
Contents
- Familial periodic paralysis
- Hypokalemic periodic paralysis
- Hypokalemic periodic paralysis causes
- Hypokalemic periodic paralysis inheritance
- Hypokalemic periodic paralysis pathophysiology
- Hypokalemic periodic paralysis prevention
- Hypokalemic periodic paralysis symptoms
- Hypokalemic periodic paralysis complications
- Hypokalemic periodic paralysis diagnosis
- Hypokalemic periodic paralysis treatment
- Hypokalemic periodic paralysis prognosis
- Hyperkalemic periodic paralysis
- Hyperkalemic periodic paralysis cause
- Hyperkalemic periodic paralysis triggers
- Hyperkalemic periodic paralysis inheritance pattern
- Hyperkalemic periodic paralysis pathophysiology
- Hyperkalemic periodic paralysis prevention
- Hyperkalemic periodic paralysis symptoms
- Hyperkalemic periodic paralysis complications
- Hyperkalemic periodic paralysis diagnosis
- Hyperkalemic periodic paralysis treatment
- Hyperkalemic periodic paralysis life expectancy
- Andersen-Tawil syndrome
- Andersen-Tawil syndrome causes
- Andersen Tawil syndrome inheritance pattern
- Andersen-Tawil syndrome signs and symptoms
- Andersen-Tawil syndrome diagnosis
- Andersen-Tawil syndrome treatment
- Episodic weakness or paralysis treatment
- Prevention of Andersen Tawil syndrome attacks
- Prevention of secondary complications
- Pregnancy management
- Paramyotonia Congenita
- Thyrotoxic periodic paralysis
Familial periodic paralysis
Periodic Paralysis or Familial Periodic Paralysis is a group of rare genetic disorders that cause short-term episodes (also known as “attacks”) of muscle weakness, paralysis or stiffness (also known as myotonia). The episodes of muscle weakness, paralysis (periodic paralysis or transient attacks of severe weakness) or muscle stiffness (myotonia) may affect the whole body or just 1 or 2 limbs. There are several types of Periodic Paralysis, all of which involve defects in voltage-gate ion channels like sodium, potassium, and calcium channels, in the muscle membrane 1, 2. Ion channels are openings that pierce the muscle membrane and act as gates which let charged minerals, such as sodium ions (Na+) and potassium ions (K+), to flow in and out of cells, which allows muscles to work. The movement of sodium ions (Na+) and potassium ions (K+) from one side of the muscle membrane to the other creates an electrical current. For the muscles to work properly the sodium ions (Na+) and potassium ions (K+) must be kept in the correct ratio both inside and outside the cell. In periodic paralysis the ion channels fail to regulate the flow of sodium ions (Na+) and potassium ions (K+) properly when potassium levels in the blood fluctuate. The ratio of sodium ions (Na+) and potassium ions (K+) inside and outside the cell become unbalanced. The muscle responds less when asked to move, which is felt as weakness. If the imbalance of sodium ions (Na+) and potassium ions (K+) inside and outside the muscle cell becomes pronounced the muscle quits responding at all, i.e. becomes paralysed. The episodes of muscle weakness or paralysis (attacks) may begin in childhood or adulthood and may happen after hard exercise. The ion channel which is affected determines the type of periodic paralysis. The most common genes involved in pathogenesis of periodic paralysis are CACN1S, SCN4A and KCNJ2, encoding calcium, sodium, and potassium channels (see Table 1) 3. Moreover, paralysis related to serum potassium values may also occur in thyrotoxicosis (too much thyroid hormone), Liddle syndrome, Gitelman syndrome, primary hyperaldosteronism (too much aldosterone), and acid-base balance disorders 3, 4.
The types of periodic paralysis or familial periodic paralysis include 5:
- Hypokalemic periodic paralysis also called HypoPP or Westphall disease,
- Hyperkalemic periodic paralysis also known as HyperPP or HyperKPP, Familial hyperkalemic periodic paralysis or Primary hyperkalemic periodic paralysis,
- Thyrotoxic periodic paralysis also called TPP or Thyrotoxic Hypokalemic Periodic Paralysis (Thyrotoxic HypoKPP),
- Paramyotonia Congenita (PMC). Paramyotonia Congenita (PMC) is a form of periodic paralysis that results from a mutation in the sodium channel and produces muscle stiffness or rigidity and weakness in response to cold or exercise. Paramyotonia Congenita (PMC) comes in two forms, one in which attacks are always associated with a rise in potassium (hyperkalemia) and a form called Paramyotonia von Eulenburg in which attacks can be associated with a fall in blood potassium levels or hypokalemia. Both result from mutations in the sodium channel. Both can accompany hyperkalemic periodic paralysis (HyperPP) or can occur alone.
- Potassium Aggravated Myotonias (PAMs). Potassium Aggravated Myotonias (PAMs) are forms of paramyotonia congenita (PMC) in which potassium intake triggers an attack of muscle stiffness (myotonia) and do not cause muscle weakness. Potassium Aggravated Myotonias (PAMs) include:
- Myotonia Fluctuans, which includes short attacks of varying degrees of mild muscle stiffness brought on by exercise.
- Myotonia Permanens, which causes severe and constant muscle stiffness and is made worse by eating potassium-rich food and exercise.
- Acetazolamide-responsive Myotonia Congenita, which causes severe and painful muscle stiffness. The muscles appear very well-developed in Acetazolamide-responsive Myotonia Congenita.
- Andersen-Tawil Syndrome (ATS) also known as Long QT syndrome 7 (LQT7). Andersen-Tawil syndrome (ATS) is a disorder that causes episodes of muscle weakness (periodic paralysis), changes in heart rhythm (arrhythmia), and developmental abnormalities 6. The episodes of muscle weakness (periodic paralysis) begins early in life, and episodes last from hours to days. These episodes may occur after exercise or long periods of rest, but they often have no obvious trigger. Muscle strength usually returns to normal between episodes. However, mild muscle weakness may eventually become permanent. In Andersen-Tawil Syndrome (ATS) the potassium shifts during attacks of paralysis are inconsistent. Potassium may rise during one attack (hyperkalemia) and fall during another (hypokalemia). The traditional classifications of Hypokalemic Periodic Paralysis or Hyperkalemic Periodic Paralysis cannot be applied. Patients also tend to have generalized weakness between attacks. In addition, Andersen-Tawil Syndrome patients experience irregular heart rhythms including a prolonged QT interval 6. Some have unusual facial and hand characteristics, such as short stature, clinodactyly (an inward curvature of the 5th fingers), fused or webbed second and third toes, scoliosis (crooked spine), widely spaced eyes, low-set ears, a broad forehead, and a small jaw 6. These signs may be absent or very subtle, or they may exist in other family members who do not experience muscle weakness or paralysis 6. Andersen-Tawil Syndrome is inherited in an autosomal dominant pattern. All Andersen-Tawil Syndrome mutations identified so far have been on the potassium channel.
Periodic paralysis is caused by mutations (changes) in the genes that control the function of certain ion channels in the muscle membrane. Mutations in the CACNA1S and SCN4A genes cause hypokalemic periodic paralysis (HypoPP). The hyperkalemic periodic paralysis (HyperPP) is due to mutations in SCN4A gene. The underlying cause of the thyrotoxic periodic paralysis (TPP) is unknown. Mutations in the KCNJ2 gene cause Andersen-Tawil syndrome. As of December 2021 approximately 70 different gene mutations have been identified, affecting sodium, calcium, or potassium channels 5. However, DNA testing still cannot be relied on entirely for diagnosis. At least 20% of clinically diagnosed patients have mutations that have not yet been identified. A negative DNA test does not rule out a diagnosis of periodic paralysis.
Most cases of periodic paralysis are inherited, but a family history may not be obvious 5. Only one parent need carry the mutated gene, and that parent may not have any symptoms 5.
Depending on the type of periodic paralysis, symptoms may be mild or severe, and may last for minutes or days. Some patients have their first attack within minutes of birth, but a few don’t have symptoms until they are in their 60’s or 70’s. Periodic Paralysis is found in all races and genders 5.
In the hypokalemic periodic paralysis (HypoPP), the paralysis is caused by low levels of potassium (hypokalemia). In the hyperkalemic periodic paralysis (HyperPP), the paralysis is caused by high levels of potassium in the blood (hyperkalemia). In the thyrotoxic periodic paralysis (TPP), the paralysis is caused by low levels of potassium in the blood (hypokalemia) and an overactive thyroid gland (hyperthyroidism). In Andersen-Tawil syndrome, potassium levels can be high, low, or normal.
Symptoms may include attacks of muscle weakness that may last for minutes to days, muscle pain in muscles after exercise, muscle cramping, feeling tingles, and permanent weakness. Attacks may last only a few moments or go on for days, depending on the type of periodic paralysis the person has. Some forms of periodic paralysis include muscle stiffness (myotonia) as part of the attacks. Some forms include permanent muscle weakness which develops over a period of many years. Some older patients have both paralytic episodes and long-lasting episodes of fatigue and weakness called Abortive Attacks. Some patients have no recognizable attacks of weakness but develop permanent muscle weakness that begins in their 40s or 50s and grows disabling over time.
Periodic paralysis treatment depends on the type and severity of symptoms with the focus on correcting the levels of potassium in the blood and preventing episodes with lifestyle changes.
- Hypokalemic periodic paralysis. Episodes of paralysis are managed by giving potassium chloride 2 to 10 g in an unsweetened oral solution or giving potassium chloride IV. Following a low-carbohydrate, low-sodium diet, avoiding strenuous activity, avoiding alcohol after periods of rest, and taking acetazolamide 250 mg orally 2 times a day may help prevent hypokalemic episodes.
- Hyperkalemic periodic paralysis. Episodes of paralysis, if mild, can be aborted at onset by light exercise and a 2-g/kg oral carbohydrate load. Established episodes require thiazides, acetazolamide, or inhaled beta-agonists. Severe episodes require calcium gluconate and insulin and dextrose IV (see also treatment of severe hyperkalemia). Regularly ingesting carbohydrate-rich, low-potassium meals and avoiding fasting, strenuous activity after meals, and cold exposure help prevent hyperkalemic episodes.
- Thyrotoxic periodic paralysis. Acute episodes are treated with potassium chloride, and serum potassium levels are closely monitored. Episodes are prevented by maintaining a normal thyroid (euthyroid) state and giving beta-blockers (eg, propranolol).
- Andersen-Tawil syndrome. In addition to lifestyle changes, including tightly controlled levels of exercise or activity, episodes may be prevented by giving a carbonic anhydrase inhibitor (eg, acetazolamide). The major complication of Andersen-Tawil syndrome is sudden death resulting from cardiac arrhythmias, and a cardiac pacemaker or implantable cardioverter-defibrillator may be required to control cardiac symptoms.
Figure 1. Periodic paralysis types
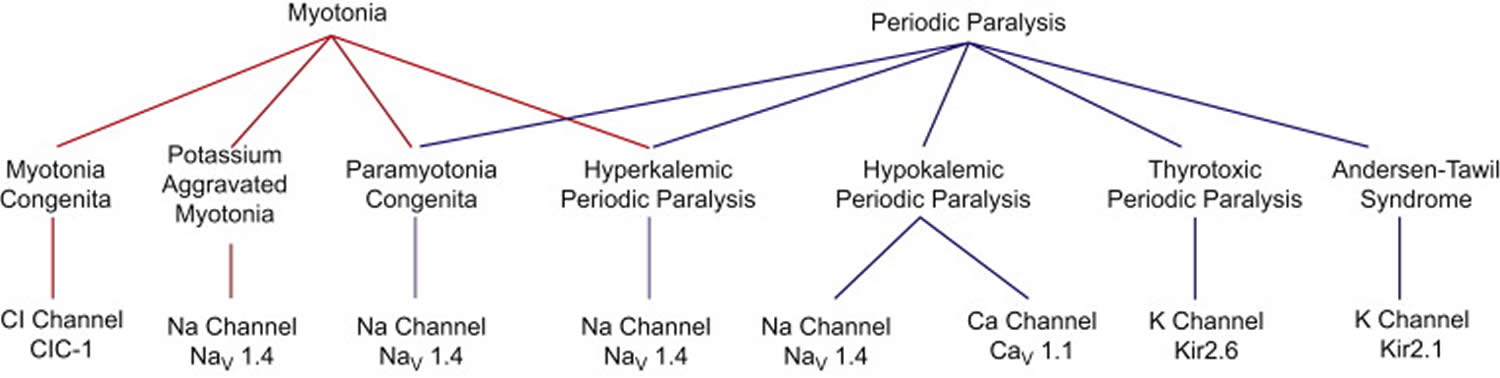
Table 1. Periodic paralysis types
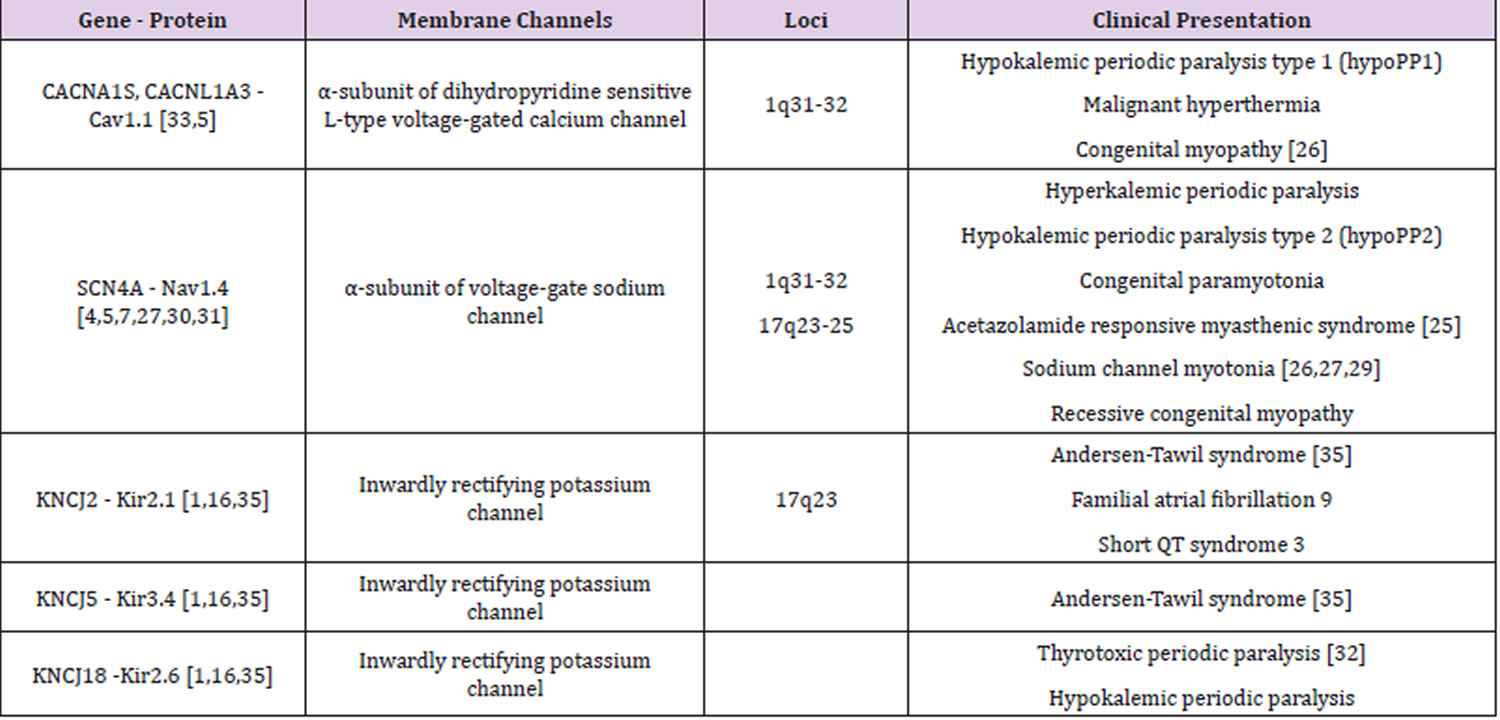
Footnotes: The table describes the main ion channel disorders and their specific gene loci mutations involvement. In the calcium, sodium and potassium channels, the gene loci involved are CACN1AS (calcium channel) located on chromosome 1, SCN4A and KCNJ2 (sodium and potassium channels, respectively) located on chromosome 17.
[Source 3 ]Hypokalemic periodic paralysis
Hypokalemic periodic paralysis also called HypoPP, HypoKPP or Westphall disease, is a rare inherited neuromuscular disorder that causes temporary episodes of muscle weakness or paralysis that is associated with a fall in blood potassium levels or hypokalemia (serum potassium less than 3.5 mmol/L) 8, 9, 10, 11, 12, 13, 14. People with hypokalemic periodic paralysis (HypoPP) typically have reduced levels of potassium in their blood (hypokalemia) during episodes of muscle weakness. Researchers are investigating how low potassium levels or hypokalemia may be related to the muscle abnormalities in hypokalemic periodic paralysis (HypoPP).
The episodes of muscle weakness or paralysis typically involve a temporary inability to move muscles in the arms and legs. The first attack usually occurs in childhood or adolescence and are triggered by strenuous exercise, high carbohydrate meals, injection of insulin, glucose, or epinephrine 8. Attacks can last for hours or days, and the frequency of attacks varies among people with hypokalemic periodic paralysis (HypoPP). The frequency is usually highest between the ages of 15 and 35, and then decreases with age. Some people with hypokalemic periodic paralysis (HypoPP) also develop chronic muscle weakness later in life or late-onset proximal myopathy affecting muscles of the trunk, shoulders, and thighs. Patients with late-onset proximal myopathy will have difficulty combing hair, difficulty climbing up the stairs, difficulty standing from a sitting position, and/or difficulty in getting up from bed 8, 15.
Hypokalemic periodic paralysis (HypoPP) is the most common type of periodic paralysis, and may be primary hypokalemic periodic paralysis (familial or idiopathic HypoPP) or secondary hypokalemic periodic paralysis (acquired HypoKPP) 16, 17, 18.
Primary hypokalemic periodic paralysis (familial or idiopathic HypoPP) is mainly caused by genetic changes in the CACNA1S or SCN4A gene where the genetic mutations occur in muscle ion channels producing potassium intracellular translocation 19, 20, 21. Both are voltage-dependent channels of the skeletal muscle fiber membrane. A mutation-induced aberrant current leads to a paradoxical membrane depolarization that renders muscle fibers unexcitable 22. Primary hypokalemic periodic paralysis (familial HypoPP) inheritance pattern is autosomal dominant.
Secondary hypokalemic periodic paralysis (acquired HypoKPP) is caused by the loss of potassium from kidneys, gastrointestinal tract or skin 12, 23, 24, 25, 26, 27. Hypokalemic periodic paralysis (HypoPP) cases related to thyroid disorders, more frequently thyrotoxicosis, and several autoimmune diseases have been previously reported 16, 23, 17. Thyrotoxicosis happens when you have too much thyroid hormone in your body in general. You could have too much thyroid hormone from taking too much thyroid medication, for example.
Although its exact prevalence is unknown, hypokalemic periodic paralysis (HypoPP) is estimated to affect 1 in 100,000 people 28, 29. However, a demographic survey in England, relying on the data of the national specialist channelopathy service, reported a minimum point prevalence of 0.13 per 100,000 people 1. Furthermore, men tend to experience symptoms of hypokalemic periodic paralysis (HypoPP) more often than women 30.
Hypokalemic periodic paralysis (HypoPP) treatment varies depending on the intensity and duration of the paralytic attacks. The goals of treatment are to relieve symptoms and prevent further attacks. Minor attacks may go away on their own 13. Moderate attacks may be self-treated in a non-medical setting by ingestion of oral potassium salts 13. But if weakness is severe, potassium may need to be given through a vein (IV). Muscle weakness that involves the breathing or swallowing muscles is an emergency situation. Dangerous irregular heartbeats (heart arrhythmias) may also occur during attacks. Any of these must be treated right away. Severe attacks typically require more intensive medical management with intravenous (IV) potassium infusion, serial measurement of serum potassium concentration, clinical evaluation of possible respiratory involvement, and continuous electrocardiogram (ECG) monitoring 13. There is no known curative treatment for hypokalemic periodic paralysis-related myopathy; physiotherapy may help to maintain strength and motor skills 13.
Hypokalemic periodic paralysis causes
Both hereditary or familial and acquired causes of hypokalemic periodic paralysis (HypoPP) have been identified 13, 14. Acquired hypokalemic periodic paralysis has been associated with thyrotoxicosis. Periodic muscle weakness can also result from hypokalemia secondary to renal and gastrointestinal potassium loss as in renal tubular acidosis, gastroenteritis, or secondary to endocrine causes.
Familial hypokalemic periodic paralysis is caused by a mutation in either of two genes, calcium or sodium ion channel gene mutation. Over the last few decades, several mutations in CACNA1S, SCN4A, and KCNJ2 genes have been identified, which underlie almost 70% to 80% of cases of hypokalemic periodic paralysis, while rest remain genetically undetermined 31, 32. The most common familial hypokalemic periodic paralysis, Hypokalemic periodic paralysis 1 or type 1 HypoPP, has a mutation in the dihydropyridine-sensitive, skeletal muscle calcium voltage-gated channel subunit alpha1 S (CACNA1S) gene 20, 21. While the other familial hypokalemic periodic paralysis, Hypokalemic periodic paralysis 2 or type 2 HypoPP, has mutations in the skeletal muscle sodium voltage-gated channel alpha subunit 4 (SCN4A) gene 20, 21. Disease-causing mutations in the gene KCNJ2 and KCNJ18, code for inward rectifier potassium (Kir) channel, have also been identified 32, 33, 34, 35, 31. The familial hypokalemic periodic paralysis and thyrotoxic hypokalemic periodic paralysis constitute the primary hypokalemic periodic paralysis (HypoPP) 14.
- The CACNA1S and SCN4A genes provide instructions for making proteins that play essential roles in muscles used for movement (skeletal muscles). For the body to move normally, skeletal muscles must tense (contract) and relax in a coordinated way. Muscle contractions are triggered by the flow of certain positively charged atoms (ions) into muscle cells. The CACNA1S and SCN4A proteins form channels that control the flow of these ions. The channel formed by the CACNA1S protein transports calcium ions (Ca2+) into cells (hypokalemic periodic paralysis 1), while the channel formed by the SCN4A protein transports sodium ions (Na+) (hypokalemic periodic paralysis 2). Mutations in the CACNA1S or SCN4A gene alter the usual structure and function of calcium or sodium channels. The altered channels are “leaky,” allowing ions to flow slowly but continually into muscle cells, which reduces the ability of skeletal muscles to contract. Because muscle contraction is needed for movement, a disruption in normal ion transport leads to episodes of severe muscle weakness or paralysis.
- CACNA1S gene provides instructions for making the main piece (subunit) of a structure called voltage-gated calcium channel Cav1.1 (Hypokalemic periodic paralysis 1) 36, 37, 38. Channels containing the CACNA1S protein are found in muscles used for movement (skeletal muscles). These skeletal muscle calcium channels play a key role in a process called excitation-contraction coupling, by which electrical signals (excitation) trigger muscle tensing (contraction). Calcium channels made with the CACNA1S subunit are located in the outer membrane of muscle cells, so they can transmit electrical signals from the cell surface to inside the cell. The channels interact with another type of calcium channel called ryanodine receptor 1 (RYR1) channels (produced from the RYR1 gene). RYR1 channels are located in the membrane of a structure inside the cell that stores calcium ions. Signals transmitted by CACNA1S-containing channels turn on (activate) RYR1 channels, which then release calcium ions inside the cells. The resulting increase in calcium ion concentration within muscle cells stimulates muscles to contract, allowing the body to move.
- SCN4A gene belongs to a family of genes that provide instructions for making voltage-gated sodium channel Nav1.4 (Hypokalemic periodic paralysis 2) 37, 38. These sodium channels, which transport positively charged sodium ions (Na+) into cells, play a key role in a cell’s ability to generate and transmit electrical signals 39. The SCN4A gene provides instructions for making a critical part (the alpha subunit) of sodium channels that are abundant in muscles used for movement (skeletal muscles). For the body to move, these muscles must tense (contract) and relax in a coordinated way. Muscle contractions are triggered by the flow of ions, including sodium, into skeletal muscle cells. Channels made with the SCN4A protein control the flow of sodium ions into these cells.
- Almost all mutations neutralize a positively charged amino acid in one of the outermost arginines or lysines of a voltage sensor. The voltage-gated sodium channel Nav1.4 mutations (hypokalemic periodic paralysis 2) are situated in the voltage sensors of repeats I, II and III 40, 37. In vivo, the muscles from these patients exhibited an intracellular sodium accumulation and edema 33.
- In most cases, hypokalemic periodic paralysis is passed down through families (inherited) as an autosomal dominant disorder. In other words, only one parent needs to pass the defective gene related to hypokalemic periodic paralysis on to their child in order for the child to be affected.
A small percentage of people with the characteristic features of hypokalemic periodic paralysis do not have identified mutations in the CACNA1S or SCN4A gene. In these cases, the cause of the condition is unknown. These minor cases of hypokalemic periodic paralysis may be the result of a genetic problem that is not inherited.
Unlike other forms of periodic paralysis, people with hypokalemic periodic paralysis have normal thyroid function. But they have a very low blood level of potassium during episodes of weakness. This results from potassium moving from the blood into muscle cells in an abnormal way.
Risk factors include having other family members with periodic paralysis. The risk is slightly higher in Asian men who also have thyroid disorders.
Hypokalemic periodic paralysis inheritance
Most familial hypokalemic periodic paralysis is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder. Familial hypokalemic periodic paralysis has lower clinical expression in females because of the lower penetrance and attack rate compared to males 41. And also women tend to have fewer attacks of muscle weakness than men.
A small percentage of people with the characteristic features of hypokalemic periodic paralysis do not have identified mutations in the CACNA1S or SCN4A gene (sporadic cases). In these sporadic cases, the cause of the condition is unknown. These minor cases of hypokalemic periodic paralysis may be the result of a genetic problem that is not inherited or represents new mutations 31, 42.
Most cases of thyrotoxic hypokalemic periodic paralysis have been identified as sporadic and is more prevalent among Asian descents with a male predominance of 9 to 1 29.
Figure 2. Hypokalemic periodic paralysis autosomal dominant inheritance pattern
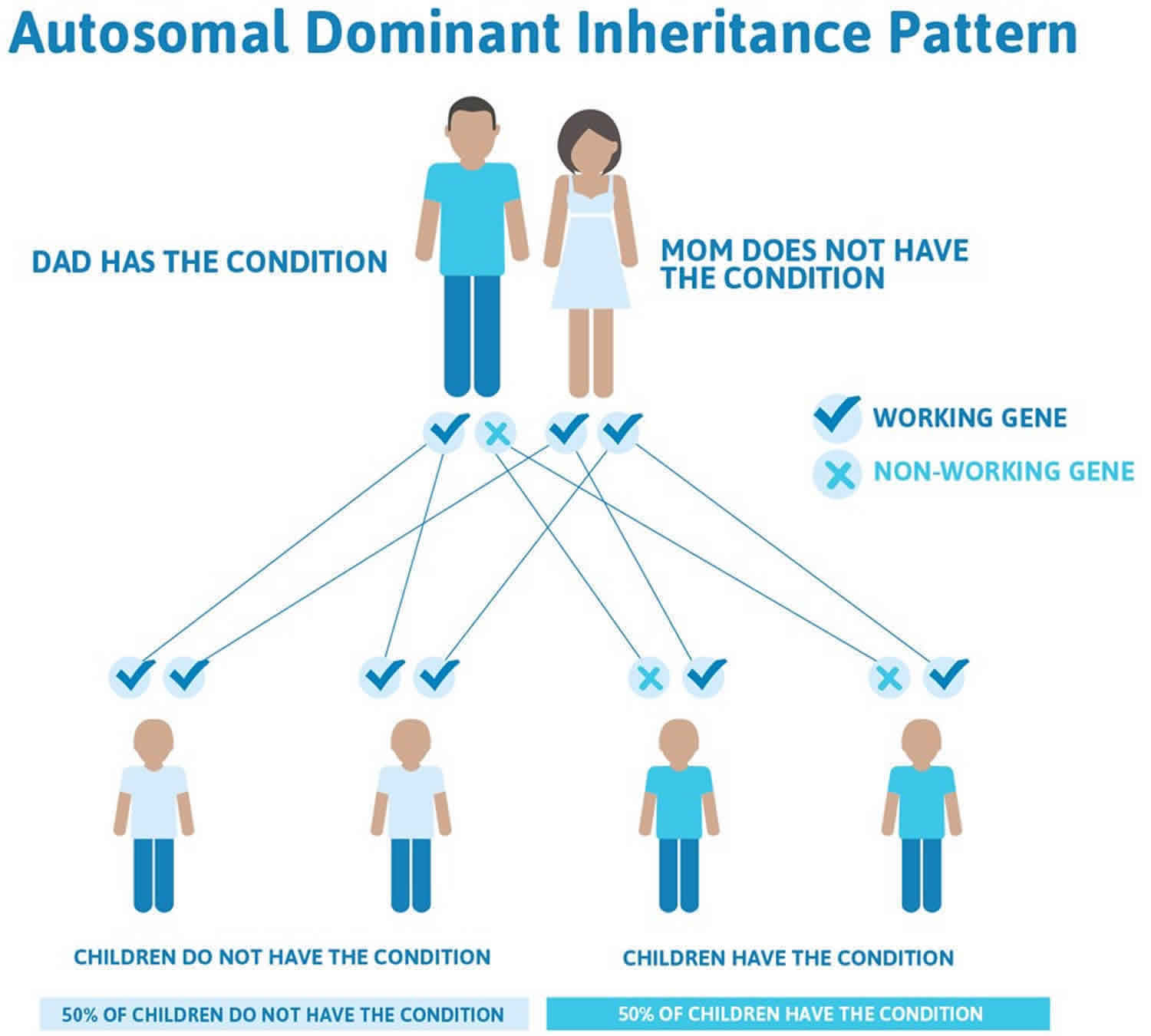
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Hypokalemic periodic paralysis pathophysiology
The most common genetic abnormality in hypokalemic periodic paralysis is the missense mutation in the positively charged residues, i.e., arginine, in the S4 domain of the alpha subunit (voltage sensor domain) of the skeletal muscle ion channel, most commonly L-type calcium channel (Cav1.1) and less commonly voltage-gated sodium channel (Nav1.4) 20, 21. Disease-causing mutations in the gene KCNJ2 and KCNJ18, code for inward rectifier potassium (Kir) channel, have also been identified 32, 33, 34, 35, 31. The final common mechanism for all mutations is the formation of anomalous gating pore current itself through the voltage sensor domain of ion channel that makes sarcolemmal muscle inexcitable, resulting in failure of muscle action potential and occurrence of subsequent attacks of flaccid paralysis 35, 32, 43, 44. In 90% of identified cases, arginine mutation in the S4 segment remains the primary cause 34. The other possible hypokalemic periodic paralysis mutations are yet to be determined.
The presence of gating pore current is mostly studied and understood in sodium channels. Many experiments demonstrated the presence of anomalous gating pore current in the setting of SCN4A mutation in sodium channels during the resting phase. The anomalous gating pore current results inward nonselective cation leak causing aberrant depolarization, which is sufficient to make the resting potential of the muscle fibers unstable 35, 32. And when serum potassium level drops below 3.0 mM, the affected muscle fibers paradoxically undergo sustained depolarization making muscle electrically inexcitable, whereas normal muscle fibers undergo hyperpolarization at this level of drop in serum potassium. Normally inward rectifying potassium (Kir) channel and membrane Na-K-ATPase maintains the normal negative resting membrane potential. In the presence of CACNA1S and SCN4A mutations, the depolarization induced by the gating pore current, at the modest drop of serum potassium level to around 3.0 mM, counterbalance the Kir current leading to sustained depolarization 32, 33, 28.
There are fewer experimental studies to demonstrate the evidence of gating pore current in calcium channels. But as the phenotypic expression of hypokalemic periodic paralysis in sodium and calcium channel mutations are similar, it is believed that the gating pore current does exist in calcium channel. While it is still unclear, there are numerous observations from different experimental studies to explain the possible underlying mechanisms behind muscle weakness with underlying calcium channel defects:
- The calcium channel mutations manifest as loss of function. Electrophysiologic studies have demonstrated slower activation of calcium channels and diminished calcium current density 31, 45. However, this observation does not correlate with episodes of depolarization, hypokalemia, and attacks of muscle weakness.
- In an experimental study, muscle biopsies taken from three hypokalemic periodic paralysis patients having R528H mutation of calcium channel (Cav1.1) showed the abnormal function of sarcolemmal ATP sensitive K+ (KATP) channel, supported by the fact that magnesium adenosine diphosphate (MgADP) did not stimulate the channel. The KATP channel showed reduced opening and reduced conductance state, i.e., reduced K current 31. The reduced K current is more likely related to depolarization with hypokalemia. Altered Ca2+ homeostasis resulting from the calcium channel mutation is likely the reason behind the altered function of the KATP channel. This observation hints toward the presence of a possible secondary channelopathy in patients with hypokalemic periodic paralysis.
Hypokalemic periodic paralysis prevention
Hypokalemic periodic paralysis (HypoPP) cannot be prevented. Because hypokalemic periodic paralysis (HypoPP) can be inherited, genetic counseling may be advised for couples at risk of the disorder. Treatment prevents attacks of weakness. Before an attack, there may be leg stiffness or heaviness in the legs. Doing mild exercise when these symptoms start may help prevent a full-blown attack. Taking potassium supplements may help prevent muscle weakness.
A diet rather low in sodium and carbohydrate and rich in potassium is recommended and may help decrease symptoms.
- Oral intake of potassium salts (10-20 mmol/dose, 3 doses/day) can prevent attacks, especially if the dose of potassium is taken some hours before the usual time of the attack (i.e., a nocturnal dose if crises occur at awakening).
- For individuals receiving chronic potassium supplementation for hypokalemic periodic paralysis, magnesium might be added, which can be helpful to promote renal retention of potassium and, therefore, reduce the potassium dose 28.
A carbonic anhydrase inhibitors medicine called acetazolamide and diclorphenamide may be prescribed to prevent attacks. Your doctor may tell you to also take potassium supplements because acetazolamide may cause your body to lose potassium. There is no standardized treatment regimen and no consensus as to when to start treatment with acetazolamide.
- Typical dosage for acetazolamide in adults is between 125 mg/day and 1000 mg/day (usually 250-500 mg/day), divided into three doses and taken with meals; in children a dose of 5-10 mg/kg/day, divided into three doses and taken with meals, is used.
- Acetazolamide treatment:
- Is beneficial in approximately 50% of individuals with hypokalemic periodic paralysis
- Has no effect in 30% of affected individuals
- May worsens hypokalemic periodic paralysis in individuals with who have a pathogenic variant in SCN4A
- In some affected persons, long-lasting interictal weakness may be partly reversed and muscle strength may be improved by acetazolamide treatment 46
- Whether acetazolamide treatment prevents or treats myopathy and the resulting fixed weakness that occurs with age is unknown.
- Further studies are needed to evaluate the effect of preventive acetazolamide treatment on attack rate, severity-weighted attack rate, long-lasting interictal weakness, and myopathy.
- Common side effects of carbonic anhydrase inhibitors include paresthesia, fatigue, mild, reversible cognitive disturbances and an increased risk of kidney stone (nephrolithiasis).
Dichlorphenamide was recently approved by the FDA for the treatment of periodic paralysis. Dichlorphenamide has been evaluated in four randomized, placebo- controlled studies, two each in patients with hypokalemic periodic paralysis (HypoPP) and hyperkalemic periodic paralysis (HyperPP) 47
- While randomized controlled trials of dichlorphenamide were performed in adults, the same approach is taken for children; dose adjustments may be required based on age.
- These studies demonstrated a significant reduction in the frequency and severity of the attacks. During a 52-week extension, in which all remaining participants received open-label dichlorphenamide, continued improvement in outcomes was observed in both placebo and dichlorphenamide groups.
- The dose of dichlorphenamide was 50 mg twice daily for treatment-naıve patients.
- Individuals already on dichlorphenamide before the study continued on the same dose during the study.
- In those taking acetazolamide before the study, the dose of dichlorphenamide was set at 20% of the acetazolamide dose.
- Dose reduction for tolerability was permitted.
- The mean dose of dichlorphenamide at week 9 was 82 mg/day.
- The most common side effects with dichlorphenamide were paresthesias, cognitive disorder, dysgeusia, headache, fatigue, hypoesthesia, and muscle spasms, generally not requiring discontinuation of dichlorphenamide, and reversible with drug discontinuation.
In the recent study of diclorphenamide 47, quality of life was assessed at 9 weeks and significant improvement was reported for the physical component and physical functioning, working time, bodily pain, vitality, and social functioning in those with hypokalemic periodic paralysis (HypoPP).
If carbonic anhydrase inhibitors are not tolerated or not effective after prolonged use, alternatives include potassium sparing diuretics like triamterene 50–150 mg/day, spironolactone 25–100 mg/day or eplerenone 50– 100mg daily.
- Because spironolactone is associated with a long half-life for substrate degradation, the individual can become hyperkalemic and weaker, develop cardiac arrhythmias, and suffer from hair loss. Additionally, spironolactone has androgenic side effects.
- The modern spironolactone derivate Eplerenone may be preferred because it causes fewer androgenic side effects. In addition, it has a very high repolarizing power, the parameter considered as most relevant for a beneficial effect.
- For individuals with hypokalemic periodic paralysis, potassium supplementation and a potassium-sparing diuretic may be used concomitantly, but potassium levels should be routinely monitored.
Avoid anything that can trigger paralytic attacks in the individual case, including the following:
- Unusually strenuous effort
- Excess of carbohydrate-rich meals or sweets
- Cold
- Stress/excitement/fear
- High salt intake
- Prolonged immobility
- Oral or intravenous glucosteroids
- Use of cooling, glucose and/or mannitol infusion, excessive sodium- containing fluids and certain anesthetics such as succinylcholine during anesthesia
- Use of alcohol
Hypokalemic periodic paralysis symptoms
Hypokalemic periodic paralysis can have its onset anywhere from early childhood to adulthood, the mean age of presentation of attacks is the first or second decade of life, usually the late childhood or teenage years 48. The age of onset of the first attack ranges from two to 30 years 14. Attacks usually begin in the teen years, but they can occur before age 10. However, in the case of thyrotoxic hypokalemic periodic paralysis, the onset is usually after age 20. Hypokalemic periodic paralysis symptoms include attacks of muscle weakness or loss of muscle movement (paralysis) that come and go (periodic paralysis). The paralytic attacks are characterized by decreased muscle tone (flaccidity) more marked proximally than distally with normal to decreased deep tendon reflexes 13. The paralytic episodes develop over minutes to hours and last several minutes to several days (paralytic episodes ranges from one to 72 hours with an average of nearly 24 hours) with spontaneous recovery. There is normal muscle strength between attacks. Attacks occur suddenly and are episodic. Some individuals may experience a milder form of muscle weakness between attacks that fluctuates and improves with mild exercise 29.
How often the attacks occur varies. Some people have attacks every day. Others have them once a year. During attacks the person remains alert. The frequency of attacks tends to decrease with age 31, 28. Long-lasting interval between episodes of muscle weakness may occur in some affected individuals and in some stages of the disease and in myopathic muscle changes. A myopathy may occur independent of paralytic symptoms and may be the sole manifestation of hypokalemic periodic paralysis.
The muscle weakness or paralysis:
- Most commonly occurs at the shoulders and hips
- May also affect the arms, legs, muscles of the eyes, and muscles that help with breathing and swallowing
- Occurs off and on
- Most commonly occurs on awakening or after sleep or rest
- Is rare during exercise, but may be triggered by resting after exercise
- May be triggered by high-carbohydrate, high-salt meals, stress, pregnancy, heavy exercise, and cold
- An attack usually lasts for several hours up to a day
Another symptom may include eyelid myotonia (a condition in which after opening and closing the eyes, they cannot be opened for a short time).
Patients usually present with attacks of generalized severe muscle weakness, with proximal muscle involvement more marked than distal and a profound decrease in serum potassium level (serum potassium less than 2.5 mmol/L) 28. Usually, patients go to bed in the normal state of health and wake up in the middle of the night or the morning, experiencing an attack of muscle weakness 31. Many patients also experience prodromal symptoms like fatigue, paresthesias, behavioral changes a day before an attack of muscle weakness 31. However, when incomplete, it predominantly involves lower limbs than the upper limbs. Bulbar, ocular, and respiratory muscles are usually spared, but respiratory muscle involvement can prove fatal when involved in severe cases 48, 29. The pattern of muscle weakness is similar in both familial and thyrotoxic hypokalemic periodic paralysis, and signs of overactive thyroid (hyperthyroidism) are clinically obvious in most cases of thyrotoxic hypokalemic periodic paralysis but are not always present. And attacks of muscle weakness occur during the state of hyperthyroidism and never when the thyroid function is normal.
The frequency of attacks of muscle weakness is very variable and infrequent. Some individuals have only one episode in a lifetime; more commonly, attacks occur repeatedly: daily, weekly, monthly, or less often 31, 13. And the duration of each attack also varies, ranging from minutes to days and can last up to several hours before they resolve spontaneously. Women tend to have fewer attacks than men.
The major triggering factors are rest following strenuous exercise and consumption of carbohydrate-rich meals 13, 31, 28. It is hypothesized that these triggering factors cause a rise in plasma epinephrine level or insulin level, causing an intracellular shift of potassium, resulting in lower serum potassium level, thus triggering the episode of weakness 29.
Additional triggers can include cold, stress, excitement, fear, salt intake, prolonged immobility, glucocorticoids use, alcohol, and anesthetic procedures 31, 13.
Hypokalemic periodic paralysis complications
Health problems that may be due to hypokalemic periodic paralysis include:
- Irregular heartbeat during attacks or life-threatening cardiac arrhythmias due to hypokalemia (are uncommon but have been reported during attacks of muscle weakness) 49
- Respiratory insufficiency due to respiratory muscle paralysis
- Difficulty breathing, speaking, or swallowing during attacks (rare)
- Muscle weakness that worsens over time. Many patients can have muscle weakness during the interictal period (i.e., between paralytic attacks), but its frequency and the risk for long-lasting weakness are unknown 28. It is believed that it is the result of permanent sodium intake, which results from the cation leak through the gating pore current 13. This may respond to potassium administration or acetazolamide 48.
- Myopathy. Most patients develop progressive proximal myopathy; however, the frequency is unknown. Myopathy usually manifests after age 50, is less fluctuating, and less sensitive to medications, which suggest there is muscle degeneration, a fixed myopathy 48, 50, 28. It may be evident early on muscle biopsy before manifesting clinically. The myopathy is more profound in pelvic girdle muscles and proximal upper and lower limbs 48, 50. The severity of myopathy varies among individuals, and some develop only mild weakness, which does not affect normal daily activities, while some may develop severe myopathy enough to make them wheelchair-bound. There is little evidence to support the correlation between the development of myopathy and the frequency or severity of paralytic attacks 50, 29.
- Kidney stones (a side effect of acetazolamide). A report showed an occurrence of renal stones in up to 15% of patients taking acetazolamide for the long term. The treatment of acetazolamide induced renal stones is the removal of stone without stopping acetazolamide therapy 28
Hypokalemic periodic paralysis diagnosis
Your doctor may suspect hypokalemic periodic paralysis (HypoPP) based on a positive family history or previous personal history of similar attacks of muscle weakness. Other clues to hypokalemic periodic paralysis (HypoPP) are muscle weakness symptoms that come and go with normal or low results of a potassium on blood test (hypokalemia). When there is an established family history of hypokalemic periodic paralysis (HypoPP) , no further diagnostic investigations are required to confirm the diagnosis of an episode of a paralytic attack. Otherwise, a low serum potassium level (hypokalemia) during a typical attack of weakness establishes the diagnosis.
During an attack, muscle reflexes are decreased (hyporeflexia) or absent (areflexia). And muscles go limp rather than staying stiff. Muscle groups near the body, such as the shoulders and hips, are involved more often than the arms and legs. During an attack of muscle weakness, blood potassium level is low (hypokalemia) and this confirms the diagnosis. There is no decrease in total body potassium.
Between attacks, a physical examination shows nothing abnormal. Before an attack, there may be leg stiffness or heaviness in the legs. Blood potassium level is normal (normokalemia) between attacks.
Tests that may be done include:
- Electrocardiogram (ECG), which may be abnormal during attacks
- Electromyography (EMG), which is usually normal between attacks and abnormal during attacks.
- During attacks of weakness, electromyography (EMG) may demonstrate reduced amplitude of compound muscle action potential (CAMP) and may show electrical silence based on the degree of muscle weakness 31, 51.
- Between attacks, EMG techniques can be used to demonstrate the change in excitability of muscle fibers due to channelopathy, called the “exercise test.” In the long exercise test, an attack of focal muscle weakness is induced by vigorously exercising a single muscle for 2-5 minutes, and the change in postexercise compound muscle action potential (CMAP) in muscle fibers is measured by the EMG. The reduction of 40% or more in compound muscle action potential (CMAP) is considered abnormal and typical for periodic paralysis. The study showed no false-positive results when the reduction is more than 40% or more, and this change was present in greater than 70% of patients 28, 52, 53. The abduction range of the little finger measured postexercise, can be a possible alternative parameter to compound muscle action potential (CMAP) in a long exercise test for diagnosis of hypokalemic periodic paralysis between attacks of muscle weakness 54.
- Muscle biopsy, which may show abnormalities. Interattack muscle biopsy is usually not performed to confirm the diagnosis. It may show the presence of vacuolar changes or tubular aggregates, but are nonspecific findings to all periodic paralysis 31.
- Exercise test. In the exercise test, the patient vigorously exercises a single muscle for 2 to 5 minutes in an attempt to cause focal muscle weakness. Weakness is assessed by an electrophysiologic study called the compound muscle action potential (CMAP), which is done before and after exercise. A ≥ 40% decrease postexercise compound muscle action potential (CMAP) is abnormal and consistent with periodic paralysis.
- Genetic testing to identify heterozygous pathogenic variant in CACNA1S or SCN4A gene. However approximately 30% do not have a pathogenic variant identified in either of these known genes.
- Other tests may be ordered to rule out other causes. These include thyroid function test (TSH, T3, T4 level) to rule out hyperthyroidism, an electrocardiogram (ECG) to look for ECG changes consistent with hypokalemia, and an ECG may also show the feature of Andersen-Tawil syndrome, long QT interval 28.
- Provocative test. Administration of potassium or insulin and glucose can be used as a provocative test to diagnose hypokalemic periodic paralysis (HypoPP). However, provocative testing with potassium or glucose and insulin administration might be potentially dangerous as it can precipitate life-threatening arrhythmia or hypoglycemia. Thus they require intensive monitoring in a hospital setting and not necessary to establish the diagnosis 55. They have been largely replaced by the exercise test, which is relatively safer.
The diagnostic criteria for hypokalemic periodic paralysis (HypoPP) include the following 56:
- 2 or more episodes of muscle weakness with serum potassium less than 3.5 mmol/L or one relative had a similar attack.
- 3 or more of the following features should be present:
- Onset in the first or second decade, onset time longer than two hours,
- The presence of triggers (previous carbohydrate-rich meal, onset during rest after exercise, stress),
- Symptomatic relief with potassium intake,
- A family history of skeletal calcium or sodium channel mutation, and
- Positive long exercise tests.
- Other causes of hypokalemia (renal and adrenal disease, thyroid dysfunction, drug abuse) are excluded.
Neurological examination of the patient during attack shows generalized muscle weakness, usually proximal muscle involvement more than distal and when incomplete legs are more often involved than arms. Hyporeflexia (decreased deep tendon reflexes) or areflexia (absent deep tendon reflexes) is typical. Neurological examination findings are usually normal between attacks. Myotonia is uncommon, unlike in hyperkalemic periodic paralysis (HyperPP), where myotonic is a common finding 31, 42, 57.
The best diagnostic indicator is a history of typical episodes. If measured during an episode, serum potassium may be abnormal.
Previously, for diagnosis, an attempt was made to provoke episodes by giving dextrose and insulin (to cause the hypokalemic periodic paralysis) or potassium chloride (to cause the hyperkalemic periodic paralysis), but because these tests may cause respiratory paralysis or cardiac conduction abnormalities and are not needed to make the diagnosis, they have been replaced by a safer exercise test.
Diagnosis of the hyperkalemic periodic paralysis is based on clinical findings and/or the identification of a heterozygous pathogenic genetic variant in the alpha-subunit of the skeletal muscle sodium channel.
In a case series of 71 diagnosed patients of hypokalemic periodic paralysis, patients without genetic mutations, compared to patients with genetic mutations, were found to have disease presentation at old age, absence of diet as a precipitating factor, and muscle biopsy showed no vacuolar myopathy 42. Phenotypic variations were also noted in patients having mutations in this case series. Patients with sodium channel mutations had attacks of shorter durations, and vacuolar changes were more common on calcium channel mutation, while tubular aggregates were seen more in sodium channel mutations 42.
Hypokalemic periodic paralysis treatment
The primary goal of treatment is to alleviate the symptoms of acute attacks, prevention and management of immediate complications, and prevention of late complications and future attacks.
Table 2. Treatment principles for individuals with hypokalemic periodic paralysis
| Goal | Means | Practical Details |
|---|---|---|
| To avoid triggering or aggravating factors for paralytic attacks | Avoid: Strenuous effort; Prolonged immobility; Carbohydrate-rich diet; High sodium diet. | Monitor episodes of weakness noting time of day & specific triggers. Provide dietary review/counseling. |
| Treatment of paralytic attack: Shorten/prevent aggravation of the weakness episode. Normalize kalemia. | Provide potassium supplementation (oral, or IV if oral impossible or if potassium very low). Avoid glucose intake. | Do not use slow-release forms of potassium. Oral potassium: initially, 1 mEq/kg; add 0.3 mEq/kg after 30 min if no improvement IV potassium: 0.3 mEq/kg/h |
| Preventive treatment for paralytic attacks | Daily potassium supplementation | Slow-release forms of potassium may be used. |
| Acetazolamide | ||
| Dichlorphenamide | ||
| Potassium-sparing diuretics | ||
| Preventive treatment for late-onset myopathy | Acetazolamide? | |
| Medical precautions | Avoid corticosteroids if possible. Use alpha- or beta adrenergic drugs w/caution, even in local anesthesia or ophthalmology. | |
| Other elements of management | Kinesiotherapy in case of long-lasting pelvic deficit Adaptive measures: (1) at school & especially for sports; (2) in work setting |
Treatment of paralytic attack
The goal is to normalize the serum potassium level by administering oral potassium chloride, which is believed to be more readily absorbed compared to other oral potassium solutions, alleviates the symptoms of muscle weakness 14. Oral potassium chloride is administered in incremental dose, starting initially with 0.5 to 1 mEq/kg (i.e., 60 to 120 mEq of potassium for a 60 kg individual) is reasonable 14. If patients do not respond to the initial dose, then 30% of the initial dose (i.e., 0.3 mEq/kg) is repeated every 30 min 13, 58, 40, 55. If the patient requires the addition of more than 100 mEq of oral potassium, then close monitoring of serum potassium is needed, and the total dose of oral potassium should not be more than 200 mEq within the 24 hours of starting of the treatment 55. The starting dose of oral potassium may vary according to the severity of hypokalemia. Patients should be kept on ECG monitoring, and muscle strength should be examined periodically. Serum potassium level should be monitored for 24 hours after treatment as the posttreatment rise in serum potassium level can have an adverse effect on patients.
Intravenous (IV) potassium is not preferred initially and is reserved for cardiac arrhythmias due to hypokalemia or if the patient has swallowing difficulties or respiratory muscle paralysis 14. Intravenous (IV) potassium is preferentially administered with the mannitol, not with dextrose or saline as both carbohydrate and salt can itself trigger the muscle paralysis and thus may worsen the weakness 55, 59. IV potassium therapy requires inpatient, continuous ECG monitoring. 40 mEq/L in 5% of mannitol solution of IV potassium is infused at a rate not more than 20 mEq/hour, not exceeding 200 mEq in 24 hours 28.
Individuals having a milder form of attacks can also benefit from low-level exercises 28, 29.
Preventive treatment for paralytic attacks
Both pharmacological and nonpharmacological interventions can be used to prevent recurrent future attacks. Nonpharmacological interventions include educating patients about triggering factors and lifestyle modifications to avoid these factors. Pharmacologic interventions include medications like chronic potassium supplementation, carbonic anhydrase inhibitors, potassium-sparing diuretics that are used when lifestyle modifications become insufficient in reducing attack rates 14. The favored approach is to add one of the diuretics with the chronic potassium supplementation. The initial choice of diuretics is carbonic anhydrase inhibitor acetazolamide 14.
Carbonic anhydrase inhibitors seem to be potent in decreasing future attacks of muscle weakness, though the mechanism of carbonic anhydrase inhibitors in hypokalemic periodic paralysis is still unclear 14. Carbonic anhydrase inhibitors promote urine potassium loss and non-anion gap metabolic acidosis, which reduce the patient’s susceptibility to muscle paralysis. It is also suggested that carbonic anhydrase inhibitors increase the opening of the calcium-activated potassium channels. Furthermore, carbonic anhydrase inhibitors also reduce intracellular sodium accumulation, thus reducing the cellular toxicity and prevent muscle degeneration, which may be effective in the treatment of permanent weakness 28. 250 mg twice daily dose of acetazolamide has been effective in lessening the frequency of attacks 29, 31.
The genetic variation in response to acetazolamide treatment had been reported. Patients with SCN4A mutations show less response compared to patients with CACNA1S mutations 14. In a study of 74 identified cases of hypokalemic periodic paralysis, 56% (31/55) of patients with CACNA1S mutations, and only 16% (3/19) of patients with SCN4A mutations showed a response to acetazolamide therapy.[12] Patients with SCN4A mutations had reported the exacerbation of the hypokalemic periodic paralysis with acetazolamide therapy.[9][12] Overall, almost half of the hypokalemic periodic paralysis patients respond to treatment with acetazolamide 28.
FDA recently approved dichlorphenamide for the treatment of hypokalemic periodic paralysis. 50 mg twice daily dose of dichlorphenamide has been more effective than a placebo in reducing the occurrence, severity, and duration of future attacks 28, 55, 60, 61. Dichlorphenamide can be used as the first choice or as a substitute for patients who do not respond or are refractory to acetazolamide 55. Some patients also benefitted from the addition of a potassium-sparing diuretic, either spironolactone (100 mg daily) or triamterene (150 mg daily), to carbonic anhydrase inhibitors or when used as monotherapy 31. Electrolytes need to be monitored regularly in patients who are on diuretics therapy.
While no definitive therapy for the late-onset myopathy has been proven to date, but it is believed that reducing the attacks of muscle weakness helps to mitigate the resulting myopathy 62, 50.
A study also reported the improvement in severity and frequency of attacks with topiramate therapy in 11 years old twins with hypokalemic periodic paralysis, thus necessitates further study regarding the efficacy of topiramate in hypokalemic periodic paralysis 63.
Myopathy treatment
No curative treatment is known for fixed myopathy in hypokalemic periodic paralysis. The effects of muscle weakness are managed as in other disorders with similar manifestations.
- Physiotherapy may help to maintain strength and motor abilities, especially after 40 years of age, when long-lasting muscle weakness is more often seen.
- The physiotherapist must be aware of the following peculiarity of periodic paralysis: that sustained effort results in exacerbation of weakness. Therefore, self-managed exercise should be preferred to superimposed physiotherapy 64.
Pre- or postoperative paralysis
Because of the risk for paralysis preceding or following anesthesia, precautions should be taken during administration of anesthesia to individuals with hypokalemic periodic paralysis. Individuals with hypokalemic periodic paralysis with CACNA1S mutation are susceptible to malignant hyperthermia, as the CACNA1S gene is allelic to the gene that increases susceptibility to malignant hyperthermia 55. Individuals with hypokalemic periodic paralysis with CACNA1S mutation should be managed with a non-triggering anesthetic technique – although general anesthesia using volatile anesthetics and succinylcholine has been reported as safe in a small number of individuals with hypokalemic periodic paralysis.
Surgeons and anesthesiologists must be aware of this circumstance while using the inhalational anesthetics and muscle relaxants like succinylcholine during surgery and be ready to deal with it. Furthermore, the cold environment and the use of saline and dextrose during surgery, and stress due to surgery itself can act as a trigger and result in muscle weakness 55. Potassium monitoring is important in such patients during the peri-surgical period.
General guidelines for perioperative care include the following 13:
- Strict control of serum potassium concentration
- Avoidance of large glucose and salt loads
- Low-carbohydrate diet
- Maintenance of body temperature and acid-base balance
- Careful use of neuromuscular blocking agents and no depolarizing muscle relaxants
Pregnancy and hypokalemic periodic paralysis
During pregnancy, potassium management during the attacks should not differ from the pre-pregnancy state 14. However, drugs like acetazolamide and dichlorphenamide are FDA pregnancy category C so, their use during pregnancy is quite challenging, and risks and benefits of drug use should be weighed in them 14. Some pregnant women prefer not to take these medicines during pregnancy 55.
Hypokalemic periodic paralysis prognosis
The prognosis of hypokalemic periodic paralysis (HypoPP) varies among individuals. The attacks of muscle weakness responds well to oral potassium administration 14. Treatment may prevent, and even reverse, progressive muscle weakness. Although muscle strength starts out normal between attacks, repeated attacks may eventually cause worsening and permanent muscle weakness between attacks, which can cause significant sickness, increase hospital admissions, and therefore can affect the patient’s social and professional life 14. Slowly progressive, permanent weakness in the legs often develops after age 50. Men and women are equally affected. The deaths related due to muscle attacks are rare, but several deaths due to aspiration pneumonia have been reported 48.
Hyperkalemic periodic paralysis
Hyperkalemic periodic paralysis also known as Familial hyperkalemic periodic paralysis, Primary hyperkalemic periodic paralysis, HyperPP or HyperKPP, is a rare, autosomal dominant condition that causes episodes of extreme muscle weakness (adynamia) or paralysis, usually beginning in infancy or early childhood (onset before age 20 years) caused by a mutation in the SCN4A gene that codes for voltage-gated sodium channel causing potassium to shift into the extracellular space due to impaired sodium channel function in skeletal muscle 65, 66, 67, 68, 69. However, in 30 to 40 percent of cases, the cause of hyperkalemic periodic paralysis is unknown 70. Changes in other genes, which have not been identified, likely cause hyperkalemic periodic paralysis in these cases 71. Hyperkalemic periodic paralysis (hyperPP) is characterized by attacks of generalized or focal flaccid muscle weakness, which may also include weakness of the muscles of the eyes, throat, breathing muscles, and trunk. In addition, most people with hyperkalemic periodic paralysis have increased levels of potassium in their blood (hyperkalemia) during attacks with serum potassium concentration greater than 5 mmol/L or an increase of serum potassium concentration of at least 1.5 mmol/L during an attack of weakness 28, 66. Hyperkalemia results when the weak or paralyzed muscles release potassium ions into the bloodstream. In other cases, attacks are associated with normal blood potassium levels (normokalemia). Ingesting potassium can trigger attacks in affected individuals, even if blood potassium levels do not go up. Most often, these episodes involve a temporary inability to move muscles in the arms and legs. Episodes tend to increase in frequency until mid-adulthood, after which they occur less frequently in many people with hyperkalemic periodic paralysis 71. Factors that can trigger attacks include rest after exercise, potassium-rich foods such as bananas and potatoes, stress, fatigue, alcohol, pregnancy, exposure to hot or cold temperatures, certain medications, and periods without food (fasting) 71. Muscle strength usually returns to normal between attacks, although many affected people continue to experience mild stiffness (myotonia), particularly in muscles of the face and hands 71.
Hyperkalemic periodic paralysis affects an estimated 1 in 200,000 to and 1 in 500 000 people 72, 73, 71. In approximately half of affected individuals, attacks of flaccid muscle weakness begin in the first decade of life, with 25% reporting their first attack at age 10 years or older 66. Initially infrequent, the attacks then increase in frequency and severity over time until approximately age 50 years, after which the frequency of attacks declines considerably 66. The major attack trigger is eating potassium-rich foods; other triggers include: cold environment; rest after exercise, stress, or fatigue; alcohol; hunger; and changes in activity level. A spontaneous attack commonly starts in the morning before breakfast, lasts for 15 minutes to one hour, and then passes. Individuals with hyperPP frequently have myotonia (muscle stiffness), especially around the time of an episode of weakness. Paramyotonia (muscle stiffness aggravated by cold and exercise) is present in about 45% of affected individuals. Although most affected individuals have normal muscle strength between attacks, some develop fixed or chronic progressive weakness, independently of the presence of episodic attacks 68, 74, 42. More than 80% of individuals with hyperkalemic periodic paralysis (hyperPP) older than age 40 years with a long disease duration report permanent muscle weakness and about one third develop a chronic progressive myopathy 66, 68, 74. It has been proposed that sodium (Na+) overload in muscle fibers can lead to muscle degeneration that increases with age 75. However, little is known about the development of permanent muscle weakness in hyperKPP, including the pattern of muscle involvement 76.
Hyperkalemic periodic paralysis (hyperPP) is caused by a mutation in the SCN4A gene that codes for voltage-gated sodium channel Na1.4 65. Diagnosis is based on clinical symptoms including the increase of blood potassium level during an episode, but normal levels of blood potassium level in between episodes. Genetic testing can confirm the diagnosis, although they are not always definitive. In case of diagnostic uncertainty, a provocative test such as the potassium challenge test can be employed, although the availability of genetic testing and electrophysiologic studies largely obviates the need for such dangerous tests 66, 77, 78.
Treatment for hyperkalemic periodic paralysis (hyperPP) is focused on avoiding triggers, decreasing the severity of an episode, preventing further attacks and relieving symptoms. Attacks are seldom severe enough to require emergency treatment. At the first sign of muscle weakness, attacks may be prevented or aborted with mild exercise and/or eating carbohydrates (sugars), intravenously injected glucocorticoids, inhalation of salbutamol, or intravenous calcium gluconate 66. Hyperkalemic attacks of weakness can be prevented by frequent meals rich in carbohydrates; continuous use of a thiazide diuretic or a carbonic anhydrase inhibitor; and avoidance of potassium-rich medications and foods, fasting, strenuous work, and exposure to cold 66. But irregular heartbeats (heart arrhythmias) may also occur during attacks, for which emergency treatment is needed. Muscle weakness can become worse with repeated attacks, so treatment to prevent the attacks should occur as soon as possible.
Hyperkalemic periodic paralysis cause
Hyperkalemic periodic paralysis is caused by mutations in the sodium voltage-gated channel alpha subunit 4 gene or SCN4A gene on chromosome 17q23-25 (long arm of chromosome 17) 79, 80, 39, 71. The SCN4A gene belongs to a family of genes that provide instructions for making a critical part (the alpha subunit) sodium channels (Nav1.4) that are abundant in muscles that plays an essential role in muscles used for movement (skeletal muscles) 81, 81. These voltage-gated sodium channel type 4 alpha, which transport positively charged sodium atoms (sodium ions Na+) into cells, play a key role in a cell’s ability to generate and transmit electrical signals. For your body to move normally, your skeletal muscles must tense (contract) and relax in a coordinated way. One of the changes that helps trigger muscle contractions is the flow of positively charged atoms (cations), including sodium ions (Na+), into muscle cells. The SCN4A protein forms channels that control the flow of sodium ions (Na+) into these cells. Mutations in the SCN4A gene alter the usual structure and function of sodium channels. The altered sodium channels stay open too long or do not stay closed long enough, allowing more sodium ions to flow into muscle cells 71. This increase in sodium ions triggers the release of potassium from muscle cells, which causes more sodium channels to open and stimulates the flow of even more sodium ions into these cells. These changes in ion transport reduce the ability of skeletal muscles to contract, leading to episodes of muscle weakness or paralysis.
In 30 to 40 percent of cases, the cause of hyperkalemic periodic paralysis is unknown. Changes in other genes, which have not been identified, likely cause hyperkalemic periodic paralysis in these cases.
Hyperkalemic periodic paralysis triggers
The major attack trigger is eating potassium-rich foods such as bananas and potatoes in affected individuals, even if blood potassium levels do not go up (see Table 3). Fruits high in potassium include cantaloupes, apricots (fresh and dried) dried figs, kiwi fruit, peaches, raisins, banana and prunes. Fruit juices are high in potassium, especially orange and pineapple juice and apricot and peach nectars. High potassium vegetables include artichoke, parsnip, potato, pumpkin, spinach, broccoli, Brussel sprouts, cauliflower, tomato juice, tomato paste and V-8 juice. Lentils and beans are high in potassium. Other foods that are high in potassium include nuts, peanut butter and chocolate.
The other major attack triggers include: exposure to hot or cold temperatures; changes in humidity; potassium supplements; rest after exercise; stress, or fatigue; alcohol; hunger or periods without food (fasting); specific foods or beverages; extra sleep; pregnancy; certain medications; illness of any type; menstruation; and changes in activity level 71, 82, 74. A spontaneous attack commonly starts in the morning before breakfast, lasts for 15 minutes to one hour, and then passes. Individuals with hyperPP frequently have myotonia (muscle stiffness), especially around the time of an episode of weakness. Paramyotonia (muscle stiffness aggravated by cold and exercise) is present in about 45% of affected individuals. Muscle strength usually returns to normal between attacks, although many affected people continue to experience mild stiffness (myotonia), particularly in muscles of the face and hands 71. More than 80% of individuals with hyperkalemic periodic paralysis (hyperPP) older than age 40 years report permanent muscle weakness and about one third develop a chronic progressive myopathy 66.
Of note, attacks occur more frequently on holidays and weekends when people rest in bed longer than usual 66.
Table 3. Potassium rich foods
| Food | Milligrams (mg) per serving | Percent DV* |
|---|---|---|
| Apricots, dried, ½ cup | 755 | 16 |
| Lentils, cooked, 1 cup | 731 | 16 |
| Squash, acorn, mashed, 1 cup | 644 | 14 |
| Prunes, dried, ½ cup | 635 | 14 |
| Raisins, ½ cup | 618 | 13 |
| Potato, baked, flesh only, 1 medium | 610 | 13 |
| Kidney beans, canned, 1 cup | 607 | 13 |
| Orange juice, 1 cup | 496 | 11 |
| Soybeans, mature seeds, boiled, ½ cup | 443 | 9 |
| Banana, 1 medium | 422 | 9 |
| Milk, 1%, 1 cup | 366 | 8 |
| Spinach, raw, 2 cups | 334 | 7 |
| Chicken breast, boneless, grilled, 3 ounces | 332 | 7 |
| Yogurt, fruit variety, nonfat, 6 ounces | 330 | 7 |
| Salmon, Atlantic, farmed, cooked, 3 ounces | 326 | 7 |
| Beef, top sirloin, grilled, 3 ounces | 315 | 7 |
| Molasses, 1 tablespoon | 308 | 7 |
| Tomato, raw, 1 medium | 292 | 6 |
| Soymilk, 1 cup | 287 | 6 |
| Yogurt, Greek, plain, nonfat, 6 ounces | 240 | 5 |
| Broccoli, cooked, chopped, ½ cup | 229 | 5 |
| Cantaloupe, cubed, ½ cup | 214 | 5 |
| Turkey breast, roasted, 3 ounces | 212 | 5 |
| Asparagus, cooked, ½ cup | 202 | 4 |
| Apple, with skin, 1 medium | 195 | 4 |
| Cashew nuts, 1 ounce | 187 | 4 |
| Rice, brown, medium-grain, cooked, 1 cup | 154 | 3 |
| Tuna, light, canned in water, drained, 3 ounces | 153 | 3 |
| Coffee, brewed, 1 cup | 116 | 2 |
| Lettuce, iceberg, shredded, 1 cup | 102 | 2 |
| Peanut butter, 1 tablespoon | 90 | 2 |
| Tea, black, brewed, 1 cup | 88 | 2 |
| Flaxseed, whole, 1 tablespoon | 84 | 2 |
| Bread, whole-wheat, 1 slice | 81 | 2 |
| Egg, 1 large | 69 | 1 |
| Rice, white, medium-grain, cooked, 1 cup | 54 | 1 |
| Bread, white, 1 slice | 37 | 1 |
| Cheese, mozzarella, part skim, 1½ ounces | 36 | 1 |
| Oil (olive, corn, canola, or soybean), 1 tablespoon | 0 | 0 |
Hyperkalemic periodic paralysis inheritance pattern
Hyperkalemic periodic paralysis is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder. In most cases, an affected person inherits the SCN4A gene mutation from one affected parent.
In 30 to 40 percent of cases, the cause of hyperkalemic periodic paralysis is unknown. Changes in other genes, which have not been identified, likely cause hyperkalemic periodic paralysis in these cases.
Figure 3. Hyperkalemic periodic paralysis autosomal dominant inheritance pattern
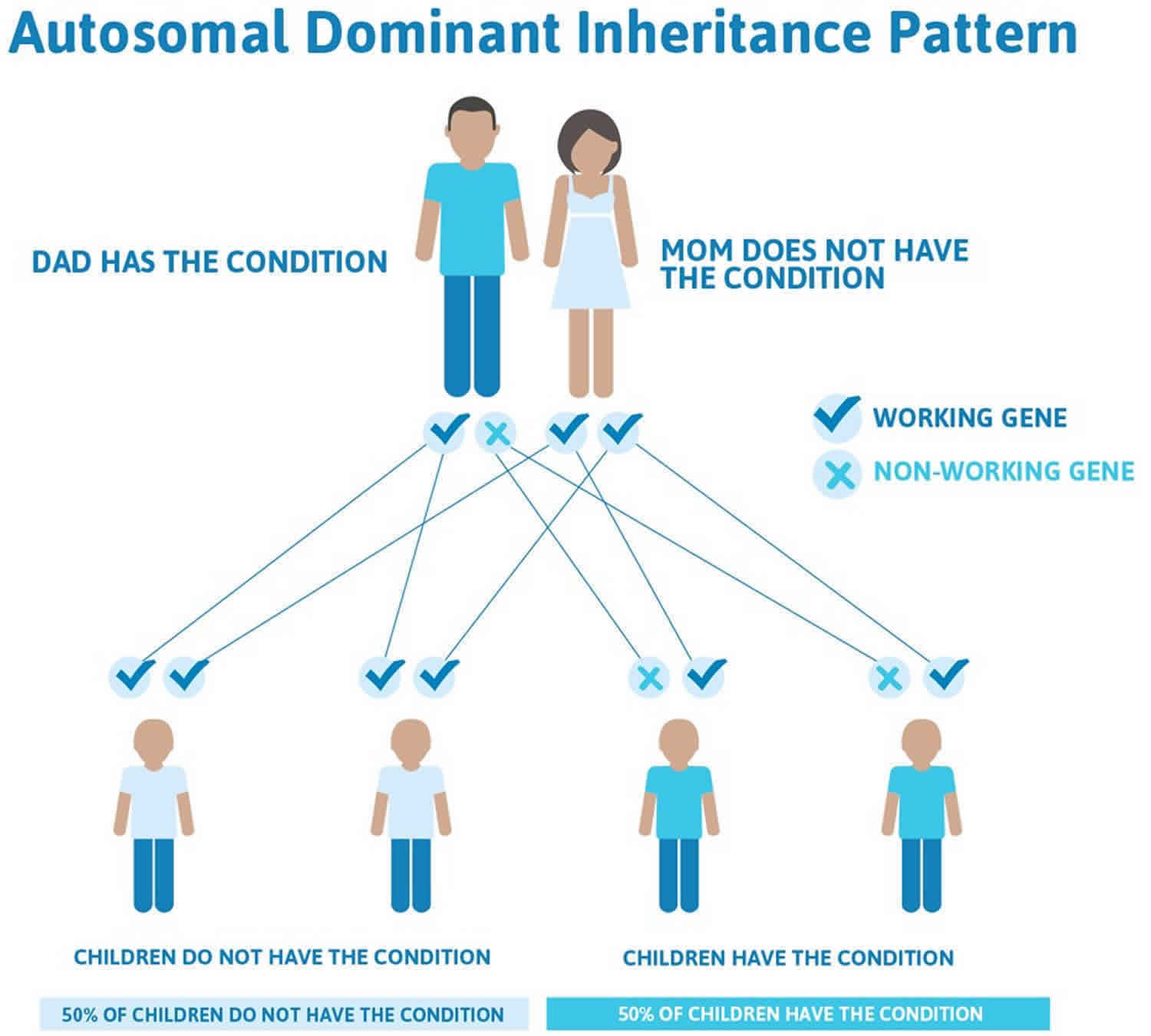
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Hyperkalemic periodic paralysis pathophysiology
Hyperkalemic periodic paralysis (HyperPP) is an autosomal dominantly inherited disease characterized by episodic paralytic attacks with hyperkalemia, and is caused by mutations of the SCN4A gene encoding the skeletal muscle type voltage-gated sodium channel Nav1.4, which is composed of four repeated domains (Domain I–IV) consisting of six transmembrane segments (S1–S6), with S1–S4 forming the voltage-sensing domain (VSD) and S5 and S6 forming the pore domain (PD) 84. The skeletal muscle type voltage-gated sodium channel Nav1.4 is co-expressed with voltage-gated sodium channel beta subunit 1 encoded by the SCN1B gene 85, and a recent study revealed the structure of the Nav1.4 complex by cryo-electron microscopy 86.
Nav1.4 has three major functional states; the closed state, the open state, and the inactivated state 84. The ion conductive pore of Nav1.4 remains closed at the resting potentials of the myofibers (the closed state). Depolarization of the membrane potential induces the conformational change of the Nav1.4 proteins mainly in the voltage-sensing domain (VSD), resulting in the pore gate opening which allows the passage of sodium ions (Na+) (the open state). Prolonged depolarizing stimulus induces another conformational state, the inactivated state, which prevents the passage of sodium ions (Na+) through the pore by additional structural rearrangement. The inactivated state is divided into two types according to the time course; fast inactivation induced by sustained depolarization for hundreds of milliseconds and slow inactivation induced by sustained depolarization for seconds to minutes. Fast inactivation has been shown to be related to the structural rearrangement in voltage-sensing domain (VSD) of Domain IV followed by that in the cytosolic linker between Domain III and Domain IV 87, 88. Although the structure–function relationship in slow inactivation has not been fully elucidated, previous reports indicated that the structural rearrangement in pore domain (PD) would be associated with it 2.
The pathological mechanism of hyperkalemic periodic paralysis was suggested to be associated with gain-of-function changes for Nav1.4 gating, some of which are defects of slow inactivation. Excitable cells at the motor endplate have a resting membrane potential of −70 mV and a threshold potential of −55 mV. These gross voltages are predominantly due to the resting potentials of potassium (Ek −75 mV) and sodium (Ena +55 mV). HyperKPP pathophysiology is triggered by a slight increase in extracellular potassium (K+), most commonly resulting from the ingestion of potassium-rich food, rest after a heavy workout, periods of fasting, emotional stress, pregnancy, exposure to cold, surgery and anaesthesia 74. This slight potassium (K+) increase may still be within normal laboratory values but causes a minor membrane depolarisation. If an impulse is generated, an unknown percentage of mutated NaV 1.4 channels may fail to inactivate, leading to a prolonged increase in intracellular sodium ion (Na+) and persistent cell depolarisation 89. If intervention occurs at this point, often paralysis can be avoided, and weakness will be transient. If this continues, the cycle undergoes a pathologic feedback loop with worsening membrane excitability at baseline from (1) the higher intracellular Na+ concentration driving K+ extracellularly and (2) continuation of the initial hyperkalemic trigger event. The net result of this cycle is the subsequent loss of electrical excitability and thus paralysis 90.
The sodium channel has an alpha subunit and a beta subunit. The alpha subunit of the sodium channel is a 260-kd glycoprotein comprising about 1800-2000 amino acids. This channel is highly conserved evolutionarily from Drosophila to human. It has 4 homologous domains (I-IV) that fold to form a central pore, each with 225-325 amino acids. Each domain consists of 6 hydrophobic segments (S1-S6) traversing the cell membrane. The main functions of the sodium channel include voltage-sensitive gating, inactivation, and ion selectivity. The extracellular loop between S5 and S6 dips into the plasma membrane and participates in the formation of the pore. The S4 segment contains positively charged amino acids at every third position and functions as a voltage sensor. Conformation changes may occur during depolarization, resulting in activation and inactivation of the channel. The cellular loop between domain III-S6 and domain IV-S1 acts as an inactivating gate.
Many SCN4A mutations associated with diseases have been found in functional studies using cultured cell lines and Xenopus oocytes expressing mutant Nav1.4 channels 84, and most of them showed gain-of-function mutations in Nav1.4. For paramyotonia congenita or sodium channel myotonia, the mutant channels represent defective fast inactivation and/or enhancement of activation, leading to pathological myotonia 84. For HyperPP, two representative variants have been reported so far, T704M and M1592V, and they are located in the transmembrane segments of the PD and represent defective slow inactivation 91, 92.
Two populations of channels exist, mutant and wild-type; the impaired fast-inactivation results in prolonged depolarization of the mutant muscle fiber membranes and can explain the 2 cardinal symptoms of these disorders, myotonia and weakness. In hyperkalemic periodic paralysis, a gain of function occurs in mutant channel gating, resulting in an increased sodium current excessively depolarizing the affected muscle. Mild depolarization (5-10 mV) of the myofiber membrane, which may be caused by increased extracellular potassium concentrations, results in the mutant channels being maintained in the noninactivated mode. The persistent inward sodium current causes repetitive firing of the wild-type sodium channels, which is perceived as stiffness (ie, myotonia).
If a more severe depolarization (20-30 mV) is present, both normal and abnormal channels are fixed in a state of inactivation, causing weakness or paralysis. Thus, subtle differences in severity of membrane depolarization may make the difference between myotonia and paralysis. Temperature sensitivity is a hallmark of paramyotonia congenita. Cold exacerbates myotonia and induces weakness. A number of mutations are associated with this condition, 3 of them at the same site in the S4 segment. These mutations replace arginine with other amino acids and neutralize this highly conserved S4 positive charge. Mutations of these residues are the most common cause of paramyotonia congenita. Some of the possible mechanisms responsible for temperature sensitivity include the following:
- Temperature may differentially affect the conformational change in the mutant channel.
- Lower temperatures may stabilize the mutant channels in an abnormal state.
- Mutations may alter the sensitivity of the channel to other cellular processes, such as phosphorylation or second messengers.
Most cases of hyperkalemic periodic paralysis are due to 2 mutations in SCN4A, T704M, and M1592V. Mutations in the sodium channel, especially at residues 1448 and 1313, are responsible for paramyotonia congenita.
Hyperkalemic periodic paralysis prevention
Preventive measures for individuals with hyperkalemic periodic paralysis (hyperPP) consist of frequent meals rich in carbohydrates and avoidance of the following 66:
- Potassium-rich medications and foods (e.g., fruits, fruit juices)
- Fasting
- Strenuous work
- Exposure to cold
- Opioids or depolarizing agents such as potassium, anticholinesterases, and succinylcholine as part of general anesthesia. These can aggravate a myotonic reaction and induce masseter spasms and stiffness of respiratory muscles, which may impair intubation; mechanical ventilation may also be impaired.
- Drugs known as ACE-inhibitors for the treatment of arterial hypertension. These may lead to hyperkalemia as a side effect, especially if they are combined with potassium-sparing diuretics (e.g., spironolactone) and/or renal function is impaired.
- Alterations of serum osmolarity, pH, and hypothermia-induced muscle shivering and mechanical stimuli during general anesthesia 93. These can exacerbate the myotonic reaction in individuals with hyperkalemic periodic paralysis.
A low potassium, high carbohydrate diet, and light exercise may help prevent attacks. Avoiding fasting, strenuous activity, or cold temperatures also may help.
Early start to the day
As attacks occur more frequently on holidays and weekends when people rest in bed longer than usual, individuals are advised to rise early and have a full breakfast.
Lifestyle
Individuals should prioritize avoidance or minimization of triggers whenever possible by keeping stress levels low and avoiding exercise that is overly intense (as rest after such exercise is a trigger).
Diuretics (water pills)
It is often advisable to prevent hyperkalemic attacks of weakness by the continuous use of a thiazide diuretic or a carbonic anhydrase inhibitor, such as acetazolamide or dichlorphenamide.
Note: In a trial of dichlorphenamide, the median attack rate was lower in participants with hyperkalemic periodic paralysis on dichlorphenamide than in participants with hyperkalemic periodic paralysis on placebo (0.9 vs 4.8), but the difference in median attack rate was not significant 47. Diuretics are used in modest dosages at intervals from twice daily to twice weekly.
- Thiazide diuretics are preferable because they have fewer side effects than either acetazolamide or dichlorphenamide therapy.
- The dosage should be kept as low as possible (e.g., 25 mg hydrochlorothiazide daily or every other day). In severe cases, 50 mg or 75 mg of hydrochlorothiazide should be taken daily very early in the morning.
- Individuals should be monitored so that the serum potassium concentration does not fall below 3.3 mmol/L or the serum sodium concentration below 135 mmol/L 94. Thiazides may be helpful even if the serum potassium concentration is in the normal range 95.
- Four weeks after start of diuretic treatment, effects should be evaluated by muscle strength measurement and MRI of proximal leg muscles.
Evaluation of relatives
It is appropriate to evaluate apparently asymptomatic older and younger at-risk relatives of an individual with hyperkalemic periodic paralysis in order to identify as early as possible those who would benefit from initiation of preventive measures, particularly those that would decrease the risk of unexpected acute paralysis or anesthetic events. Evaluations include:
- Molecular genetic testing if the pathogenic variant in the family is known;
- Full neurologic examination to rule out muscular weakness and electromyogram (EMG) to rule out myotonia if the pathogenic variant in the family is not known.
At-risk relatives who have not undergone molecular genetic testing or clinical evaluation (i.e., neurologic examination and electromyogram (EMG)) must be considered at risk for hyperkalemic periodic paralysis-related complications and precautions are indicated – particularly during anesthesia.
Hyperkalemic periodic paralysis symptoms
Hyperkalemic periodic paralysis symptoms include attacks of muscle weakness or loss of muscle movement (paralysis) that come and go. There is normal muscle strength between attacks. Attacks usually begin in childhood. How often the attacks occur varies. Some people have several attacks a day. They are usually not severe enough to need therapy. Some people have associated myotonia, in which they cannot immediately relax their muscles after use.
The muscle weakness or paralysis:
- Most commonly occurs at the shoulders, back, and hips
- May also involve the arms and legs, but does not affect muscles of the eyes and muscles that help with breathing and swallowing
- Most commonly occurs while resting after activity or exercise
- May occur on awakening
- Occurs on and off
- Usually lasts 15 minutes to 1 hour, but may last up to an entire day
Triggers may include:
- Eating potassium-rich foods or taking medicines that contain potassium
- Rest after exercise
- Exposure to cold
- Skipping meals
- Stress
Hyperkalemic periodic paralysis (hyperPP) has three clinically distinct signs and symptoms:
- Without myotonia (muscle stiffness),
- With clinical or electromyographic (EMG) myotonia, or
- With paramyotonia congenita (muscle stiffness aggravated by cold and exercise beginning in infancy or early childhood).
In all three forms, the course of the paralytic attacks is the same 96, 77. Electrical myotonia (muscle stiffness) can be demonstrated on EMG in 50 to 75% of patients with hyperPP, while less than 20% manifest clinically 31. In those with muscle stiffness (myotonia), i.e., a tonic spasm of muscle 97, the myotonia is often mild and can be provoked with percussion or activity in the face, tongue, forearms, and thenar eminence 74. The myotonia eases with repetitive activity 77. From birth onwards, those with paramyotonia congenita (muscle stiffness aggravated by cold and exercise beginning in infancy or early childhood) experience muscle stiffness that increases with continued activity (paramyotonia) and is cold-induced 72. Paralytic attacks can occur at any time, though often occur spontaneously in the morning prior to breakfast, last up to an hour, and unpredictably subside 77, 66. Attacks may be provoked or worsened by anesthesia 78, rest after exercise, potassium loading, cold environments, hunger, emotional stress, glucocorticoids, or pregnancy. During attacks, individuals may be hyperkalemic (high potassium levels) or normal serum potassium levels (normokalemic) 71. The concomitant rise in serum potassium levels can range from upper normal values to those in the cardiotoxic spectrum 70. After an attack, serum potassium levels may be transiently low due to the elimination of potassium from the kidneys and the reuptake of potassium by muscle 96. Usually, individuals do not experience cardiac arrhythmias or respiratory insufficiency during attacks 66. After an attack, individuals may feel pain for up to several days in the involved muscle groups 77. Between attacks, affected individuals have normal serum potassium levels (normokalemia), normal sensation and muscle stretch reflexes, and normal muscle strength, although they may experience minor myotonia that does not hinder voluntary movement 74. “Lid lag” secondary to eyelid myotonia may be the only clinical sign present between attacks 77, 66.
The attacks of flaccid muscle weakness associated with hyperkalemic periodic paralysis (hyperPP) usually begin in the first decade of life and increase in frequency and severity over time, with 25% experiencing their sentinel attack in the second decade of life. Initially infrequent, the attacks increase in frequency and severity over time until approximately age 50 years, after which the frequency declines considerably.
Pattern of attacks
A spontaneous attack commonly starts in the morning before breakfast, lasts for 15 minutes to an hour, and then passes and may not happen often, though some patients have several episodes per day 66. In about 20% of affected individuals the attacks last considerably longer, from more than two days to more than a week 66.
In some individuals, a burning or prickling sensation or “pins-and-needles” sensation that is usually felt in the hands, arms, legs, or feet (paresthesias) probably induced by the hyperkalemia herald the weakness. During an attack of weakness, the muscle stretch reflexes are abnormally diminished or absent. Difficulty swallowing (dysphagia) during an attack of weakness has also been described 98. The strength of the attacks is not always consistent; sometimes the patient only feels fatigued, but can still move around slowly. Other times the patients are completely paralyzed. Sometimes attacks may come very suddenly.
Individuals most commonly describe their attacks as stiffness followed by weakness, although many have described their attacks as some other permutation of weakness and/or stiffness. The arms and hands are just as frequently affected as the thighs and calves 74.
Frequency of attacks can vary greatly among individuals. Some have attacks every day, others several times a month; others have them every few months or less often.
Usually, cardiac arrhythmia or respiratory insufficiency does not occur during the attacks. When present, respiratory insufficiency manifests as shortness of breath. In a study by Charles et al 74, 26% of subjects reported that their breathing musculature was affected and 62% reported that their face was affected during attacks. The mouse model has demonstrated a resistance to weakness triggered by hyperkalemia in diaphragmatic muscle as compared to skeletal muscle 99.
Individuals learn that they have to stay warm, avoid unaccustomed heavy exercise, and not sit still too long at a time. Many find they can abort a developing episode by drinking a sweet beverage or eating some hard candy at the first sign of an attack. Unfortunately this can lead to weight gain.
Between paralytic attacks findings
After an attack, affected individuals report clumsiness, weakness, and irritability, and in 62% muscle pain secondary to the attack 66. One observational study identified fibromyalgia in half of the individuals surveyed who had hyperPP 100. Between attacks, the majority report no or mild symptoms. However, 12% report severe symptoms between attacks that impair activities of daily living 100.
Muscle issues
Individuals with hyperPP frequently (i.e., >50% of the time) have myotonia (muscle stiffness), especially around the time of an episode of weakness. Mild myotonia (muscle stiffness) that does not impede voluntary movements is often present between attacks 66. Myotonia is most readily observed in the facial, lingual, thenar, and finger extensor muscles; eyelid myotonia (lid lag myotonia) has been rarely reported. Paramyotonia (muscle stiffness aggravated by cold and exercise) is present in about 45% of affected individuals 66. Of individuals with myotonia, 37% have experienced progressive myopathy, while of those reporting absence of myotonia, 33% have experienced progressive myopathy 74.
Bradley et al 68 reported more than 80% of the affected individuals older than 40 years to have permanent muscle weakness and approximately one third of older affected individuals developed a chronic progressive myopathy. The myopathy mainly affects the pelvic girdle and proximal and distal lower-limb muscles. A more recent study using MRI reveals an even earlier onset of progressive myopathy: progressive myopathy was observed even in individuals at the second and third decades of life with myopathic findings prominent in the gastrocnemius muscle. Muscle atrophy, edematous change, and fatty change were prominent in the superficial posterior compartment of the lower leg 101.
Thyroid dysfunction
As shown by an observational study, individuals with hyperPP appear to be at higher risk for thyroid dysfunction than those in the general population 74.
Hyperkalemic periodic paralysis complications
Health problems that may be due to hyperkalemic periodic paralysis include:
- Kidney stones (a side effect of medicine used to treat the condition)
- Irregular heart beat
- Muscle weakness that slowly continues to get worse
Hyperkalemic periodic paralysis diagnosis
The diagnosis of hyperkalemic periodic paralysis is established based on the clinical presentation of muscle weakness or paralysis, hyperkalemia and a positive family history and sometimes with the use of molecular genetic testing in cases of diagnostic uncertainty 66, 74. The diagnosis is suggested by a history of attacks of weakness or paralysis, a positive family history, and the presence of myotonia (muscle stiffness) or paramyotonia (muscle stiffness aggravated by cold and exercise). Serum creatine kinase (CK) values may be elevated, and some individuals exhibit calf hypertrophy 74. The muscles are typically well-developed 70, 77; however, a large proportion of individuals with hyperkalemic periodic paralysis develop a chronic progressive proximal myopathy as they age 102, 31. Individuals without muscle stiffness (myotonia) between attacks are much more susceptible to developing this progressive myopathy than are individuals with muscle stiffness (myotonia) 72, 66.
Muscle biopsy is non-specific, though will frequently reveal muscle fiber atrophy with vacuoles 77, 78. Genetic testing is positive in approximately 60% of individuals who meet clinical diagnostic criteria. An electromyogram (EMG) may show myotonic signs, which strongly support the diagnosis, although approximately half of those with the most common mutation show no such signs 66. Provocative tests, such as the potassium challenge test, pose obvious risks to the patient but may be done to support the diagnosis 74. The availability of genetic testing and electrophysiologic studies largely obviates the need for such dangerous tests 66, 77, 78.
Clinical findings
- History of at least two attacks of flaccid limb weakness (which may also include weakness of the muscles of the eyes, throat, breathing muscles, and trunk)
- Onset or worsening of an attack as a result of oral potassium intake
- Disease manifestations before age 20 years
- Absence of cardiac arrhythmia between attacks
- Normal psychomotor development
Family history
- Typically, at least one affected first-degree relative
- Note: Absence of a family history suggestive of hyperkalemic periodic paralysis does not preclude the diagnosis.
Electromyogram (EMG)
- During the attack, EMG demonstrates a reduced number of motor units or may be silent (no insertional or voluntary activity).
- In the intervals between attacks, EMG may reveal myotonic activity (bursts of muscle fiber action potentials with amplitude and frequency modulation, firing rate generally between 20 and 150 Hz), even though myotonic stiffness may not be clinically present.
- In some individuals, especially in those with permanent weakness, a myopathic pattern may be visible.
- Note: Approximately 50% of affected individuals have no detectable electric myotonia.
Suggestive laboratory findings during attacks
- Hyperkalemia (serum potassium concentration >5 mmol/L) or an increase of serum potassium concentration of at least 1.5 mmol/L.
- Note: Serum potassium concentration seldom reaches cardiotoxic levels, but changes in the EKG (increased amplitude of T waves) may occur.
- Elevated serum creatine kinase (CK) concentration (sometimes 5-10x the normal range)
Suggestive laboratory findings between attacks
- Normal serum potassium concentration and muscle strength between attacks
- Note: At the end of an attack of weakness, elimination of potassium via the kidney and reuptake of potassium by the muscle can cause transient hypokalemia that may lead to the misdiagnosis of hypokalemic periodic paralysis.
- Elevated serum creatine kinase (CK) concentration with normal serum sodium concentration
Genetic testing
The diagnosis of hyperkalemic periodic paralysis (hyperPP) is established in affected individual with suggestive clinical and lab findings and a heterozygous pathogenic variant in SCN4A ionic channel gene identified by molecular genetic testing, but up to 20% of the cases would be tested negative for the gene mutation 66. In cases of diagnostic uncertainty, provocative tests such as a potassium challenge test can be employed but simultaneously carrying risks of triggering a severe attack 66. Muscle biopsy is generally not advisable as the findings are non-specific 66.
Provocative testing
In case of diagnostic uncertainty (i.e., in the absence of a measurement of during an attack serum potassium concentration and normal molecular genetic studies), a provocative test may be employed to ensure the diagnosis 66. The availability of genetic testing and electrophysiologic studies largely obviates the need for such dangerous tests 66. Systemic provocative testing carries the risk of inducing a severe attack; therefore, such testing must be performed by an experienced physician and a stand-by anesthetist, with close monitoring of the EKG and serum concentration of potassium 66.
The classic provocative test consists of the administration of 2-10 g potassium under clinical surveillance with serum potassium concentration and strength measured at 20-minute intervals 66. Usually, an attack is induced within an hour and lasts approximately 30 to 60 minutes, accompanied by an increase in serum potassium concentration, similar to spontaneously occurring attacks of weakness.
Note: This test is contraindicated in individuals who already have hyperkalemia and in those individuals who do not have adequate renal or adrenal function.
Hyperkalemic periodic paralysis treatment
Treatment for hyperkalemic periodic paralysis is symptomatic, as there is no curative treatment for hyperPP. Hyperkalemic periodic paralysis treatment is focused on correcting the levels of potassium in your blood and preventing episodes with lifestyle changes.
A tablespoonful of calcium gluconate syrup stirred into a glass of sweet beverage may stop a mild episode in the early stages. Calcium gluconate syrup is a mineral supplement available off the shelf and is found in most pharmacies. Mild exercise with intake of carbohydrates may be helpful to relieve symptoms. Some patients find that drinking 1/4 cup of tonic water daily (available at the grocer’s) helps ease symptoms. Albuterol, administered by nebulizer or inhaler (puffer), is an effective treatment for some patients. For those who have frequent episodes more aggressive treatment is advisable, especially since some patients with hyperkalemic periodic paralysis may develop permanent muscle weakness after years of episodes.
Preventive measures such as avoidance of potassium-rich foods and medications, fasting, strenuous work and exposure to cold are equally important 74, 66.
The carbonic anhydrase inhibitors ‘Diamox’ (acetazolomide) and ‘Keveyis’ (dichlorphenamide) are often prescribed for hyperkalemic periodic paralysis patients. About 25% of patients do not respond to carbonic anhydrase inhibitors and must take other medication. Other medications used to treat hyperkalemic periodic paralysis include the potassium-wasting diuretics (thiazide diuretics) – Lasix (furosemide), Hydrodiuril (hydrochlorothiazide), etc. For those who have significant myotonia, the anti-arrhythmic drug Mexitil (mexiletine) is used in low doses; 50-150 mg daily is reported to be adequate by patients. Very low doses of Paxil (paroxetine) (10-20 mg daily) have also been reported to help relieve myotonia.
The individuals’ serum electrolytes should be monitored, and their serum potassium and sodium levels should not fall below 3.3 mmol/L and 135 mmol/L, respectively 103.
Individuals with hyperPP are at risk of complications from surgery and anaesthetics 74. Non-anaesthetic surgical complications were reported in 22.8% of cases, including temporary complete paralysis postoperatively, respiratory difficulties and hyperthermia. Up to 29.9% of patients experienced attacks during general anaesthesia. During anaesthesia, non-depolarizing muscle relaxants are preferred 74, 66. The use of depolarizing muscle relaxants such as suxamethonium as well as anticholinesterase types of reversal agents should be avoided as there will be a risk of masseter spasm and respiratory muscle stiffness rendering endotracheal intubation and mechanical ventilation difficult 104. Perioperatively, maintaining normoglycemia (normal blood sugar level) and normal body temperature are crucial to prevent attacks. Postoperatively, the affected patient needs to be closely monitored for signs of respiratory distress attributable to the disease itself, anesthetic drugs that depress respiration and hypothermia 66. There is currently no agreed recommendation for the choice of regional or general anaesthesia for patients with hyperPP.
Table 4. Treatment of Hyperkalemic Periodic Paralysis (hyperPP)
| Signs and symptoms | Treatment | Considerations/Other |
|---|---|---|
| Attacks of flaccid muscle weakness | Continuing mild exercise &/or oral ingestion of carbohydrates (2 g glucose per kg body weight) at onset of weakness | May prevent or abort attacks |
| Intravenous (IV) glucocorticoids or inhalation of 2 puffs of 0.1 mg salbutamol | May abort or attenuate attacks | |
| Calcium gluconate (0.5-2 g taken intravenously) 1 | May terminate attacks in some persons | |
| Myotonia associated with general anesthesia 2 | An induction sequence incorporating inhalation of oxygen, cricoid pressure, thiopental, & 2x the ED95 dose of an intermediate or short-action non-depolarizing muscle relaxant, followed by intubation, is a reasonable approach to securing the airway in persons with hyperPP. | Avoid opioids or depolarizing agents incl potassium, anticholinesterases, & succinylcholine, which can aggravate a myotonic reaction & induce masseter spasms & stiffness of respiratory muscles. Alterations of serum osmolarity, pH, & hypothermia-induced muscle shivering & mechanical stimuli can exacerbate myotonic reaction. |
| Inhalational induction may also be possible for hyperPP & is well tolerated in those undergoing elective surgery. | ||
| Post anesthesia general & respiratory muscle weakness | To prevent this complication the following are recommended: 3 Glucose infusion Maintain normal body temperature. Maintain serum potassium at low level. | |
| Myotonia | Mexiletine has been used to treat myotonia in this disorder 105. |
Footnotes:
- One case report suggested that intravenous magnesium is beneficial as well 106.
- Because the generalized muscle spasms associated with such attacks may lead to an increase in body temperature, individuals with hyperkalemic periodic paralysis have been considered to be susceptible to malignant hyperthermia. Most likely, anesthesia-related complications suggestive of a malignant hyperthermia crisis result from severe myotonic reactions 94, 107.
- Klingler et al 107, Mackenzie et al 108, Jurkat-Rott & Lehmann-Horn 109, Barker 110
Table 5. Recommended monitoring of individuals with Hyperkalemic Periodic Paralysis (hyperPP)
| System/Concern | Evaluation | Frequency |
|---|---|---|
| Diuretic-induced hypokalemia 1 | Serum potassium concentration (target: between 3.0 & 3.5 mmol/L) | Every 6 mos |
| Muscle weakness | Neurologic exam w/focus on muscle strength in the legs in order to detect permanent weakness | Annually |
| MRI of the leg muscles 2 | Every 1-3 yrs | |
| Thyroid dysfunction | Thyroid function testing | Annually |
Footnote:
- If on continuous prophylactic diuretic treatment
- To judge how much normal muscle tissue is preserved and whether edema is present. MRI is ideal, but only investigational at this time.
Pregnancy management
More than 90% of affected women report an increase in attack frequency during pregnancy. While approximately 80% reported improved muscle weakness during attacks, 75% also reported worse muscle stiffness during attacks 74, 104. In contrast, however, are case reports describing improvement during pregnancy [Finsterer et al 2017, Huang et al 2019].
Women who are chronically treated with a diuretic may continue treatment in pregnancy. Human data on prenatal exposure to acetazolamide have not demonstrated an increased risk of fetal malformations. Human data on the use of oral dichlorphenamide therapy during pregnancy – and whether it leads to an increased risk of malformations in exposed fetuses – are limited.
Hyperkalemic periodic paralysis life expectancy
Most people with hyperkalemic periodic paralysis lead reasonably normal lives. Many cases of hyperKPP are mild, and even those who are severely affected can have their symptoms eased through medication and attention to diet and lifestyle issues. There is no cure, but most people manage to lead well-rounded and fulfilling lives. Several people with hyperkalemic periodic paralysis have lived well into their 80’s, in complete control of their mental faculties and capable of independent life. However, some people with hyperKPP have physically-compromised lives, especially patients with frequent and severe attacks which may lead to permanent muscle weakness. But many are quite active. Furthermore, permanent muscle weakness is a fact of life for many older patients, and activities like climbing stairs and walking long distances may become very fatiguing. Some old people with hyperkalemic periodic paralysis use a wheelchair or ‘scooter’ outside their home, but are mobile and on their feet inside the house. Very few rely on a wheelchair full time unless their condition is complicated by other problems.
Andersen-Tawil syndrome
Andersen-Tawil syndrome also called Andersen syndrome, long QT syndrome 7 or LQTS7, is a rare genetic disorder that causes episodes of muscle weakness (periodic paralysis), abnormalities affecting the electrical system of the heart that can cause abnormal life-threatening heart rhythms (arrhythmias), and a variety of distinctive facial and skeletal features, such as short stature, clinodactyly (an inward curvature of the 5th fingers), short index fingers, fused or webbed second and third toes (syndactyly), scoliosis (crooked spine), widely spaced eyes (ocular hypertelorism), low-set ears, broad nose, a broad forehead, and a small jaw (micrognathia) 111, 112, 113, 114, 115, 6. The specific signs and severity can vary greatly from one person to another, even among members of the same family or they may exist in other family members who do not experience muscle weakness or paralysis. Some individuals will not develop all of the facial and skeletal features. Distinctive facial features may be so mild as to go unnoticed 116. Periodic paralysis begins early in life, and episodes last from hours to days. These episodes may occur after exercise or long periods of rest, but they often have no obvious trigger. Muscle strength usually returns to normal between episodes. However, mild muscle weakness may eventually become permanent.
The relationship between Andersen-Tawil syndrome and potassium is inconsistent, and it varies between patients 117. It’s also possible for one patient to have different experiences at different times. Some attacks are caused by a rise in blood potassium levels (hyperkalemia) or fall in blood potassium levels (hypokalemia) but can also happen with normal blood potassium levels (normokalemia). The levels of rise or fall in potassium levels in Andersen Tawil syndrome are not dramatic as in other forms of periodic paralysis and so treatment does not usually include lowering or raising the blood levels of potassium.
Andersen-Tawil syndrome is sometimes referred to as long QT syndrome 7 because some individuals in early reports of the disorder had a prolonged QT interval, which is measured on an electrocardiogram (ECG) and indicates that the heart muscle is taking longer than usual to recharge between beats. Long QT means that there is a longer than normal period of time between the start of the Q wave and the end of the T wave as seen on an ECG. The prolongation of this period tends to trigger irregular heart rhythms. In people with Andersen-Tawil syndrome, the most common changes affecting the heart are ventricular arrhythmia, which is a disruption in the rhythm of the heart’s lower chambers (the ventricles), and long QT syndrome. However, subsequent clinical reports have shown the QT interval is not prolonged or only mildly prolonged in most cases. Instead, the Q-U interval is markedly prolonged. In addition, unlike most forms of long QT syndrome, Andersen-Tawil syndrome is associated with symptoms in addition to disturbances of the electrical system of the heart. Although still sub-classified as a form of long QT syndrome, Andersen-Tawil syndrome is recognized as separate from traditional long QT syndromes. Long QT syndrome is a heart condition that causes the heart (cardiac) muscle to take longer than usual to recharge between beats. The irregular heartbeats can lead to discomfort, such as the feeling that the heart is skipping beats (palpitations). Uncommonly, the irregular heartbeats can cause fainting (syncope), and even more rarely, sudden death due to cardiac arrest 118.
Physical abnormalities associated with Andersen-Tawil syndrome typically affect the face, other parts of the head, and the limbs. These features often include a very small lower jaw (micrognathia), dental abnormalities (such as crowded teeth), low-set ears, widely spaced eyes, fusion (syndactyly) of the second and third toes, and unusual curving of the fingers or toes (clinodactyly). Some affected people also have short stature and an abnormal side-to-side curvature of the spine (scoliosis).
The signs and symptoms of Andersen-Tawil syndrome vary widely, and they can be different even among affected members of the same family. About 60 percent of affected individuals have all three major features (periodic paralysis, cardiac arrhythmia, and physical abnormalities) and up to 80% of the cases express two of the three major features 119.
About 60% of cases of Andersen-Tawil syndrome are caused by mutations in the KCNJ2 gene, a gene encoding the inward rectifier potassium channel 2 protein (Kir2.1) 120, 121. The cause of the remaining 40% of Andersen Tawil syndrome cases remains unknown. Andersen-Tawil syndrome is inherited in an autosomal dominant pattern 122.
The terms Andersen-Tawil syndrome type 1 or type 2 are also used in the medical literature. Andersen-Tawil syndrome type 1 (ATS1) refers to cases caused by a known KCNJ2 gene mutation; Andersen-Tawil syndrome type 2 (ATS2) refers to cases without an identified KCNJ2 mutation 123.
In a study involving 57 Andersen-Tawil syndrome affected individuals (82% female; age at first visit 20 ± 15 years, range 1.5 years to 51 years) and their 61 affected relatives (54% female; age 24 ± 21 years, range 1 month to 69 years) 124. Overall, 97% of patients with Andersen-Tawil Syndrome type 1 (ATS1) exhibited heart symptoms (more frequently a large U-wave and ventricular bigeminy), and 75% of patients presented dysmorphic features (more often a small mandible) (see Figure 4 and 5) 124. A total of 77 patients with Andersen-Tawil Syndrome type 1 (ATS1) had the documentation of ventricular tachycardia (VT) and/or nonsustained VT during their clinical evaluation, and in 58 (75%) of 77 of them episodes of bidirectional VT (BidVT). In most cases, the episodes of bidirectional VT (BidVT) were observed as brief “interludes” that interrupted prolonged episodes of polymorphic VT (Figure 6). A family history of sudden death was present in 11 (19%) of 57 kindreds, with a total of 19 individuals (68% female), who died suddenly at 29 ± 18 years of age 124. All sudden death cases occurred in families in which the KCNJ2 gene mutation segregated with the Andersen-Tawil syndrome type 1 (ATS1) phenotype 124. The highest incidence of life-threatening arrhythmic events in that population occurred between the third and the fifth decades 124. Four cases, the arrhythmic events occurred within the first decade of life, suggesting that Andersen-Tawil syndrome may manifest with life-threatening arrhythmic event during childhood 124.
Andersen-Tawil syndrome exact prevalence is unknown, although it is estimated to affect 1 in 1 million people worldwide 6, 115. About 200 affected individuals have been described in the medical literature. Researchers believe that Andersen-Tawil syndrome accounts for less than 10 percent of all cases of periodic paralysis. Consequently information on the clinical course derive from case series that are either small 125 or devoid of outcome data 126.
Andersen-Tawil syndrome needs to be distinguished from other forms of periodic paralysis including hypokalemic periodic paralysis (HypoPP), hyperkalemic period paralysis (HyperPP), and thyrotoxic periodic paralysis (TPP). Other conditions that cause a prolonged QT interval or syncope should be ruled out as well including vasovagal syncope, orthostatic hypotension, hypertrophic cardiomyopathy, arrhythmogenic right ventricular cardiomyopathy, and catecholamingergic polymorphic ventricular tachycardia (CPVT).
Treatment for Andersen Tawil syndrome is complicated because different patients have different symptoms, and what helps one patient might hurt another 117. For this reason, it is crucial that doctors come up with an individualized treatment plan for each patient. It is also essential that the patient is followed by a neurologist experienced with managing patients with periodic paralysis as well as a cardiologist for regular follow-up to determine if and when they may need to be on heart medications to prevent irregularities in the heart rhythm 117. Medications which regulate the heart are often prescribed, other treatment depends on the individual and their reaction to potassium. Patients with severe heart rhythm problems may require a pacemaker-like device.
Beta-blockers such as propranolol, anti-arrhythmics such as flecainide, and calcium-channel blockers such as verapamil are possible options for heart rhythm problems, but some of these heart medications could make muscle weakness worse, so caution is needed. Also, people with Andersen Tawil syndrome should avoid medications that can make their heart’s long QT interval even longer. See CredibleMeds (https://crediblemeds.org) for a complete and updated list (free registration required). Salbutamol inhalers, which may be used in the treatment of primary hyperkalemic periodic paralysis, should be avoided because of the potential for exacerbation of cardiac arrhythmias. Thiazide and other potassium-wasting diuretics may provoke drug-induced hypokalemia and could aggravate the QT interval prolongation.
Carbonic anhydrase inhibitors drug such as acetazolamide is likely to help with muscle weakness attacks, as well as dichlorphenamide (brand name KEVEYIS, the only FDA-approved periodic paralysis treatment). Taking potassium supplements can help patients whose attacks are triggered by not having enough potassium in their blood. On the other hand, patients whose attacks are triggered by high potassium (hyperkalemia) may be able to shorten the length of attacks by having a high-carbohydrate food or beverage (sugary foods and drinks) during an attack.
Lifestyle changes are important, too. Andersen Tawil syndrome patients should try to avoid being in cold environments, and they should also be careful when exercising so that they are not overexerting themselves. Stress management also plays a big role in reducing the chances of an attack. It’s helpful to have healthy ways to cope with stressful situations. Changes in diet may be necessary, because certain foods may trigger attacks.
Figure 4. Andersen Tawil syndrome
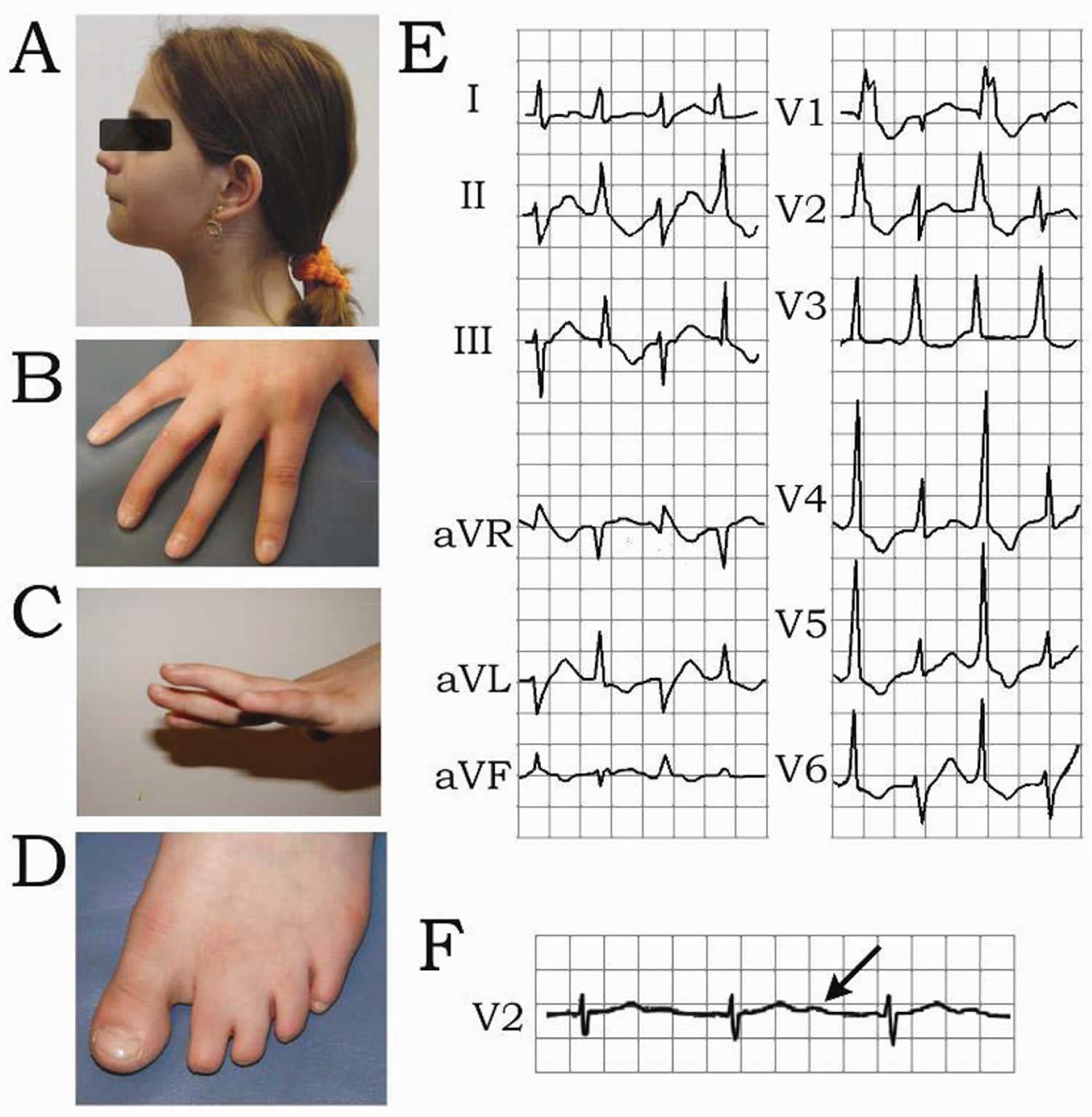
Footnote: Andersen-Tawil syndrome is characterized by dysmorphic features and cardiac arrhythmias. Patient exhibited micrognathia (small chin), retrognathia and hypertelorism (A), clinodactyly (B and C) and syndactyly (D). Electrocardiogram rhythm strip shows bidirectional ventricular tachycardia (BiVT) (E) and prolonged U wave (QUc=626 ms) (F).
[Source 127 ]Figure 5. Andersen Tawil syndrome symptoms
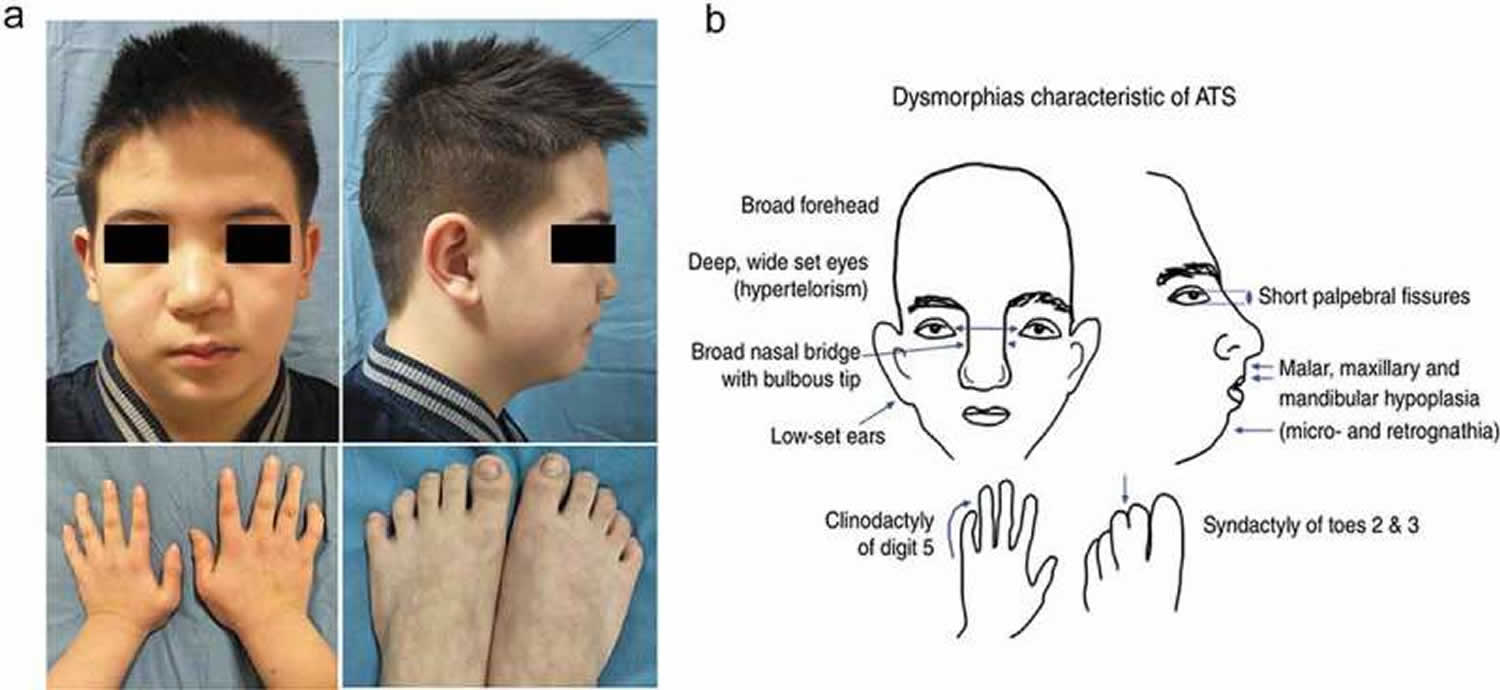
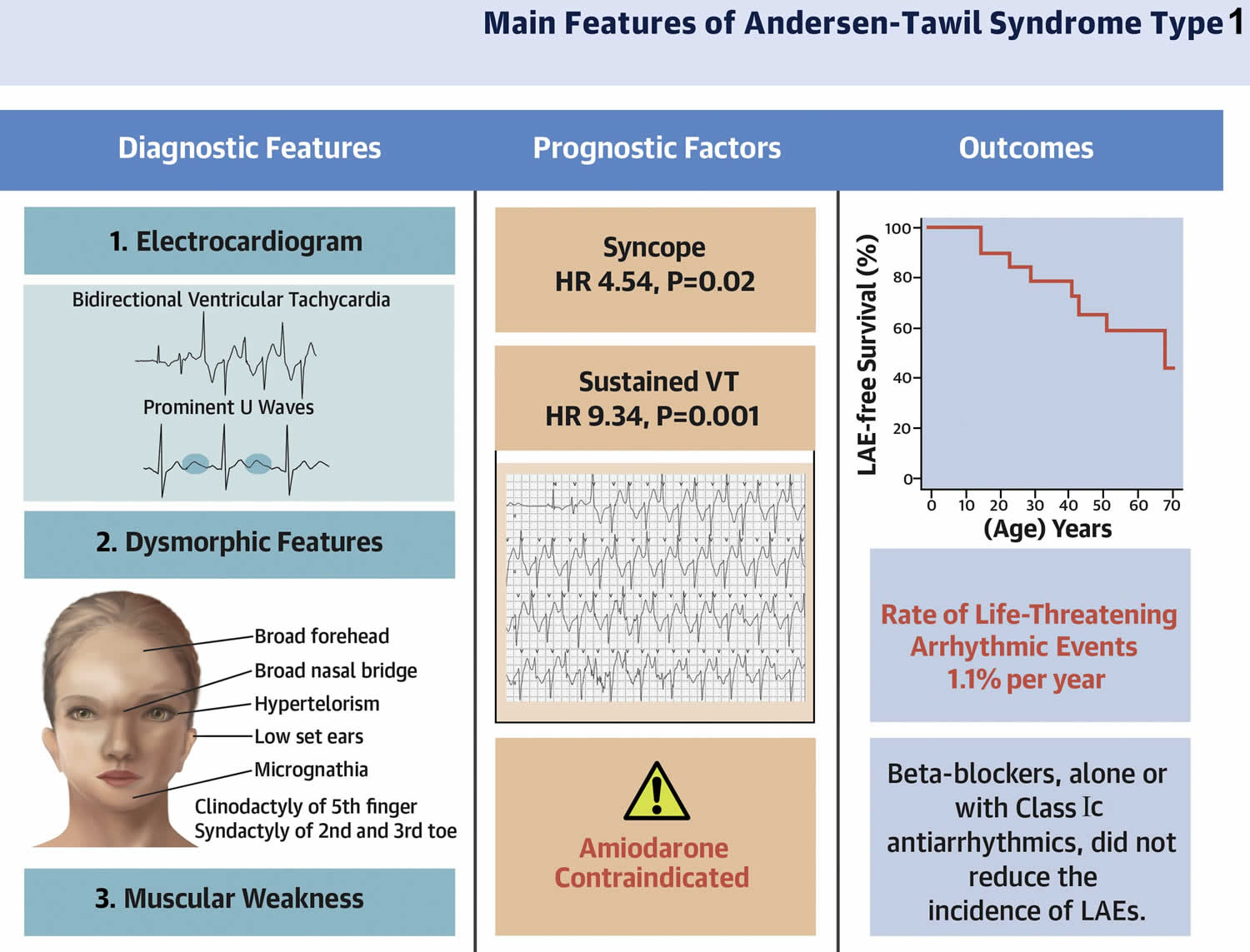
Footnotes: (a) Widely spaced eyes, small mandible, low-set ears, and fifth-digit clinodactyly. (b) Craniofacial anomalies characteristic of Andersen‐Tawil syndrome, one of the three features used for diagnosis of Andersen‐Tawil syndrome, craniofacial anomalies are seen in a majority of patients. These features are variably penetrant, however, with different patients showing different degrees of severity and different subsets of those shown 128, 129.
Abbreviation: LAEs = life-threatening arrhythmic events
[Sources 124, 130 ]Figure 6. Bidirectional ventricular tachycardia in Andersen Tawil syndrome
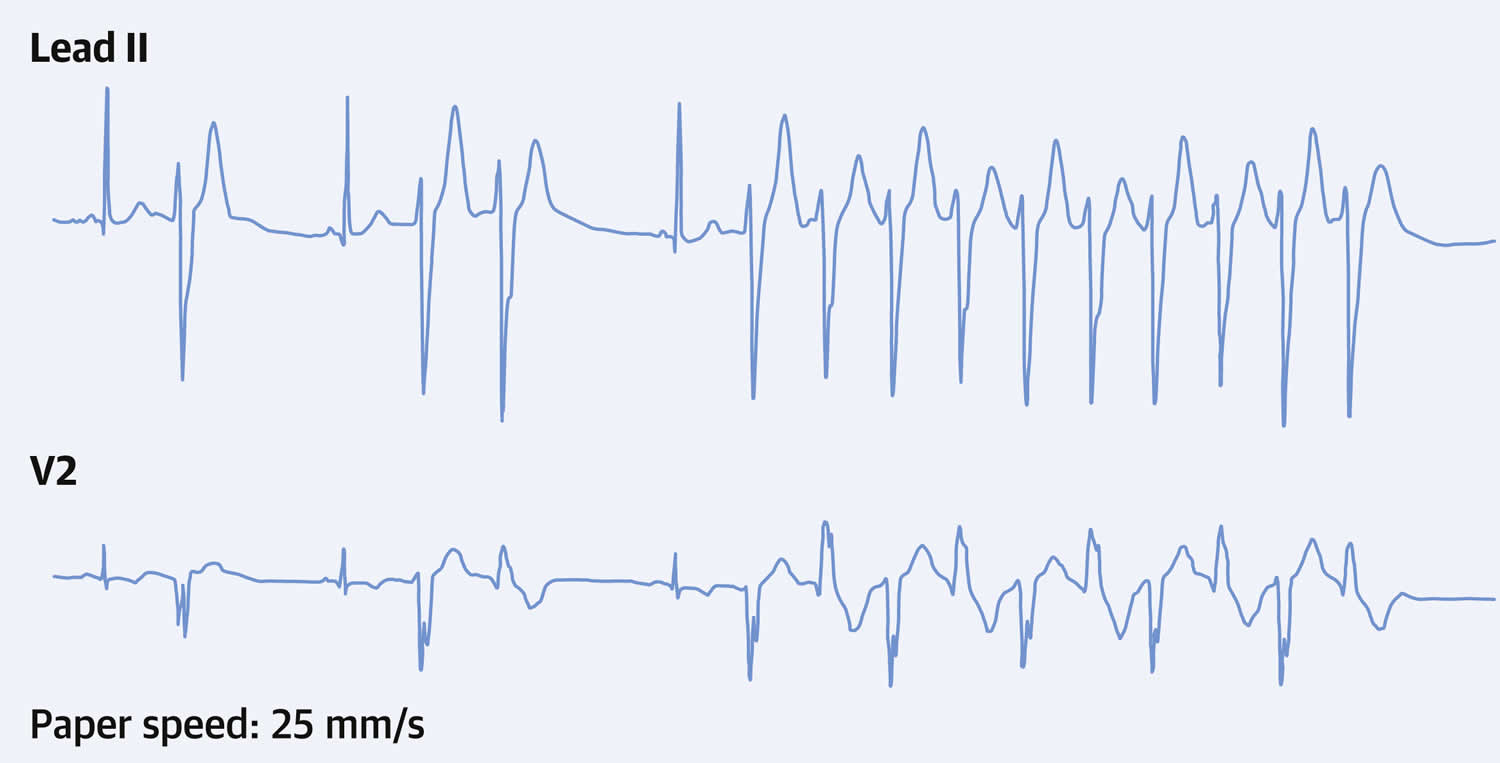
Will I pass Andersen Tawil syndrome on to my children?
Andersen Tawil syndrome is inherited in an autosomal dominant pattern (see Figure 5 below). The chances that a child of an affected person will inherit the defective KCNJ2 gene is ‘statistically’ 50%, but not all who inherit the gene will have symptoms. The degree to which children are affected may vary from one child to the next. One child may be mildly affected, the next seriously affected and the next completely unaffected.
However, in approximately 50% of cases, Andersen Tawil syndrome results from a new (de novo) mutation in the KCNJ2 gene and not inherited from either parent, which means that in those specific cases the gene mutation has occurred at the time of the formation of the egg or sperm for that child only and no other family member will be affected. These cases occur in people with no history of Andersen Tawil syndrome in their family and the disorder is usually not inherited from or “carried” by a healthy parent. Speak with a doctor or genetic counselor if you or someone in your family has been diagnosed with Andersen Tawil syndrome.
Andersen-Tawil syndrome causes
Mutations in the potassium inwardly rectifying channel subfamily J member 2 gene or KCNJ2 gene cause about 60 percent of all cases of Andersen-Tawil syndrome and more than 60 mutations in the KCNJ2 gene have been found to cause Andersen-Tawil syndrome 131. When the disorder is caused by mutations in KCNJ2 gene, it is classified as Andersen-Tawil syndrome type 1 (ATS1). In the other 40% of Andersen Tawil syndrome cases (Andersen-Tawil syndrome type 2), the underlying genetic mutation is unknown, suggesting that additional as-yet-unidentified genes also cause the disorder.
The KCNJ2 gene provides instructions for making the inward rectifier potassium (Kir2.1) channels that transport positively charged potassium ions across the membrane of skeletal muscle and cardiac muscle cells (Figure 7) 132, 133, 134, 135. Most of the KCNJ2 gene mutations change a single protein building block (amino acid) in the KCNJ2 protein. The movement of potassium ions through these channels is critical for maintaining the normal function of muscles used for movement (skeletal muscles) and cardiac muscle. Mutations in the KCNJ2 gene alter the usual structure and function of these potassium channels. These changes disrupt the flow of potassium ions in skeletal and cardiac muscle, leading to the periodic paralysis and irregular heart rhythm characteristic of Andersen-Tawil syndrome.
Many KCNJ2 mutations prevent phosphatidylinositol 4,5-bisphosphate (PIP2) from effectively binding to and activating potassium channels 111, 112. If the KCNJ2 protein is unable to bind to phosphatidylinositol 4,5-bisphosphate (PIP2), the channels remain closed and potassium ions are unable to flow across the cell membrane. Researchers believe that problems with PIP2 binding are a major cause of Andersen-Tawil syndrome.
A loss of inward rectifier potassium (Kir2.1) channel’s function in skeletal and cardiac muscle cells disrupts the normal flow of potassium ions out of these cells, resulting in periodic paralysis and an irregular heart rhythm. It is not known how mutations in the KCNJ2 gene contribute to the skeletal changes and other physical abnormalities often found in Andersen-Tawil syndrome 131.
In the 40 percent of cases not caused by KCNJ2 gene mutations, the cause of Andersen-Tawil syndrome is usually unknown. These cases are classified as Andersen-Tawil syndrome type 2 (ATS2). Studies suggest that variations in at least one other potassium channel gene may underlie the disorder in some of these affected individuals.
Figure 7. Structure of Kir2.1 Channel in Andersen Tawil Syndrome type 1 (ATS1)
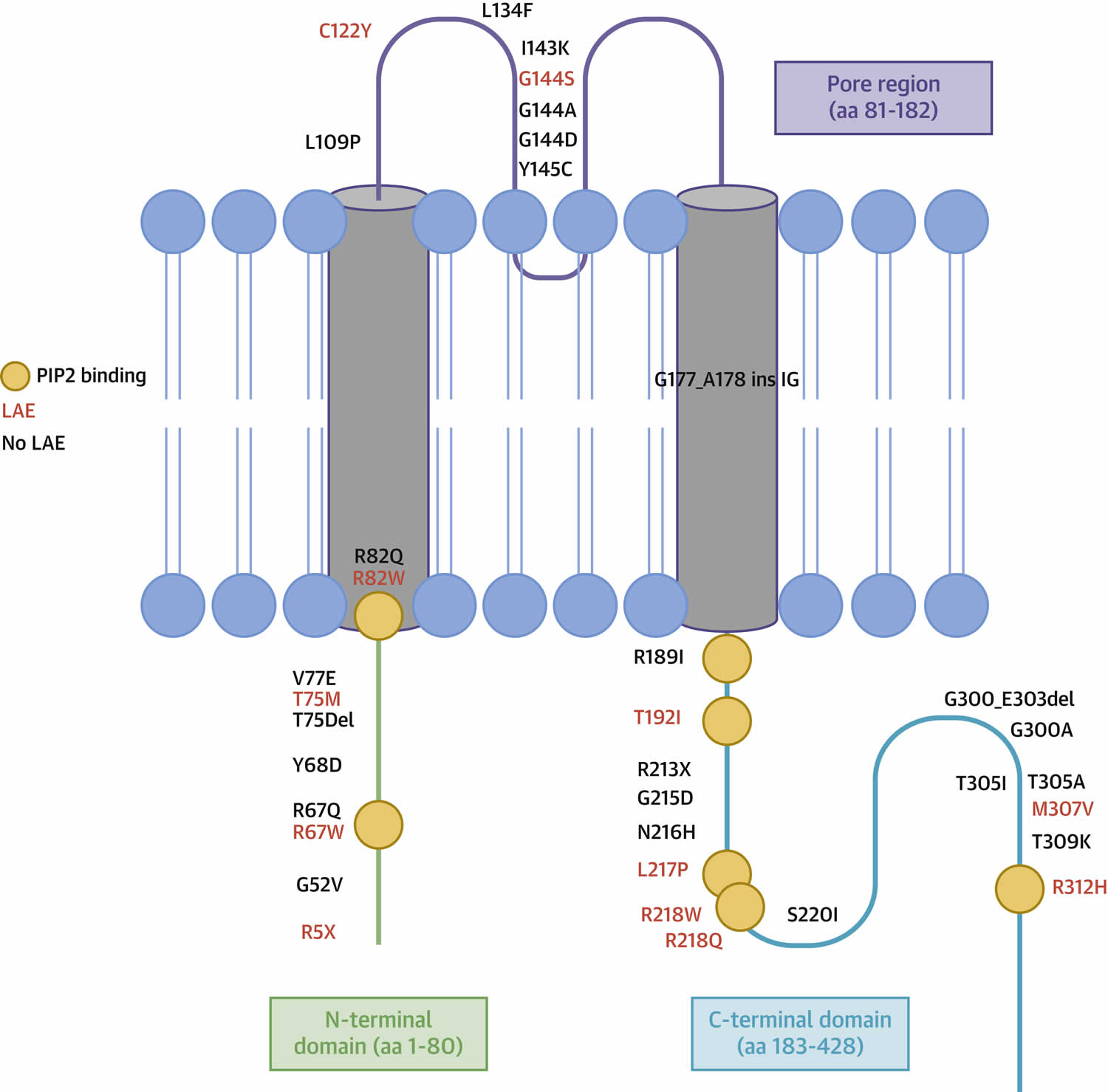
Footnote: Amino acid location of the 35 different KCNJ2 mutations. N-terminal domain is highlighted in green, transmembrane and pore domains in purple, and C-terminal domain in light blue. Mutations associated with life-threatening arrhythmic events (LAEs) are reported in red. Orange circles indicate phosphatidylinositol (4, 5)-bisphosphate (PIP2) binding residues. Blue circles and tails represent the phospholipid bilayer of the cell membrane.
Abbreviation: LAE = life-threatening arrhythmic event
[Source 124 ]Andersen Tawil syndrome triggers
Triggers may vary from person to person. For example food triggers for Andersen Tawil syndrome are not as easily recognized as they are in other types of periodic paralysis, where it is possible to say potassium-rich foods or carbohydrate-rich foods trigger episodes. Attacks can be triggered by high blood potassium levels (hyperkalemia) or low blood potassium levels (hypokalemia), and during attacks, potassium may rise, fall, or remain normal (normokalemia). Since the person’s potassium level may vary from episode to episode it may take real attention to one’s body’s signals to recognize food triggers. In some Andersen Tawil syndrome patients food does not seem to be a trigger at all. Because Andersen Tawil syndrome causes attacks that vary in severity and triggers, it is recommended that patients keep a diary to help them determine what triggers their attacks of weakness. Here’s how: After you have an attack, you should write down what happened, how you felt, what you were doing before the attack, and how you were able to recover from it. After doing this for a while, you’ll be able to look back on your previous diary entries and possibly notice certain patterns. Even if there are no clear patterns, the diary will be useful to discuss with your doctor.
Common triggers are rest after activity and periods of inactivity (for example; sitting through a long class or church service, especially in a cool room). Sleep is a potent trigger. Going too long without eating is another trigger, as is eating a large meal. Getting chilled is a trigger. Emotional events may trigger attacks. Gasoline or paint fumes and car exhaust have been reported as triggers by some patients, and are probably best avoided when possible.
Andersen Tawil syndrome inheritance pattern
Andersen Tawil syndrome is inherited (passed through families) in an autosomal dominant pattern, which means one copy of an altered KCNJ2 gene in each cell is sufficient to cause the condition (see Figure 8). When Andersen Tawil syndrome results from a mutation in the KCNJ2 gene, an affected individual may inherit the mutation from one affected parent. If an individual has Andersen Tawil syndrome, there is a one in two (50%) chance that they will pass it on to each of their children. However, in approximately 50% of cases, Andersen Tawil syndrome results from a new (de novo) mutation in the KCNJ2 gene and not inherited from either parent, which means that in those specific cases the gene mutation has occurred at the time of the formation of the egg or sperm for that child only and no other family member will be affected. These cases occur in people with no history of Andersen Tawil syndrome in their family and the disorder is usually not inherited from or “carried” by a healthy parent. Speak with a doctor or genetic counselor if you or someone in your family has been diagnosed with Andersen Tawil syndrome.
Often autosomal dominant conditions can be seen in multiple generations within the family. If one looks back through their family history they notice their mother, grandfather, aunt/uncle, etc., all had the same condition. In cases where the autosomal dominant condition does run in the family, the chance for an affected person to have a child with the same condition is 50% regardless of whether it is a boy or a girl. These possible outcomes occur randomly. The chance remains the same in every pregnancy and is the same for boys and girls.
- When one parent has the abnormal gene, they will pass on either their normal gene or their abnormal gene to their child. Each of their children therefore has a 50% (1 in 2) chance of inheriting the changed gene and being affected by the condition.
- There is also a 50% (1 in 2) chance that a child will inherit the normal copy of the gene. If this happens the child will not be affected by the disorder and cannot pass it on to any of his or her children.
Figure 8 illustrates autosomal dominant inheritance. The example below shows what happens when dad has the condition, but the chances of having a child with the condition would be the same if mom had the condition.
Figure 8. Andersen Tawil syndrome autosomal dominant inheritance pattern
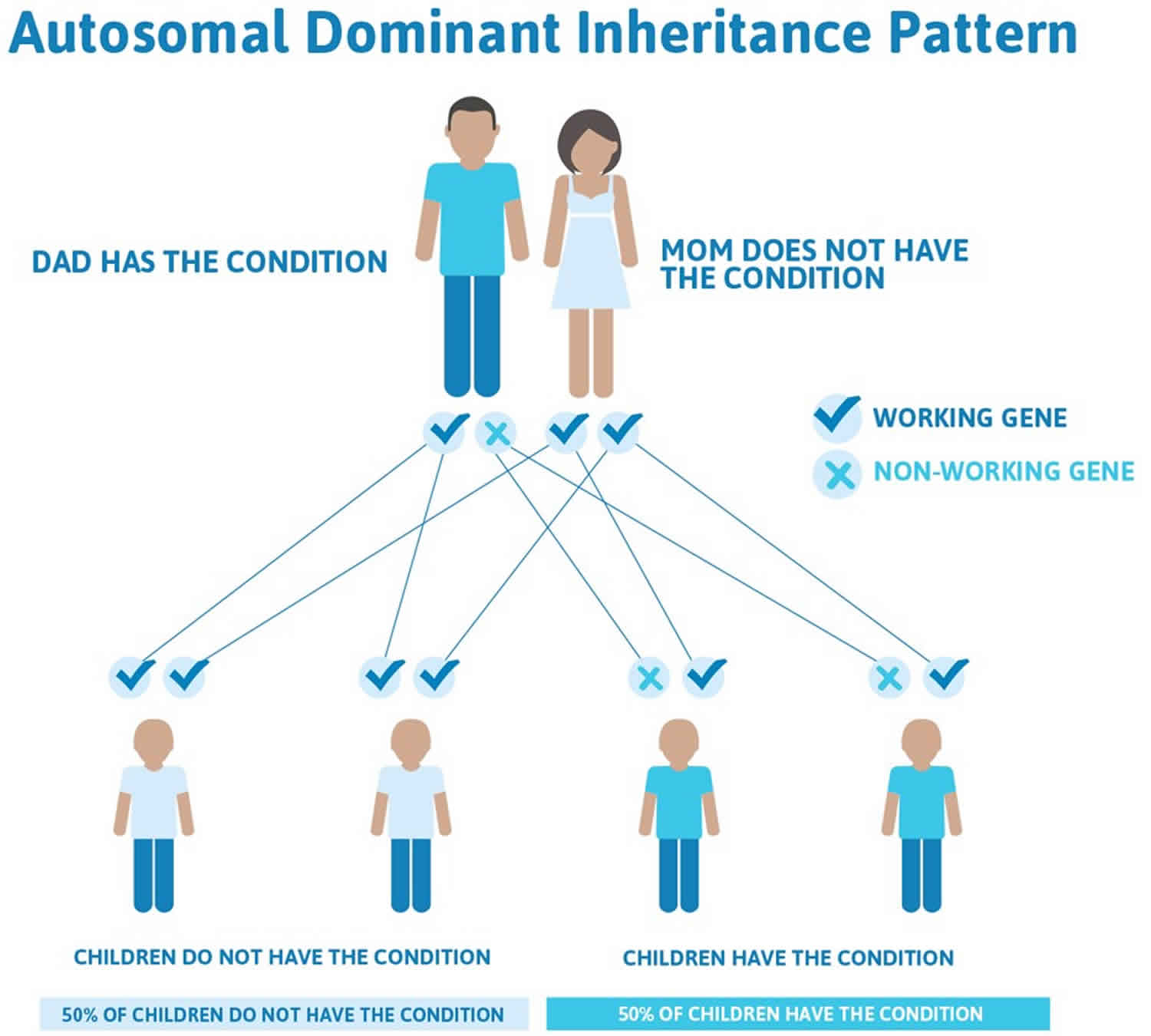
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Andersen-Tawil syndrome signs and symptoms
Andersen-Tawil syndrome is defined by three main features (i.e. a clinical triad), specifically:
- Episodic flaccid muscle weakness (periodic paralysis): Intermittent weakness occurs spontaneously, or alternatively may be triggered by prolonged rest or rest following exertion. The attack frequency, duration, and severity are variable between and within affected individuals. Mild permanent weakness is common 136. Affected individuals can develop fixed proximal weakness over time.
- Arrhythmias and heart abnormalities (ventricular arrhythmias, prolonged QTc or QUc intervals, and prominent U waves):
- Ventricular arrhythmias including bidirectional ventricular tachycardia (VT), polymorphic VT, and multifocal premature ventricular contractions may be asymptomatic, or may manifest (most commonly) as palpitations. Less common symptomatic presentations include syncope, cardiac arrest, or sudden death 137. While the ECG may reveal a long QTc (LQT) interval, characteristic T-U patterns including enlarged U waves, a wide T-U junction, and prolonged terminal T-wave downslope distinguish Andersen Tawil syndrome from other LQT syndromes 138. A large case series found no significant difference in the incidence of ventricular tachyarrhythmias between individuals with typical and atypical presentations of Andersen Tawil syndrome 139. A retrospective multicenter French study of 36 individuals with Andersen Tawil syndrome followed for an average of 9.5 years reported no deaths in follow up; four individuals experienced syncope and one individual had a non-fatal cardiac arrest 140.
- Dilated cardiomyopathy was observed in two of three affected individuals in a single kindred with pathogenic variant p.Arg218Trp 141. Additionally, cardiomyopathy was documented and reversed by treatment with flecainide in an individual with pathogenic variant p.Arg67Trp 142 and reversed by bisoprolol in another individual with typical clinical features of Andersen Tawil syndrome and novel variant p.Leu222Ser 143. These reports suggest that dilated cardiomyopathy is a secondary phenotype as a consequence of chronic tachycardia rather than a primary phenotypic manifestation. Additional study is needed to further delineate this clinical feature.
- Distinctive physical features. Distinctive physical features recognized initially included low-set ears, widely spaced eyes, small mandible, fifth-digit clinodactyly, second and third toe syndactyly, short stature, broad nasal root, and scoliosis 137. Dental enamel discoloration was noted in two kindreds with the p.Gly300Asp and p.Arg218Trp pathogenic variants 144. Detailed, prospectively collected data in ten individuals with confirmed KCNJ2 pathogenic variants have expanded the phenotype to include a characteristic facies and dental and skeletal anomalies 145. Characteristic facies include broad forehead, short palpebral fissures, wide nasal bridge with bulbous nose, hypoplasia of maxilla and mandible, thin upper lip, and a triangular face.
- Dental findings include (among others) persistent primary dentition, multiple missing teeth (oligodontia), and dental crowding.
- Skeletal findings include mild syndactyly of toes two and three as well as fifth-digit clinodactyly.
- Novel findings include small hands and feet (<10th centile for age) and joint laxity.
However, the disorder is highly variable and not all affected individuals will develop all three of these characteristic symptoms. Andersen-Tawil syndrome can vary greatly in expression and severity from one person to another, even among members of the same family.
Most people who have Andersen-Tawil syndrome start experiencing symptoms before they turn 20. Some will begin to have heart palpitations and fainting spells, while others may start having episodes of transient muscle weakness. Severity and frequency can vary greatly, and there are many possible triggers, so patients may not be able to pinpoint what triggers the episodes of weakness.
Although researchers have established a clear syndrome with characteristic or “core” symptoms, much about the disorder is not fully understood. Several factors including the small number of identified cases, the lack of large clinical studies, and the possibility of other genes influencing the disorder prevent physicians from developing a complete picture of associated symptoms and prognosis. Therefore, it is important to note that affected individuals may not have all of the symptoms discussed below. Parents should talk to their children’s physician and medical team about their specific case, associated symptoms and overall prognosis.
Affected individuals may experience temporary episodes of flaccid, muscle weakness or paralysis, known as periodic paralysis. The legs are most often affected and the severity of muscle weakness can range from mild weakness to an inability to walk unassisted. The arms, hands, legs and feet are also commonly affected. The frequency and duration of episodes varies from one person to another and from one episode to the next for the same person. Some episodes may last only minutes to hours; others can go on for days. Episodes can occur without warning (spontaneously), but can also occur following prolonged exercise, prolonged rest (e.g. upon awaking in the morning), rest after exercise, going too long without eating, eating a large meal, or emotional stress. Episodes can range in frequency from once per day to once per year. In some cases, a mild, but permanent weakness, present even between episodes, can develop with age and progress slowly over time.
In most cases, periodic paralysis may be associated with low levels of potassium in the blood (hypokalemia), a common finding with other forms of periodic paralysis. However, some individuals who experience periodic paralysis have had normal potassium levels or even elevated levels (hyperkalemia). Low potassium levels can also impact the function of heart muscle cells.
Affected individuals may experience disturbances of the normal rhythm of the heartbeat (arrhythmias), which can include abnormally fast heartbeats that originate in the lower chamber of the heart (ventricular tachycardia). Generally, this may not cause any symptoms (asymptomatic) or may cause shortness of breath or palpitations. In some cases, these arrhythmias may cause episodes of fainting or loss of consciousness (syncope). In severe cases, the possibility of cardiac arrest and sudden death exists. Although sudden death due to the cardiac abnormalities has occurred in Andersen-Tawil syndrome, it is extremely rare.
Some affected individuals also have characteristic physical features including distinctive facial features, which are often mild in expression. Such features include a broad forehead, low-set ears, eyes that are spaced apart wider than usual (hypertelorism), and a small jaw (micrognathia). Additional facial features include a round (bulbous) nose, a thin upper lip, a triangular-shaped face, highly-arched roof of the mouth (palate), a cleft palate, and underdevelopment of the cheek bones (malar hypoplasia). Common physical features include webbing (syndactyly) of the second and/or third toes, pinkies that are fixed in a bent or crooked position (clinodactyly), and disproportionately small fingers and toes (brachydactyly). Additional findings include small hands and feet, loose joints, and abnormal sideways curvature of the spine (scoliosis). Dental anomalies have also been reported including delayed loss of primary or ‘baby’ teeth (persistent primary dentition), multiple missing teeth (oligodontia), and teeth that are abnormally crowded together.
As affected children grow into adulthood, short stature may become evident. Short stature refers to an individual whose height is much shorter than would otherwise be expected based upon age and gender.
Some individuals with Andersen-Tawil syndrome have experienced neuropsychiatric abnormalities including mild learning disabilities, depression, and deficits in executive functioning and abstract reasoning. Some infants experience seizures without fever (afebrile seizures). Afebrile seizures occurring only in infancy were reported in 4/23 (17%) of a Japanese cohort with molecularly confirmed Andersen-Tawil syndrome 138.
Isolated reports of renal anomalies include unilateral hypoplastic kidney 146 and renal tubular defect 144.
Mild learning difficulties have been described 144. A distinct neurocognitive phenotype (i.e., deficits in executive function and abstract reasoning) has been recognized in individuals with a KCNJ2 pathogenic variant despite IQ levels similar to those of their unaffected sibs 147. Growth restriction and developmental delay have been described as well 148.
Andersen-Tawil syndrome diagnosis
A diagnosis of Andersen-Tawil syndrome is based upon identification of characteristic symptoms (e.g. periodic paralysis, symptomatic arrhythmias, and/or distinctive facial and skeletal features), a detailed family and patient history, a thorough clinical evaluation and a variety of specialized tests.
The diagnosis of Andersen-Tawil syndrome might be suspected in individuals with either A or B 149:
- A. Presence of two of the following three criteria:
- Periodic paralysis
- Symptomatic cardiac arrhythmias or evidence of enlarged U-waves, ventricular ectopy, or a prolonged QTc or QUc interval on electrocardiogram (ECG)
- Characteristic facial features, dental abnormalities, small hands and feet, AND at least two of the following:
- Low-set ears
- Widely spaced eyes
- Small lower jaw (mandible)
- Fifth-digit clinodactyly (curved pinky finger)
- Syndactyly of toes 2 and 3
OR
- B. One of the above three criteria in addition to at least one other family member who meets two of the three criteria 28.
The presence of a mutation in the KCNJ2 gene confirms the diagnosis of Andersen-Tawil syndrome 122.
Table 6. Recommended evaluations following initial diagnosis in individuals with Andersen Tawil syndrome
| Organ System | Evaluation | Comment |
|---|---|---|
| Cardiovascular | Baseline assessment | Performed by cardiologist familiar with long QT management |
| 12-lead EKG & 24-hour Holter monitor | ||
| Serum potassium concentrations | Performed at baseline & during attacks of weakness | |
| Neurologic | Baseline assessment | Performed by neurologist familiar w/periodic paralysis |
| Electrophysiologic studies incl long exercise protocol | ||
| Dental | Baseline assessment for dental abnormalities assoc with Andersen-Tawil syndrome | Follow up as needed |
| Musculoskeletal | Baseline assessment & to establish care with orthopedist or spine surgeon if scoliosis identified | Follow up as needed |
| Miscellaneous/ Other | Serum TSH concentration | Verification that serum TSH concentration is w/in normal limits |
| Consultation with clinical geneticist and/or genetic counselor |
Abbreviation: TSH = thyroid-stimulating hormone
[Source 149 ]Clinical testing and workup
Because potassium levels may be reduced during an episode of periodic paralysis, a blood test to determine the serum potassium levels during an episode can be helpful in diagnosing the disorder in some cases.
Long exercise nerve conduction studies have been used to help diagnose individuals with Andersen-Tawil syndrome. During this test, an affected individual will perform voluntary muscle contractions of a small muscle on the ulnar side of the palm of the hand for approximately 2-5 minutes. This test allows physician to evaluate muscle function and specific results can be indicative of periodic paralysis.
An electrocardiogram or EKG records the heart’s electrical impulses and may reveal abnormal electrical patterns or activity commonly associated with Andersen-Tawil syndrome including prominent U waves, prolonged QU intervals, prolonged QT intervals, premature ventricular contractions, or polymorphic ventricular tachycardia.
Some individuals may undergo 24-Holter monitoring, during which an affected individual wears a small device for 24 hours. Through electrodes attached to the chest, this device continuously records the rhythm of the heart in order to detect the presence, frequency and duration of ventricular tachycardia and other symptoms.
Molecular genetic testing can confirm a diagnosis of Andersen-Tawil syndrome in some cases. Molecular genetic testing can detect mutations in the KCNJ2 gene known to cause the disorder, but is available only as a diagnostic service at specialized laboratories.
Supportive findings
Individuals with either episodic weakness or cardiac symptoms require careful evaluation by a neurologist and/or cardiologist as well as measurement of serum potassium concentration (baseline and during attacks of flaccid paralysis), a 12-lead ECG, a 24-hour Holter monitor, and possibly the long exercise protocol.
- Serum potassium concentration during episodes of weakness may be elevated, normal, or, most commonly, reduced (<3.5 mmol/L) 28, 135.
- Routine nerve conduction electrophysiology is normal between episodes. A more sensitive electrophysiologic study, the long exercise protocol, may reveal an immediate post-exercise increment followed by an abnormal decrement in the compound motor action potential (CMAP) amplitude (>40%) 150 or area (>50%) 20-40 minutes post exercise 151, 152. In a study of 11 individuals with Andersen-Tawil syndrome, 82% met long-exercise amplitude decrement criteria for abnormal testing 153.
- Electrocardiogram (ECG) may reveal characteristic abnormalities including prominent U waves, prolonged Q-U intervals, premature ventricular contractions, polymorphic ventricular tachycardia, and bidirectional ventricular tachycardia 28, 126, 125, 154.
- 24-hour Holter monitoring is important to document the presence, frequency, and duration of ventricular tachycardia (VT) and the presence or absence of associated symptoms.
Andersen-Tawil syndrome treatment
The treatment of Andersen-Tawil syndrome is directed toward the specific symptoms that are apparent in each individual. Management of individuals with Andersen Tawil syndrome requires the coordinated efforts of a team of specialists. Pediatricians, neurologists experienced in the treatment of periodic paralysis, cardiologists experienced in the treatment of long QT syndrome, and other healthcare professionals may need to systematically and comprehensively plan an affect child’s treatment.
There are no standardized treatment protocols or guidelines for affected individuals. Due to the rarity of Andersen Tawil syndrome, there are no randomized clinical therapeutic trials that have been tested on a large group of patients. Various treatments have been reported in the medical literature as part of single case reports or small series of patients. Treatment trials would be very helpful to determine the long-term safety and effectiveness of specific medications and treatments for individuals with Andersen-Tawil syndrome.
Affected individuals are encouraged to avoid potential triggers of periodic paralysis (e.g. rest following exercise or prolonged exercise). Avoidance of drugs that can prolong the QT interval is also recommended.
When periodic paralysis is associated with low potassium levels, treatment with oral supplemental potassium can be beneficial. In individuals prone to low potassium levels, daily potassium supplementation can be considered. If serum potassium concentration is low (<3.0 mmol/L), administration of oral potassium (20-30 mEq/L) every 15-30 minutes (not to exceed 200 mEq in a 12-hour period) until the serum concentration normalizes; if a relative drop in serum potassium within the normal range causes episodic paralysis, an individual potassium replacement regimen with a goal of maintaining serum potassium levels in the high range of normal can be considered; if serum potassium concentration is high, ingesting carbohydrates may lower serum potassium levels. Mild exercise may shorten or reduce the severity of the attack. Potassium supplementation may also shorten the QT interval, which can be of benefit for individuals who also experience a long QT interval.
A periodic paralysis episode that occurs when potassium levels are high usually resolve on their own within 60 minutes. However, eating carbohydrates or continuing mild exercise can shorten the duration of the episode.
Specific drugs known as carbonic anhydrase inhibitors, such as acetazolamide and dichlorpenamide, are used to treat periodic paralysis in individuals with Andersen-Tawil syndrome. Clinical trials in other forms of periodic paralysis showed that dichlorphenamide reduces the frequency and severity of attacks of periodic paralysis and is now an FDA approved for the treatment of periodic paralysis.
Despite a high frequency of ventricular arrhythmias in some individuals with Andersen-Tawil syndrome, they rarely degenerate into life-threatening arrhythmias. Many arrhythmias do not cause symptoms and go away on their own without problems (self-terminate). Various different drugs have been used, but no standard, effective therapy has been established. Beta-adrenergic blocking drugs (beta blockers), drugs that suppress abnormal heart rhythms (anti-arrhythmics) such as flecainide or amiodarone, or calcium-channel blocking drugs such as verapamil have all shown some effect. Beta blockers are commonly used to treat abnormal heart rhythms. These drugs, which include propranolol, atenolol, metroprolol, and nadolol, reduce the workload of the heart by decreasing the electrical stimulation of the heart, thereby slowing the heartbeat and preventing symptoms. Beta blockers have been used in conjunction with flecainide. Some anti-arrhythmic drugs can worsen neuromuscular symptoms and should be used with caution in individuals with Andersen-Tawil syndrome.
Treatment with an implantable automatic cardioverter-defibrillator (ICD) is necessary in rare cases. Implantable automatic cardioverter-defibrillators (ICDs) are considered for individuals in whom cardiac arrhythmias are severe and symptomatic. These small devices are implanted under the skin of the chest. The device detects the abnormal heartbeat automatically and selectively delivers an electrical impulse to restore the proper heartbeat. Opting for an ICD is a lifelong therapy that carries significant implications including the potential for complications, especially in younger individuals, and should be undertaken only after consultation with appropriate medical personnel and a careful risk vs. benefit evaluation.
Genetic counseling is recommended for affected individuals and their families.
Episodic weakness or paralysis treatment
Management of attacks of episodic weakness depends on the associated serum potassium concentration 149:
- If the serum potassium concentration is low (<3.0 mmol/L), administer oral potassium (20-30 mEq/L) every 15-30 minutes (not to exceed 200 mEq in a 12-hour period) until the serum concentration normalizes often shortens the attack. As dysphagia is almost never a problem during attacks of paralysis, oral potassium replacement is the safest route. If intravenous potassium replacement is needed, a 5% mannitol solution instead of a saline or glucose solution (both of which may exacerbate weakness) is recommended. Close monitoring of serum potassium concentrations and ECG is necessary during potassium replacement therapy in an emergency setting to avoid secondary hyperkalemia.
- Whether a relative drop in serum potassium within the normal range causes episodic paralysis is not clear. If such cases are suspected, affected individuals can work with their physician to devise an individual potassium replacement regimen, with a goal of maintaining serum potassium levels in the high range of normal.
- Attacks of weakness when serum potassium concentration is high usually resolve within 60 minutes. Episodes may be shortened by ingesting carbohydrates or continuing mild exercise. Intravenous calcium gluconate is rarely necessary for management in an individual seen in an emergency setting.
Vasovagal syncope in individuals with Andersen Tawil syndrome mandates a careful cardiology assessment 155.
Prevention of Andersen Tawil syndrome attacks
Prophylactic treatment aimed at reduction of attack frequency and severity can be achieved, as in other forms of periodic paralysis, with the following 149:
- Lifestyle and dietary modification to avoid known triggers
- Use of carbonic anhydrase inhibitors (acetazolamide: adults 125-1,000 mg daily and children 5-10 mg/kg/day divided 1-2x/day or dichlorphenamide 50-200 mg/1-2x/day). Use of potassium-sparing diuretic should be individualized based on patient needs.
- Daily use of slow-release potassium supplements, which may also be helpful in controlling attack rates in individuals prone to hypokalemia. Elevating the serum potassium concentration (>4 mEq/L) has the added benefit of narrowing the QT interval, thus reducing the risk of long QT-associated arrhythmias.
- An implantable cardioverter-defibrillator in individuals with tachycardia-induced syncope 156
- Empiric treatment with flecainide should be considered for significant, frequent ventricular arrhythmias in the setting of reduced left ventricular function 157, 158, 159, 160. A prospective open label study in ten individuals with Andersen Tawil syndrome and a confirmed KCNJ2 pathogenic variant tested the effect of flecainide, a type 1c antiarrhythmic, for the prevention of cardiac arrhythmias 161. Outcomes included a 24-hour Holter monitor before and after treatment and a treadmill exercise test. Flecainide was found to significantly reduce the number of ventricular arrhythmias seen on Holter monitor and to suppress exercise-induced ventricular arrhythmias. Individuals were then followed for a mean of 23 months and no syncope or cardiac arrest was documented 161. Other case studies have reported beneficial effects with flecainide 28. A recent study showed that fleicainide suppresses arrhythmogenicity through Na+/Ca2+ exchanger flux in induced pluripotent stem cells derived from patients with Andersen Tawil syndrome 162. Threfore, flecainide may reduce cardiac arrhythmias in Andersen Tawil syndrome.
Prevention of secondary complications
Cardiologists should be aware that some antiarrhythmic drugs (e.g., lidocaine, mexiletine, propafenone, quinidine), particularly Class 1 drugs, may paradoxically exacerbate the neuromuscular symptoms and should be used cautiously in individuals with Andersen Tawil syndrome 149.
Although malignant hyperthermia has not been reported in Andersen Tawil syndrome, appropriate anesthetic precautions should be undertaken, as with individuals with other forms of periodic paralysis 149.
Pregnancy management
The rarity of Andersen Tawil syndrome and the paucity of reports pertaining to pregnancy in women with Andersen Tawil syndrome make an evidence-based approach to pregnancy management difficult to formulate 149. One case study reported an uneventful pregnancy, with increased episodes of weakness but reduced ventricular ectopy compared to the pre-pregnancy period 163. However, as data are limited, a multidisciplinary approach to individual care and anticipation of increased risk (as can be seen in those with long QT syndrome) seems reasonable 149.
Paramyotonia Congenita
Paramyotonia congenita also called PMC, Eulenburg disease, Von Eulenburg’s disease, paralysis periodica paramyotonica, paramyotonia congenita of von Eulenburg, is a form of periodic paralysis that results from a mutation in the sodium channel and produces muscle stiffness (myotonia) or rigidity and weakness in response to cold or exercise 164, 165, 166, 167, 168, 169, 170, 171, 172, 173, 174. Myotonia is a disorder that resulted in the delayed relaxation of skeletal muscles after voluntary contraction 175. Beginning in infancy or early childhood, people with paramyotonia congenita experience bouts of sustained muscle stiffness (myotonia) that prevent muscles from relaxing normally. Paramyotonia congenita causes muscle stiffness that typically appears after exercise and can also be induced by muscle cooling. Exposure to cold initially causes muscle stiffness (myotonia) in these individuals, and prolonged cold exposure leads to temporary episodes of mild to severe muscle weakness that may last for several hours at a time. Some older people with paramyotonia congenita develop permanent muscle weakness that can be disabling. Paramyotonia congenita can make small everyday activities difficult, such as letting go of small objects (e.g. pens or door knobs).
The muscle stiffness (myotonia) mainly affects muscles in the eyelids, face, tongue, neck, arms, and hands, although it can also affect muscles used for breathing and muscles in the lower limbs 42. Unlike many other forms of myotonia, the muscle stiffness associated with paramyotonia congenita tends to worsen with repeated movements. Paramyotonia Congenita patients may complain of hand stiffness while shoveling snow or in the frozen food section of the supermarket 176. Parents will occasionally report that affected infants are unable to open their eyes after a crying spell, presumably due to the eyelids being “exercised” while crying 176.
Paramyotonia Congenita symptoms can begin during infancy, and are always apparent by the teenage years. Paramyotonia Congenita patients characteristically present in their childhood complaining of inability to open their eyes following rapid, forceful successive closures. Weakness and myotonia (muscle stiffness) last for minutes to hours. Even after the immediate rewarding of the muscles, cold-induced weakness usually persists for several hours. The disease is non-progressive, does not cause muscle wasting or hypertrophy.
Paramyotonia congenita is caused by mutations in the sodium channel gene SCN4A that codes for the alpha-subunit of the skeletal muscle sodium channels, i.e., voltage sensor domain 48. Paramyotonia congenita is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder. In many cases, an affected person has one parent with the condition.
Paramyotonia congenita is a very rare disorder that affects males and females in equal numbers. It is estimated to affect fewer than 1 in 250,000 people 177, 174.
Paramyotonia Congenita (PMC) comes in two forms, one in which attacks are always associated with a rise in potassium (hyperkalemia) and a form called Paramyotonia von Eulenburg in which attacks can be associated with a fall in blood potassium levels or hypokalemia. Both result from mutations in the sodium channel. Both can accompany hyperkalemic periodic paralysis (HyperPP) or can occur alone.
When paramyotonia congenita is suspected, a test is administered to test the capacity of muscles to conduct electricity called electromyography (EMG). During the EMG (electromyography) test, the muscles are chilled and electrical signals are recorded before and after the muscle is cooled. The electromyography (EMG) taken during cooling of a muscle shows profuse myotonic discharges and reduced compound muscle action potential (CMAP) amplitudes. However, EMG cannot always diagnose paramyotonia congenita definitively, and further testing may be necessary.
Genetic testing on a blood sample will result in a definitive diagnosis by showing the presence of a characteristic mutation in the SCN4A gene.
The treatment of paramyotonia congenita is based on the individual’s symptoms. Paramyotonia congenita can be handled on a day-to-day basis and many patients can lead normal lives. Individuals must be cautious to sudden exposures to very cold weather, as well as avoiding sudden heavy physical activity.
Muscle stiffness could also be triggered or enhanced by potassium-rich foods. Patients will need to learn how to manage their potassium-intake. They should avoid potassium-rich foods, avoid skipping meals and take carbohydrate rich snacks in between meals.
The aim of treatment is to reduce the intensity of acute symptoms and to prevent, as far as possible, further attacks. Some attacks are so mild that treatment is not necessary. However, in other instances drug therapy is required.
Treatment with medications that block the sodium channels such as mexiletine and lamotrigine may help reduce the stiffness related to myotonia. Some patients with paramyotonia congenita may benefit from acetazolamide or thiazide diuretic drugs to reduce the number of paralytic attacks.
Genetic counseling is recommended for patients and their families.
There is no cure to paramyotonia congenita. Paramyotonia congenita patients usually live a normal life and the condition does not affect their lifespan 178. With the proper management of diet that avoid potassium-rich foods, avoidance of cold exposure and physical overactivity and medication, patients can lead normal lives 48.
Thyrotoxic periodic paralysis
Thyrotoxic periodic paralysis also called TPP or Thyrotoxic Hypokalemic Periodic Paralysis (Thyrotoxic HypoKPP) is a very rare life-threatening complication of thyrotoxicosis (too much thyroid hormone) or hyperthyroidism (overactive thyroid) that is characterized by episodes (also known as “attacks”) of muscle weakness and muscle paralysis (periodic paralysis) due to hypokalemia (low blood potassium levels) caused by massive intracellular shift of potassium by the excessive thyroid hormones 179, 180, 181, 182, 183, 184, 185, 186, 187. (Kalemic refers to potassium; hypo means too little; hypokalemia means too little potassium in your blood). The attack is often precipitated by exercise, high carbohydrate meals, and occasionally fasting 188. The attacks may occur daily to yearly. Episodes of muscle weakness may last for a few hours or several days. During the attacks, you will be alert and can answer questions. Normal muscle strength returns between attacks. With repeat attacks, you may develop muscle weakness. The cause of thyrotoxicosis is most often attributable to Graves disease and patients typically have symptoms of thyrotoxicosis for months leading up to the episodes of paralysis 188.
Your thyroid gland is part of the endocrine system. Your thyroid gland is located in your neck and produces several hormones (triiodothyronine [T3], and thyroxine [T4]) that help control growth, digestion, and metabolism. A complex set of mechanisms control the rate of thyroid gland activity. Too much thyroid hormone called hyperthyroidism or thyrotoxicosis is due to an overactive thyroid gland. Hyperthyroidism is not a specific disease, but a symptom of an underlying condition or disease. The causes of hyperthyroidism include Graves’ disease; tumors of the thyroid or other endocrine glands; inflammation or infection of the thyroid (thyroiditis); taking too much thyroid hormone (thyrotoxicosis factitia); and taking too much iodine. Graves’ disease accounts for 85% of all cases of hyperthyroidism. Graves disease (like Hashimoto’s thyroiditis) sometimes occurs with other autoimmune disorders, including type 1 diabetes mellitus, vitiligo, premature graying of hair, pernicious anemia, connective tissue disorders, and polyglandular deficiency syndrome. Heredity increases the risk of Graves disease, although the genes involved are unknown.
Thyroid hormones regulate the sodium-potassium pump (Na+-K+-ATPase pump) at a transcriptional and post-transcriptional level and also induce the release of catecholamines via beta 2 receptors, furthermore stimulating the Na+-K+-ATP pump 189. Thyrotoxic periodic paralysis patients were shown to have 80% more Na+-K+-ATPase pump activity than other thyrotoxic patients 184. The Na+ K+-ATPase pump maintains the gradient of a higher concentration of sodium ion (Na+) extracellularly and a higher level of potassium ion (K+) intracellularly 190. The Na+ K+ ATPase pumps 3 sodium ions (Na+) out of the cell and 2 potassium ions (K+) that into the cell, for every single ATP consumed 190. The sustained concentration gradient of a higher concentration of sodium extracellularly and a higher level of potassium intracellularly is crucial for physiological processes in many organs and has an ongoing role in stabilizing the resting membrane potential of the cell, regulating the cell volume, and cell signal transduction 191. The overactivity of Na+-K+-ATP pump drives serum potassium into the intracellular compartment causing hyperpolarization, hypokalemia and muscle weakness 192. In addition, insulin and testosterone increase the activity of the Na+-K+-ATP pump, which might explain the higher prevalence of thyrotoxic periodic paralysis in males and the manifestation of symptoms after an intense workout and a high-carbohydrate meal 184.
Thyrotoxic periodic paralysis in most cases are hereditary (some cases may occur sporadically) and is commonly reported in Asian men (Japanese, Chinese, Vietnamese, Korean, and Filipino) between 20–40 years of age, but it is rare in children and adolescents 193, 194, 183. Most people who develop high thyroid hormone levels are not at risk of thyrotoxic periodic paralysis and the incidence has been reported at a rate of approximately 2% in hyperthyroidism patients 195. Thyrotoxic periodic paralysis also occurs more frequently in those of Native American and Latin American descent, but only occasionally in those of European descent (0.1 to 0.2% in American Caucasians) 196, 197.
Although thyrotoxic periodic paralysis is rare in children and adolescents, some cases of thyrotoxic periodic paralysis in adolescents have been reported from China, Korea, and in other ethnic groups 198. Most recently, the reported cases of thyrotoxic periodic paralysis have been attributable to Graves’ disease, although autoimmune thyroiditis can be a very rare cause of thyrotoxic periodic paralysis in children and adolescents 183.
To date, genes that have been studied in relation to thyrotoxic periodic paralysis include the KCNJ18 and KCNJ2 genes. The KCNJ18 gene (also called potassium inwardly rectifying channel subfamily J member 18) located in chromosome 17p11.2 (short arm of chromosome 17), which encodes for the inwardly rectifying potassium (Kir) channel Kir2.6, is known to be more common in Western countries, and has been reported at rates of 33.3% in Caucasian populations, primarily among individuals with Brazilian heritage, 25.9% in Singaporeans, and 1.2% in people from Hong Kong 199, 200. In contrast, in recent studies, genetic variants of the KCNJ2 gene have been reported to affect the development of thyrotoxic periodic paralysis in Korean and Chinese populations 201, 202. These ethnic genetic differences might explain the difference in the incidence of thyrotoxic periodic paralysis.
The most common cause of thyrotoxic periodic paralysis is Graves disease (toxic diffuse goiter) affecting 96% of thyrotoxic periodic paralysis patients. However, thyrotoxic periodic paralysis can occur in any type of thyrotoxicosis (including administration of excessive amounts of exogenous thyroid hormone or thyrotoxicosis factitia [hyperthyroidism resulting from intentional or accidental over ingestion of thyroid hormone]) or hyperthyroidism (e.g., multinodular goiter, thyroiditis, single autonomous hyperfunctioning “hot” nodule [toxic nodular adenoma], excess iodine ingestion) can trigger attacks of thyrotoxic periodic paralysis in susceptible subjects 203, 204, 205, 206, 207, 208, 209, 210. In general, autoimmune thyroid diseases such as Graves’ disease are more common in pubertal girls than boys. However, the incidence of thyrotoxic periodic paralysis is higher in boys than in girls 211. Unusual is also the disproportional occurrence of thyrotoxic periodic paralysis in men compared to women (26: 1), despite Graves’ disease being predominantly encountered in women 203.
Only 17 to 50% of thyrotoxic periodic paralysis patients exhibit clinical signs of hyperthyroidism, making the diagnosis even more challenging 203. The sudden onset muscle paralysis due to hypokalemia (due to acute potassium ions (K+) shift into the intracellular space) in the context of thyrotoxicosis can lead to cardiac arrhythmias and respiratory failure if not recognized and treated on time 184.
The diagnosis of thyrotoxic periodic paralysis is often delayed, possibly due to a lack of awareness and the rarity of the condition; approximately half of the patients have had history of an episode before the diagnosis was made 212. The diagnosis is delayed on average for 14 months 213.
Early diagnosis of thyrotoxic periodic paralysis is essential, as the condition is potentially reversible by oral or intravenous potassium treatment, leading to rapid resolution without lasting weakness 205, 213. Overlooking the diagnosis may result in respiratory failure and cardiac arrhythmias including QT prolongation, Torsades de points, and ventricular arrhythmias 180. Potassium chloride (KCl) supplementation is essential but often not enough to control thyrotoxic periodic paralysis 214. The management of thyrotoxic periodic paralysis is further complicated by the thin line between refractory hypokalemia and rebound hyperkalemia, both predisposing to serious cardiac events 215.
Treatment of thyrotoxic periodic paralysis includes correction of hypokalemia and maintenance of an normal thyroid (euthyroid) status. Immediate intravenous (IV) infusion with potassium chloride (KCl) is needed to prevent major heart arrhythmias and to foster muscle paralysis recovery 200. However, another study has reported that there is lack of correlation between recovery time of muscle paralysis and the dose of infused potassium chloride (KCl) 216. Despite intravascular hypokalemia, the total body potassium level is normal. Therefore, potassium chloride (KCl) infusion should be administered with caution. Potassium chloride (KCl) infusion causes rebound hyperkalemia in up to 70% of cases, and as a result, fatal arrhythmias may occur 183. Rebound hyperkalemia can be prevented by lower doses of potassium chloride (KCl) and close cardiac monitoring is necessary 217.
A beta blocker like propranolol is an alternative treatment that can reduce paralysis without rebound hyperkalemia. Because thyrotoxic periodic paralysis does not usually recur once an normal thyroid (euthyroid) state has been attained, adequate control of hyperthyroidism is important after the acute phase 211. Currently, antithyroid drugs remain the mainstay treatment of the overactive thyroid (hyperthyroidism). Once the underlying thyroid problem is corrected with medication, radiation or surgery, the symptoms of thyrotoxic periodic paralysis usually disappear.
What triggers attacks of thyrotoxic periodic paralysis?
The same factors which trigger attacks of hypokalemic periodic paralysis will trigger attacks of thyrotoxic periodic paralysis if thyroid levels are too high. Meals high in starchy and sweet foods may trigger an attack. Taking thyroid hormones may trigger an attack. Sleep or resting after vigorous exercise may trigger an attack. For a more complete discussion on triggers see Hypokalemic Periodic Paralysis.
How do I avoid having attacks?
Your doctor will treat the underlying thyroid disorder, which will eventually cure your thyrotoxic periodic paralysis. In the meantime you should determine what triggers your attacks and avoid those triggers. Medication is usually necessary until the thyroid problem is brought under control.
Thyrotoxic periodic paralysis causes
Thyrotoxic periodic paralysis may be caused by any causes of hyperthyroidism, including Graves disease (most common), toxic multinodular goiter, toxic adenoma, thyroiditis, amiodarone-induced thyrotoxicosis, iodine-induced thyrotoxicosis, excess exogenous thyroxine use and thyrotropin (TSH) producing pituitary adenoma 218, 181, 219, 220. A rare cause of thyrotoxic periodic paralysis is thyrotoxicosis factitia or exogenous thyrotoxicosis from surreptitious use of thyroid hormone. Ragesh et al. 221 described a case of thyrotoxic periodic paralysis secondary to consumption of nutraceuticals containing triiodothyronine (T3).
To date, genes that have been studied in relation to thyrotoxic periodic paralysis include the KCNJ18 and KCNJ2 genes. The KCNJ18 gene (also called potassium inwardly rectifying channel subfamily J member 18) located in chromosome 17p11.2 (short arm of chromosome 17), which encodes for the inwardly rectifying potassium (Kir) channel Kir2.6, is known to be more common in Western countries, and has been reported at rates of 33.3% in Caucasian populations, primarily among individuals with Brazilian heritage, 25.9% in Singaporeans, and 1.2% in people from Hong Kong 199, 200. In contrast, in recent studies, genetic variants of the KCNJ2 gene have been reported to affect the development of thyrotoxic periodic paralysis in Korean and Chinese populations 201, 202. These ethnic genetic differences might explain the difference in the incidence of thyrotoxic periodic paralysis.
Common factors triggering attacks of periodic paralysis include the consumption of carbohydrate-rich foods, strenuous physical activity, high salt or sodium intake, stresses (surgical, infectious, psychological), trauma, and drugs (diuretics, estrogens, acetazolamide, epinephrine, laxatives, corticosteroids, non-steroidal anti-inflammatory drugs, licorice, fluoroquinolones, aminoglycosides, and ecstasy) 219, 222, 49, 223, 224, 225, 226.
Thyrotoxic periodic paralysis pathophysiology
The underlying pathophysiology of thyrotoxic periodic paralysis is poorly understood, however the mechanism of thyrotoxic periodic paralysis involves two major factors: the occurrence of hypokalemia and associated muscle paralysis 227. The hypokalemia in thyrotoxic periodic paralysis results from a rapid and massive shift of potassium from the extracellular to the intracellular compartment, mainly into muscles induced by an increased adrenergic sensitization of Na + /K + –ATPase pump. Potassium is the most abundant cation (positively charged ion) in the intracellular fluid (ICF). Potassium (K+) is actively transported into the intracellular fluid (ICF) by the Na + /K + –ATPase pump, which also transports the sodium (Na+) in to the extracellular fluid (ECF). This is expressed in various tissues including the liver, muscle and kidney 228, 229. The Na-K-ATP pump is under the influence of various hormones, which can modulate its activity. The outward flow of potassium is regulated by the inward rectifying K channels (Kir) (4). These two channels working in tandem, keep the potassium levels tightly controlled.
Thyroid hormones stimulate the Na+–K+ ATPase pump in skeletal muscle by binding to the thyroid response elements (TRE) which are upstream of the genes for Na-K pump, and increase its activity by both transcriptional and post transcriptional modification 230, 231, 232. Thyrotoxic periodic paralysis patients have 80% more Na+–K+ ATPase pump activity as compared to thyrotoxic patients 233. Therefore, there is no pure potassium depletion in the body 200.
Catecholamines can also act on stimulating the activity of Na+–K+ ATPase pump through the beta-adrenergic receptors. Insulin, a high carbohydrate meal, exercise and testosterone can increase the activity of Na+–K+ ATPase pump while estrogen decreases it, this may be partly responsible for the male preponderance of thyrotoxic periodic paralysis 234, 235, 236, 237. In addition, insulin and catecholamines can inhibit the inwardly rectifying potassium (Kir) channels decreasing the transport of potassium into extracellular fluid (ECF). Mutations in the KCNJ 18 gene, which encode for the Kir channels, have been found in approximately 33% of patients with thyrotoxic periodic paralysis 238. This massive influx of potassium along with decreased outflow leads to hypokalemia and thyrotoxic periodic paralysis.
Although serum potassium levels are usually decreased in thyrotoxic periodic paralysis, in exceptionally rare circumstances normal serum potassium levels (normokalemia) can lead to normokalemic thyrotoxic periodic paralysis 239. Patients who initially showed normokalemic thyrotoxic periodic paralysis also eventually progressed to hypokalemia 239. However, the pathogenesis of normokalemic thyrotoxic periodic paralysis remains unclear 183.
Genetic and environmental factors may possibly play a role 240, 200. Thyrotoxic periodic paralysis is known to be a channelopathy caused by mutations of the ion channel of the cell membrane and masked under normal thyroid (euthyroid) conditions 187.
Figure 9. Pathogenesis of hypokalemia in thyrotoxicosis
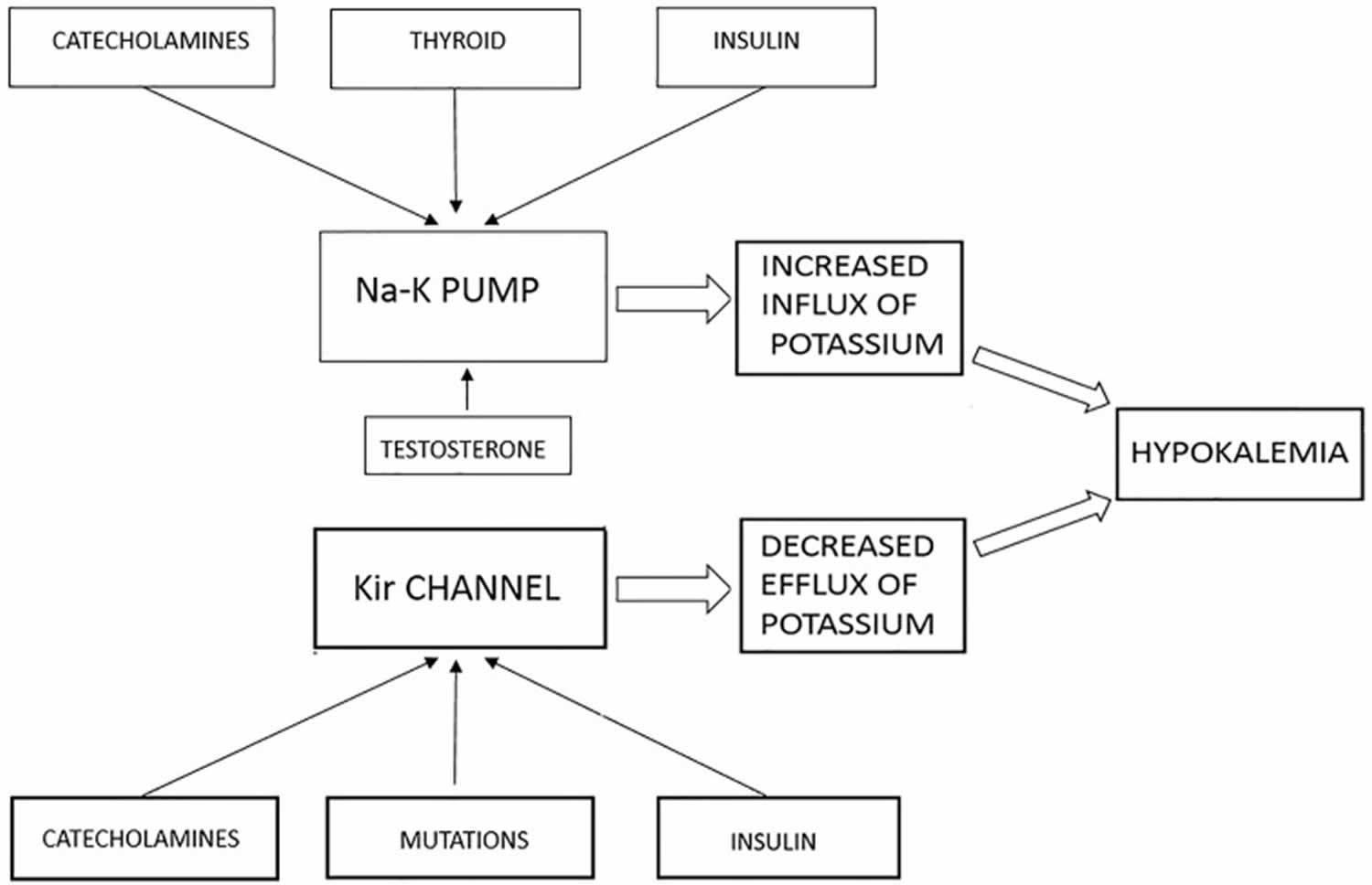
Thyrotoxic periodic paralysis symptoms
Thyrotoxic periodic paralysis symptoms involve attacks of muscle weakness or paralysis. Between attacks, normal muscle function returns. Attacks often begin after symptoms of hyperthyroidism have developed. Hyperthyroid symptoms may be subtle. The attacks may occur daily to yearly. Episodes of muscle weakness may last for a few hours or several days.
The muscle weakness or paralysis 241:
- Comes and goes
- Lasts from a few hours up to several days (rare)
- Occurs more often in the legs than the arms
- Is most common in the shoulders and hips
- Is triggered by heavy, high-carbohydrate, high-salt meals
- Is triggered during rest after exercise
Other rare symptoms may include any of the following 241:
- Trouble breathing
- Speech problems
- Trouble swallowing
- Vision changes
Thyrotoxic periodic paralysis is characterized by episodes of sudden onset of muscle weakness. The severity of the episodes can range from weakness to complete paralysis and the duration can range from a few hours to 3 days 242. Eye muscles, muscles in the head and neck and respiratory muscle weakness is rare, but has been reported 243, 200. During the attacks, you will be alert and can answer questions. Typically, the weakness starts in the proximal muscles of the lower extremities; however, it involves all four extremities in 80% of cases 244. Normal muscle strength returns between attacks. With repeat attacks, you may develop muscle weakness.
Heavy meals, alcohol, exercise, high salt diet, stress, infections, menstruation and glucocorticoids have all been known to precipitate the paralytic episodes 181. Most of these attacks happen at night, likely related to prior alcohol or heavy meals and thereby was given the old name “nocturnal palsy.” Manoukian et al. 216 noted that 84% of thyrotoxic periodic paralysis patients presented to the emergency room between 1 am to 8 am. Thyrotoxic periodic paralysis is seen mostly in males aged 20–40 years, although it can be seen in adolescents and children 245, 243, 224, 246.
The severity of muscle paralysis is correlated with the degree of hypokalemia, but not with clinical signs and symptoms of hyperthyroidism or thyroid hormone levels 211. However, in approximately 75% of patients with thyrotoxic periodic paralysis have it as the presenting symptom of their hyperthyroidism 247.
Symptoms of hyperthyroidism include 241:
- Excessive sweating
- Fast heart rate
- Fatigue
- Headache
- Heat intolerance
- Increased appetite
- Insomnia
- Having bowel movements more often
- Feeling strong heartbeat (palpitations)
- Tremors of the hand
- Warm, moist skin
- Weight loss
Signs of hyperthyroidism may include:
- warm, moist skin,
- tremor,
- tachycardia,
- widened pulse pressure, and
- atrial fibrillation.
Many common symptoms of hyperthyroidism are due to enhanced sensitivity to adrenergic hormones, such as nervousness, palpitations, hyperactivity, increased sweating, heat hypersensitivity, fatigue, increased appetite, weight loss, insomnia, weakness, and frequent bowel movements (occasionally diarrhea). Hypomenorrhea (infrequent menstruation or abnormally low bleeding, less than 30 ml per menstrual cycle) may be present.
Older patients, particularly those with toxic nodular goiter, may present atypically (apathetic or masked hyperthyroidism) with symptoms more akin to depression or dementia. Most do not have exophthalmos or tremor. Atrial fibrillation, syncope, altered sensorium, heart failure, and weakness are more likely. Symptoms and signs may involve only a single organ system.
Eye signs include stare, eyelid lag, eyelid retraction, and mild conjunctival injection and are largely due to excessive adrenergic stimulation. They usually remit with successful treatment. Infiltrative ophthalmopathy, a more serious development, is specific to Graves disease and can occur years before or after hyperthyroidism. It is characterized by orbital pain, lacrimation, irritation, photophobia, increased retro-orbital tissue, exophthalmos, and lymphocytic infiltration of the extraocular muscles, causing ocular muscle weakness that frequently leads to double vision.
Infiltrative dermopathy, also called pretibial myxedema (a confusing term, because myxedema suggests hypothyroidism), is characterized by nonpitting infiltration by proteinaceous ground substance, usually in the pretibial area. It rarely occurs in the absence of Graves ophthalmopathy. The lesion is often itchy and reddish in its early stages and subsequently becomes brawny. Infiltrative dermopathy may appear years before or after hyperthyroidism.
Thyrotoxic periodic paralysis is very rare in children and adolescents and is even more unusual in girls. In addition, although there are aggravating factors, such as a high carbohydrate meal and exercise, that are associated with the occurrence of thyrotoxic periodic paralysis, thyrotoxic periodic paralysis can occur without such factors.
Thyrotoxic periodic paralysis complications
Untreated thyrotoxic periodic paralysis can lead to:
- Difficulty breathing, speaking, or swallowing during attacks (rare)
- Heart arrhythmias during attacks
- Muscle weakness that gets worse over time
Thyrotoxic periodic paralysis diagnosis
Diagnosis of thyrotoxic periodic paralysis is based on recurrent clinical features of thyrotoxic periodic paralysis and blood test results suggestive of hyperthyroidism (abnormal thyroid hormone levels) and hypokalemia (low potassium level during attacks). Your doctor will do blood tests to check the levels of various thyroid hormones including:
- TSH (thyroid stimulating hormone) levels,
- T3 (triiodothyronine),
- T3 resin uptake (T3RU) and
- T4 (thyroxine).
Other test results:
- Abnormal electrocardiogram (ECG) might be present during attacks. The most common electrocardiogram (EKG) changes include ST depression, sinus tachycardia, U waves, and AV blocks 248. Very rarely, patients can present with prolonged QT interval or ventricular tachyarrhythmias.
- Abnormal electromyogram (EMG) during attacks. Electromyograms (EMG) might show a myopathic pattern with decreased duration of muscle action potentials, reduced amplitude, and an increase in polyphasic potentials. These changes might completely disappear during remission 249.
- Low serum potassium during attacks, but normal between attacks. During an attack of weakness your doctor will do a blood test to check the level of potassium. In thyrotoxic periodic paralysis, the level of potassium is low during attacks but normal between attacks. Diagnosis also involves ruling out disorders caused by low potassium.
- A muscle biopsy may sometimes be taken.
Your doctor may try to trigger an attack by giving you insulin and sugar. The sugar is glucose, which reduces potassium level. Or you may be given thyroid hormone. The following signs may be seen during the attack:
- Decreased or no reflexes
- Heart arrhythmias
- Low potassium in the bloodstream (potassium levels are normal between attacks)
Between attacks, the examination is normal. Or, there may be signs of hyperthyroidism. These include an enlarged thyroid changes in the eyes, tremor, or hair and nail changes.
Previously reported cases with thyrotoxic periodic paralysis had only mildly elevated serum thyroid hormone levels. Ko et al. 250 have reported that only 10% of patients have mild thyrotoxic symptoms. Therefore, thyrotoxic periodic paralysis should be distinguished from other causes of acute paralysis such as familial hypokalemic periodic paralysis, Guillain-Barré syndrome, myasthenic crisis, and conditions that produce spinal cord compression 217.
Hypokalemia is the hallmark feature of thyrotoxic periodic paralysis, most of the time, potassium less than 3 mmol/L 251. As the hypokalemia is the result of transcellular shift, urinary potassium is < 20 mmol/lt and urine potassium creatinine ratio is <2 mmol/lt, consistent with renal conservation of potassium (5, 29). Other electrolyte disorders like hypomagnesemia and hypophosphatemia due to transcellular shift are observed. Urine calcium is high due to increased filtration and decreased reabsorption, whereas urine phosphate excretion is decreased. Lin et al. found that a urine calcium/phosphate ratio of >1.4 detected thyrotoxic periodic paralysis with a sensitivity of 100% and specificity of 96% (7). thyrotoxic periodic paralysis occurs in the hyperthyroid state and thyroid studies show a high thyroxine (T4) and suppressed TSH. However, the severity of the paralysis is not directly related to the degree of severity of hyperthyroidism (31). Electrocardiograms (EKG) are abnormal in 83–100% of patients with thyrotoxic periodic paralysis (29, 32). Besides signs of hypokalemia, (U waves, ST segment depression and T wave flattening), atrioventricular block and arrhythmias (supraventricular arrhythmias, atrial fibrillation and ventricular arrhythmias) have also been reported. thyrotoxic periodic paralysis needs to be differentiated from familial hypokalemic periodic paralysis (FHPP), which presents with episodic muscular weakness as well. However, it affects Caucasians with equal sex distribution. FHPP is autosomal dominant and a family history of hypokalemic paralysis is often positive (33). Thyroid studies in thyrotoxic periodic paralysis show hyperthyroidism, whereas in FHPP they are normal.
In one study of 19 men with thyrotoxic periodic paralysis initial serum potassium (K) levels upon admittance to hospital ranged from 1.1 to 3.4 mmol/L (mean, 1.90.5 mmol/L). Serum magnesium (Mg) level was measured in 18 episodes during paralysis and in 13 episodes after paralysis. During paralysis episodes, all patients had low or low-normal magnesium (Mg) levels (0.60-0.80 mmol/L 1.5-1.9 mg/dL). Only two patients received supplemental magnesium sulphate, but magnesium (Mg) levels increased by 0.1 mmol/L or more (0.24 mg/dL) in all patients who had it checked. Serum creatine phosphokinase levels were obtained in 18 episodes during paralysis. Twelve patients had elevated creatine phosphokinase values, 5 of which were of 1000 U/L or more. Serum alkaline phosphatase levels were mildly elevated in 12 of 16 patients, ranging from 118 to 268 U/L (normal, 39-117 U/L).
Diagnosis of hyperthyroidism is based on history, physical examination, and thyroid function tests. Serum TSH (thyroid stimulating hormone) measurement is the best test because TSH is suppressed in hyperthyroid patients except in the rare instance when the cause is a TSH-secreting pituitary adenoma or pituitary resistance to the normal inhibition by thyroid hormone.
Free T4 (thyroxine) is increased in hyperthyroidism. However, T4 can be falsely normal in true hyperthyroidism in patients with a severe systemic illness (similar to the falsely low levels that occur in euthyroid sick syndrome) and in T3 (triiodothyronine) toxicosis. If free T4 level is normal and TSH is low in a patient with subtle symptoms and signs of hyperthyroidism, then serum T3 should be measured to detect T3 toxicosis; an elevated level confirms that diagnosis.
The cause can often be diagnosed clinically (eg, the presence of signs specific to Graves disease). If not, radioactive iodine uptake by the thyroid may be measured by using iodine-123. When hyperthyroidism is due to hormone overproduction, radioactive iodine uptake by the thyroid is usually elevated. When hyperthyroidism is due to thyroiditis, iodine ingestion, or overtreatment with thyroid hormones, radioactive iodine uptake is low.
TSH receptor antibodies can be measured to evaluate for Graves disease. Measurement is done in pregnant women with a history of Graves disease during the 3rd trimester of pregnancy to assess the risk of neonatal Graves disease; TSH receptor antibodies readily cross the placenta to stimulate the fetal thyroid. Most patients with Graves disease have circulating antithyroid peroxidase antibodies, and fewer have antithyroglobulin antibodies.
Inappropriate TSH secretion is uncommon. The diagnosis is confirmed when hyperthyroidism occurs with elevated circulating free T4 and T3 concentrations and normal or elevated serum TSH.
If thyrotoxicosis factitia is suspected, serum thyroglobulin can be measured; it is usually low or low-normal—unlike in all other causes of hyperthyroidism.
Thyrotoxic periodic paralysis treatment
Potassium should also be given during the attack, most often by mouth, however not all patients respond to potassium alone and recent evidence suggests that combining potassium and propranolol is a more effective therapy. If weakness is severe, you may need to get potassium through a vein (IV). Note: You should only get intravenous (IV) potassium if your kidney function is normal and you are monitored in the hospital. The ultimate goal of treatment is to reduce your thyroid hormone levels and restore normal thyroid status.
Weakness that involves the muscles used for breathing or swallowing is an emergency. You must be taken to a hospital. Serious irregularity of heartbeat also occur during attacks.
Acute treatment recommended that 30 mEq of potassium chloride (KCl) be given every 2 hours orally for 6 hours and then every 4 hours with careful monitoring until recovery begins, with a maximum oral dose of 90 mEq in 24 hours 227, 252. Some experts suggestions include replacing at a rate of less than 10 mEq orally. Because thyrotoxic periodic paralysis patients may develop rebound hyperkalemia, Manoukian et al 216 recommends that potassium replacement therapy should be cautious and should not exceed 90 mEq of potassium chloride (KCl) per 24 hours unless there is a reason for potassium (K) loss, such as diarrhea, vomiting, or diuretic use.
In the Manoukian study (19 patients) all patients remained attack free as long as they took methimazole and propranolol hydrochloride or after radioiodine (iodine-131) treatment 216. Eighteen patients were eventually treated with radioiodine (iodine-131) therapy. None of the patients had paralytic episodes after a euthyroid state was achieved. Nonselective beta-blockers such as propranolol may be useful to prevent attacks of paralysis once patients begin taking antithyroid medications but are not yet euthyroid.
Your doctor may also recommend a diet low in carbohydrates and salt to prevent attacks. Precipitating factors like strenuous exercise and high carbohydrate meals should be avoided 253. Glucose infusions need to be avoided as they can raise insulin levels and worsen hypokalemia. Prophylactic administration of potassium chloride (KCl) to prevent thyrotoxic periodic paralysis is not effective and therefore not recommended 247.
You may be given beta-blocker medicines to reduce the number and severity of attacks while your hyperthyroidism is brought under control. Non-selective beta-blockers have been shown to improve neuromuscular symptoms by reducing the intracellular shift of phosphate and potassium 254. Intravenous propranolol 1 mg every 10 minutes up to 3 doses can be given in patients unresponsive to potassium replacement 255, 256, 257. Oral propranolol (40 mg four time daily) is also effective as prophylaxis, preventing further episodes of thyrotoxic periodic paralysis 258. However, propranolol needs to be administered with caution in case of a heart block, as it can result in severe bradycardia and cardiovascular collapse 259.
Definitive treatment of hyperthyroidism, either with radioiodine ablation or with thyroidectomy, results in resolution of thyrotoxic periodic paralysis. Use of antithyroid medications alone resulted in a relapse attack in 56% patients within 7 months 212.
Treatment of hyperthyroidism
Treatment of hyperthyroidism depends on cause but may include 260:
- Radioactive iodine
- Methimazole or propylthiouracil
- Beta-blockers
- Iodine
- Surgery
Radioactive sodium iodine (iodine-131, radioiodine)
In the United States, iodine-131 is the most common treatment for hyperthyroidism. Radioiodine is often recommended as the treatment of choice for Graves disease and toxic nodular goiter in all patients, including children. Dosage of iodine-131 is difficult to adjust because the response of the gland cannot be predicted; some physicians give a standard dose of 8 to 15 millicurie. Others adjust the dose based on estimated thyroid size and the 24-hour uptake to provide a dose of 80 to 120 microcurie/g thyroid tissue.
When sufficient iodine-131 is given to cause euthyroidism, about 25 to 50% of patients become hypothyroid 1 year later, and the incidence continues to increase yearly. Thus, most patients eventually become hypothyroid. However, if smaller doses are used, incidence of recurrence is higher. Larger doses, such as 10 to 20 millicurie, often cause hypothyroidism within 6 months, and thus ablative therapy (ie, iodine-131) has become the preferred approach.
Radioactive iodine is not used during lactation because it can enter breast milk and cause hypothyroidism in the infant. It is not used during pregnancy because it crosses the placenta and can cause severe fetal hypothyroidism. There is no proof that radioiodine increases the incidence of tumors, leukemia, thyroid cancer, or birth defects in children born to previously hyperthyroid women who become pregnant later in life.
Methimazole and propylthiouracil
These antithyroid drugs block thyroid peroxidase, decreasing the organification of iodide, and impair the coupling reaction. Propylthiouracil in high doses also inhibits the peripheral conversion of T4 to T3.
Methimazole is the preferred drug. The usual starting dosage of methimazole is 5 to 20 mg orally 2 or 3 times a day. Normalization of TSH lags normalization of T4 and T3 levels by one or more weeks. Therefore, when T4 and T3 levels normalize, the dosage is decreased to the lowest effective amount, usually methimazole 2.5 to 10 mg once a day in order to avoid inducing hypothyroidism. Control generally is achieved in 2 to 3 months. Maintenance doses of methimazole may be continued for one or many years depending on the clinical circumstances. Carbimazole, which is used widely in Europe but is unavailable in the US, is rapidly converted to methimazole. The usual starting dose is similar to that of methimazole; maintenance dosage is 2.5 to 10 mg orally once a day or 2.5 to 5 mg twice a day.
Because of severe liver failure in some patients < age 40, especially children, propylthiouracil is recommended only in special situations (eg, in the 1st trimester of pregnancy, in thyroid storm). The usual starting dose of propylthiouracil is 100 to 150 mg orally every 8 hours. Rapid control can be achieved by increasing the dosage of propylthiouracil to 150 to 200 mg every 8 hours. Such dosages or higher ones (up to 400 mg every 8 hours) are generally reserved for severely ill patients, including those with thyroid storm, to block the conversion of T4 to T3. Maintenance dosing with propylthiouracil is 50 mg twice a day or 3 times a day
About 20 to 50% of patients with Graves disease remain in remission after a 1- to 2-year course of either drug. The return to normal or a marked decrease in gland size, the restoration of a normal serum TSH level, and less severe hyperthyroidism before therapy are good prognostic signs of long-term remission. The concomitant use of antithyroid drug therapy and levothyroxine does not improve the remission rate in patients with Graves disease. Because toxic nodular goiter rarely goes into remission, antithyroid drug therapy is given only in preparation for surgical treatment or iodine-131 therapy.
Adverse effects include rash, allergic reactions, abnormal liver function (including hepatic failure with propylthiouracil), and, in about 0.1% of patients, reversible agranulocytosis. Patients allergic to one drug can be switched to the other, but cross-sensitivity may occur. If agranulocytosis occurs, the patient cannot be switched to the other drug; other therapy (eg, radioiodine, surgery) should be used.
- If agranulocytosis occurs with one of the antithyroid peroxidase drugs (methimazole or propylthiouracil), avoid using another drug in the same class; use another therapy (eg, radioiodine, surgery) instead.
Potential adverse effects or other characteristics vary between the two drugs and guide the indications for each. Methimazole need only be given once a day, which improves adherence. Furthermore, when methimazole is used in dosages of < 20 mg a day, agranulocytosis is less common; with propylthiouracil, agranulocytosis may occur at any dosage.
Methimazole has been used successfully in pregnant and nursing women without fetal or infant complications, but rarely methimazole has been associated with scalp and gastrointestinal defects in neonates and with a rare embryopathy. Because of these complications, propylthiouracil is used in the 1st trimester of pregnancy.
Propylthiouracil is preferred for the treatment of thyroid storm, because the high dosages used (over 800 mg a day) partially block the peripheral conversion of T4 to T3 in addition to decreasing production in the thyroid.
The combination of high-dose propylthiouracil and dexamethasone, also a potent inhibitor of T4 to T3 conversion, can relieve symptoms of severe hyperthyroidism as seen in patients with thyroid storm and restore the serum T3 level to normal within a week.
Beta-blockers
Symptoms and signs of hyperthyroidism due to adrenergic stimulation may respond to beta-blockers; propranolol has had the greatest use, but atenolol or metoprolol may be preferable.
Other manifestations typically do not respond.
Manifestations typically responding to beta-blockers: Tachycardia, tremor, mental symptoms, eyelid lag; occasionally heat intolerance and sweating, diarrhea, proximal myopathy
Manifestations typically not responding to beta-blockers: goiter, exophthalmos, weight loss, bruit, increased oxygen consumption, and increased circulating thyroxine levels
Propranolol is indicated in thyroid storm. It rapidly decreases heart rate, usually within 2 to 3 hours when given orally and within minutes when given intravenously. Esmolol should be used only in the intensive care unit because it requires careful titration and monitoring. Beta-blockers are also indicated for tachycardia with hyperthyroidism, especially in older patients, because antithyroid drugs usually take several weeks to become fully effective. Calcium channel blockers may control tachyarrhythmias in patients in whom beta-blockers are contraindicated.
Iodine
Iodine in pharmacologic doses inhibits the release of T3 and T4 within hours and inhibits the organification of iodine, a transitory effect lasting from a few days to a week, after which inhibition usually ceases. Iodine is used for emergency management of thyroid storm, for hyperthyroid patients undergoing emergency nonthyroid surgery, and (because it also decreases the vascularity of the thyroid) for preoperative preparation of hyperthyroid patients undergoing thyroidectomy. Iodine generally is not used for routine treatment of hyperthyroidism. The usual dosage is 2 to 3 drops (100 to 150 mg) of a saturated potassium iodide solution orally 3 times a day or 4 times a day or sodium iodide in 1 L 0.9% saline solution 0.5 to 1 g IV given slowly once a day.
Complications of iodine therapy include inflammation of the salivary glands, conjunctivitis, and rash.
Surgery
Surgery is indicated for patients with Graves disease whose hyperthyroidism has recurred after courses of antithyroid drugs and who refuse iodine-131 therapy, patients who cannot tolerate antithyroid drugs, patients with very large goiters, and in some younger patients with toxic adenoma and multinodular goiter. Surgery may be done in older patients with giant nodular goiters.
Surgery usually restores normal function. Postoperative recurrences vary between 2 and 16%; risk of hypothyroidism is directly related to the extent of surgery. Vocal cord paralysis and hypoparathyroidism are uncommon complications. Saturated solution of potassium iodide 3 drops (about 100 to 150 mg) orally 3 times a day should be given for 10 days before surgery to reduce the vascularity of the gland. Methimazole must be given first because the patient should be euthyroid before iodide is given. Dexamethasone can be added to rapidly restore euthyroidism. Surgical procedures on the anterior neck are more difficult in patients who previously underwent thyroidectomy or radioiodine therapy.
Treatment of infiltrative dermopathy and ophthalmopathy
In infiltrative dermopathy in Graves disease, topical corticosteroids or corticosteroid injections into the lesions may decrease the dermopathy. Dermopathy sometimes remits spontaneously after months or years.
Ophthalmopathy should be treated jointly by the endocrinologist and ophthalmologist and may require selenium, corticosteroids, orbital radiation, and surgery. Surgical thyroidectomy may help resolve or prevent progression of ophthalmopathy. Teprotumumab, an insulin-like growth factor 1 (IGF-1) receptor inhibitor, is very effective therapy for moderately severe ophthalmopathy 261. Radioiodine therapy may accelerate progression of ophthalmopathy when ophthalmopathy is active, and is thus contraindicated in this active phase.
Thyrotoxic periodic paralysis prognosis
Thyrotoxic periodic paralysis responds well to treatment. Treating hyperthyroidism will prevent attacks. It may even reverse muscle weakness.
If an attack is not treated and your breathing muscles are affected, death can occur.
Chronic attacks over time can lead to muscle weakness. This weakness can continue even between attacks if the thyrotoxicosis is not treated.
- Horga A, Raja Rayan DL, Matthews E, Sud R, Fialho D, Durran SC, Burge JA, Portaro S, Davis MB, Haworth A, Hanna MG. Prevalence study of genetically defined skeletal muscle channelopathies in England. Neurology. 2013 Apr 16;80(16):1472-5. doi: 10.1212/WNL.0b013e31828cf8d0[↩][↩]
- Cannon SC. Channelopathies of skeletal muscle excitability. Compr Physiol. 2015 Apr;5(2):761-90. doi: 10.1002/cphy.c140062[↩][↩]
- Lucio C, Roberta P, Daniela I, Miriana G, Francesco C, et al., Periodic Paralysis: A Case Report of Normokalemic Presentation in a Young Girl. Biomed J Sci & Tech Res 35(2)-2021. BJSTR. MS.ID.005672. https://biomedres.us/pdfs/BJSTR.MS.ID.005673.pdf[↩][↩][↩]
- Thor, M.G., Vivekanandam, V., Sampedro-Castañeda, M. et al. Myotonia in a patient with a mutation in an S4 arginine residue associated with hypokalaemic periodic paralysis and a concomitant synonymous CLCN1 mutation. Sci Rep 9, 17560 (2019). https://doi.org/10.1038/s41598-019-54041-0[↩]
- What is Periodic Paralysis? https://hkpp.org/basic-information[↩][↩][↩][↩][↩]
- Andersen-Tawil syndrome. https://medlineplus.gov/genetics/condition/andersen-tawil-syndrome[↩][↩][↩][↩][↩][↩]
- Channelopathies of skeletal muscle excitability. Muscle – Fundamental Biology and Mechanisms of Disease, Volume 2, 2012, Pages 1013-1022. https://doi.org/10.1016/B978-0-12-381510-1.00073-9[↩]
- Hypokalemic periodic paralysis. https://rarediseases.info.nih.gov/diseases/6729/hypokalemic-periodic-paralysis[↩][↩][↩]
- Venkatesan PE, Gnanashanmugham G, Balakrishnan R, Ramadoss K. Hypokalemic periodic paralysis: Three rare secondary causes. Med J DY Patil Univ 2015;8:760-2. https://journals.lww.com/mjdy/pages/default.aspx/text.asp?2015/8/6/760/169918[↩]
- Velarde-Mejía Y, Gamboa-Cárdenas R, Ugarte-Gil M, et al.: Hypokalemic Paralysis: A Hidden Card of Several Autoimmune Diseases. Clin Med Insights Arthritis Musculoskelet Disord. 2017;10: 1179544117722763. 10.1177/1179544117722763[↩]
- Dormohammadi Toosi T, Naderi N, Movassaghi S, Seradj MH, Khalvat A, Shahbazi F. Secondary Sjogren’s syndrome presenting with hypokalemic periodic paralysis. Oxf Med Case Reports. 2014 Nov 3;2014(8):135-7. doi: 10.1093/omcr/omu052[↩]
- Hypokalemic periodic paralysis. https://www.uptodate.com/contents/hypokalemic-periodic-paralysis[↩][↩]
- Weber F, Lehmann-Horn F. Hypokalemic Periodic Paralysis. 2002 Apr 30 [Updated 2018 Jul 26]. In: Adam MP, Mirzaa GM, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2023. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1338[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Phuyal P, Nagalli S. Hypokalemic Periodic Paralysis. [Updated 2023 Feb 7]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK559178[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Familial Periodic Paralyses. https://www.ninds.nih.gov/health-information/disorders/familial-periodic-paralyses[↩]
- Velarde-Mejía Y, Gamboa-Cárdenas R, Ugarte-Gil M, Asurza CP. Hypokalemic Paralysis: A Hidden Card of Several Autoimmune Diseases. Clin Med Insights Arthritis Musculoskelet Disord. 2017 Aug 3;10:1179544117722763. doi: 10.1177/1179544117722763[↩][↩]
- Koul PA, Wahid A. Distal renal tubular acidosis and hypokalemic paralysis in a patient with hypothyroidism. Saudi J Kidney Dis Transpl. 2011 Sep;22(5):1014-6.[↩][↩]
- Basak RC, Sharkawi KM, Rahman MM, Swar MM. Distal renal tubular acidosis, hypokalemic paralysis, nephrocalcinosis, primary hypothyroidism, growth retardation, osteomalacia and osteoporosis leading to pathological fracture: a case report. Oman Med J. 2011 Jul;26(4):271-4. doi: 10.5001/omj.2011.66[↩]
- Meregildo-Rodríguez ED, Failoc-Rojas VE. Case Report: Recurrent hypokalemic periodic paralysis associated with distal renal tubular acidosis (type 1) and hypothyroidism secondary to Hashimoto’s thyroiditis. F1000Res. 2018 Jul 30;7:1154. doi: 10.12688/f1000research.15662.3[↩]
- Meola G, Hanna MG, Fontaine B. Diagnosis and new treatment in muscle channelopathies. J Neurol Neurosurg Psychiatry 2009;80:360–5. doi:10.1136/jnnp.2008.164046[↩][↩][↩][↩]
- Jurkat-Rott K, Lehmann-Horn F. Muscle channelopathies and critical points in functional and genetic studies. J Clin Invest 2005;115:2000–9. doi:10.1172/JCI25525[↩][↩][↩][↩]
- Jurkat-Rott K, Weber MA, Fauler M, et al. K+-dependent paradoxical membrane depolarization and Na+ overload, major and reversible contributors to weakness by ion channel leaks. Proc Natl Acad Sci USA 2009;106:4036–41. doi:10.1073/pnas.0811277106[↩]
- Goyal G, Kant R, Vajpayee A: Hypokalemic respiratory paralysis due to distal renal tubular acidosis as the presenting manifestation of Sjögren’s syndrome. Journal of Acute Medicine. 2014;4(1):49–52. 10.1016/j.jacme.2014.01.002[↩][↩]
- Sinha U, Sengupta N, Sinharay K, et al.: Recurrent hypokalemic paralysis: An atypical presentation of hypothyroidism. Indian J Endocrinol Metab. 2013;17(1):174–176. 10.4103/2230-8210.107880[↩]
- Singh ThK, Sangme N, Singh LN, et al.: Hypokalaemic Periodic Paralysis Associated with Hypothyroidism. Indian J Phys Med Rehabil. 2012;23(2):79–81.[↩]
- Gündüz E, Zengin Y, Dursun R, et al.: Hypokalemic Periodic Paralysis Due To Distal Renal Tubular Acidosis. Eur J Gen Med. 2015;12(2):164–166. 10.15197/sabad.1.12.33[↩]
- Finn BC, Young P, Bruetman JE, Forrester M, Lombi F, Campolo Girard V, Pereyra H, Trimarchi H. Hipokalemia, acidosis tubular distal y tiroiditis de Hashimoto [Hypokalemia, distal renal tubular acidosis, and Hashimoto’s thyroiditis]. Nefrologia. 2008;28(5):569-70. https://www.revistanefrologia.com/en-hypokalemia-distal-renal-tubular-acidosis-articulo-X2013251408004065[↩]
- Statland JM, Fontaine B, Hanna MG, Johnson NE, Kissel JT, Sansone VA, Shieh PB, Tawil RN, Trivedi J, Cannon SC, Griggs RC. Review of the Diagnosis and Treatment of Periodic Paralysis. Muscle Nerve. 2018 Apr;57(4):522-530. doi: 10.1002/mus.26009[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Fontaine B. Periodic paralysis. Adv Genet. 2008;63:3-23. doi: 10.1016/S0065-2660(08)01001-8[↩][↩][↩][↩][↩][↩][↩][↩]
- Hypokalemic periodic paralysis. https://medlineplus.gov/genetics/condition/hypokalemic-periodic-paralysis[↩]
- Venance SL, Cannon SC, Fialho D, Fontaine B, Hanna MG, Ptacek LJ, Tristani-Firouzi M, Tawil R, Griggs RC; CINCH investigators. The primary periodic paralyses: diagnosis, pathogenesis and treatment. Brain. 2006 Jan;129(Pt 1):8-17. doi: 10.1093/brain/awh639[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Groome JR, Moreau A, Delemotte L. Gating Pore Currents in Sodium Channels. Handb Exp Pharmacol. 2018;246:371-399. doi: 10.1007/164_2017_54[↩][↩][↩][↩][↩][↩]
- Jurkat-Rott K, Weber MA, Fauler M, Guo XH, Holzherr BD, Paczulla A, Nordsborg N, Joechle W, Lehmann-Horn F. K+-dependent paradoxical membrane depolarization and Na+ overload, major and reversible contributors to weakness by ion channel leaks. Proc Natl Acad Sci U S A. 2009 Mar 10;106(10):4036-41. doi: 10.1073/pnas.0811277106[↩][↩][↩][↩]
- Matthews E, Labrum R, Sweeney MG, Sud R, Haworth A, Chinnery PF, Meola G, Schorge S, Kullmann DM, Davis MB, Hanna MG. Voltage sensor charge loss accounts for most cases of hypokalemic periodic paralysis. Neurology. 2009 May 5;72(18):1544-7. doi: 10.1212/01.wnl.0000342387.65477.46[↩][↩][↩]
- Francis DG, Rybalchenko V, Struyk A, Cannon SC. Leaky sodium channels from voltage sensor mutations in periodic paralysis, but not paramyotonia. Neurology. 2011 May 10;76(19):1635-41. doi: 10.1212/WNL.0b013e318219fb57[↩][↩][↩][↩]
- CACNA1S gene. https://medlineplus.gov/genetics/gene/cacna1s[↩]
- Jurkat-Rott K, Holzherr B, Fauler M, Lehmann-Horn F. Sodium channelopathies of skeletal muscle result from gain or loss of function. Pflugers Arch. 2010 Jul;460(2):239-48. doi: 10.1007/s00424-010-0814-4[↩][↩][↩]
- Jurkat-Rott K, Mitrovic N, Hang C, Kouzmekine A, Iaizzo P, Herzog J, Lerche H, Nicole S, Vale-Santos J, Chauveau D, Fontaine B, Lehmann-Horn F. Voltage-sensor sodium channel mutations cause hypokalemic periodic paralysis type 2 by enhanced inactivation and reduced current. Proc Natl Acad Sci U S A. 2000 Aug 15;97(17):9549-54. doi: 10.1073/pnas.97.17.9549[↩][↩]
- SCN4A gene. https://medlineplus.gov/genetics/gene/scn4a[↩][↩]
- Jurkat-Rott K, Lehmann-Horn F. State of the art in hereditary muscle channelopathies. Acta Myol. 2010 Oct;29(2):343-50. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3040592[↩][↩]
- Ke Q, Luo B, Qi M, Du Y, Wu W. Gender differences in penetrance and phenotype in hypokalemic periodic paralysis. Muscle Nerve. 2013 Jan;47(1):41-5. doi: 10.1002/mus.23460[↩]
- Miller TM, Dias da Silva MR, Miller HA, Kwiecinski H, Mendell JR, Tawil R, McManis P, Griggs RC, Angelini C, Servidei S, Petajan J, Dalakas MC, Ranum LP, Fu YH, Ptácek LJ. Correlating phenotype and genotype in the periodic paralyses. Neurology. 2004 Nov 9;63(9):1647-55. doi: 10.1212/01.wnl.0000143383.91137.00[↩][↩][↩][↩][↩][↩]
- Jiang D, Gamal El-Din TM, Ing C, Lu P, Pomès R, Zheng N, Catterall WA. Structural basis for gating pore current in periodic paralysis. Nature. 2018 May;557(7706):590-594. doi: 10.1038/s41586-018-0120-4[↩]
- Sokolov S, Scheuer T, Catterall WA. Gating pore current in an inherited ion channelopathy. Nature. 2007 Mar 1;446(7131):76-8. doi: 10.1038/nature05598[↩]
- Lapie P, Goudet C, Nargeot J, Fontaine B, Lory P. Electrophysiological properties of the hypokalaemic periodic paralysis mutation (R528H) of the skeletal muscle alpha 1s subunit as expressed in mouse L cells. FEBS Lett. 1996 Mar 18;382(3):244-8. doi: 10.1016/0014-5793(96)00173-1[↩]
- Lehmann-Horn F, Jurkat-Rott K, Rüdel R; Ulm Muscle Centre. Diagnostics and therapy of muscle channelopathies–Guidelines of the Ulm Muscle Centre. Acta Myol. 2008 Dec;27(3):98-113. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2858942[↩]
- Sansone VA, Burge J, McDermott MP, Smith PC, Herr B, Tawil R, Pandya S, Kissel J, Ciafaloni E, Shieh P, Ralph JW, Amato A, Cannon SC, Trivedi J, Barohn R, Crum B, Mitsumoto H, Pestronk A, Meola G, Conwit R, Hanna MG, Griggs RC; Muscle Study Group. Randomized, placebo-controlled trials of dichlorphenamide in periodic paralysis. Neurology. 2016 Apr 12;86(15):1408-1416. doi: 10.1212/WNL.0000000000002416[↩][↩][↩]
- Finsterer J. Primary periodic paralyses. Acta Neurol Scand. 2008 Mar;117(3):145-58. doi: 10.1111/j.1600-0404.2007.00963.x[↩][↩][↩][↩][↩][↩][↩][↩]
- Ober KP. Thyrotoxic periodic paralysis in the United States. Report of 7 cases and review of the literature. Medicine (Baltimore). 1992 May;71(3):109-20. doi: 10.1097/00005792-199205000-00001[↩][↩]
- Links TP, Zwarts MJ, Wilmink JT, Molenaar WM, Oosterhuis HJ. Permanent muscle weakness in familial hypokalaemic periodic paralysis. Clinical, radiological and pathological aspects. Brain. 1990 Dec;113 ( Pt 6):1873-89. doi: 10.1093/brain/113.6.1873[↩][↩][↩][↩]
- Sharma CM, Nath K, Parekh J. Reversible electrophysiological abnormalities in hypokalemic paralysis: Case report of two cases. Ann Indian Acad Neurol. 2014 Jan;17(1):100-2. doi: 10.4103/0972-2327.128566[↩]
- McManis PG, Lambert EH, Daube JR. The exercise test in periodic paralysis. Muscle Nerve. 1986 Oct;9(8):704-10. doi: 10.1002/mus.880090805[↩]
- Tengan CH, Antunes AC, Gabbai AA, Manzano GM. The exercise test as a monitor of disease status in hypokalaemic periodic paralysis. J Neurol Neurosurg Psychiatry. 2004 Mar;75(3):497-9. doi: 10.1136/jnnp.2003.013870[↩]
- Zhang L, Niu J, Li Y, Guan Y, Cui L, Liu M. Abduction range: A potential parameter for the long exercise test in hypokalemic periodic paralysis during inter-attack periods. Muscle Nerve. 2020 Jan;61(1):104-107. doi: 10.1002/mus.26721[↩]
- Levitt JO. Practical aspects in the management of hypokalemic periodic paralysis. J Transl Med. 2008 Apr 21;6:18. doi: 10.1186/1479-5876-6-18. Erratum in: J Transl Med. 2014;12:198. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2374768[↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Li J. Hypokalemic Periodic Paralysis Secondary to Medullary Sponge Kidney Complicated With Renal Tubular Acidosis. Cureus. 2022 Oct 10;14(10):e30160. doi: 10.7759/cureus.30160[↩]
- Sugiura Y, Makita N, Li L, Noble PJ, Kimura J, Kumagai Y, Soeda T, Yamamoto T. Cold induces shifts of voltage dependence in mutant SCN4A, causing hypokalemic periodic paralysis. Neurology. 2003 Oct 14;61(7):914-8. doi: 10.1212/01.wnl.0000086820.54065.a0[↩]
- Greig SL. Dichlorphenamide: A Review in Primary Periodic Paralyses. Drugs. 2016 Mar;76(4):501-7. doi: 10.1007/s40265-016-0559-2[↩]
- Griggs RC, Resnick J, Engel WK. Intravenous treatment of hypokalemic periodic paralysis. Arch Neurol. 1983 Sep;40(9):539-40. doi: 10.1001/archneur.1983.04050080039005[↩]
- Sansone V, Meola G, Links TP, Panzeri M, Rose MR. Treatment for periodic paralysis. Cochrane Database Syst Rev. 2008 Jan 23;(1):CD005045. doi: 10.1002/14651858.CD005045.pub2[↩]
- Tawil, R., McDermott, M.P., Brown, R., Jr, Shapiro, B.C., Ptacek, L.J., McManis, P.G., Dalakas, M.C., Spector, S.A., Mendell, J.R., Hahn, A.F., Griggs, R.C. and (2000), Randomized trials of dichlorphenamide in the periodic paralyses. Ann Neurol., 47: 46-53. https://doi.org/10.1002/1531-8249(200001)47:1<46::AID-ANA9>3.0.CO;2-H[↩]
- Dalakas MC, Engel WK. Treatment of “permanent” muscle weakness in familial Hypokalemic Periodic Paralysis. Muscle Nerve. 1983 Mar-Apr;6(3):182-6. doi: 10.1002/mus.880060303[↩]
- Fiore DM, Strober JB. Treatment of hypokalemic periodic paralysis with topiramate. Muscle Nerve. 2011 Jan;43(1):127-9. doi: 10.1002/mus.21854[↩]
- Cavel-Greant D, Lehmann-Horn F, Jurkat-Rott K. The impact of permanent muscle weakness on quality of life in periodic paralysis: a survey of 66 patients. Acta Myol. 2012 Oct;31(2):126-33. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3476862[↩]
- Sekhon DS, Vaqar S, Gupta V. Hyperkalemic Periodic Paralysis. [Updated 2023 May 8]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK564319[↩][↩]
- Weber F. Hyperkalemic Periodic Paralysis. 2003 Jul 18 [Updated 2021 Jul 1]. In: Adam MP, Mirzaa GM, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2023. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1496[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Gamstorp, I. (1963), ADYNAMIA EPISODICA HEREDITARIA AND MYOTONIA. Acta Neurologica Scandinavica, 39: 41-58. https://doi.org/10.1111/j.1600-0404.1963.tb05386.x[↩]
- Bradley WG, Taylor R, Rice DR, Hausmanowa-Petruzewicz I, Adelman LS, Jenkison M, Jedrzejowska H, Drac H, Pendlebury WW. Progressive myopathy in hyperkalemic periodic paralysis. Arch Neurol. 1990 Sep;47(9):1013-7. doi: 10.1001/archneur.1990.00530090091018[↩][↩][↩][↩]
- CARL M. PEARSON, THE PERIODIC PARALYSES: DIFFERENTIAL FEATURES AND PATHOLOGICAL OBSERVATIONS IN PERMANENT MYOPATHIC WEAKNESS, Brain, Volume 87, Issue 2, June 1964, Pages 341–354, https://doi.org/10.1093/brain/87.2.341[↩]
- Schapira A, Griggs R. Muscle disease: blue books of practical neurology. New York: Elsevier; 1991.[↩][↩][↩]
- Hyperkalemic periodic paralysis. https://medlineplus.gov/genetics/condition/hyperkalemic-periodic-paralysis[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Lehmann-Horn F, Rüdel R, Jurkat-Rott K. Nondystrophic myotonias and periodic paralyses. In: Engel A, Franzini-Armstrong C, editors. Myology. 3. New York: McGraw-Hill Professional; 2004. pp. 1257–1300.[↩][↩][↩]
- Jurkat-Rott K, Lehmann-Horn F. Genotype-Phenotype correlation and therapeutic rationale in hyperkalemic periodic paralysis. Neurotherapeutics 2007;4:216–24. 10.1016/j.nurt.2007.02.001[↩]
- Charles G, Zheng C, Lehmann-Horn F, Jurkat-Rott K, Levitt J. Characterization of hyperkalemic periodic paralysis: a survey of genetically diagnosed individuals. J Neurol. 2013 Oct;260(10):2606-13. doi: 10.1007/s00415-013-7025-9[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Amarteifio E, Nagel AM, Weber MA, Jurkat-Rott K, Lehmann-Horn F. Hyperkalemic periodic paralysis and permanent weakness: 3-T MR imaging depicts intracellular 23Na overload–initial results. Radiology. 2012 Jul;264(1):154-63. doi: 10.1148/radiol.12110980[↩]
- Lee YH, Lee HS, Lee HE, Hahn S, Nam TS, Shin HY, Choi YC, Kim SM. Whole-Body Muscle MRI in Patients with Hyperkalemic Periodic Paralysis Carrying the SCN4A Mutation T704M: Evidence for Chronic Progressive Myopathy with Selective Muscle Involvement. J Clin Neurol. 2015 Oct;11(4):331-8. doi: 10.3988/jcn.2015.11.4.331[↩]
- Amato A, Russell J. Neuromuscular disorders. New York: McGraw Hill Professional; 2008.[↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Hyperkalemic periodic paralysis. https://www.orpha.net/consor/cgi-bin/Disease_Search.php?lng=EN&data_id=212[↩][↩][↩][↩]
- George AL Jr, Ledbetter DH, Kallen RG, Barchi RL. Assignment of a human skeletal muscle sodium channel alpha-subunit gene (SCN4A) to 17q23.1-25.3. Genomics. 1991 Mar;9(3):555-6. https://doi.org/10.1016/0888-7543(91)90425-E[↩]
- Han JY, Kim JB. Familial hyperkalemic periodic paralysis caused by a de novo mutation in the sodium channel gene SCN4A. Korean J Pediatr. 2011 Nov;54(11):470-2. doi: 10.3345/kjp.2011.54.11.470[↩]
- Lee SC, Kim HS, Park YE, Choi YC, Park KH, Kim DS. Clinical Diversity of SCN4A-Mutation-Associated Skeletal Muscle Sodium Channelopathy. J Clin Neurol. 2009 Dec;5(4):186-91. doi: 10.3988/jcn.2009.5.4.186[↩][↩]
- Hyperkalemic periodic paralysis. https://rarediseases.info.nih.gov/diseases/195/hyperkalemic-periodic-paralysis[↩]
- U.S. Department of Agriculture, Agricultural Research Service. USDA National Nutrient Database for Standard Reference, Release 28. https://ndb.nal.usda.gov/ndb/[↩]
- Segawa K, Nishiyama M, Mori I, Kubota T, Takahashi MP. Hyperkalemic periodic paralysis associated with a novel missense variant located in the inner pore of Nav1.4. Brain Dev. 2023 Apr;45(4):205-211. doi: 10.1016/j.braindev.2022.12.003[↩][↩][↩][↩]
- Kokunai Y, Goto K, Kubota T, Fukuoka T, Sakoda S, Ibi T, Doyu M, Mochizuki H, Sahashi K, Takahashi MP. A sodium channel myotonia due to a novel SCN4A mutation accompanied by acquired autoimmune myasthenia gravis. Neurosci Lett. 2012 Jun 21;519(1):67-72. doi: 10.1016/j.neulet.2012.05.023[↩]
- Cummins TR, Sigworth FJ. Impaired slow inactivation in mutant sodium channels. Biophys J. 1996 Jul;71(1):227-36. doi: 10.1016/S0006-3495(96)79219-6[↩]
- Takahashi MP, Cannon SC. Enhanced slow inactivation by V445M: a sodium channel mutation associated with myotonia. Biophys J. 1999 Feb;76(2):861-8. doi: 10.1016/S0006-3495(99)77249-8[↩]
- von Stein RT, Soderlund DM. Compound-specific effects of mutations at Val787 in DII-S6 of Nav 1.4 sodium channels on the action of sodium channel inhibitor insecticides. Neurotoxicology. 2012 Oct;33(5):1381-9. doi: 10.1016/j.neuro.2012.09.003[↩]
- Lehmann-Horn F, Jurkat-Rott K. Voltage-Gated ion channels and hereditary disease. Physiol Rev 1999;79:1317–72. 10.1152/physrev.1999.79.4.1317[↩]
- Spier SJ. Hyperkalemic periodic paralysis: 14 years later. AAEP Proceedings 2006;52:347–50.[↩]
- Lehmann-Horn F, Küther G, Ricker K, Grafe P, Ballanyi K, Rüdel R. Adynamia episodica hereditaria with myotonia: a non-inactivating sodium current and the effect of extracellular pH. Muscle Nerve. 1987 May;10(4):363-74. doi: 10.1002/mus.880100414[↩]
- Lehmann-Horn F, Iaizzo PA, Hatt H, Franke C. Altered gating and conductance of Na+ channels in hyperkalemic periodic paralysis. Pflugers Arch. 1991 Apr;418(3):297-9. doi: 10.1007/BF00370530[↩]
- Sender D, Doyal A. Hyperkalemic periodic paralysis with paramyotonia and the anaesthetic implications. BMJ Case Rep. 2023 Jan 3;16(1):e251699. doi: 10.1136/bcr-2022-251699[↩]
- Lehmann-Horn F, Rudel R, Jurkat-Rott K. Non-dystrophic myotonias and periodic paralyses. In: Engel AG, Franzini-Armstrong C, eds. Myology: Basic and Clinical. 3 ed. New York, NY: McGraw-Hill. 2004:1257-300.[↩][↩]
- Akaba Y, Takahashi S, Sasaki Y, Kajino H. Successful treatment of normokalemic periodic paralysis with hydrochlorothiazide. Brain Dev. 2018 Oct;40(9):833-836. doi: 10.1016/j.braindev.2018.05.011[↩]
- Lehmann-Horn F, Jurkat-Rott K. Genotype-phenotype correlation and therapeutic rationale in hyperkalemic periodic paralysis. Neurotherapeutics. 2007;4:216–224. doi: 10.1016/j.nurt.2007.02.001[↩][↩]
- Dorland’s Illustrated Medical Dictionary. W.B. Saunders Company, Philadelphia 1974[↩]
- Benhammou JN, Phan J, Lee H, Ghassemi K, Parsons W, Grody WW, Pisegna JR. A Sodium Channel Myotonia Presenting with Intermittent Dysphagia as a Manifestation of a Rare SCN4A Variant. J Mol Neurosci. 2017 Mar;61(3):312-314. doi: 10.1007/s12031-016-0878-5[↩]
- Ammar T, Lin W, Higgins A, Hayward LJ, Renaud JM. Understanding the physiology of the asymptomatic diaphragm of the M1592V hyperkalemic periodic paralysis mouse. J Gen Physiol. 2015 Dec;146(6):509-25. doi: 10.1085/jgp.201511476[↩]
- Giacobbe A, Subramony S, Chuquilin M. Pain as a significant symptom in patients with periodic paralysis-A cross-sectional survey. Muscle Nerve. 2021 Jun;63(6):897-901. doi: 10.1002/mus.27241[↩][↩]
- Jeong HN, Yi JS, Lee YH, Lee JH, Shin HY, Choi YC, Kim SM. Lower-extremity magnetic resonance imaging in patients with hyperkalemic periodic paralysis carrying the SCN4A mutation T704M: 30-month follow-up of seven patients. Neuromuscul Disord. 2018 Oct;28(10):837-845. doi: 10.1016/j.nmd.2018.06.008[↩]
- Amarteifio E, Nagel AM, Weber MA, Jurkat-Rott K, Lehmann-Horn F. Hyperkalemic periodic paralysis and permanent weakness: 3-T MR imaging depicts intracellular 23Na overload—initial results. Radiology. 2012;264:154–163. doi: 10.1148/radiol.12110980[↩]
- Lehmann-Horn F, Rudel R, Jurkat-Rott K. Non-dystrophic myotonias and periodic paralyses : Engel AG, Franzini-Armstrong C, Myology: Basic and Clinical. 3 ed New York, NY: McGraw-Hill, 2004:1257–300.[↩]
- Yong SL, Sin TH, Tang EB, Chai MC. Hyperkalaemic periodic paralysis in pregnancy. BMJ Case Rep. 2018 Jun 4;2018:bcr2017223588. doi: 10.1136/bcr-2017-223588[↩][↩]
- Modoni A, D’Amico A, Primiano G, Capozzoli F, Desaphy JF, Lo Monaco M. Long-Term Safety and Usefulness of Mexiletine in a Large Cohort of Patients Affected by Non-dystrophic Myotonias. Front Neurol. 2020 May 20;11:300. doi: 10.3389/fneur.2020.00300[↩]
- Mankodi A, Grunseich C, Skov M, Cook L, Aue G, Purev E, Bakar D, Lehky T, Jurkat-Rott K, Pedersen TH, Childs RW. Divalent cation-responsive myotonia and muscle paralysis in skeletal muscle sodium channelopathy. Neuromuscul Disord. 2015 Nov;25(11):908-12. doi: 10.1016/j.nmd.2015.08.007[↩]
- Klingler W, Lehmann-Horn F, Jurkat-Rott K. Complications of anaesthesia in neuromuscular disorders. Neuromuscul Disord. 2005 Mar;15(3):195-206. doi: 10.1016/j.nmd.2004.10.017[↩][↩]
- Mackenzie MJ, Pickering E, Yentis SM. Anaesthetic management of labour and caesarean delivery of a patient with hyperkalaemic periodic paralysis. Int J Obstet Anesth. 2006 Oct;15(4):329-31. doi: 10.1016/j.ijoa.2006.01.003[↩]
- Jurkat-Rott K, Lehmann-Horn F. Genotype-phenotype correlation and therapeutic rationale in hyperkalemic periodic paralysis. Neurotherapeutics. 2007 Apr;4(2):216-24. doi: 10.1016/j.nurt.2007.02.001[↩]
- Barker MC. Combined spinal/general anesthesia with postoperative femoral nerve block for total knee replacement in a patient with familial hyperkalemic periodic paralysis: a case report. AANA J. 2010 Jun;78(3):191-4.[↩]
- Moreno-Manuel AI, Gutiérrez LK, Vera-Pedrosa ML, Cruz FM, Bermúdez-Jiménez FJ, Martínez-Carrascoso I, Sánchez-Pérez P, Macías Á, Jalife J. Molecular stratification of arrhythmogenic mechanisms in the Andersen Tawil syndrome. Cardiovasc Res. 2023 May 2;119(4):919-932. doi: 10.1093/cvr/cvac118[↩][↩]
- Yim J, Kim KB, Kim M, Lee GD, Kim M. Andersen-Tawil Syndrome With Novel Mutation in KCNJ2: Case Report. Front Pediatr. 2022 Jan 31;9:790075. doi: 10.3389/fped.2021.790075[↩][↩]
- Andersen ED, Krasilnikoff PA, Overvad H. Intermittent muscular weakness, extrasystoles, and multiple developmental anomalies. A new syndrome? Acta Paediatr Scand. 1971 Sep;60(5):559-64. doi: 10.1111/j.1651-2227.1971.tb06990.x[↩]
- Tawil R, Ptacek LJ, Pavlakis SG, DeVivo DC, Penn AS, Ozdemir C, Griggs RC. Andersen’s syndrome: potassium-sensitive periodic paralysis, ventricular ectopy, and dysmorphic features. Ann Neurol. 1994 Mar;35(3):326-30. doi: 10.1002/ana.410350313[↩]
- Stunnenberg BC, Raaphorst J, Deenen JCW, Links TP, Wilde AA, Verbove DJ, Kamsteeg EJ, van den Wijngaard A, Faber CG, van der Wilt GJ, van Engelen BGM, Drost G, Ginjaar HB. Prevalence and mutation spectrum of skeletal muscle channelopathies in the Netherlands. Neuromuscul Disord. 2018 May;28(5):402-407. doi: 10.1016/j.nmd.2018.03.006[↩][↩]
- Andersen-Tawil Syndrome. https://rarediseases.org/rare-diseases/andersen-tawil-syndrome[↩]
- Andersen-Tawil Syndrome. https://periodicparalysis.org/andersen-tawil-syndrome[↩][↩][↩]
- Mazzanti A, Guz D, Trancuccio A, et al. Natural History and Risk Stratification in Andersen-Tawil Syndrome Type 1. J Am Coll Cardiol. 2020 Apr 21;75(15):1772-1784. doi: 10.1016/j.jacc.2020.02.033[↩]
- van der Werf-‘t Lam AS, van Haeringen A, Rinnen T, Robles de Medina RM, Wilde AAM, Hennekam RC, Barge-Schaapveld DQCM. Andersen-Tawil syndrome: Overlapping clinical features with Noonan syndrome? Eur J Med Genet. 2022 Jan;65(1):104382. doi: 10.1016/j.ejmg.2021.104382[↩]
- Plaster NM, Tawil R, Tristani-Firouzi M, Canún S, Bendahhou S, Tsunoda A, Donaldson MR, Iannaccone ST, Brunt E, Barohn R, Clark J, Deymeer F, George AL Jr, Fish FA, Hahn A, Nitu A, Ozdemir C, Serdaroglu P, Subramony SH, Wolfe G, Fu YH, Ptácek LJ. Mutations in Kir2.1 cause the developmental and episodic electrical phenotypes of Andersen’s syndrome. Cell. 2001 May 18;105(4):511-9. doi: 10.1016/s0092-8674(01)00342-7[↩]
- Sakmann B, Trube G. Conductance properties of single inwardly rectifying potassium channels in ventricular cells from guinea-pig heart. J Physiol. 1984 Feb;347:641-57. doi: 10.1113/jphysiol.1984.sp015088[↩]
- Veerapandiyan A, Statland JM, Tawil R. Andersen-Tawil Syndrome. 2004 Nov 22 [Updated 2018 Jun 7]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1264[↩][↩]
- Donaldson MR, Yoon G, Fu YH, Ptacek LJ. Andersen-Tawil syndrome: a model of clinical variability, pleiotropy, and genetic heterogeneity. Ann Med. 2004;36 Suppl 1:92-7. doi: 10.1080/17431380410032490[↩]
- Mazzanti A, Guz D, Trancuccio A, et al. Natural History and Risk Stratification in Andersen-Tawil Syndrome Type 1. J Am Coll Cardiol. 2020 Apr 21;75(15):1772-1784. https://www.jacc.org/doi/10.1016/j.jacc.2020.02.033[↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Delannoy E, Sacher F, Maury P, Mabo P, Mansourati J, Magnin I, Camous JP, Tournant G, Rendu E, Kyndt F, Haïssaguerre M, Bézieau S, Guyomarch B, Le Marec H, Fressart V, Denjoy I, Probst V. Cardiac characteristics and long-term outcome in Andersen-Tawil syndrome patients related to KCNJ2 mutation. Europace. 2013 Dec;15(12):1805-11. doi: 10.1093/europace/eut160[↩][↩]
- Zhang L, Benson DW, Tristani-Firouzi M, Ptacek LJ, Tawil R, Schwartz PJ, George AL, Horie M, Andelfinger G, Snow GL, Fu YH, Ackerman MJ, Vincent GM. Electrocardiographic features in Andersen-Tawil syndrome patients with KCNJ2 mutations: characteristic T-U-wave patterns predict the KCNJ2 genotype. Circulation. 2005 May 31;111(21):2720-6. doi: 10.1161/CIRCULATIONAHA.104.472498[↩][↩]
- Barajas-Martinez H, Hu D, Ontiveros G, et al. Biophysical and molecular characterization of a novel de novo KCNJ2 mutation associated with Andersen-Tawil syndrome and catecholaminergic polymorphic ventricular tachycardia mimicry. Circ Cardiovasc Genet. 2011;4(1):51-57. doi:10.1161/CIRCGENETICS.110.957696 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3041844[↩]
- Adams DS, Uzel SG, Akagi J, Wlodkowic D, Andreeva V, Yelick PC, Devitt-Lee A, Pare JF, Levin M. Bioelectric signalling via potassium channels: a mechanism for craniofacial dysmorphogenesis in KCNJ2-associated Andersen-Tawil Syndrome. J Physiol. 2016 Jun 15;594(12):3245-70. doi: 10.1113/JP271930[↩]
- Yoon G, Oberoi S, Tristani-Firouzi M, Etheridge SP, Quitania L, Kramer JH, Miller BL, Fu YH, Ptácek LJ. Andersen-Tawil syndrome: prospective cohort analysis and expansion of the phenotype. Am J Med Genet A. 2006 Feb 15;140(4):312-21. doi: 10.1002/ajmg.a.31092[↩]
- Wang Q, Zhao Z, Shen H, Bing Q, Li N, Hu J. The clinical and genetic heterogeneity analysis of five families with primary periodic paralysis. Channels (Austin). 2021 Dec;15(1):20-30. doi: 10.1080/19336950.2020.1857980[↩]
- KCNJ2 gene. https://medlineplus.gov/genetics/gene/kcnj2[↩][↩]
- Lopes CM, Zhang H, Rohacs T, Jin T, Yang J, Logothetis DE. Alterations in conserved Kir channel-PIP2 interactions underlie channelopathies. Neuron. 2002 Jun 13;34(6):933-44. doi: 10.1016/s0896-6273(02)00725-0[↩]
- Ai T, Fujiwara Y, Tsuji K, Otani H, Nakano S, Kubo Y, Horie M. Novel KCNJ2 mutation in familial periodic paralysis with ventricular dysrhythmia. Circulation. 2002 Jun 4;105(22):2592-4. doi: 10.1161/01.cir.0000019906.35135.a3[↩]
- Davies NP, Imbrici P, Fialho D, Herd C, Bilsland LG, Weber A, Mueller R, Hilton-Jones D, Ealing J, Boothman BR, Giunti P, Parsons LM, Thomas M, Manzur AY, Jurkat-Rott K, Lehmann-Horn F, Chinnery PF, Rose M, Kullmann DM, Hanna MG. Andersen-Tawil syndrome: new potassium channel mutations and possible phenotypic variation. Neurology. 2005 Oct 11;65(7):1083-9. doi: 10.1212/01.wnl.0000178888.03767.74[↩]
- Sansone V, Tawil R. Management and treatment of Andersen-Tawil syndrome (ATS). Neurotherapeutics. 2007 Apr;4(2):233-7. doi: 10.1016/j.nurt.2007.01.005[↩][↩]
- Tristani-Firouzi M, Jensen JL, Donaldson MR, Sansone V, Meola G, Hahn A, Bendahhou S, Kwiecinski H, Fidzianska A, Plaster N, Fu YH, Ptacek LJ, Tawil R. Functional and clinical characterization of KCNJ2 mutations associated with LQT7 (Andersen syndrome). J Clin Invest. 2002;110:381–8.[↩]
- Donaldson MR, Jensen JL, Tristani-Firouzi M, Tawil R, Bendahhou S, Suarez WA, Cobo AM, Poza JJ, Behr E, Wagstaff J, Szepetowski P, Pereira S, Mozaffar T, Escolar DM, Fu YH, Ptácek LJ. PIP2 binding residues of Kir2.1 are common targets of mutations causing Andersen syndrome. Neurology. 2003;60:1811–6.[↩][↩]
- Haruna Y, Kobori A, Makiyama T, Yoshida H, Akao M, Doi T, Tsuji K, Ono S, Nishio Y, Shimizu W, Inoue T, Murakami T, Tsuboi N, Yamanouchi H, Ushinohama H, Nakamura Y, Yoshinaga M, Horigome H, Aizawa Y, Kita T, Horie M. Genotype-phenotype correlations of KCNJ2 mutations in Japanese patients with Andersen-Tawil syndrome. Hum Mutat. 2007;28:208.[↩][↩]
- Kimura H, Zhou J, Kawamura M, Itoh H, Mizusawa Y, Ding WG, Wu J, Ohno S, Makiyama T, Miyamoto A, Naiki N, Wang Q, Xie Y, Suzuki T, Tateno S, Nakamura Y, Zang WJ, Ito M, Matsuura H, Horie M. Phenotype variability in patients carrying KCNJ2 mutations. Circ Cardiovasc Genet. 2012;5:344–53.[↩]
- Delannoy E, Sacher F, Maury P, Mabo P, Mansourati J, Magnin I, Camous JP, Tournant G, Rendu E, Kyndt F, Haïssaguerre M, Bézieau S, Guyomarch B, Le Marec H, Fressart V, Denjoy I, Probst V. Cardiac characteristics and long-term outcome in Andersen-Tawil syndrome patients related to KCNJ2 mutation. Europace. 2013;15:1805–11.[↩]
- Schoonderwoerd BA, Wiesfeld AC, Wilde AA, van den Heuvel F, Van Tintelen JP, van den Berg MP, Van Veldhuisen DJ, Van Gelder IC. A family with Andersen-Tawil syndrome and dilated cardiomyopathy. Heart Rhythm. 2006;3:1346–50.[↩]
- Pellizzón OA, Kalaizich L, Ptácek LJ, Tristani-Firouzi M, Gonzalez MD. Flecainide suppresses bidirectional ventricular tachycardia and reverses tachycardia-induced cardiomyopathy in Andersen-Tawil syndrome. J Cardiovasc Electrophysiol. 2008;19:95–7.[↩]
- Rezazadeh S, Guo J, Duff HJ, Ferrier RA, Gerull B. Reversible dilated cardiomyopathy caused by a high burden of ventricular arrhythmias in Andersen-Tawil syndrome. Can J Cardiol. 2016;32:1576.e15–e18.[↩]
- Davies NP, Imbrici P, Fialho D, Herd C, Bilsland LG, Weber A, Mueller R, Hilton-Jones D, Ealing J, Boothman BR, Giunti P, Parsons LM, Thomas M, Manzur AY, Jurkat-Rott K, Lehmann-Horn F, Chinnery PF, Rose M, Kullmann DM, Hanna MG. Andersen-Tawil syndrome: new potassium channel mutations and possible phenotypic variation. Neurology. 2005;65:1083–9.[↩][↩][↩]
- Yoon G, Oberoi S, Tristani-Firouzi M, Etheridge SP, Quitania L, Kramer JH, Miller BL, Fu YH, Ptácek LJ. Andersen-Tawil syndrome: prospective cohort analysis and expansion of the phenotype. Am J Med Genet A. 2006a;140:312–21.[↩]
- Andelfinger G, Tapper AR, Welch RC, Vanoye CG, George AL Jr, Benson DW. KCNJ2 mutation results in Andersen syndrome with sex-specific cardiac and skeletal muscle phenotypes. Am J Hum Genet. 2002;71:663–8.[↩]
- Yoon G, Quitania L, Kramer JH, Fu YH, Miller BL, Ptácek LJ. Andersen-Tawil syndrome: definition of a neurocognitive phenotype. Neurology. 2006b;66:1703–10.[↩]
- Kim NR, Jang J, Jean GW, Cho E, Sin JB. Identification of the KCNJ2 mutation in a Korean family with Andersen-Tawil syndrome and developmental delay. Ann Clin Lab Sci. 2016;46:110–3.[↩]
- Veerapandiyan A, Statland JM, Tawil R. Andersen-Tawil Syndrome. 2004 Nov 22 [Updated 2018 Jun 7]. In: Adam MP, Mirzaa GM, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2023. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1264[↩][↩][↩][↩][↩][↩][↩][↩]
- Katz JS, Wolfe GI, Iannaccone S, Bryan WW, Barohn RJ. The exercise test in Andersen syndrome. Arch Neurol. 1999 Mar;56(3):352-6. doi: 10.1001/archneur.56.3.352[↩]
- Fournier E, Arzel M, Sternberg D, Vicart S, Laforet P, Eymard B, Willer JC, Tabti N, Fontaine B. Electromyography guides toward subgroups of mutations in muscle channelopathies. Ann Neurol. 2004 Nov;56(5):650-61. doi: 10.1002/ana.20241[↩]
- Kuntzer T, Flocard F, Vial C, Kohler A, Magistris M, Labarre-Vila A, Gonnaud PM, Ochsner F, Soichot P, Chan V, Monnier G. Exercise test in muscle channelopathies and other muscle disorders. Muscle Nerve. 2000 Jul;23(7):1089-94. doi: 10.1002/1097-4598(200007)23:7<1089::aid-mus12>3.0.co;2-q[↩]
- Tan SV, Matthews E, Barber M, Burge JA, Rajakulendran S, Fialho D, Sud R, Haworth A, Koltzenburg M, Hanna MG. Refined exercise testing can aid DNA-based diagnosis in muscle channelopathies. Ann Neurol. 2011 Feb;69(2):328-40. doi: 10.1002/ana.22238[↩]
- Koppikar S, Barbosa-Barros R, Baranchuk A. A Practical Approach to the Investigation of an rSr’ Pattern in Leads V1-V2. Can J Cardiol. 2015 Dec;31(12):1493-6. doi: 10.1016/j.cjca.2015.04.008[↩]
- Airey KJ, Etheridge SP, Tawil R, Tristani-Firouzi M. Resuscitated sudden cardiac death in Andersen-Tawil syndrome. Heart Rhythm. 2009 Dec;6(12):1814-7. doi: 10.1016/j.hrthm.2009.08.032[↩]
- Chun TU, Epstein MR, Dick M 2nd, Andelfinger G, Ballester L, Vanoye CG, George AL Jr, Benson DW. Polymorphic ventricular tachycardia and KCNJ2 mutations. Heart Rhythm. 2004 Jul;1(2):235-41. doi: 10.1016/j.hrthm.2004.02.017[↩]
- Bökenkamp R, Wilde AA, Schalij MJ, Blom NA. Flecainide for recurrent malignant ventricular arrhythmias in two siblings with Andersen-Tawil syndrome. Heart Rhythm. 2007 Apr;4(4):508-11. doi: 10.1016/j.hrthm.2006.12.031[↩]
- Fox DJ, Klein GJ, Hahn A, Skanes AC, Gula LJ, Yee RK, Subbiah RN, Krahn AD. Reduction of complex ventricular ectopy and improvement in exercise capacity with flecainide therapy in Andersen-Tawil syndrome. Europace. 2008 Aug;10(8):1006-8. doi: 10.1093/europace/eun180[↩]
- Pellizzón OA, Kalaizich L, Ptácek LJ, Tristani-Firouzi M, Gonzalez MD. Flecainide suppresses bidirectional ventricular tachycardia and reverses tachycardia-induced cardiomyopathy in Andersen-Tawil syndrome. J Cardiovasc Electrophysiol. 2008 Jan;19(1):95-7. doi: 10.1111/j.1540-8167.2007.00910.x[↩]
- Tristani-Firouzi M, Etheridge SP. Kir 2.1 channelopathies: the Andersen-Tawil syndrome. Pflugers Arch. 2010 Jul;460(2):289-94. doi: 10.1007/s00424-010-0820-6[↩]
- Miyamoto K, Aiba T, Kimura H, Hayashi H, Ohno S, Yasuoka C, Tanioka Y, Tsuchiya T, Yoshida Y, Hayashi H, Tsuboi I, Nakajima I, Ishibashi K, Okamura H, Noda T, Ishihara M, Anzai T, Yasuda S, Miyamoto Y, Kamakura S, Kusano K, Ogawa H, Horie M, Shimizu W. Efficacy and safety of flecainide for ventricular arrhythmias in patients with Andersen-Tawil syndrome with KCNJ2 mutations. Heart Rhythm. 2015 Mar;12(3):596-603. doi: 10.1016/j.hrthm.2014.12.009[↩][↩]
- Kuroda Y, Yuasa S, Watanabe Y, Ito S, Egashira T, Seki T, Hattori T, Ohno S, Kodaira M, Suzuki T, Hashimoto H, Okata S, Tanaka A, Aizawa Y, Murata M, Aiba T, Makita N, Furukawa T, Shimizu W, Kodama I, Ogawa S, Kokubun N, Horigome H, Horie M, Kamiya K, Fukuda K. Flecainide ameliorates arrhythmogenicity through NCX flux in Andersen-Tawil syndrome-iPS cell-derived cardiomyocytes. Biochem Biophys Rep. 2017 Jan 11;9:245-256. doi: 10.1016/j.bbrep.2017.01.002[↩]
- Subbiah RN, Gula LJ, Skanes AC, Krahn AD. Andersen-Tawil syndrome: management challenges during pregnancy, labor, and delivery. J Cardiovasc Electrophysiol. 2008 Sep;19(9):987-9. doi: 10.1111/j.1540-8167.2008.01216.x[↩]
- Lee MJ, Lin PC, Lin MH, Chiou HC, Wang K, Huang CW. Kinetic Alterations in Resurgent Sodium Currents of Mutant Nav1.4 Channel in Two Patients Affected by Paramyotonia Congenita. Biology (Basel). 2022 Apr 18;11(4):613. doi: 10.3390/biology11040613[↩]
- Featherstone DE, Fujimoto E, Ruben PC. A defect in skeletal muscle sodium channel deactivation exacerbates hyperexcitability in human paramyotonia congenita. J Physiol. 1998 Feb 1;506 ( Pt 3)(Pt 3):627-38. doi: 10.1111/j.1469-7793.1998.627bv.x[↩]
- Huang CW, Lai HJ, Lin PC, Lee MJ. Changes in Resurgent Sodium Current Contribute to the Hyperexcitability of Muscles in Patients with Paramyotonia Congenita. Biomedicines. 2021 Jan 8;9(1):51. doi: 10.3390/biomedicines9010051[↩]
- Siow WS, Chiew WA. Anaesthesia challenges of a parturient with paramyotonia congenita and terminal filum lipoma presenting for labour and caesarean section under epidural anaesthesia – a case report. BMC Anesthesiol. 2021 Feb 18;21(1):57. doi: 10.1186/s12871-021-01262-4[↩]
- Wang Q, Zhao Z, Shen H, Bing Q, Li N, Hu J. The Clinical, Myopathological, and Genetic Analysis of 20 Patients With Non-dystrophic Myotonia. Front Neurol. 2022 Mar 8;13:830707. doi: 10.3389/fneur.2022.830707[↩]
- Brooks EK, Schweitzer D, Robinson HL. A case of paramyotonia congenita in pregnancy. Obstet Med. 2020 Dec;13(4):192-194. doi: 10.1177/1753495X18816171[↩]
- Huang S, Zhang W, Chang X, Guo J. Overlap of periodic paralysis and paramyotonia congenita caused by SCN4A gene mutations two family reports and literature review. Channels (Austin). 2019 Dec;13(1):110-119. doi: 10.1080/19336950.2019.1600967[↩]
- Najid NM, Razak TA, Günaydın DB. Analgesia and Anaesthesia Management of Labour and Caesarean Delivery for a Parturient with Paramyotonia Congenita. Turk J Anaesthesiol Reanim. 2019 Aug;47(4):345-347. doi: 10.5152/TJAR.2019.69094[↩]
- Trivedi JR, Bundy B, Statland J, Salajegheh M, Rayan DR, Venance SL, et al. Non-dystrophic myotonia: prospective study of objective and patient reported outcomes. Brain. 2013;136(Pt 7):2189–200. doi: 10.1093/brain/awt133[↩]
- Matthews E, Fialho D, Tan SV, Venance SL, Cannon SC, Sternberg D, Fontaine B, Amato AA, Barohn RJ, Griggs RC, Hanna MG; CINCH Investigators. The non-dystrophic myotonias: molecular pathogenesis, diagnosis and treatment. Brain. 2010 Jan;133(Pt 1):9-22. doi: 10.1093/brain/awp294[↩]
- Ptacek LJ, Trimmer JS, Agnew WS, Roberts JW, Petajan JH, Leppert M. Paramyotonia congenita and hyperkalemic periodic paralysis map to the same sodium-channel gene locus. Am J Hum Genet. 1991 Oct;49(4):851-4. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1683172/pdf/ajhg00081-0159.pdf[↩][↩]
- Roberts K, Kentris M. Myotonia. [Updated 2023 May 1]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK559272[↩]
- Hahn C, Salajegheh MK. Myotonic disorders: A review article. Iran J Neurol. 2016 Jan 5;15(1):46-53. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4852070[↩][↩]
- Phillips L, Trivedi JR. Skeletal Muscle Channelopathies. Neurotherapeutics. 2018 Oct;15(4):954-965. doi: 10.1007/s13311-018-00678-0[↩]
- Péréon Y, Lande G, Demolombe S, Nguyen The Tich S, Sternberg D, Le Marec H, David A. Paramyotonia congenita with an SCN4A mutation affecting cardiac repolarization. Neurology. 2003 Jan 28;60(2):340-2. doi: 10.1212/01.wnl.0000042093.96309.5a[↩]
- Patti RK, Kaur A, Somal N, Dalsania N, Lu T, Kupfer Y. Thyrotoxic periodic paralysis-still a diagnostic challenge. Proc (Bayl Univ Med Cent). 2022 Jul 13;35(6):863-865. doi: 10.1080/08998280.2022.2095144[↩]
- Heydorn A, Bertelsen B, Nolsöe RLM, Eiken P, Kristensen PL. Thyrotoxic periodic paralysis in a Caucasian man without identifiable genetic predisposition: a case report. Thyroid Res. 2023 May 1;16(1):10. doi: 10.1186/s13044-023-00152-w[↩][↩]
- Garla VV, Gunturu M, Kovvuru KR, Salim SA. Thyrotoxic periodic paralysis: case report and review of the literature. Electron Physician. 2018 Aug 25;10(8):7174-7179. doi: 10.19082/7174[↩][↩][↩][↩]
- Chinyere Agu, MD and others, ODP535 Thyrotoxic Periodic Paralysis (TPP) in A Young African American Male: Post Exercise, Journal of the Endocrine Society, Volume 6, Issue Supplement_1, November-December 2022, Pages A790–A791, https://doi.org/10.1210/jendso/bvac150.1634[↩]
- Roh JG, Park KJ, Lee HS, Hwang JS. Thyrotoxic hypokalemic periodic paralysis due to Graves’ disease in 2 adolescents. Ann Pediatr Endocrinol Metab. 2019 Jun;24(2):133-136. doi: 10.6065/apem.2019.24.2.133[↩][↩][↩][↩][↩]
- Bilha S, Mitu O, Teodoriu L, Haba C, Preda C. Thyrotoxic Periodic Paralysis-A Misleading Challenge in the Emergency Department. Diagnostics (Basel). 2020 May 18;10(5):316. doi: 10.3390/diagnostics10050316[↩][↩][↩][↩]
- Lin C, Lin CS, Lee DJ, Lee CC, Chen SJ, Tsai SH, Kuo FC, Chau T, Lin SH. Artificial Intelligence-Assisted Electrocardiography for Early Diagnosis of Thyrotoxic Periodic Paralysis. J Endocr Soc. 2021 Jun 29;5(9):bvab120. doi: 10.1210/jendso/bvab120[↩]
- Maciel RMB, Lindsey SC, Dias da Silva MR. Novel etiopathophysiological aspects of thyrotoxic periodic paralysis. Nat Rev Endocrinol. 2011;7(11):-. doi: 10.1038/nrendo.2011.58[↩]
- Shinsuke Noso and others, Contribution of Asian Haplotype of KCNJ18 to Susceptibility to and Ethnic Differences in Thyrotoxic Periodic Paralysis, The Journal of Clinical Endocrinology & Metabolism, Volume 104, Issue 12, December 2019, Pages 6338–6344, https://doi.org/10.1210/jc.2019-00672[↩][↩]
- Karolina Anderson, MD , Melissa Wellons, MD, ODP525 Thyrotoxic Periodic Paralysis, Journal of the Endocrine Society, Volume 6, Issue Supplement_1, November-December 2022, Pages A785–A786, https://doi.org/10.1210/jendso/bvac150.1624[↩][↩]
- Falhammar H., Thorén M., Calissendorff J. Thyrotoxic periodic paralysis: Clinical and molecular aspects. Endocrine. 2013;43:274–284. doi: 10.1007/s12020-012-9777-x[↩]
- Pirahanchi Y, Jessu R, Aeddula NR. Physiology, Sodium Potassium Pump. [Updated 2023 Mar 13]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK537088[↩][↩]
- Pivovarov AS, Calahorro F, Walker RJ. Na+/K+-pump and neurotransmitter membrane receptors. Invert Neurosci. 2018 Nov 28;19(1):1. doi: 10.1007/s10158-018-0221-7[↩]
- Kalita J, Goyal G, Bhoi SK, Chandra S, Misra UK. Comparative study of thyrotoxic periodic paralysis from idiopathic hypokalemic periodic paralysis: An experience from India. Ann Indian Acad Neurol. 2012 Jul;15(3):186-90. doi: 10.4103/0972-2327.99708[↩]
- Shizume K, Shishiba Y, Kuma K, Noguchi S, Tajiri J, Ito K, Noh JY. Comparison of the incidence of association of periodic paralysis and hyperthyroidism in Japan in 1957 and 1991. Endocrinol Jpn. 1992 Jun;39(3):315-8. doi: 10.1507/endocrj1954.39.315[↩]
- Cheung CL, Lau KS, Ho AY, Lee KK, Tiu SC, Lau EY, Leung J, Tsang MW, Chan KW, Yeung CY, Woo YC, Cheung EY, Hung VH, Pang HK, Hung CS, Sham PC, Kung AW. Genome-wide association study identifies a susceptibility locus for thyrotoxic periodic paralysis at 17q24.3. Nat Genet. 2012 Sep;44(9):1026-9. doi: 10.1038/ng.2367[↩]
- Kelley DE, Gharib H, Kennedy FP, Duda RJ Jr, McManis PG. Thyrotoxic periodic paralysis. Report of 10 cases and review of electromyographic findings. Arch Intern Med. 1989 Nov;149(11):2597-600. doi: 10.1001/archinte.149.11.2597[↩]
- Thethi T.K., Parks R., Katalenich B., Chhabra P., Mccaw J., Syu S., Nguyen T., Larrazolo J., Munshi K., Waddadar J., et al. Case Report Case of Thyrotoxic Periodic Paralysis in a Caucasian Male and Review of Literature. Case Rep. Med. 2014;2014:314262. doi: 10.1155/2014/314262[↩]
- Thyrotoxic HypoKPP FAQs for Patients. https://hkpp.org/articles/thyrotoxic-hypokpp-faqs-for-patients[↩]
- Jung SY, Song KC, Shin JI, Chae HW, Kim HS, Kwon AR. A case of thyrotoxic periodic paralysis as initial manifestation of Graves’ disease in a 16-year-old Korean adolescent. Ann Pediatr Endocrinol Metab. 2014 Sep;19(3):169-73. doi: 10.6065/apem.2014.19.3.169[↩]
- Ryan DP, da Silva MR, Soong TW, Fontaine B, Donaldson MR, Kung AW, Jongjaroenprasert W, Liang MC, Khoo DH, Cheah JS, Ho SC, Bernstein HS, Maciel RM, Brown RH Jr, Ptácek LJ. Mutations in potassium channel Kir2.6 cause susceptibility to thyrotoxic hypokalemic periodic paralysis. Cell. 2010 Jan 8;140(1):88-98. doi: 10.1016/j.cell.2009.12.024[↩][↩]
- Falhammar H, Thorén M, Calissendorff J. Thyrotoxic periodic paralysis: clinical and molecular aspects. Endocrine. 2013 Apr;43(2):274-84. doi: 10.1007/s12020-012-9777-x[↩][↩][↩][↩][↩][↩]
- Li X, Yao S, Xiang Y, Zhang X, Wu X, Luo L, Huang H, Zhu M, Wan H, Hong D. The clinical and genetic features in a cohort of mainland Chinese patients with thyrotoxic periodic paralysis. BMC Neurol. 2015 Mar 21;15:38. doi: 10.1186/s12883-015-0290-8[↩][↩]
- Park S, Kim TY, Sim S, Oh HS, Song E, Kim M, Kwon H, Choi YM, Jeon MJ, Kim WG, Shong YK, Kim WB. Association of KCNJ2 Genetic Variants with Susceptibility to Thyrotoxic Periodic Paralysis in Patients with Graves’ Disease. Exp Clin Endocrinol Diabetes. 2017 Feb;125(2):75-78. doi: 10.1055/s-0042-119527[↩][↩]
- Chang CC, Cheng CJ, Sung CC, Chiueh TS, Lee CH, Chau T, Lin SH. A 10-year analysis of thyrotoxic periodic paralysis in 135 patients: focus on symptomatology and precipitants. Eur J Endocrinol. 2013 Oct 1;169(5):529-36. doi: 10.1530/EJE-13-0381[↩][↩][↩]
- Oh SB, Ahn J, Oh MY, Choi BG, Kang JH, Jeon YK, Kim SS, Kim BH, Kim YK, Kim IJ. Thyrotoxic periodic paralysis associated with transient thyrotoxicosis due to painless thyroiditis. J Korean Med Sci. 2012 Jul;27(7):822-6. doi: 10.3346/jkms.2012.27.7.822[↩]
- Lin SH, Chu P, Cheng CJ, Chu SJ, Hung YJ, Lin YF. Early diagnosis of thyrotoxic periodic paralysis: spot urine calcium to phosphate ratio. Crit Care Med. 2006 Dec;34(12):2984-9. doi: 10.1097/01.CCM.0000242249.10856.49[↩][↩]
- Ozaki H, Mori K, Nakagawa Y, Hoshikawa S, Ito S, Yoshida K. Autonomously functioning thyroid nodule associated with thyrotoxic periodic paralysis. Endocr J. 2008;55(1):113–9. doi: 10.1507/endocrj.K07E-017[↩]
- Alings AM, Fliers E, de Herder WW, Hofland LJ, Sluiter HE, Links TP, et al. A thyrotropin-secreting pituitary adenoma as a cause of thyrotoxic periodic paralysis. J Endocrinol Invest. 1998;21(10):703–6. doi: 10.1007/BF03350802[↩]
- Lee JI, Sohn TS, Son HS, Oh SJ, Kwon HS, Chang SA, et al. Thyrotoxic periodic paralysis presenting as polymorphic ventricular tachycardia induced by painless thyroiditis. Thyroid. 2009;19(12):1433–4. doi: 10.1089/thy.2009.0253[↩]
- Hannon MJ, Behan LA, Agha A. Thyrotoxic periodic paralysis due to excessive L-thyroxine replacement in a Caucasian man. Ann Clin Biochem. 2009;46(Pt 5):423–5. doi: 10.1258/acb.2009.009012[↩]
- Laroia ST, Zaw KM, Ganti AK, Newman W, Akinwande AO. Amiodarone-induced thyrotoxicosis presenting as hypokalemic periodic paralysis. South Med J. 2002;95(11):1326–8. doi: 10.1097/00007611-200295110-00018[↩]
- Kung AW. Clinical review: Thyrotoxic periodic paralysis: a diagnostic challenge. J Clin Endocrinol Metab. 2006 Jul;91(7):2490-5. doi: 10.1210/jc.2006-0356[↩][↩][↩]
- Abbas MT, Khan FY, Errayes M, Baidaa AD, Haleem AH. Thyrotoxic periodic paralysis admitted to the medical department in Qatar. Neth J Med. 2008 Oct;66(9):384-8.[↩][↩]
- Maciel RM, Lindsey SC, Dias da Silva MR. Novel etiopathophysiological aspects of thyrotoxic periodic paralysis. Nat Rev Endocrinol. 2011 May 10;7(11):657-67. doi: 10.1038/nrendo.2011.58[↩][↩]
- Vijayakumar A., Ashwath G., Thimmappa D. Thyrotoxic periodic paralysis: Clinical challenges. J. Thyroid Res. 2014;2014:649502. doi: 10.1155/2014/649502[↩]
- Shiang J.C., Cheng C.J., Tsai M.K., Hung Y.J., Hsu Y.J., Yang S.S., Chu S.J., Lin S.H. Therapeutic analysis in Chinese patients with thyrotoxic periodic paralysis over 6 years. Eur. J. Endocrinol. 2009;161:911–916. doi: 10.1530/EJE-09-0553[↩]
- Manoukian MA, Foote JA, Crapo LM. Clinical and metabolic features of thyrotoxic periodic paralysis in 24 episodes. Arch Intern Med. 1999 Mar 22;159(6):601-6. doi: 10.1001/archinte.159.6.601[↩][↩][↩][↩]
- Pothiwala P, Levine SN. Analytic review: thyrotoxic periodic paralysis: a review. J Intensive Care Med. 2010 Mar-Apr;25(2):71-7. doi: 10.1177/0885066609358849[↩][↩]
- Patel AJ, Tejera S, Klek SP, Rothberger GD. THYROTOXIC PERIODIC PARALYSIS IN A COMPETITIVE BODYBUILDER WITH THYROTOXICOSIS FACTITIA. AACE Clin Case Rep. 2020 Sep 21;6(5):e252-e256. doi: 10.4158/ACCR-2020-0154[↩]
- Lin SH. Thyrotoxic periodic paralysis. Mayo Clin Proc. 2005 Jan;80(1):99-105. doi: 10.1016/S0025-6196(11)62965-0[↩][↩]
- Chou HK, Tsao YT, Lin SH. An unusual cause of thyrotoxic periodic paralysis: triiodothyronine-containing weight reducing agents. Am J Med Sci. 2009 Jan;337(1):71-3. doi: 10.1097/01.MAJ.0000310783.66897.b6[↩]
- Panikkath R, Nugent K. I lost weight, but I became weak and cannot walk—a case of nutraceutical (T3)-induced thyrotoxic periodic paralysis. Am J Ther. 2014;21(6):e211–4. doi: 10.1097/MJT.0b013e318288a460[↩]
- Lin SH, Lin YF, Chen DT, Chu P, Hsu CW, Halperin ML. Laboratory tests to determine the cause of hypokalemia and paralysis. Arch Intern Med. 2004 Jul 26;164(14):1561-6. doi: 10.1001/archinte.164.14.1561[↩]
- Liu Z, Braverman LE, Malabanan A. Thyrotoxic periodic paralysis in a Hispanic man after the administration of prednisone. Endocr Pract. 2006 Jul-Aug;12(4):427-31. doi: 10.4158/EP.12.4.427[↩]
- McFadzean AJ, Yeung R. Periodic paralysis complicating thyrotoxicosis in Chinese. Br Med J. 1967 Feb 25;1(5538):451-5. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1840834/pdf/brmedj02124-0025.pdf[↩][↩]
- Hsieh MJ, Lyu RK, Chang WN, Chang KH, Chen CM, Chang HS, Wu YR, Chen ST, Ro LS. Hypokalemic thyrotoxic periodic paralysis: clinical characteristics and predictors of recurrent paralytic attacks. Eur J Neurol. 2008 Jun;15(6):559-64. doi: 10.1111/j.1468-1331.2008.02132.x[↩]
- Forrest L, Platts J. Ecstasy-induced thyrotoxic periodic paralysis. BMJ Case Rep. 2009;2009:bcr09.2009.2280. doi: 10.1136/bcr.09.2009.2280[↩]
- Siddamreddy S, Dandu VH. Thyrotoxic Periodic Paralysis. [Updated 2022 Jul 25]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK560670[↩][↩]
- Liberman UA, Asano Y, Lo CS, Edelman IS. Relationship between Na+-dependent respiration and Na+ + K+-adenosine triphosphatase activity in the action of thyroid hormone on rat jejunal mucosa. Biophys J. 1979;27(1):127–44. doi: 10.1016/S0006-3495(79)85207-8[↩]
- Lo CS, Edelman IS. Effect of triiodothyronine on the synthesis and degradation of renal cortical (Na+ + k+)-adenosine triphosphatase. J Biol Chem. 1976 Dec 25;251(24):7834-40.[↩]
- Barahona MJ, Vinagre I, Sojo L, Cubero JM, Pérez A. Thyrotoxic periodic paralysis: a case report and literature review. Clin Med Res. 2009 Sep;7(3):96-8. doi: 10.3121/cmr.2009.816[↩]
- Desai-Yajnik V, Zeng J, Omori K, Sherman J, Morimoto T. The effect of thyroid hormone treatment on the gene expression and enzyme activity of rat liver sodium-potassium dependent adenosine triphosphatase. Endocrinology. 1995;136(2):629–39. doi: 10.1210/endo.136.2.7835297[↩]
- Blanco G, Mercer RW. Isozymes of the Na-K-ATPase: heterogeneity in structure, diversity in function. Am J Physiol. 1998 Nov;275(5):F633-50. doi: 10.1152/ajprenal.1998.275.5.F633[↩]
- Mulder JE. Thyroid disease in women. Med Clin North Am. 1998;82(1):103–25. doi: 10.1016/S0025-7125(05)70596-4[↩]
- Guerra M, Rodriguez del Castillo A, Battaner E, Mas M. Androgens stimulate preoptic area Na+,K+-ATPase activity in male rats. Neurosci Lett. 1987 Jul 9;78(1):97-100. doi: 10.1016/0304-3940(87)90568-4[↩]
- Chan A, Shinde R, Chow CC, Cockram CS, Swaminathan R. Hyperinsulinaemia and Na+, K+ -ATPase activity in thyrotoxic periodic paralysis. Clinical Endocrinolog. 1994;41:213–6. doi: 10.1111/j.1365-2265.1994.tb02532.x[↩]
- Azzarolo AM, Mircheff AK, Kaswan RL, Stanczyk FZ, Gentschein E, Becker L, et al. Androgen support of lacrimal gland function. Endocrine. 1997;6(1):39–45. doi: 10.1007/BF02738800[↩]
- Aggarwal R, Chugh P. Thyrotoxic periodic paralysis: a short clinical review. Int J Res Med Sci. 2015;3:539–42. doi: 10.5455/2320-6012.ijrms20150302[↩]
- Lin SH, Huang CL. Mechanism of Thyrotoxic Periodic Paralysis. J Am Soc Nephrol. 2012;23:985–8. doi: 10.1681/ASN.2012010046[↩]
- Chakrabarti S. Normokalemic thyrotoxic periodic paralysis with preserved reflexes- a unique case report. J Clin Diagn Res. 2015 Feb;9(2):OD05-6. doi: 10.7860/JCDR/2015/11034.5538[↩][↩]
- Mohamed Ghoweba, MD and others, ODP526 Thyrotoxic Periodic Paralysis in Graves’ Disease: A rare presentation in an African American male, Journal of the Endocrine Society, Volume 6, Issue Supplement_1, November-December 2022, Page A786, https://doi.org/10.1210/jendso/bvac150.1625[↩]
- Thyrotoxic periodic paralysis. https://medlineplus.gov/ency/article/000319.htm[↩][↩][↩]
- Shizume K, Shishiba Y, Kuma K, Noguchi S, Tajiri J, Ito K, Noh JY. Comparison of the incidence of association of periodic paralysis and hyperthyroidism in Japan in 1957 and 1991. Endocrinol Jpn. 1992;39(3):315–8. doi: 10.1507/endocrj1954.39.315[↩]
- Satam N, More V, Shanbag P, Kalgutkar A. Fatal thyrotoxic periodic paralysis with normokalemia. Indian J Pediatr. 2007;74(11):1041–3. doi: 10.1007/s12098-007-0194-8[↩][↩]
- Hsieh MJ, Lyu RK, Chang WN, Chang KH, Chen CM, Chang HS, et al. Hypokalemic thyrotoxic periodic paralysis: clinical characteristics and predictors of recurrent paralytic attacks. Eur J Neurol. 2008;15(6):559–64. doi: 10.1111/j.1468-1331.2008.02132.x[↩]
- Okinaka S, Shizume K, Iino S, Watanabe A, Irie M, Noguchi A, et al. The association of periodic paralysis and hyperthyroidism in Japan. J Clin Endocrinol Metab. 1957;17(12):1454–9. doi: 10.1210/jcem-17-12-1454[↩]
- Wong GW, Leung TF, Lo AF, Ahuja AT, Cheng PS. Thyrotoxic periodic paralysis in a 14-year-old boy. Eur J Pediatr. 2000;159(12):934. doi: 10.1007/PL00008375[↩]
- Kung AW. Clinical review: Thyrotoxic periodic paralysis: a diagnostic challenge. J Clin Endocrinol Metab. 2006;91(7):2490–5. doi: 10.1210/jc.2006-0356[↩][↩]
- Lopez S, Henderson SO. Electrocardiogram changes in Thyrotoxic Periodic Paralysis. West J Emerg Med. 2012 Dec;13(6):512-3. doi: 10.5811/westjem.2011.11.12127[↩]
- Puvanendran K, Cheah JS, Wong PK. Electromyographic (EMG) study in thyrotoxic periodic paralysis. Aust N Z J Med. 1977 Oct;7(5):507-10. doi: 10.1111/j.1445-5994.1977.tb03372.x[↩]
- Ko GT, Chow CC, Yeung VT, Chan HH, Li JK, Cockram CS. Thyrotoxic periodic paralysis in a Chinese population. QJM. 1996 Jun;89(6):463-8. doi: 10.1093/qjmed/89.6.463[↩]
- Garla VV, Gunturu M, Kovvuru KR, Salim SA. Thyrotoxic periodic paralysis: case report and review of the literature. Electron Physician. 2018 Aug 25;10(8):7174-7179. doi: 10.19082/717[↩]
- Lu KC, Hsu YJ, Chiu JS, Hsu YD, Lin SH. Effects of potassium supplementation on the recovery of thyrotoxic periodic paralysis. Am J Emerg Med. 2004;22(7):544–7. doi: 10.1016/j.ajem.2004.09.016[↩]
- Tessier JJ, Neu SK, Horning KK. Thyrotoxic periodic paralysis (TPP) in a 28-year-old sudanese man started on prednisone. J Am Board Fam Med. 2010 Jul-Aug;23(4):551-4. doi: 10.3122/jabfm.2010.04.090220[↩]
- Lin SH, Lin YF. Propranolol rapidly reverses paralysis, hypokalemia, and hypophosphatemia in thyrotoxic periodic paralysis. Am J Kidney Dis. 2001 Mar;37(3):620-3.[↩]
- Yu TS, Tseng CF, Chuang YY, Yeung LK, Lu KC. Potassium chloride supplementation alone may not improve hypokalemia in thyrotoxic hypokalemic periodic paralysis. J Emerg Med. 2007 Apr;32(3):263-5. doi: 10.1016/j.jemermed.2006.06.009[↩]
- Birkhahn RH, Gaeta TJ, Melniker L. Thyrotoxic periodic paralysis and intravenous propranolol in the emergency setting. J Emerg Med. 2000;18(2):199–202. doi: 10.1016/S0736-4679(99)00194-8[↩]
- Shayne P, Hart A. Thyrotoxic periodic paralysis terminated with intravenous propranolol. Ann Emerg Med. 1994;24(4):736–40. doi: 10.1016/S0196-0644(94)70286-1[↩]
- Yeung RT, Tse TF. Thyrotoxic periodic paralysis. Effect of propranolol. Am J Med. 1974;57(4):584–90. doi: 10.1016/0002-9343(74)90010-2[↩]
- Dalan R, Leow MK. Cardiovascular collapse associated with beta blockade in thyroid storm. Exp Clin Endocrinol Diabetes. 2007;115(6):392–6. doi: 10.1055/s-2007-971065[↩]
- Ross DS, Burch HB, Cooper DS, Greenlee MC, Laurberg P, Maia AL, Rivkees SA, Samuels M, Sosa JA, Stan MN, Walter MA. 2016 American Thyroid Association Guidelines for Diagnosis and Management of Hyperthyroidism and Other Causes of Thyrotoxicosis. Thyroid. 2016 Oct;26(10):1343-1421. doi: 10.1089/thy.2016.0229. Erratum in: Thyroid. 2017 Nov;27(11):1462.[↩]
- Douglas RS, Kahaly GJ, Patel A, et al: Teprotumumab for the treatment of active thyroid eye disease. N Engl J Med 382(4):341–352. 2020. https://www.nejm.org/doi/pdf/10.1056/NEJMoa1910434[↩]





{kind=link}