Contents
What is topoisomerase
DNA topoisomerases are essential enzymes that control the topological state of DNA replication during mitosis 1. DNA topoisomerases catalyze the interconversion of different topological forms of DNA, such as the knotting and unknotting of DNA and catenation and decatenation of DNA rings 2. All organisms contain at least two classes of DNA topoisomerases, namely type 1 and type 2 DNA topoisomerases in which the enzymes are classified based on their mechanisms and physical properties. Type 1 topoisomerases are able to unknot or decatenate single strand of the DNA duplex and catenanes, but are unable to carry out these reactions on intact double-stranded DNA 3, 4, 5, 6, 5. Type 2 topoisomerases are able to catalize the knotting/unknotting of intact double-stranded DNA and the catenation/decatenation of intact double-stranded DNA 3, 4, 5, 6, 5. Both type 1 and type 2 topoisomerases can relax supercoiled DNA 3, 4, 5, 6, 5. Because type 1 topoisomerases cleave only a single strand DNA, they can modulate DNA supercoiling, but cannot remove knots or tangles from intact duplex DNA 7, 8, 9. In contrast, because type 2 topoisomerases cleave both strands of the double helix DNA, they can regulate DNA under- and overwinding and can also remove tangles and knots from the genome 7, 8, 9, 10, 11.
The DNA topoisomerase subtypes (A, B or C) are differentiated by amino acid sequences or structures 12. For example, enzymes that cleave only one strand of DNA are defined as type 1 with a further classification of type 1-A subtypes for proteins linked to a 5‟-phosphate and type 1-B subtypes for proteins attached to a 3‟-phosphate during the relaxation of the cleavable DNA. The enzymes are further divided into subfamilies based on the structural changes induced by gene duplication, such as DNA topoisomerase IIα and IIβ 13.
DNA topoisomerase enzymes are classified based on their mechanisms and physical properties. During mitosis, superhelical DNA must be unwound or relaxed by DNA topoisomerases prior to a decoding step by DNA processing enzymes, such as DNA polymerase and RNA polymerase. By blocking the reaction of resealing the breaks in the DNA ultimately can result in cellular death. Compounds that inhibit the catalytic function of DNA topoisomerase enzymes represent highly relevant targets for the treatment of various types of cancer and DNA topoisomerases are under continuous investigation as targets for novel classes of inhibitory agents. While nearly all clinically marketed topoisomerase inhibitors used to treat cancer fall within the topoisomerase poison mechanistic category, several very promising catalytic inhibitors have been disclosed recently and offer promise as potentially novel anti-cancer agents with significantly reduced toxicity profiles. Of note are the novel chemical classes with activity against type I topoisomerases, as there is currently only one marketed class of inhibitors in clinical use.
DNA topoisomerases are essential enzymes that control the topological state of DNA during cellular processes in both prokaryotic and eukaryotic cells. The cellular processes include DNA replication, transcription, recombination and chromatin segregation, to control the synthesis of proteins and to facilitate DNA replication during mitosis 14.
Studies in eukaryotes have shown that type 1 DNA topoisomerase is associated with actively transcribed genes, whereas type 2 DNA topoisomerase is required for DNA replication and the successful traverse of mitosis 15. Type 1 and type 2α DNA topoisomerases are well known targets for chemotherapy of advanced or recurrent human cancers.
DNA topoisomerase types
The DNA topoisomerases can be categorized into two general subfamilies, type I and type II topoisomerases. Further sub-divisions of the topoisomerases, types IA, IB, and IIA are categorized based upon the polarity of the DNA-protein bond (i.e., tyrosine attachment to the 5′- or 3′-phosphate), the mechanism (strand passage or rotation), and the number of overhanging DNA bases in the staggered double strand cleavage of the type II topoisomerases 16. Structurally, eukaryotic type I topoisomerases are monomeric protein, single-gene products, while eukaryotic type II topoisomerases are homodimers, composed of the products of a distinct gene. Functionally, type I topoisomerases are not dependent on ATP hydrolysis to power the energy required for their reaction, instead they derive the energy required from the intrinsic strain energy of the supercoiled DNA itself 17. Type II topoisomerases are typically ATP-dependent and possess an ATP-binding domain separate from the DNA-binding domain. Additionally, the DNA topoisomerases can also be distinguished by their dependence, or lack of dependence, on Mg2+ as a requirement for catalytic activity. The type IA and all type II topoisomerases appear to require Mg2+, while the type IB topoisomerases are catalytically active in the absence of Mg2+ 18. The targets of the currently marketed cancer chemotherapeutic agents are topoisomerase I, IIα, and IIβ 16.
Topoisomerase 1
DNA topoisomerase 1 or type 1 DNA topoisomerases are monomeric proteins and ATP-independent enzymes that induce a single strand DNA break via a phosphodiester bond between the tyrosine group of the enzyme and the phosphate group of the DNA (Figure 1). DNA topoisomerase III is an example of a type I-A enzyme that is more active as a decatenating enzyme than as a DNA-relaxing enzyme 19.
Figure 1. DNA topoisomerase type 1
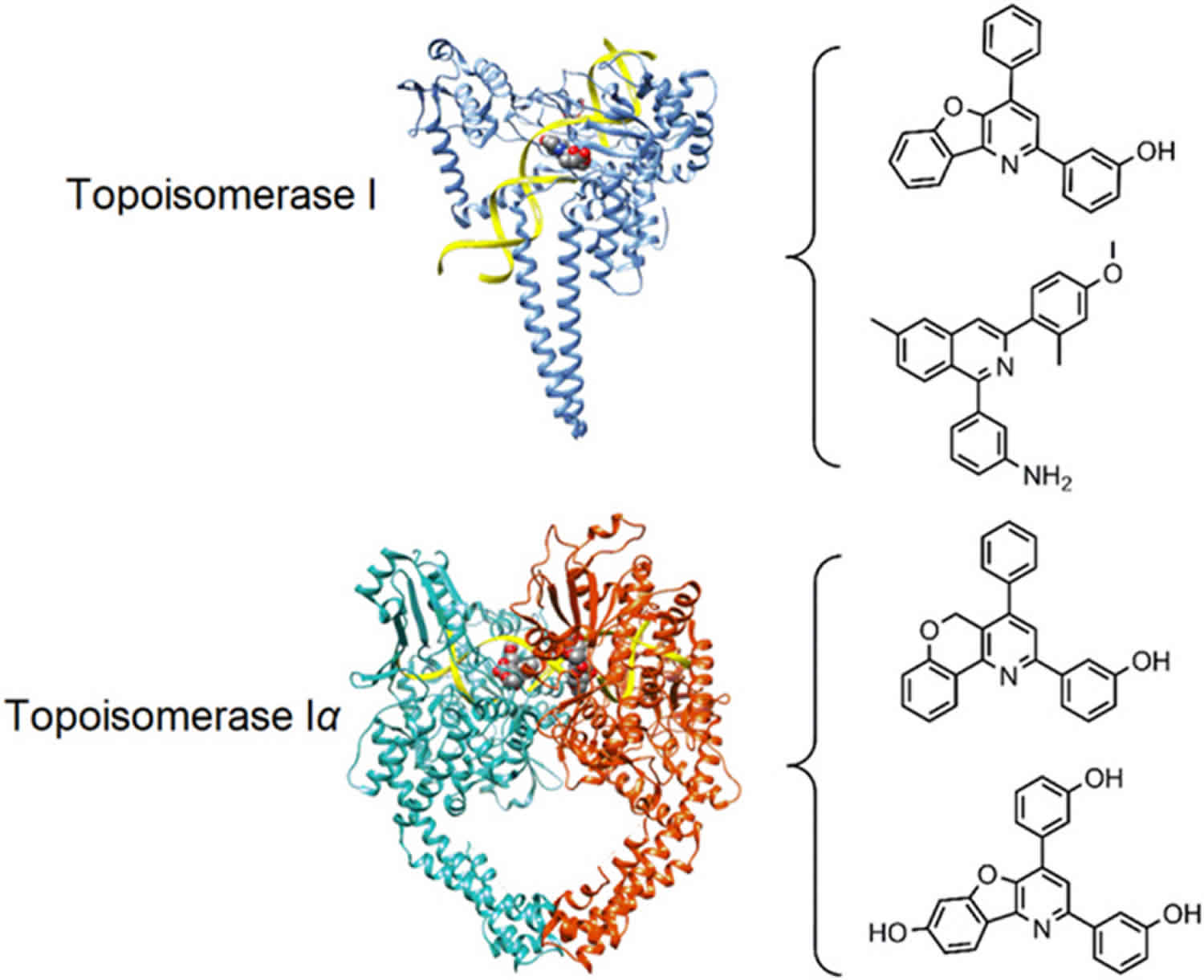
Topoisomerase 2
Type 2 DNA topoisomerases are tetrameric proteins formed by two different subunits, GyrA2GyrB2 for gyrase and ParC2ParE2 for DNA topoisomerase 4 20. Requiring ATP, these enzymes act by making a transient break on the double stranded DNA, passing through an intact duplex DNA via the broken strand followed by a resealing of the transient break 21.
Figure 2. Human DNA topoisomerases type 1 and type 2 structure
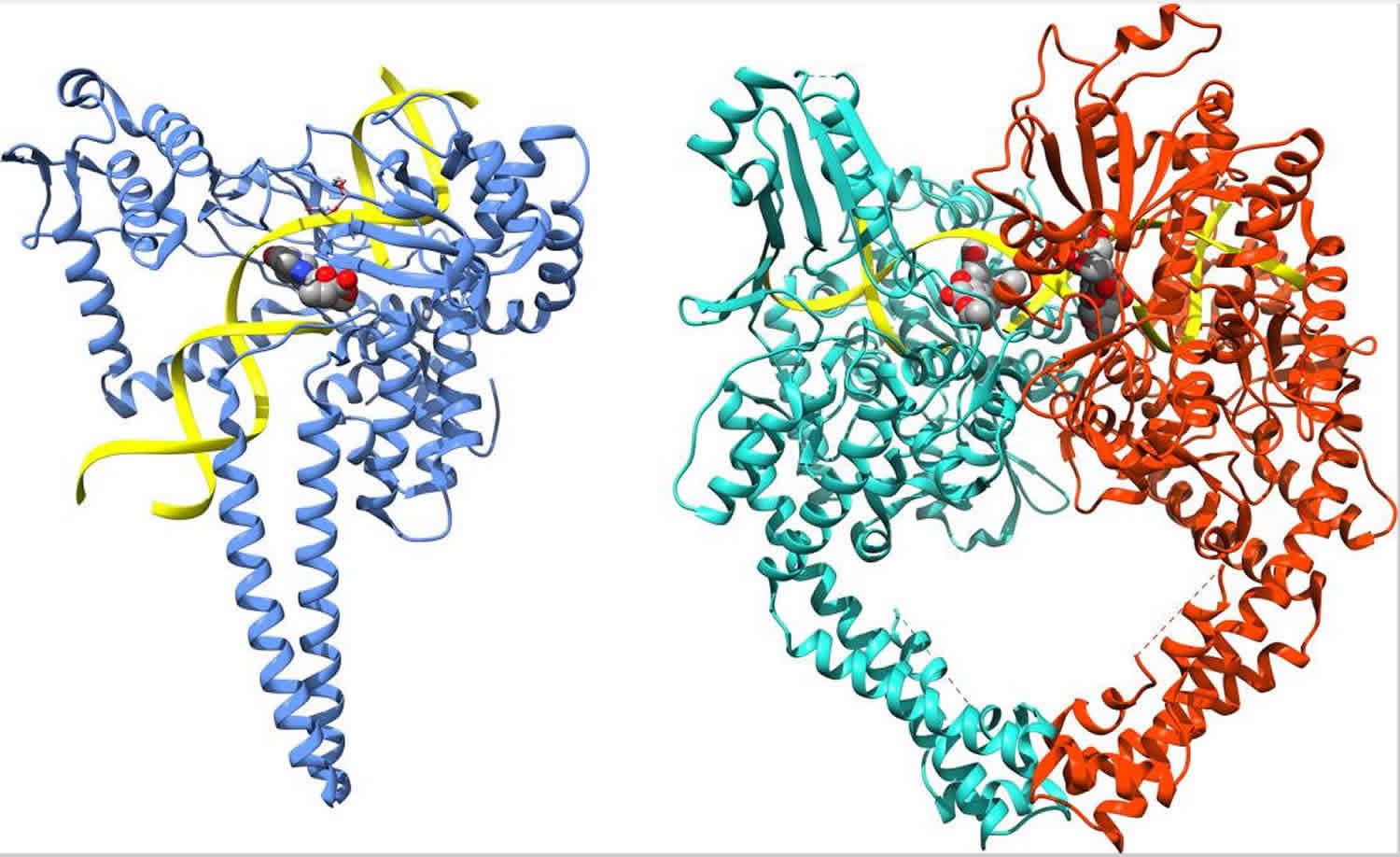
Footnote: Structures of human topoisomerases. Shown are the structures of the full length human topoisomerase 1 (left, PDB ID 1k4t) and topoisomerase 2α (right, PDB ID 5qwk) enzymes with bound DNA, representative of the overall structure and domains of the two sub-family types (DNA ribbon is colored yellow; chain A of topoisomerase 1 is blue; chain A of topoisomerase IIα is green; chain B of topoisomerase 2α is red).
[Source 16 ]Topoisomerase function
The various topoisomerases enzymes deal with the topological issues related to DNA transcription and replication and are capable of relaxing positive or negatively supercoiled DNA, introducing negative or positive supercoils into DNA, and catenating or decatenating (disentangling) circular and linear DNA 22.
Table 1. Summary of eukaryotic DNA topoisomerase enzyme function and mechanism
| Type | Subtype | Protein | Gene | Function | Mechanism | Multimericity | Metal dependence | Cleavage polarity |
|---|---|---|---|---|---|---|---|---|
| 1 | A | Topoisomerase 3α | top3A | (—) Supercoil relaxation | Strand passage | Monomer | Yes (Mg2+) | 5′ |
| Topoisomerase 3β | top3B | Unknown | ||||||
| B | Topoisomerase 1 | top1 | (—) and (+) supercoil relaxation | Strand rotation | Monomer | No | 3′ | |
| 2 | A | Topoisomerase 2α | top2A | Decatenation during replication | Strand passage (ATPase) | Homodimer | Yes (Mg2+) | 5′ |
| Topoisomerase 2β | top2B | Various, neuronal transcription |
The type IA topoisomerases represented by eukaryotic topoisomerase IIIα and IIIβ consist of four domains that coordinate DNA binding, cleavage, and strand passage 23. Domain I contains the so-called TOPRIM fold, a Rossman-fold like structure that can bind magnesium ions 23. Domain II consists primarily of β-strands that forms the central core and links domain III, which contains the catalytic tyrosine residue, to domain IV. The type IB topoisomerases, represented by eukaryotic topoisomerase I, is also composed of four domains including an N-terminal domain, a linker domain, a core domain, and a C-terminal domain 24. The core domain is responsible for DNA binding and appears to be highly conserved. The catalytic tyrosine is in the C-terminal domain and operates as part of a catalytic pentad with four residues located in the core domain. Type IIA topoisomerases, represented by eukaryotic topoisomerase IIα and IIβ, consist of a three-domain structure spanning the A and B subunits that form the homodimer (or heterotetramer in prokaryotes). The N-terminal domain contains the ATB-binding region (ATPase domain), a core domain that contains a TOPRIM fold and DNA-binding region, and a C-terminal domain with unclear function 25.
The sub-types of topoisomerases are mechanistically distinct with respect to how they act on their DNA substrates. Type IA topoisomerases operate using a “strand-passage” mechanism, whereby a single strand of DNA is passed through a break in a second single DNA stand. In this mechanism, the first DNA single strand binds to domain I and III of the topoisomerase and is cleaved by the catalytic tyrosine residue located on domain III, creating a 5′-phosphodiester bond between the enzyme and the DNA. Domain II is then believed to act as a hinge, separating the cleaved DNA strand and permitting a second strand to pass through, after which domains I and III come back together and the cleaved DNA strand is re-joined. The type IB topoisomerases operate using a “hindered rotation mechanism”, whereby the enzyme binds to DNA and cleaves a single strand via a 3′-phosphodiester bond using an active site tyrosine residue. The 5′-end then rotates about the second DNA strand, relaxing the DNA. As discussed above, both type IA and type IB topoisomerases use the DNA torsional strain (torque) to drive the uncoiling process rather than ATP hydrolysis. Type IA topoisomerases relax negatively supercoiled DNA and perform DNA decatenation, while type IB topoisomerases can relax both negatively and positively supercoiled DNA. Like the type IA topoisomerases, the type IIA topoisomerases exert their effect through a strand passage mechanism, in this case using what is known as a “two-gate” mechanism24. Double-stranded DNA, called the “G-segment”, binds to the topoisomerase in a central DNA-binding region called the “DNA-gate”. A second double-stranded DNA, the “T-segment”, binds to the N-terminal ATPase domain facilitated by ATP binding and domain dimerization. ATP hydrolysis and the release of inorganic phosphate causes a double-stranded break with 4-base overhang in the G-segment DNA facilitated by the formation of 5′-phosphodiester bonds between two tyrosine residues and individual strands of the G-segment DNA. The DNA-binding gate separates, and the T-segment is passed through the G-segment into the C-terminal region, or “C-gate”. The C-gate opens and releases the T-segment DNA, and ADP product is released, resetting the enzyme for another catalytic cycle.
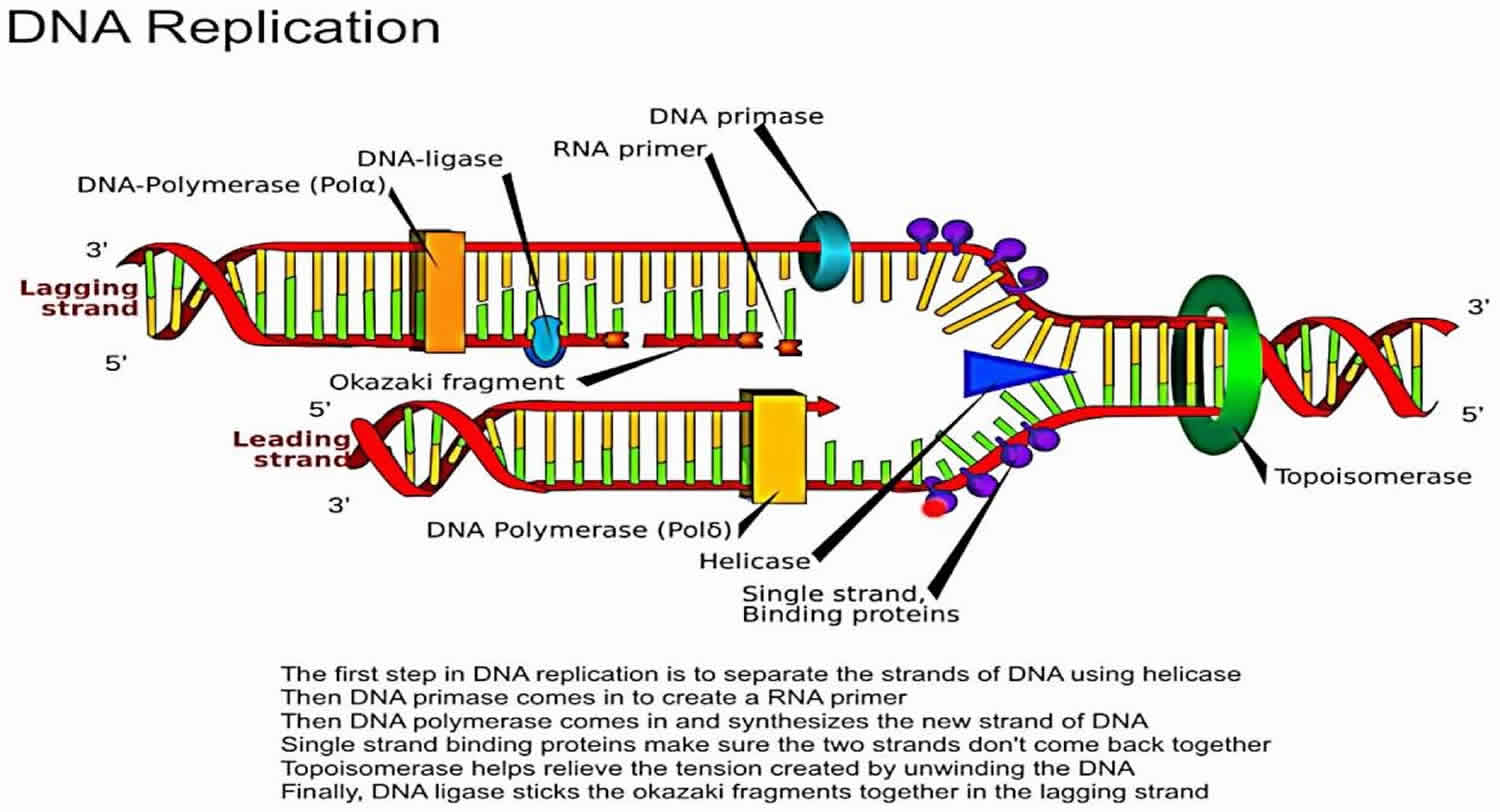
Topoisomerase in DNA replication
In general, DNA is wound around a basic protein called histone in a tightly coiled or supercoiled formation 14. Sequences of DNA must be unwound or relaxed prior to being read by DNA processing enzymes, such as DNA polymerase and RNA polymerase. Type II DNA topoisomerases or DNA topoisomerase II temporarily break the DNA strand, allowing both ends to rotate within the enzyme freely. This mechanism allows the DNA to unwind. The enzyme then reconnects the two ends, leaving a part of the relaxed DNA ready for processing. DNA topoisomerase II inhibitors, such as etoposide, stabilize the enzyme with the DNA strand cut in the enzyme-DNA complex, leaving a permanent break in the double strand of the DNA. This DNA strand break is capped by the remnants of the enzyme and is difficult to repair.
The expressions of DNA topoisomerases in cancer tissues and cells
DNA topoisomerases are over-expressed at different levels in both normal and cancerous tissues. The expression of DNA topoisomerase I in colorectal cancer, as detected by immunohistochemical methods, was a biological marker for the chemosensitivity of tumour against DNA topoisomerase I inhibitors 26. Using the investigation with reverse transcription PCR, differential expression studies in small cell lung cancer (SCLC) and non-small cell lung cancer (NSCLC) demonstrated that the DNA topoisomerase IIα gene in tumors was expressed at significantly higher level than in normal lung tissues, and the relative expression in small cell lung cancer was significantly higher than in non-small cell lung cancer 27. Meanwhile, immunostaining for topoisomerase IIIβ was found in 33.9% of breast carcinoma and the immunopositivity was correlated with distant metastasis and death 28. Decreased expression of topoisomerase IIIβ was correlated with low expression of Ki67, positivity to BRCA1, but negativity for HER2 and p53 expressions in the study.
The highly invasive human cancer cell lines HT-29 and MDA-MB-231 expressed higher levels of DNA topoisomerases than the levels measured in HeLa and MCF-7. Both DNA topoisomerases I and II were not detected in HeLa and MCF-7 by conventional PCR. These results indicated that both HT-29 and MDA-MB-231 may be sensitive to DNA topoisomerase inhibitors whereas HeLa and MCF-7 may not be sensitive to these compounds. Expression levels of DNA topoisomerase II were observed to be higher than DNA topoisomerase I in both HT-29 and MDA-MB-231, indicating that DNA topoisomerase II plays a more important role than DNA topoisomerase I in these highly invasive human cancer cells. In addition, these cells might be more sensitive to DNA topoisomerase II inhibitors than DNA topoisomerase inhibitor I. Decreased expression of DNA topoisomerase I and II had been demonstrated to indicate a resistance against certain anticancer agents 29, suggesting that these enzymes are promising targets for anticancer agents.
Topoisomerase inhibitors
The first known class of topoisomerase inhibitors used for the treatment of cancer was the anthracycline agents. Table 2 summarizes the currently marketed topoisomerase active agents used for cancer chemotherapy. The anthracyclines were first extracted from bacterial Streptomyces species and discovered to possess antibiotic and antitumor activity 30. The clinically marketed anthracycline derivatives include doxorubicin, epirubicin, valrubicin, daunorubicin, and idarubicin. Their structures are shown in Figure 3. Doxorubicin has several current indications including treatment of breast cancer, various types of leukemia, lymphoma, sarcomas, carcinomas, and other tumors. Other anthracyclines which have indications for treatment of leukemia include daunorubicin and idarubicin. Epirubicin is indicated in breast cancer following resection, while valrubicin is indicated in urinary bladder carcinoma. The anthracycline agents affect their cytotoxic activity by acting as topoisomerase “poisons”, as discussed in the previous section. They primarily affect type IIa topoisomerases, topoisomerase IIα and topoisomerase IIβ indiscriminately, by intercalation into the bound and cleaved DNA, stabilizing the DNA and topoisomerase complex and preventing DNA re-ligation by the topoisomerase. Interestingly, this appears to be one of the mechanisms that the anthracyclines induce cell death 31. The production of free radical species in an iron-dependent manner appears to be another mechanism behind the anthracyclines׳ cytotoxic effects in cancer cells. Unfortunately, this mechanism also appear to be responsible for additional toxicities associated with this class of agents 32. Anthracyclines are well known for their cardiotoxic properties, causing both acute and chronic cardiac complications in a dose-dependent manner. Menna and colleagues 33 discuss the mechanisms behind this toxicity, including anthracyclines׳ production of superoxide anions and hydrogen peroxide in cardiac muscle cells, which contribute to oxidative stress and eventually apoptosis. Anthracyclines also disrupt calcium and iron levels in cardiac cells, which leads to even more free-radical generation 33. Some recent studies have implied another potential mechanism of the cardiotoxicity 34. These studies suggest that inhibition of topoisomerase IIβ, which is overexpressed in the heart, by the anthracycline agents may result cardiotoxicity from apoptosis and ROS. Dexrazoxane, discussed above, is an FDA-approved agent indicated to reduce adverse cardiac affects in women who have received a total dose of 300 mg/m² of doxorubicin and are continuing to receive doxorubicin 35. Interestingly, the mechanism of cardiotoxicity mitigation appears to be related, at least partially, to the ability of the drug to chelate iron. The drug is also known to catalytically inhibit topoisomerase IIβ, as discussed above, which may reduce cardiotoxicity related to generation of the topoisomerase IIβ poisons formed by anthracyclines. Other investigational measures to prevent cardiotoxicity include the use of the beta blocker carvedilol. Spallarossa and colleagues found a decrease in free radical production and apoptosis in heart muscle cells when treating cells with carvedilol prior to treatment with doxorubicin, due to carvedilol׳s unique antioxidant effect 36.
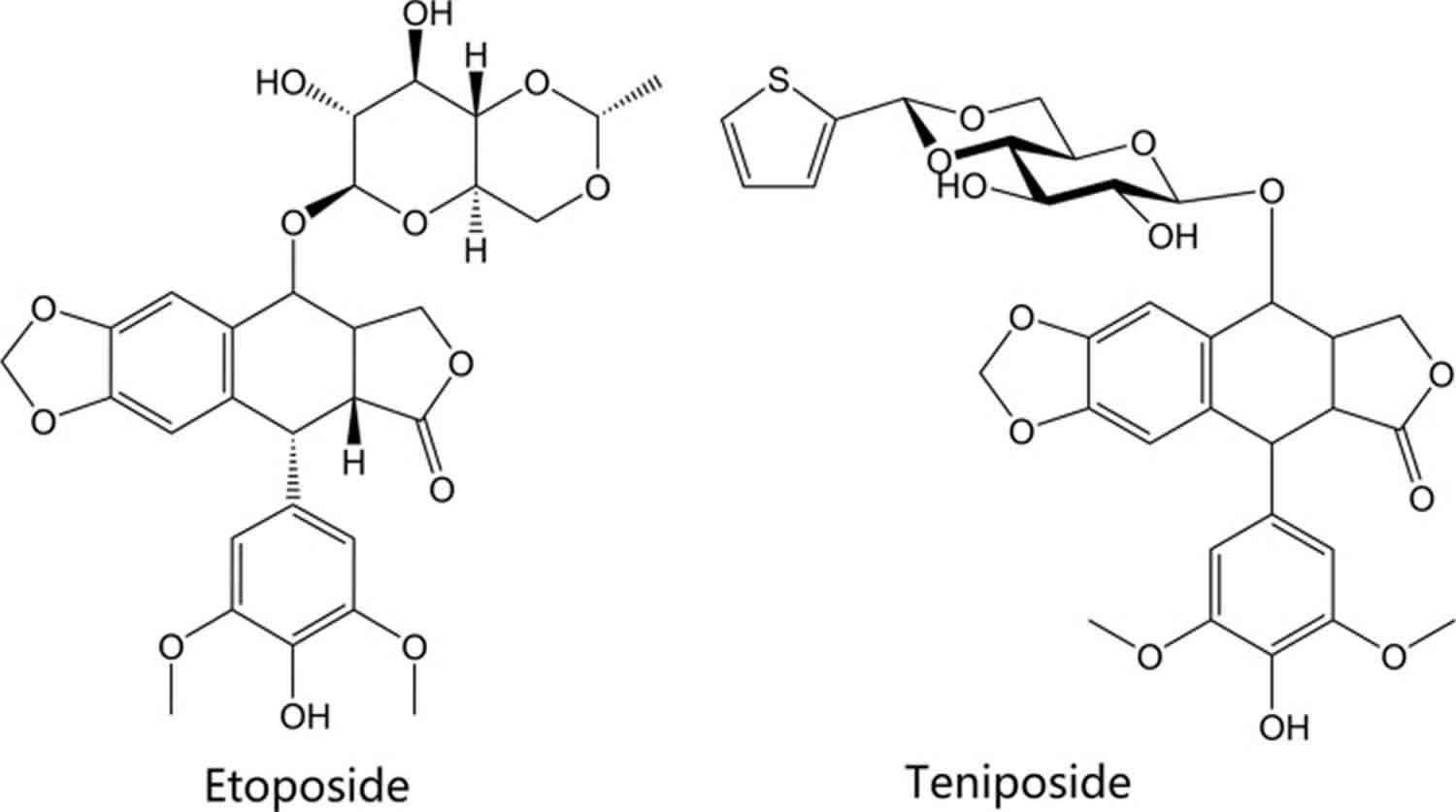
The anthracenedione agents include the drugs mitoxantrone and pixantrone. The anthracenedione (anthraquinone) agents are synthetic agents, designed to act similarly to the anthracycline drugs, with fewer adverse effects and toxicities. Mitoxantrone was approved in 1987 in the U.S. and pixantrone, a newer agent, was approved in 2012 in Europe. The structures of these agents are shown in Figure 4. Mitoxantrone can cross the blood brain barrier and is indicated for the reduction of the frequency and intensity of multiple sclerosis relapses in addition to its indication as a chemotherapeutic agent in leukemia and prostate cancer 37. Similar to the anthracyclines, the anthracenedione agents are topoisomerase poisons, primarily affecting type II topoisomerases. Like the previously discussed agents, mitoxantrone intercalates topoisomerase-bound DNA, preventing DNA re-ligations, and ultimately resulting in DNA strand breakage and disruption of DNA repair. Pixantrone, an aza-anthracenedione approved in treatment of non-hodgkin B-cell lymphoma, exhibits its cytotoxic effects by intercalating into DNA like the anthracyclines, but also causes long term cell damage and eventual death by causing errors in mitosis and segregation of chromosomes 38. Pixantrone has shown less toxicity than doxorubicin in cardiac muscle cells because of its inability to bind iron and contribute to free radical production in the heart; as a result, animal models showed decreased heart weight in animals treated with doxorubicin as compared to pixantrone 39.
Clinically marketed camptothecin derivatives include topotecan, irinotecan, and belotecan. Their structures are shown in Figure 5. The camptothecin alkaloid was first derived from the Chinese tree, Camptotheca acuminata. The camptothecin-derived agents are topoisomerase poisons which primarily affect type I topoisomerases. In vitro studies showed camptothecin, a cytotoxic alkaloid, was capable of inhibiting topoisomerase I and causing DNA strand breaks, thus preventing DNA replication 40. The water-soluble forms of camptothecin include the clinically marketed irinotecan and topotecan, which reversibly bind and form a ternary complex with topoisomerase I and DNA 41. Topotecan is approved as second-line small cell lung cancer and, in combination with cisplatin, for patients with stage IV-B cervical carcinoma not treated by surgery or radiation. Irinotecan is approved following failure or progression following treatment with fluorouracil or in combination with 5-fluorouracil and leucovorin for patients with metastatic colon or rectal carcinoma. Belotecan is a relatively new camptothecin derivative agent, approved in South Korea for treatment of non-small-cell lung cancer and ovarian cancer in 2003 42. The mechanism of action is the same as other agents in this class. Compared with older camptothecin agents, belotecan is reported to have a similar efficacy profile, with reduced toxicities 43.
The drugs etoposide and teniposide are epipodophyllotoxin-derived agents. Epipodophyllotoxins are natural substances derived from the Mayapple plant (wild mandrake), Podophyllum peltatum 44. The drugs have been available in the U.S. and other countries since the early 1980s. Their structures are shown in Figure 6. Both agents act as topoisomerase poisons and cause DNA strand breaks by binding to type II topoisomerases, similar to the agents described above 45. Etoposide is indicated as part of a multi-drug chemotherapy regimen for refractory testicular tumors and in combination with cisplatin to treat small-cell lung cancer. Teniposide is approved in patients with refractory childhood acute lymphoblastic leukemia in combination with other chemotherapy drugs.
The sole marketed agent it its chemical class, amsacrine (m-AMSA, Figure 4), is a synthetic agent composed of a planar, acridine ring system. Like the agents discussed above, amsacrine is a topoisomerase poison targeting the type II topoisomerases. Interestingly, amsacrine was the first drug proven to poison eukaryotic topoisomerase II 46. The acridine ring system is the component of the drug that intercalates DNA and contributes to the activity of the drug, while the non-intercalative 4′-amino-methane-sulfon-m-anisidide (m-AMSA) headgroup imparts specificity for the DNA-topoisomerase cleavage complex 47. Amsacrine is approved in Canada to induce remission in adults with acute leukemia resistant to conventional therapy 48.
Table 2. Summary of marketed topoisomerase inhibitors and indications
| Drug | Class | Approval date | Mechanism/Target | Indication |
|---|---|---|---|---|
| Doxorubicin | Anthracycline | 8/7/1974 (U.S.) | Type IIA poison | In combination with other chemotherapy agents to treat women after surgical resection of breast cancer with axillary lymph node involvement; acute lymphoblastic and myeloblastic leukemias; Hodgkin and non-Hodgkin lymphomas; metastatic neuroblastoma, Wilms′ tumor, cancers of the breast, soft tissue sarcoma, and bone sarcomas; metastatic ovarian, transitional cell bladder, thyroid, gastric, and bronchogenic carcinomas |
| Epirubicin | Anthracycline | 9/15/1999 (U.S.) | Type IIA poison | Combination with other chemotherapy agents to treat women after surgical resection of breast cancer with axillary lymph node involvement |
| Valrubicin | Anthracycline | 9/25/1998 (U.S.) | Type IIA poison | Intravesical administration for urinary bladder carcinoma refractory to BCG therapy in patients who are not candidates for cystectomy |
| Daunorubicin | Anthracycline | 12/19/1979 (U.S.) | Type IIA poison | In combination with other approved chemotherapy agents to induce remission in acute myelogenous, monocytic, and erythroid lymphocytic leukemias in adults and in acute lymphocytic leukemia in children |
| Idarubicin | Anthracycline | 9/27/1990 (U.S.) | Type IIA poison | In combination with other approved agents to treat adults with acute myeloid leukemia, including French-American-British M1—M7 classifications |
| Mitoxantrone | Anthracenedione | 12/23/1987 (U.S.) | Type IIA poison | For patients with secondary (chronic) multiple sclerosis with significantly abnormal neurologic status between relapses to reduce neurologic disability and or/the frequency of relapses; in combination with corticosteroids as initial chemotherapy to treat pain related to advanced hormone-refractory prostate cancer; in combination with other approved agents to as initial therapy of acute nonlymphocytic anemia in adults |
| Pixantrone | Anthracenedione | 2/16/2012 (E.U.) | Type IIA poison | Monotherapy for the treatment of adult patients with multiply relapsed or refractory aggressive Non-Hodgkin B-cell lymphomas |
| Etoposide | Epipodophyllotoxin | 11/10/1983 (U.S.) | Type IIA poison | Refractory testicular tumors in combination with other chemotherapy agents; first-line treatment in combination with cisplatin for small-cell lung cancer |
| Teniposide | Epipodophyllotoxin | 7/14/1992 (U.S.) | Type IIA poison | Induction of remission in patients with refractory childhood acute lymphoblastic leukemia |
| Amsacrine | Miscellaneous | 12/31/1983 (Canada) | Type IIA poison | Induction of remission in acute adult leukemia refractory to conventional therapy |
| Topotecan | Camptothecin | 5/28/1996 (U.S.) | Type IB poison | Small-cell lung cancer after failure of first-line chemotherapy; combination with cisplatin for persistent or recurrent stage IV-B carcinoma of the cervix not cured by surgery and/or radiation |
| Irinotecan | Camptothecin | 6/14/1996 (U.S.) | Type IB poison | First-line chemotherapy in combination with 5-fluorouracil and leucovorin in patients with metastatic carcinoma of the colon or rectum; recurrent or progressive metastatic carcinoma of the colon or rectum following initial fluorouracil-based therapy |
| Belotecan | Camptothecin | 12/10/2003 (S. Korea) | Type IB poison | Non-small-cell lung cancer; ovarian cancer |
Figure 3. Anthracycline topoisomerase inhibitors
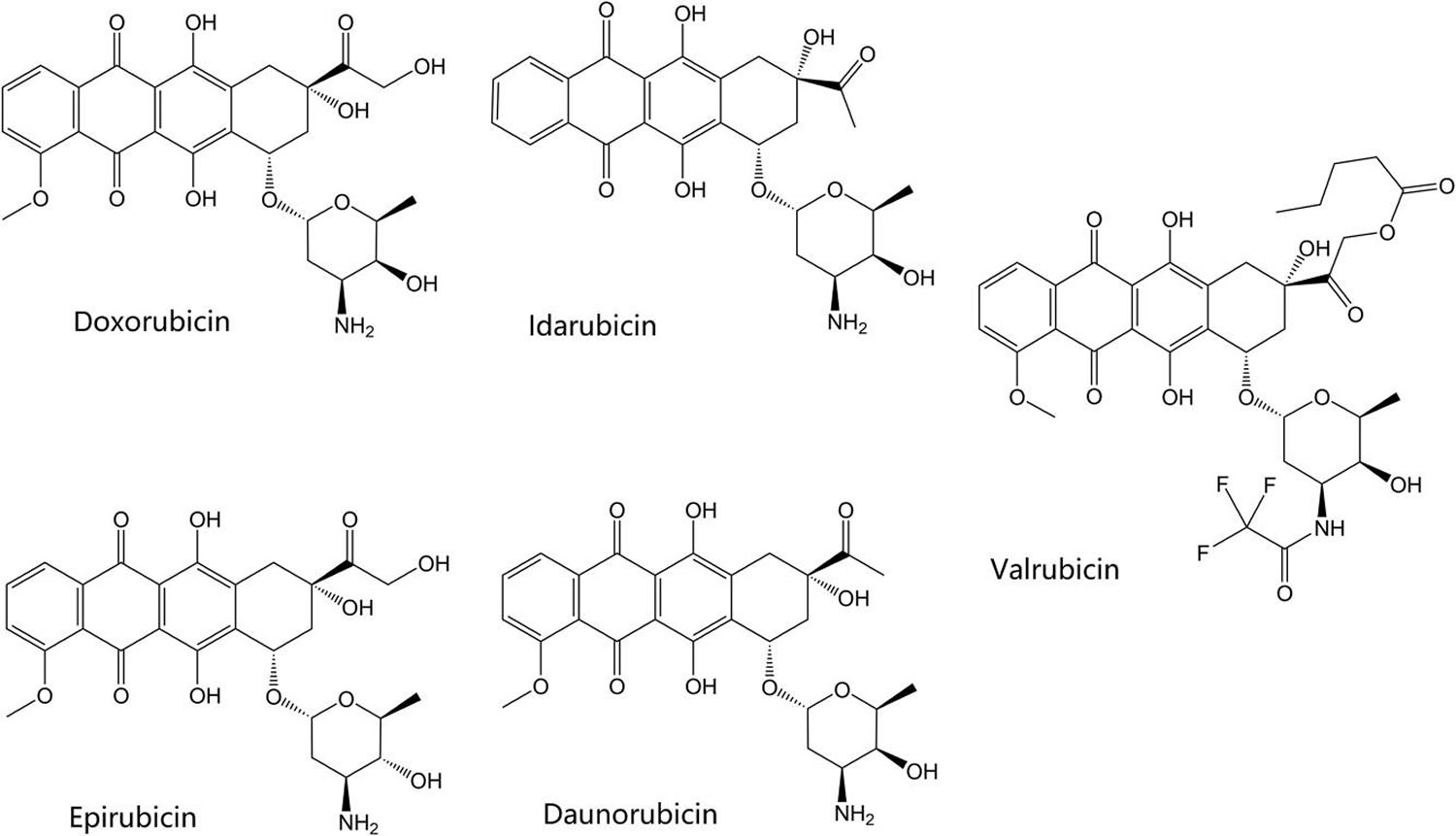
Figure 4. Anthracenedione and acridine-derived topoisomerase inhibitors
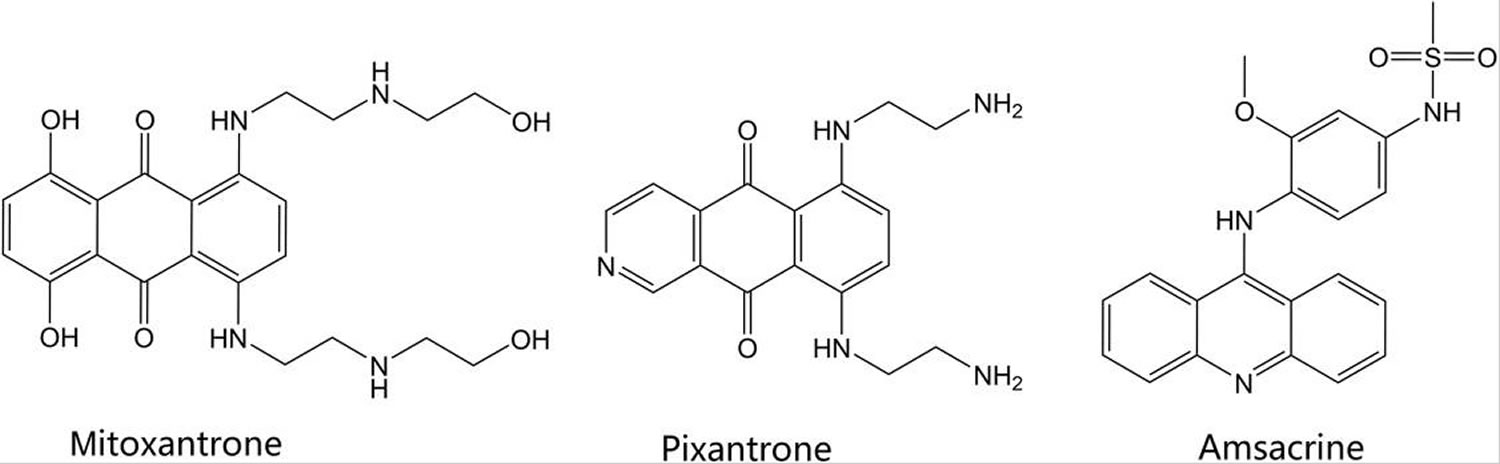
Figure 5. Camptothecin-derived topoisomerase inhibitors
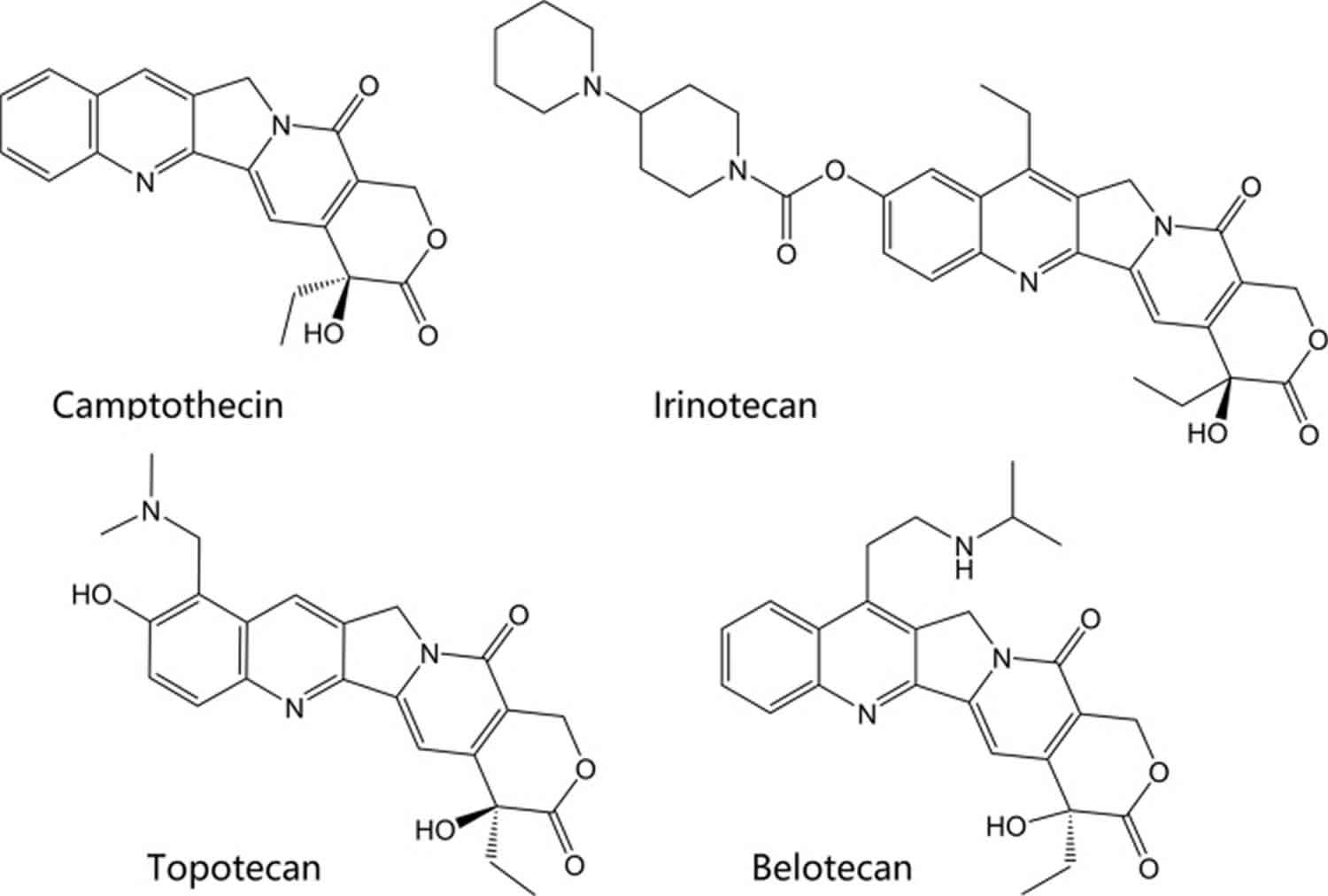
Figure 6. Epipodophyllotoxin-derived topoisomerase inhibitors
- Mooi Kwai, Chan & Adila Fadzil, Nur & Lan, Ai & Boon Yin, Khoo. (2013). New molecular biologist perspective and insight: DNA topoisomerases production by recombinant DNA technology for medical laboratory application and pharmaceutical industry. Electronic Journal of Biotechnology. 16. 18-18. 10.2225/vol16-issue6-fulltext-6[↩]
- Nitiss JL, Kiianitsa K, Sun Y, Nitiss KC, Maizels N. Topoisomerase Assays. Curr Protoc. 2021 Oct;1(10):e250. doi: 10.1002/cpz1.250[↩]
- Pommier Y, Sun Y, Huang SN, & Nitiss JL (2016). Roles of eukaryotic topoisomerases in transcription, replication and genomic stability. Nature reviews. Molecular cell biology, 17(11), 703–721. doi: 10.1038/nrm.2016.111[↩][↩][↩]
- McKie SJ, Neuman KC, & Maxwell A (2021). DNA topoisomerases: Advances in understanding of cellular roles and multi-protein complexes via structure-function analysis. Bioessays, 43(4), e2000286. doi: 10.1002/bies.202000286[↩][↩][↩]
- Vos SM, Tretter EM, Schmidt BH, & Berger JM (2011). All tangled up: how cells direct, manage and exploit topoisomerase function. Nat Rev Mol Cell Biol, 12(12), 827–841. doi: 10.1038/nrm3228[↩][↩][↩][↩][↩][↩]
- Nitiss JL (2009a). DNA topoisomerase II and its growing repertoire of biological functions. Nature reviews. Cancer, 9(5), 327–337. doi: 10.1038/nrc2608[↩][↩][↩]
- Ashley RE, and Osheroff N (2016) Regulation of DNA topology by topoisomerases: mathematics at the molecular level, In Knots, Low-Dimensional Topology and Applications (Adams CC, Gordon CM, Jones VFR, Kauffman LH, Lambropoulou S, Millett KC, Przytycki JH, Ricca R, and Sazadanovic R, Eds.), Springer, Greece.[↩][↩]
- Liu Z, Deibler RW, Chan HS, Zechiedrich L. The why and how of DNA unlinking. Nucleic Acids Res. 2009 Feb;37(3):661-71. doi: 10.1093/nar/gkp041[↩][↩]
- Deweese JE, Osheroff MA, Osheroff N. DNA Topology and Topoisomerases: Teaching a “Knotty” Subject. Biochem Mol Biol Educ. 2008;37(1):2-10. doi: 10.1002/bmb.20244[↩][↩]
- Dalvie ED, and Osheroff N (2021) DNA topoisomerases: type II, In The Encyclopedia of Biological Chemistry (Jez JM, Ed.) in press ed., Elsevier, Inc.[↩]
- Deweese JE, Osheroff N. The DNA cleavage reaction of topoisomerase II: wolf in sheep’s clothing. Nucleic Acids Res. 2009 Feb;37(3):738-48. doi: 10.1093/nar/gkn937[↩]
- VOS, S.M.; TRETTER, E.M.; SCHMIDT, B.H. and BERGER, J.M. (2011). All tangled up: How cells direct, manage and exploit topoisomerase function. Nature Reviews Molecular Cell Biology, vol. 12, no. 12, p. 827-841[↩]
- CHAMPOUX, J.J. (2001). DNA topoisomerases: Structure, function, and mechanism. Annual Review of Biochemistry, vol. 70, no. 1, p. 369-413[↩]
- Durand-Dubief M.; SVENSSON, J.P.; PERSSON, J. and EKWALL, K. (2011). Topoisomerases, chromatin and transcription termination. Transcription, vol. 2, no. 2, p. 66-70[↩][↩]
- WANG, J.C. (2002). Cellular roles of DNA topoisomerases: A molecular perspective. Nature Reviews Molecular Cell Biology, vol. 3, no. 6, p. 430-440.[↩]
- Hevener K, Verstak TA, Lutat KE, Riggsbee DL, Mooney JW. Recent developments in topoisomerase-targeted cancer chemotherapy. Acta Pharm Sin B. 2018;8(6):844–861. doi:10.1016/j.apsb.2018.07.008 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6251812[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Capranico G., Marinello J., Chillemi G. Type I DNA topoisomerases. J Med Chem. 2017;60:2169–2192[↩]
- Sissi C., Palumbo M. Effects of magnesium and related divalent metal ions in topoisomerase structure and function. Nucleic Acids Res. 2009;37:702–711[↩]
- DIGATE, R.J. and MARIANS, K.J. (1988). Identification of a potent decatenating enzyme from Escherichia coli. Journal of Biological Chemistry, vol. 263, no. 26, p. 13366-13373.[↩]
- PENG, H. and MARIANS, K.J. (1993). Escherichia coli topoisomerase IV. Purification, characterization, subunit structure, and subunit interactions. Journal of Biological Chemistry, vol. 268, no. 32, p. 24481-24490[↩]
- WANG, J.C. (1996). DNA Topoisomerases. Annual Review of Biochemistry, vol. 65, no. 1, p. 635-692.[↩]
- Bush N.G., Evans-Roberts K., Maxwell A. DNA topoisomerases. EcoSal. 2015;6:2[↩]
- Garnier F., Debat H., Nadal M., Type I.A. DNA topoisomerases: a universal core and multiple activities. Methods Mol Biol. 2018;1703:1–20[↩][↩]
- Staker B.L., Hjerrild K., Feese M.D., Behnke C.A., Burgin A.B., Jr, Stewart L. The mechanism of topoisomerase I poisoning by a camptothecin analog. Proc Natl Acad Sci U S A. 2002;99:15387–15392[↩]
- Dong K.C., Berger J.M. Structural basis for gate-DNA recognition and bending by type IIA topoisomerases. Nature. 2007;450:1201–1205[↩]
- ATAKA, M.; IKEGUCHI, M.; YAMAMOTO, M.; INOUE, M.; TANIDA, T.; OKA, S.I. and KATANO, K. (2007). Topoisomerase I protein expression and prognosis of patients with colorectal cancer. Yonago Acta medica, vol. 50, no. 4, p. 81-87[↩]
- SYAHRUDDIN, E.; OGURI, T.; TAKAHASHI, T.; ISOBE, T.; FUJIWARA, Y. and YAMAKIDO, M. (1998). Differential expression of DNA topoisomerase IIα and IIβ genes between small cell and non-small cell lung cancer. Cancer Science, vol. 89, no. 8, p. 855-861.[↩]
- OLIVEIRA-COSTA, J.P.; ZANETTI, J.; OLIVEIRA, L.R.; SOARES, F.A.; ZAMBELLI RAMALHO, L.; SILVA RAMALHO, F.; BRITTO GARCIA, S. and RIBEIRO-SILVA, A. (2010). Significance of topoisomerase IIIβ expression in breast ductal carcinomas: Strong associations with disease-specific survival and metastasis. Human Pathology, vol. 41, no. 11, p. 1624-1630[↩]
- SUGIMOTO, Y.; TSUKAHARA, S.; OH-HARA, T.; LIU, L.F. and TSURUO, T. (1990b). Elevated expression of DNA topoisomerase II in camptothecin-resistant human tumor cell lines. Cancer Research, vol. 50, no. 24, p. 7962-7965.[↩]
- Oki T. Recent developments in the process improvement of production of antitumor anthracycline antibiotics. Adv Biotechnol Process. 1984;3:163–196[↩]
- Bodley A., Liu L.F., Israel M., Seshadri R., Koseki Y., Giuliani F.C. DNA topoisomerase II-mediated interaction of doxorubicin and daunorubicin congeners with DNA. Cancer Res. 1989;49:5969–5978[↩]
- Bredehorst R., Panneerselvam M., Vogel C.W. Doxorubicin enhances complement susceptibility of human melanoma cells by extracellular oxygen radical formation. J Biol Chem. 1987;262:2034–2041[↩]
- Menna P., Salvatorelli E., Minotti G. Cardiotoxicity of antitumor drugs. Chem Res Toxicol. 2008;21:978–989[↩][↩]
- Sawyer D.B. Anthracyclines and heart failure. N Engl J Med. 2013;368:1154–1156.[↩]
- Cvetkovic R.S., Scott L.J. Dexrazoxane: a review of its use for cardioprotection during anthracycline chemotherapy. Drugs. 2005;65:1005–1024[↩]
- Spallarossa P., Garibaldi S., Altieri P., Fabbi P., Manca V., Nasti S. Carvedilol prevents doxorubicin-induced free radical release and apoptosis in cardiomyocytes in vitro. J Mol Cell Cardiol. 2004;37:837–846[↩]
- Fox E.J. Mechanism of action of mitoxantrone. Neurology. 2004;63(12 Suppl 6):S15–S18[↩]
- Beeharry N., Di Rora A.G., Smith M.R., Yen T.J. Pixantrone induces cell death through mitotic perturbations and subsequent aberrant cell divisions. Cancer Biol Ther. 2015;16:1397–1406[↩]
- Longo M., Della Torre P., Allievi C., Morisetti A., Al-Fayoumi S., Singer J.W. Tolerability and toxicological profile of pixantrone (Pixuvri®) in juvenile mice. Comparative study with doxorubicin. Reprod Toxicol. 2014;46:20–30[↩]
- Hsiang Y.H., Hertzberg R., Hecht S., Liu L.F. Camptothecin induces protein-linked DNA breaks via mammalian DNA topoisomerase I. J Biol Chem. 1985;260:14873–14878[↩]
- Pommier Y. Topoisomerase I inhibitors: camptothecins and beyond. Nat Rev Cancer. 2006;6:789–802[↩]
- Liu Y.Q., Li W.Q., Morris-Natschke S.L., Qian K., Yang L., Zhu G.X. Perspectives on biologically active camptothecin derivatives. Med Res Rev. 2015;35:753–789[↩]
- Park Y.H., Chung C.U., Park B.M., Park M.R., Park D.I., Moon J.Y. Lesser toxicities of belotecan in patients with small cell lung cancer: a retrospective single-center study of camptothecin analogs. Can Respir J. 2016;(2016):3576201.[↩]
- Zhang X., Rakesh K.P., Shantharam C.S., Manukumar H.M., Asiri A.M., Marwani H.M. Podophyllotoxin derivatives as an excellent anticancer aspirant for future chemotherapy: a key current imminent needs. Bioorg Med Chem. 2018;26:340–355[↩]
- Ross W., Rowe T., Glisson B., Yalowich J., Liu L. Role of topoisomerase II in mediating epipodophyllotoxin-induced DNA cleavage. Cancer Res. 1984;44:5857–5860.[↩]
- Nelson E.M., Tewey K.M., Liu L.F. Mechanism of antitumor drug action: poisoning of mammalian DNA topoisomerase II on DNA by 4′-(9-acridinylamino)-methanesulfon-m-anisidide. Proc Natl Acad Sci U S A. 1984;81:1361–1365[↩]
- Ketron A.C., Denny W.A., Graves D.E., Osheroff N. Amsacrine as a topoisomerase II poison: importance of drug-DNA interactions. Biochemistry. 2012;51:1730–1739[↩]
- Jehn U., Heinemann V. New drugs in the treatment of acute and chronic leukemia with some emphasis on m-AMSA. Anticancer Res. 1991;11:705–711[↩]





{kind=link}