive safely. Car crashes are one of the most common causes of spinal cord injuries. Wear a seat belt every time you drive or ride in a car.
- Make sure that your children wear a seat belt or use an age- and weight-appropriate child safety seat. To protect them from air bag injuries, children under age 12 should always ride in the back seat.
- Check water depth before diving. To make sure you don’t dive into shallow water, don’t dive into a pool unless it’s 12 feet (about 3.7 meters) or deeper, don’t dive into an aboveground pool, and don’t dive into any water of which you don’t know the depth.
- Prevent falls. Use a step stool with a grab bar to reach objects in high places. Add handrails along stairways. Put nonslip mats on tile floors and in the tub or shower. For young children, use safety gates to block stairs and consider installing window guards.
- Take precautions when playing sports. Always wear recommended safety gear. Avoid leading with your head in sports. For example, don’t slide headfirst in baseball, and don’t tackle using the top of your helmet in football. Use a spotter for new moves in gymnastics.
- Don’t drink and drive. Don’t drive while intoxicated or under the influence of drugs. Don’t ride with a driver who’s been drinking.
Paraplegia symptoms
In addition to complete loss of motor function of the trunk and lower extremities, people who survive a spinal cord injury often have medical complications resulting in bladder, bowel, and sexual dysfunction. They may also develop chronic pain, autonomic dysfunction, and spasticity (increased tone in and contractions of muscles of the arms and legs) , but this is highly variable and poorly understood. Higher levels of injury may have an increased susceptibility to respiratory and heart problems.
About 30% of persons with spinal cord injury are re-hospitalized one or more times during any given year following injury. Among those rehospitalized, the length of hospital stay averages about 22 days. Diseases of the genitourinary system are the leading cause of rehospitalization, followed by disease of the skin. Respiratory, digestive, circulatory, and musculoskeletal diseases are also common causes.
Persons enrolled in the National spinal cord injury Database since its inception in 1973 have now been followed for 40 years after injury. During that time,
the causes of death that appear to have the greatest impact on reduced life expectancy for this population are pneumonia and septicemia. Mortality rates are declining for cancer, heart disease, stroke, arterial diseases, pulmonary embolus, urinary diseases, digestive diseases, and suicide. However, these gains are being offset by increasing mortality rates for endocrine, metabolic and nutritional diseases, accidents, nervous system diseases, musculoskeletal disorders, and mental disorders. There has been no change in the mortality rate for septicemia in the past 40 years, and there has only been a slight decrease in mortality due to respiratory diseases.
Circulatory problems
Spinal cord injuries can cause a variety of changes in circulation, including blood pressure instability, abnormal heart rhythms (arrhythmias) that may appear days after the injury, and blood clots. Because the brain’s control of the cardiac nerves is cut off, the heart can beat at a dangerously slow pace, or it can pound rapidly and irregularly. Arrhythmias are more common and severe in the most serious injuries. Low blood pressure also often occurs due to changes in nervous system control of blood vessels, which then widen, causing blood to pool in the small arteries far away from the heart. Blood pressure needs to be closely monitored to keep blood and oxygen flowing through the spinal cord tissue, with the understanding that baseline blood pressure can be significantly lower than usual in people living with spinal cord injuries. Since muscle movement contributes to moving blood back to the heart, people with spinal cord injuries are at triple the usual risk for blood clots due to stagnation of blood flow in the large veins in the legs. Treatment includes anticoagulant drugs and compression stockings to increase blood flow in the lower legs and feet.
Spasticity and muscle tone
When the spinal cord is damaged, information from the brain can no longer regulate reflex activity. Reflexes may become exaggerated over time, causing muscle spasticity. Muscles may waste away or diminish due to underuse. If spasms become severe enough, they may require medical treatment. For some, spasms can be as much of a help as they are a hindrance, since spasms can tone muscles that would otherwise waste away. Some people can even learn to use the increased tone in their legs to help them turn over in bed, propel them into and out of a wheelchair, or stand.
Autonomic dysreflexia
The autonomic nervous system controls involuntary actions such as blood pressure, heartbeat, and bladder and bowel function. Autonomic dysreflexia is a life-threatening reflex action that primarily affects those with injuries to the neck or upper back. It happens when there is an irritation, pain, or stimulus to the nervous system below the level of injury. The irritated area tries to send a sensory signal to the brain, but the signal may be misdirected, causing a runaway reflex action in the spinal cord that has been disconnected from the brain’s regulation. Unlike spasms that affect muscles, autonomic dysreflexia affects blood vessels and organ systems controlled by the sympathetic nervous system. Anything that causes pain or irritation can set off autonomic dysreflexia, including a full bladder, constipation, cuts, burns, bruises, sunburn, pressure of any kind on the body, or tight clothing. Symptoms of its onset may include flushing or sweating, a pounding headache, anxiety, sudden increase in blood pressure, vision changes, or goose bumps on the arms and legs. Emptying the bladder or bowels and removing or loosening tight clothing are just a few of the possibilities that should be tried to relieve whatever is causing the irritation. If possible, the person should be kept in a sitting position, rather than lying flat, to keep blood flowing to the lower extremities and help reduce blood pressure.
Pressure sores or pressure ulcers
Pressure sores are areas of skin tissue that have broken down because of continuous pressure on the skin and reduced blood flow to the area. People with paraplegia and tetraplegia are susceptible to pressure sores because they may lose all or part of skin sensations and cannot shift their weight. As a result, individuals must be shifted periodically by a caregiver if they cannot shift positions themselves. Good nutrition and hygiene can also help prevent pressure sores by encouraging healthy skin. Special motorized rotating beds may be used to prevent and treat sores.
Pain
Some people who have spinal cord nerve are paralyzed often develop neurogenic pain—pain or an intense burning or stinging sensation may be unremitting due to hypersensitivity in some parts of the body. It can either be spontaneous or triggered by a variety of factors and can occur even in parts of the body that have lost normal sensation. Almost all people with spinal cord injury are prone to normal musculoskeletal pain as well, such as shoulder pain due to overuse of the shoulder joint from using a wheelchair. Treatments for chronic pain include medications, acupuncture, spinal or brain electrical stimulation, and surgery. However, none of these treatments are completely effective at relieving neurogenic pain.
Bladder and bowel problems
Most spinal cord injuries affect bladder and bowel functions because the nerves that control the involved organs originate in the segments near the lower end of the spinal cord and lose normal brain input. Although the kidneys continue to produce urine, bladder control may be lost and the risk of bladder and urinary tract infections increases. Some people may need to use a catheter to empty their bladders. The digestive system may be unaffected, but people recovering from a spinal cord injury may need to learn ways to empty their bowels. A change in diet may be needed to help with control.
Sexual function
Depending on the level of injury and recovery from the trauma, sexual function and fertility may be affected. A urologist and other specialists can suggest different options for sexual functioning and health.
Depression
Many people living with a spinal cord injury may develop depression as a result of lifestyle changes. Therapy and medicines may help treat depression.
Paraplegia complications
At first, changes in the way your body functions may be overwhelming. However, your rehabilitation team will help you develop the tools you need to address the changes caused by the spinal cord injury, in addition to recommending equipment and resources to promote quality of life and independence. Areas often affected include:
- Bladder control. Your bladder will continue to store urine from your kidneys. However, your brain may not be able to control your bladder as well because the message carrier (the spinal cord) has been injured. The changes in bladder control increase your risk of urinary tract infections. The changes also may cause kidney infections and kidney or bladder stones. During rehabilitation, you’ll learn new techniques to help empty your bladder.
- Bowel control. Although your stomach and intestines work much like they did before your injury, control of your bowel movements is often altered. A high-fiber diet may help regulate your bowels, and you’ll learn techniques to optimize your bowel function during rehabilitation.
- Skin sensation. Below the neurological level of your injury, you may have lost part of or all skin sensations. Therefore, your skin can’t send a message to your brain when it’s injured by certain things such as prolonged pressure, heat or cold. This can make you more susceptible to pressure sores, but changing positions frequently — with help, if needed — can help prevent these sores. You’ll learn proper skin care during rehabilitation, which can help you avoid these problems.
- Circulatory control. A spinal cord injury may cause circulatory problems ranging from low blood pressure when you rise (orthostatic hypotension) to swelling of your extremities. These circulation changes may also increase your risk of developing blood clots, such as deep vein thrombosis or a pulmonary embolus. Another problem with circulatory control is a potentially life-threatening rise in blood pressure (autonomic hyperreflexia). Your rehabilitation team will teach you how to address these problems if they affect you.
- Respiratory system. Your injury may make it more difficult to breathe and cough if your abdominal and chest muscles are affected. These include the diaphragm and the muscles in your chest wall and abdomen. Your neurological level of injury will determine what kind of breathing problems you may have. If you have a cervical and thoracic spinal cord injury, you may have an increased risk of pneumonia or other lung problems. Medications and therapy can help prevent and treat these problems.
- Muscle tone. Some people with spinal cord injuries experience one of two types of muscle tone problems: uncontrolled tightening or motion in the muscles (spasticity) or soft and limp muscles lacking muscle tone (flaccidity).
- Fitness and wellness. Weight loss and muscle atrophy are common soon after a spinal cord injury. Limited mobility may lead to a more sedentary lifestyle, placing you at risk of obesity, cardiovascular disease and diabetes. A dietitian can help you eat a nutritious diet to sustain an adequate weight. Physical and occupational therapists can help you develop a fitness and exercise program.
- Sexual health. Sexuality, fertility and sexual function may be affected by a spinal cord injury. Men may notice changes in erection and ejaculation; women may notice changes in lubrication. Physicians specializing in urology or fertility can offer options for sexual functioning and fertility.
- Pain. Some people experience pain, such as muscle or joint pain, from overuse of particular muscle groups. Nerve pain can occur after a spinal cord injury, especially in someone with an incomplete injury.
- Depression. Coping with all the changes a spinal cord injury brings and living with pain causes some people to experience depression.
Paraplegia diagnosis
Doctors in the emergency room do an exam, test for sensory function and movement, and ask questions about your accident. They may be able to rule out a spinal cord injury based on this evaluation. But emergency diagnostic tests may be needed. They should be done if the injured person has neck pain, isn’t fully awake, or has obvious weakness or neurological injury.
These tests can include:
- X-rays. X-rays can reveal damage to the bone surrounding the spinal cord, known as the vertebrae. They also can find tumors, fractures or changes in the spine.
- CT scan. A CT scan can provide a clearer image compared with an X-ray. This scan uses computers to form a series of cross-sectional images that can define bone, disk and other changes.
- MRI scan. MRI uses a strong magnetic field and radio waves to produce computer-generated images. This test is helpful for looking at the spinal cord to find herniated disks, blood clots or other masses that might compress the spinal cord.
A few days after your injury, when some of the swelling might have gone down, a more comprehensive neurological exam may be done. The exam looks at the level and completeness of the injury. This involves testing muscle strength and your ability to sense light touch and pinprick sensations.
Paraplegia treatment
Once someone has survived the injury and begins to cope psychologically and emotionally, the next concern is how to live with disabilities. Doctors are now able to predict with reasonable accuracy the likely long-term outcome of spinal cord injuries. This helps people experiencing spinal cord injury set achievable goals for themselves, and gives families and loved ones a realistic set of expectations for the future.
No two people will experience the same emotions after surviving a spinal cord injury, but almost everyone will feel frightened, anxious, or confused about what has happened. It’s common for people to have very mixed feelings: relief that they are still alive, but disbelief at the nature of their disabilities.
Rehabilitation programs combine physical therapies with skill-building activities and counseling to provide social and emotional support. The education and active involvement of the newly injured person and his or her family and friends is crucial.
A rehabilitation team is usually led by a doctor specializing in physical medicine and rehabilitation (called a physiatrist), and often includes social workers, physical and occupational therapists, recreational therapists, rehabilitation nurses, rehabilitation psychologists, vocational counselors, nutritionists, a case worker, and other specialists.
In the initial phase of rehabilitation, therapists emphasize regaining communication skills and leg and arm strength. For some individuals, mobility will only be possible with the assistance of devices such as a walker, leg braces, or a wheelchair.
Physical therapy includes exercise programs geared toward muscle strengthening. Occupational therapy helps redevelop fine motor skills, particularly those needed to perform activities of daily living such as getting in and out of a bed, self-grooming, and eating. Bladder and bowel management programs teach basic toileting routines. People acquire coping strategies for recurring episodes of spasticity, autonomic dysreflexia, and neurogenic pain.
Vocational rehabilitation includes identifying the person’s basic work skills and physical and cognitive capabilities to determine the likelihood for employment; identifying potential work places and any assistive equipment that will be needed; and arranging for a user-friendly workplace. If necessary, educational training is provided to develop skills for a new line of work that may be less dependent upon physical abilities and more dependent upon computer or communication skills. Individuals with disabilities that prevent them from returning to the workforce are encouraged to maintain productivity by participating in activities that provide a sense of satisfaction and self-esteem, such as educational classes, hobbies, memberships in special interest groups, and participation in family and community events.
Recreation therapy encourages people with spinal cord injury to participate in recreational sports or activities at their level of mobility, as well as achieve a more balanced and normal lifestyle that provides opportunities for socialization and self-expression.
Adaptive devices also may help people with spinal cord injury to regain independence and improve mobility and quality of life. Such devices may include a wheelchair, electronic stimulators, assisted gait training, neural prostheses, computer adaptations, and other computer-assisted technology.
New technologies
Inventive medical devices can help people with a spinal cord injury become more independent and more mobile. Some devices may also restore function. These include:
- Modern wheelchairs. Improved, lighter weight wheelchairs are making people with spinal cord injuries more mobile and more comfortable. For some, an electric wheelchair may be needed. Some wheelchairs can even climb stairs, travel over rough terrain and elevate a seated passenger to eye level to reach high places without help.
- Computer adaptations. For someone who has limited hand function, computers can be very powerful tools, but they’re difficult to operate. Computer adaptations range from simple to complex, such as key guards or voice recognition.
- Electronic aids to daily living. Essentially any device that uses electricity can be controlled with an electronic aid to daily living. Devices can be turned on or off by switch or voice-controlled and computer-based remotes.
- Electrical stimulation devices. These sophisticated devices use electrical stimulation to produce actions. They’re often called functional electrical stimulation systems, and they use electrical stimulators to control arm and leg muscles to allow people with spinal cord injuries to stand, walk, reach and grip.
- Robotic gait training. This emerging technology is used for retraining walking ability after a spinal cord injury.
Coping and support
An accident that results in paralysis is a life-changing event. Suddenly having a disability can be frightening and confusing, and adapting is no easy task. You may wonder how your spinal cord injury will affect your everyday activities, job, relationships and long-term happiness.
Recovery from such an event takes time, but many people who are paralyzed progress to lead productive and fulfilling lives. It’s essential to stay motivated and get the support you need.
Grieving
If you’re newly injured, you and your family will likely experience a period of mourning and grief. Although the grieving process is different for everyone, it’s common to experience denial or disbelief, followed by sadness, anger, bargaining and, finally, acceptance.
The grieving process is a common, healthy part of your recovery. It’s natural — and important — to grieve the loss of the way you were. But it’s also necessary to set new goals and find a way to move forward with your life.
You’ll probably have concerns about how your injury will affect your lifestyle, your financial situation and your relationships. Grieving and emotional stress are normal and common.
However, if your grief and sadness are affecting your care, causing you to isolate yourself from others, or prompting you to abuse alcohol or other drugs, you may want to consider talking to a social worker, psychologist or psychiatrist. Or you might find a support group of people with spinal cord injuries helpful.
Talking with others who understand what you’re going through can be encouraging, and members of the group may have good advice on adapting areas of your home or work space to better accommodate your current needs. Ask your doctor or rehabilitation specialist if there are any support groups in your area.
Taking control
One of the best ways to regain control of your life is to educate yourself about your injury and your options for reclaiming an independent life. A range of driving equipment and vehicle modifications is available today.
The same is true of home modification products. Ramps, wider doors, special sinks, grab bars and easy-to-turn doorknobs make it possible for you to live more autonomously.
Because the costs of a spinal cord injury can be overwhelming, you may want to find out if you’re eligible for economic assistance or support services from the state or federal government or from charitable organizations. Your rehabilitation team can help you identify resources in your area.
Talking about your disability
Your friends and family may respond to your disability in different ways. Some may be uncomfortable and unsure if they’re saying or doing the right thing.
Being educated about your spinal cord injury and willing to educate others is helpful. Children are naturally curious and sometimes adjust rather quickly if their questions are answered in a clear, straightforward way. Adults also can benefit from learning the facts.
Explain the effects of your injury and what your family and friends can do to help. At the same time, don’t hesitate to tell friends and loved ones when they’re helping too much. Although it may be uncomfortable at first, talking about your injury often strengthens your relationships with family and friends.
Dealing with intimacy, sexuality and sexual activity
Your spinal cord injury may affect your body’s response to sexual stimuli. However, you’re a sexual being with sexual desires. A fulfilling emotional and physical relationship is possible but requires communication, experimentation and patience.
A professional counselor can help you and your partner communicate your needs and feelings. Your doctor can provide the medical information you need regarding sexual health. You can have a satisfying future complete with intimacy and sexual pleasure.
Looking ahead
By nature, a spinal cord injury has a sudden impact on your life and the lives of those closest to you. When you first hear your diagnosis, you may start making a mental list of all of the things you can’t do anymore. However, as you learn more about your injury and your treatment options, you may be surprised by all you can do.
Thanks to new technologies, treatments and devices, people with spinal cord injuries play basketball and participate in track meets. They paint and take photographs. They get married, have and raise children, and have rewarding jobs.
Today, advances in stem cell research and nerve cell regeneration give hope for a greater recovery for people with spinal cord injuries. At the same time, new treatments are being investigated for people with long-standing spinal cord injuries.
No one knows when new treatments will be available, but you can remain hopeful about the future of spinal cord research while living your life to the fullest today.
Hereditary spastic paraplegia
Hereditary spastic paraplegia (HSP) also known as hereditary spastic paraparesis, familial spastic paraplegia, familial spastic paraparesis or Strümpell-Lorrain syndrome is a group of rare inherited neurodegenerative disorders that cause weakness and stiffness in the leg muscles 3, 4, 5, 6, 7, 8, 9, 10, 11. There are more than 80 different genetic types of hereditary spastic paraplegia. Hereditary spastic paraplegia primary symptoms are difficulty walking due to muscle weakness and muscle tightness (spasticity) in the legs. The clinical manifestation of hereditary spastic paraplegia starts in infancy and continues into adulthood with slow progression 12, 13. The symptoms gradually get worse over time.
Hereditary spastic paraplegia is associated with significant disability and a negative impact on quality of life 14.
There may be significant variation in the severity of leg weakness (varying from none to marked), the degree of spasticity (varying from minimal to severe), and the occurrence of other neurologic symptoms between different genetic types of hereditary spastic paraplegia; as well differences in the nature and severity of symptoms between individuals who have exactly the same genetic type of hereditary spastic paraplegia 15.
Various types of hereditary spastic paraplegia are classified according to 16, 17, 15:
- A) Mode of inheritance (autosomal dominant, autosomal recessive, X-linked recessive, mitochondrial, or maternal inheritance);
- B) The gene in which the mutation occurs; and
- C) The clinical syndrome (pattern of symptoms and neurological findings).
Clinically, hereditary spastic paraplegia can be categorized into “uncomplicated” or “pure” hereditary spastic paraplegia when symptoms are confined to leg weakness, tightness (spasticity), impaired vibration sense, and urinary urgency. Hereditary spastic paraplegia syndromes are classified as “complicated” or “complex” hereditary spastic paraplegia when leg weakness and tightness (spasticity) are accompanied by other neurological disturbance such as peripheral nerve impairment, muscle atrophy, or intellectual impairment 18.
Hereditary spastic paraplegia prevalence ranges from 0.1 to 9.6 per 100,000 individuals reported around the world 19, 20, 21, 22, 23, 24. Hereditary spastic paraplegia affects males and females of all ethnic groups from around the world. It’s difficult to know exactly how many people have hereditary spastic paraplegia because hereditary spastic paraplegia has high clinical and genetic heterogeneity and is prone to misdiagnosis.
Hereditary spastic paraplegia is mainly observed in autosomal dominant pure form in about 80% of the North American and north European hereditary spastic paraplegia populations, with SPG4/SPAST mutations in 40%, SPG3A/ATL1 mutations in 10% at hereditary spastic paraplegias’ early beginning, about 10% SPG31/REEP1 mutations, and almost 3% SPG10/KIF5A mutations 19, 25. The mutation mentioned above is seen in complex hereditary spastic paraplegias and other neuropathies involving motor and sensory neurons 26.
On the other hand, autosomal recessive hereditary spastic paraplegias are more complex and seen in a high degree of the consanguineous marriages (marriage between individuals who are closely related) population, with nearly 30% registered hereditary spastic paraplegias from the Middle East and northern Africa. SPG11 and SPG15 constitute a significant chunk of autosomal recessive forms of hereditary spastic paraplegias 27. The common phenotypic characters include thinning corpus callosum, periventricular white matter change in the ear lynx, early development of parkinsonism, cognitive ability slacking, moderate ataxia, retina abnormalities, and prominent paraplegia 19. In addition, SPG35/FA2H and SPG45/C19orf12 are prevalent forms of autosomal recessive hereditary spastic paraplegias 28, 29. SPG5A shows CYP7B1 pathological variant in 7.3% autosomal recessive hereditary spastic paraplegia and 3% sporadic pure hereditary spastic paraplegia 30. 5–12% of autosomal recessive hereditary spastic paraplegia cases account for SPG7 31 and about 3–5% of hereditary spastic paraplegia individuals show SPG11 variants in autosomal recessive mode.
X-linked hereditary hereditary spastic paraplegias shows complex phenotypes with few cases, and five hereditary spastic paraplegias are known to date with three genes identified as SPG22/SLC16A2, SPG1/L1CAM, and SPG2/PLP1.
Late spastic paraplegia-like symptoms are due to alterations (m.9176T>C) in the ATP6 of mitochondrial DNA 32. Similarly, with their colleagues, Sánchez-Ferrero et al. 33 reported alterations in mitochondrial MT-CO3 and MT-T1 causing hereditary spastic paraplegia. About 1–2% of cases show X and mitochondrial chromosomes mutations.
Testing for hereditary spastic paraplegia genes is available and performed for individual hereditary spastic paraplegia genes, for panels containing dozens of hereditary spastic paraplegia genes, and by analysis of all genes (whole exome and whole genome analysis). Genetic testing is often helpful to confirm the clinical diagnosis of hereditary spastic paraplegia. Genetic testing is most often able to find causative gene mutations for subjects with hereditary spastic paraplegia who have a family history of a similarly affected first-degree relative.
Despite discovery of more than 80 genes in which mutations cause various types of hereditary spastic paraplegia, many individuals with hereditary spastic paraplegia do not have an identified gene mutation. This is because: a) genes for all types of hereditary spastic paraplegia have not been discovered and furthermore, some discovered genes are not yet included in clinical testing panels; b) methods of gene sequencing typically used to analyze large panels of genes do not analyze all regions of genes. Furthermore, while sensitively detecting gene sequence changes, these “next generation sequencing” methods are less sensitive in detecting gene insertions and deletions that do not change the sequence of the remaining portion of the gene.
Genetic testing is expensive and not all insurance companies provide reimbursement for this analysis. Identifying a causative gene mutation can bring closure to a diagnostic odyssey, contribute insight into the prognosis and can be applied to genetic counseling and prenatal diagnosis. Nonetheless at present, genetic testing results very rarely influence treatment which is largely directed toward reducing symptoms.
Interpreting hereditary spastic paraplegia genetic test results may be straightforward. Genetic testing may identify a gene mutation that is known to be associated with hereditary spastic paraplegia in other subjects, absent in unaffected subjects, and known or predicted to change the protein function. These mutations are termed “likely pathogenic” (likely to be disease causing). On the other hand, genetic testing may also identify gene variations that are considered normal variations (for example, they may be present in subjects who do not have hereditary spastic paraplegia and may be predicted to not change the protein function). Such mutations are considered “benign” variations and are not likely to cause hereditary spastic paraplegia.
In addition to “likely pathogenic” and “likely benign” mutations”, it is not uncommon for genetic testing to identify gene variations that are “of uncertain significance”. Such mutations may not have been reported to be associated with the disorder or may not be predicted to disturb the function of the protein. By definition, it is not known if gene variations of uncertain significance cause hereditary spastic paraplegia (i.e. are pathogenic) or are actually normal variations that are of no medical consequence. Individuals seeking more information regarding results of genetic testing are recommended to consult a medical geneticist or genetic counselor.
At present, there is no specific treatment to prevent or reverse nerve degeneration in hereditary spastic paraplegia. Treatments are directed at reducing symptoms and improving balance, strength, and agility. Individuals should be evaluated periodically (annually or as needed) by a neurologist and physiatrist (medical doctor who specializes in pain management and rehabilitation) to assess progression and develop treatment strategies to maximize walking ability and reduce symptoms.
Current treatment recommendations for hereditary spastic paraplegia include:
- Daily regimen of physical therapy directed toward improving cardiovascular fitness, maintaining and improving muscle strength and gait, and reducing spasticity
- Occupational therapy, assistive walking devices, and ankle-foot orthotics as needed
- Drugs to reduce muscle spasticity (e.g., Lioresal [oral or intrathecal], tizanidine, dantrolene, botulinum A and B toxin injections [Botox, Dysport, Xeomin, or Myoblock]) and urinary urgency (e.g., oxybutynin, solifenacin, mirabegron, or intrabladder injections with Botox)
Individuals with hereditary spastic paraplegia are recommended to pursue active lifestyles including physical rehabilitation in order to maintain and improve functional abilities and cardiovascular fitness. In addition, genetic counselling in patients and their families to understand the transmission risk of hereditary spastic paraplegia in successive generations and the proband state (Table 2) are recommended.
Figure 1. Hereditary spastic paraplegia
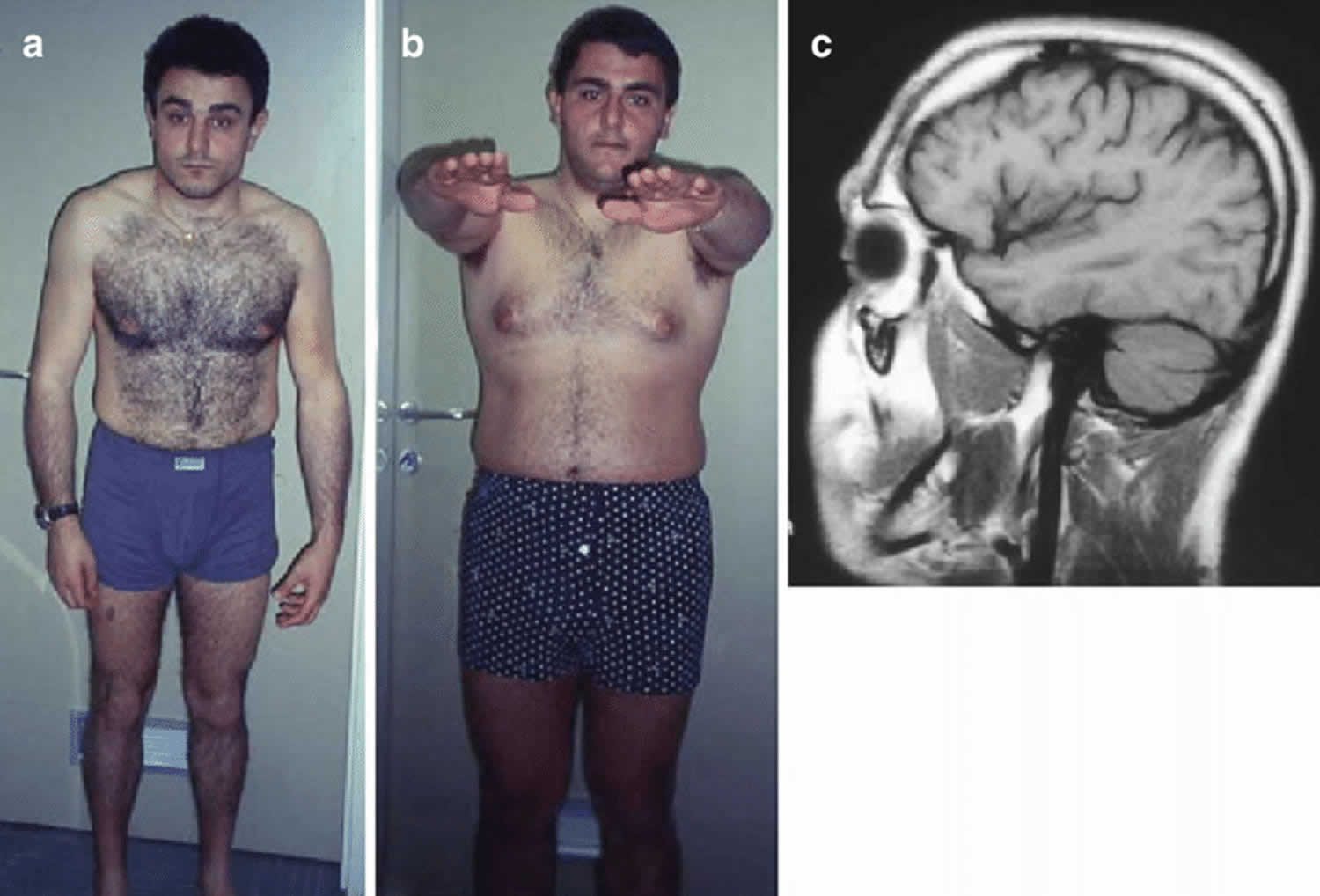
Footnotes: (a, b) Two siblings with hereditary spastic paraplegia. Note bended position of the shoulders with abnormal position of the left arm (a) and tendency to uneven position in Mingazzini I (b). (c) On brain MRI there are both cortical atrophy and cerebellar atrophy.
[Source 34 ]Figure 2. Hereditary spastic paraplegia genes
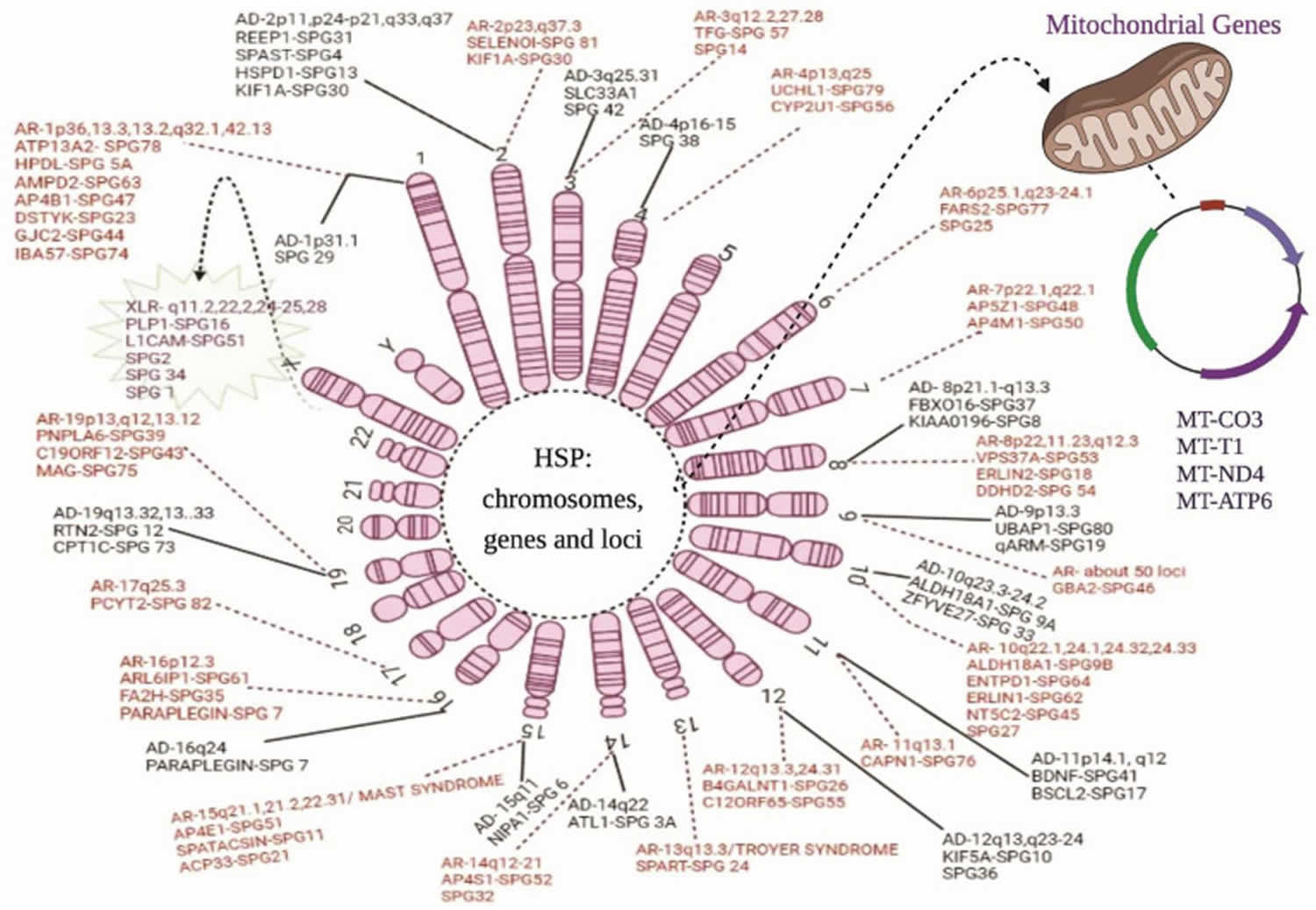
Footnotes: Chromosomal and genetic markers in hereditary spastic paraplegia. The above diagram illustrates all the genes and their location on the chromosomes related to the hereditary spastic paraplegia phenotypes: AD = autosomal dominant forms; AR = autosomal recessive forms; XLR = X-linked recessive forms; and maternal or mitochondrial inheritance. The solid line represents the autosomal dominant forms of hereditary spastic paraplegia, whereas the dotted lines show the autosomal recessive forms. The spiky green cloud with a dotted line represents the X-linked recessive hereditary spastic paraplegia form.
[Source 9 ]Figure 3. Hereditary spastic paraplegia pathophysiology
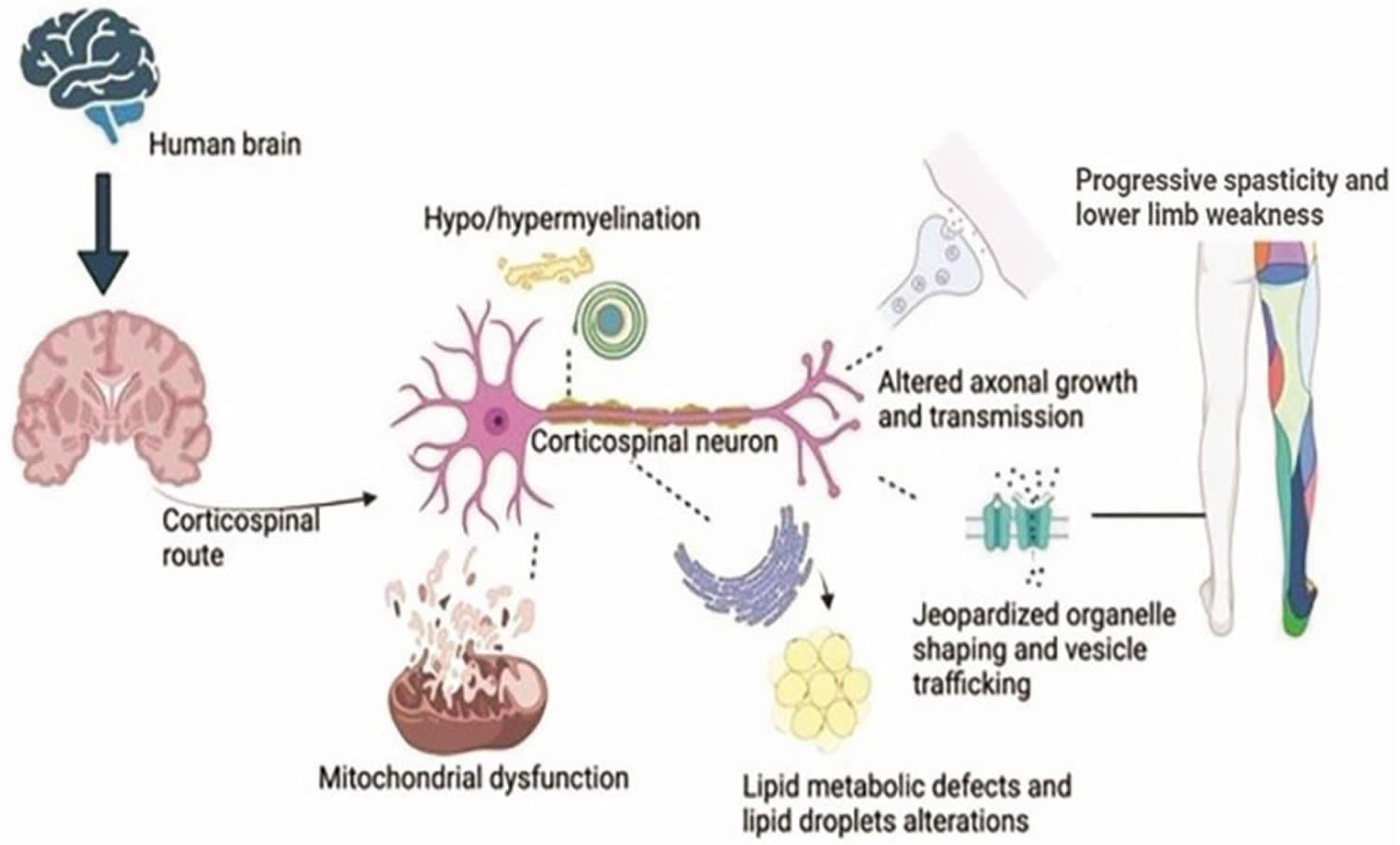
Footnotes: Hereditary spastic paraplegia pathophysiology. The neurons in the corticospinal zone of the brain undergo mutations in the genes, causing a breakdown of organelle shaping and trafficking and dysfunction in the mitochondrial cells at the neuron’s nuclear region. Few gene mutations lead to faulty transmission in the axons, and some mutations cause degeneration of the myelin sheath of the corticospinal neuron. Likewise, an endoplasmic reticulum shaping genes’ mutation causes defective metabolism, especially lipid droplet formations. All these characteristics lead to lower limb spasticity and weakness, causing hereditary spastic paraplegia phenotypes.
[Source 9 ]Table 2. Hereditary spastic paraplegia clinical classifications as pure and complex
| Pure hereditary spastic paraplegia | Complex hereditary spastic paraplegia |
|---|---|
| Impairments present in uncomplicated hereditary spastic paraplegia plus other neurologic findings such as:
|
Uncomplicated (pure) hereditary spastic paraplegia
Uncomplicated (or “pure”) hereditary spastic paraplegia is characterized by neurologic impairment limited to progressive lower-extremity spastic weakness, hypertonic urinary bladder disturbance, and mild diminution of lower-extremity vibration sensation. Individuals with uncomplicated hereditary spastic paraplegia experience the following:
- Difficulty walking (may either be non-progressive or worsen insidiously)
- Often, the need for canes, walkers, or wheelchairs
- Possible urinary urgency and lower-extremity paresthesias (the sensation of pins and needles)
- Typically, normal strength and dexterity of the upper extremities
- No involvement of speech, chewing, or swallowing
Though symptoms may be disabling, life span is not shortened.
Complex hereditary spastic paraplegia
Complex hereditary spastic paraplegia is characterized by the impairments present in uncomplicated “pure” hereditary spastic paraplegia plus other system involvement or other neurological findings including any of the following:*
- Ataxia
- Seizures
- Intellectual disability
- Dementia
- Muscle atrophy
- Extrapyramidal disturbance
- Peripheral neuropathy
Note:* In the absence of other causes for these additional features.
Hereditary spastic paraplegia causes
Hereditary spastic paraplegias are caused by gene mutations. To date, more than 80 genetic types of hereditary spastic paraplegia have been defined by genetic linkage analysis and identification of hereditary spastic paraplegia-related gene variants (Table 3). Underlying causes of hereditary spastic paraplegia: Each of the more than 80 genetic types of hereditary spastic paraplegia is due to mutations in a different gene. Each genetic type of hereditary spastic paraplegia is due to a mutation in a specific “hereditary spastic paraplegia gene”. For example, mutations in SPG3A/atlastin, SPG4/spastin, and SPG7/paraplegin genes cause SPG3A, SPG4, and SPG7 hereditary spastic paraplegia, respectively. These genes encode proteins that have diverse molecular functions including movement of chemicals from one part of the cell to another (“axon transport”), energy production (“mitochondrial disturbance”), and disorders of specific lipid metabolism, among others 35, 36.
Disturbance in some of these functions appears to lead to altered nerve cell (neuron) development. For these types of hereditary spastic paraplegia, the disorder is not a degenerative process, but rather a developmental disturbance in which the formation of selected nerve pathways during intra-uterine development was abnormal.
For other genetic types, hereditary spastic paraplegia gene mutations cause the ends of very long nerve processes (axons) to slowly degenerate within the spinal cord. This impairs nerve transmission from the brain through the spinal cord. To be clear, the entire spinal cord is not degenerating. Rather, the abnormalities in hereditary spastic paraplegia appear to selectively affect only specific nerve pathways, particularly the very long nerve processes (axons) that carry signals from the brain motor cortex to the lower part of the thoracic spinal cord. In some types, this disturbance is not limited to the spinal cord but also affects nerves in the legs (and arms, to a lesser extent). This latter process is termed “peripheral neuropathy”.
Depending on the genetic type of hereditary spastic paraplegia, hereditary spastic paraplegia may be transmitted to offspring (and inherited from parents) as Autosomal dominant, Autosomal recessive, X-Linked Inheritance, and “maternal” or mitochondrial inheritance. The various genetic types of hereditary spastic paraplegia and their inheritance patterns are summarized in the Table 3 below.
The following discussion of inheritance patterns is intended as an overview. Individuals seeking genetic counseling for hereditary spastic paraplegia are recommended to consult a genetic counselor or medical geneticist for specific information. In general, autosomal dominantly inherited forms of hereditary spastic paraplegia can be transmitted by (or inherited from) an individual who has the disorder. In general, each child of an individual who has a autosomal dominantly inherited form of hereditary spastic paraplegia has a 50% chance of inheriting the gene mutation and a similar (approximately 50% chance) of developing the condition. Occasionally, autosomal dominantly inherited hereditary spastic paraplegia “skips” a generation. (i.e. genetic penetrance is very high, exceeding 90%, but is occasionally incomplete). Although the chance of inheriting the condition can be estimated, it is difficult to predict with certainty the age at which symptoms would begin or their severity. There may be significant differences in the severity of the disorder between family members.
For autosomal recessive inherited forms of hereditary spastic paraplegia, both parents are usually carriers of the gene mutation and usually do not have symptoms (there are exceptions to this generalization: occasionally, parents who are carriers of some forms of autosomal recessive hereditary spastic paraplegia have had symptoms of hereditary spastic paraplegia). In general, if one individual in a family has a autosomal recessively inherited disorder, each of this individual’s full siblings (for example, another child in this family) has approximately a 25% chance of having the same disorder. In general, individuals who have autosomal recessive inherited disorders do not transmit the disorder to their children. There have been some reported exceptions to this however.
X-linked inheritance disorders are transmitted from women to their sons. Daughters may carry X-linked gene mutations, but like their mothers, usually do not have symptoms although they may have mild symptoms and rarely, may have more significant symptoms of the disorder.
Maternally or mitochondrial transmitted disorders are those in which the gene mutation involves a mitochondrial gene, are transmitted from mothers to sons or daughters (not transmitted from males).
Table 3. Chromosomes, loci, and genes involved in hereditary spastic paraplegias and their subtypes
| Inheritance Mode | Chromosome Number | Locus | Genes | Hereditary Spastic Paraplegia Subtype |
|---|---|---|---|---|
| Autosomal dominance (AD) | 1 | 1p31.1 | NERG1 * | SPG29 |
| 2 | 2p11.2 2p22.3 2q33.1 2q37.3 | REEP1 SPAST HSPD1 KIF1A | SPG31 SPG4 SPG13 SPG30 | |
| 3 | 3q25.31 | SLC33A1 | SPG42 | |
| 4 | 4p16-15 | JAKM1P1 * | SPG38 | |
| 8 | 8p21.1-q13.3 8q24.13 | FBXO16 KIAA0196 | SPG37 SPG8 | |
| 9 | 9p13.3 9q | UBAP1 – | SPG80 SPG19 | |
| 10 | 10q24.1 10q24.2 | ALDH18A1 ZFYVE27 | SPG9A SPG33 | |
| 11 | 11p14.1-p11.2 11q12.3 | BDNF * BSCL2 | SPG41 SPG17 | |
| 12 | 12q13.3 12q23-24 | KIF5A CKAP4 * | SPG10 SPG36 | |
| 14 | 14q22.1 | ATL1 | SPG3A | |
| 15 | 15q11.2 | NIPA1 | SPG6 | |
| 16 | 16q24.3 | PARAPLEGIN | SPG7 | |
| 19 | 19q13.32 19q13.33 | RTN2 CPT1C | SPG12 SPG73 | |
| Autosomal recessive (AR) | 1 | 1p36.13 1p34.1 1p13.3 1p13.2 1q32.1 1q42.13 1q42.13 | ATP13A2 HPDL AMPD2 AP4B1 DSTYK GJC2 IBA57 | SPG78 SPG83 SPG63 SPG47 SPG21 SPG44 SPG74 |
| 2 | 2p23.3 2q37.3 | SELENOI KIF1A | SPG81 SPG30 | |
| 3 | 3q12.2 3q27-q28 | TFG – | SPG57 SPG14 | |
| 4 | 4p13 4q25 | UCHL1 CYP2U1 | SPG79 SPG56 | |
| 6 | 6p25.1 6q23-24.1 | FARS2 – | SPG77 SPG25 | |
| 7 | 7p22.1 7q22.1 | AP5Z1 AP4M1 | SPG48 SPG50 | |
| 8 | 8p11.23 8p11.23 8p22 8q12.3 | ERLIN2 DDHD2 VPS37A CYP7B1 | SPG18 SPG54 SPG53 SPG5A | |
| 9 | 9p13.3 | GBA2 | SPG46 | |
| 10 | 10q22.1-q24.1 10q24.1 10q24.1 10q24.31 10q24.31-10q24.33 | – ALDH18A1 ENTPD1 ERLIN1 NT5C2 | SPG27 SPG9B SPG64 SPG62 SPG45 | |
| 11 | 11q13.1 | CAPN1 | SPG76 | |
| 12 | 12q13.3 12q24.31 | B4GALNT1 C12ORF65 | SPG26 SPG55 | |
| 13 | 13q13.3 (TROYER SYNDROME) 13q14 | SPART – | SPG20 SPG24 | |
| 14 | 14q22.1 14q24.1 14q32.31 | DDHD1 ZFYVE26 TECPR2 | SPG28 SPG15 SPG49 | |
| 15 | 15q21.1 15q21.2 15q22.31 (MAST SYNDROME) | KIAA1840 AP4E1 ACP33 | SPG11 SPG51 SPG21 | |
| 16 | 16p12.3 16q23.1 16q24.3 | ARL6IP1 FA2H PARAPLEGIN | SPG61 SPG35 SPG7 | |
| 17 | 17q25.3 | PCYT2 | SPG 82 | |
| 19 | 19p13.2 19q12 19q13.12 | PNPLA6 C19ORF12 MAG | SPG39 SPG43 SPG75 | |
| X-Linked Inheritance (XLR) | X | Xq11.2 Xq11.2 Xq22.2 Xq24-25 Xq28 | MTMR8 * ZC4H2 * PLP1 GLUD2 L1CAM | SPG16 SPG16 SPG2 SPG34 SPG1 |
| Mitochondrial or Maternal Inheritance | Mitochondria | – – – – | MT-CO3 MT-T1 MT-ND4 MT-ATP6 | – – – – |
Footnotes: * The predicted genes that might induce hereditary spastic paraplegia phenotypes are in bold letters.
[Source 9 ]Hereditary spastic paraplegia symptoms
The primary symptom of hereditary spastic paraplegia is difficulty walking due to weakness and tightness (spasticity) in the legs. Both legs are affected, usually to a relatively similar degree.
The term “paraplegia” means severe weakness in both legs including paralysis. “Paraparesis” indicates weakness in both legs of lesser severity than paraplegia. Although HSP is typically referred to as hereditary spastic paraplegia the degree of weakness is variable and ranges from no weakness (full strength) to marked weakness (paraplegia).
When present, weakness does not affect all leg muscles, but rather is most obvious in muscles of hip flexion (iliopsoas), hip abduction (gluteus medius), knee flexion (hamstrings), and foot dorsiflexsion (bending the foot back toward the shin via tibialis anterior muscle). In contrast, muscles of leg extension (quadriceps) and foot extension (gastrocnemius-soleus) usually are not affected in uncomplicated hereditary spastic paraplegia.
Spasticity primarily affects muscles of leg extension (quadriceps), knee flexion (hamstrings), hip adduction (bringing the knees together, thigh adductor muscles), and muscles that extend the feet (gastrocnemius-soleus [Achilles tendon]).
Walking pattern described as “spastic gait” occurs in which the following elements are present, each to variable degree in different individuals:
- a) heel strike is shifted forward (landing on the mid-foot or even further forward on the balls of the feet);
- b) there is reduced foot dorsiflexion (not bending the toes up, but instead tending to drag the toes, often catching them on carpet or when stepping over curbs, and causing the toes of the shoes to be worn out);
- c) stride length may become shorter;
- d) there may be “circumduction” or “scissoring”, with one leg crossing into the path of the other;
- e) there is a tendency for the knees to be maintained flexed (not fully extended in mid-stride),
- f) for thighs to be close together (adductor tightness), and
- g) hip flexion (knee lifting) to be reduced.
Balance difficulty, often worse when walking in the dark or on uneven surfaces is not uncommon in individuals with hereditary spastic paraplegia.
Tightness in the legs and leg muscle spasm (often at night) are common.
The consequences of abnormal walking pattern cause strain on the ankles, knees, hips, and back and often cause pain in these areas.
Urinary urgency, the symptom of experiencing a very short interval between the sensation of need to urinate and difficulty remaining continent, is very common in hereditary spastic paraplegia and occasionally may be an early symptom. Bowel urgency is less common but may occur. Medications such as oxybutynin may reduce urinary urgency. If urinary urgency is severe or accompanied by difficulty initiating urination, consultation with a urologist is recommended.
Additional symptoms
Some genetic types of hereditary spastic paraplegia tend to cause only spastic weakness in the legs and urinary urgency. These syndromes are referred to as “uncomplicated hereditary spastic paraplegia”. Other genetic types of hereditary spastic paraplegia tend to be associated with additional symptoms (“complicated hereditary spastic paraplegia”) including difficulty with coordination (“ataxia”), impaired vision, seizures (epilepsy), muscle atrophy, disturbance of the nerves in the arms and legs (neuropathy), and disturbance cognitive ability (intellectual impairment and dementia). Previously, it was considered that hereditary spastic paraplegia caused symptoms only in the legs, and therefore, did not affect the strength or coordination of the arms and hands, or speech or swallowing. As the number of hereditary spastic paraplegia types has grown, it is now recognized that the arms, hands, and speech and swallowing may be affected in some genetic types of complicated hereditary spastic paraplegia
Pattern of symptom progression
When hereditary spastic paraplegia begins in very early childhood (before age two years, for example), symptoms may not worsen even over many years or decades. Individuals with this “non-progressive” (non-worsening) pattern may resemble subjects with spastic cerebral palsy, a life-long disorder that also remains relatively stable. One caveat however: although early childhood-onset forms of hereditary spastic paraplegia may be “non-progressive”, the degree of spasticity may increase slowly if adequate range-of-motion is not maintained through stretching exercises and muscle spasticity reduction.
In contrast, when hereditary spastic paraplegia symptoms begin after early childhood (in adolescence or adulthood), symptoms usually worsen very slowly over a number of years. Sudden onset or rapid worsening over weeks or months is not typical of hereditary spastic paraplegia and suggests an alternate disorder or co-existing condition. After a number of years of very gradual worsening, the rate of worsening appears to slow down for many (not all) subjects. These subjects seem to reach a “functional plateau” beyond which the degree of worsening seems to be similar to that expected for age and similar degrees of physical exercise. Nevertheless, not all patients reach an apparent “leveling off” or functional plateau but instead experience continuous worsening of walking ability due to very slowly progressive muscle weakness and tightness .
Variability in the type of symptoms and their severity
There may be significant variability in the type of symptoms and their severity. For example, symptoms may remain mild in some patients or become quite severe in others patients. This variability may occur between different genetic types of hereditary spastic paraplegia as well as in between individuals with the same genetic type of hereditary spastic paraplegia including family members who share not only the same genetic type of hereditary spastic paraplegia but also precisely the same genetic mutation.
There is not a perfect correlation between the genetic type of hereditary spastic paraplegia and the pattern of symptoms. For example, while some genetic types of hereditary spastic paraplegia (e.g. dominantly inherited hereditary spastic paraplegia due to SPG4 or spastin mutation) usually are associated with “uncomplicated” hereditary spastic paraplegia syndromes, some patients with these types of hereditary spastic paraplegia develop additional neurologic symptoms. As another example, although SPG7 and SPG11 typically are associated with additional neurologic symptoms (ataxia, neuropathy, cognitive impairment, for example), some subjects with mutations in these genes have uncomplicated hereditary spastic paraplegia (only spastic weakness in the legs). There also may be variation in severity and the nature of symptoms between affected family members. Therefore, it is generally not possible to predict with certainty the severity or exact nature of symptoms associated with given genetic type of hereditary spastic paraplegia. A cautious, “wait and see” approach, combined with pro-active, individualized physical therapy is recommended.
Hereditary spastic paraplegia complications
Possible complications of hereditary spastic paraplegia include:
- shortening and hardening of the calf muscles – having regular physiotherapy may help prevent this
- cold feet – this is fairly common and occurs as a result of the deterioration of the nerves in the spine
- extreme tiredness (fatigue) – this may be because of the extra effort needed for walking and the symptoms interrupting sleep
- back and knee pain – caused by the muscle weakness and walking problems
- stress and depression
Hereditary spastic paraplegia diagnosis
Hereditary spastic paraplegia is diagnosed by the following 37:
- Typical symptoms lower extremity spastic weakness that may be non-worsening (early childhood onset) or slowly progressive over many years. The clinical manifestations are spastic paraplegia of both lower limbs, but cases with increased tendon reflexes, slight impairment of rapid alternating movements or mild distal amyotrophy in the upper limbs are not excluded; the cranial nerves and language are not affected.
- Findings on neurologic examination lower extremity hyperreflexia usually accompanied by some degree of spasticity and sometimes a specific pattern of muscle weakness; and
- The exclusion of alternate disorders by history, examination, neuroimaging, and laboratory studies as needed.
- Laboratory testing: Common biomarkers are normal.
- Imaging: Brain and spinal cord magnetic resonance imaging (MRI) are usually normal, but some patients may present with dysgenesis of the corpus callosum or tapering of the spinal cord.
The occurrence of similarly affected family members is helpful in recognizing hereditary spastic paraplegia but is not required for the diagnosis of hereditary spastic paraplegia. Many individuals with hereditary spastic paraplegia do not have similarly affected family members. Such individuals could represent the first occurrence of a genetic mutation (“de novo mutation”). Depending on the genetic type of hereditary spastic paraplegia (dominant, recessive, X-linked, or maternal transmission), there may be a possibility that the disorder could be transmitted to the offspring of these individuals. Genetic testing is often helpful in confirming the clinical diagnosis of hereditary spastic paraplegia and in determining the genetic type of hereditary spastic paraplegia. Results of genetic testing can be used, together with clinical information, to provide genetic counseling.
Hereditary spastic paraplegia diagnostic tests
Neurologic examination is important for patients with symptoms of hereditary spastic paraplegia. First, this establishes the diagnosis and excludes alternative and co-existing disorders, some of which may have specific treatments. Second, neurologic examination helps identify the specific features of an individual’s walking disturbance. Knowing which specific muscles need strengthening, which specific muscles need spasticity-reduction (through medication, Botox injection, and stretching), and the degree of impairment of balance, speed, and precision of movement helps neurologists and physiatrists develop a proactive therapy approach to improve and maintain the ability to walk; and limit the cumulative impact of abnormal walking patterns on ankles, knees, hips, and spine.
Laboratory tests, neurophysiologic testing, and neuroimaging: Routine laboratory studies (such as blood counts, serum electrolytes, and tests of kidney, liver, and endocrine functions) including analysis of cerebrospinal fluid (obtained by “spinal tap”) are normal in most types of hereditary spastic paraplegia. The primary role of such testing is to help exclude alternate and co-existing diagnoses.
Magnetic resonance imaging (MRI) scans of the brain and spinal cord are important in diagnosing hereditary spastic paraplegia because they help exclude other disorders such as multiple sclerosis and structural abnormalities of the brain and spinal cord. Routine magnetic resonance imaging (MRI) of the brain is usually normal in uncomplicated hereditary spastic paraplegia, and, depending on the genetic type and its neurologic features, in many forms of complicated hereditary spastic paraplegia. In contrast to routine brain MRI, which is usually normal in uncomplicated hereditary spastic paraplegia, special MRI techniques such as diffusion tensor imaging, reserved primarily for research purposes often show more widespread nerve pathway abnormalities in uncomplicated and complicated hereditary spastic paraplegia.
In contrast to the typically normal brain MRI in subjects with uncomplicated hereditary spastic paraplegia, there are many types of complicated hereditary spastic paraplegia in which brain MRI demonstrates specific abnormalities including reduced size of the corpus callosum (a structure containing nerve fibers that transit from one brain hemisphere to the other).Thin corpus callosum is a frequent (but not constant) feature of SPG11 and SPG15 and has also been present in many other types of hereditary spastic paraplegia (including SPG3A, SPG4, SPG7, SPG15, SPG21, SPG32, SPG47, PG49, SPG54, and SPG56) 38, 39. In addition to thin corpus callosum, many genetic types of hereditary spastic paraplegia have abnormal appearing brain white matter (e.g. due SPG5/CYPB7, SPG7/paraplegin, SPG21/maspardin, and SPG35/FA2H gene mutations) 38, 39.
Spinal cord MRI scan in hereditary spastic paraplegia is usually normal although may show somewhat smaller diameter of the thoracic spinal cord 40.
Genetic testing
There are different approaches available to test hereditary spastic paraplegia genetically, and the most cost-effective and widely available one is the next generation sequencing (NGS), as it comprises of screening the whole exons to find the number of genes linked to hereditary spastic paraplegia phenotypes but still possess limitations for differentiating the variants of large deletions, duplications, alterations in the promotor or intronic regions, and cases of triplet repeat disorders 41. To obtain normal results, the multiplex ligation-dependent probe amplification is utilized for genes such as SPAST with exon deletions 42. In a few places, first-generation sequencing is initially carried out with a set of targeted genes, and next generation sequencing is considered later 20. The algorithm varies from clinician to clinician; some centres prefer next generation sequencing (NGS) panel sequencing as a first-line investigatory protocol, even in the absence of finding a pathogenic variant.
The clinician must conclude to find the gene for multiple ligation-dependent probe amplification or relate the uncertain variant with the phenotype. In addition, clinicians should have knowledge on the other monogenic diseases with the same phenotype of slowly progressing spasticity on lower limbs with no spinal cord imaging abnormalities not to be categorized in spastic paraplegia gene (SPG). The next generation sequencing (NGS) panel does not comprehensively cover hereditary spastic paraplegia alone, and they include the panels for ataxias (Spinocerebellar, AR, spastic), myelination defects, and other neurometabolic disorders 20.
Hereditary spastic paraplegia treatment
Despite encouraging progress in many research laboratories, treatment for hereditary spastic paraplegia is presently limited to reducing symptoms of muscle weakness, improving balance, strength, agility, spasticity, and urinary urgency 43. Individuals should be evaluated periodically (annually or as needed) by a neurologist and physiatrist (medical doctor who specializes in pain management and rehabilitation) to assess progression and develop treatment strategies to maximize walking ability and reduce symptoms.
Current treatment recommendations for hereditary spastic paraplegia include:
- Daily regimen of physical therapy directed toward improving cardiovascular fitness, maintaining and improving muscle strength and gait, and reducing spasticity
- Occupational therapy, assistive walking devices, and ankle-foot orthotics as needed
- Drugs to reduce muscle spasticity (e.g., Lioresal [oral or intrathecal], tizanidine, dantrolene, botulinum A and B toxin injections [Botox, Dysport, Xeomin, or Myoblock]) and urinary urgency (e.g., oxybutynin, solifenacin, mirabegron, or intrabladder injections with Botox)
Individuals with hereditary spastic paraplegia are recommended to pursue active lifestyles including physical rehabilitation in order to maintain and improve functional abilities and cardiovascular fitness.
Hereditary spastic paraplegia physical therapy
Physiotherapy with daily physical exercise program especially foot and ankle orthotics and peroneal nerve stimulation guided by physical therapist or personal trainer and developed for each patient’s unique type of symptoms is recommended for lower limb building and strength, reducing toe dragging, and enhancing the cardiovascular system’s functioning 44. This recommendation is based not on peer-reviewed scientific publications but rather on the reports of large numbers of hereditary spastic paraplegia subjects who state that exercise helps and that periods of reduced exercise are associated with increased symptoms. Hereditary spastic paraplegia symptoms are variable. One type of exercise may not benefit all individuals. Exercise programs should be developed by a neurologist, physiatrist, physical therapist or personal trainer who is experienced with hereditary spastic paraplegia or similar disorders and should focus on the specific factors that make walking difficult for the specific individual. Individuals are advised to consult their primary care physician before beginning exercise programs, to begin with low intensity, increase slowly, set small goals, keep records of their progress, add variety, and be creative.
Exercise goals are to:
- Improve and maintain cardiovascular fitness;
- Reverse the reduced functional capacity, stiffness, and weakness due to relatively sedentary lifestyle that often accompanies chronic gait disorders and which are superimposed on walking disturbance due to hereditary spastic paraplegia;
- Improve the mechanics of walking, facilitate neurologic circuits underlying walking reflexes, reduce falling, and maintain bone and joint fitness; and
- Maximize an individual’s independence and sense of control.
For some patients, weakness in certain muscles is the most significant factor making walking difficult. For these patients, daily exercise programs should focus on resistance exercises designed to maintain and very gradually increase the strength of these weak muscles. For others, spasticity is a more significant factor than weakness in limiting the ability to walk. For these patients, stretching exercises and muscle relaxant medication may be more beneficial than muscle strengthening exercises alone. A variety of exercises is recommended including walking, for example in shallow swimming pool, water aerobics, swimming, bicycling (including pedaling in reverse to exercise the strength and speed of hip flexion), yoga, dance, core exercises, balance exercises, and therapeutic horseback riding.
Muscle relaxing medication
Patients with significant degrees of spasticity may benefit from antispasmodic (skeletal muscle relaxants) medications such as tizanidine and baclofen. In general, reducing spasticity through medication improves walking primarily when spasticity, not weakness is the primary factor limiting the ability to walk. Early intrathecal baclofen improves gait in severe spastic patients wheelchair-ridden by declining pain, disability, and muscle tone 45. When weakness is the major factor, markedly reducing spasticity (e.g. through intrathecal baclofen pump) may make the legs very relaxed (even hypotonic or “floppy”) but actually make walking and standing more difficult. Patients considering intrathecal baclofen pumps should undergo at least one trial in which the important criteria is not simply if spasticity reduction occurred, but rather if spasticity reduction resulted in improved walking ability. Botulinum toxin (“Botox” or botulinum toxin type-A) injection may be helpful when muscle tightness particular affects a limited number of muscles (e.g. adductors or ankles), with stretching exercises of ankle, hips, and knees, increases gait velocity by reducing calf muscle tone to maintain strength and balance 46. In addition, the Botulinum toxin efficacy has not been accessed in the common nonmotor manifestations observed in hereditary spastic paraplegia cases such as pain, fatigue, depression, and excessive daytime sleepiness, but studies by Servelhere et al. 47 found significant improvement for fatigue after Botox treatment in hereditary spastic paraplegia patients by relieving spasticity and improving gait biomechanics to reduce fatigue. Yet studies are required to identify the analgesic response of the Botox in more hereditary spastic paraplegia cases to obtain conclusive knowledge.
Oxybutynin and related medications may reduce urinary urgency and infection 20. Individuals with more advanced bladder or bowel symptoms are recommended to consult urology or gastroenterology specialists, respectively.
A positive response is observed in a few to 4-aminpyridine (Dalfampridine), which is awaiting conclusive results from larger group studies 48.
Orthotics
Ankle-foot orthotics and heel raise orthotics may be useful to reduce the tendency for the feet to be extended (toes down) causing toe dragging and tripping 20. Ankle-foot orthotics are often used in combination with medications (e.g. Lioresal or Botox) that reduce muscle spasticity.
Potential future treatments
Treatment based on the genotype has paved its way in neurological diseases in general for Huntington’s disease or spinal muscular atrophy. However, hereditary spastic paraplegia is lagging due to its complex genetics, mechanism diversity, various subtypes with rare forms, and slower disease progression 20. Through gene therapy, microtubule loss can be corrected in SPG4 phenotypes 49. Havlicek et al. 50 showed that spastin-mutated human-induced pluripotent stem cells of the patient develop neutrino membrane again with branching and increasing the length to reduce neuron swelling. Cholesterol breakdown is controlled by oxysterol hydroxylase-7α encoding the CYP7B1, and this gene is linked with AR SPG5. Primarily lowering cholesterol levels is recommended for managing hereditary spastic paraplegia phenotypes using atorvastatin. Deeper studies are required to obtain reliable results for utilising combined therapies 51.
The paraplegin gene mediates the fast opening of the transition pore, and it can be modulated using medications 20. One study showed that intramuscular paraplegin administration in hereditary spastic paraplegia mice model prevents pathology progression in neurons and protects mitochondrial structure in the peripheral nerves 52. Gene therapy for the recessive hereditary spastic paraplegia forms such as SPG11, SPG15, and SPG7 can allow editing genes at their target location or for replacement. Currently, the collaboration between world researchers and randomized control studies on large groups is required to obtain efficacious treatment for this neuromuscular pathology 20.
Hereditary spastic paraplegia prognosis
There is significant variation in hereditary spastic paraplegia symptoms and their severity. This limits the certainty of making predictions. In general however, some genetic types of hereditary spastic paraplegia are usually associated with only leg weakness, spasticity, and urinary urgency (“uncomplicated hereditary spastic paraplegia”). Other types of hereditary spastic paraplegia are usually associated with other neurological disturbances in addition to these symptoms (“complicated hereditary spastic paraplegia”). Although there are exceptions, an individual with pure hereditary spastic paraplegia (uncomplicated hereditary spastic paraplegia) patient survival are unaffected. Complicated hereditary spastic paraplegia or complex hereditary spastic paraplegia can also be associated with extrapyramidal symptoms, cerebellar or cognitive dysfunction and peripheral neuropathy. The survival of complex hereditary spastic paraplegia patients is often related to the severity of other non-limb symptoms 53, 12, 54.
Symptoms of hereditary spastic paraplegia vary from mild to severe. Individuals with severe symptoms may be unable to walk independently. In general, hereditary spastic paraplegia does not shorten lifespan.
- Spinal Cord Injury (SCI): Condition Information. https://www.nichd.nih.gov/health/topics/spinalinjury/conditioninfo/default[↩][↩]
- Levels of Injury. http://www.spinalinjury101.org/details/levels-of-injury[↩]
- Elsayed LEO, Eltazi IZ, Ahmed AE, Stevanin G. Insights into Clinical, Genetic, and Pathological Aspects of Hereditary Spastic Paraplegias: A Comprehensive Overview. Front Mol Biosci. 2021 Nov 26;8:690899. doi: 10.3389/fmolb.2021.690899[↩]
- Yu W, He J, Liu X, Wu J, Cai X, Zhang Y, Liu X, Fan D. Clinical features and genetic spectrum of Chinese patients with hereditary spastic paraplegia: A 14-year study. Front Genet. 2023 Feb 27;14:1085442. doi: 10.3389/fgene.2023.1085442[↩]
- van Gelder LMA, Bonci T, Buckley EE, Price K, Salis F, Hadjivassiliou M, Mazzà C, Hewamadduma C. A Single-Sensor Approach to Quantify Gait in Patients with Hereditary Spastic Paraplegia. Sensors (Basel). 2023 Jul 20;23(14):6563. doi: 10.3390/s23146563[↩]
- Regensburger M, Spatz IT, Ollenschläger M, Martindale CF, Lindeburg P, Kohl Z, Eskofier B, Klucken J, Schüle R, Klebe S, Winkler J, Gaßner H. Inertial Gait Sensors to Measure Mobility and Functioning in Hereditary Spastic Paraplegia: A Cross-Sectional Multicenter Clinical Study. Neurology. 2022 Jun 6;99(10):e1079–89. doi: 10.1212/WNL.0000000000200819[↩]
- Siow SF, Yeow D, Rudaks LI, Jia F, Wali G, Sue CM, Kumar KR. Outcome Measures and Biomarkers for Clinical Trials in Hereditary Spastic Paraplegia: A Scoping Review. Genes (Basel). 2023 Sep 3;14(9):1756. doi: 10.3390/genes14091756[↩]
- Toupenet Marchesi L, Leblanc M, Stevanin G. Current Knowledge of Endolysosomal and Autophagy Defects in Hereditary Spastic Paraplegia. Cells. 2021 Jul 2;10(7):1678. doi: 10.3390/cells10071678[↩]
- Meyyazhagan A, Orlacchio A. Hereditary Spastic Paraplegia: An Update. Int J Mol Sci. 2022 Feb 1;23(3):1697. doi: 10.3390/ijms23031697[↩][↩][↩][↩][↩]
- De Beukelaer N., Bar-On L., Hanssen B., Peeters N., Prinsen S., Ortibus E., Desloovere K., Van Campenhout A. Muscle characteristics in pediatric hereditary spastic paraplegia vs. bilateral spastic cerebral palsy: An exploratory study. Front. Neurol. 2021;12:635032. doi: 10.3389/fneur.2021.635032[↩]
- Hedera P. Hereditary Spastic Paraplegia Overview. 2000 Aug 15 [Updated 2021 Feb 11]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2023. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1509[↩]
- Fink J. K. (2013). Hereditary spastic paraplegia: Clinico-pathologic features and emerging molecular mechanisms. ACTA NEUROPATHOL. 126 (3), 307–328. 10.1007/s00401-013-1115-8[↩][↩]
- Shi Y., Wang A., Chen B., Wang X., Niu S., Li W., et al. (2022). Clinical features and genetic spectrum of patients with clinically suspected hereditary progressive spastic paraplegia. Front. Neurol. 13, 872927. 10.3389/fneur.2022.872927[↩]
- Kerstens H.C.J.W., Lith B.J.H.v., Nijkrake M.J., Swart B.J.M.d., Bemd L.A.C.v.d., Smeets R., Klemens F., Warrenburg B.P.C.v.d., Wees P.J.v.d., Geurts A.C.H. Healthcare needs, expectations, utilization, and experienced treatment effects in patients with hereditary spastic paraplegia: A web-based survey in the Netherlands. Orphanet J. Rare Dis. 2021;16:1–283. doi: 10.1186/s13023-021-01915-0[↩]
- Hereditary Spastic Paraplegia. https://rarediseases.org/rare-diseases/hereditary-spastic-paraplegia[↩][↩]
- Koh K., Ishiura H., Tsuji S., Takiyama Y. JASPAC: Japan spastic paraplegia research consortium. Brain Sci. 2018;8:153. doi: 10.3390/brainsci8080153[↩]
- Schüle R., Wiethoff S., Martus P., Karle K.N., Otto S., Klebe S., Klimpe S., Gallenmüller C., Kurzwelly D., Henkel D., et al. Hereditary spastic paraplegia: Clinicogenetic lessons from 608 patients. Ann. Neurol. 2016;79:646–658. doi: 10.1002/ana.24611[↩]
- Harding A.E. Classification of the hereditary ataxias and paraplegias. Lancet. 1983;1:1151–1155. doi: 10.1016/S0140-6736(83)92879-9[↩]
- Blackstone C. Hereditary spastic paraplegia. Handb Clin Neurol. 2018;148:633-652. doi: 10.1016/B978-0-444-64076-5.00041-7[↩][↩][↩]
- Shribman S., Reid E., Crosby A.H., Houlden H., Warner T.T. Hereditary spastic paraplegia: From diagnosis to emerging therapeutic approaches. Lancet Neurol. 2019;18:1136–1146. doi: 10.1016/S1474-4422(19)30235-2[↩][↩][↩][↩][↩][↩][↩][↩]
- Ruano L., Melo C., Silva M.C., Coutinho P. The Global Epidemiology of Hereditary Ataxia and Spastic Paraplegia: A Systematic Review of Prevalence Studies. Neuroepidemiology. 2014;42:174–183. doi: 10.1159/000358801[↩]
- Erichsen A.K., Koht J., Stray-Pedersen A., Abdelnoor M., Tallaksen C.M.E. Prevalence of hereditary ataxia and spastic paraplegia in southeast Norway: A population-based study. Brain. 2009;132:1577–1588. doi: 10.1093/brain/awp056[↩]
- Coutinho P., Ruano L., Loureiro J.L., Cruz V.T., Barros J., Tuna A., Barbot C., Guimarães J., Alonso I., Silveira I., et al. Hereditary ataxia and spastic paraplegia in Portugal: A population-based prevalence study. JAMA Neurol. 2013;70:746–755. doi: 10.1001/jamaneurol.2013.1707[↩]
- Vander Stichele G., Durr A., Yoon G., Schüle R., Blackstone C., Esposito G., Buffel C., Oliveira I., Freitag C., van Rooijen S., et al. An integrated modelling methodology for estimating global incidence and prevalence of hereditary spastic paraplegia subtypes SPG4, SPG7, SPG11, and SPG15. BMC Neurol. 2022;22:115. doi: 10.1186/s12883-022-02595-4[↩]
- Tesson C., Koht J., Stevanin G. Delving into the complexity of hereditary spastic paraplegias: How unexpected phenotypes and inheritance modes are revolutionizing their nosology. Hum. Genet. 2015;134:511–538. doi: 10.1007/s00439-015-1536-7[↩]
- Beetz C., Pieber T.R., Hertel N., Schabhüttl M., Fischer C., Trajanoski S., Graf E., Keiner S., Kurth I., Wieland T., et al. Exome sequencing identifies a REEP1 mutation involved in distal hereditary motor neuropathy type v. Am. J. Hum. Genet. 2012;91:139–145. doi: 10.1016/j.ajhg.2012.05.007[↩]
- Ruano L., Melo C., Silva M.C., Coutinho P. The global epidemiology of hereditary ataxia and spastic paraplegia: A systematic review of prevalence studies. Neuroepidemiology. 2014;42:174–183. doi: 10.1159/000358801[↩]
- Novarino G., Fenstermaker A.G., Zaki M.S., Hofree M., Silhavy J.L., Heiberg A.D., Abdellateef M., Rosti B., Scott E., Mansour L., et al. Exome sequencing links corticospinal motor neuron disease to common neurodegenerative disorders. Science. 2014;343:506–511. doi: 10.1126/science.1247363[↩]
- Klebe S., Stevanin G., Depienne C. Clinical and genetic heterogeneity in hereditary spastic paraplegias: From SPG1 to SPG72 and still counting. Rev. Neurol. 2015;171:505–530. doi: 10.1016/j.neurol.2015.02.017[↩]
- Schüle R., Siddique S., Deng X., Yang Y., Donkervoort S., Hansson M., Madrid R.E., Siddique N., Schöls L., Björkhem I. Marked accumulation of 27-hydroxycholesterol in SPG5 patients with hereditary spastic paresis. J. Lipid Res. 2010;51:819–823. doi: 10.1194/jlr.M002543[↩]
- Casari G., Marconi R. Spastic Paraplegia. Br. Med. J. 2018;1:642. doi: 10.1136/bmj.1.3456.642[↩]
- Verny C., Guegen N., Desquiret V., Chevrollier A., Prundean A., Dubas F., Cassereau J., Ferre M., Amati-Bonneau P., Bonneau D., et al. Hereditary spastic paraplegia-like disorder due to a mitochondrial ATP6 gene point mutation. Mitochondrion. 2011;11:70–75. doi: 10.1016/j.mito.2010.07.006[↩]
- Sánchez-Ferrero E., Coto E., Corao A.I., Díaz M., Gámez J., Esteban J., Gonzalo J.F., Pascual-Pascual S.I., De Munaín A.L., Morís G., et al. Mitochondrial DNA polymorphisms/haplogroups in hereditary spastic paraplegia. J. Neurol. 2012;259:246–250. doi: 10.1007/s00415-011-6155-1[↩]
- Angelini, Corrado. (2014). Spastic Paraparesis Type 7. 10.1007/978-3-319-07500-6_82[↩]
- Fink JK. Hereditary spastic paraplegia: clinico-pathologic features and emerging molecular mechanisms. Acta Neuropathol. 2013 Sep;126(3):307-28. doi: 10.1007/s00401-013-1115-8[↩]
- Novarino G, Fenstermaker AG, Zaki MS, et al. Exome sequencing links corticospinal motor neuron disease to common neurodegenerative disorders. Science. 2014 Jan 31;343(6170):506-511. doi: 10.1126/science.1247363[↩]
- Harding A. E. (1983). Classification of the hereditary ataxias and paraplegias. LANCET 1 (8334), 1151–1155. 10.1016/s0140-6736(83)92879-9[↩]
- Fink JK. Upper Motor Neuron Disorders: Hereditary Spastic Paraplegia and Primary Lateral Sclerosis. In: Johnson MV, Adams HP, Fatemi A, eds. Neurobiology of Disease.Oxford University Press, 2016.[↩][↩]
- Fink JK. The Hereditary Spastic Paraplegias. In: Rosenberg R, ed. Molecular and Genetic Basis of Neurologic and Psychiatric Disease, 5th edition ed Lippincott Williams & Wilkins, 2014.[↩][↩]
- Hedera P, Eldevik OP, Maly P, Rainier S, Fink JK. Spinal cord magnetic resonance imaging in autosomal dominant hereditary spastic paraplegia. Neuroradiology. 2005 Oct;47(10):730-4. doi: 10.1007/s00234-005-1415-3[↩]
- Hensiek A., Kirker S., Reid E. Diagnosis, investigation and management of hereditary spastic paraplegias in the era of next-generation sequencing. J. Neurol. 2015;272:1601–1612. doi: 10.1007/s00415-014-7598-y[↩]
- Boone P.M., Liu P., Zhang F., Carvalho C.M., Towne C.F., Batish S.D., Lupski J.R. Alu-specific microhomology-mediated deletion of the final exon of SPAST in three unrelated subjects with hereditary spastic paraplegia. Genet. Med. 2011;13:582–592. doi: 10.1097/GIM.0b013e3182106775[↩]
- Julien C, Lissouba A, Madabattula S, Fardghassemi Y, Rosenfelt C, Androschuk A, Strautman J, Wong C, Bysice A, O’sullivan J, Rouleau GA, Drapeau P, Parker JA, Bolduc FV. Conserved pharmacological rescue of hereditary spastic paraplegia-related phenotypes across model organisms. Hum Mol Genet. 2016 Mar 15;25(6):1088-99. doi: 10.1093/hmg/ddv632[↩]
- Fink J.K. Hereditary spastic paraplegia: Clinico-pathologic features and emerging molecular mechanisms. Acta Neuropathol. 2013;126:307–328. doi: 10.1007/s00401-013-1115-8[↩]
- Margetis K., Korfias S., Boutos N., Gatzonis S., Themistocleous M., Siatouni A., Dalivigka Z., Flaskas T., Stranjalis G., Boviatsis E., et al. Intrathecal baclofen therapy for the symptomatic treatment of hereditary spastic paraplegia. Clin. Neurol. Neurosurg. 2014;123:142–145. doi: 10.1016/j.clineuro.2014.05.024[↩]
- De Niet M., De Bot S.T., Van De Warrenburg B.P.C., Weerdesteyn V., Geurts A.C. Functional efects of botulinum toxin type-A treatment and subsequent stretching of spastic calf muscles: A study in patients with hereditary spastic paraplegia. J. Rehabil. Med. 2015;47:147–153. doi: 10.2340/16501977-1909[↩]
- Servelhere K.R., Faber I., Martinez A., Nickel R., Moro A., Germiniani F.M.B., Moscovich M., Blume T.R., Munhoz R.P., Teive H.A.G., et al. Botulinum toxin for hereditary spastic paraplegia: Effects on motor and non-motor manifestations. Arq. Neuropsiquiatr. 2018;76:183–188. doi: 10.1590/0004-282×20180013[↩]
- Béreau M., Anheim M., Chanson J.B., Tio G., Echaniz-Laguna A., Depienne C., Collongues N., de Sèze J. Dalfampridine in hereditary spastic paraplegia: A prospective, open study. J. Neurol. 2015;262:1285–1288. doi: 10.1007/s00415-015-7707-6[↩]
- Mohan N., Qiang L., Morfini G., Baas P.W. Therapeutic Strategies for Mutant SPAST-Based Hereditary Spastic Paraplegia. Brain Sci. 2021;11:1081. doi: 10.3390/brainsci11081081[↩]
- Havlicek S., Kohl Z., Mishra H.K., Prots I., Eberhardt E., Denguir N., Wend H., Plötz S., Boyer L., Marchetto M.C.N., et al. Gene dosage-dependent rescue of HSP neurite defects in SPG4 patients’ neurons. Hum. Mol. Genet. 2014;23:2527–2541. doi: 10.1093/hmg/ddt644[↩]
- Xue H., Yu A., Chen X., Lin N., Lin M., Huang H., Xu L. Prenatal diagnosis of PLP1 duplication by single nucleotide polymorphism array in a family with Pelizaeus-Merzbacher disease. Aging. 2021;13:1488–1497. doi: 10.18632/aging.202477[↩]
- Pirozzi M., Quattrini A., Andolfi G., Dina G., Malaguti M.C., Auricchio A., Rugarli E.I. Intramuscular viral delivery of paraplegin rescues peripheral axonopathy in a model of hereditary spastic paraplegia. J. Clin. Investig. 2006;116:202–208. doi: 10.1172/JCI26210[↩]
- Coutinho P., Ruano L., Loureiro J. L., Cruz V. T., Barros J., Tuna A., et al. (2013). Hereditary ataxia and spastic paraplegia in Portugal: A population-based prevalence study. JAMA NEUROL. 70 (6), 746–755. 10.1001/jamaneurol.2013.1707[↩]
- Dong E., Wang C., Wu S., Lu Y. Q., Lin X. H., Su H. Z., et al. (2018). Clinical spectrum and genetic landscape for hereditary spastic paraplegias in China. Mol. Neurodegener. 13 (1), 36. 10.1186/s13024-018-0269-1[↩]


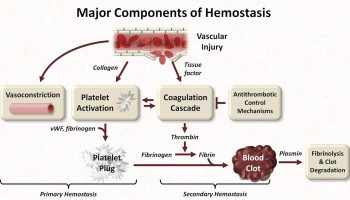
{kind=link}