Contents
- Secondary hemophagocytic lymphohistiocytosis
- Secondary hemophagocytic lymphohistiocytosis causes
- Hemophagocytic lymphohistiocytosis disease triggers
- Infection-associated hemophagocytic lymphohistiocytosis (infection-associated HLH)
- Rheumatologic hemophagocytic lymphohistiocytosis (R-HLH) or Macrophage Activation Syndrome (MAS-HLH)
- Hemophagocytic lymphohistiocytosis in patients with cancer (M-HLH)
- Hemophagocytic lymphohistiocytosis during chemotherapy (Ch-HLH)
- Hemophagocytic lymphohistiocytosis associated with receiving immune activating therapies (Rx-HLH)
- Transplant-related hemophagocytic lymphohistiocytosis (PT-HLH)
- Hemophagocytic lymphohistiocytosis in patients who are immune compromised (IC-HLH)
- Hemophagocytic lymphohistiocytosis and metabolic disorders
- Hemophagocytic lymphohistiocytosis of unknown or uncertain origin
- Secondary hemophagocytic lymphohistiocytosis pathophysiology
- Secondary hemophagocytic lymphohistiocytosis signs and symptoms
- Secondary hemophagocytic lymphohistiocytosis diagnosis
- Secondary hemophagocytic lymphohistiocytosis treatment
- Secondary hemophagocytic lymphohistiocytosis prognosis
- Secondary hemophagocytic lymphohistiocytosis causes
Secondary hemophagocytic lymphohistiocytosis
Secondary hemophagocytic lymphohistiocytosis (secondary HLH) also known as acquired hemophagocytic lymphohistiocytosis (acquired HLH) is a rare life-threatening intense immune activation syndrome in which your body makes too many activated immune cells called macrophages (histiocytes) and lymphocytes (T cells, natural killer cells and B cells) in the setting of infections, autoimmune disorders or cancer triggers 1, 2, 3, 4, 5, 6. In secondary hemophagocytic lymphohistiocytosis (sHLH) disease the immune system responds to a stimulus or ‘trigger’, often an infection, cancer or rheumatologic disease, but the response is ineffective and abnormal. This ineffective, abnormal response, causes a variety of signs and symptoms, which, if not treated, can potentially become life-threatening.
Secondary HLH is associated with a wide variety of diseases such as viral, bacterial and fungical infections, lymphomas and other malignancies, as well as autoimmune and metabolic diseases 7. The two major causes of secondary HLH are infection and lymphoproliferative diseases. Infectious disease causes are mainly represented by viruses and, less frequently, by bacterial or even fungal infections. Among the herpes viruses, Epstein-Barr virus (EBV) is a frequent inducer of HLH, but other viruses such as cytomegalovirus (CMV), parvovirus B19, human herpesvirus 6 (HHV-6), adenovirus and varicella-zoster virus (VZV), may also act as inducers 8.
Although hemophagocytic lymphohistiocytosis (HLH) has various underlying causes, all subtypes of HLH results from an ineffective, abnormal response of the immune system to a stimulus or ‘trigger’. The underlying mechanisms that cause signs and symptoms to develop are complex. There is overproduction and overactivity of immune system cells called histiocytes (also called macrophages) and T cells (also called T-lymphocytes). These are types of white blood cells, which are the primary cell of the immune system and help the body to fight off infection. Hemophagocytic lymphohistiocytosis (HLH) are related to immune dysregulation that leads to hypercytokinemia and an accumulation of activated macrophages in organs and tissues 9. In HLH, when the white blood cells act in a dysregulated manner, they often over-produce cytokines, and the abnormally functioning white blood cells and cytokines can damage organs including the liver, spleen, bone marrow and brain. Without prompt diagnosis and treatment, HLH can lead to severe organ damage and death. Therefore, early diagnosis and prompt treatment is essential.
HLH is rare, from the institution of the first international HLH registry in 1989 to the HLH-2004 therapeutic study, only approximately 700 patients have been formally evaluated worldwide 10. In Europe and Japan an HLH incidence of 1 to 2 per million was reported in 2005 11, however, there is the possibility that the diagnosis is under-reported, especially in developing countries 12. Therefore a high grade of suspicion is essential for diagnosis.
HLH should be suspected in all patients with prolonged high-grade fever associated with abnormally large spleen (splenomegaly) and multiple organ involvement 13. The clinical spectrum of hemophagocytic lymphohistiocytosis disease is wide, ranging from mild organ dysfunction to multiorgan failure requiring intensive care. Central nervous system (CNS) involvement is frequent and often severe, even though it is not included in the official diagnostic criteria 13.
Hemophagocytic lymphohistiocytosis most often affects infants or young children, but can affect individuals of any age. It affects boys and girls in equal numbers. In adults, it affects men slightly more often than women. The exact incidence and prevalence of hemophagocytic lymphohistiocytosis is unknown. Rare disorders often go misdiagnosed or undiagnosed making it difficult to determine the true frequency in the general population. About 25% of the people with hemophagocytic lymphohistiocytosis, have the familial form.
The typical patient is an infant under 1 year of age, ill-appearing (toxic aspect), occasionally with a critical sepsis-like aspect 4. A younger age at onset suggests an underlying genetic basis, as seen in familial hemophagocytic lymphohistiocytosis (F-HLH) or in HLH forms arising from genetic primary immunodeficiencies; however, familial HLH can present at any age, including adulthood 14.
Progressive splenomegaly (abnormally large spleen) is typically observed in patients, and can be associated with liver involvement, neurological signs, respiratory and kidney failure 15. Skin rashes, reddening of the skin because of inflammation (erythroderma), swelling (edema) or tiny spots on the skin due to bleeding under the skin (petechiae) have been reported 15. Atypical forms, usually seen in children older than 1 year, include isolated fever of unknown origin (FUOs), isolated central nervous system (CNS) involvement 16 or isolated acute liver failure 17, 18. Enlarged lymph node (lymphadenopathy) is uncommon in patients and indicates a potential underlying lymphoma 19.
Typical laboratory findings include cytopenias (low levels of red blood cells [anemia], white blood cells [leukopenia] or platelets [thrombocytopenia]), high level of triglycerides in blood (hypertriglyceridemia), low fibrinogen in blood (hypofibrinogenemia) are suggestive of HLH in the context of general inflammation and hyperferritinemia. Liver function tests are frequently altered 13. Leukocytosis (elevated white blood cell (WBC) count) is not typical of HLH except in HLH-associated with defined rheumatological conditions or macrophage activation syndrome-HLH (MAS-HLH) 13. Hemophagocytosis describes the pathognomonic findings where highly activated macrophages taking up different cells including lymphocytes, erythrocytes, leukocytes, and platelets in different tissues, producing excessive cytokines and an uncontrolled inflammatory reaction 20, 13, 21.
Despite great improvement in HLH diagnosis and treatment, it still represents a challenge in clinical management, with poor prognosis in the absence of an aggressive therapeutic approach 10, 13.
If HLH is diagnosed quickly and accurately at a center equipped to manage this complicated disease, a cure is possible. Often, due to the severity of the clinical picture, it is necessary to start an aggressive treatment before having clarified the primary or secondary nature of HLH syndrome 22.
Secondary hemophagocytic lymphohistiocytosis prognosis widely varies from a 55% survival at 3 years with the standard treatment protocols 23 to 100% survival in specific conditions such as Leishmania-triggered HLH when treated appropriately 24.
Figure 1. Blood cell development
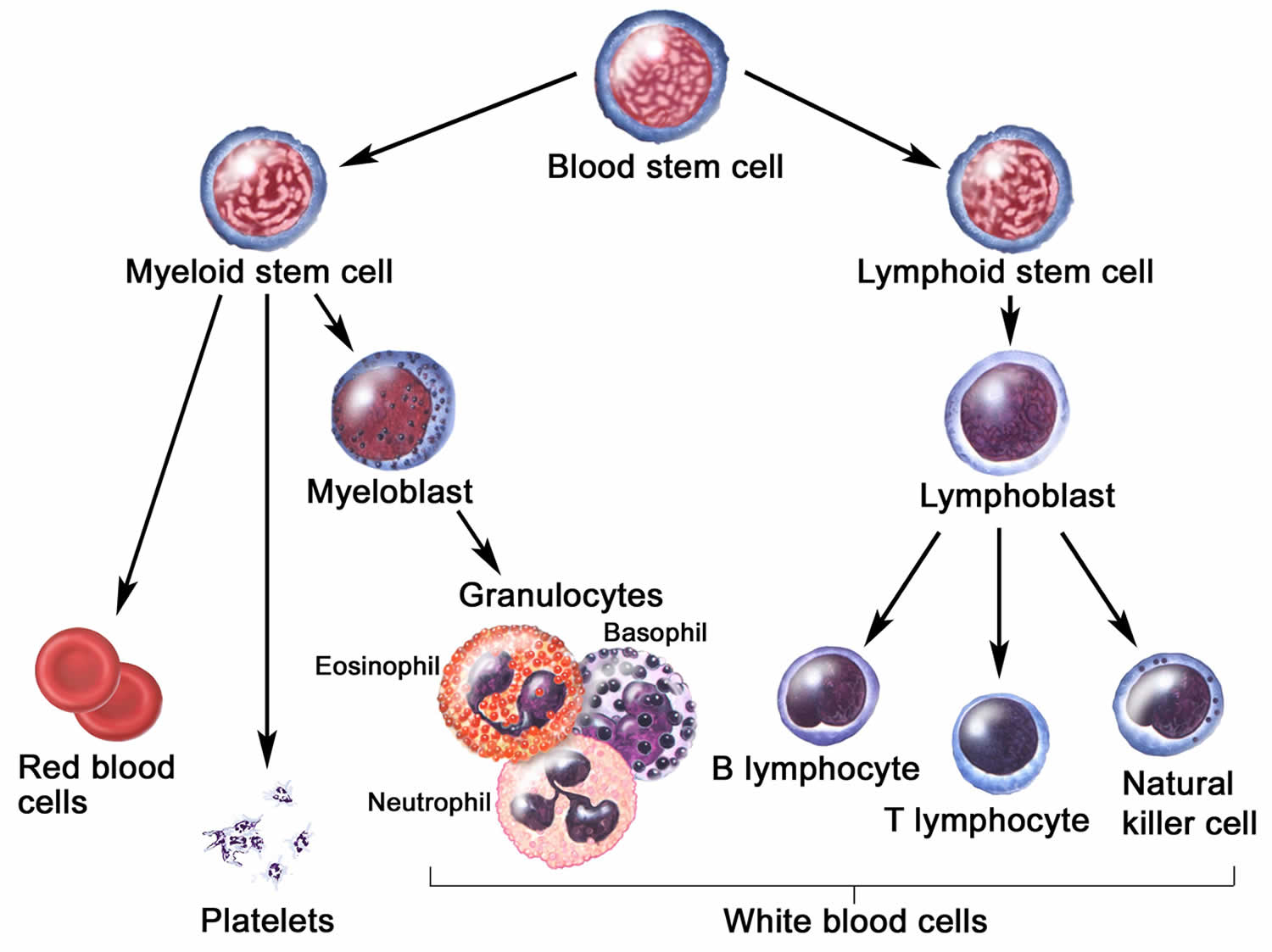
Figure 2. Hemophagocytic lymphohistiocytosis syndrome (HLH syndrome)
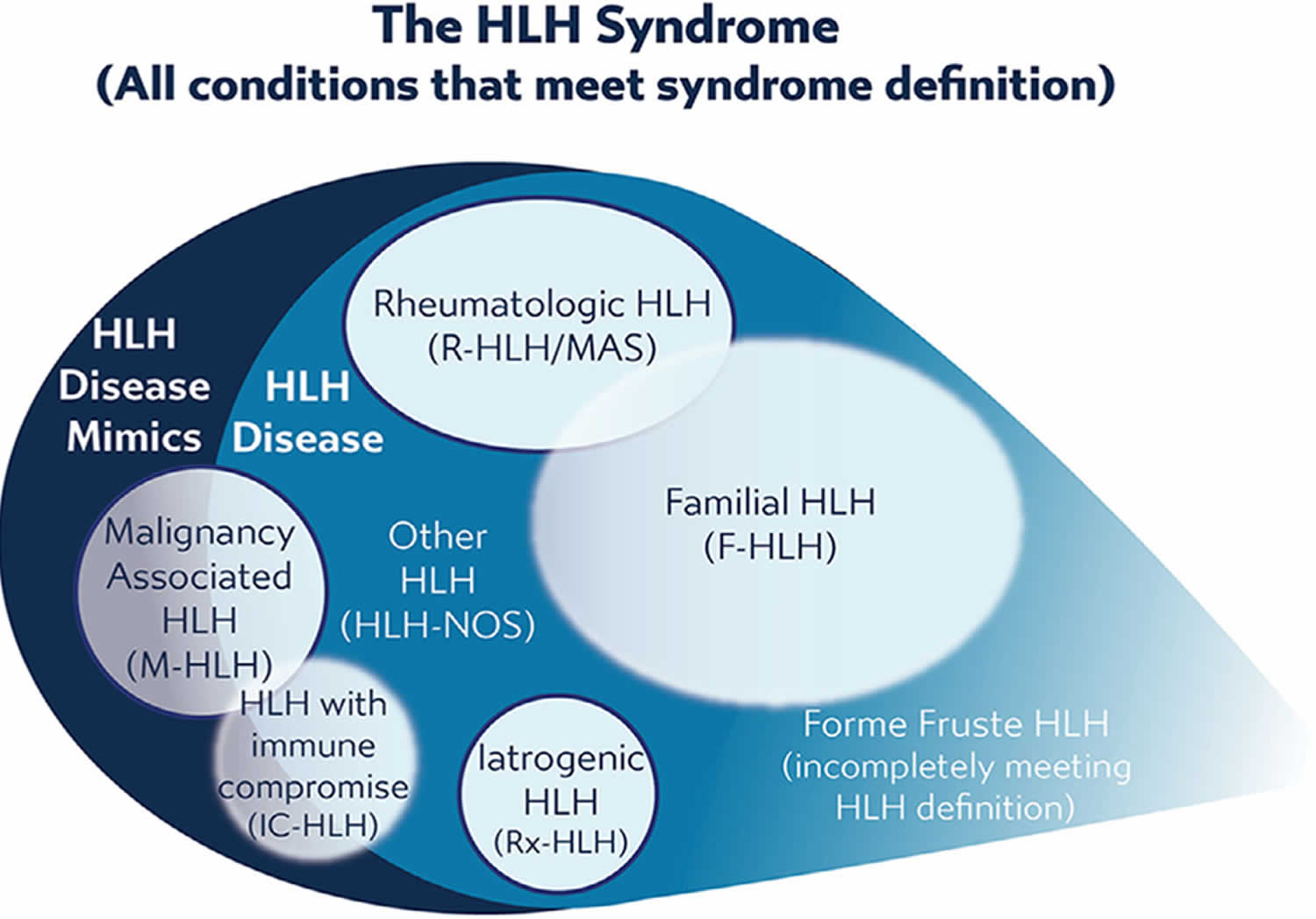
Footnote: Hemophagocytic lymphohistiocytosis syndrome includes all conditions meeting consensus diagnostic criteria. HLH syndrome includes conditions that would benefit from HLH-directed immunosuppressive therapies, which are termed “HLH disease” and those conditions that would not benefit from immunosuppressive therapy or require entirely different treatments, termed as “HLH disease mimics.” HLH disease includes recognizable subgroups: familial HLH with clear genetic origin, HLH associated with malignancy, HLH associated with rheumatologic conditions (macrophage activation syndrome [MAS]), HLH observed after immune activating therapies (iatrogenic HLH, also called cytokine release syndrome), HLH associated with immune compromise (either primary immune deficiency or treatment-related immune suppression), and HLH not associated with other specific conditions. Recognition of these subcategories is valuable as this may alter treatment, though some categories overlap with each other or have indistinct borders and may include examples of both HLH disease and HLH disease mimics. Use of these category-specific terms is preferred over the historical terms of “primary” and “secondary” because the older concepts are ambiguous due to increasing understanding of genetic complexity, involvement of infection in triggering multiple distinct variations of HLH, and varied application. Incomplete, forme fruste episodes of HLH (similar but not completely fulfilling diagnostic criteria) are also well recognized in patients with familial HLH and may occur in others, such as “mild MAS” in patients with systemic onset juvenile arthritis (soJIA).
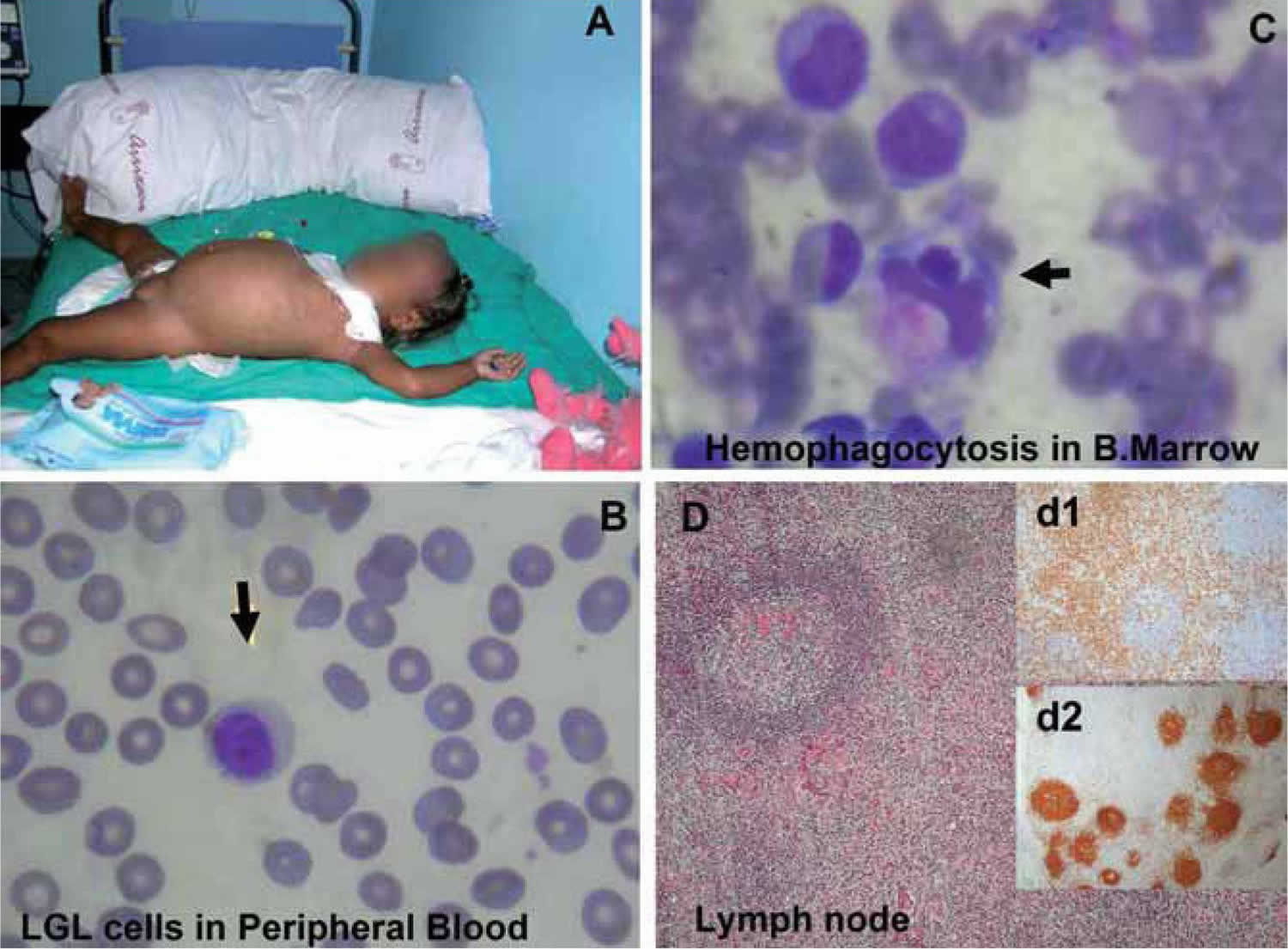
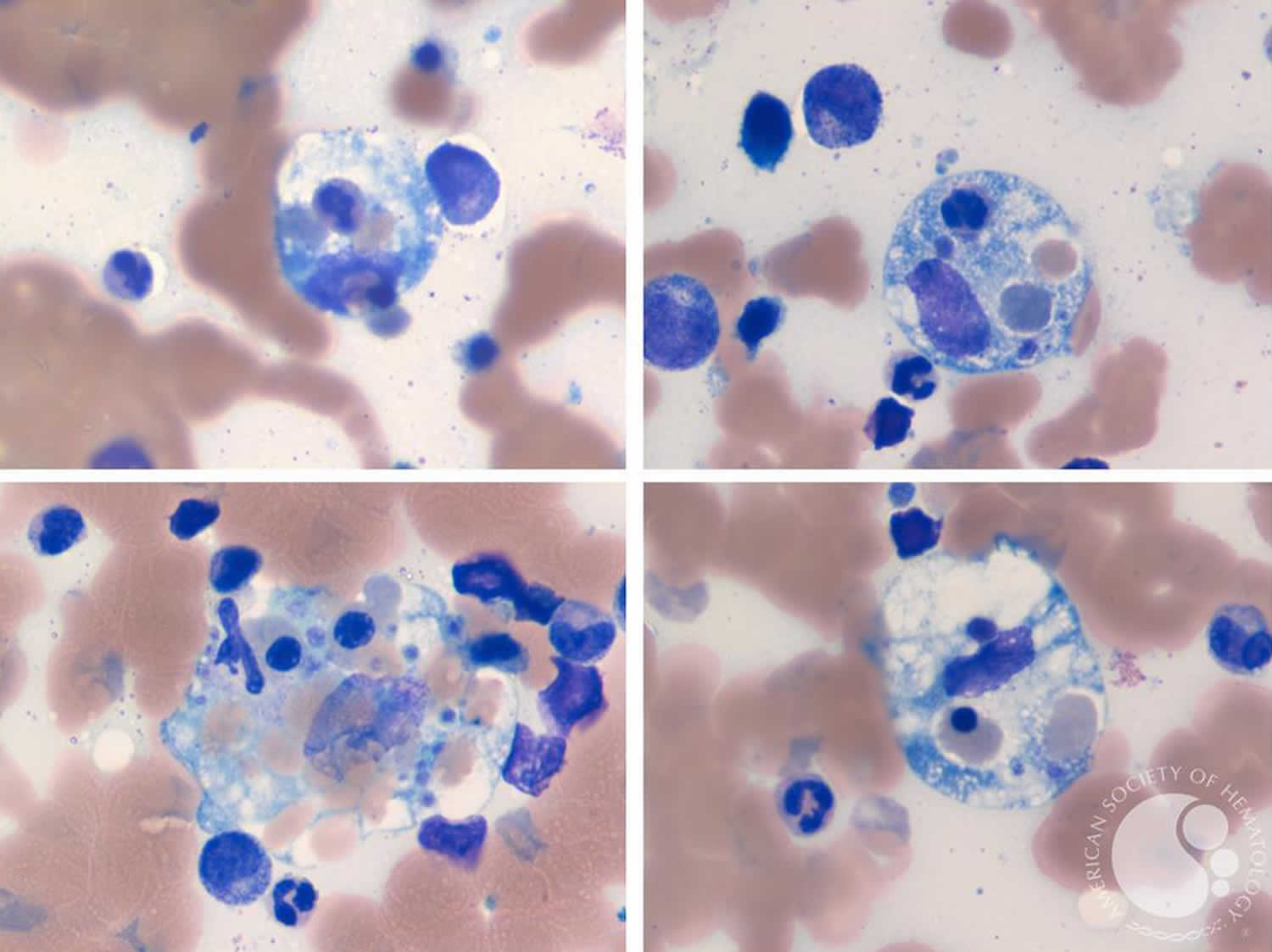
[Source 4 ]Figure 3. Hemophagocytic lymphohistiocytosis
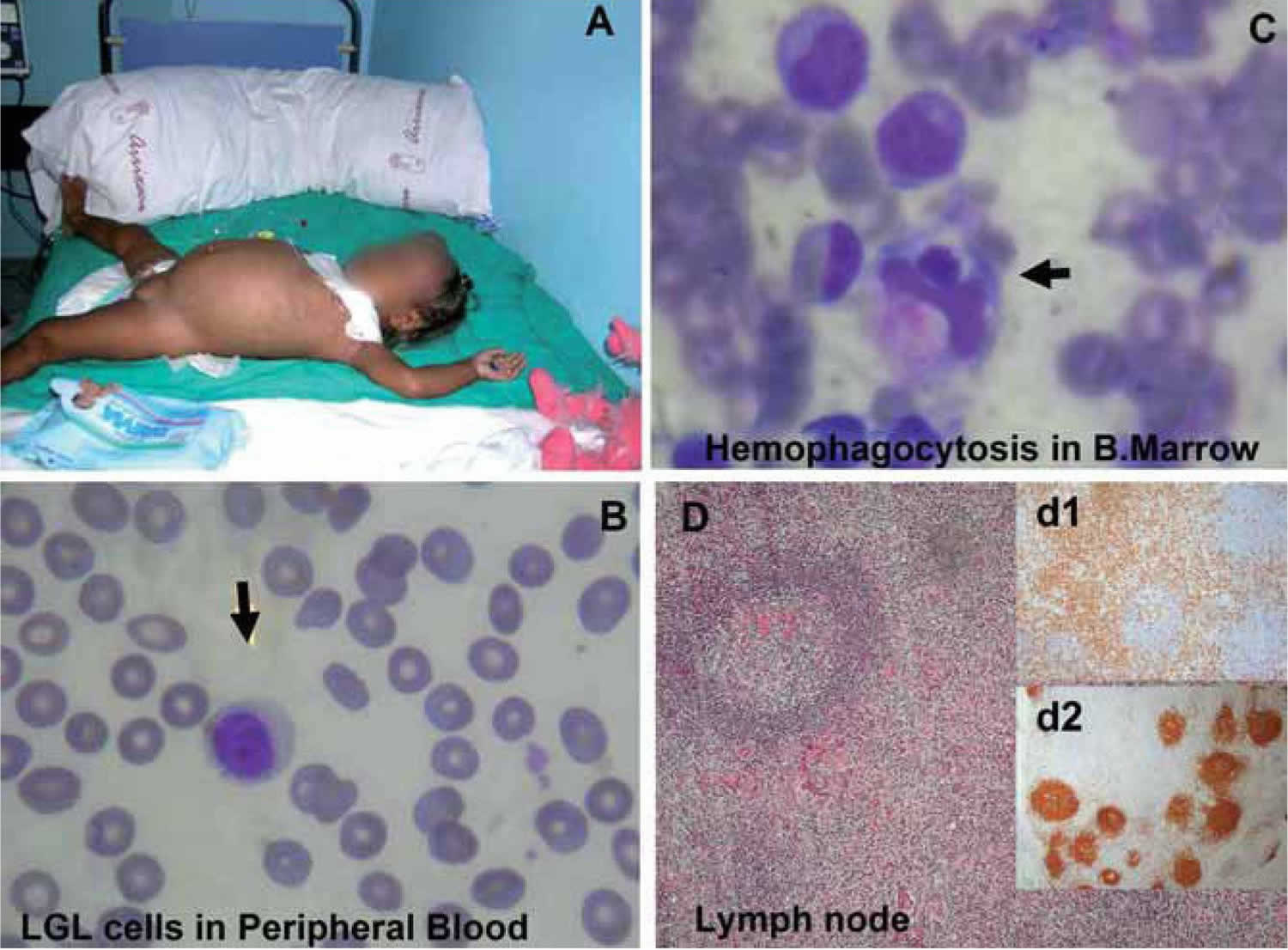
Footnotes: (A) 17-month-old Ecuatorian girl showing partial albinism and suffering from fever and cytopenias persisting over 3 weeks, splenomegaly (10 cm), hepatomegaly (5 cm), múltiple adenopathies, purpura and edema; (B) cell expansion of large granular lymphocytes (LGL cells) in peripheral blood; (C) bone marrow with erithroid, megakaryocytic, and mononuclear-phagocyte hyperplasia and scarce signs of hemophagocytosis. Arrows indicate hemophagocytosis. (D) Paracortical diffuse hyperplasia in a lymph node biopsy with no signs of hemophagocytosis or lymphoproliferative disease; d1 and d2 inserts, interfollicular/paracortical and follicular details of anti-CD3 and anti-CD20 immunohistochemistry analysis, respectively.
[Source 25 ]Figure 4. Hemophagocytic lymphohistiocytosis pathology
Footnotes: Hemophagocytic lymphohistiocytosis (HLH) primarily involves histiocytes (macrophages) which engulf and destroy red blood cells, a process known as erythrophagocytosis and depicted in the images. Hematologic analysis will also likely reveal increased histiocytes, hypercellular bone marrow, and pancytopenia. Cellular damage in HLH results in stimulation of inflammatory cytokines including TNF-α and IFN-γ, which suppress hematopoiesis, induce apoptosis in hematopoietic cells, and further activate histiocytes 26. If HLH is suspected, it is critical to evaluate serum ferritin, triglyceride, and fibrinogen levels. Work-up revealed positive Epstein-Barr virus (EBV) virology in this case, without morphologic or immunophenotypic evidence of lymphoma or leukemia involvement. Secondary HLH in adults is typically caused by virus or lymphoma, more commonly T cell lymphoma. Therefore, monitoring and further testing for an occult secondary lymphoma could be beneficial.
- Elevated ferritin is expected because destruction of red blood cells results in the release of iron into the serum. Additionally, histiocytes increase ferritin production secondary to increased levels of heme-oxygenase due to the inflammatory cytokines 26.
- Hypertriglyceridemia is expected in HLH, because TNF-α and IFN-γ also inhibit lipoprotein lipase activity, which decreases the breakdown of triglycerides for uptake and storage by tissues.1
- Plasmin will also be elevated due to histiocyte secretion of plasminogen activator, leading to fibrin breakdown, which decreases fibrinogen1 and increases D-dimer 27.
- In addition to elevated ferritin and triglycerides with lowered fibrinogen as key laboratory findings for HLH, elevated CD25 marks activated lymphocytes, and is the alpha subunit of the interleukin 2 receptor (IL-2R) 26.
Secondary hemophagocytic lymphohistiocytosis causes
Secondary HLH or acquired hemophagocytic lymphohistiocytosis is usually triggered by underlying infections, cancer, or autoimmune diseases. There is no family history of the disorder and no known genetic factors can be identified. Conditions that can lead to secondary hemophagocytic lymphohistiocytosis include viral infections especially Epstein-Barr virus (EBV), other infections including bacterial, viral and fungal infections, a weakened or depressed immune system, autoimmune diseases, autoinflammatory diseases, rheumatological diseases such as juvenile idiopathic arthritis, metabolic disorders, and cancer such as non-Hodgkin lymphoma. Epstein-Barr virus (EBV) is considered a common infectious inducer of secondary HLH (EBV-HLH) 5, 29.
More than 50% of pediatric HLH in eastern countries may be associated with Epstein-Barr virus (EBV) infection. Interestingly, most EBV-HLH cases are characterized by mono- or oligo-clonal proliferation of EBV-infected NK or T cells 8. HLH is also seen in patients with peripheral T or NK cell leukemias or lymphomas 30. Epstein-Barr virus (EBV) infection plays a critical role in the pathogenesis of these leukemias or lymphomas, in particular, nasal-type NK/T-cell lymphoma and aggressive NK cell leukemia. NK cell leukemias usually possess a single episomal form of EBV, indicating that this type of leukemia is of clonal origin. Like nasal-type NK/T-cell lymphoma, most patients with NK cell leukemia exhibit an aggressive clinical course. NK cell leukemia is characterized by a systemic proliferation of NK cells, predominantly involving peripheral blood and bone marrow. Besides, it has been reported that EBV-NK leukemia cells constitutively secrete interferon gamma (IFN-γ) 30, suggesting that interferon gamma (IFN-γ) may constitute an autocrine survival signal important for the progression of NK cell leukemia, that eventually, might lead to the occurrence of secondary HLH.
Recently, some cases of “secondary” HLH have been linked to mutations that confer a partial impairment of cytolytic function 31. These findings make the distinction between primary and secondary HLH increasingly difficult. Symptoms of HLH developing in a patient with an underlying rheumatic condition are historically called Macrophage Activation Syndrome (MAS) (Ravelli A. Macrophage activation syndrome. Curr Opin Rheumatol. 2002 Sep;14(5):548-52. doi: 10.1097/00002281-200209000-00012)), 32. Suggestions to replace the term Macrophage Activation Syndrome (MAS) with “secondary HLH” first appeared in the literature in 2002 33, 34. Emile et al 35 suggest using the term MAS-HLH for this subset of HLH.
Secondary hemophagocytic lymphohistiocytosis causes 35:
- Infection-associated HLH
- Virus-associated HLH
- Epstein-Barr virus (EBV)-associated HLH
- Cytomegalovirus (CMV)-associated HLH
- HLH associated with other defined herpes virus infections
- HIV-associated HLH
- Influenza-associated HLH
- HLH associated with other defined virus infections
- Bacteria-associated HLH
- Parasite-associated HLH
- Fungal-associated HLH
- Virus-associated HLH
- Malignancy-associated HLH
- Malignancy-triggered HLH (HLH at onset of malignancy)
- Hematological malignancies
- T-cell lymphoblastic lymphoma/leukemia
- T-cell non-lymphoblastic lymphomas
- B-cell leukemias
- B-cell lymphomas (non-Hodgkin)
- Hodgkin lymphomas
- Natural killer (NK)-cell lymphomas/leukemias
- Myeloid neoplasia
- Other hematological malignancies
- Solid tumors
- Unclassified malignancies
- Hematological malignancies
- HLH occurring during chemotherapy (not associated with initial diagnosis of malignancy)
- HLH associated with a malignancy, but not further defined
- Malignancy-triggered HLH (HLH at onset of malignancy)
- HLH associated with defined rheumatologic conditions (R-HLH or MAS-HLH)
- HLH associated with systemic-onset juvenile idiopathic arthritis (SoJIA)
- HLH associated with adult-onset Still disease
- HLH associated with SLE
- HLH associated with vasculitis
- HLH associated with other defined autoimmune conditions
- HLH associated with a not defined autoimmune condition
- Transplant-related HLH
- HLH associated with iatrogenic immune activation
- HLH associated with iatrogenic immune suppression
- HLH associated with other apparently non-Mendelian conditions
- HLH of unknown/uncertain origin
The exact manner that these predisposing conditions cause the signs and symptoms, and specifically how they cause an ineffective, abnormal immune response, in hemophagocytic lymphohistiocytosis are not fully understood and continue to be investigated.
Hemophagocytic lymphohistiocytosis (HLH) is caused by an overactive and abnormal immune response resulting in an uncontrolled excessive inflammatory and ineffective immune response resulting in progressive multi-organ damage and potentially death if untreated 20, 36. In hemophagocytic lymphohistiocytosis (HLH) there is overactivation of T cells, natural killer (NK) cells and macrophages (histiocytes) causing an uninhibited release of pro-inflammatory cytokines (cytokine storm) resulting in a clinical presentation of fever, abnormally large spleen (splenomegaly), low fibrinogen in blood (hypofibrinogenemia) and impaired blood clotting ability (coagulopathy) 5, 6.
Your immune system is complex and has many components. White blood cells comprise an important part of this system; these cells communicate with each other and other cells of the body by making chemical “messengers” called cytokines. Your immune system is your body’s natural defense system against foreign or invading organisms or substances. The immune system is a complex network of cells, tissues, organs, and proteins that work together to keep your body healthy. Macrophages are a type of white blood cells involved in the detection, phagocytosis and destruction of bacteria and other harmful organisms. Macrophages are effector cells of the innate immune system that phagocytose (a process by which certain living cells ingest or engulf other cells or particles) bacteria and secrete both pro-inflammatory and antimicrobial mediators 37. In addition, macrophages play an important role in eliminating diseased and damaged cells through their programmed cell death. Generally, macrophages ingest and degrade dead cells, debris, tumor cells, and foreign materials. Macrophages promote homeostasis by responding to internal and external changes within the body, not only as phagocytes, but also through trophic, regulatory, and repair functions 37. Furthermore, macrophages can also present antigens to T lymphocytes (T cells) and initiate inflammation by releasing molecules known as cytokines that activate other cells. On the other hand, lymphocytes are mature, infection-fighting cells that develop from lymphoblasts, a type of blood stem cell in your bone marrow (Figure 1). Lymphocytes are the main cells that make up lymphoid tissue and your lymphoid tissue is found in lymph nodes, the thymus gland, the spleen, the tonsils, and adenoids. Lymphocytes consist of several subtypes with different functions:
- B lymphocytes also called B-cells make antibodies to help fight infection. B lymphocytes (B cells) protect the body from invading germs by developing (maturing) into plasma cells, which make proteins called antibodies. The antibodies attach to the germs (bacteria, viruses, and fungi), which helps other white blood cells called granulocytes recognize and destroy them.
- T lymphocytes also called T-cells that help B lymphocytes make the antibodies that help fight infection. T lymphocytes (T cells) can also recognize cells infected by viruses and directly destroy these cells. They also help regulate your immune system.
- Natural killer cells also called NK cells that attack cancer cells and viruses. Natural killer cells serve as pivotal “watchers” or “guards” within the immune system as they respond quickly to a pathogenic infiltration and alert the host about infections. Natural killer cells are able to recognize and break down tumor cells and virus-infected cells, even without any previous sensitization. NK cells originate in the bone marrow and are released into the bloodstream upon maturation, thus being able to respond to stimuli such as pathogen molecules, cytokines, or by the interaction with any target cell that expresses ligands for activating NK cell receptors.
Histiocytes are large phagocytic cells that normally play a role in responding to infection and injury. A phagocytic cell is any “scavenger” cell that engulfs and destroys invading microorganisms or cellular debris. Macrophages also secrete cytokines, which are proteins that stimulate or inhibit other immune system cells and promote inflammation in response to disease. Excessive cytokine production will eventually cause severe tissue damage. Macrophages may also mistakenly engulf and destroy healthy tissue including healthy blood cells, which is called hemophagocytosis. Cytotoxic lymphocytes, which include T cells and natural killer cells, do not function properly. These cells eliminate other cells that are damaged, stressed, or infected. In HLH, cytotoxic lymphocytes fail to eliminate activated macrophages allowing them to abnormally build up in the organs and tissues of the body, which further activates this ineffective immune response. These immune system abnormalities cause the excessive inflammation and tissue destruction that characterizes the condition.
Hemophagocytic lymphohistiocytosis disease triggers
Infection and cancer, acting alone or in concert with the above-mentioned genetic susceptibility factors, are common HLH triggers 38, 39. HLH that occurs in a patient who has a strong immunologic trigger such as infection or cancer, in the absence of a genetic disease which features HLH, are often said to have secondary HLH (acquired HLH).
Hemophagocytic lymphohistiocytosis can occur with virtually any infection or cancer. More common infections include DNA viruses (Epstein-Barr virus (EBV), cytomegalovirus, and adenovirus) and intracellular pathogens (eg, Leishmania), but the list of infections that have been reported to occur with HLH is extensive and influenced by geographic region (leishmaniasis and tick-borne illnesses), season (influenza viruses, tick-borne illnesses), and socioeconomic status (tuberculosis) 5. Immune reconstitution inflammatory syndrome occurring when newly treated HIV patients are coinfected with (usually) tuberculosis bears striking similarity to HLH and has been associated with high interleukin-18 (IL-18) 40. Features of HLH should be considered in all serious infections, and infectious disease consultation may be appropriate in many/most HLH patients.
Lymphoma and leukemia are common malignancies associated with HLH, particularly T-cell and NK-cell lymphomas or leukemias, diffuse large B-cell lymphoma, and Hodgkin lymphoma 41. Solid tumors can also trigger HLH. It is important to realize that hemophagocytic lymphohistiocytosis is due to an underlying cancer in >50% of adult cases 42, 43. Aggressive evaluations should be performed to evaluate for cancer in adult patients with HLH.
Hemophagocytic lymphohistiocytosis can also occur during the course of chemotherapy and is often associated with an infection 41. Treatment with chimeric antigen receptor–modified T cells called CAR T-cell therapy or bispecific T-cell–engaging antibodies can be associated with a cytokine release syndrome that mimics hemophagocytic lymphohistiocytosis 44, 45.
Infection-associated hemophagocytic lymphohistiocytosis (infection-associated HLH)
Infection-associated hemophagocytic lymphohistiocytosis (infection-associated HLH) represents a challenge in classification 1. This is because infectious diseases can trigger HLH in both primary and secondary forms, and because septic shock can either be a presentation of HLH syndrome or may mimic it, with great implications in therapeutic approach. A thorough infectious screening is thus highly recommended when facing an HLH syndrome 13, 46.
Among the case reports the most frequent infectious triggers were viruses, such as the herpetic viruses, dengue virus, Crimean Congo hemorrhagic fever virus, Eastern equine encephalitis virus, hepatitis A virus (HAV), hepatitis B virus (HBV), human immunodeficiency virus (HIV), adenovirus, parvovirus B19, measles, influenza virus and severe acute respiratory syndrome coronavirus 2 (COVID-19).
Epstein-Barr virus (EBV) has been frequently reported from all across the world, and especially from the Asian Countries 47, 48, 49, 50, 51, 52, 53, 54, 55, 56. The reason for this geographical heterogeneity may involve a higher virulence in EBV viral strains circulating in Asia 57 or a different immune predisposition in the Asian patients, but this issue has not been clarified yet. EBV typically targets B cells, but in a subset of patients with EBV-HLH, of prevalent Asian origin again, it infects T or NK cells leading to oligoclonal or monoclonal proliferation and massive cytokine production 58, 59. In extreme cases, EBV-HLH clinical picture can be difficult to differentiate from T cell lymphoproliferative disorder 59. EBV-hemophagocytic lymphohistiocytosis is usually quite aggressive, with a frequent involvement of central nervous system (CNS). Once EBV is demonstrated by serology tests or molecular biology methods, a targeted approach is recommended 57, 60. Clinical scores to differentiate low risk vs. high-risk patients have been proposed 60, but have not achieved conclusive results yet.
Cytomegalovirus (CMV) is typical of newborns and immunocompromised patients 55, 61, 62, 63, 64, 65, 56 and so is the varicella zoster virus 66. Dengue virus plays a significant role in tropical countries 56, 4, 67, 68, 69, 70, 71. Tick-borne diseases include the Crimean-Congo hemorrhagic fever nairovirus 72, 73 and the Eastern equine encephalitis virus 74. Sporadic case reports include HLH related to hepatitis A virus 71, 75, hepatitis B virus 76, human immunodeficiency virus (HIV) 70, 77, Adenovirus 55, 70, 78, 79, Parvovirus B19 55, Measles 80 and Influenza virus 81, 82, 83. Sars-Cov2 infection, causative of the recent COVID19 pandemic, has been reported to also be a HLH trigger, mostly in adults 84, 85.
Bacterial infections associated to HLH have frequently been reported in tropical countries and include Brucella 55, 86, 87, 88, Salmonella enteritidis 71, 89, 56, Tuberculosis 56, 71, 90, sepsis by streptococcal infection [group B streptococcus 91 and Streptococcus suis 92 ], by Listeria 55 or without identified pathogen 71. Orientia tsutsugamushi, the causing agent of scrub typhus, has been reported in India 70, 71, 56 and South Korea 93, 94, 95, 96. Ehrlichia chaffeensis, the agent of human monocytic ehrlichiosis, has been reported from the US 97, 98, 99, 100, 101. A case of Strep pneumoniae 23A, serotype that is not included in the pneumococcal 13-valent conjugated vaccine (PCV-13) has been reported from Japan 102.
HLH in kidney transplant recipients has been associated with Ehrlichiosis in one case 100, and with Bartonella henselae in one other case 103. A case associated with Serratia marcescens has been reported in a preterm newborn 104.
Fungal infection-associated HLH has been reported in immunocompromised hosts (being treated for aplastic anemia and preB-acute lymphoblastic leukemia, respectively), caused by Trichosporon asahii 105.
Leishmania 54, 55, 106, 107, 108, 24 and Plasmodium falciparum 109 and Plasmodium vivax 110 have been reported as parasitic triggers of HLH 71.
Rheumatologic hemophagocytic lymphohistiocytosis (R-HLH) or Macrophage Activation Syndrome (MAS-HLH)
Macrophage Activation Syndrome (MAS) is the term most commonly used to refer to an HLH or HLH-like syndrome occurring in the context of rheumatologic disorders (R-HLH) 111. While MAS and HLH are very similar and should be viewed as the same disease, regardless of differences in presentation and treatment 112, 113, 114, 115. Consensus criteria for recognizing MAS in the context of systemic onset juvenile arthritis (soJIA) have been developed, and in general, these patients with MAS are older than patients with familial hemophagocytic lymphohistiocytosis and they present with substantially higher platelet and neutrophil counts as well as higher fibrinogen levels 116 These laboratory indices are typically elevated in patients with juvenile idiopathic arthritis (JIA), so “falling” to near-normal levels is often a substantial change indicating a more severe process than would ordinarily be implied by such values. The frequency of MAS is ∼10% of patients with systemic onset juvenile arthritis (soJIA), but 30–40% may have an incomplete presentation of MAS 116, 117. MAS in adults is most commonly seen with adult-onset Still’s disease, reported in ∼15% of adult-onset Still’s disease cases 118, 119. MAS has also been reported in systemic lupus (estimated incidence of MAS in 4% of cases), dermatomyositis, Kawasaki’s disease, and rheumatoid arthritis 120, 31. Obviously, a pre-existing diagnosis of a rheumatologic disorder would define R-HLH, but unexplained rash, recurrent fever, arthralgia/arthritis in an undiagnosed patient presenting with HLH may suggest R-HLH.
Hemophagocytic lymphohistiocytosis in patients with cancer (M-HLH)
The association of hemophagocytic lymphohistiocytosis in patients with cancer (M-HLH) has been recognized for decades and it can be the initial manifestation of an underlying malignancy, it may develop during therapy as the consequence of chemotherapy or of treatment-induced mutations [treatment-induced HLH (Ch-HLH)], or it can develop as a consequence of an intercurrent infection 121, 122, 123, 124. Hemophagocytic lymphohistiocytosis in patients with cancer (M-HLH) is common in adults, where it accounts for ~50% of HLH cases 19, while it is significantly rarer in children, with a prevalence of 8-11% 121, 125.
Patients may present with the clinical syndrome of HLH associated with undiagnosed underlying malignancy (new onset), or they may develop HLH during treatment for known malignancy (on therapy), usually in the context of infection. With new onset M-HLH, this HLH syndrome is typically a “HLH disease mimic”, as the disease features appear to be driven directly by the cancer, and antineoplastic (instead of anti-HLH) treatment should be the priority, though pathophysiology is not well defined and therapy of malignancy may overlap with HLH treatment. “On therapy” M-HLH should most often be considered to be a manifestation of HLH due to infection in the context of immune compromise (IC-HLH), though it may rarely be associated with occult relapse. When considering hemophagocytic lymphohistiocytosis in patients with cancer (M-HLH), it is important to note that the presence of EBV viremia does not rule out malignancy (including B- or T-cell lymphomas) and sCD25 may be disproportionately elevated compared to other features of HLH in patients with occult lymphoma 126, 127.
The excess of proinflammatory cytokines produced by activated T cells infiltrating or surrounding the tumor or by the neoplastic T-cells in T/NK cell lymphoma is likely to cause hemophagocytic lymphohistiocytosis in patients with cancer (M-HLH) 6, 128. In rare cases, the diagnosis of HLH anticipates that of malignancy by several weeks 121, 129.
Lymphoma deserves special mention as it is the most common malignancy associated with HLH at its initial presentation. Lymphomas, though relatively uncommon at pediatric age, are frequently reported in association with HLH 121. Among the cases reviewed, HLH has been associated with Hodgkin lymphoma (73,121,140,144), anaplastic large cell lymphoma 56, 130, 131, peripheral T-cell lymphoma 130, 132, post-transplant lymphoproliferative disorder-lymphoma 133, subcutaneous panniculitis-like T-cell lymphoma 54, extranodal NK/T cell lymphoma, hepatosplenic T-cell lymphoma, systemic EBV-positive T-cell lymphoma of childhood 130. In addition, HLH and lymphoma can be alternative diagnoses as well 134, and extreme caution is required in differentiating the two conditions before starting steroid therapy. Acute leukemia (both myeloblastic and lymphoblastic) presenting as HLH syndrome has also been reported 55, 100, 130.
Because of the difficulty of distinguishing lymphoma from familial hemophagocytic lymphohistiocytosis (F-HLH) or rheumatologic disorders associated hemophagocytic lymphohistiocytosis (R-HLH), thorough imaging and aggressive biopsy, often guided by PET-CT, should be pursued or at least considered before starting corticosteroids and other therapies that may obscure diagnosis 135.
Hemophagocytic lymphohistiocytosis during chemotherapy (Ch-HLH)
Among the cases of hemophagocytic lymphohistiocytosis during chemotherapy (Ch-HLH), HLH developed on therapy or after treatment for juvenile myelomonocytic leukemia 136, acute monocytic leukemia 137, Langherhans cell histiocytosis 138 and solid tumors such as Wilms tumor 139, neuroblastoma 55, 140, rhabdomyosarcoma 123. Langerhans cell histiocytosis has been associated to HLH at presentation, during therapy or as a consequence of viral infection 123, 129, 141.
The immunosuppression induced by treatment frequently causes viral infections or reactivations, that in turn can trigger HLH. Among the cases reviewed, viral reactivation included EBV, CMV, respiratory syncytial virus, BK virus, human herpes virus 6, adenovirus and parvovirus B19 124, 142.
Hemophagocytic lymphohistiocytosis associated with receiving immune activating therapies (Rx-HLH)
Various emerging immune activating therapies such as chimeric antigen receptor T cells (CAR-T cells), immune checkpoint inhibitors and Blinatumomab a bispecific T-cell engager (BiTE, an antibody therapy that binds to two target proteins on different cells), have been associated with cytokine release syndrome (CRS) 143. Cytokine release syndrome shows major overlaps with HLH, so numerous authors classify it as a form of secondary hemophagocytic lymphohistiocytosis 144, 145, 146; some other authors, conversely, restrict the diagnosis of HLH following CAR-T infusion (car-HLH) to the cases in which a severe cytokine release syndrome is associated to the typical HLH laboratory findings, often including hemophagocitosis 147, 148.
CAR-T cells were officially licensed for use in children and young adults by the U.S. Food and Drug Administration in 2017 for the treatment of refractory B-cell acute lymphoblastic leukemia 149. Cytokine release syndrome is a common severe adverse reaction following such therapies, especially in the acute phase 143. It probably results from the high amount of IFN-γ and IL-6 produced by the constitutionally activated CAR-T cells 150 or by the subsequently activated macrophages 151, 152, 153 and by the targeted tumor cell lysis 154. Development of car-HLH seems associated with pre-infusion NK lymphopenia (although it is not associated with impaired NK function) that is further amplified by treatment 147.
The St. Jude Children’s Research Hospital group recently described a cohort of 27 pediatric patients treated with CD-19 CAR-T cells 148: 12/27 patients developed cytokine release syndrome alone, while four progressed to car-HLH despite appropriate therapy. The car-HLH subgroup showed higher and more sustained inflammation parameters and a poorer antileukemic response and survival. Blinatumomab, as well, has been associated with cytokine release syndrome and/or HLH in adults 143 and children 155.
Patients treated with T-activating therapies should be closely monitored for hemophagocytic lymphohistiocytosis, in order to optimize diagnosis and therapeutic approach.
Additional iatrogenic causes of cytokine storm include rituximab 156, gene therapies, immune checkpoint inhibitors, allogeneic hematopoietic stem cell transplantation (allo-HSCT) and heart transplantation 143.
Transplant-related hemophagocytic lymphohistiocytosis (PT-HLH) has been described in the context of hematopoietic stem cell transplantation (HSCT) 157, 158 and, more rarely, in kidney 103, 151 and liver transplant recipients 159, 160, 161. Post-transplant HLH (PT-HLH) might be triggered by a combination of tissue damage, immunosuppressive therapy 162, alloimmune response, residual malignancies or by infections 157. Viral infections or reactivations, such as EBV and CMV but also gastrointestinal viruses, represent the most frequent trigger, but bacterial and fungal infections have also occasionally been described 103, 151, 158, 163, 164, 165.
HLH post-hematopoietic stem cell transplantation (post-HSCT HLH) can occur within the first 30 days after transplantation (early onset) or later (late onset) 157. Late onset is usually related to infectious events, while for early onset the causes are not fully understood. In some cases the triggering factor may be the residual disease 157. Post transplant-related hemophagocytic lymphohistiocytosis (PT-HLH) diagnosis is extremely challenging because of the complex clinical and laboratory picture of the affected patients, for whom specific diagnostic criteria do not exist yet, and a high suspicion rate is essential 166.
In early onset post-hematopoietic stem cell transplantation (post-HSCT HLH), aspects of differential diagnosis may occur with engraftment syndrome and acute graft-vs.-host disease 157. The treatment of post-transplant HLH (PT-HLH) is also particularly complex, since we are dealing with immunosuppressed patients, often with transplant-related toxicity 157. In EBV-related forms, the use of rituximab plays an important role 49, 167.
Hemophagocytic lymphohistiocytosis in patients who are immune compromised (IC-HLH)
HLH has been reported to occur in a variety of patients who are immune compromised, including those receiving chemotherapy or those with primary immune deficiency (PID). As noted above, HLH may occur in patients with cancer who have often received chemotherapy for an extended period of time and who typically have a recent triggering infection. Similarly, a number of children (and adults) with inflammatory bowel disease (IBD), typically treated with azathioprine or mercaptopurine have been reported to develop HLH after primary infection with EBV or CMV 168. In both contexts, pathophysiology is not well understood and while it may be related to the underlying disease, HLH appears to be a dysregulated response to infection which is related to immunosuppression. It is difficult to generalize about whether IC-HLH may benefit from significant immune suppression. For patients with IBD, withdrawal of mercaptopurine, treatment of infection, supportive care, and moderate dose corticosteroids often suffice. “On therapy” M-HLH does not appear to respond well to any treatment, though corticosteroids ± etoposide are usually tried. Thus, IC-HLH ambiguously straddles the categories of HLH disease and HLH disease mimics.
A variety of PIDs have been reported to present with HLH 169, 170: severe combined immunodeficiency (SCID), Omenns syndrome, severe DiGeorge syndrome, Wiskott-Aldrich syndrome, chronic granulomatous disease (CGD), X-linked agammaglobulinemia, and autoimmune lymphoproliferative syndrome. Patients with primary immune deficiency and HLH often have unresolved, severe infections. Patients with SCID most often have viral infections, while those with chronic granulomatous disease present with bacterial infections. Thus, the presence of HLH associated with unusual or unusually severe infection should suggest undiagnosed immune deficiency. For patients with severe combined immunodeficiency (SCID) and infection, immunosuppressive therapy is generally not helpful and this condition should be considered to be a mimic of HLH disease 4. HLH in patients with chronic granulomatous disease is less clear, though typical treatment for HLH beyond corticosteroids is usually not indicated. Therefore, the constellation of HLH symptoms in the context of primary immune deficiency should usually be considered a mimic of HLH disease, though some patients may require immunosuppressive therapy 4.
Hemophagocytic lymphohistiocytosis and metabolic disorders
Among the cases reviewed in the present study, several patients were diagnosed with secondary hemophagocytic lymphohistiocytosis or HLH syndrome in the context of an inherited condition associated with metabolic diseases, such as Wolman’s disease 171, galactosemia, Gaucher disease 172, Niemann-Pick disease, methylmalonic acidemia and propionic acidemia 173. Long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency 174, type 1 glycogen storage disease 175, lysosomal acid lipase deficiency 176, mucopolysaccharidosis-plus syndrome 177.
These patients fulfilled the HLH 2004 criteria, but it is possible that some HLH features, such as splenomegaly and cytopenias, were caused by the metabolic disease itself rather than by immune hyperactivation 1. On the other hand, it is possible that metabolites accumulation activated macrophages and triggered a proper secondary hemophagocytic lymphohistiocytosis. The link between metabolic disorders and HLH needs to be clarified, but an extensive screening for underlying metabolic diseases should be performed in patients presenting with HLH 171, 172.
Hemophagocytic lymphohistiocytosis of unknown or uncertain origin
HLH has recently been described in the context of febrile infection related epilepsy syndrome (FIRES) 178, an epileptic encephalopathy characterized by refractory status epilepticus following unspecific febrile illnesses. The pathophysiology of febrile infection related epilepsy syndrome (FIRES) has not been completely understood, but it likely to depend, once more, on dysfunctional activation of the innate immune system 178. Moreover, though rare, this association stresses the need of screening for patients with HLH presenting with severe CNS symptoms.
A curious case of HLH following spider bite (Loxosceles reclusa) has been reported from the US 179.
Drug reaction with eosinophilia and systemic symptoms induced by vancomycin, carbamazepine and levetiracetam has been described as an occasional trigger of HLH 55.
Secondary hemophagocytic lymphohistiocytosis pathophysiology
The cellular and molecular mechanisms underlying HLH are incompletely understood. HLH is not one disease, but rather a distinct state of sustained immune system activation that may be engaged through a number of different pathways, depending on an individual’s predispositions and environmental triggers. Three major observations can be distilled from decades of clinical observation, genetic analysis, and basic science research 4:
- Disease in HLH is driven primarily by the aberrant immune response, not underlying triggers;
- Immune responses in HLH do not appear to target self-antigens, as seen in autoimmune diseases; and
- Uncontrolled activation of T cells (especially CD8+ cytotoxic T cells), not macrophages, is the root abnormality in most forms of HLH.
Genetic susceptibility to HLH clusters around genes whose products are involved in cell-mediated cytotoxicity and lymphocyte activation/survival. Immunosuppression and chronic inflammation also predispose to HLH; in these settings, HLH is often triggered by infection, most often viruses, but also other intracellular pathogens. Certain hematological malignancies and cancer-related therapies may trigger and drive HLH in the absence of identifiable genetic or infectious factors. Clinical and laboratory features of HLH reflect tissue infiltration by activated immune cells (especially T cells, macrophages, and neutrophils) and the local and systemic effects of inflammatory cytokines such as IFN-γ, tumor necrosis factor (TNF)-α, IL-1β, IL-6, IL-10, and IL-18 180, 181. Perpetuation of HLH immunopathology appears to involve feed-forward amplification loops whereby activated CD8+ T cells and macrophages stimulate each other in a so-called “cytokine storm.”
The pathogenesis of HLH has mostly been studied in familial hemophagocytic lymphohistiocytosis (FHL). An inherited defect in the perforin/granzyme pathway or in the fusion of cytotoxic lytic granules with the surface of natural killer (NK) cells causes, in the presence of an external trigger, an over-response by cytotoxic CD8+ T lymphocytes 1. Viral infections are the most common triggers 182.
Cytotoxic CD8+ T cells produce large amounts of interferon gamma (IFN-γ), which in turn activates macrophages 1. Overstimulated macrophages release large amounts of inflammatory cytokines, such as interleukin-1 beta (IL-1β), interleukin-6 (IL-6), interleukin-12 (IL-12), interleukin-18 (IL-18) and tumor necrosis factor alpha (TNFα). There is also an increased production of interleukin-10 (IL-10), with inhibitory activity, but not sufficient to limit the phenomenon 1. IL-12 and IL-18 produced by macrophages in turn stimulate CD8+ T cells, amplifying the inflammatory response 6. The resulting tissue damage causes a release of interleukin-33 (IL-33) and IL-1β, which further activates the macrophages. Activated macrophages engulf blood cells and produce large amounts of ferritin. The ‘cytokine storm’ causes all the clinical manifestations of HLH, from endothelial damage to coagulopathy and multi-organ failure 6, 14, 183, 184, 185. The central role of interferon gamma (IFN-γ) in the pathogenesis of familial hemophagocytic lymphohistiocytosis has been demonstrated in a perforin-deficient mouse model 186.
A similar pathogenesis of HLH can also be observed in patients with primary immunodeficiencies involving granule trafficking or exocytosis, such as Hermansky-Pudlak syndrome type 2, Griscelli syndrome type 2 and Chediak-Higashi syndrome 2, all with reduced cytotoxic T lymphocytes (CTL) cytotoxicity 187, 188. Advances in genetic diagnosis suggest that cell killing by cytotoxic T lymphocytes (CTLs) and NK cells can be affected from mildly to severely, thus explaining the different HLH phenotypes as a continuum 189. The known mutations in genes related to granule-mediated killing account for familial hemophagocytic lymphohistiocytosis (FHL) and primary forms in general 13. Minor alterations of the cytotoxic T lymphocytes (CTL) and NK cells activity, together with an external trigger, account for secondary HLH, severe sepsis, multi-organ failure and HLH in the context of rheumatologic diseases 189.
In all cases, a cytokine storm causing a devastating inflammation is the primal agent of the multiorgan failure, regardless of the underlying defect 190, 189, 191, 192, 193, 143 and HLH should be considered a clinical syndrome of hyperinflammation with different phenotypes 194. Moreover, this finding likely explains the numerous similarities and overlaps between HLH and other systemic inflammatory syndromes, such as septic shock, cytokine release syndrome following viral infections 143 and acute liver failure 195.
The case of malignancy-associated HLH includes two completely heterogeneous mechanisms. Firstly, when HLH is a manifestation of the underlying malignancy, particularly in the case of lymphomas, the overproduction of inflammatory cytokines such as INF-γ and TNF-α is likely caused by neoplastic cells 6, 128. Secondly, when HLH develops as the consequence of chemotherapy, the process is more likely to be caused by the association of drug-induced cytotoxic T lymphocytes (CTL) suppression and infection 196.
Figure 5. Hemophagocytic lymphohistiocytosis pathophysiology
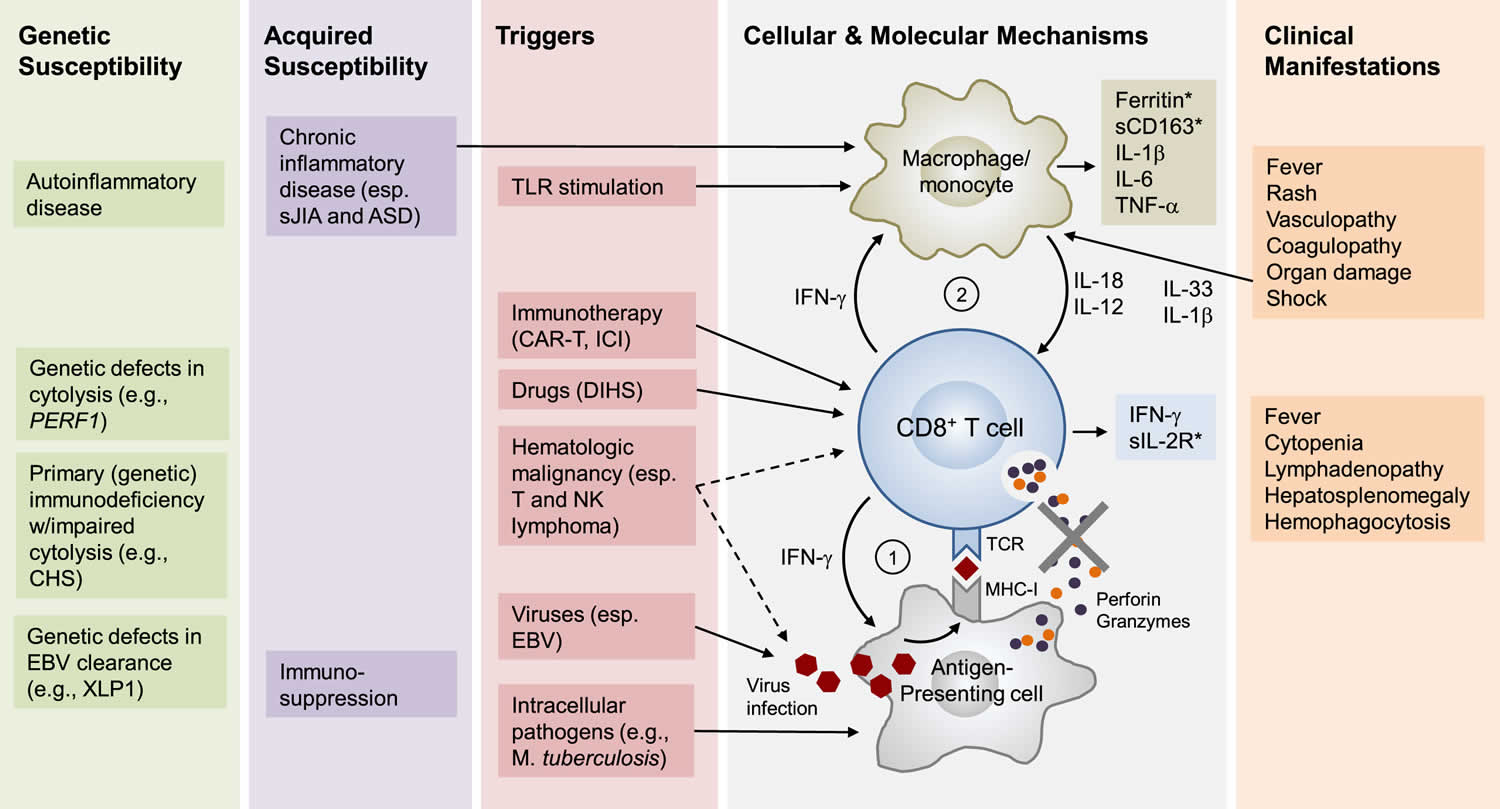
Footnotes: HLH is not one disease but rather a highly distinct state of immunological hyperactivation that has many different causes. A sustained and uncontrolled activation of CD8+ T cells is a core immunological feature of HLH; and in most forms of HLH, this activation is sustained by amplification loops involving antigen-presenting cells (APCs) and macrophages/monocytes. Depending on the individual, the development of HLH involves genetic and/or acquired risk factors. In familial HLH (F-HLH), genetic defects in cytolysis are the primary determinants of risk. In HLH associated with systemic juvenile idiopathic arthritis (sJIA), risk is determined by genetic and acquired factors, particularly chronic inflammation. In adults, HLH most often occurs in the settings of untreated hematological malignancy, chronic rheumatic disease, or immunosuppression. In many patients with HLH, a proximate inciting event or agent (trigger) is identified, most often an infection (particularly viral), occult hematological malignancy (particularly T or NK lymphoma), recent/ongoing immune-activating therapy (e.g., CAR-T therapy), or certain drugs associated with drug-induced hypersensitivity syndromes (DIHS). In F-HLH and certain forms of HLH associated with primary EBV infection, genetic defects in the killing of virally infected cells (e.g., PERF1 deficiency) results in sustained antigen presentation to, and activation of, CD8+ T cells, which is amplified by IFN-g release and subsequent MHC-I upregulation (Loop #1). Sustained IFN-g release from activated CD8+ T cells is believed to drive the activation of tissue macrophages/monocytes, which results in the release of inflammatory mediators such as IL-1b, IL-6, IL-18, and IL-12. IL-18 and IL-12 may serve to enhance IFN-g production by CD8+ T cells (Loop #2). Macrophages/monocytes are further activated by chronic inflammation, tissue injury (through Il-1b and IL-33), and TLR stimulation from infection. In malignancy-associated HLH, transformed cells may drive HLH through autonomous cytokine release and/or sustained presentation of EBV antigen. HLH after CAR-T therapy likely reflects exuberant activation of therapeutic CD8+ CAR-T cells. In HLH associated with DIHS, drug-MHC-I complexes may drive CD8+ T cell hyperactivation. Important clinical and laboratory abnormalities appear to be driven by macrophage/monocyte- and T-cell-derived molecules, several of which serve as important disease biomarkers (∗). Abbreviations: ASD, adult onset Stills disease; CAR-T, chimeric antigen receptor T cell; CHS, Chédiak-Higashi syndrome; DIHS, drug-induced hypersensitivity syndrome; EBV, Epstein-Barr virus; ICI, immune checkpoint inhibitor; MHC-I, major histocompatibility complex I; PERF-1, perforin-1; sIL-2 R, soluble Il-2 receptor; sJIA, systemic juvenile idiopathic arthritis; TCR, T cell antigen receptor; TLR, toll-like receptor; XLP1, and X-linked lymphoproliferative disorder type 1.
[Source 197 ]Secondary hemophagocytic lymphohistiocytosis signs and symptoms
Secondary HLH may occur at any age, and the first clinical symptoms are usually associated with an infectious episode, rheumatic condition, or cancer 31. People with hemophagocytic lymphohistiocytosis (HLH) usually develop symptoms within the first months or years of life. Hemophagocytic lymphohistiocytosis most often affects infants from birth to 18 months, but can affect individuals of any age from childhood to adulthood 198.
The specific symptoms that develop can also vary greatly, although HLH often causes multiorgan involvement. Secondary HLH symptoms may include fevers, a rash, abnormally large liver (hepatomegaly) and abnormally large spleen (splenomegaly), decreased number of blood cells (cytopenia), and neurological abnormalities such as seizures, changes in mental status and irritability, paralysis (palsy) of certain cranial nerves, and problems coordinating voluntary movements (ataxia) 199, 187, 188. Fevers of unknown origin may be prolonged and persistent, often failing to respond to antibiotics 200. Sometimes, the lymph nodes are also abnormally large (lymphadenopathy). Lymph nodes are part of your lymphatic system, a circulatory network of vessels, ducts, and nodes that filter and distribute certain protein-rich (lymph) and blood cells throughout the body. Lymph nodes are small structures, found in groups throughout the body, that help to filter or drain out harmful substances from the body.
These initial sign and symptoms are described as nonspecific. This means that these signs and symptoms are common to many other different disorders or conditions, which can make getting a correct diagnosis difficult.
Affected individuals may also have low levels circulating red blood cells (anemia) and low levels of circulating platelets (thrombocytopenia). Red blood cells deliver oxygen to the body and platelets allow the body to form clots to stop bleeding. Individuals with anemia may experience tiredness, increased need for sleep, weakness, lightheadedness, dizziness, irritability, headaches, pale skin color, difficulty breathing (dyspnea), and cardiac symptoms. Individuals with thrombocytopenia (low levels of circulating platelets) are more susceptible to excessive bruising following minimal injury and to spontaneous bleeding from the mucous membranes, especially those of the gums and nose.
Some affected individuals may develop neurological symptoms including seizures, changes in mental status and irritability, paralysis (palsy) of certain cranial nerves, and problems coordinating voluntary movements (ataxia). Affected individuals are at risk of developing posterior reversible encephalopathy syndrome, which causes a rapid onset of headaches, altered consciousness, seizures, and disturbances in vision. Neurological problems are most common with familial hemophagocytic lymphohistiocytosis.
Additional symptoms can occur depending upon the specific organ system involved in an individual. These symptoms can include significant problems breathing (lung dysfunction), severe low blood pressure (hypotension), liver inflammation (hepatitis), kidney dysfunction, yellowing of the skin and whites of the eyes (jaundice), swelling due to fluid accumulation (edema), abdominal swelling due to fluid accumulation (ascites), and a variety of skin problems including widespread, reddening of the skin because of inflammation (erythroderma), rashes, blood spots (purpura), and tiny spots on the skin due to bleeding under the skin (petechiae).
Secondary hemophagocytic lymphohistiocytosis diagnosis
Diagnosing hemophagocytic lymphohistiocytosis can be challenging because many of its initial symptoms mimic other common conditions. Symptoms such as persistent fevers, respiratory issues, rash, anemia, seizures and enlarged liver, spleen or lymph nodes may be cause other conditions. Because the symptoms of hemophagocytic lymphohistiocytosis are nonspecific and may overlap with other inflammatory or hematopoietic diseases, affected individuals may often have been through a prolonged illness and be hospitalized before a diagnosis is made.
Figure 6 displays a hemophagocytic lymphohistiocytosis diagnostic algorithm in newly presenting patients.
Hemophagocytic lymphohistiocytosis diagnosis is based upon identification of characteristic symptoms, a detailed patient history, a thorough clinical evaluation and a variety of specialized tests. Guidelines have been published that detail the criteria necessary for a diagnosis of hemophagocytic lymphohistiocytosis (see Table 1).
HLH is diagnosed when an individual meets established HLH-2004 diagnostic criteria 201. HLH-2004 criteria for diagnosis of HLH requires either 1 or 2.
- The presence of a known HLH-causing genetic mutation, OR
- Has five or more of the clinical or laboratory findings listed below:
- High and often prolonged periods of fever (more than 7 days);
- Rash, irritability and/or seizures;
- An abnormally large spleen (splenomegaly);
- Low blood cells (cytopenias): red blood cells (hemoglobin less than 9 g/dL in adults, hemoglobin less than 10 g/dL in infants <4 wk), white blood cells (neutrophils less than 1 × 109/L), or platelets (platelets count less than 100 × 109/L);
- Abnormally high levels of a type of fat called a triglyceride in the blood (hypertriglyceridemia) triglyceride ≥ 3.0 mmol/L (≥ 265 mg/dl);
- Abnormally low levels of fibrinogen (a specific blood clotting protein) (hypofibrinogenemia) serum fibrinogen < 150 mg/dL;
- Destruction of blood cells by macrophages (hemophagocytosis) in the bone marrow, spleen, or lymph nodes and/or cerebrospinal fluid with no evidence of malignancy;
- Low or absent natural killer (NK) cell activity;
- Abnormal high levels of ferritin (a protein that binds to iron) in the blood (ferritin ≥ 500mcg/L); and
- Elevated soluble interleukin-2 receptor alpha chain (sCD25 ≥ 2400 U/mL), a specialized protein that builds up in the blood when the immune system is stimulated.
- Reduced expression of proteins such as perforin, signaling lymphocytic activation molecule-associated protein (SAP), X-linked inhibitor of apoptosis protein (XIAP), and/or depressed cell functions
On occasion, HLH may be strongly considered, and HLH-directed therapy may be initiated, even though the 5 HLH-2004 diagnostic criteria are not fulfilled 202.
If hemophagocytic activity is not proven at the time of presentation, further search for hemophagocytic activity is encouraged. If the bone marrow specimen is not conclusive, material may be obtained from other organs. Serial marrow aspirates over time may also be helpful. The following findings may provide strong supportive evidence for the diagnosis: spinal fluid pleocytosis (mononuclear cells) and/or elevated spinal fluid protein and histological picture in the liver resembling chronic persistent hepatitis (biopsy). Other abnormal clinical and laboratory findings consistent with the diagnosis are cerebromeningeal symptoms, lymph node enlargement, jaundice, edema, skin rash, hepatic enzyme abnormalities, hypoproteinemia, hyponatremia, and elevated very low-density lipoprotein (VLDL↑)/low high-density lipoprotein (HDL↓).
Figure 6. Hemophagocytic lymphohistiocytosis diagnostic algorithm
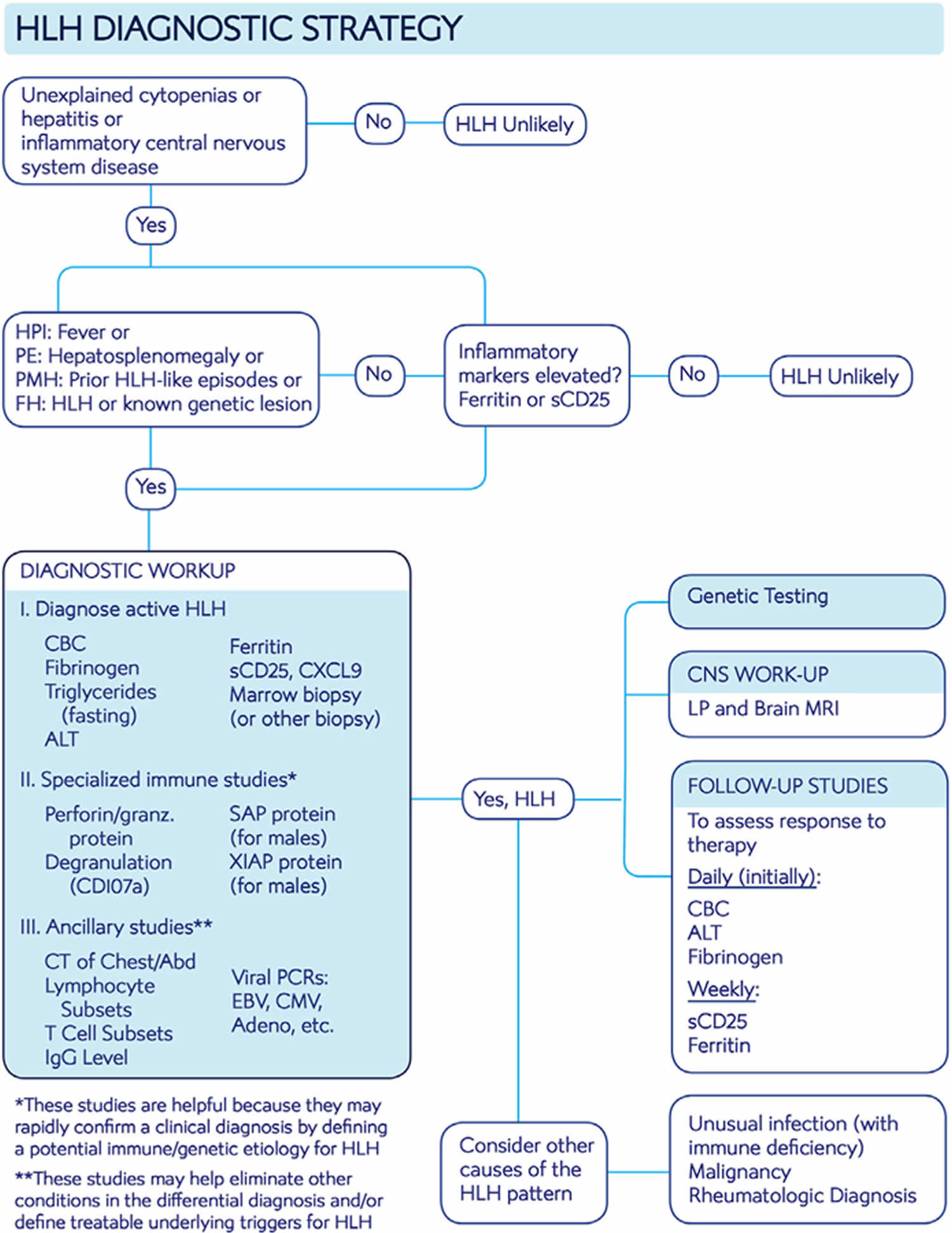
Table 1. Secondary HLH diagnostic criteria diagnostic criteria
| HLH 2004 criteria | Comment |
|---|---|
| A. Molecular diagnosis consistent with HLH: Pathologic mutations of PRF1, UNC13D, STXBP2, Rab27a, STX11, SH2D1A, or XIAP or | In a patient with known genetic defects, treatment before full development of HLH may be appropriate, but genetic studies usually just help to define HLH recurrence risk, not the presence of an active disease state. |
| B. Five of the eight criteria listed below are fulfilled | |
| 1. Fever ≥38.3°C | Nearly universal in untreated HLH. |
| 2. Splenomegaly | While splenomegaly and hepatomegaly are very common in HLH, adenopathy is not. |
| 3. Cytopenias (affecting at least two of three lineages in the peripheral blood): Hemoglobin <9 g/dL (in infants <4 weeks: hemoglobin <10 g/dL) Platelets <100 × 10³/mL Neutrophils < 1 × 10³/mL | Cytopenias are ubiquitous in HLH. Lack of cytopenias should make one doubt a diagnosis of HLH, except in the special case of isolated, CNS-only disease. |
| 4. Hypertriglyceridemia (>265 mg/dL) and/or hypofibrinogenemia (<150 mg/dL) | Low fibrinogen in the context of inflammation is paradoxical and one of the more distinctive features of HLH. |
| 5. Hemophagocytosis in bone marrow or spleen or lymph nodes or liver | Not specific to HLH, or essential for the diagnosis, but helpful as a disease marker. Of note, it is often not evident early after disease onset. |
| 6. Low or absent NK-cell activity | More modern and robust assays measuring perforin levels and its degranulation should replace this assay for reliable diagnosis of HLH. This assay is not specific for primary HLH. |
| 7. Ferritin >500 ng/mL | Most patients have much higher levels than this threshold suggests. |
| 8. Elevated Soluble CD25 (soluble IL-2 receptor alpha) | As HLH is a T-cell driven disease, this assay is extremely informative for diagnosis and response to therapy. See Clinical Testing and Workup for patients with suspected HLH disease for more information. |
Clinical testing and workup for patients with suspected HLH disease
The initial workup is two-pronged and aims (a) to establish the diagnosis of HLH promptly and (b) to identify mimickers of HLH disease (if present), or potentially treatable underlying HLH disease triggers.
Common laboratory studies
- Complete blood count (CBC), hepatic panel, fibrinogen, triglycerides. Essential to define typical features of HLH. Though not part of diagnostic criteria, hepatitis is extremely common in HLH.
- Ferritin. Usually (but not always) very elevated
- Search for triggering infection with cultures, viral PCRs: EBV, CMV, adenovirus
Specialized laboratory studies
- Markers of immune activation: soluble interleukin-2 receptor alpha chain (sCD25), granzyme B expression and CXCL9. Essentially always elevated in untreated HLH; very sensitive, but not specific. sCD25, and perhaps CXCL9, are also useful for monitoring response to therapy
- Interlukin-18 (IL-18) level. Elevated in HLH associated with inflammasome-opathies (XIAP, NLRC4, systemic onset juvenile arthritis)
- Measurement of proteins affected in familial HLH: perforin, SAP, XIAP. Helpful to quickly confirm a suspected familial HLH diagnosis
- Functional studies: lymphocyte degranulation (CD107a mobilization), NK cell function. May help to fulfill diagnostic criteria. Degranulation is the preferred functional assay over NK cell function 204.
- Lymphocyte subsets, T-cell subsets, immune globulin levels. Screening studies for primary immune deficiency
- Genetic testing (multigene panel or whole exome). Essential for defining HLH recurrence risk
Imaging studies (should be routinely performed to help rule out malignancy, or unusual infections if trigger is unknown)
- Body cavity CT’s (chest/abdomen/pelvis)
- Consider 18F-fluorodeoxyglucose (FDG) positron emission tomography-computed tomography (PET-CT) if suspicion of lymphoma
- MRI of brain. Brain MRI complements lumbar puncture for central nervous system (CNS) assessment
Tissue sampling (essential to rule out malignancy, identify hemophagocytosis, and identify central nervous system (CNS) involvement)
- Bone marrow biopsy
- Lumbar puncture
- Other tissue biopsies as appropriate: liver, lymph node, masses
Physicians may order blood tests to take a complete blood cell count, which will measure the levels of red cells, white cells and platelets. Blood tests can also reveal abnormally high ferritin levels, or abnormal high levels of triglycerides. Physicians may also use blood tests to look for signs of infection in the blood and conduct tests to determine how well the blood clots (coagulation studies). Physicians may also order tests that can assess the health and function of the liver.
Sometimes, a bone marrow biopsy (the surgical removal and microscopic examination of a tissue sample) may be taken and studied for signs of hemophagocytosis, signs of infection or infectious organisms, and the accumulation of macrophages.
Molecular genetic testing can confirm a diagnosis of hemophagocytic lymphohistiocytosis in certain people. Molecular genetic testing can detect mutations in one of the four specific genes known to cause familial forms of this disorder, but is available only as a diagnostic service at specialized laboratories.
It should be emphasized that since T-cell activation is central to HLH pathogenesis, elevated soluble IL2 receptor alpha chain (sCD25) should always be observed in untreated HLH 127, 205, 206. If sCD25 is not elevated, then one should doubt a diagnosis of HLH 4. Similarly, though not as well established because HLH appears to be largely driven by interferon gamma (IFN-γ), elevations of CXCL9 (a sensitive indicator of IFN-γ bioactivity) should be seen in untreated cases of HLH disease 114, 207, 208. Additionally, elevated expression of granzyme B in NK cells has been shown to be similarly ubiquitous in HLH; normal expression levels would suggest that HLH is not the correct diagnosis 209. Ferritin levels above 10,000 ng/mL appear to be relatively specific for HLH but are not very sensitive 210. While specialized immunologic testing may facilitate diagnosis, if a diagnosis can be made without them then treatment should not be delayed pending these results. Likewise, treatment should not be delayed for assessment of central nervous system (CNS) involvement, though this should always be conducted (once a lumbar puncture may be safely performed).
Practical examples of when to not diagnose HLH disease
Certain clinical features may suggest mimics of HLH disease 4:
- Despite the common presence of splenomegaly, prominent or abnormal lymphadenopathy is not typically seen in HLH. This finding strongly suggests lymphoma, Castleman disease, or unusual infection such as HIV, histoplasmosis or mycobacterial disease 135. Similarly, leukocytosis is not typical of HLH (except in R-HLH/MAS) and should prompt a search for an alternative diagnosis.
- An extremely elevated sCD25 (>10- to 20-fold above normal) in a noninfantile patient suggests undiagnosed lymphoma, especially when ferritin is not similarly elevated 135, 211.
- The early occurrence of isolated and aggressive “CNS relapse” during treatment of systemic HLH may suggest an undiagnosed malignant disorder relapsing in the CNS.
- A normal or only modestly elevated sCD25, despite extremely elevated ferritin, suggests disseminated infection in the context of primary immune deficiency, especially in an infant 212.
Other diagnostic tools
Other features supporting an HLH diagnosis that are not part of the HLH-2004 diagnostic criteria include hyperbilirubinemia, hepatomegaly, transaminitis (present in the vast majority of patients with HLH), and elevated lactate dehydrogenase and d-dimer levels, with the latter usually elevated even when international normalized ratio, partial thromboplastin time, and fibrinogen are normal 213. These findings may help to discriminate HLH from septic shock and conditions such as autoimmune hemolytic anemia, and they are also useful in assessing response to therapy.
H Score
The HLH-probability calculator (HScore), with graded clinical and laboratory parameters, is a Web-based online calculator developed retrospectively in adult patients that may be a helpful diagnostic tool (Table 3) 214. The pattern of inflammatory cytokines (elevated levels of interferon-γ [IFN-γ] and IL-10, with only modestly elevated IL-6 levels) has high diagnostic accuracy for secondary hemophagocytic lymphohistiocytosis and may be a useful approach to differentiate HLH from infection and to monitor patients; however, the utility of this pattern of changes needs to be verified in children and adults outside of China 215, 216, 217.
Table 2. Parameters and points in the HScore
| Parameter | No. of points (criteria for scoring) |
|---|---|
| Known underlying immunosuppression* | 0 (no) or 18 (yes) |
| Temperature (°C) | 0 (<38.4), 33 (38.4–39.4), or 49 (>39.4) |
| Organomegaly | 0 (no), 23 (hepatomegaly or splenomegaly), or 38 (hepatomegaly and splenomegaly) |
| No. of cytopenias† | 0 (1 lineage), 24 (2 lineages), or 34 (3 lineages) |
| Ferritin (μg/L) | 0 (<2000), 35 (2000-6000), or 50 (>6000) |
| Triglyceride (mmol/L) | 0 (<1.5), 44 (1.5-4), or 64 (>4) |
| Fibrinogen (g/L) | 0 (>2.5) or 30 (≤2.5) |
| Aspartate aminotransferase (U/L) | 0 (<30) or 19 (≥30) |
| Hemophagocytosis on bone marrow aspirate | 0 (no) or 35 (yes) |
Footnote:
* HIV positive or receiving long-term immunosuppressive therapy (ie, glucocorticoids, cyclosporine A, azathioprine).
† Defined as a hemoglobin level of 9.2 g/L and/or a leukocyte count ≤5 × 109/L and/or a platelet count ≤110 × 109/L.
Secondary hemophagocytic lymphohistiocytosis treatment
The treatment of hemophagocytic lymphohistiocytosis is directed toward the specific symptoms that are apparent in each individual. Treatment may require the coordinated efforts of a team of specialists. Pediatricians, specialists in diagnosing and treating blood disorders (hematologists), specialists in diagnosing and treating cancer (oncologists), specialists in diagnosing and treating immune system diseases (immunologists), geneticists (for familial forms), social workers, and other healthcare professionals may need to systematically and comprehensively plan treatment. Psychosocial support for the entire family is essential as well. Genetic counseling may be of benefit for affected individuals and their families. A useful algorithm for medical managements of adults with HLH is presented in Figure 7 below.
Treatment for secondary HLH is determined more by the severity of the disorder and its symptoms, rather than its classification. The aim of hemophagocytic lymphohistiocytosis treatment is to suppress the life-threatening inflammation that leads to organ damage 185. Specific therapeutic procedures and interventions may vary, depending upon numerous factors, such as the underlying cause; the presence or absence of certain symptoms; the overall severity of the symptoms and the disorder; an individual’s age and general health; and/or other elements. Decisions concerning the use of particular drug regimens and/or other treatments should be made by physicians and other members of the health care team in careful consultation with the patient based upon the specifics of his or her case; a thorough discussion of the potential benefits and risks, including possible side effects and long-term effects; patient preference; and other appropriate factors.
Affected individuals whose overall health is strong enough may undergo treatment for the underlying condition such as medications to treat an underlying infection, or appropriate treatment for autoimmune disorders or cancer. Treating the underlying condition may remove the “trigger” that has led to the abnormal immune system response.
The first goal is to induce disease remission by controlling the hyper-activated T cells and the cytokine storm they generate 13, 218. The second step, instead, may differ according to the underlying condition: in familial hemophagocytic lymphohistiocytosis and HLH cases, due to primary immunodeficiency, allogeneic hematopoietic stem cell transplantation (allo-HSCT) is the only curative treatment that is currently known, while there is no consensus on secondary hemophagocytic lymphohistiocytosis best treatment 1.
For children with secondary hemophagocytic lymphohistiocytosis, the aim is to identify and treat the underlying cause of HLH such as an infection, cancer or rheumatologic disease. In many cases, by treating the underlying cause, HLH will go into remission. In some cases, however, it will be necessary to use steroids and/or chemotherapy to treat the condition – similar to treatment for familial hemophagocytic lymphohistiocytosis.
Figure 7. Hemophagocytic lymphohistiocytosis treatment algorithm
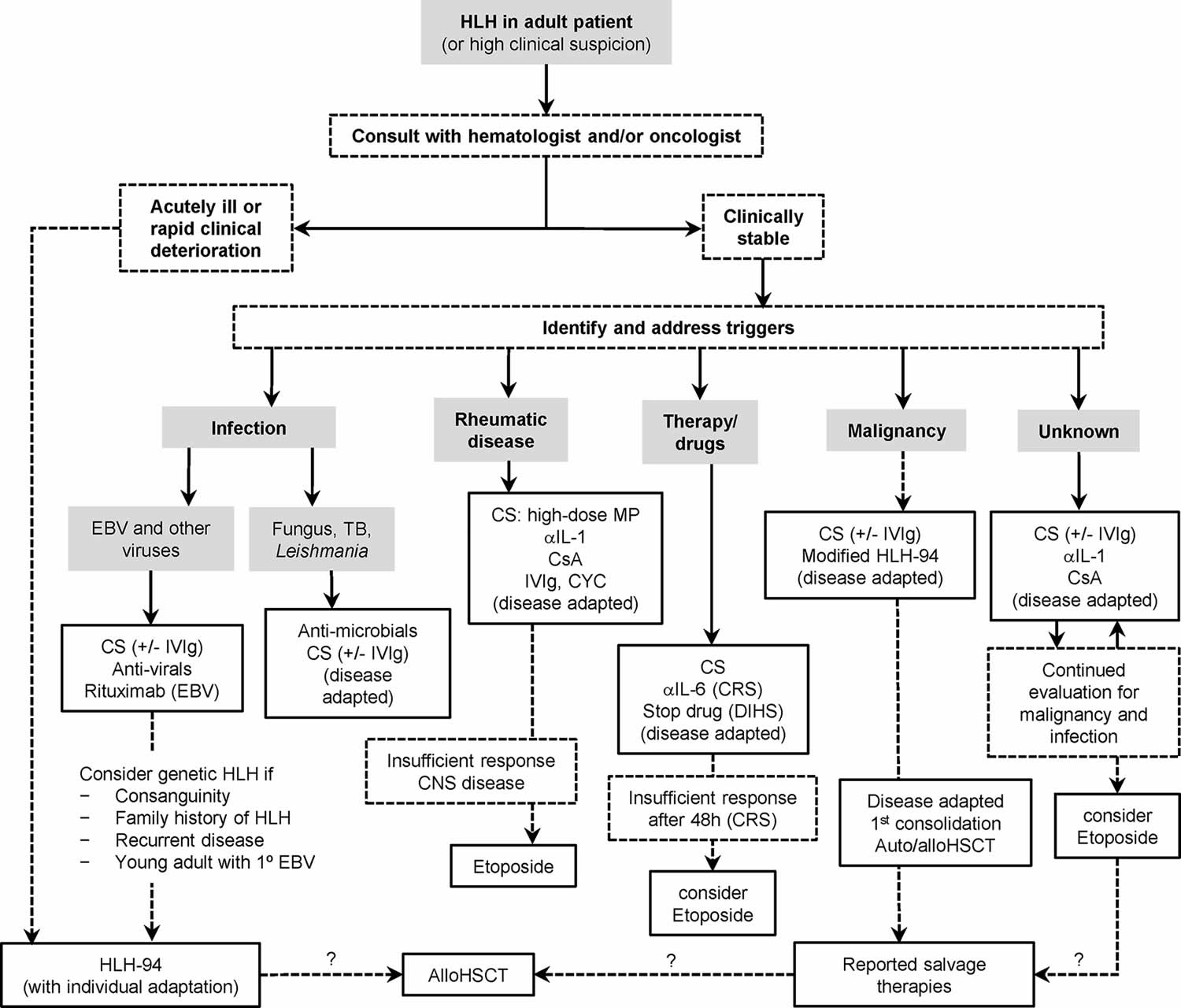
Footnotes: Management algorithm for adults with HLH or suspected HLH. The HLH-94 treatment protocol is consensus therapy for familial hemophagocytic lymphohistiocytosis (F-HLH). Medical treatment of adults with HLH is not standardized and should be adapted to severity of illness and underlying triggers (infection, rheumatic disease, drugs, and malignancy). Hematologists/oncologists should be involved early in care, because the treatment of severe, refractory, or relapsed disease involves chemotherapy (dashed lines) and case-by-case consideration of allogenic stem cell transplant or highly experimental therapies (question marks). Shown here are frequently encountered clinical scenarios (shaded boxes), major assessment nodes (dashed line boxes), and treatment options (solid line boxes).
Abbreviations: aIL-1, anti-IL-1 (e.g., anakinra); aIL-6, anti-IL-6 (e.g., tocilizumab); Allo-HSCT = allogeneic hematopoietic stem cell transplant; Auto-HSCT = autologous hematopoietic stem cell transplant; CNS = central nervous system; CRS = cytokine release syndrome; CS = corticosteroids; CsA = cyclosporine A; CYC = cyclophosphamide; DIHS = drug-induced hypersensitivity syndrome; EBV = Epstein-Barr virus; IVIg = intravenous immunoglobulin; MP = methylprednisolone; TB = Mycobacterium tuberculosis.
[Source 197 ]Figure 8. HLH-94 protocol
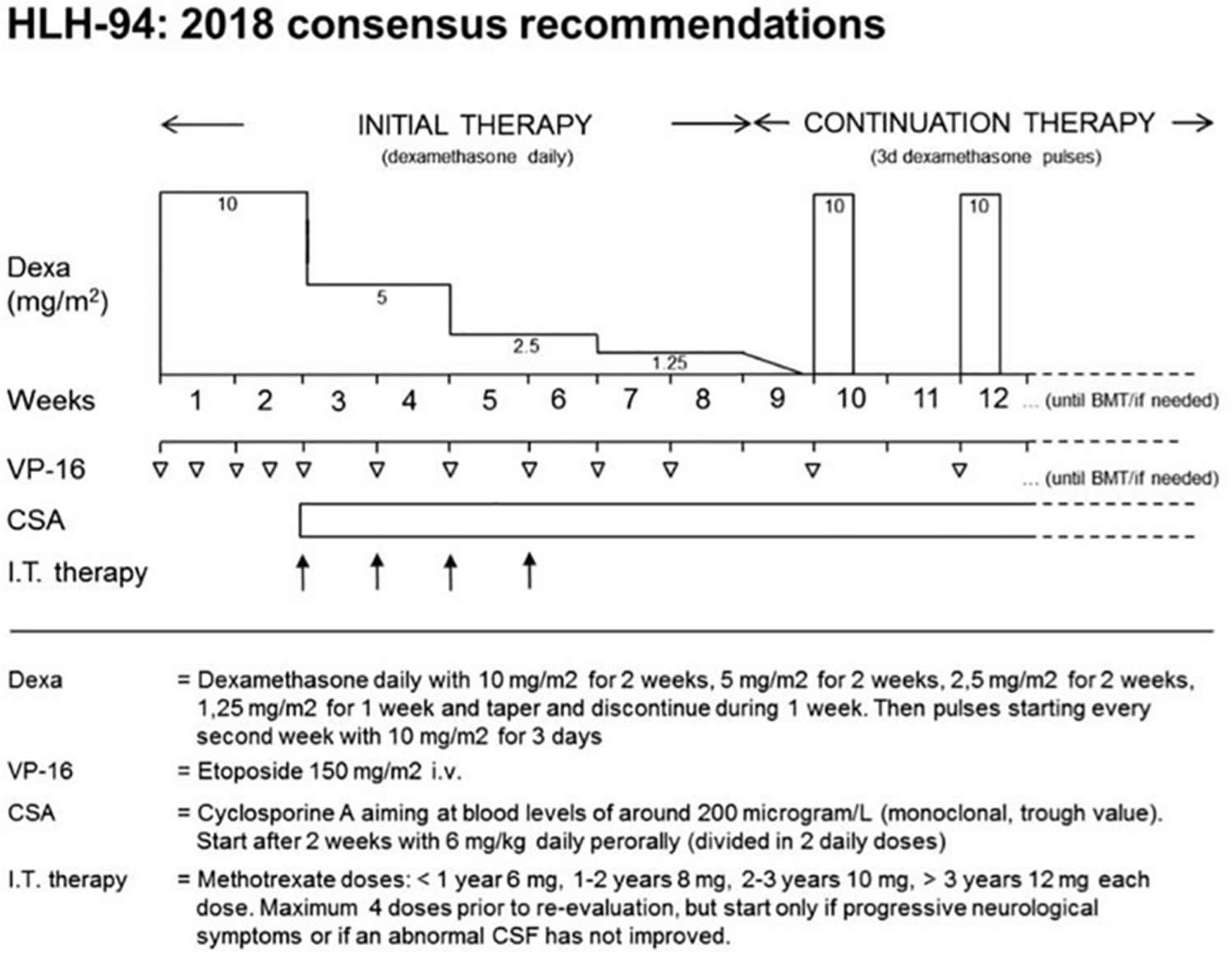
Footnotes: The HLH-94 protocol is based on immunochemotherapy including dexamethasone, etoposide, and cyclosporine A (CSA). After an intensive phase of 2 weeks with high doses of dexamethasone and twice weekly administration of etoposide, dexamethasone is tapered until week nine. Cyclosporine A (CSA) is used from week three onward to prevent reactivation. Intrathecal therapy with methotrexate is recommended in patients with central nervous system (CNS) involvement. Immunological testing and genetic confirmation of the underlying genetic disease is required in all patients and should provide the basis for hematopoietic stem cell transplantation (HSCT) within 8 weeks.
[Source 219 ]HLH-94 protocol
2018 consensus statements by the HLH Steering Committee of the Histiocyte Society recommending the use of HLH-94. The HLH-94 protocol is based on immuno-chemotherapy including dexamethasone, etoposide and cyclosporine A to achieve remission of the hyperinflammatory state and to maintain remission until hematopoietic stem cell transplantation (HSCT) can be performed 220. Functionally, all agents target lymphocytes, macrophages and antigen presenting cells. The cytostatic agent etoposide induces cell death mainly in activated T cells 221, but also in macrophages and dendritic cells. The use of the calcineurin inhibitor cyclosporine A leads to an inhibition of the transcription factor NFAT (nuclear factor of activated T-cells) and thus to a reduced activation and proliferation of T cells. Steroids slow down inflammation by reducing cytokine secretion, but in addition, they have a moderate cytotoxic effect on activated T cells. It is recommended to treat patients with central nervous system (CNS) involvement also with intrathecal methotrexate 222, although there is no clear evidence of benefit. The protocol has a 2-week intensive phase with dexamethasone and twice weekly administration of etoposide, followed by 6 weeks of weekly etoposide and steroid tapering. In this second phase, cyclosporine A is used to prevent reactivation 223. Rapid immunological testing followed by genetic confirmation of the underlying genetic disease is required in all patients and should provide the basis for hematopoietic stem cell transplantation (HSCT) within these 8 weeks. In the international multicenter registry-based HLH-2004 study, 5-year probability of survival for children with genetically verified familial HLH treated with this protocol was 59% 224. Dexamethasone/etoposide-based protocols have been successfully used in X-linked lymphoproliferative disorder or patients with TIM3 deficiency. It remains an ultimate choice also in other inborn errors of immunity, but the toxicity and immunosuppression associated with etoposide asks for more targeted therapies in these conditions.
Treatment of infection-associated HLH depends on the underlying infection, since some forms respond to anti-infectious treatment such as Leishmania 225, 107, 108, Ehrlichiosis 101, scrub typhus 70, 71 and other tropical diseases-associated forms, while reaction to some other pathogens displays intermediate features and benefits of standard aggressive immunosuppression.
Several studies 225, 108, 226, 227 have reported complete response of Leishmania-associated HLH to amphotericin B therapy +/- steroids, with an excellent prognosis (3 year overall survival rate 24% in pHLH vs. 100% in visceral leishmaniasis-associated HLH in Iranian cohort of 60 children 107. A Chinese retrospective study 24 reported good results with antiparasitic therapy associated with chemotherapy according to the HLH94 protocol in patients with visceral leishmaniosis-HLH; however, the cohort includes children and adults and it is impossible to discriminate whether treatment intensification depends on age.
Human monocytic ehrlichiosis, can also trigger an HLH form that has proved to respond to antimicrobial therapy alone (doxycycline) 101.
Scrub typhus, dengue fever and tropical infections in general respond to steroids alone or to regimens without cytotoxic drugs in cases with mild to moderate disease activity 70, 71.
The hepatitis-associated HLH cases reviewed in the present study have experienced complete recovery on antiviral therapy and ruxolitinib in the case of a patient with hepatitis B virus infection 76 and after IVIG-infusion in the cases of patients with hepatitis A virus infection 75.
A patient developing HLH after HIV diagnosis has required both antiretroviral therapy and chemotherapy according to HLH94 protocol 77.
The Crimean Congo hemorrhagic fever-associated HLH has been resolved using antiviral therapy combined with IVIG therapy and plasma exchange 72.
The case of EBV-HLH is more complex. EBV-HLH standard treatment is by the HLH94 protocol, and early initiation of etoposide-based regimens improves survival in severe EBV-HLH 46, 57, 228. Chemotherapy can be combined with targeted therapies such as Rituximab 49, 167 or Ruxolitinib 229. Treatment intensification with the L-DEP regimen (PEG-asparaginase in combination with liposomal doxorubicin, etoposide and high-dose methylprednisolone) has been reported as salvage therapy and bridge to allo-HSCT for children with refractory disease 230. Blood purification techniques (plasma exchange and continuous renal replacement therapy) have proved to be safe and effective in addition to standard chemotherapy in severe cases 231. On the other hand, patients presenting with mild-to-moderate disease activity may only require immunomodulating therapies, such as IVIGs, cyclosporine A and steroids 59. A Japanese group 232 described a cohort of 22 patients aged 6 months to 41 years in which >60% recovered after immunotherapy (IVIG, cyclosporine A 6 mg/kg/day, prednisone); they suggest that early immunotherapy may modulate T-cell activation and reduce the chance of unnecessary chemotherapy. Sporadic cases of spontaneous resolution have been reported: An American group 233 reported the cases of two adolescents who fulfilled the HLH2004 diagnostic criteria in the context of an acute EBV infection. In both cases, treatment was planned according to HLH94 protocol, but the patients spontaneously improved before the initiation of therapy. The authors recommend caution in starting aggressive treatment, especially when patients experience mild-to-moderate symptoms. These differences in treatment approaches and outcomes underline the need for evidence-based risk stratification criteria in patients with EBV-HLH.
Cancer associated HLH (M-HLH) treatment
Malignancy-triggered forms should be distinguished from the forms that occur after chemotherapy (Ch-HLH) to optimize the therapeutic approach.
Hemophagocytic lymphohistiocytosis during chemotherapy (Ch-HLH) is often caused by infections related to immunosuppression, and benefits of anti-infectious therapy, steroids and IVIGs.
HLH as a manifestation of underlying malignancy. Cancer associated HLH (M-HLH) is associated with increased mortality and the early detection of malignancy can favorably influence the prognosis. Treatment is not homogeneous, and can address the underlying malignancy first, be focused on HLH or a combination of the two 234, 121, 130, 235, 236, 237. The published series refer to small numbers and suggest an individualized therapeutic approach. Aggressive HLH-directed therapy at the onset of malignancy-associated HLH can delay or complicate anticancer treatment 234, 196.
Rheumatologic HLH (MAS-HLH) treatment
MAS-HLH usually responds to steroid therapy or to anakinra (recombinant human IL-1 receptor antagonist), especially when given in the early course of therapy 238, 239. The present study refers to specific international guidelines for more detailed considerations on MAS-HLH 111, 240, 241.
HLH associated with iatrogenic immune activation treatment
The cytokine release syndrome following CAR-T or Blinatumomab administration usually responds to Tocilizumab, steroids, Anakinra or to a combination of such molecules 143, 151, 155. Whether carHLH should be differentiated from a severe form of cytokine-release syndrome is still an open question, and a more precise definition of both conditions is essential in order to guide therapeutic choices in the most complex cases 148, 242.
Post-transplant HLH requires prompt and aggressive treatment because of the high risk of graft failure, but no international consensus exists on treatment protocol. Reported therapeutic approaches range from corticosteroids, IVIGs and low-dose etoposide 243, 244, 245 to standard HLH-94 protocol with eventual rescue HSCT 158. Ruxolitinib was recently reported as salvage therapy in 2 children, with alternating results 246. Outcome is unclear, usually compromised by graft failure.
Secondary hemophagocytic lymphohistiocytosis prognosis
Secondary hemophagocytic lymphohistiocytosis clinical course may be severe, and the mortality is still significant 247. Secondary hemophagocytic lymphohistiocytosis prognosis widely varies from a 55% survival at 3 years with the standard treatment protocols 23 to 100% survival in specific conditions such as Leishmania-triggered HLH when treated appropriately 24. Cancer associated HLH (M-HLH) has the worst outcomes in secondary hemophagocytic lymphohistiocytosis prognosis 1. In the 1980s, long-term survival in hemophagocytic lymphohistiocytosis was less than 5% 248.
- Benevenuta C, Mussinatto I, Orsi C, Timeus FS. Secondary hemophagocytic lymphohistiocytosis in children (Review). Exp Ther Med. 2023 Jul 17;26(3):423. doi: 10.3892/etm.2023.12122[↩][↩][↩][↩][↩][↩][↩][↩]
- Janka GE. Familial and acquired hemophagocytic lymphohistiocytosis. Annu Rev Med. 2012;63:233-46. doi: 10.1146/annurev-med-041610-134208[↩]
- Bergsten E, Horne A, Aricó M, Astigarraga I, Egeler RM, Filipovich AH, Ishii E, Janka G, Ladisch S, Lehmberg K, McClain KL, Minkov M, Montgomery S, Nanduri V, Rosso D, Henter JI. Confirmed efficacy of etoposide and dexamethasone in HLH treatment: long-term results of the cooperative HLH-2004 study. Blood. 2017 Dec 21;130(25):2728-2738. doi: 10.1182/blood-2017-06-788349[↩]
- Jordan MB, Allen CE, Greenberg J, Henry M, Hermiston ML, Kumar A, Hines M, Eckstein O, Ladisch S, Nichols KE, Rodriguez-Galindo C, Wistinghausen B, McClain KL. Challenges in the diagnosis of hemophagocytic lymphohistiocytosis: Recommendations from the North American Consortium for Histiocytosis (NACHO). Pediatr Blood Cancer. 2019 Nov;66(11):e27929. doi: 10.1002/pbc.27929[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Canna SW, Marsh RA. Pediatric hemophagocytic lymphohistiocytosis. Blood. 2020 Apr 16;135(16):1332-1343. doi: 10.1182/blood.2019000936[↩][↩][↩][↩]
- Griffin G, Shenoi S, Hughes GC. Hemophagocytic lymphohistiocytosis: An update on pathogenesis, diagnosis, and therapy. Best Pract Res Clin Rheumatol. 2020;34(101515) doi: 10.1016/j.berh.2020.101515[↩][↩][↩][↩][↩][↩]
- Janka G, Imashuku S, Elinder G, Schneider M, Henter JI. Infection- and malignancy-associated hemophagocytic syndromes. Secondary hemophagocytic lymphohistiocytosis. Hematol Oncol Clin North Am. 1998 Apr;12(2):435-44. doi: 10.1016/s0889-8588(05)70521-9[↩]
- Imashuku S. Advances in the management of hemophagocytic lymphohistiocytosis. Int J Hematol. 2000 Jul;72(1):1-11.[↩][↩]
- Henter JI, Elinder G, Söder O, Hansson M, Andersson B, Andersson U. Hypercytokinemia in familial hemophagocytic lymphohistiocytosis. Blood. 1991 Dec 1;78(11):2918-22. https://doi.org/10.1182/blood.V78.11.2918.2918[↩]
- Bergsten E, Horne A, Aricó M, Astigarraga I, Egeler RM, Filipovich AH, Ishii E, Janka G, Ladisch S, Lehmberg K, et al. Confirmed efficacy of etoposide and dexamethasone in HLH treatment: Long-term results of the cooperative HLH-2004 study. Blood. 2017;130:2728–2738. doi: 10.1182/blood-2017-06-788349[↩][↩]
- Ishii E, Ohga S, Imashuku S, Yasukawa M, Tsuda H, Miura I, Yamamoto K, Horiuchi H, Takada K, Ohshima K, et al. Nationwide survey of hemophagocytic lymphohistiocytosis in Japan. Int J Hematol. 2007;86:58–65. doi: 10.1532/IJH97.07012[↩]
- Cleves D, Lotero V, Medina D, Perez PM, Patiño JA, Torres-Canchala L, Olaya M. Pediatric hemophagocytic lymphohistiocytosis: A rarely diagnosed entity in a developing country. BMC Pediatr. 2021;21(411) doi: 10.1186/s12887-021-02879-7[↩]
- Henter JI, Horne A, Aricó M, Egeler RM, Filipovich AH, Imashuku S, Ladisch S, McClain K, Webb D, Winiarski J, Janka G. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48:124–131. doi: 10.1002/pbc.21039[↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Sieni E, Cetica V, Piccin A, Gherlinzoni F, Sasso FC, Rabusin M, Attard L, Bosi A, Pende D, Moretta L, Aricò M. Familial hemophagocytic lymphohistiocytosis may present during adulthood: Clinical and genetic features of a small series. PLoS One. 2012;7(e44649) doi: 10.1371/journal.pone.0044649[↩][↩]
- Malinowska I, Machaczka M, Popko K, Siwicka A, Salamonowicz M, Nasiłowska-Adamska B. Hemophagocytic syndrome in children and adults. Arch Immunol Ther Exp (Warsz) 2014;62:385–394. doi: 10.1007/s00005-014-0274-1[↩][↩]
- Benson LA, Li H, Henderson LA, Solomon IH, Soldatos A, Murphy J, Bielekova B, Kennedy AL, Rivkin MJ, Davies KJ, et al. Pediatric CNS-isolated hemophagocytic lymphohistiocytosis. Neurol Neuroimmunol Neuroinflamm. 2019;6(e560) doi: 10.1212/NXI.0000000000000560[↩]
- de Kerguenec C, Hillaire S, Molinié V, Gardin C, Degott C, Erlinger S, Valla D. Hepatic manifestations of hemophagocytic syndrome: A study of 30 cases. Am J Gastroenterol. 2001;96:852–857. doi: 10.1111/j.1572-0241.2001.03632.x[↩]
- Amir AZ, Ling SC, Naqvi A, Weitzman S, Fecteau A, Grant D, Ghanekar A, Cattral M, Nalli N, Cutz E, et al. Liver transplantation for children with acute liver failure associated with secondary hemophagocytic lymphohistiocytosis. Liver Transpl. 2016;22:1245–1253. doi: 10.1002/lt.24485[↩]
- Ramos-Casals M, Brito-Zerón P, López-Guillermo A, Khamashta MA, Bosch X. Adult haemophagocytic syndrome. Lancet. 2014;383:1503–1516. doi: 10.1016/S0140-6736(13)61048-X[↩][↩]
- Janka GE, Lehmberg K. Hemophagocytic syndromes – An update. Blood Rev. 2014;28(4):135–42. doi: 10.1016/j.blre.2014.03.002[↩][↩]
- Macheta M, Will AM, Houghton JB, Wynn RF. Prominent dyserythropoiesis in four cases of haemophagocytic lymphohistiocytosis. J Clin Pathol. 2001;54:961–963. doi: 10.1136/jcp.54.12.961[↩]
- Ehl S, Astigarraga I, von Bahr Greenwood T, Hines M, Horne A, Ishii E, Janka G, Jordan MB, La Rosée P, Lehmberg K, et al. Recommendations for the use of etoposide-based therapy and bone marrow transplantation for the treatment of HLH: Consensus statements by the HLH steering committee of the histiocyte society. J Allergy Clin Immunol Pract. 2018;6:1508–1517. doi: 10.1016/j.jaip.2018.05.031[↩]
- Henter JI, Samuelsson-Horne A, Aricò M, Egeler RM, Elinder G, Filipovich AH, Gadner H, Imashuku S, Komp D, Ladisch S, et al. Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunochemotherapy and bone marrow transplantation. Blood. 2002;100:2367–2373. doi: 10.1182/blood-2002-01-0172[↩][↩]
- Shi Q, Huang M, Li X, Zheng X, Wang F, Zou Y, Wang L, Jia J. Clinical and laboratory characteristics of hemophagocytic lymphohistiocytosis induced by Leishmania infantum infection. PLoS Negl Trop Dis. 2021;15(e0009944) doi: 10.1371/journal.pntd.0009944[↩][↩][↩][↩]
- Hemophagocytic lymphohistiocytosis: Overview and diagnostic procedure. A case induced by an expansion of monoclonal EBV-negative NK cells. Inmunología Vol. 28, Núm 3, Julio-Septiembre 2009: 135-146. https://www.elsevier.es/es-revista-inmunologia-322-pdf-S021396260970037X[↩]
- Rosado FG, Kim AS. Hemophagocytic lymphohistiocytosis: an update on diagnosis and pathogenesis. Am J Clin Pathol. 2013 Jun;139(6):713-27. doi: 10.1309/AJCP4ZDKJ4ICOUAT[↩][↩][↩]
- Grzybowski B, Vishwanath VA. Hemophagocytic Lymphohistiocytosis: A Diagnostic Conundrum. J Pediatr Neurosci. 2017 Jan-Mar;12(1):55-60. doi: 10.4103/jpn.JPN_140_16[↩]
- Hemophagocytic Lymphohistiocytosis. https://imagebank.hematology.org/image/63563/hemophagocytic-lymphohistiocytosis[↩]
- Chellapandian D, Das R, Zelley K, Wiener SJ, Zhao H, Teachey DT, et al. Treatment of Epstein Barr virus-induced haemophagocytic lymphohistiocytosis with rituximab-containing chemo-immunotherapeutic regimens. Br J Haematol. 2013;162(3):376–382. doi: 10.1111/bjh.12386[↩]
- Akashi K, Mizuno S. Epstein-Barr virus-infected natural killer cell leukemia. Leuk Lymphoma. 2000 Dec;40(1-2):57-66. doi: 10.3109/10428190009054881[↩][↩]
- Ramos-Casals M, Brito-Zerón P, López-Guillermo A, Khamashta MA, Bosch X. Adult haemophagocytic syndrome. Lancet. 2014 Apr 26;383(9927):1503-1516. doi: 10.1016/S0140-6736(13)61048-X. Epub 2013 Nov 27. Erratum in: Lancet. 2014 Apr 26;383(9927):1464.[↩][↩][↩]
- Sawhney S, Woo P, Murray KJ. Macrophage activation syndrome: a potentially fatal complication of rheumatic disorders. Arch Dis Child. 2001 Nov;85(5):421-6. doi: 10.1136/adc.85.5.421[↩]
- Athreya BH. Is macrophage activation syndrome a new entity? Clin Exp Rheumatol. 2002 Mar-Apr;20(2):121-3.[↩]
- Ramanan AV, Baildam EM. Macrophage activation syndrome is hemophagocytic lymphohistiocytosis–need for the right terminology. J Rheumatol. 2002 May;29(5):1105; author reply 1105.[↩]
- Emile JF, Abla O, Fraitag S, Horne A, Haroche J, Donadieu J, Requena-Caballero L, Jordan MB, Abdel-Wahab O, Allen CE, Charlotte F, Diamond EL, Egeler RM, Fischer A, Herrera JG, Henter JI, Janku F, Merad M, Picarsic J, Rodriguez-Galindo C, Rollins BJ, Tazi A, Vassallo R, Weiss LM; Histiocyte Society. Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood. 2016 Jun 2;127(22):2672-81. doi: 10.1182/blood-2016-01-690636[↩][↩]
- Madkaikar M, Shabrish S, Desai M. Current Updates on Classification, Diagnosis and Treatment of Hemophagocytic Lymphohistiocytosis (HLH) Indian J Pediatr. 2016;83(5):434–43. doi: 10.1007/s12098-016-2037-y[↩]
- Hirayama D, Iida T, Nakase H. The Phagocytic Function of Macrophage-Enforcing Innate Immunity and Tissue Homeostasis. Int J Mol Sci. 2017 Dec 29;19(1):92. doi: 10.3390/ijms19010092[↩][↩]
- Rouphael NG, Talati NJ, Vaughan C, Cunningham K, Moreira R, Gould C. Infections associated with haemophagocytic syndrome. Lancet Infect Dis. 2007 Dec;7(12):814-22. doi: 10.1016/S1473-3099(07)70290-6[↩]
- Chesshyre E, Ramanan AV, Roderick MR. Hemophagocytic Lymphohistiocytosis and Infections: An Update. Pediatr Infect Dis J. 2019 Mar;38(3):e54-e56. doi: 10.1097/INF.0000000000002248[↩]
- Tan HY, Yong YK, Andrade BB, Shankar EM, Ponnampalavanar S, Omar SF, Narendran G, Kamarulzaman A, Swaminathan S, Sereti I, Crowe SM, French MA. Plasma interleukin-18 levels are a biomarker of innate immune responses that predict and characterize tuberculosis-associated immune reconstitution inflammatory syndrome. AIDS. 2015 Feb 20;29(4):421-31. doi: 10.1097/QAD.0000000000000557[↩]
- Lehmberg K, Sprekels B, Nichols KE, Woessmann W, Müller I, Suttorp M, Bernig T, Beutel K, Bode SF, Kentouche K, Kolb R, Längler A, Minkov M, Schilling FH, Schmid I, Vieth S, Ehl S, Zur Stadt U, Janka GE. Malignancy-associated haemophagocytic lymphohistiocytosis in children and adolescents. Br J Haematol. 2015 Aug;170(4):539-49. doi: 10.1111/bjh.13462[↩][↩]
- Rivière S, Galicier L, Coppo P, Marzac C, Aumont C, Lambotte O, Fardet L. Reactive hemophagocytic syndrome in adults: a retrospective analysis of 162 patients. Am J Med. 2014 Nov;127(11):1118-1125. doi: 10.1016/j.amjmed.2014.04.034[↩]
- Parikh SA, Kapoor P, Letendre L, Kumar S, Wolanskyj AP. Prognostic factors and outcomes of adults with hemophagocytic lymphohistiocytosis. Mayo Clin Proc. 2014 Apr;89(4):484-92. doi: 10.1016/j.mayocp.2013.12.012[↩]
- Namuduri M, Brentjens RJ. Medical management of side effects related to CAR T cell therapy in hematologic malignancies. Expert Rev Hematol. 2016 Jun;9(6):511-3. doi: 10.1080/17474086.2016.1183479[↩]
- Teachey DT, Rheingold SR, Maude SL, Zugmaier G, Barrett DM, Seif AE, Nichols KE, Suppa EK, Kalos M, Berg RA, Fitzgerald JC, Aplenc R, Gore L, Grupp SA. Cytokine release syndrome after blinatumomab treatment related to abnormal macrophage activation and ameliorated with cytokine-directed therapy. Blood. 2013 Jun 27;121(26):5154-7. doi: 10.1182/blood-2013-02-485623[↩]
- Jordan MB, Allen CE, Greenberg J, Henry M, Hermiston ML, Kumar A, Hines M, Eckstein O, Ladisch S, Nichols KE, et al. Challenges in the diagnosis of hemophagocytic lymphohistiocytosis: Recommendations from the North American consortium for histiocytosis (NACHO) Pediatr Blood Cancer. 2019;66(e27929) doi: 10.1002/pbc.27929[↩][↩]
- Kawa K, Sawada A, Sato M, Okamura T, Sakata N, Kondo O, Kimoto T, Yamada K, Tokimasa S, Yasui M, Inoue M. Excellent outcome of allogeneic hematopoietic SCT with reduced-intensity conditioning for the treatment of chronic active EBV infection. Bone Marrow Transplant. 2011 Jan;46(1):77-83. doi: 10.1038/bmt.2010.122[↩]
- Fujiwara S, Kimura H, Imadome K, Arai A, Kodama E, Morio T, Shimizu N, Wakiguchi H. Current research on chronic active Epstein-Barr virus infection in Japan. Pediatr Int. 2014 Apr;56(2):159-66. doi: 10.1111/ped.12314[↩]
- Balamuth NJ, Nichols KE, Paessler M, Teachey DT. Use of rituximab in conjunction with immunosuppressive chemotherapy as a novel therapy for Epstein Barr virus-associated hemophagocytic lymphohistiocytosis. J Pediatr Hematol Oncol. 2007;29:569–573. doi: 10.1097/MPH.0b013e3180f61be3[↩][↩][↩]
- Mărginean MO, Molnar E, Chinceşan MI. Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis in a small child: A case report. Medicine (Baltimore) 2020;99(e18759) doi: 10.1097/MD.0000000000018759[↩]
- Fałkowska A, Prądzyńska K, Drabko K. Difficult balance between EBV treatment and posttransplant immunosuppression: A successful transplant in a child with recurrent Epstein-Barr virus-induced hemophagocytic lymphohistiocytosis. Transplant Proc. 2021;53:2035–2039. doi: 10.1016/j.transproceed.2021.03.044[↩]
- Pan H, Feng DN, Song L, Sun LR. Acute myeloid leukemia following etoposide therapy for EBV-associated hemophagocytic lymphohistiocytosis: A case report and a brief review of the literature. BMC Pediatr. 2016;16(116) doi: 10.1186/s12887-016-0649-z[↩]
- DomInguez-Pinilla N, Baro-Fernández M, González-Granado LI. Hemophagocytic lymphohistiocytosis secondary to Epstein Barr virus and Leishmania co-infection in a toddler. J Postgrad Med. 2015;61:44–45. doi: 10.4103/0022-3859.147052[↩]
- Gera A, Misra A, Tiwari A, Singh A, Mehndiratta S. A hungry Histiocyte, altered immunity and myriad of problems: Diagnostic challenges for Pediatric HLH. Int J Lab Hematol. 2021;43:1443–1450. doi: 10.1111/ijlh.13626[↩][↩][↩]
- Oguz MM, Sahin G, Altinel Acoglu E, Polat E, Yucel H, Oztek Celebi FZ, Unsal H, Akcaboy M, Sari E, Senel S. Secondary hemophagocytic lymphohistiocytosis in pediatric patients: A single center experience and factors that influenced patient prognosis. Pediatr Hematol Oncol. 2019;36:1–16. doi: 10.1080/08880018.2019.1572253[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Paul T, Kalra M, Danewa A, Sachdeva P, Thatikonda KB, Sachdeva D, Sachdeva A. Pediatric Hemophagocytic Lymphohistiocytosis – A Single Center Study. Indian Pediatr. 2022 Apr 15;59(4):283-286. https://www.indianpediatrics.net/apr2022/283.pdf[↩][↩][↩][↩][↩][↩][↩]
- Imashuku S, Tabata Y, Teramura T, Hibi S. Treatment strategies for Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis (EBV-HLH) Leuk Lymphoma. 2000;39:37–49. doi: 10.3109/10428190009053537[↩][↩][↩]
- Kawaguchi H, Miyashita T, Herbst H, Niedobitek G, Asada M, Tsuchida M, Hanada R, Kinoshita A, Sakurai M, Kobayashi N, et al. Epstein-Barr virus-infected T lymphocytes in Epstein-Barr virus-associated hemophagocytic syndrome. J Clin Invest. 1993;92:1444–1450. doi: 10.1172/JCI116721[↩]
- Lin MT, Chang HM, Huang CJ, Chen WL, Lin CY, Lin CY, Chuang SS. Massive expansion of EBV+ monoclonal T cells with CD5 down regulation in EBV-associated haemophagocytic lymphohistiocytosis. J Clin Pathol. 2007;60:101–103. doi: 10.1136/jcp.2005.034371[↩][↩][↩]
- Imashuku S. Clinical features and treatment strategies of Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis. Crit Rev Oncol Hematol. 2002;44:259–272. doi: 10.1016/s1040-8428(02)00117-8[↩][↩]
- Lohana C, Jhatial MA, Bokhari SWI. A challenging case of haemophagocytic lymphohistiocytosis (HLH) secondary to CMV infection in an adolescent with B-acute lymphoblastic leukemia. J Coll Physicians Surg Pak. 2021;31:1011–1012. doi: 10.29271/jcpsp.2021.08.1011[↩]
- Pessoa FS, Gonçalves VC, Lacerda EMDCB. Haemophagocytic lymphohistiocytosis secondary to intrauterine cytomegalovirus infection. Rev Inst Med Trop Sao Paulo. 2021;63(e15) doi: 10.1590/S1678-9946202163015[↩]
- Eldem İ, Al-Rahawan MM, Levent F. How to treat cytomegalovirus-induced hemophagocytic lymphohistiocytosis in a child with leukemia. J Pediatr Hematol Oncol. 2020;42:313–315. doi: 10.1097/MPH.0000000000001547[↩]
- Silwedel C, Frieauff E, Thomas W, Liese JG, Speer CP. Secondary haemophagocytic lymphohistiocytosis triggered by postnatally acquired cytomegalovirus infection in a late preterm infant. Infection. 2017;45:355–359. doi: 10.1007/s15010-016-0970-3[↩]
- Hasan MR, Sundaram MS, Sundararaju S, Tsui KM, Karim MY, Roscoe D, Imam O, Janahi MA, Thomas E, Dobson S, et al. Unusual accumulation of a wide array of antimicrobial resistance mechanisms in a patient with cytomegalovirus-associated hemophagocytic lymphohistiocytosis: A case report. BMC Infect Dis. 2020;20(237) doi: 10.1186/s12879-020-04966-z[↩]
- Gur G, Cakar N, Uncu N, Ayar G, Basaran O, Taktak A, Koksoy AY, Acar B, Caycı FS. Hemophagocytic lymphohistiocytosis secondary to Varicella zoster infection in a child with Henoch-Schönlein purpura. Pediatr Int. 2015;57:e37–e38. doi: 10.1111/ped.12523[↩]
- Agrawal G, Wazir S, Sachdeva A, Kumar S. Primary dengue infection triggered haemophagocytic lymphohistiocytosis in a neonate. BMJ Case Rep. 2020;13(e236881) doi: 10.1136/bcr-2020-236881[↩]
- Krithika MV, Amboiram P, Latha SM, Ninan B, Suman FR, Scott J. Neonate with haemophagocytic lymphohistiocytosis secondary to dengue infection: A case report. Trop Doct. 2017;47:253–255. doi: 10.1177/0049475516644102[↩]
- Jasmine YSY, Lee SL, Kan FK. Infection associated haemophagocytic syndrome in severe dengue infection-a case series in a district hospital. Med J Malaysia. 2017;72:62–64.[↩]
- Nandhakumar D, Loganatha A, Sivasankaran M, Sivabalan S, Munirathnam D. Hemophagocytic lymphohistiocytosis in children. Indian J Pediatr. 2020;87:526–531. doi: 10.1007/s12098-020-03190-6[↩][↩][↩][↩][↩][↩]
- Simon AC, Delhi Kumar CG, Basu D, Ramesh Kumar R. Hemophagocytic lymphohistiocytosis in children: Clinical profile and outcome. J Pediatr Hematol Oncol. 2020;42:e281–e285. doi: 10.1097/MPH.0000000000001791[↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Gayretli Aydin ZG, Yesilbas O, Reis GP, Guven B. The first pediatric case of hemophagocytic lymphohistiocytosis secondary to Crimean-Congo haemorrhagic fever successfully treated with therapeutic plasma exchange accompanying ribavirin and intravenous immunoglobulin. J Clin Apher. 2021;36:780–784. doi: 10.1002/jca.21915[↩][↩]
- Bıçakçı Z, Tavil B, Tezer H, Olcay L. Hemophagocytosis in a case with Crimean-Congo hemorrhagic fever and an overview of possible pathogenesis with current evidence. Turk J Pediatr. 2013 May-Jun;55(3):344-8.[↩]
- Mancao MY, Imran H, Chandra S, Estrada B, Figarola M, Sosnowski J, Vidal R. Eastern equine encephalitis virus infection and hemophagocytic lymphohistiocytosis in a 5-month-old infant. Pediatr Infect Dis J. 2009;28:543–545. doi: 10.1097/INF.0b013e3181950e99[↩]
- Bay A, Bosnak V, Leblebisatan G, Yavuz S, Yilmaz F, Hizli S. Hemophagocytic lymphohistiocytosis in 2 pediatric patients secondary to hepatitis A virus infection. Pediatr Hematol Oncol. 2012;29:211–214. doi: 10.3109/08880018.2012.666783[↩][↩]
- Jianguo L, Zhixuan Z, Rong L, Xiaodong S. Ruxolitinib in alleviating the cytokine storm of hemophagocytic lymphohistiocytosis. Pediatrics. 2020;146(e20191301) doi: 10.1542/peds.2019-1301[↩][↩]
- Chiperi LE, Ionescu AD, Marcu CT, Itu-Mureşan C, Pantelemon C. Hemophagocytic lymphohistiocytosis in a child with human immunodeficiency virus infection-a case report. Rom J Morphol Embryol. 2021;62:279–282. doi: 10.47162/RJME.62.1.29[↩][↩]
- Censoplano N, Gorga S, Waldeck K, Stillwell T, Rabah-Hammad R, Flori H. Neonatal adenovirus infection complicated by hemophagocytic lymphohistiocytosis syndrome. Pediatrics. 2018;141 (Suppl 5):S475–S480. doi: 10.1542/peds.2017-2061[↩]
- Otto WR, Behrens EM, Teachey DT, Lamson DM, Barrett DM, Bassiri H, Lambert MP, Mount S, Petrosa WL, Romberg N, et al. Human adenovirus 7-associated hemophagocytic lymphohistiocytosis-like illness: Clinical and virological characteristics in a cluster of five pediatric cases. Clin Infect Dis. 2021;73:e1532–e1538. doi: 10.1093/cid/ciaa1277[↩]
- Joshi R, Phatarpekar A, Currimbhoy Z, Desai M. Haemophagocytic lymphohistiocytosis: A case series from Mumbai. Ann Trop Paediatr. 2011;31:135–140. doi: 10.1179/1465328111Y.0000000009[↩]
- Zhang XY, Ye XW, Feng DX, Han J, Li D, Zhang C. Hemophagocytic lymphohistiocytosis induced by severe pandemic influenza A (H1N1) 2009 virus infection: A case report. Case Rep Med. 2011;2011(951910) doi: 10.1155/2011/951910[↩]
- Shrestha B, Omran A, Rong P, Wang W. Report of a fatal pediatric case of hemophagocytic lymphohistiocytosis associated with pandemic influenza A (H1N1) infection in 2009. Pediatr Neonatol. 2015;56:189–192. doi: 10.1016/j.pedneo.2013.03.006[↩]
- Ozdemir H, Çiftçi E, Ince EU, Ertem M, Ince E, Doğru U. Hemophagocytic lymphohistiocytosis associated with 2009 pandemic influenza A (H1N1) virus infection. J Pediatr Hematol Oncol. 2011;33:135–137. doi: 10.1097/MPH.0b013e3181f46baf[↩]
- Tholin B, Hauge MT, Aukrust P, Fehrle L, Tvedt TH. Hemophagocytic lymphohistiocytosis in a patient with COVID-19 treated with tocilizumab: A case report. J Med Case Rep. 2020;14(187) doi: 10.1186/s13256-020-02503-9[↩]
- Hakim NN, Chi J, Olazagasti C, Liu JM. Secondary hemophagocytic lymphohistiocytosis versus cytokine release syndrome in severe COVID-19 patients. Exp Biol Med (Maywood) 2021;246:5–9. doi: 10.1177/1535370220962043[↩]
- Mittal J, Kumar P, Goyal JP, Purohit A. Haemophagocytic lymphohistiocytosis secondary to brucellosis in a young child. BMJ Case Rep. 2021;14(e240759) doi: 10.1136/bcr-2020-240759[↩]
- Pekpak E, Sirvan Cetin B. Secondary hemophagocytic lymphohistocytosis in a child with brucellosis. J Pediatr Hematol Oncol. 2017;39:e501–e503. doi: 10.1097/MPH.0000000000000849[↩]
- Yaman Y, Gözmen S, Özkaya AK, Oymak Y, Apa H, Vergin C, Devrim İ. Secondary hemophagocytic lymphohistiocytosis in children with brucellosis: Report of three cases. J Infect Dev Ctries. 2015;9:1172–1176. doi: 10.3855/jidc.6090[↩]
- Banday AZ, Mehta R, Vignesh P, Kanaujia R, Durgadevi S, Angrup A, Kumar N, Ray P, Suri D. Case Report: Ceftriaxone-resistant invasive Salmonella enteritidis infection with secondary hemophagocytic lymphohistiocytosis: A contrast with enteric fever. Am J Trop Med Hyg. 2020;103:2515–2517. doi: 10.4269/ajtmh.20-0566[↩]
- Maheshwari P, Chhabra R, Yadav P. Perinatal tuberculosis associated hemophagocytic lymphohistiocytosis. Indian J Pediatr. 2012;79:1228–1229. doi: 10.1007/s12098-011-0675-7[↩]
- Choi YB, Yi DY. Fatal case of hemophagocytic lymphohistiocytosis associated with group B streptococcus sepsis: A case report. Medicine (Baltimore) 2018;97(e12210) doi: 10.1097/MD.0000000000012210[↩]
- Liu SS, Wang Y, Xue L, Ma C, Li CH. Hemophagocytic lymphohistiocytosis due to Streptococcus suis in a 12-year-old girl: A case report. Medicine (Baltimore) 2019;98(e15136) doi: 10.1097/MD.0000000000015136[↩]
- Sahu SK, Behera JR, Yadav SK. Scrub typhus with secondary hemophagocytic lymphohistiocytosis in a 3-month-old child from a tertiary care hospital of Odisha. Indian J Public Health. 2021;65:85–86. doi: 10.4103/ijph.IJPH_565_20[↩]
- Han DK, Baek HJ, Shin MG, Kim JW, Kook H, Hwang TJ. Scrub typhus-associated severe hemophagocytic lymphohistiocytosis with encephalomyelitis leading to permanent sequelae: A case report and review of the literature. J Pediatr Hematol Oncol. 2012;34:531–533. doi: 10.1097/MPH.0b013e318257a442[↩]
- Agrwal S, Dabas A, Mantan M, Yadav S. Hemophagocytic lymphohistiocytosis with neurological manifestations in an infant with scrub typhus: A rare fatal occurrence. Trop Doct. 2019;49:52–53. doi: 10.1177/0049475518804696[↩]
- Pazhaniyandi S, Lenin R, Sivathanu S. Hemophagocytic lymphohistiocytosis with a leukemoid reaction in an infant with scrub typhus. J Infect Public Health. 2015;8:626–629. doi: 10.1016/j.jiph.2015.05.012[↩]
- Otrock ZK, Gonzalez MD, Eby CS. Ehrlichia-induced hemophagocytic lymphohistiocytosis: A case series and review of literature. Blood Cells Mol Dis. 2015;55:191–193. doi: 10.1016/j.bcmd.2015.06.009[↩]
- Cheng A, Williams F, Fortenberry J, Preissig C, Salinas S, Kamat P. Use of extracorporeal support in hemophagocytic lymphohistiocytosis secondary to ehrlichiosis. Pediatrics. 2016;138(e20154176) doi: 10.1542/peds.2015-4176[↩]
- Burns S, Saylors R, Mian A. Hemophagocytic lymphohistiocytosis secondary to Ehrlichia chaffeensis infection: A case report. J Pediatr Hematol Oncol. 2010;32:e142–143. doi: 10.1097/MPH.0b013e3181c80ab9[↩]
- Singh NS, Pagano AL, Hays AJ, Kats A, Dahl SM, Warady BA, Beins NT, Yin DE. Ehrlichia-induced hemophagocytic lymphohistiocytosis in a pediatric kidney transplant recipient. Pediatr Transplant. 2022;26(e14134) doi: 10.1111/petr.14134[↩][↩][↩]
- Cabler SS, Hogan PG, Fritz SA, Bednarski JJ, Hunstad DA. Incidence and treatment of hemophagocytic lymphohistiocytosis in hospitalized children with Ehrlichia infection. Pediatr Blood Cancer. 2020;67(e28436) doi: 10.1002/pbc.28436[↩][↩][↩]
- Tsuge M, Miyamoto M, Miyawaki R, Kondo Y, Tsukahara H. Hemophagocytic lymphohistiocytosis complicating invasive pneumococcal disease: A pediatric case report. BMC Pediatr. 2020;20(15) doi: 10.1186/s12887-020-1915-7[↩]
- Poudel A, Lew J, Slayton W, Dharnidharka VR. Bartonella henselae infection inducing hemophagocytic lymphohistiocytosis in a kidney transplant recipient. Pediatr Transplant. 2014;18:E83–E87. doi: 10.1111/petr.12235[↩][↩][↩]
- Edner J, Rudd E, Zheng C, Dahlander A, Eksborg S, Schneider EM, Edner A, Henter JI. Severe bacteria-associated hemophagocytic lymphohistiocytosis in an extremely premature infant. Acta Paediatr. 2007;96:1703–1706. doi: 10.1111/j.1651-2227.2007.00505.x[↩]
- Karapinar DY, Karadaş N, Yazici P, Polat SH, Karapinar B. Trichosporon asahii, sepsis, and secondary hemophagocytic lymphohistiocytosis in children with hematologic malignancy. Pediatr Hematol Oncol. 2014;31:282–284. doi: 10.3109/08880018.2013.851754[↩]
- Guo F, Kang L, Xu M. A case of pediatric visceral leishmaniasis-related hemophagocytic lymphohistiocytosis diagnosed by mNGS. Int J Infect Dis. 2020;97:27–29. doi: 10.1016/j.ijid.2020.05.056[↩]
- Mottaghipisheh H, Kalantar K, Amanati A, Shokripour M, Shahriari M, Zekavat OR, Zareifar S, Karimi M, Haghpanah S, Bordbar M. Comparison of the clinical features and outcome of children with hemophagocytic lymphohistiocytosis (HLH) secondary to visceral leishmaniasis and primary HLH: A single-center study. BMC Infect Dis. 2021;21(732) doi: 10.1186/s12879-021-06408-w[↩][↩][↩]
- de Carvalho FHG, Lula JF, Teles LF, Caldeira AP, de Carvalho SFG. Hemophagocytic lymphohistiocytosis secondary to visceral leishmaniasis in an endemic area in the north of Minas Gerais, Brazil. Rev Soc Bras Med Trop. 2020;53(e20190491) doi: 10.1590/0037-8682-0491-2019[↩][↩][↩]
- Srivatsav S, Mahalingam S, Ramineni P, Manya S. Dengue and Plasmodium falciparum coinfection with secondary hemophagocytic lymphohistiocytosis in a 3-year-old boy: A clinical conundrum. J Pediatr Hematol Oncol. 2022;44:e253–e254. doi: 10.1097/MPH.0000000000002018[↩]
- Padhi S, Varghese RGB, Ramdas A, Phansalkar MD, Sarangi R. Hemophagocytic lymphohistiocytosis: Critical reappraisal of a potentially under-recognized condition. Front Med. 2013;7:492–498. doi: 10.1007/s11684-013-0292-0[↩]
- Henderson LA, Cron RQ. Macrophage activation syndrome and secondary hemophagocytic lymphohistiocytosis in childhood inflammatory disorders: Diagnosis and management. Paediatr Drugs. 2020;22:29–44. doi: 10.1007/s40272-019-00367-1[↩][↩]
- Emile JF, Abla O, Fraitag S, Horne A, Haroche J, Donadieu J, Requena-Caballero L, Jordan MB, Abdel-Wahab O, Allen CE, et al. Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood. 2016;127:2672–2681. doi: 10.1182/blood-2016-01-690636[↩]
- Vastert SJ, van Wijk R, D’Urbano LE, de Vooght KM, de Jager W, Ravelli A, Magni-Manzoni S, Insalaco A, Cortis E, van Solinge WW, Prakken BJ, Wulffraat NM, de Benedetti F, Kuis W. Mutations in the perforin gene can be linked to macrophage activation syndrome in patients with systemic onset juvenile idiopathic arthritis. Rheumatology (Oxford). 2010 Mar;49(3):441-9. doi: 10.1093/rheumatology/kep418[↩]
- Bracaglia C, de Graaf K, Pires Marafon D, Guilhot F, Ferlin W, Prencipe G, Caiello I, Davì S, Schulert G, Ravelli A, Grom AA, de Min C, De Benedetti F. Elevated circulating levels of interferon-γ and interferon-γ-induced chemokines characterise patients with macrophage activation syndrome complicating systemic juvenile idiopathic arthritis. Ann Rheum Dis. 2017 Jan;76(1):166-172. doi: 10.1136/annrheumdis-2015-209020[↩][↩]
- Bleesing J, Prada A, Siegel DM, Villanueva J, Olson J, Ilowite NT, Brunner HI, Griffin T, Graham TB, Sherry DD, Passo MH, Ramanan AV, Filipovich A, Grom AA. The diagnostic significance of soluble CD163 and soluble interleukin-2 receptor alpha-chain in macrophage activation syndrome and untreated new-onset systemic juvenile idiopathic arthritis. Arthritis Rheum. 2007 Mar;56(3):965-71. doi: 10.1002/art.22416[↩]
- Minoia F, Bovis F, Davì S, et al. Pediatric Rheumatology International Trials Organization, the Childhood Arthritis and Rheumatology Research Alliance, the Pediatric Rheumatology Collaborative Study Group, and the Histiocyte Society. Development and Initial Validation of the Macrophage Activation Syndrome/Primary Hemophagocytic Lymphohistiocytosis Score, a Diagnostic Tool that Differentiates Primary Hemophagocytic Lymphohistiocytosis from Macrophage Activation Syndrome. J Pediatr. 2017 Oct;189:72-78.e3. doi: 10.1016/j.jpeds.2017.06.005[↩][↩]
- Ravelli A, Davì S, Minoia F, Martini A, Cron RQ. Macrophage Activation Syndrome. Hematol Oncol Clin North Am. 2015 Oct;29(5):927-41. doi: 10.1016/j.hoc.2015.06.010[↩]
- Efthimiou P, Kadavath S, Mehta B. Life-threatening complications of adult-onset Still’s disease. Clin Rheumatol. 2014 Mar;33(3):305-14. doi: 10.1007/s10067-014-2487-4[↩]
- Lenert A, Yao Q. Macrophage activation syndrome complicating adult onset Still’s disease: A single center case series and comparison with literature. Semin Arthritis Rheum. 2016 Jun;45(6):711-6. doi: 10.1016/j.semarthrit.2015.11.002[↩]
- Ravelli A, Minoia F, Davì S, et al. Paediatric Rheumatology International Trials Organisation; Childhood Arthritis and Rheumatology Research Alliance; Pediatric Rheumatology Collaborative Study Group; Histiocyte Society. 2016 Classification Criteria for Macrophage Activation Syndrome Complicating Systemic Juvenile Idiopathic Arthritis: A European League Against Rheumatism/American College of Rheumatology/Paediatric Rheumatology International Trials Organisation Collaborative Initiative. Ann Rheum Dis. 2016 Mar;75(3):481-9. doi: 10.1136/annrheumdis-2015-208982[↩]
- Lehmberg K, Sprekels B, Nichols KE, Woessmann W, Müller I, Suttorp M, Bernig T, Beutel K, Bode SF, Kentouche K, et al. Malignancy-associated haemophagocytic lymphohistiocytosis in children and adolescents. Br J Haematol. 2015;170:539–549. doi: 10.1111/bjh.13462[↩][↩][↩][↩][↩]
- O’Brien MM, Lee-Kim Y, George TI, McClain KL, Twist CJ, Jeng M. Precursor B-cell acute lymphoblastic leukemia presenting with hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2008;50:381–383. doi: 10.1002/pbc.20950[↩]
- Celkan T, Berrak S, Kazanci E, Ozyürek E, Unal S, Uçar C, Yilmaz S, Gürgey A. Malignancy-associated hemophagocytic lymphohistiocytosis in pediatric cases: A multicenter study from Turkey. Turk J Pediatr. 2009;51:207–213.[↩][↩][↩]
- Strenger V, Merth G, Lackner H, Aberle SW, Kessler HH, Seidel MG, Schwinger W, Sperl D, Sovinz P, Karastaneva A, et al. Malignancy and chemotherapy induced haemophagocytic lymphohistiocytosis in children and adolescents-a single centre experience of 20 years. Ann Hematol. 2018;97:989–998. doi: 10.1007/s00277-018-3254-4[↩][↩]
- Chinn IK, Eckstein OS, Peckham-Gregory EC, Goldberg BR, Forbes LR, Nicholas SK, Mace EM, Vogel TP, Abhyankar HA, Diaz MI, et al. Genetic and mechanistic diversity in pediatric hemophagocytic lymphohistiocytosis. Blood. 2018;132:89–100. doi: 10.1182/blood-2017-11-814244[↩]
- Tabata C, Tabata R. Possible prediction of underlying lymphoma by high sIL-2R/ferritin ratio in hemophagocytic syndrome. Ann Hematol. 2012 Jan;91(1):63-71. doi: 10.1007/s00277-011-1239-7[↩]
- Hayden A, Lin M, Park S, Pudek M, Schneider M, Jordan MB, Mattman A, Chen LYC. Soluble interleukin-2 receptor is a sensitive diagnostic test in adult HLH. Blood Adv. 2017 Dec 6;1(26):2529-2534. doi: 10.1182/bloodadvances.2017012310[↩][↩]
- Daver N, McClain K, Allen CE, Parikh SA, Otrock Z, Rojas-Hernandez C, Blechacz B, Wang S, Minkov M, Jordan MB, et al. A consensus review on malignancy-associated hemophagocytic lymphohistiocytosis in adults. Cancer. 2017;123:3229–3240. doi: 10.1002/cncr.30826[↩][↩]
- Chellapandian D, Hines MR, Zhang R, Jeng M, van den Bos C, Santa-María López V, Lehmberg K, Sieni E, Wang Y, Nakano T, et al. A multicenter study of patients with multisystem Langerhans cell histiocytosis who develop secondary hemophagocytic lymphohistiocytosis. Cancer. 2019;125:963–971. doi: 10.1002/cncr.31893[↩][↩]
- Huang Z, Jia Y, Zuo Y, Wu J, Lu A, Zhang L. Malignancy-associated hemophagocytic lymphohistiocytosis in children: A 10-year experience of a single pediatric hematology center. Hematology. 2020;25:389–399. doi: 10.1080/16078454.2020.1833505[↩][↩][↩][↩][↩]
- Sakamoto K, Osumi T, Yoshimura S, Shimizu S, Kato M, Tomizawa D, Fukuda A, Sakamoto S, Nakano N, Yoshioka T, et al. Living-donor liver transplantation providing an adequate chemotherapy for a pediatric patient with anaplastic large cell lymphoma complicated with liver failure due to the aggravation of biliary hepatopathy by secondary hemophagocytic lymphohistiocytosis. Int J Hematol. 2020;112:900–905. doi: 10.1007/s12185-020-02949-z[↩]
- Teshigawara S, Katada Y, Maeda Y, Yoshimura M, Kudo-Tanaka E, Tsuji S, Harada Y, Matsushita M, Ohshima S, Watanabe K, et al. Hemophagocytic lymphohistiocytosis with leukoencephalopathy in a patient with dermatomyositis accompanied with peripheral T-cell lymphoma: A case report. J Med Case Rep. 2016;10(212) doi: 10.1186/s13256-016-0986-4[↩]
- Sinno MG, Rosen D, Wittler R. Concomitant presentation of hemophagocytic lymphohistiocytosis and posttransplant lymphoproliferative disease-like lymphoma in a mildly immunosuppressed leukemia patient: An unusual association. Pediatr Blood Cancer. 2016;63:1474–1476. doi: 10.1002/pbc.26033[↩]
- McCall CM, Mudali S, Arceci RJ, Small D, Fuller S, Gocke CD, Vuica-Ross M, Burns KH, Borowitz MJ, Duffield AS. Flow cytometric findings in hemophagocytic lymphohistiocytosis. Am J Clin Pathol. 2012;137:786–794. doi: 10.1309/AJCPP40MEXWYRLPN[↩]
- Gurunathan A, Boucher AA, Mark M, Prus KM, O’Brien MM, Breese EH, Mizukawa BE, Absalon MJ, Nelson AS, Jordan MB, Grimley MS, Lorsbach RB, Rotz SJ, Mathanda R, Kumar AR. Limitations of HLH-2004 criteria in distinguishing malignancy-associated hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2018 Dec;65(12):e27400. doi: 10.1002/pbc.27400[↩][↩][↩]
- Zheng F, Jia Y, Zhang L, Qin J. Hemophagocytic lymphohistiocytosis secondary to juvenile myelomonocytic leukemia: A case report and review of the literature. J Pediatr Hematol Oncol. 2022;44:e580–e584. doi: 10.1097/MPH.0000000000002273[↩]
- Farias MG, Freitas PAC, Spagnol F, Souza MV, Alegretti AP, Riegel M, Taniguchi ANR, Daudt LE. Hemophagocytosis by blasts in a child with acute monocytic leukemia after chemotherapy. Rev Paul Pediatr. 2021;39(e2019290) doi: 10.1590/1984-0462/2021/39/2019290[↩]
- Dokmanovic L, Krstovski N, Jankovic S, Janic D. Hemophagocytic lymphohistiocytosis arising in a child with Langerhans cell histiocytosis. Turk J Pediatr. 2014;56:452–457.[↩]
- Murphy EP, Mo J, Yoon JM. Secondary hemophagocytic lymphohistiocytosis in a patient with favorable histology wilms tumor. J Pediatr Hematol Oncol. 2015;37:e494–e496. doi: 10.1097/MPH.0000000000000433[↩]
- Nascimento FA, Nery J, Trennepohl J, Pianovski MAD. Hemophagocytic lymphohistiocytosis after initiation of chemotherapy for bilateral adrenal neuroblastoma. J Pediatr Hematol Oncol. 2016;38:e13–e15. doi: 10.1097/MPH.0000000000000469[↩]
- Tang X, Guo X, Gao J, Sun JJ, Wan Z. Hemophagocytic lymphohistiocytosis in langerhans cell histiocytosis: A case series and literature review. J Pediatr Hematol Oncol. 2022;44:e20–e25. doi: 10.1097/MPH.0000000000002212[↩]
- Devecioglu O, Anak S, Atay D, Aktan P, Devecioglu E, Ozalp B, Saribeyoglu E. Pediatric acute lymphoblastic leukemia complicated by secondary hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2009;53:491–492. doi: 10.1002/pbc.22064[↩]
- Fajgenbaum DC, June CH. Cytokine storm. N Engl J Med. 2020;383:2255–2273. doi: 10.1056/NEJMra2026131[↩][↩][↩][↩][↩][↩][↩]
- Sandler RD, Tattersall RS, Schoemans H, Greco R, Badoglio M, Labopin M, Alexander T, Kirgizov K, Rovira M, Saif M, et al. Diagnosis and management of secondary HLH/MAS following HSCT and CAR-T cell therapy in adults; A review of the literature and a survey of practice within EBMT centres on behalf of the autoimmune diseases working party (ADWP) and transplant complications working party (TCWP) Front Immunol. 2020;11(524) doi: 10.3389/fimmu.2020.00524[↩]
- Teachey DT, Lacey SF, Shaw PA, Melenhorst JJ, Maude SL, Frey N, Pequignot E, Gonzalez VE, Chen F, Finklestein J, et al. Identification of predictive biomarkers for cytokine release syndrome after chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Cancer Discov. 2016;6:664–679. doi: 10.1158/2159-8290.CD-16-0040[↩]
- Neelapu SS, Tummala S, Kebriaei P, Wierda W, Gutierrez C, Locke FL, Komanduri KV, Lin Y, Jain N, Daver N, et al. Chimeric antigen receptor T-cell therapy-assessment and management of toxicities. Nat Rev Clin Oncol. 2018;15:47–62. doi: 10.1038/nrclinonc.2017.148[↩]
- Lichtenstein DA, Schischlik F, Shao L, Steinberg SM, Yates B, Wang HW, Wang Y, Inglefield J, Dulau-Florea A, Ceppi F, et al. Characterization of HLH-like manifestations as a CRS variant in patients receiving CD22 CAR T cells. Blood. 2021;138:2469–2484. doi: 10.1182/blood.2021011898[↩][↩]
- Hines MR, Keenan C, Maron Alfaro G, Cheng C, Zhou Y, Sharma A, Hurley C, Nichols KE, Gottschalk S, Triplett BM, Talleur AC. Hemophagocytic lymphohistiocytosis-like toxicity (carHLH) after CD19-specific CAR T-cell therapy. Br J Haematol. 2021;194:701–707. doi: 10.1111/bjh.17662[↩][↩][↩]
- Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, Bader P, Verneris MR, Stefanski HE, Myers GD, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med. 2018;378:439–448. doi: 10.1056/NEJMoa1709866[↩]
- Xu XJ, Tang YM. Cytokine release syndrome in cancer immunotherapy with chimeric antigen receptor engineered T cells. Cancer Lett. 2014;343:172–178. doi: 10.1016/j.canlet.2013.10.004[↩]
- Singh N, Hofmann TJ, Gershenson Z, Levine BL, Grupp SA, Teachey DT, Barrett DM. Monocyte lineage-derived IL-6 does not affect chimeric antigen receptor T-cell function. Cytotherapy. 2017;19:867–880. doi: 10.1016/j.jcyt.2017.04.001[↩][↩][↩][↩]
- Giavridis T, van der Stegen SJC, Eyquem J, Hamieh M, Piersigilli A, Sadelain M. CAR T cell-induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat Med. 2018;24:731–738. doi: 10.1038/s41591-018-0041-7[↩]
- Norelli M, Camisa B, Barbiera G, Falcone L, Purevdorj A, Genua M, Sanvito F, Ponzoni M, Doglioni C, Cristofori P, et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat Med. 2018;24:739–748. doi: 10.1038/s41591-018-0036-4[↩]
- Liu Y, Fang Y, Chen X, Wang Z, Liang X, Zhang T, Liu M, Zhou N, Lv J, Tang K, et al. Gasdermin E-mediated target cell pyroptosis by CAR T cells triggers cytokine release syndrome. Sci Immunol. 2020;5(eaax7969) doi: 10.1126/sciimmunol.aax7969[↩]
- Teachey DT, Rheingold SR, Maude SL, Zugmaier G, Barrett DM, Seif AE, Nichols KE, Suppa EK, Kalos M, Berg RA, et al. Cytokine release syndrome after blinatumomab treatment related to abnormal macrophage activation and ameliorated with cytokine-directed therapy. Blood. 2013;121:5154–5157. doi: 10.1182/blood-2013-02-485623[↩][↩]
- Winkler U, Jensen M, Manzke O, Schulz H, Diehl V, Engert A. Cytokine-release syndrome in patients with B-cell chronic lymphocytic leukemia and high lymphocyte counts after treatment with an anti-CD20 monoclonal antibody (rituximab, IDEC-C2B8). Blood. 1999 Oct 1;94(7):2217-24.[↩]
- Asano T, Kogawa K, Morimoto A, Ishida Y, Suzuki N, Ohga S, Kudo K, Ohta S, Wakiguchi H, Tabuchi K, et al. Hemophagocytic lymphohistiocytosis after hematopoietic stem cell transplantation in children: A nationwide survey in Japan. Pediatr Blood Cancer. 2012;59:110–114. doi: 10.1002/pbc.23384[↩][↩][↩][↩][↩][↩]
- Noguchi M, Inagaki J. Hemophagocytic lymphohistiocytosis and graft failure following unrelated umbilical cord blood transplantation in children. J Pediatr Hematol Oncol. 2020;42:e440–e444. doi: 10.1097/MPH.0000000000001795[↩][↩][↩]
- Jarchin L, Chu J, Januska M, Merola P, Arnon R. Autoimmune hemolytic anemia: An unusual presentation of hemophagocytic lymphohistiocytosis in a pediatric post-liver transplant patient. Pediatr Transplant. 2018;22(e13281) doi: 10.1111/petr.13281[↩]
- Nakajima K, Hiejima E, Nihira H, Kato K, Honda Y, Izawa K, Kawabata N, Kato I, Ogawa E, Sonoda M, et al. Case report: A case of Epstein-Barr virus-associated acute liver failure requiring hematopoietic cell transplantation after emergent liver transplantation. Front Immunol. 2022;13(825806) doi: 10.3389/fimmu.2022.825806[↩]
- Jha B, Mohan N, Gajendra S, Sachdev R, Goel S, Sahni T, Raina V, Soin A. Prompt diagnosis and management of Epstein-Barr virus-associated post-transplant lymphoproliferative disorder and hemophagocytosis: A dreaded complication in a post-liver transplant child. Pediatr Transplant. 2015;19:E177–E180. doi: 10.1111/petr.12558[↩]
- Matsuda K, Toyama K, Toya T, Ikemura M, Nakamura F, Kurokawa M. Reactivation of hemophagocytic lymphohistiocytosis triggered by antithymocyte globulin. Intern Med. 2018;57:583–586. doi: 10.2169/internalmedicine.9226-17[↩]
- Ali S, AlThubaiti S, Renzi S, Krueger J, Chiang KY, Naqvi A, Schechter T, Punnett A, Ali M. Hemophagocytic lymphohistiocytosis is a sign of poor outcome in pediatric Epstein-Barr virus-associated post-transplant lymphoproliferative disease after allogeneic hematopoietic stem cell transplantation. Pediatr Transplant. 2019;23(e13319) doi: 10.1111/petr.13319[↩]
- Park M, Yun YJ, Woo SI, Lee JW, Chung NG, Cho B. Rotavirus-associated hemophagocytic lymphohistiocytosis (HLH) after hematopoietic stem cell transplantation for familial HLH. Pediatr Int. 2015;57:e77–e80. doi: 10.1111/ped.12567[↩]
- Salvador C, Meister B, Larcher H, Crazzolara R, Kropshofer G. Hemophagocytic lymphohistiocytosis after allogeneic bone marrow transplantation during chronic norovirus infection. Hematol Oncol. 2014;32:102–106. doi: 10.1002/hon.2052[↩]
- Hattori N, Sato M, Uesugi Y, Nakata A, Sasaki Y, Shimada S, Watanuki M, Fujiwara S, Kawaguchi Y, Arai N, et al. Characteristics and predictors of post-transplant-associated hemophagocytic lymphohistiocytosis in adults. Int J Hematol. 2021;113:693–702. doi: 10.1007/s12185-020-03067-6[↩]
- Stefanou C, Tzortzi C, Georgiou F, Timiliotou C. Combining an antiviral with rituximab in EBV-related haemophagocytic lymphohistiocytosis led to rapid viral clearance; and a comprehensive review. BMJ Case Rep. 2016;2016(bcr2016216488) doi: 10.1136/bcr-2016-216488[↩][↩]
- Fitzgerald MP, Armstrong L, Hague R, Russell RK. A case of EBV driven haemophagocytic lymphohistiocytosis complicating a teenage Crohn’s disease patient on azathioprine, successfully treated with rituximab. J Crohns Colitis. 2013 May;7(4):314-7. doi: 10.1016/j.crohns.2012.05.002[↩]
- Bode SF, Ammann S, Al-Herz W, Bataneant M, Dvorak CC, Gehring S, Gennery A, Gilmour KC, Gonzalez-Granado LI, Groß-Wieltsch U, Ifversen M, Lingman-Framme J, Matthes-Martin S, Mesters R, Meyts I, van Montfrans JM, Pachlopnik Schmid J, Pai SY, Soler-Palacin P, Schuermann U, Schuster V, Seidel MG, Speckmann C, Stepensky P, Sykora KW, Tesi B, Vraetz T, Waruiru C, Bryceson YT, Moshous D, Lehmberg K, Jordan MB, Ehl S; Inborn Errors Working Party of the EBMT. The syndrome of hemophagocytic lymphohistiocytosis in primary immunodeficiencies: implications for differential diagnosis and pathogenesis. Haematologica. 2015 Jul;100(7):978-88. doi: 10.3324/haematol.2014.121608[↩]
- Schultz KA, Neglia JP, Smith AR, Ochs HD, Torgerson TR, Kumar A. Familial hemophagocytic lymphohistiocytosis in two brothers with X-linked agammaglobulinemia. Pediatr Blood Cancer. 2008 Aug;51(2):293-5. doi: 10.1002/pbc.21573[↩]
- Alabbas F, Elyamany G, Alanzi T, Ali TB, Albatniji F, Alfaraidi H. Wolman’s disease presenting with secondary hemophagocytic lymphohistiocytosis: A case report from Saudi Arabia and literature review. BMC Pediatr. 2021;21(72) doi: 10.1186/s12887-021-02541-2[↩][↩]
- Schüller S, Attarbaschi A, Berger A, Hutter C, Klebermass-Schrehof K, Steiner M. Hemophagocytic lymphohistiocytosis triggered by Gaucher disease in a preterm neonate. Pediatr Hematol Oncol. 2016;33:462–467. doi: 10.1080/08880018.2016.1234011[↩][↩]
- Gokce M, Unal O, Hismi B, Gumruk F, Coskun T, Balta G, Unal S, Cetin M, Kalkanoglu-Sivri HS, Dursun A, Tokatlı A. Secondary hemophagocytosis in 3 patients with organic acidemia involving propionate metabolism. Pediatr Hematol Oncol. 2012;29:92–98. doi: 10.3109/08880018.2011.601402[↩]
- Erdol S, Ture M, Baytan B, Yakut T, Saglam H. An unusual case of LCHAD deficiency presenting with a clinical picture of hemophagocytic lymphohistiocytosis: Secondary HLH or coincidence? J Pediatr Hematol Oncol. 2016;38:661–662. doi: 10.1097/MPH.0000000000000626[↩]
- Düzenli Kar Y, Özdemir ZC, Kiral E, Kiliç Yildirim G, Dinleyici EÇ, Bör Ö. Hemophagocytic lymphohystiocytosis associated with type Ia glycogen storage disease. J Pediatr Hematol Oncol. 2019;41:e260–e262. doi: 10.1097/MPH.0000000000001208[↩]
- Santos Silva E, Klaudel-Dreszler M, Bakuła A, Oliva T, Sousa T, Fernandes PC, Tylki-Szymańska A, Kamenets E, Martins E, Socha P. Early onset lysosomal acid lipase deficiency presenting as secondary hemophagocytic lymphohistiocytosis: Two infants treated with sebelipase alfa. Clin Res Hepatol Gastroenterol. 2018;42:e77–e82. doi: 10.1016/j.clinre.2018.03.012[↩]
- Faraguna MC, Musto F, Crescitelli V, Iascone M, Spaccini L, Tonduti D, Fedeli T, Kullmann G, Canonico F, Cattoni A, et al. Mucopolysaccharidosis-plus syndrome, a rapidly progressive disease: Favorable impact of a very prolonged steroid treatment on the clinical course in a child. Genes (Basel) 2022;13(442) doi: 10.3390/genes13030442[↩]
- Farias-Moeller R, LaFrance-Corey R, Bartolini L, Wells EM, Baker M, Doslea A, Suslovic W, Greenberg J, Carpenter JL, Howe CL. Fueling the FIRES: Hemophagocytic lymphohistiocytosis in febrile infection-related epilepsy syndrome. Epilepsia. 2018;59:1753–1763. doi: 10.1111/epi.14524[↩][↩]
- Dandoy C, Grimley M. Secondary hemophagocytic lymphohistiocytosis (HLH) from a presumed brown recluse spider bite. J Clin Immunol. 2014;34:544–547. doi: 10.1007/s10875-014-0036-1[↩]
- Al-Samkari H, Berliner N. Hemophagocytic Lymphohistiocytosis. Annu Rev Pathol. 2018 Jan 24;13:27-49. doi: 10.1146/annurev-pathol-020117-043625[↩]
- Crayne CB, Albeituni S, Nichols KE, Cron RQ. The Immunology of Macrophage Activation Syndrome. Front Immunol. 2019 Feb 1;10:119. doi: 10.3389/fimmu.2019.00119[↩]
- Al-Samkari H, Berliner N. Hemophagocytic lymphohistiocytosis. Annu Rev Pathol. 2018;13:27–49. doi: 10.1146/annurev-pathol-020117-043625[↩]
- Ishii E. Hemophagocytic lymphohistiocytosis in children: Pathogenesis and treatment. Front Pediatr. 2016;4(47) doi: 10.3389/fped.2016.00047[↩]
- Skinner J, Yankey B, Shelton BK. Hemophagocytic lymphohistiocytosis. AACN Adv Crit Care. 2019;30:151–164. doi: 10.4037/aacnacc2019463[↩]
- Morimoto A, Nakazawa Y, Ishii E. Hemophagocytic lymphohistiocytosis: Pathogenesis, diagnosis, and management. Pediatr Int. 2016;58:817–825. doi: 10.1111/ped.13064[↩][↩]
- Jordan MB, Hildeman D, Kappler J, Marrack P. An animal model of hemophagocytic lymphohistiocytosis (HLH): CD8+ T cells and interferon gamma are essential for the disorder. Blood. 2004;104:735–743. doi: 10.1182/blood-2003-10-3413[↩]
- Brisse E, Wouters CH, Matthys P. Advances in the pathogenesis of primary and secondary haemophagocytic lymphohistiocytosis: Differences and similarities. Br J Haematol. 2016;174:203–217. doi: 10.1111/bjh.14147[↩][↩]
- Brisse E, Matthys P, Wouters CH. Understanding the spectrum of haemophagocytic lymphohistiocytosis: Update on diagnostic challenges and therapeutic options. Br J Haematol. 2016;174:175–187. doi: 10.1111/bjh.14144[↩][↩]
- Castillo L, Carcillo J. Secondary hemophagocytic lymphohistiocytosis and severe sepsis/systemic inflammatory response syndrome/multiorgan dysfunction syndrome/macrophage activation syndrome share common intermediate phenotypes on a spectrum of inflammation. Pediatr Crit Care Med. 2009;10:387–392. doi: 10.1097/PCC.0b013e3181a1ae08[↩][↩][↩]
- Jordan MB, Hildeman D, Kappler J, Marrack P. An animal model of hemophagocytic lymphohistiocytosis (HLH): CD8+ T cells and interferon gamma are essential for the disorder. Blood. 2004 Aug 1;104(3):735-43. doi: 10.1182/blood-2003-10-3413[↩]
- Grom AA, Horne A, De Benedetti F. Macrophage activation syndrome in the era of biologic therapy. Nat Rev Rheumatol. 2016;12:259–268. doi: 10.1038/nrrheum.2015.179[↩]
- Canna SW, Behrens EM. Making sense of the cytokine storm: A conceptual framework for understanding, diagnosing, and treating hemophagocytic syndromes. Pediatr Clin North Am. 2012;59:329–344. doi: 10.1016/j.pcl.2012.03.002[↩]
- Jordan MB. Emergence of targeted therapy for hemophagocytic lymphohistiocytosis. Hematologist. 2018;15[↩]
- Freeman HR, Ramanan AV. Review of haemophagocytic lymphohistiocytosis. Arch Dis Child. 2011;96:688–693. doi: 10.1136/adc.2009.176610[↩]
- Alonso EM, Horslen SP, Behrens EM, Doo E. Pediatric acute liver failure of undetermined cause: A research workshop. Hepatology. 2017;65:1026–1037. doi: 10.1002/hep.28944[↩]
- Lehmberg K, Nichols KE, Henter JI, Girschikofsky M, Greenwood T, Jordan M, Kumar A, Minkov M, La Rosée P, Weitzman S; Study Group on Hemophagocytic Lymphohistiocytosis Subtypes of the Histiocyte Society. Consensus recommendations for the diagnosis and management of hemophagocytic lymphohistiocytosis associated with malignancies. Haematologica. 2015 Aug;100(8):997-1004. doi: 10.3324/haematol.2015.123562[↩][↩]
- Griffin G, Shenoi S, Hughes GC. Hemophagocytic lymphohistiocytosis: An update on pathogenesis, diagnosis, and therapy. Best Pract Res Clin Rheumatol. 2020 Aug;34(4):101515. https://doi.org/10.1016/j.berh.2020.101515[↩][↩]
- Farquhar JW, Claireaux AE. Familial haemophagocytic reticulosis. Arch Dis Child. 1952;27:519–525. doi: 10.1136/adc.27.136.519[↩]
- Price B, Lines J, Lewis D, Holland N. Haemophagocytic lymphohistiocytosis: A fulminant syndrome associated with multiorgan failure and high mortality that frequently masquerades as sepsis and shock. S Afr Med J. 2014;104:401–406. doi: 10.7196/samj.7810[↩]
- Hemophagocytic Lymphohistiocytosis. https://rarediseases.org/rare-diseases/hemophagocytic-lymphohistiocytosis[↩]
- Henter JI, Horne A, Aricó M, Egeler RM, Filipovich AH, Imashuku S, Ladisch S, McClain K, Webb D, Winiarski J, Janka G. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007 Feb;48(2):124-31. doi: 10.1002/pbc.21039[↩]
- Ehl S, Astigarraga I, von Bahr Greenwood T, Hines M, Horne A, Ishii E, Janka G, Jordan MB, La Rosée P, Lehmberg K, Machowicz R, Nichols KE, Sieni E, Wang Z, Henter JI. Recommendations for the Use of Etoposide-Based Therapy and Bone Marrow Transplantation for the Treatment of HLH: Consensus Statements by the HLH Steering Committee of the Histiocyte Society. J Allergy Clin Immunol Pract. 2018 Sep-Oct;6(5):1508-1517. doi: 10.1016/j.jaip.2018.05.031[↩]
- Henter, J.-I., Horne, A., Aricó, M., Egeler, R.M., Filipovich, A.H., Imashuku, S., Ladisch, S., McClain, K., Webb, D., Winiarski, J. and Janka, G. (2007), HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr. Blood Cancer, 48: 124-131. https://doi.org/10.1002/pbc.21039[↩]
- Rubin TS, Zhang K, Gifford C, Lane A, Choo S, Bleesing JJ, Marsh RA. Perforin and CD107a testing is superior to NK cell function testing for screening patients for genetic HLH. Blood. 2017 Jun 1;129(22):2993-2999. doi: 10.1182/blood-2016-12-753830[↩]
- Lin M, Park S, Hayden A, Giustini D, Trinkaus M, Pudek M, Mattman A, Schneider M, Chen LYC. Clinical utility of soluble interleukin-2 receptor in hemophagocytic syndromes: a systematic scoping review. Ann Hematol. 2017 Aug;96(8):1241-1251. doi: 10.1007/s00277-017-2993-y[↩]
- Meeths M, Horne A, Sabel M, Bryceson YT, Henter JI. Incidence and clinical presentation of primary hemophagocytic lymphohistiocytosis in Sweden. Pediatr Blood Cancer. 2015 Feb;62(2):346-352. doi: 10.1002/pbc.25308[↩]
- Buatois V, Chatel L, Cons L, Lory S, Richard F, Guilhot F, Johnson Z, Bracaglia C, De Benedetti F, de Min C, Kosco-Vilbois MH, Ferlin WG. Use of a mouse model to identify a blood biomarker for IFNγ activity in pediatric secondary hemophagocytic lymphohistiocytosis. Transl Res. 2017 Feb;180:37-52.e2. doi: 10.1016/j.trsl.2016.07.023[↩]
- Prencipe G, Caiello I, Pascarella A, Grom AA, Bracaglia C, Chatel L, Ferlin WG, Marasco E, Strippoli R, de Min C, De Benedetti F. Neutralization of IFN-γ reverts clinical and laboratory features in a mouse model of macrophage activation syndrome. J Allergy Clin Immunol. 2018 Apr;141(4):1439-1449. doi: 10.1016/j.jaci.2017.07.021[↩]
- Mellor-Heineke S, Villanueva J, Jordan MB, Marsh R, Zhang K, Bleesing JJ, Filipovich AH, Risma KA. Elevated Granzyme B in Cytotoxic Lymphocytes is a Signature of Immune Activation in Hemophagocytic Lymphohistiocytosis. Front Immunol. 2013 Mar 22;4:72. doi: 10.3389/fimmu.2013.00072[↩]
- Allen CE, Yu X, Kozinetz CA, McClain KL. Highly elevated ferritin levels and the diagnosis of hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2008 Jun;50(6):1227-35. doi: 10.1002/pbc.21423[↩]
- Tsuji T, Hirano T, Yamasaki H, Tsuji M, Tsuda H. A high sIL-2R/ferritin ratio is a useful marker for the diagnosis of lymphoma-associated hemophagocytic syndrome. Ann Hematol. 2014 May;93(5):821-6. doi: 10.1007/s00277-013-1925-8[↩]
- Bode SF, Ammann S, Al-Herz W, et al. Inborn Errors Working Party of the EBMT. The syndrome of hemophagocytic lymphohistiocytosis in primary immunodeficiencies: implications for differential diagnosis and pathogenesis. Haematologica. 2015 Jul;100(7):978-88. doi: 10.3324/haematol.2014.121608[↩]
- La Rosée P, Horne A, Hines M, von Bahr Greenwood T, Machowicz R, Berliner N, Birndt S, Gil-Herrera J, Girschikofsky M, Jordan MB, Kumar A, van Laar JAM, Lachmann G, Nichols KE, Ramanan AV, Wang Y, Wang Z, Janka G, Henter JI. Recommendations for the management of hemophagocytic lymphohistiocytosis in adults. Blood. 2019 Jun 6;133(23):2465-2477. https://doi.org/10.1182/blood.2018894618[↩][↩]
- Fardet L, Galicier L, Lambotte O, Marzac C, Aumont C, Chahwan D, Coppo P, Hejblum G. Development and validation of the HScore, a score for the diagnosis of reactive hemophagocytic syndrome. Arthritis Rheumatol. 2014 Sep;66(9):2613-20. doi: 10.1002/art.38690[↩]
- Xu XJ, Tang YM, Song H, Yang SL, Xu WQ, Zhao N, Shi SW, Shen HP, Mao JQ, Zhang LY, Pan BH. Diagnostic accuracy of a specific cytokine pattern in hemophagocytic lymphohistiocytosis in children. J Pediatr. 2012 Jun;160(6):984-90.e1. doi: 10.1016/j.jpeds.2011.11.046[↩]
- Han XC, Ye Q, Zhang WY, Tang YM, Xu XJ, Zhang T. Cytokine profiles as novel diagnostic markers of Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis in children. J Crit Care. 2017 Jun;39:72-77. doi: 10.1016/j.jcrc.2017.02.018[↩]
- Zondag TC, Rokx C, van Lom K, van den Berg AR, Sonneveld P, Dik WA, van Doornum GJ, Lam KH, van Laar JA. Cytokine and viral load kinetics in human herpesvirus 8-associated multicentric Castleman’s disease complicated by hemophagocytic lymphohistiocytosis. Int J Hematol. 2016 Apr;103(4):469-72. doi: 10.1007/s12185-015-1928-4[↩]
- Merli P, Quintarelli C, Strocchio L, Locatelli F. The role of interferon-gamma and its signaling pathway in pediatric hematological disorders. Pediatr Blood Cancer. 2021;68(e28900) doi: 10.1002/pbc.28900[↩]
- Wegehaupt O, Wustrau K, Lehmberg K, Ehl S. Cell Versus Cytokine – Directed Therapies for Hemophagocytic Lymphohistiocytosis (HLH) in Inborn Errors of Immunity. Front Immunol. 2020 May 8;11:808. doi: 10.3389/fimmu.2020.00808[↩]
- Henter JI, Samuelsson-Horne A, Aricò M, Egeler RM, Elinder G, Filipovich AH, et al. Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunochemotherapy and bone marrow transplantation. Blood. (2002) 100:2367–73. 10.1182/blood-2002-01-0172[↩]
- Johnson TS, Terrell CE, Millen SH, Katz JD, Hildeman DA, Jordan MB. Etoposide selectively ablates activated T cells to control the immunoregulatory disorder hemophagocytic lymphohistiocytosis. J Immunol. (2014) 192:84–91. 10.4049/jimmunol.1302282[↩]
- Horne A, Wickström R, Jordan MB, Ann Yeh E, Naqvi A, Henter JI, et al. How to treat involvement of the central nervous system in hemophagocytic lymphohistiocytosis? Curr Treat Options Neurol. (2017) 19:3 10.1007/s11940-017-0439-4[↩]
- Ehl S, Astigarraga I, von Bahr Greenwood T, Hines M, Horne A, Ishii E, et al. Recommendations for the use of etoposide-based therapy and bone marrow transplantation for the treatment of HLH: consensus statements by the HLH steering committee of the histiocyte society. J Allergy Clin Immunol Pract. (2018) 6:1508–17. 10.1016/j.jaip.2018.05.031[↩]
- Bergsten E, Horne A, Aricó M, Astigarraga I, Egeler RM, Filipovich AH, et al. Confirmed efficacy of etoposide and dexamethasone in HLH treatment: long-term results of the cooperative HLH-2004 study. Blood. (2017) 130:2728–38. 10.1182/blood-2017-06-788349[↩]
- Rajagopala S, Dutta U, Chandra KSP, Bhatia P, Varma N, Kochhar R. Visceral leishmaniasis associated hemophagocytic lymphohistiocytosis-case report and systematic review. J Infect. 2008;56:381–388. doi: 10.1016/j.jinf.2008.02.013[↩][↩]
- Cançado GGL, Freitas GG, Faria FHF, de Macedo AV, Nobre V. Hemophagocytic lymphohistiocytosis associated with visceral leishmaniasis in late adulthood. Am J Trop Med Hyg. 2013;88:575–577. doi: 10.4269/ajtmh.12-0563[↩]
- Ay Y, Yildiz B, Unver H, Karapinar DY, Vardar F. Hemophagocytic lymphohistiocytosis associated with H1N1 virus infection and visceral leishmaniasis in a 4.5-month-old infant. Rev Soc Bras Med Trop. 2012 Jun;45(3):407-9.[↩]
- Imashuku S, Kuriyama K, Teramura T, Ishii E, Kinugawa N, Kato M, Sako M, Hibi S. Requirement for etoposide in the treatment of Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis. J Clin Oncol. 2001;19:2665–2673. doi: 10.1200/JCO.2001.19.10.2665[↩]
- Huang Z, Xie J. Ruxolitinib in conjunction with the HLH-94 protocol for Epstein-Barr virus-related hemophagocytic lymphohistiocytosis in the intensive care unit: A case report. Medicine (Baltimore) 2021;100(e25188) doi: 10.1097/MD.0000000000025188[↩]
- Zhao Y, Li Z, Zhang L, Lian H, Ma H, Wang D, Zhao X, Zhang Q, Wang T, Zhang R. L-DEP regimen salvage therapy for paediatric patients with refractory Epstein-Barr virus-associated haemophagocytic lymphohistiocytosis. Br J Haematol. 2020;191:453–459. doi: 10.1111/bjh.16861[↩]
- Huang P, Huang C, Xu H, Lu J, Tian R, Wang Z, Chen Y. Early use of blood purification in severe Epstein-Barr virus-associated hemophagocytic syndrome. Pediatrics. 2020;145(e20193197) doi: 10.1542/peds.2019-3197[↩]
- Shiraishi A, Ohga S, Doi T, Ishimura M, Takimoto T, Takada H, Miyamoto T, Abe Y, Hara T. Treatment choice of immunotherapy or further chemotherapy for Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2012;59:265–270. doi: 10.1002/pbc.24039[↩]
- Belyea B, Hinson A, Moran C, Hwang E, Heath J, Barfield R. Spontaneous resolution of Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2010;55:754–756. doi: 10.1002/pbc.22618[↩]
- Gurunathan A, Boucher AA, Mark M, Prus KM, O’Brien MM, Breese EH, Mizukawa BE, Absalon MJ, Nelson AS, Jordan MB, et al. Limitations of HLH-2004 criteria in distinguishing malignancy-associated hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2018;65(e27400) doi: 10.1002/pbc.27400[↩][↩]
- Lackner H, Seidel MG, Strenger V, Sovinz P, Schwinger W, Benesch M, Sperl D, Urban C. Hemophagocytic syndrome in children with acute monoblastic leukemia-another cause of fever of unknown origin. Support Care Cancer. 2013;21:3519–3523. doi: 10.1007/s00520-013-1937-x[↩]
- Han L, Li L, Wu J, Li X, Zhang L, Wang X, Fu X, Ma W, Sun Z, Zhang X, et al. Clinical features and treatment of natural killer/T cell lymphoma associated with hemophagocytic syndrome: comparison with other T cell lymphoma associated with hemophagocytic syndrome. Leuk Lymphoma. 2014;55:2048–2055. doi: 10.3109/10428194.2013.876629[↩]
- Chi MS, Chang YC, Chi KH. In Reply to Roos et al. Int J Radiat Oncol Biol Phys. 2018 Jul 15;101(4):1003. doi: 10.1016/j.ijrobp.2018.03.042[↩]
- Eloseily EM, Weiser P, Crayne CB, Haines H, Mannion ML, Stoll ML, Beukelman T, Atkinson TP, Cron RQ. Benefit of anakinra in treating pediatric secondary hemophagocytic lymphohistiocytosis. Arthritis Rheumatol. 2020;72:326–334. doi: 10.1002/art.41103[↩]
- Bami S, Vagrecha A, Soberman D, Badawi M, Cannone D, Lipton JM, Cron RQ, Levy CF. The use of anakinra in the treatment of secondary hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2020;67(e28581) doi: 10.1002/pbc.28581[↩]
- Bagri NK, Gupta L, Sen ES, Ramanan AV. Macrophage activation syndrome in children: Diagnosis and management. Indian Pediatr. 2021;58:1155–1161. doi: 10.1007/s13312-021-2399-8[↩]
- Ravelli A, Minoia F, Davì S, Horne A, Bovis F, Pistorio A, Aricò M, Avcin T, Behrens EM, De Benedetti F, et al. 2016 Classification criteria for macrophage activation syndrome complicating systemic juvenile idiopathic arthritis: A European league against rheumatism/American college of rheumatology/paediatric rheumatology international trials organisation collaborative initiative. Arthritis Rheumatol. 2016;68:566–576. doi: 10.1002/art.39332[↩]
- Shalabi H, Gust J, Taraseviciute A, Wolters PL, Leahy AB, Sandi C, Laetsch TW, Wiener L, Gardner RA, Nussenblatt V, et al. Beyond the storm-subacute toxicities and late effects in children receiving CAR T cells. Nat Rev Clin Oncol. 2021;18:363–378. doi: 10.1038/s41571-020-00456-y[↩]
- Ishida H, Yoshida H, Yoshihara T, Ito M, Morimoto A. Origin of macrophages involved in the development of allogeneic hematopoietic stem cell transplantation-associated hemophagocytic syndrome: Observations on a patient with juvenile myelomonocytic leukemia. Bone Marrow Transplant. 2007;40:701–703. doi: 10.1038/sj.bmt.1705783[↩]
- Ostronoff M, Ostronoff F, Coutinho M, Calixto R, Souto Maior AP, Sucupira A, Florencio R, Tagliari C. Hemophagocytic syndrome after autologous peripheral blood stem cell transplantation for multiple myeloma; successful treatment with high-dose intravenous immunoglobulin. Bone Marrow Transplant. 2006;37:797–798. doi: 10.1038/sj.bmt.1705329[↩]
- Koyama M, Sawada A, Yasui M, Inoue M, Kawa K. Encouraging results of low-dose etoposide in the treatment of early-onset hemophagocytic syndrome following allogeneic hematopoietic stem cell transplantation. Int J Hematol. 2007;86:466–467. doi: 10.1007/BF02984009[↩]
- Ono R, Ashiarai M, Hirabayashi S, Mizuki K, Hosoya Y, Yoshihara H, Ohtake J, Mori S, Manabe A, Hasegawa D. Ruxolitinib for hematopoietic cell transplantation-associated hemophagocytic lymphohistiocytosis. Int J Hematol. 2021;113:297–301. doi: 10.1007/s12185-020-02999-3[↩]
- Imashuku S. Treatment of Epstein-Barr virus-related hemophagocytic lymphohistiocytosis (EBV-HLH); update 2010. J Pediatr Hematol Oncol. 2011 Jan;33(1):35-9. doi: 10.1097/MPH.0b013e3181f84a52[↩]
- Janka GE. Familial hemophagocytic lymphohistiocytosis. Eur J Pediatr. 1983;140:221–230. doi: 10.1007/BF00443367[↩]

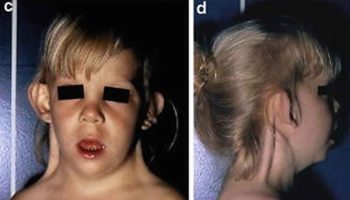



{kind=link}