Contents
What is parkinsonism
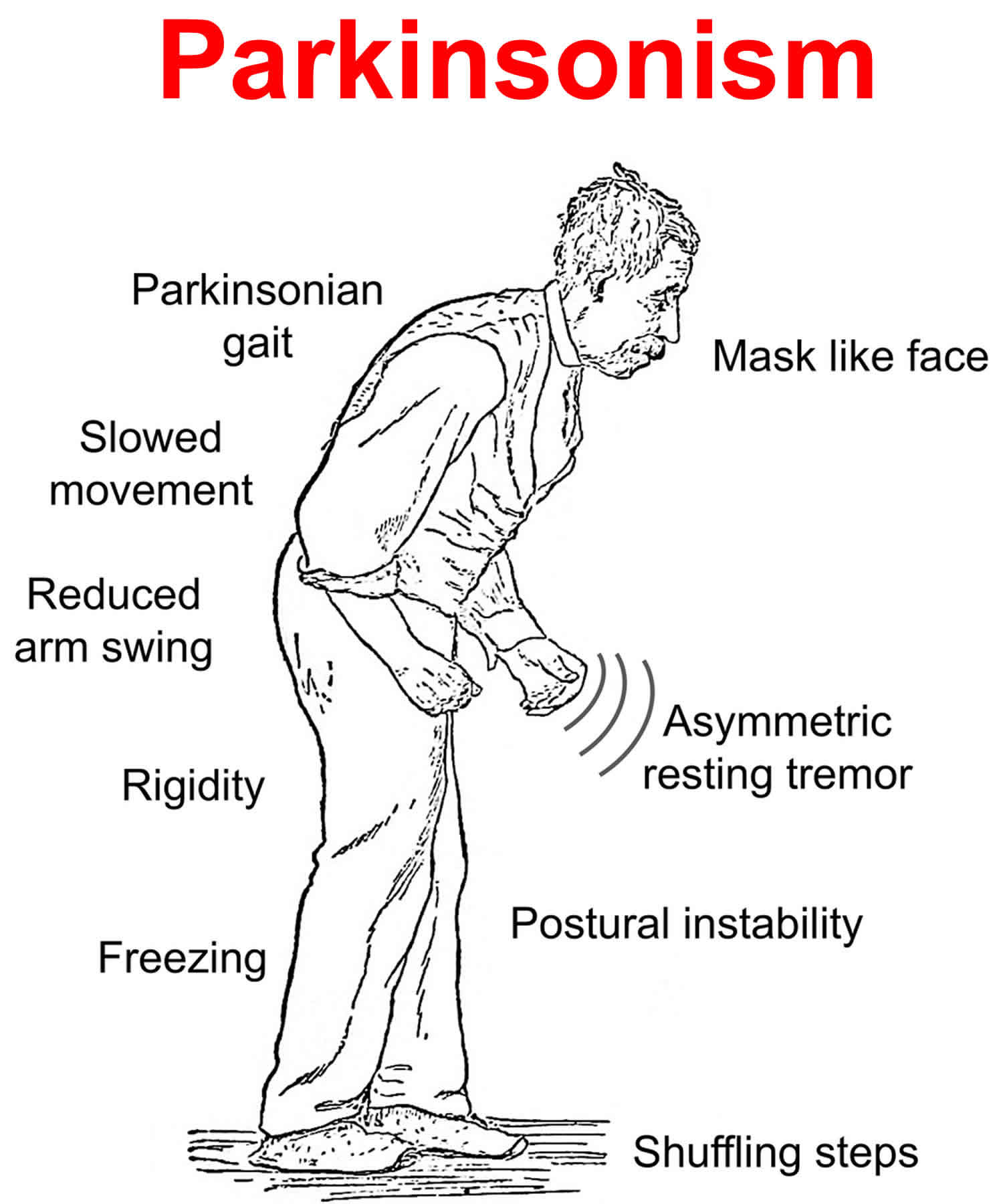
Parkinsonism refers to a group of movement abnormalities — such as impaired speech or muscle stiffness, slow movement (bradykinesia), shuffling of the feet and often tremor — that are classic features of Parkinson’s disease but that can also be caused by medications and other disorders with overlapping symptoms. Parkinsonism also known as atypical Parkinson’s disease or Parkinson’s plus, represent about 10-15% of all diagnosed cases of parkinsonism. These syndromes tend to progress more rapidly than Parkinson’s disease, present with additional symptoms such as early falling, dementia or hallucinations, and do not respond or respond only for a short time to levodopa therapy.
Types of parkinsonisms:
- Corticobasal syndrome
- Dementia with Lewy bodies
- Drug-induced Parkinsonism
- Multiple System Atrophy (MSA)
- Progressive Supranuclear Palsy (PSP)
- Vascular Parkinsonism
Following are descriptions of some of the most common parkinsonism disorders. It is important to remember that many people will not exhibit the cardinal symptoms necessary for a diagnosis of a specific disorder and will simply be labeled “parkinsonism”. For these people a definite diagnosis will only come if the family requests a brain autopsy at time of death.
Drug-Induced Parkinsonism
Drug-induced parkinsonism also known as secondary parkinsonism, is the second-most-common cause of parkinsonism in the elderly after Parkinson’s disease 1. Many patients with drug-induced parkinsonism may be misdiagnosed with Parkinson’s disease because the clinical features of these two conditions are indistinguishable, though the tremors and postural instability may be less severe 2. Moreover, neurological deficits in patients with drug-induced parkinsonism may be severe enough to affect daily activities and may persist for long periods of time after the cessation of drug taking. Drug-induced parkinsonism is usually the side effect of drugs that affect dopamine levels in the brain, such as atypical antipsychotics, gastrointestinal prokinetics, some calcium channel blockers and stimulants like amphetamines and cocaine and antiepileptic drugs. If the affected person stops taking the drug(s), symptoms usually subside over time, but may take as long as 18 months to do so.
The clinical manifestations of drug-induced parkinsonism are classically described as bilateral and symmetric parkinsonism without tremor at rest. However, about half of drug-induced parkinsonism patients show asymmetrical parkinsonism and tremor at rest, making it difficult to differentiate drug-induced parkinsonism from Parkinson’s disease. The pathophysiology of drug-induced parkinsonism is related to drug-induced changes in the basal ganglia motor circuit secondary to dopaminergic receptor blockade. Since these effects are limited to postsynaptic dopaminergic receptors, it is expected that presynaptic dopaminergic neurons in the striatum will be intact. Dopamine transporter (DAT) imaging is useful for diagnosing presynaptic parkinsonism. Dopamine transporter uptake in the striatum is significantly decreased even in the early stage of Parkinson’s disease, and this characteristic may help in differentiating Parkinson’s disease from drug-induced parkinsonism. Drug-induced parkinsonism may have a significant and longstanding effect on patients’ daily lives, and so physicians should be cautious when prescribing dopaminergic receptor blockers and should monitor patients’ neurological signs, especially for parkinsonism and other movement disorders.
Drug-induced parkinsonism causes
Typical antipsychotics, also known as neuroleptics, are the most common causes of drug-induced parkinsonism. However, atypical antipsychotics, which were thought to be free from extrapyramidal side effects, can also induce parkinsonism. In addition to antipsychotics, gastrointestinal (GI) motility drugs, calcium channel blockers, and antiepileptic drugs have been found to induce drug-induced parkinsonism (Table 1).
Table 1. Common offending drugs of drug-induced parkinsonism
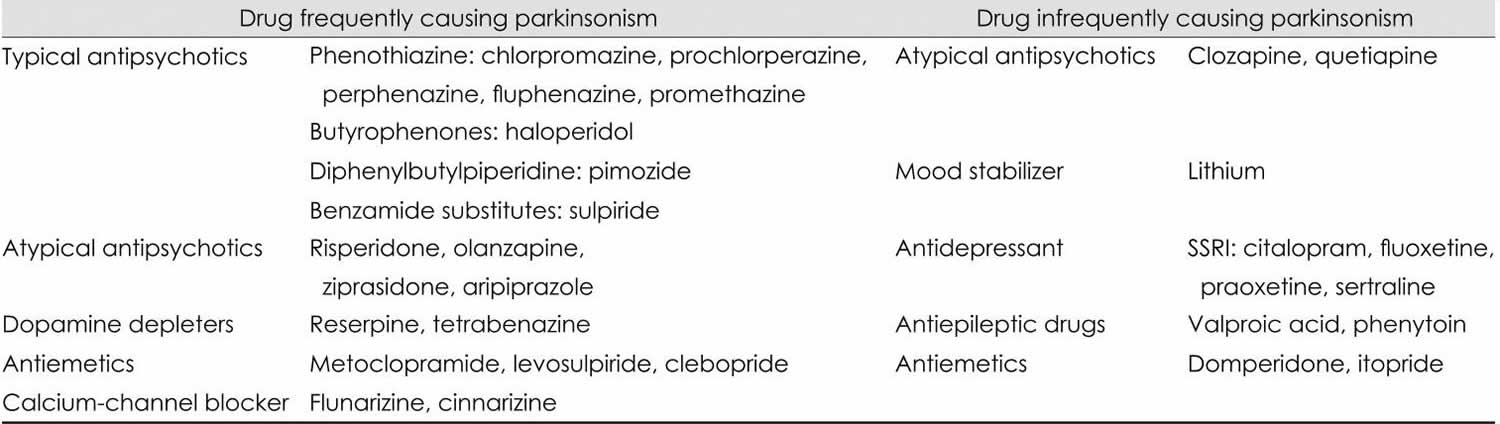
Abbreviation: SSRI = Selective serotonin reuptake inhibitor.
[Source 1 ]Drug-induced parkinsonism symptoms
Drug-induced parkinsonism is generally characterized clinically as bilateral and symmetric parkinsonism, with more prominent bradykinesia and rigidity than in patients with Parkinson’s disease 3. However, it has been suggested in many studies that clinical manifestations alone cannot be used to differentiate drug-induced parkinsonism from Parkinson’s disease 1.
Typically 30-50% of patients with drug-induced parkinsonism show asymmetric parkinsonism and tremor at rest; these characteristics are considered to support a Parkinson’s disease diagnosis 4. Interestingly, drug-induced parkinsonism patients with typical tremor at rest tremors usually also have postural tremors. The similar clinical manifestations of drug-induced parkinsonism and Parkinson’s disease indicate that patients with drug-induced parkinsonism may have been in a preclinical stage of Parkinson’s disease and that their parkinsonism may have been unmasked by the offending drugs. This is supported by findings that parkinsonism persists or even progresses after cessation of the drug in many drug-induced parkinsonism patients 4. In a long-term follow-up study, parkinsonism reappeared several months after the complete resolution from drug-induced parkinsonism in 7% of patients 4; drug-induced parkinsonism might have anteceded Parkinson’s disease in those patients.
The mean age at symptom onset is greater for drug-induced parkinsonism than for Parkinson’s disease patients. In addition, drug-induced parkinsonism is more common in females, wh-ereas males are more likely to have Parkinson’s disease. While the duration of symptoms is similar, neurological deficits are more severe in drug-induced parkinsonism than in Parkinson’s disease. Orolingual dyskinesia in drug-naïve Parkinson’s disease patients is rare, whereas it is frequently combined with parkinsonism in drug-induced parkinsonism patients. The symmetry and type of tremor are not useful in distinguishing drug-induced parkinsonism from Parkinson’s disease.
Drug-induced parkinsonism diagnosis
The clinical diagnostic criteria for drug-induced parkinsonism are defined as 1) the presence of parkinsonism, 2) no history of parkinsonism before the use of the offending drug, and 3) onset of parkinsonian symptoms during use of the offending drug 1. Since asymmetrical rest tremors are common in many drug-induced parkinsonism patients and symptoms persist or progress after cessation of the offending drug, patients clinically diagnosed with drug-induced parkinsonism may include individuals in the preclinical stage of Parkinson’s disease whose symptoms were unmasked by the drug 4.
Dopamine transporters (DATs) are presynaptic proteins in the membrane on terminals of dopaminergic neurons. They take up dopamine from the synaptic cleft projections that extend from the substantia nigra to the striatum. These transporters control dopaminergic transmission by spatial and temporal buffering, rendering the molecule an imaging target in diseases affecting the dopaminergic nigrostriatal pathway. Single-photon-emission computed tomography (SPECT) and positron-emission tomography (PET) scans are available using several dopamine transporter ligands 5. SPECT radioligands include 123I-N-3-fluoropropyl-2β-carbomethoxy-3β-(4-iodophenyl)nortropane (123I-FP-CIT or 123I-β-CIT-FP), 123I-ioflupane, DaTSCAN, and 123I-2β-carbomethoxy-3β-(4-iodophenyl)tropane (123I-β-CIT) 6. PET scans may be superior to SPECT for imaging DATs, in that the lower energy of positrons provides higher resolution, resulting in better image quality with widespread clinical applications 7. However, most DAT imaging studies, including those in patients with drug-induced parkinsonism, have utilized SPECT 8.
DAT uptake in the striatum is significantly decreased in Parkinson’s disease patients, even during the early stages of the disease, because the motor symptoms of Parkinson’s disease do not appear until 60-80% of dopaminergic neurons degenerate 9. In addition, drugs causing parkinsonism, such as DRBAs, have negligible affinity for dopamine transporter (DAT) 10. Dopamine transporter (DAT) scans may show symmetric uptake of radiotracer in the bilateral striatum in patients with pure drug-induced parkinsonism, even if they have significant parkinsonism. Parkinson’s disease can be diagnosed in drug-induced parkinsonism patients whose DAT uptake decreases asymmetrically in the striatum. Therefore, dopamine transporter (DAT) scans may be useful for differentiating Parkinson’s disease unmasked by drugs from pure drug-induced parkinsonism. Follow-up dopamine transporter (DAT) scans of drug-induced parkinsonism patients with initially normal DAT scans exhibited normal uptake in the striatum, whereas scans of patients with initially decreased DAT scans in the striatum exhibited progressive reduction of striatal tracer uptake 8. These findings suggest that DAT imaging provides useful information for diagnosing drug-induced parkinsonism and may help in the design of a therapeutic plan. Further studies involving larger numbers of patients are needed to determine the sensitivity and specificity of DAT scans in drug-induced parkinsonism diagnosis.
Drug-induced parkinsonism treatment
Drug-induced parkinsonism is generally treated by cessation of the offending drugs. Patients who cannot stop taking antipsychotic drugs because of their psychiatric diseases, such as those with schizophrenia or major depressive disorders, may be switched to atypical antipsychotics that have a lower risk of EPS. People who are prescribed dopamine antagonists due to simple GI disturbance, headache, dizziness, or insomnia should stop taking the offending drugs as soon as possible. Anticholinergics including trihexyphenidyl, benztropine, amantadine, and levodopa have been empirically tested for their ability to relieve symptoms of drug-induced parkinsonism, but this has produced no clear evidence of their effects in drug-induced parkinsonism patients 11.
Drug-induced parkinsonism usually resolves within weeks to months after stopping the offending drug; however, parkinsonism may persist or progress in 10-50% of patients. The prognosis of patients with drug-induced parkinsonism can be classified into the following types: 1) full and long-lasting recovery from drug-induced parkinsonism with no subsequent development of parkinsonism, 2) persistence but not progression of parkinsonism, 3) persistence and eventual worsening of parkinsonism, and 4) full remission of parkinsonism but later reappearance after discontinuation of the offending drug. Only patients classified as type 1 can be defined as having ‘pure drug-induced parkinsonism’, whereas those classified as type 3 or 4 may be in the preclinical stages of Parkinson’s disease 10. The finding of Lewy bodies in two drug-induced parkinsonism patients who had completely recovered after stopping the offending drugs 12 suggests that clinical course alone cannot distinguish ‘pure drug-induced parkinsonism’ among patients clinically diagnosed with drug-induced parkinsonism. In drug-induced parkinsonism patients classified as type 2, the persistence of parkinsonism may be due to permanent dopamine receptor blocking agent-induced damage to dopamine receptors 13. Dopamine transporter (DAT) imaging may be useful in diagnosing drug-induced parkinsonism patients and predicting their clinical course. A large prospective study may provide more information on the exact prognosis and factors related to recovery in drug-induced parkinsonism patients.
Progressive Supranuclear Palsy
Progressive supranuclear palsy (PSP) is an uncommon neurological disorder that affects movement, gait, balance, speech, swallowing, vision, eye movements, mood, behavior, and cognition. Steele, Richardson, and Olszewski described the syndrome in 1964 as an unusual constellation of supranuclear gaze palsy, progressive axial rigidity, pseudobulbar palsy, and mild dementia 14. This disease is now a well-recognized atypical parkinsonian syndrome or Parkinson-plus disorder 15.
In 1972, Steele predicted clinical variants of the syndrome were likely to occur as the disease affected different brainstem nuclei at different times and to different degrees 16 Since then, different phenotypes have been characterized and have been linked to the severity of abnormal tau accumulation and neuronal loss in various brain regions 17. Different progressive supranuclear palsy, regardless of clinical characteristics, share similar neuropathologic features 18.
Progressive supranuclear palsy key facts
- Unlike typical Parkinson disease, falls begin within the first year of progressive supranuclear palsy, and by year 3, they are common unless precautions are taken to prevent them.
- Downgaze is affected before upgaze; whereas; lateral eye movements are usually preserved in progressive supranuclear palsy.
- Apathy seems to predominate over other neurobehavioral abnormalities in progressive supranuclear palsy.
- Patients with progressive supranuclear palsy develop frontal cognitive impairment.
- The pathology of progressive supranuclear palsy is characterized by widespread neurodegeneration associated with tau protein deposition in subcortical regions.
- The first therapeutic step in progressive supranuclear palsy is identifying and prioritizing the symptoms that can be treated.
Progressive supranuclear palsy (PSP) is slightly more common than amyotrophic lateral sclerosis (also called ALS or Lou Gehrig disease). Progressive supranuclear palsy symptoms usually begin in the early 60’s. Common early symptoms include loss of balance while walking that results in unexplained falls, forgetfulness and personality changes. The visual problems associated with progressive supranuclear palsy generally occur 3 to 5 years after the walking
problems and involve the inability to aim the eyes properly because of weakness or paralysis of the muscles that move the eyeballs. Individuals with progressive supranuclear palsy (PSP) may have some response to dopaminergic treatment but may require higher doses than patients with Parkinson’s disease.
The cause of progressive supranuclear palsy is unknown. Advanced age 19 and environmental factors such as exposure to toxins 20.
The defining histopathologic feature of progressive supranuclear palsy is an intracerebral aggregation of the microtubule-associated protein tau with preferential involvement of the subthalamic nucleus, pallidum, striatum, red nucleus, substantia nigra, pontine tegmentum, oculomotor nucleus, medulla, and dentate nucleus 17. The aggregates predominantly contain tau isoforms with four microtubule-binding repeats (4R-tau) in neurofibrillary tangles, oligodendrocytic coils, and astrocytic tufts 21. Normally, tau is phosphorylated on a series of serine and threonine residues, regulated by numerous kinases and phosphatases. In progressive supranuclear palsy and other tauopathies, the tau protein is hyperphosphorylated, which causes it to lose its affinity for microtubules and become resistant to proteolysis. This results in the accumulation of tau and the formation of neurofibrillary tangles 22. Definite diagnosis of progressive supranuclear palsy currently requires neuropathological examination 18.
Macroscopic pathology
The gross examination of the brain in progressive supranuclear palsy often shows atrophy of the frontal lobe (especially the precentral gyrus), midbrain, (especially the tectum), and to a lesser degree, the pons. The subthalamic nucleus is smaller than expected and may be discolored gray 23. The superior cerebellar peduncle and the hilum of the cerebellar dentate nucleus are usually atrophic and have a gray discoloration due to myelinated fiber loss.
Natural history of progressive supranuclear palsy
The progression of disease and accumulation of disability in progressive supranuclear palsy is more rapid and severe than in Parkinson’s disease, even given the variability described among the different clinical subtypes. The mean age at diagnosis is 65 years 24. Frequent falls, causing fractures and head injuries, contribute substantially to morbidity and may be minimized by physical therapy, use of a weighted walker, and eventually wheelchair use. The terminal stages of disease are characterized by severe communication difficulties, immobility, severe axial rigidity, severe dysphagia and complete ophthalmoplegia (particularly in progressive supranuclear palsy-Richardson syndrome). As in Parkinson’s disease, the mode of death is most often related to respiratory compromise in the setting of bronchopneumonia.
Progressive supranuclear palsy treatment
The management of the cognitive, motor and gait aspects of progressive supranuclear palsy is challenging and the treatment for individuals suspected to have progressive supranuclear palsy remains symptomatic and supportive, with ongoing clinical trials striving to identify disease-modifying therapies often targeting the underlying tau pathology. The active treatment of progressive supranuclear palsy is directed at optimizing function and alleviating suffering. Supportive therapy using pharmacologic approaches and rehabilitation by a multidisciplinary team is usual. For motor (parkinsonian) symptoms, levodopa combined with a dopa decarboxylase inhibitor (e.g., carbidopa) is generally tried, with typically modest to no success in most progressive supranuclear palsy phenotypes but potential benefit in the progressive supranuclear palsy-parkinsonism predominance type. Overall, evidence for mild to moderate benefits with levodopa is low 25, but given limited therapeutic options, levodopa is generally tried at doses of up to 1000 mg daily. Other dopaminergic agents are rarely of benefit; amantadine is sometimes tried with limited supportive evidence 26. Botulinum toxin injections can be used for focal dystonias including apraxia of eyelid opening 26.
The multidisciplinary team should include the following:
- Physiotherapist-to assess mobility, with a view to prescribing gait aides when necessary, and instruct on techniques for safe transfers
- Occupational therapist-to perform an environmental assessment, including the need for lifting devices or wheeled mobility aides, and optimize upper limb function
- Speech pathologist-to treat difficulties with communication (eg, speech amplifiers or communication boards) and monitor swallowing (modified barium swallow test), with a view to prescribing increased consistencies of food when required
- Nurse or social worker-to provide support and liaison for strategies to manage medication delivery and coordinate the provision of home care and support as required
Other aspects of care that should be considered include counseling (to assist in coming to terms with the diagnosis of a neurodegenerative condition and end-of-life decision making) and palliative care (for appropriate nursing near the end of disease).
The potential value of physical therapy is of increasing interest particularly given evidence of benefit for individuals with Parkinson disease, and a recent trial showed improvement in the Progressive Supranuclear Palsy Rating Scale 27. Another study also suggested potential benefit of the Lee Silverman Voice Treatment in individuals with progressive supranuclear palsy, though benefits in progressive supranuclear palsy were less frequently significant than those observed in Parkinson disease patients 28. Physical and occupational therapy with particular attention to balance training and “learning to fall” can minimize the potential for injury and provide patients with tools for maneuvering after a fall has occurred. Counseling on food preparation and swallowing to avoid aspiration is indicated in many. The use of a walker is often necessary for safe ambulation and transfers. At late stages, even using a walker is not enough to maintain balance and patients may need to use a wheelchair exclusively.
While case reports and series suggest promising experiences with unilateral or bilateral pedunculopontine nucleus deep brain stimulation (DBS) in patients with suspected progressive supranuclear palsy, a recently published randomized controlled trial of unilateral pedunculopontine nucleus deep brain stimulation (DBS) in 8 individuals with PSP-Richardson’s syndrome showed no benefit in gait, postural stability, and fall PSP-Richardson’s syndrome subitems when comparing ON and OFF stimulation conditions at 6- and 12-month follow-up 29. Deep brain stimulation (DBS) is currently not recommended for progressive supranuclear palsy outside of research settings 25.
There are no accepted treatments for cognitive symptoms in individuals with suspected progressive supranuclear palsy, with small trials and case series of cholinesterase inhibitors suggesting that these drugs may help cognition but worsen motor function 25. It is critical to address potentially treatable symptoms in progressive supranuclear palsy such as depression, but no progressive supranuclear palsy-specific recommendations for such symptomatic management exist.
To date, studies of potentially disease-modifying therapies have failed to demonstrate efficacy in individuals suspected to have progressive supranuclear palsy. Randomized, placebo-controlled trials of riluzole 30, davunetide 31 tideglusib 32, sodium valproate 33 and rasagiline 34 showed no impact on primary endpoints tracking disease progression, though study limitations include sample size (for some studies) and lack of evidence that the agents had the intended effect through theorized mechanisms. Though there have been negative double-blind studies of CoQ10 35, there has been a positive double-blind study using the liposomal form 36.
Regardless of investigational and symptomatic treatment approaches used through the disease course, palliative care is an important component of progressive supranuclear palsy treatment with hospice as a valuable resource in late stages.
Current investigations of tau-focused progressive supranuclear palsy therapies include TPI-287, a microtubule stabilizer, C2N-8E12/ABBV-8E12 and BMS-986168/BIIB092, both anti-tau monoclonal antibodies, and salsalate, a tau acetylation inhibitor. Microtubule stabilizers are hoped to compensate for microtubule dysfunction associated with loss of tau function; anti-tau monoclonal antibodies are hoped to impede the spread of pathogenic tau, and tau acetylation inhibitors are hoped to inhibit acetylation of soluble tau and thus limit hyperphosphorylation.
Progressive supranuclear palsy prognosis
Disease progression in progressive supranuclear palsy usually occurs fairly rapidly and relentlessly 37. Most patients become dependent on care within 3 or 4 years from presentation. The disorder culminates in death at a median of 6 to 9 years after the diagnosis 38. In addition, quality of life is significantly reduced 27.
In a 2017 systematic review and meta-analysis, predictors of shorter survival included the progressive supranuclear palsy-Richardson syndrome (PSP-RS) phenotype compared with the progressive supranuclear palsy-parkinsonism (PSP-P) phenotype, early falls, and early cognitive symptoms (both more common in the progressive supranuclear palsy-Richardson syndrome phenotype) 25. Early onset of dysphagia, which is seen in both the progressive supranuclear palsy-Richardson syndrome and progressive supranuclear palsy-parkinsonism phenotypes, was also predictive of shorter survival. The prognostic effect of the presence of supranuclear gaze palsy was inconsistent across studies. Levodopa response, as often seen in the early stages of the progressive supranuclear palsy-parkinsonism phenotype, did not predict longer survival.
Multiple System Atrophy
Multiple System Atrophy (MSA) also referred to as Shy-Drager syndrome, is the term for a group of disorders in which one or more systems in the body stop working. It is not unusual for a gastroenterologist, cardiologist, sleep medicine physician, and urologist to be involved in the care of multiple system atrophy (MSA) patients by the time the neurologic diagnosis is made. In multiple system atrophy (MSA) the autonomic nervous system is often severely affected early in the course of the disease. Multiple system atrophy (MSA) symptoms include bladder problems resulting in urgency, hesitancy or incontinence and orthostatic hypotension (postural hypotension). In orthostatic hypotension (postural hypotension) the blood pressure drops so low when standing that fainting or near fainting can occur. When lying down, the patient’s blood pressure can be quite high. For men, the earliest sign may be loss of erectile function. Other symptoms that may develop include impaired speech, difficulties with breathing and swallowing, and inability to sweat.
People with multiple system atrophy typically live for six to nine years after their symptoms start and may deteriorate quickly during this time. Some people may live for more than 10 years after being diagnosed.
The classic and striking clinical characteristic of multiple system atrophy is the progressive autonomic dysfunction that often dominates the early clinical picture and precedes the evolution of motor symptoms by up to several years 39. The diagnosis of multiple system atrophy is usually considered in patients who develop parkinsonism in the presence of increasing urinary urgency, constipation, postural hypotension, and erectile dysfunction in men 40. A neuroimaging feature supportive of multiple system atrophy-parkinsonism is the subtle slitlike signal abnormality of the posterolateral putamen, bilaterally or only contralateral to the more affected side, due to atrophy and excessive iron deposition at the putamen. A proportion of patients with multiple system atrophy develop a predominantly cerebellar phenotype with no or onlyvery subtle parkinsonism (multiple system atrophy-cerebellar), usually in association with autonomic dysfunction 40.
The parkinsonism of multiple system atrophy is usually symmetrical and classically responds poorly to dopaminergic therapies (althoughin approximately 30% of patients the response is good), and drug-induced dyskinesias can develop, particularly axially. Bradykinesia and rigidity progress somewhat faster than in Parkinson’s disease, and as a consequence, postural instability and falls usually emerge within the first 3 years of disease onset. Rest tremor can be present, but stimulus-sensitive myoclonus is more frequent. The gait disturbance of multiple system atrophy may be purely parkinsonian, purely cerebellar (broad based, unsteady, with truncal and upper limb ataxia giving an appearance of flailing), or a combination of both.
To aid in the clinical diagnosis, in addition to the bedside assessment of parkinsonism, postural blood pressure recordings should be taken. The patient should be assessed after lying supine for several minutes. The blood pressure and pulse rate should be taken, after which the patient stands and blood pressure and pulse is recorded after 2 to 3 minutes of standing. When autonomic failure is present, the blood pressure will fall by more than 20 mm Hg systolic and/or 10 mm Hg diastolic on standing, with no reactive increase in pulse rate 40.
Like the other parkinsonisms, multiple system atrophy (MSA) symptoms either don’t respond very much or don’t respond at all to Parkinson’s medications.
Multiple system atrophy (MSA) tends to advance rapidly over the course of 5 to 10 years, with progressive loss of motor skills, eventual confinement to bed, and death. There is no remission from the disease. There is currently no cure.
Multiple system atrophy causes
The causes of multiple system atrophy aren’t well understood. It doesn’t appear to be inherited – there’s no evidence that an affected person’s children will develop it.
However, it’s possible that both genetic and environmental factors may contribute, so research is currently looking at whether there’s a genetic tendency (predisposition) to develop it.
The brain cells of a person with multiple system atrophy contain a protein called alpha-synuclein. A build-up of abnormal alpha-synuclein is thought to be responsible for damaging areas of the brain that control balance, movement and the body’s autonomic functions.
Multiple system atrophy symptoms
Symptoms of multiple system atrophy usually start when someone is between 50 and 60 years of age, but they can come at any time after 30.
The symptoms are wide-ranging and include muscle control problems, similar to those of Parkinson’s disease.
Many different functions of the body can be affected, including the urinary system, blood pressure control and muscle movement.
Although there are many different possible symptoms of multiple system atrophy, not everyone who’s affected will have all of them.
Bladder problems
Men and women with multiple system atrophy will usually have one or more of the following bladder symptoms:
- constantly feeling the need to pee
- peeing more frequently
- loss of bladder control
- being unable to empty the bladder properly
- being unable to pee
Erection problems
Men with multiple system atrophy will usually experience erectile dysfunction (the inability to get and maintain an erection), although this is a common problem that many men without the condition develop.
Low blood pressure when standing up
Someone with multiple system atrophy will often feel lightheaded, dizzy and faint after standing up. This is known as postural hypotension or orthostatic hypotension and is caused by a drop in blood pressure when they stand upright.
When you stand up after lying down, your blood vessels usually narrow quickly and your heart rate increases slightly to prevent your blood pressure from dropping and decreasing blood flow to your brain.
This function is carried out automatically by the autonomic nervous system; however, this system doesn’t work properly in people with multiple system atrophy, so the control is lost.
Problems with co-ordination, balance and speech
In multiple system atrophy, a part of the brain called the cerebellum is damaged. This can make the person clumsy and unsteady when walking, and can also cause slurred speech. These problems are collectively known as cerebellar ataxia.
Slowness of movement and feeling stiff
A person with multiple system atrophy has much slower movements than normal (bradykinesia). This can make it difficult to carry out everyday tasks. Movement is hard to initiate, and the person will often have a distinctive slow, shuffling walk with very small steps.
Some people may also have stiff, tense muscles. This can make it even more difficult to move around and cause painful muscle cramps (dystonia).
The above symptoms are typical of Parkinson’s disease but, unfortunately, the medication used to relieve them in people with Parkinson’s disease (levodopa) isn’t very effective for people with multiple system atrophy.
Other signs and symptoms
People with multiple system atrophy may also have:
- shoulder pain and neck pain
- constipation
- cold hands and feet
- problems controlling sweating
- muscle weakness in the body and limbs – it may be more pronounced in one arm or leg
- uncontrollable laughing or crying
- sleep problems – insomnia, snoring, restless legs or nightmares
- noisy breathing and unintentional sighing
- a weak, quiet voice
- swallowing problems
- blurred vision
- depression
- dementia (although this is uncommon)
Multiple system atrophy diagnosis
Multiple system atrophy or Parkinson’s disease?
A person is more likely to have multiple system atrophy rather than Parkinson’s disease if:
- their symptoms have progressed rapidly – a person with Parkinson’s disease deteriorates more slowly
- they’ve experienced falls in the early stages of the condition – this isn’t a typical symptom of Parkinson’s
- they don’t respond well to levodopa therapy – levodopa can significantly improve symptoms of Parkinson’s disease
- their speech is severely affected – this isn’t a typical symptom of Parkinson’s disease
- they gasp and breathe noisily – this isn’t a typical symptom of Parkinson’s disease
Further tests
If multiple system atrophy is suspected, a doctor (usually a neurologist) will test the person’s reflexes and “automatic” bodily functions, such as their bladder function.
A brain scan is often needed – usually an MRI scan or a SPECT scan – to detect any loss of brain cells.
More detailed assessments of autonomic function may also be carried out – for example, recording blood pressure changes when lying down and standing.
Multiple system atrophy treatment
There is no cure for multiple system atrophy and no way of slowing its progression. Currently, there are no treatments to delay the progress of neurodegeneration in the brain. But there are treatments available to help people cope with some of the more disabling symptoms of multiple system atrophy. In some individuals, levodopa may improve motor function, but the benefit may not continue as the disease progresses.
The appropriate management of patients with multiple system atrophy requires a multidisciplinary team approach, as for patients with progressive supranuclear palsy (PSP).
Dopaminergic medications. Approximately one-third of patients with multiple system atrophy-parkinsonism benefit from dopaminergic medication, and 10% may improve more than 50% of their motor symptoms after levodopa therapy. However, in general, the benefits of levodopa in multiple system atrophy are less gratifying than in PD because they are not as significant and long-lasting. Despite this, an adequate trial of levodopa should be attempted in multiple system atrophy patients who exhibit parkinsonism. The treatment should beinitiated with carbidopa/levodopa 25/100 and could be started at a lower dose than usual (eg, one-half tablet with an increase in dose every other day as tolerated to 3 times a day, and thereafter a weekly increase of one-half tablet per dose to a total dose of up to 1200 mg/d, according to best response or emergent side effects. The use of dopaminergic medications requires caution, as it may worsen orthostatic hypotension and rapid eye movement (REM) sleep behavior disorder.
Autonomic dysfunction. Monitoring and management of comorbid orthostatic hypotension is critical in multiple system atrophy. It is important to reduce or discontinue any concurrent antihypertensive medications. Other strategies are increasing dietary salt and noncaffeinated fluids, getting up slowly, and wearing thigh-high compression stockings.
Pharmacologic strategies are necessary when the above measures fail. These include increasing the patient’s blood volume through the use of fludrocortisone or increasing the peripheral vascular resistance via midodrine or, if midodrine is ineffective, indomethacin or pyridostigmine.
Urologic symptoms. Urinary incontinence or retention are usually present early in multiple system atrophy. Urodynamic studies are helpful to determine the type of neurogenic bladder. The principal problem is often bladder spasticity, which responds to peripherally acting anticholinergic agents or botulinum toxin. Occasionally, intermittent catheterization or transcutaneous suprapubic catheterization may be required.
Supportive therapy including allied health care team. Considering the limited pharmacologic benefit, it is crucial that multiple system atrophy patients be treated by a multidisciplinary team as described above for progressive supranuclear palsy (PSP). Because of the occurrence of cervical, laryngeal, and pharyngeal dystonia that can cause upper airway obstruction, tracheostomy and gastrostomy tube may be considered in selected patients. The advisability of either gastrostomy or tracheostomy should be approached on an individual basis with a realistic appraisal of the patient’s general quality of life.
Multiple system atrophy prognosis
The median survival of patients with multiple system atrophy with either the parkinsonism or cerebellar phenotype is approximately 8 years, but the range is large 41. Motor and autonomic features lead to major disability. Early autonomic failure; older age of onset; short interval from disease onset to frequent falling, cognitive disability, unintelligible speech, severe dysphagia, dependence on wheelchair for mobility, and urinary catheter use; and lack of admission to a nursing home facility independently predict short disease survival 42.
Vascular parkinsonism
Vascular parkinsonism is usually caused by clotting in the brain from multiple small strokes. People with vascular parkinsonism tend to have more problems with gait than tremor and have more problems in the lower body. Vascular parkinsonism progresses very slowly in comparison to other types of parkinsonism. People might report an abrupt onset of symptoms or step-wise deterioration (symptoms get worse then plateau for a while). Symptoms in vascular parkinsonism may or may not respond to levodopa, depending on the location of vascular disease in the brain.
No specific clinical features or diagnostic tests reliably differentiate Parkinson’s disease and vascular parkinsonism, though some features may suggest vascular parkinsonism. A severe onset of parkinsonism immediately following or progressively occurring within a year of a stroke may indicate vascular parkinsonism.
Other signs that can indicate vascular parkinsonism include: evidence of vascular disease on an MRI (magnetic resonance imaging) of the brain in combination with varying levels of deterioration, prominent early cognitive problems and lower body issues, such as early gait and balance problems.
Vascular parkinsonism is suspected in the setting of a lower body–predominant parkinsonism and a brain MRI showing extensivesubcortical white matter lesions 43. Only around half of patients with vascular parkinsonism develop pyramidal signs, but when present, according to diagnostic criteria, they exclude PD. In a minority of cases, a history of stepwise deterioration will be present, which suggests a large-vessel infarct, and in these patients MRI should demonstrate a vascular lesion involving the globus pallidus externa, substantia nigra, ventrolateral nucleus of the thalamus, or frontal lobe contralateral to the side of parkinsonian symptoms.
Lewy body dementia
Dementia with Lewy Bodies also called Lewy Body dementia, is second only to Alzheimer’s as the most common cause of dementia in the elderly. Lewy body dementia encompasses two clinical entities, namely dementia with Lewy bodies and Parkinson disease dementia 44. Lewy body dementia is a progressive degenerative brain disorder characterized by dementia, psychosis, and features of parkinsonism. Symptoms fluctuate with time and vary among different individuals. Lewy body dementia causes progressive intellectual and functional deterioration. In addition to the signs and symptoms of Parkinson’s disease, people with dementia with Lewy Bodies tend to have frequent changes in thinking ability, level of attention or alertness and visual hallucinations. They usually do not have a tremor or have only a slight tremor. The parkinsonian symptoms may or may not respond to levodopa.
Lewy body dementia is an under-diagnosed condition as it is poorly understood and its clinical features overlap with other more common disorders, like Parkinson’s disease and Alzheimer’s disease 45. Studies have shown, however, that it accounts for up to 20% to 30% of all dementia cases. It is more common in men, and incidence increases with advancing age. It is prevalent in Asian, African, and European races. A family history of Lewy body dementia and Parkinson disease increases a patient’s risk.
Diagnosis of Lewy body dementia requires thorough clinical examination as many of its features overlap with other dementia disorders. It is the third most common type of dementia after Alzheimer disease and vascular dementia. It characterizes by the deposition of Lewy bodies in the brain that are intraneuronal cytoplasmic inclusion bodies having aggregates of alpha-synuclein and ubiquitin 46.
Lewy body dementia causes
The cause of Lewy body dementia is still unknown. Genetics, environmental factors, and changes linked to aging, however, may have a role and still require further research.
Like Alzheimer disease, Lewy body dementia presents with acetylcholine deficiency, but it is more pronounced in Lewy body dementia. Decreased levels of acetylcholine in temporal and parietal cortex result in visual hallucinations (a prominent feature of Lewy body dementia), while up-regulation of muscarinic M1 receptors in the temporal lobe results in delusions. Dopamine levels also diminish.
The pathology of Lewy body dementia overlaps that of Parkinson’s disease and Alzheimer’s disease. Neuronal cytoplasmic inclusion bodies, called Lewy bodies (comprising aggregates of ubiquitin and alpha-synuclein) and found within brain parenchyma (mainly in the brainstem, limbic system, and cerebral cortex), are characteristic of Lewy body dementia 47. Genetic mutations, environmental toxins, and the aging process can lead to misfolding of alpha-synuclein and its accumulation in the form of Lewy bodies via oxidative stress and mitochondrial dysfunction. The location of Lewy bodies determines the clinical presentation. If Lewy bodies develop initially in the brainstem and cerebral cortex, then dementia sets on early, and we call it dementia with Lewy bodies; however, if Lewy bodies develop initially in the brain stem only and extend to cerebral cortex later, then dementia occurs late in the disease process and we call it Parkinson disease dementia. Other features include Lewy neuritis, senile plaques, neurofibrillary tangles, and neuronal loss in the substantia nigra, locus coeruleus, and Meynert nucleus.
Lewy body dementia symptoms
There is a large variation in symptoms manifested among patients with Lewy bodies dementia and their time of onset. The main features include progressive dementia (mainly affects attention and executive function, memory loss is not common but can occur later in the disease process), fluctuation cognitive function (with variation in attention and episodes of drowsiness), visual hallucinations (detailed and recurrent), and features of parkinsonism like muscular rigidity, tremors, and bradykinesia 48.
Dementia with Lewy bodies causes problems with mental abilities and a number of other difficulties.
People with dementia with Lewy bodies may have:
- problems with understanding, thinking, memory and judgement – this is similar to Alzheimer’s disease, although memory may be less affected in people with dementia with Lewy bodies
- periods of fluctuating alertness alternating with periods of confusion or sleepiness – this can change over hours or days
- slow movement, stiff limbs and tremors (uncontrollable shaking)
- hallucinations (usually seeing or sometimes hearing things that aren’t there)
- disturbed sleep, often with violent movements and shouting out
- fainting spells, unsteadiness and falls
Problems with mental abilities
As with other types of dementia, dementia with Lewy bodies typically causes problems with:
- thinking speed
- understanding
- judgement
- visual perception
- language
- memory (but significant memory loss may not occur until later on)
These problems may be constant but typically tend to come and go. These problems can make daily activities increasingly difficult and someone with the condition may eventually be unable to look after themselves.
Other less common features include rapid eye movement (REM) sleep behavior disorder, autonomic dysfunction, unexplained falls, depression, and sensitivity to antipsychotic medication.
There are also other symptoms of dementia with Lewy bodies that can help distinguish it from other types of dementia, such as:
- marked swings between alertness and confusion or sleepiness – this can happen unexpectedly and change over hours or days
- slow movement, stiff limbs, tremors (uncontrollable shaking) and shuffling when walking – similar to Parkinson’s disease
- seeing or sometimes hearing things that aren’t there (hallucinations) – these can range from pleasant to distressing
- fainting, unsteadiness and falls
- disturbed sleep – this could be talking in sleep, acting out dreams or sleepiness during the day
- difficulty swallowing
- depression
Daily activities become increasingly difficult and there may be further health problems, such as an injury after a fall or a chest infection caused by accidentally inhaling food.
Lewy body dementia diagnosis
There’s no single test for dementia with Lewy bodies. Due to the incomplete specificity in the clinical diagnosis and the pathological definition of the disease, a postmortem biopsy or autopsy is the only method to secure a definite diagnosis 44.
Diagnostic Criteria 44
- Probable Lewy body dementia: progressive dementia + 2 main features.
- Possible Lewy body dementia: progressive dementia + 1 main feature.
Types 44
- Dementia with Lewy Bodies: dementia occurring first or within one year of movement disorder.
- Parkinson Disease Dementia: dementia occurring in a patient who receives a diagnosed of Parkinson’s disease and then develops dementia symptoms after one year or more of the diagnosis.
The following may be needed to make a diagnosis:
- an assessment of symptoms – for example, whether there are typical symptoms of dementia with Lewy bodies
- an assessment of mental abilities – this will usually involve a number of tasks and questions
- blood tests (e.g., vitamin B12 levels, chemistry panel, thyroid profile, syphilis, HIV) to rule out other causes of dementia
- brain scans, such as an MRI scan, CT scan or a single photon-emission computed tomography (SPECT) dopamine transporter scan – these can detect signs of dementia or other problems with the brain
- cerebrospinal fluid examinations have no significant role when conducted in these patients
- sleep evaluation for rapid eye movement (REM) sleep behavior disorder
Lewy body dementia treatment
There’s currently no cure for dementia with Lewy bodies or any treatment that will slow it down.
But there are treatments that can help control some of the symptoms, possibly for several years.
Treatments include:
- medicines to reduce hallucinations, confusion, drowsiness, movement problems and disturbed sleep
- therapies such as physiotherapy, occupational therapy and speech and language therapy for problems with movement, everyday tasks, and communication
- psychological therapies, such as cognitive stimulation (activities and exercises designed to improve memory, problem-solving skills and language ability)
- dementia activities, such as memory cafés (drop-in sessions for people with memory problems and their carers to get support and advice)
Medications
Medication can’t stop dementia with Lewy bodies getting worse, but for some people it can help reduce some of the symptoms.
- Acetylcholinesterase (AChE) inhibitors: Used to treat the cognitive symptoms of Lewy body dementia and are the mainstay of treatment. Initially developed for Alzheimer disease treatment, they are probably more effective in patients with Lewy body dementia. Acetylcholinesterase (AChE) inhibitors, such as donepezil (Aricept), rivastigmine (Exelon) and galantamine (Reminyl), may help improve hallucinations, confusion and sleepiness in some people. These work by increasing levels of a chemical called acetylcholine in the brain, which improves the ability of the brain cells to send signals to each other. Common side effects include feeling and being sick, diarrhea, headaches, tiredness and muscle cramps.
- Memantine: Memantine works by blocking the effects of an excessive amount of a chemical in the brain called glutamate. Memantine is used for moderate or severe dementia with Lewy bodies. It’s suitable for those who cannot take or are unable to tolerate acetylcholinesterase (AChE) inhibitors. Side effects can include headaches, dizziness and constipation, but these are usually only temporary. For more information about the possible side effects of your specific medication, read the patient information leaflet that comes with it and speak to your doctor.
- Carbidopa-Levodopa: Used to treat movement symptoms.; however, it has serious side effects and can lead to delusions, hallucinations, and confusion and practitioners should use them with caution in these patients, and they should start with low doses if required.
- Atypical antipsychotics: These may help with severely challenging behaviour and hallucinations that’s putting you or others at risk of harm that cause significant distress in patients not responding to standard acetylcholinesterase inhibitors. Commonly used drugs include clozapine, quetiapine, and aripiprazole. Use with caution due to neuroleptic sensitivity in these patients.
- Clonazepam: Used for rapid eye movement (REM) sleep behavior disorder.
- Selective serotonin reuptake inhibitors (SSRIs): Depression is common in patients with Lewy body dementia and often requires antidepressant therapy.
Support and therapies
In addition to medication, there are a number of therapies and practical measures that can help make everyday living easier for someone with dementia. Understanding the disease helps the caregivers cope with everyday challenges. They require home modifications occasionally and individual patient needs should specifically guide them 49.
Patients can take part in different therapies to improve their quality of life, including:
- Physiotherapy to help with movement difficulties
- Occupational therapy to identify problem areas in everyday life, such as getting dressed, and help work out practical solutions
- Speech and language therapy to help improve any communication or swallowing problems
- Support groups
- Individual and family psychotherapies. Psychological therapies, such as cognitive stimulation (activities and exercises designed to improve memory, problem-solving skills and language ability)
- Relaxation techniques, such as massage, and music or dance therapy
- Social interaction, leisure activities and other dementia activities, such as memory cafes (drop-in sessions for people with memory problems and their carers to get support and advice)
- Home modifications, such as removing loose carpets and potential trip hazards, ensuring the home is well lit, and adding grab bars and handrails.
The outcomes for most patients is guarded and the quality of life is poor. Most patients eventually end up in a long term care facility, where optimal treatment for basic living activities is not always provided 50.
Lewy body dementia prognosis
The prognosis of Lewy body dementia is fair to poor. Patients die from multiple complications like falls, immobility, cardiac complications, medication side effects, pneumonia, swallowing problems, and depression leading to suicide. The average life expectancy is only five to eight years after the initial diagnosis. This also can be due to a lack of knowledge regarding Lewy body dementia among physicians and the population and difficulty in differentiating it from other similar conditions which leads to a delay in diagnosis which delays the onset of specific therapy.
Corticobasal degeneration
Corticobasal degeneration is the least common atypical parkinsonism. Corticobasal degeneration usually develops in the fifth to seventh decades of life and presents with various phenotypes that include corticobasal syndrome, frontotemporal dementia, progressive nonfluent aphasia, and Richardson syndrome, which make it a very challenging disorder to diagnose 39. None of these phenotypes is sufficiently specific to unequivocally diagnose corticobasal degeneration. New diagnostic criteria have been recently developed that include all these phenotypes 51. These criteria are a step forward regarding identification of possible corticobasal degeneration, but they will need to be validated and may require further refinement. Definite diagnosis of corticobasal degeneration requires autopsy confirmation.
Corticobasal degeneration symptoms include a loss of function on one side of the body, involuntary and jerky movements of a limb and speech problems. It may become difficult or impossible to use the affected limb although there is no weakness or sensory loss. The individual may feel as if the limb is not under his/her voluntary control.
There is no specific treatment at this time for corticobasal degeneration.
Corticobasal degeneration–corticobasal syndrome
Corticobasal syndrome is the classical presentation of corticobasal degeneration; however, corticobasal syndrome can be due to PSP (as described above), a focal form of Alzheimer disease, or frontotemporal dementia 52. The corticobasal syndrome usually presents with an asymmetric progressive ideomotor apraxia that frequently affects the hand and is associated with rigidity, myoclonus, and dystonia. These symptoms spread to the lower extremity and eventually affect all four extremities but remain asymmetric. The myoclonus is frequently stimulus sensitive but not always present. Dystonia and myoclonus are less frequent than the akinetic-rigid syndrome and apraxia 53. Alien-limb phenomenon is seen in some patients and identified by involuntary grasping, purposeless movements, or levitation in an apraxic limb. When corticobasal syndrome affects the right extremities, it is more likely to be associated with a nonfluent aphasia, whereas, when affecting left extremities, it may associate at onset with visuospatial and visuoconstructive deficits. Eventually, patients may develop both language and visuospatial deficits, as well as a cortical sensory syndrome and an alien limb. corticobasal syndrome less frequently affects lower extremities first.
Corticobasal degeneration–progressive supranuclear palsy
Infrequently, corticobasal degeneration can present with a progressive supranuclear palsy phenotype that is hard to differentiate from progressive supranuclear palsy-Richardson syndrome, but incorticobasal degeneration usually there are more cognitive and behavioral frontal disturbances 54. Corticobasal degeneration-progressive supranuclear palsy patients tend to be more disinhibited than progressive supranuclear palsy-Richardson syndrome patients.
Corticobasal degeneration treatment
Pharmacologic therapies are usually of no benefit. The parkinsonism usually does not benefit from dopaminergic therapy, although it is always useful to attempt treatment with levodopa for at least 1 month with the maximum tolerated dose (900 to 1200 mg/d). The most useful symptomatic therapies are those targeting myoclonus (eg, valproic acid, clonazepam, levetiracetam, and piracetam) and dystonia (eg, botulinum toxin) when they affectthe patient’s quality of life. Treatment of dystonia is indicated when the contractures cause pain or impede hygiene.
Supportive therapy including an allied health care team is important and should follow the principles described above. Patients with corticobasal degeneration benefit from rehabilitation services more than pharmacologic approaches.
Corticobasal degeneration prognosis
Patients with corticobasal degeneration may exhibit more than one phenotype during life. Symptoms are relentless and survival is usually 7 to 8 years 39.
Parkinson’s disease vs Parkinsonism
Parkinson’s disease is a neurodegenerative brain disorder that progresses slowly in most people. Parkinson’s disease symptoms can take years to develop, and most people live for many years with Parkinson’s disease. The symptoms caused by Parkinson’s disease include an ongoing loss of motor control (resting tremors, stiffness, slow movement, postural instability) as well as a wide range of non-motor symptoms (such as depression, loss of sense of smell, gastric problems, cognitive changes and many others).
Parkinsonism also known as atypical Parkinson’s disease or Parkinson’s plus, is a general term that refers to a group of neurological disorders that cause movement problems similar to those seen in Parkinson’s disease such as tremors, slow movement and stiffness. Under the category of parkinsonism there are a number of disorders, some of which have yet to be clearly defined or named. Early in the disease process, it is often hard to know whether a person has idiopathic (meaning “of unknown origins”) Parkinson’s disease or a syndrome that mimics it.
While Parkinson’s disease represents 85-90% of all cases of parkinsonism, a definitive diagnosis for atypical parkinsonism may never be made while the person is alive.
Parkinsonism causes
Possible causes of parkinsonism
Not everyone who has parkinsonism has Parkinson’s disease. There are many other causes of parkinsonism (secondary parkinsonism), including:
- Medications, such as those used to treat psychosis, major psychiatric disorders and nausea
- Repeated head trauma, such as injuries sustained in boxing
- Certain neurodegenerative disorders, such as multiple system atrophy, Lewy body dementia and progressive supranuclear palsy
- Exposure to toxins, such as carbon monoxide, cyanide and organic solvents
- Certain brain lesions, such as tumors, or fluid buildup
- Metabolic and other disorders, such as chronic liver failure or Wilson’s disease
Parkinsonism diagnosis
The diagnosis of the parkinsonian syndromes is entirely clinical, as at the present time no imaging, biochemical, or genetic tests definitively diagnose or separate the different diseases 55. For diagnosis, doctors rely on taking a complete medical history that includes timeline of symptoms, recognition of the important clinical signs, and consideration of the differential diagnoses. Individuals’ diagnostic acumen is substantially influenced by clinical experience, and even among movement disorder specialists, the clinical diagnosis can change over time because of emerging clinical signs 56.
Because of the observational nature of the diagnosis, Parkinson’s disease can sometimes be confused with parkinsonism, and the diagnosis may need to be revised over time based on speed of disease progression, response to medications and other factors. All the parkinsonisms have a loss of dopamine, so a DatScan cannot be used to differentiate between them and idiopathic Parkinson’s disease.
While the identification of parkinsonism is an important first step for the consideration of therapeutic options, the differential diagnosis between Parkinson’s disease, progressive supranuclear palsy (PSP), multiple system atrophy (MSA), corticobasal degeneration (CBD), and vascular Parkinsonism (VaP) does not often provide for further specific disease-modifying therapy 39. However, a definitive diagnosis serves to inform patients, caregivers, family, and the broader multidisciplinary care team about prognosis, expected clinical progression, disease course, and potentially useful therapeutic modalities. Furthermore, a definitive clinical diagnosis gives a name for the disease, which is regarded by patients as very important and helpful when coming to terms with a chronic disease 39. Where possible, it is not recommended to diagnose “atypical parkinsonism” or a “parkinsonian syndrome,” as these terms are meaningless for patients and their care team and provide no further information about management or prognosis. If a definitive diagnosis cannot be reached, then a hierarchical list of diagnostic possibilities should be discussed. In the case of progressive supranuclear palsy (PSP), multiple system atrophy (MSA) and corticobasal degeneration (CBD), diagnostic criteria allow for possible and probable diagnostic categories, according to levels of diagnostic certainty.
The clinical manifestations of progressive supranuclear palsy-tau pathology are variable, and diagnosis can be difficult at times because of the subtle early signs that may be difficult to discern from other physical or psychological symptoms. The diagnosis of progressive supranuclear palsy should be considered in all patients presenting with parkinsonism not responding to levodopa therapy; postural instability with falls; executive dysfunction; slowing of vertical saccades/supranuclear vertical gaze palsy; or dysarthria/dysphagia 57.
Parkinsonism treatment
There is overlap in treatment for Parkinson’s disease and parkinsonisms. Dopaminergic therapy (the first line treatment for Parkinson’s disease) can be effective in some parkinsonisms. A regular daily exercise program is vital for maintaining muscle tone, strength and flexibility. Other common treatments for both Parkinson’s disease and parkinsonisms include physical, occupational and speech therapy; antidepressants and botulinum toxin (Botox) for dystonia. For all the conditions, health care providers aim to treat the symptoms that most affect a person’s quality of life.
Managing parkinsonism with medications
- For drug-induced parkinsonism, discontinuing the medications that cause the condition may reverse it.
- For other forms of parkinsonism, taking Parkinson’s disease medications — typically a carbidopa-levodopa combination drug (Sinemet, Duopa, Stalevo) — can help.
However, these drugs aren’t likely to be as effective for some forms of parkinsonism as they are for Parkinson’s disease. Levodopa — which occurs naturally in the body and is always taken as a combination drug — replenishes brain dopamine, and brain dopamine loss is fundamental to Parkinson’s disease. However, in other parkinsonian disorders, additional brain pathways may be affected.
Other steps you can take
Certain lifestyle changes also may help you cope with parkinsonism:
- Stay physically active. To the extent you’re able, try to sustain your normal daily activities, exercise regularly, and incorporate physical and occupational therapy as needed.
- Create a safe environment. If gait and balance become impaired, consider modifying your environment. For example, install grab bars next to your toilet or in your shower; remove obstacles, such as throw rugs; and keep frequently used items within reach.
- Shin HW, Chung SJ. Drug-induced parkinsonism. J Clin Neurol. 2012;8(1):15–21. doi:10.3988/jcn.2012.8.1.15 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3325428[↩][↩][↩][↩]
- Esper CD, Factor SA. Failure of recognition of drug-induced parkinsonism in the elderly. Mov Disord. 2008;23:401–404[↩]
- Thanvi B, Treadwell S. Drug induced parkinsonism: a common cause of parkinsonism in older people. Postgrad Med J. 2009;85:322–326[↩]
- Shin HW, Kim MJ, Kim JS, Lee MC, Chung SJ. Levosulpiride-induced movement disorders. Mov Disord. 2009;24:2249–2253[↩][↩][↩][↩]
- Scherfler C, Schwarz J, Antonini A, Grosset D, Valldeoriola F, Marek K, et al. Role of DAT-SPECT in the diagnostic work up of parkinsonism. Mov Disord. 2007;22:1229–1238[↩]
- Varrone A, Halldin C. Molecular imaging of the dopamine transporter. J Nucl Med. 2010;51:1331–1334[↩]
- Davis MR, Votaw JR, Bremner JD, Byas-Smith MG, Faber TL, Voll RJ, et al. Initial human PET imaging studies with the dopamine transporter ligand 18F-FECNT. J Nucl Med. 2003;44:855–861[↩]
- Diaz-Corrales FJ, Sanz-Viedma S, Garcia-Solis D, Escobar-Delgado T, Mir P. Clinical features and 123I-FP-CIT SPECT imaging in drug-induced parkinsonism and Parkinson’s disease. Eur J Nucl Med Mol Imaging. 2010;37:556–564[↩][↩]
- Poewe W, Scherfler C. Role of dopamine transporter imaging in investigation of parkinsonian syndromes in routine clinical practice. Mov Disord. 2003;18(Suppl 7):S16–S21[↩]
- Tolosa E, Coelho M, Gallardo M. DAT imaging in drug-induced and psychogenic parkinsonism. Mov Disord. 2003;18(Suppl 7):S28–S33[↩][↩]
- Sethi KD. Movement disorders induced by dopamine blocking agents. Semin Neurol. 2001;21:59–68[↩]
- Rajput AH, Rozdilsky B, Hornykiewicz O, Shannak K, Lee T, Seeman P. Reversible drug-induced parkinsonism. Clinicopathologic study of two cases. Arch Neurol. 1982;39:644–646[↩]
- Bishnoi M, Chopra K, Kulkarni SK. Protective effect of adenosine reuptake inhibitors in haloperidol-induced orofacial dyskinesia and associated behavioural, biochemical and neurochemical changes. Pharmacology. 2007;79:171–183[↩]
- STEELE JC, RICHARDSON JC, OLSZEWSKI J. PROGRESSIVE SUPRANUCLEAR PALSY. A HETEROGENEOUS DEGENERATION INVOLVING THE BRAIN STEM, BASAL GANGLIA AND CEREBELLUM WITH VERTICAL GAZE AND PSEUDOBULBAR PALSY, NUCHAL DYSTONIA AND DEMENTIA. Arch. Neurol. 1964 Apr;10:333-59.[↩]
- Agarwal S, Gilbert R. Progressive Supranuclear Palsy. [Updated 2019 Mar 27]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2019 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK526098[↩]
- Steele JC. Progressive supranuclear palsy. Brain. 1972;95(4):693-704[↩]
- Dickson DW, Ahmed Z, Algom AA, Tsuboi Y, Josephs KA. Neuropathology of variants of progressive supranuclear palsy. Curr. Opin. Neurol. 2010 Aug;23(4):394-400[↩][↩]
- Litvan I, Hauw JJ, Bartko JJ, Lantos PL, Daniel SE, Horoupian DS, McKee A, Dickson D, Bancher C, Tabaton M, Jellinger K, Anderson DW. Validity and reliability of the preliminary NINDS neuropathologic criteria for progressive supranuclear palsy and related disorders. J. Neuropathol. Exp. Neurol. 1996 Jan;55(1):97-105[↩][↩]
- Golbe LI, Davis PH, Schoenberg BS, Duvoisin RC. Prevalence and natural history of progressive supranuclear palsy. Neurology. 1988 Jul;38(7):1031-4[↩]
- Kawashima M, Miyake M, Kusumi M, Adachi Y, Nakashima K. Prevalence of progressive supranuclear palsy in Yonago, Japan. Mov. Disord. 2004 Oct;19(10):1239-40[↩]
- Litvan I, Agid Y, Calne D, Campbell G, Dubois B, Duvoisin RC, Goetz CG, Golbe LI, Grafman J, Growdon JH, Hallett M, Jankovic J, Quinn NP, Tolosa E, Zee DS. Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome): report of the NINDS-SPSP international workshop. Neurology. 1996 Jul;47(1):1-9.[↩]
- Birdi S, Rajput AH, Fenton M, Donat JR, Rozdilsky B, Robinson C, Macaulay R, George D. Progressive supranuclear palsy diagnosis and confounding features: report on 16 autopsied cases. Mov. Disord. 2002 Nov;17(6):1255-64[↩]
- Pierrot-Deseilligny C, Rivaud S, Pillon B, Fournier E, Agid Y. Lateral visually-guided saccades in progressive supranuclear palsy. Brain. 1989 Apr;112 ( Pt 2):471-87.[↩]
- Characteristics of two distinct clinical phenotypes in pathologically proven progressive supranuclear palsy: Richardson’s syndrome and PSP-parkinsonism. Williams DR, de Silva R, Paviour DC, Pittman A, Watt HC, Kilford L, Holton JL, Revesz T, Lees AJ. Brain. 2005 Jun; 128(Pt 6):1247-58.[↩]
- Glasmacher SA, Leigh PN, Saha RA. Predictors of survival in progressive supranuclear palsy and multiple system atrophy: a systematic review and meta-analysis. J. Neurol. Neurosurg. Psychiatry. 2017 May;88(5):402-411[↩][↩][↩][↩]
- Glasmacher SA, Leigh PN, Saha RA. Predictors of survival in progressive supranuclear palsy and multiple system atrophy: a systematic review and meta-analysis. J. Neurol. Neurosurg. Psychiatry. 2017 May;88(5):402-411.[↩][↩]
- Pekmezović T, Ječmenica-Lukić M, Petrović I, Špica V, Tomić A, Kostić VS. Quality of life in patients with progressive supranuclear palsy: one-year follow-up. J. Neurol. 2015 Sep;262(9):2042-8[↩][↩]
- Chu FC, Reingold DB, Cogan DG, Williams AC. The eye movement disorders of progressive supranuclear palsy. Ophthalmology. 1979 Mar;86(3):422-8[↩]
- Chen AL, Riley DE, King SA, Joshi AC, Serra A, Liao K, Cohen ML, Otero-Millan J, Martinez-Conde S, Strupp M, Leigh RJ. The disturbance of gaze in progressive supranuclear palsy: implications for pathogenesis. Front Neurol. 2010;1:147[↩]
- Stamelou M, Höglinger G. A Review of Treatment Options for Progressive Supranuclear Palsy. CNS Drugs. 2016 Jul;30(7):629-36[↩]
- Golbe LI, Ohman-Strickland PA. A clinical rating scale for progressive supranuclear palsy. Brain. 2007 Jun;130(Pt 6):1552-65[↩]
- Sale P, Castiglioni D, De Pandis MF, Torti M, Dall’armi V, Radicati FG, Stocchi F. The Lee Silverman Voice Treatment (LSVT®) speech therapy in progressive supranuclear palsy. Eur J Phys Rehabil Med. 2015 Oct;51(5):569-74[↩]
- Scelzo E, Lozano AM, Hamani C, Poon YY, Aldakheel A, Zadikoff C, Lang AE, Moro E. Peduncolopontine nucleus stimulation in progressive supranuclear palsy: a randomised trial. J. Neurol. Neurosurg. Psychiatry. 2017 Jul;88(7):613-616[↩]
- Bensimon G, Ludolph A, Agid Y, Vidailhet M, Payan C, Leigh PN., NNIPPS Study Group. Riluzole treatment, survival and diagnostic criteria in Parkinson plus disorders: the NNIPPS study. Brain. 2009 Jan;132(Pt 1):156-71[↩]
- Tolosa E, Litvan I, Höglinger GU, Burn D, Lees A, Andrés MV, Gómez-Carrillo B, León T, Del Ser T., TAUROS Investigators. A phase 2 trial of the GSK-3 inhibitor tideglusib in progressive supranuclear palsy. Mov. Disord. 2014 Apr;29(4):470-8.[↩]
- Leclair-Visonneau L, Rouaud T, Debilly B, Durif F, Houeto JL, Kreisler A, Defebvre L, Lamy E, Volteau C, Nguyen JM, Dily SL, Damier P, Boutoleau-Bretonnière C, Lejeune P, Derkinderen P. Randomized placebo-controlled trial of sodium valproate in progressive supranuclear palsy. Clin Neurol Neurosurg. 2016 Jul;146:35-9[↩]
- Apetauerova D, Scala SA, Hamill RW, Simon DK, Pathak S, Ruthazer R, Standaert DG, Yacoubian TA. CoQ10 in progressive supranuclear palsy: A randomized, placebo-controlled, double-blind trial. Neurol Neuroimmunol Neuroinflamm. 2016 Oct;3(5):e266[↩]
- dell’Aquila C, Zoccolella S, Cardinali V, de Mari M, Iliceto G, Tartaglione B, Lamberti P, Logroscino G. Predictors of survival in a series of clinically diagnosed progressive supranuclear palsy patients. Parkinsonism Relat. Disord. 2013 Nov;19(11):980-5[↩]
- Williams DR, Litvan I. Parkinsonian syndromes. Continuum (Minneap Minn). 2013;19(5 Movement Disorders):1189–1212. doi:10.1212/01.CON.0000436152.24038.e0 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4234134[↩][↩][↩][↩][↩]
- Gilman S, Wenning GK, Low PA, et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology 2008; 71 (9): 670– 676[↩][↩][↩]
- Nath U, Ben-Shlomo Y, Thomson RG, et al. Clinical features and natural history of progressive supranuclear palsy: a clinical cohort study. Neurology 2003; 60 (6): 910– 916[↩]
- O’Sullivan SS, Massey LA, Williams DR, et al. Clinical outcomes of progressive supranuclear palsy and multiple system atrophy. Brain 2008; 131 (pt 5): 1362– 1372[↩]
- Glass PG, Lees AJ, Bacellar A, et al. The clinical features of pathologically confirmed vascular parkinsonism. J Neurol Neurosurg Psychiatry 2012; 83 (10): 1027– 1029[↩]
- Haider A, Sánchez-Manso JC. Lewy Body Dementia. [Updated 2019 May 5]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2019 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK482441[↩][↩][↩][↩]
- Brenowitz WD, Keene CD, Hawes SE, Hubbard RA, Longstreth WT, Woltjer RL, Crane PK, Larson EB, Kukull WA. Alzheimer’s disease neuropathologic change, Lewy body disease, and vascular brain injury in clinic- and community-based samples. Neurobiol. Aging. 2017 May;53:83-92[↩]
- King E, O’Brien JT, Donaghy P, Morris C, Barnett N, Olsen K, Martin-Ruiz C, Taylor JP, Thomas AJ. Peripheral inflammation in mild cognitive impairment with possible and probable Lewy body disease and Alzheimer’s disease. Int Psychogeriatr. 2019 Apr;31(4):551-560[↩]
- Chen X, Kordich JK, Williams ET, Levine N, Cole-Strauss A, Marshall L, Labrie V, Ma J, Lipton JW, Moore DJ. Parkinson’s disease-linked D620N VPS35 knockin mice manifest tau neuropathology and dopaminergic neurodegeneration. Proc. Natl. Acad. Sci. U.S.A. 2019 Mar 19;116(12):5765-5774[↩]
- Chin KS, Teodorczuk A, Watson R. Dementia with Lewy bodies: Challenges in the diagnosis and management. Aust N Z J Psychiatry. 2019 Apr;53(4):291-303[↩]
- Armstrong MJ. Lewy Body Dementias. Continuum (Minneap Minn). 2019 Feb;25(1):128-146[↩]
- Armstrong MJ, Alliance S, Corsentino P, DeKosky ST, Taylor A. Cause of Death and End-of-Life Experiences in Individuals with Dementia with Lewy Bodies. J Am Geriatr Soc. 2019 Jan;67(1):67-73[↩]
- Armstrong MJ, Litvan I, Lang AE, et al. Criteria for the diagnosis of corticobasal degeneration. Neurology 2013; 80 (5): 496– 503[↩]
- Ling H, O’Sullivan SS, Holton JL, et al. Does corticobasal degeneration exist? A clinicopathological re-evaluation. Brain 2010; 133 (pt 7): 2045– 2057[↩]
- Stamelou M, Alonso-Canovas A, Bhatia KP. Dystonia in corticobasal degeneration: a review of the literature on 404 pathologically proven cases. Mov Disord 2013; 27 (6): 696– 702[↩]
- Kouri N, Murray ME, Hassan A, et al. Neuropathological features of corticobasal degeneration presenting as corticobasal syndrome or Richardson syndrome. Brain 2011; 134 (pt 11): 3264– 3275.[↩]
- EFNS/MDS-ES/ENS [corrected] recommendations for the diagnosis of Parkinson’s disease. Berardelli A, Wenning GK, Antonini A, Berg D, Bloem BR, Bonifati V, Brooks D, Burn DJ, Colosimo C, Fanciulli A, Ferreira J, Gasser T, Grandas F, Kanovsky P, Kostic V, Kulisevsky J, Oertel W, Poewe W, Reese JP, Relja M, Ruzicka E, Schrag A, Seppi K, Taba P, Vidailhet M. Eur J Neurol. 2013 Jan; 20(1):16-34.[↩]
- Hughes AJ, Daniel SE, Ben-Shlomo Y, Lees AJ. The accuracy of diagnosis of parkinsonian syndromes in a specialist movement disorder service. Brain 2002; 125 (pt 4): 861– 870[↩]
- Executive dysfunction is the primary cognitive impairment in progressive supranuclear palsy. Gerstenecker A, Mast B, Duff K, Ferman TJ, Litvan I, ENGENE-PSP Study Group. Arch Clin Neuropsychol. 2013 Mar; 28(2):104-13.[↩]





{kind=link}