Contents
What is Pfeiffer syndrome
Pfeiffer syndrome is a rare genetic disorder that affects the development of the bones in the skull, hands and feet 1. Pfeiffer syndrome signs and symptoms can include the premature fusion of certain skull bones (craniosynostosis), which prevents the skull from growing normally and affects the shape of the head and face; distinctive facial features including bulging and wide-set eyes, a high forehead, an underdeveloped upper jaw, and a beaked nose; hearing loss; and dental problems. Advanced craniosynostosis or craniosynostosis involving multiple sutures, may cause a build-up of pressure within the skull or a constriction of the growth of the brain. This may result in developmental delays, mental retardation, seizures or blindness. The sutures most commonly involved in Pfeiffer syndrome include the coronal, lambdoid and sagittal sutures. Pfeiffer syndrome also affects bones in the hands and feet. Other features may include broad and deviated thumbs and great toes; short fingers and toes (brachydactyly); and webbing or fusion between the digits (syndactyly). Pfeiffer syndrome is caused by mutations in the fibroblast growth factor receptor (FGFR 1 or FGFR2) genes and is inherited in an autosomal dominant manner. Pfeiffer syndrome affects about 1 in 100,000 individuals.
The major characteristics of Pfeiffer syndrome include craniosynostosis, along with short, broad thumbs and toes. Craniosynostosis is a process of premature fusion of the fibrous joints (soft spots) of the bones of the skull. In an unaffected child, the skull evenly expands as the brain grows. In a child with craniosynostosis, one or two of the sutures may prematurely fuse, causing abnormal and asymmetric growth of the skull and face. Abnormal growth of these bones leads to bulging and wide-set eyes, a high forehead, an underdeveloped upper jaw, and a beaked nose. More than half of all children with Pfeiffer syndrome have hearing loss; dental problems are also common.
Patients with Pfeiffer Syndrome have a disproportionately wide head with a high forehead and retruded or sunken midface (the area of the face from the middle of the eye socket to the upper jaw). The nose is frequently small with a low nasal bridge. The eyes can be widely spaced (hypertelorism) and prominent (proptotic) because of the shallow eye sockets (orbits).
Approximately 50% of children with Pfeiffer syndrome have some form of hearing loss secondary to an abnormally small ear canal and middle ear. Dental problems are also common. Visual problems can occur because of the position of the eyes or increased intracranial pressure from the premature fusion of the cranial sutures.
In people with Pfeiffer syndrome, the thumbs and first (big) toes are broad and short and they bend away from the rest of the digits. Unusually short fingers and toes (brachydactyly) are also common, and there may be some webbing or fusion between between the second and third fingers and toes (syndactyly).
Pfeiffer syndrome is divided into 3 subtypes (type 1, type 2 and type 3) based on the presence and severity of specific features.
Type 1, also known as classic Pfeiffer syndrome, is considered mild compared to types 2 and 3 and has symptoms as described above and below. Most individuals with type 1 Pfeiffer syndrome have normal intelligence and a normal life span.
Types 2 and 3 are more severe forms of Pfeiffer syndrome that often involve problems with the nervous system. The premature fusion of skull bones can limit brain growth, leading to delayed development and other neurological problems. In addition, individuals with type 2 or 3 can have fusion of the bones (ankylosis) in the elbow or other joints, limiting mobility, and abnormalities of the face and airways, which can cause life-threatening breathing problems.
Type 2 is distinguished from type 3 Pfeiffer syndrome by the presence of a cloverleaf-shaped head, which is caused by more extensive fusion of bones in the skull.
Management typically includes various surgical interventions 2.
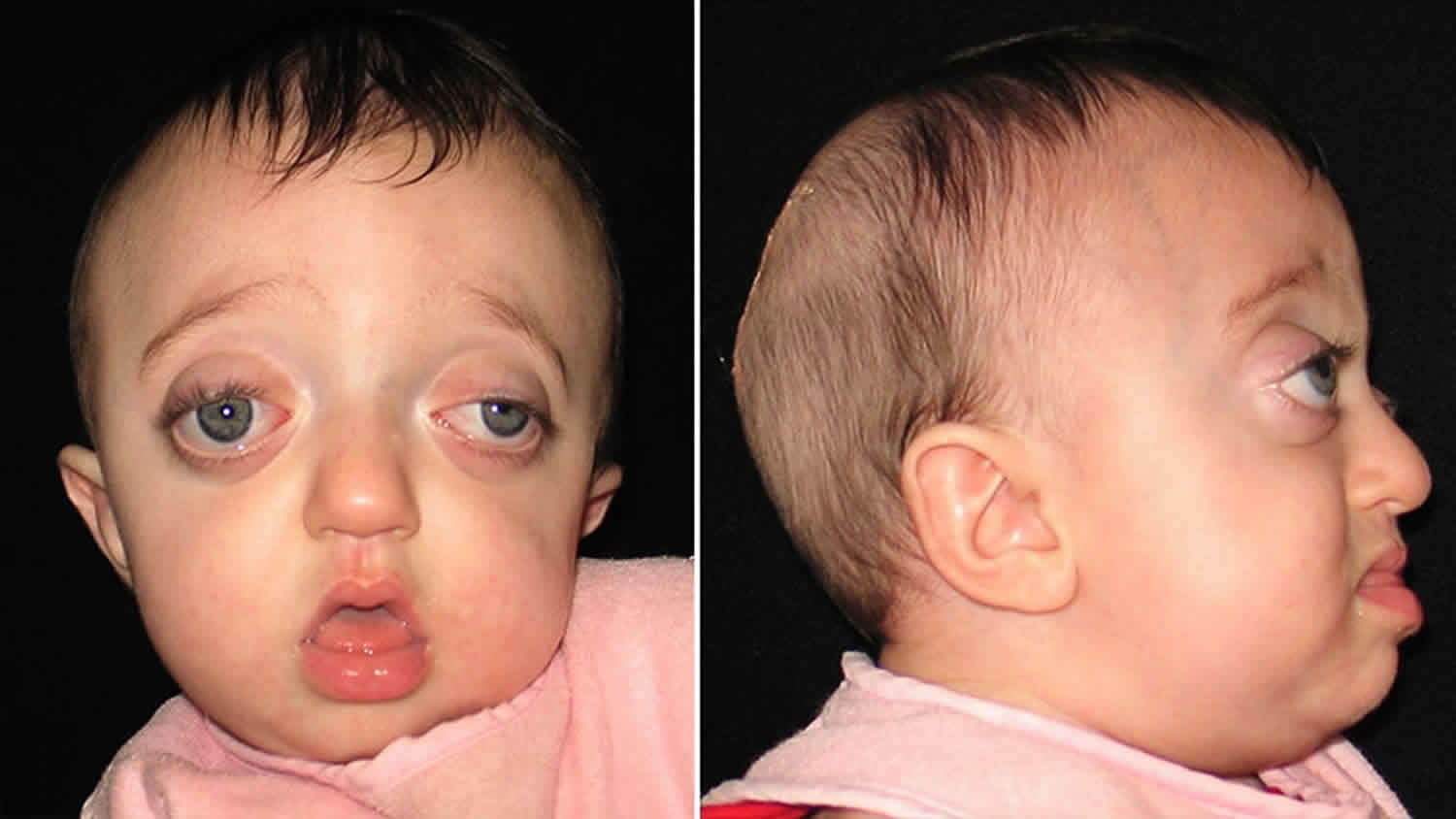
Figure 1. Pfeiffer syndrome
Footnote: 6-month-old with Pfeiffer syndrome. Note characteristic features of wide but short appearance of skull and tall appearance of forehead. The flat and low nasal bridge is also a characteristic of Pfeiffer syndrome.
[Source 3 ]Normal newborn skull anatomy and physiology
It is important to have an understanding of skull anatomy and growth in order to understand craniosynostosis. An infant’s skull has 7 bone plates that relate to each other through specialized joints called “cranial sutures”. Sutures are made of tough, elastic fibrous tissue and separate the bones from one another. Sutures meet up (intersect) at two spots on the skull called fontanelles, which are better known as an infant’s “soft spots”. Although there are several major and minor sutures, the sutures that potentially have the most clinical significance are the singular metopic and sagittal sutures, as well as the paired (right and left) coronal and lambdoid sutures (see Figure 2). The seven bones of an infant’s skull normally do not fuse together until around age two or later. The sutures normally remain flexible until this point. In infants with primary craniosynostosis, the sutures abnormally stiffen or harden causing one or more of the bones of the skull to prematurely fuse together. This in turn, may lead to asymmetric skull growth.
Cranial sutures are very unique and specialized joints (syndesmosis joints). Their primary purpose is to grow bone in response to the rapidly developing brain within the protective skull compartment (cranial cavity or calvarium). The skull not only needs to be firm to protect the brain from accidental blows, but should also be expansile to accommodate its rapid growth. The brain doubles in volume in the first year of life and almost triples in volume by the age of three. The sutures of the skull allow for this important but almost contradictory balance of protection and growth.
Each cranial suture is designed to generate growth in the skull in a very specific area and configuration, ultimately reflecting the size and shape of the underlying brain structure. The overall bone development from cranial sutures occurs in a direction perpendicular to the long axis of the suture. Understanding these two facts, it makes sense that a fusion of each suture independently would cause a unique head shape.
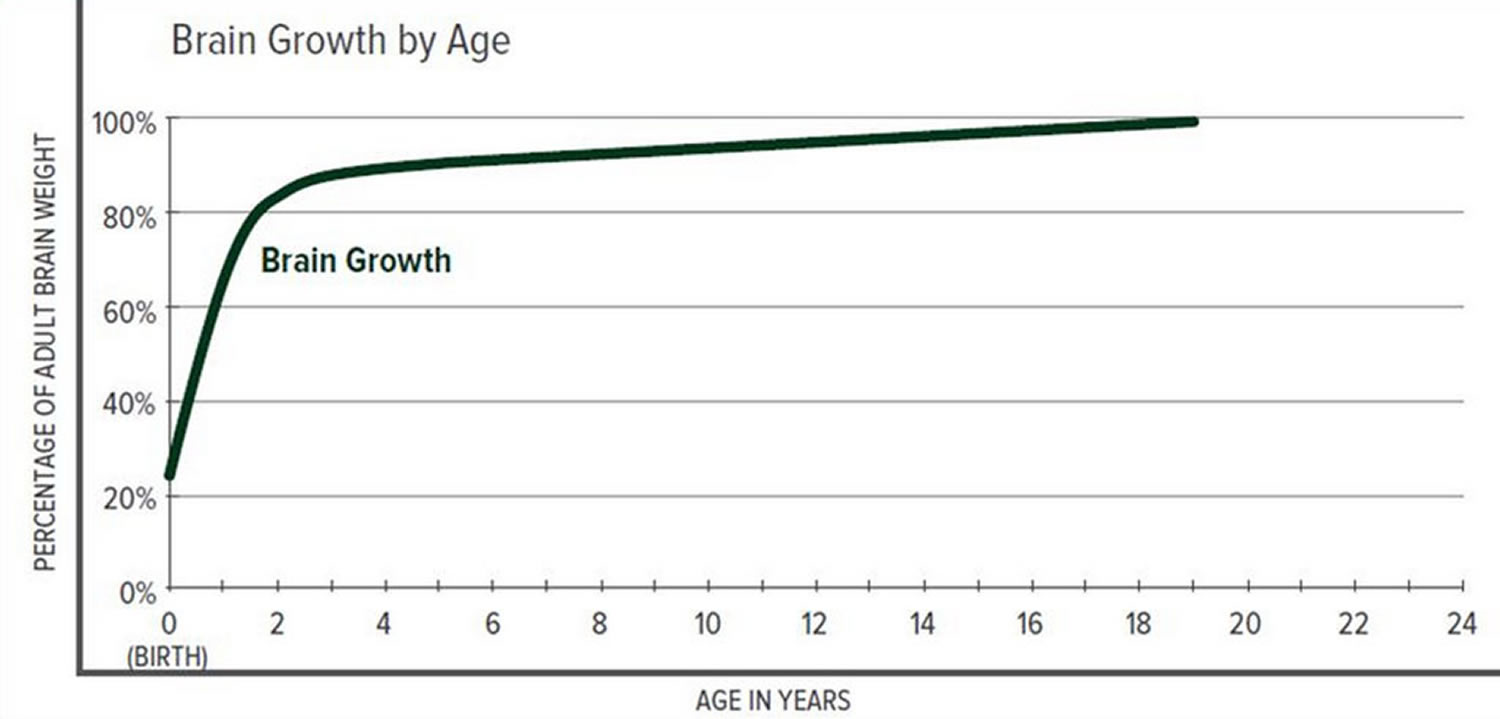
The greatest increase in brain volume (brain growth) occurs from 0 to 14 months of age. The size of a child’s brain typically reaches 80% of adult size by the age of 2. The growth in head circumference after that age is more related to growth in the thickness of the skull and scalp but not actual brain growth. “Hat size” increases but not necessarily “brain size”.
The various cranial sutures close at different ages. The metopic suture closes earliest, around 6 months to 2 years. The rest of the sutures stay open into the 20’s and 30’s. The brain and fluid cavities of the brain do continue to grow in volume as you go into early adulthood, albeit not nearly as rapidly as the first couple years of life. Since the skull is much firmer (calcified or “rock-like”) and thicker, the skull needs the sutures to grow bone for any increase in volume.
Interestingly, there is a lot of variability here. Doctors have operated on adults in their 30’s for reasons unrelated to their skull sutures and have coincidentally found open metopic sutures. They have also seen young adults with closed coronal, lambdoid, and sagittal sutures, but with normal head shapes and often, no indication or symptoms of high pressure.
Scientists have learned that a cranial suture’s purpose is to grow bone to accommodate a growing brain, and that most brain growth occurs in the first two years of life. The brain reaches 85% of adult size by age 3 years (see Figure 3. Brain size vs. age diagram).
So it makes sense that the sutures are vitally important in the first two years of life. The earlier the fusion, the more severe the restriction in growth and, consequently, volume provided to the brain.
Figure 2. Normal skull of a newborn
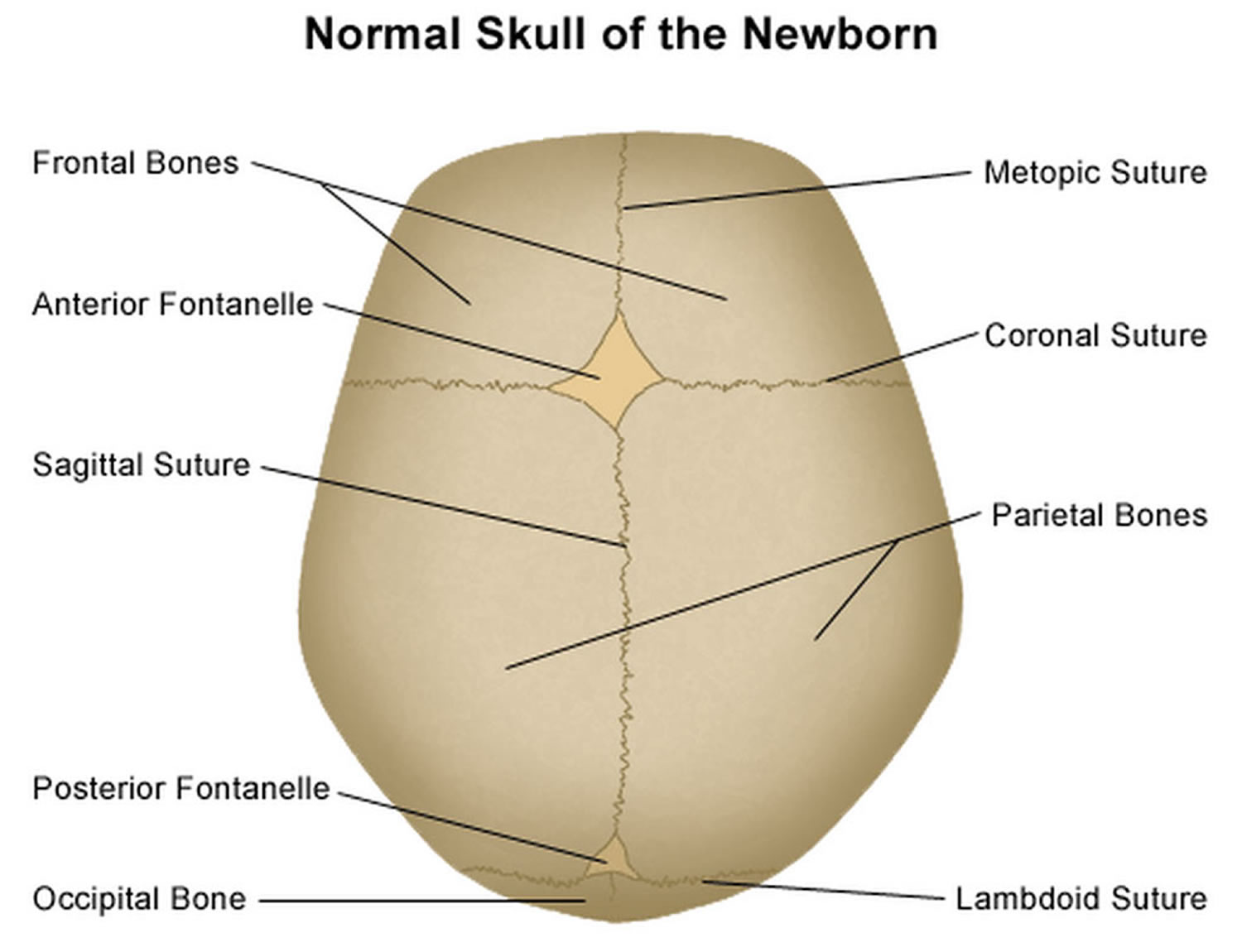
Figure 3. Brain size versus age diagram
Do individuals with Pfeiffer syndrome often have problems with circulation?
Expert were unable to locate articles that discuss the occurrence of circulation problems in individuals with Pfeiffer syndrome. It is known that Pfeiffer syndrome is caused by mutations in the FGFR1 and FGFR2 genes, which are involved in the formation of blood vessels as well as in cell division, regulation of cell growth and maturation, wound healing, and embryonic development. However, to our knowledge, there have not been published reports of circulation issues associated with Pfeiffer syndrome 4, 5.
Pfeiffer syndrome type 1
Infants with Pfeiffer syndrome type 1 have craniosynostosis that causes the head to appear short and tall (turribrachycephaly). Additional features may include a high, full forehead; underdeveloped midfacial regions (midface hypoplasia); widely spaced eyes (ocular hypertelorism); an underdeveloped upper jaw (hypoplastic maxilla), with a prominent lower jaw; and dental abnormalities. Intelligence is usually normal. Hydrocephalus (build up of cerebrospinal fluid (CSF) around the brain) and hearing loss can occur.
Pfeiffer syndrome type 2
Pfeiffer syndrome type 2 is characterized by a more severe form of craniosynostosis (Cloverleaf skull), with more severe hand and foot anomalies and additional malformations of the limbs. In infants with Pfeiffer syndrome type 2, premature closure of the fibrous joints (cranial sutures) between several bones in the skull causes the skull to have a “tri-lobed” appearance (cloverleaf skull deformity, or Kleeblattschadel type craniosynostosis). In addition, this form of craniosynostosis is often associated with hydrocephalus, a condition in which the normal flow of cerebrospinal fluid (CSF) is altered, leading to abnormal widening (dilatation) of the spaces within the brain (ventricles) causing accumulation of cerebrospinal fluid (CSF) in the skull and increased pressure on the brain. Characteristic craniofacial features associated with Pfeiffer syndrome type 2 may include an abnormally high, broad forehead; severe protrusion of the eyes (ocular proptosis); an unusually flat middle portion of the face (midface hypoplasia); a “beak-shaped” nose; and downwardly displaced ears. Affected infants may also exhibit abnormal fixation and lack of mobility (ankylosis) of the elbow joints and/or, in some cases, various malformations of certain internal organs in the abdomen (visceral anomalies). In addition, infants with Pfeiffer syndrome type 2 often experience impaired mental development (intellectual disability) and neurological problems due to severe involvement of the brain, and/or hypoxia due to problems with breathing. Without appropriate treatment, the physical abnormalities associated with the disorder may lead to life-threatening complications during infancy.
Pfeiffer syndrome type 3
Individuals with Pfeiffer syndrome type 3 have symptoms and findings similar to those present in Pfeiffer syndrome type 2, with the exception of the cloverleaf skull deformity. Additional characteristics associated with Pfeiffer syndrome type 3 include a shortened base of the skull (anterior cranial base); the abnormal presence of certain teeth at birth (natal teeth); severe protrusion of the eyes (ocular proptosis) due to abnormal shallowness of the bony cavities that accommodate the eyeballs (orbit); and/or various malformations of certain internal organs in the abdominal area (visceral anomalies). As in type 2, individuals with Pfeiffer syndrome type 3 often experience impaired mental development and severe neurological problems and may develop potentially life-threatening complications early in life without appropriate treatment.
Pfeiffer syndrome life expectancy
Most individuals with type 1 Pfeiffer syndrome have normal intelligence and a normal life span.
Pfeiffer syndrome type 2 is characterized by a more severe form of craniosynostosis (Cloverleaf skull), with more severe hand and foot anomalies and additional malformations of the limbs. Advanced craniosynostosis or craniosynostosis involving multiple sutures, may cause a build-up of pressure within the skull or a constriction of the growth of the brain. This may result in developmental delays, mental retardation, seizures or blindness. Infants with Pfeiffer syndrome type 2 often experience impaired mental development and neurological problems due to severe involvement of the brain, and/or hypoxia due to problems with breathing. Without appropriate treatment, the physical abnormalities associated with the disorder may lead to life-threatening complications during infancy.
Individuals with Pfeiffer syndrome type 3 have symptoms and findings similar to those present in Pfeiffer syndrome type 2, with the exception of the cloverleaf skull deformity. As in type 2, individuals with Pfeiffer syndrome type 3 often experience impaired mental development and severe neurological problems and may develop potentially life-threatening complications early in life without appropriate treatment.
Pfeiffer syndrome causes
Pfeiffer syndrome is most commonly caused by mutations in the FGFR2 gene. Mutations in the FGFR1 gene cause a small percentage of cases of type 1 Pfeiffer syndrome. Mutations in FGFR1 gene have not been associated with type 2 or 3.
The FGFR1 and FGFR2 genes provide instructions for making proteins known as fibroblast growth factor receptors 1 and 2, respectively. Among their multiple functions, these proteins signal immature cells to become bone cells during embryonic development. A mutation in either the FGFR1 or FGFR2 gene alters the function of the respective protein, causing prolonged signaling, which can promote the premature fusion of skull bones and affect the development of bones in the hands and feet.
Inheritance pattern
This condition is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder.
The abnormal gene can be inherited from either parent or can be the result of a new mutation (gene change) in the affected individual. Essentially all cases of Pfeiffer syndrome type 2 and type 3 have resulted from new mutations. Advanced paternal age is associated with an increased risk for new mutations for Pfeiffer syndrome. The risk of passing the abnormal gene from an affected parent to offspring is 50% for each pregnancy. The risk is the same for males and females.
Figure 4. Pfeiffer syndrome autosomal dominant inheritance pattern

If a person has the mild form of Pfeiffer syndrome, can his children still have the more severe kind?
Yes. While features and/or severity can differ among people with Pfeiffer syndrome (even within the same family), the condition is said to have variable expressivity. There have been several reports of families in which the specific features and severity have differed among family members with Pfeiffer syndrome.
In one family, one person reportedly had all of the features associated with Pfeiffer syndrome, while 8 other family members of the previous 3 generations reportedly had large halluces (large “big toes”) and partial syndactyly (webbing) of the toes. Another report described a girl with severe features of Pfeiffer syndrome, first thought to be due to a new mutation. However, after detailed examination, her mother was found to have abnormalities of the right thumb and possible mild midfacial hypoplasia, consistent with mild expression of Pfeiffer syndrome. The mother may have had a mosaic form of Pfeiffer syndrome 6. There was also a report of a three-generation family with 5 members having an FGFR1 mutation associated with Pfeiffer syndrome. Features in this family varied considerably from apparently normal skulls and limbs, to the characteristic brachycephaly and digital (finger and toe) abnormalities. In this family, the “typical” features of Pfeiffer syndrome appeared only in the third generation, then allowing recognition of the syndrome in several family members in two previous generations 7.
There have also been rare instances in which some family members had features suggestive of Pfeiffer syndrome, whereas others had features suggestive of Jackson-Weiss or Crouzon syndromes (syndromes with overlapping features that may also be caused by mutations in the FGFR2 gene) 8.
Pfeiffer syndrome diagnosis
The diagnosis of Pfeiffer syndrome is based on clinical findings. Molecular genetic testing for FGFR1 and FGFR2 is available if the diagnosis is uncertain.
Pfeiffer syndrome treatment
The treatment of Pfeiffer syndrome is directed toward the specific symptoms that are apparent in each individual. Treatment may require the coordinated efforts of a team of specialists. Pediatricians; surgeons; physicians who diagnose and treat disorders of the ears, nose, and throat (otolaryngologists); neurologists; specialists who assess and treat hearing problems (audiologists); and/or other health care professionals may need to systematically and comprehensively plan an affected child’s treatment.
Specific therapies for Pfeiffer syndrome are symptomatic and supportive. Because craniosynostosis and, in some cases, associated hydrocephalus may result in abnormally increased pressure within the skull (intracranial pressure) and on the brain, early surgery may be advised to correct craniosynostosis and, in the case of hydrocephalus, to insert a tube (shunt) to drain excess cerebrospinal fluid (CSF) away from the brain and into another part of the body where the CSF can be absorbed. Early corrective and reconstructive surgery may also be performed in infants with Pfeiffer syndrome to help correct certain associated craniofacial abnormalities (e.g., midface hypoplasia, facial asymmetry, nasal abnormalities, ocular proptosis due to shallow orbits). The results of such craniofacial surgery may vary.
Airway compromise can also occur, especially in very young children. This causes low oxygen levels that can, if unrecognized and untreated, result in brain damage.
In addition, in some cases, reconstructive surgery may be performed to help correct ear malformations and/or specialized hearing aids may be used to improve conductive hearing loss.
In some individuals with Pfeiffer syndrome, surgery may also be conducted to help correct syndactyly and/or other skeletal malformations and improve function and mobility. Physical therapy and additional orthopedic and supportive measures may also be used to help further improve an affected individual’s mobility. The surgical procedure(s) performed to correct certain craniofacial, audiological, digital, and/or skeletal abnormalities associated with the disorder will depend upon the severity and location of the anatomical abnormalities and their associated symptoms.
Early intervention may be important to ensure that children with Pfeiffer syndrome reach their potential. Special services that may be beneficial to affected children include special social support, physical therapy, and other medical, social, and/or vocational services.
Genetic counseling is recommended for affected individuals and their families. In addition, thorough clinical evaluations may be important in family members of diagnosed individuals to detect any symptoms and physical characteristics that may be potentially associated with Pfeiffer syndrome.
The following online resources can also help you find a genetics professional in your community:
- The National Society of Genetic Counselors (https://www.nsgc.org/) provides a searchable directory of US and international genetic counseling services.
- The American College of Medical Genetics (https://www.acmg.net/) has a searchable database of US genetics clinics.
- The University of Kansas Medical Center (http://www.kumc.edu/gec/prof/genecntr.html) provides a list of US and international genetic centers, clinics, and departments.
- The American Society of Human Genetics (http://www.ashg.org/membership/member_search.shtml) maintains a database of its members, which includes individuals who live outside the United States. Visit the link to obtain a list of the geneticists in your country, some of whom may be researchers that do not provide medical care.
- Pfeiffer syndrome. https://ghr.nlm.nih.gov/condition/pfeiffer-syndrome[↩]
- Nathaniel H Robin, Marni J Falk, Chad R Haldeman-Englert. FGFR-Related Craniosynostosis Syndromes. GeneReviews. June 7, 2011; https://www.ncbi.nlm.nih.gov/books/NBK1455/[↩]
- Pfeiffer syndrome. https://www.chop.edu/conditions-diseases/pfeiffer-syndrome[↩]
- http://www.bornahero.org/medical-issues/[↩]
- https://ccakids.org//assets/syndromebk_pfeiffer.pdf[↩]
- Pfeiffer syndrome. http://www.omim.org/entry/101600[↩]
- Bessenyei B, Tihanyi M, Hartwig M, Szakszon K, Oláh É. Variable expressivity of pfeiffer syndrome in a family with FGFR1 p.Pro252Arg mutation. Am J Med Genet A. December, 2014; 164(12):3176–3179[↩]
- Robin NH, Falk MJ, Haldeman-Englert CR. FGFR-Related Craniosynostosis Syndromes. 1998 Oct 20 [Updated 2011 Jun 7]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2018. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1455[↩]





{kind=link}