Contents
What is trisomy 18
Trisomy 18 syndrome also known as Edwards syndrome, is a common chromosomal disorder due to the presence of an extra chromosome 18 (instead of the usual two copies), either full, mosaic trisomy, or partial trisomy 18q 1. Edwards syndrome (trisomy 18) was first described by John Hilton Edwards (1928 – 2007), a British medical geneticist. Trisomy 18 syndrome (Edwards syndrome) is the second most common autosomal trisomy syndrome after trisomy 21 (Down Syndrome) 1. The live born prevalence of trisomy 18 syndrome (Edwards syndrome) is estimated as 1/6,000-1/8,000, but the overall prevalence is higher (1/2500-1/2600) due to the high frequency of fetal loss and pregnancy termination after prenatal diagnosis. Although women of all ages can have a child with trisomy 18, the chance of having a child with this condition increases as a woman gets older. The recurrence risk for a family with a child with full trisomy 18 is about 1% 1.
Babies born with Edwards syndrome usually do not survive for much longer than a week or within their first month due to the presence of several life-threatening medical problems. 5 to 10 percent of children with this condition live past their first year, and these children often have severe intellectual disability.
Each cell in your body normally contains 23 pairs of chromosomes, which carry the genes you inherit from your parents. But a baby with Edwards’ syndrome has three copies of chromosome number 18, instead of two.
The presence of this extra chromosome in cells severely disrupts normal development.
Edwards’ syndrome is rarely inherited and is not caused by anything the parents have done. The development of three copies of chromosome 18 usually happens at random during the formation of either the egg or sperm.
As this happens randomly, it’s extremely unlikely for parents to have more than one pregnancy affected by Edwards’ syndrome. However, the chance of having a baby with Edwards’ syndrome does increase as the mother gets older.
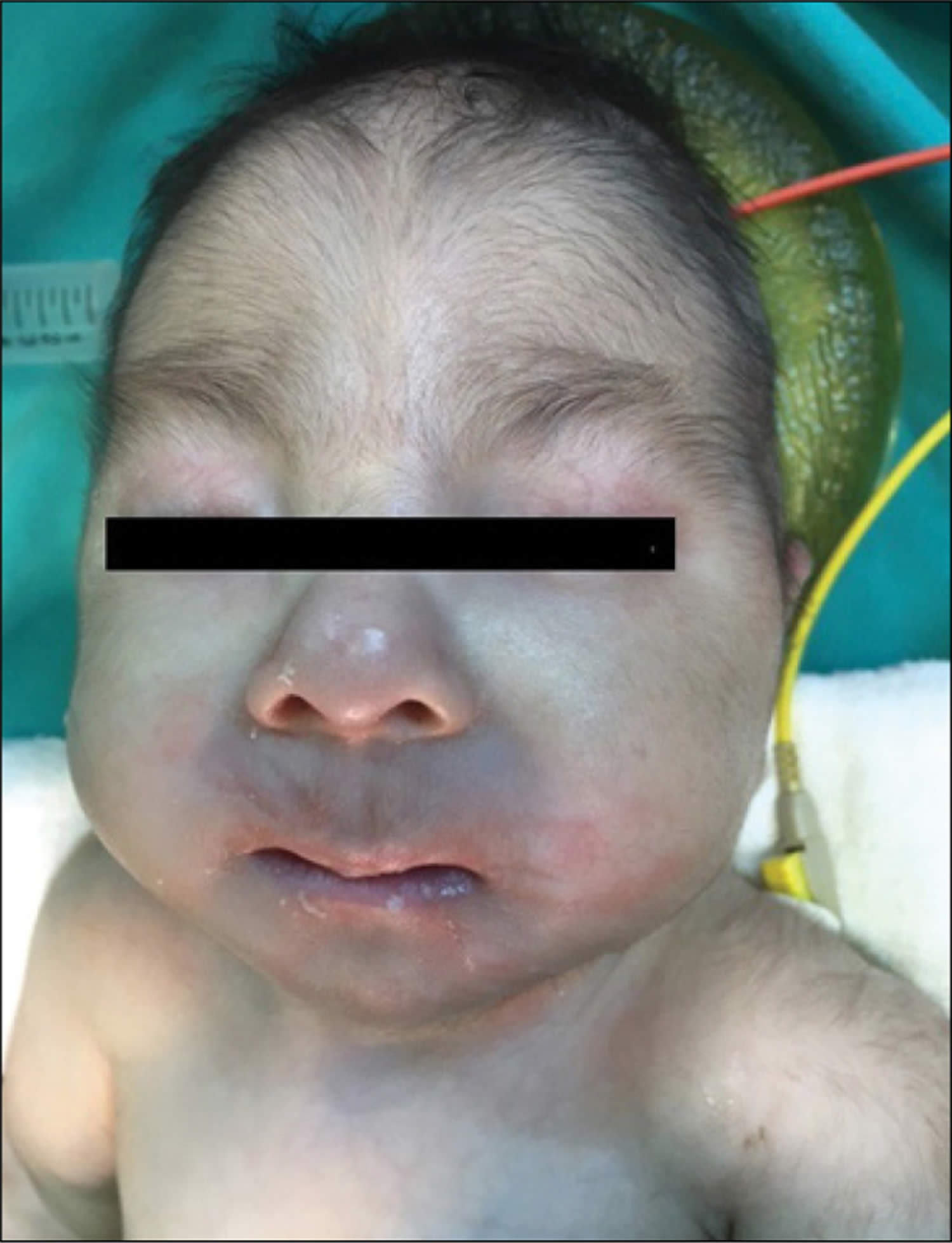
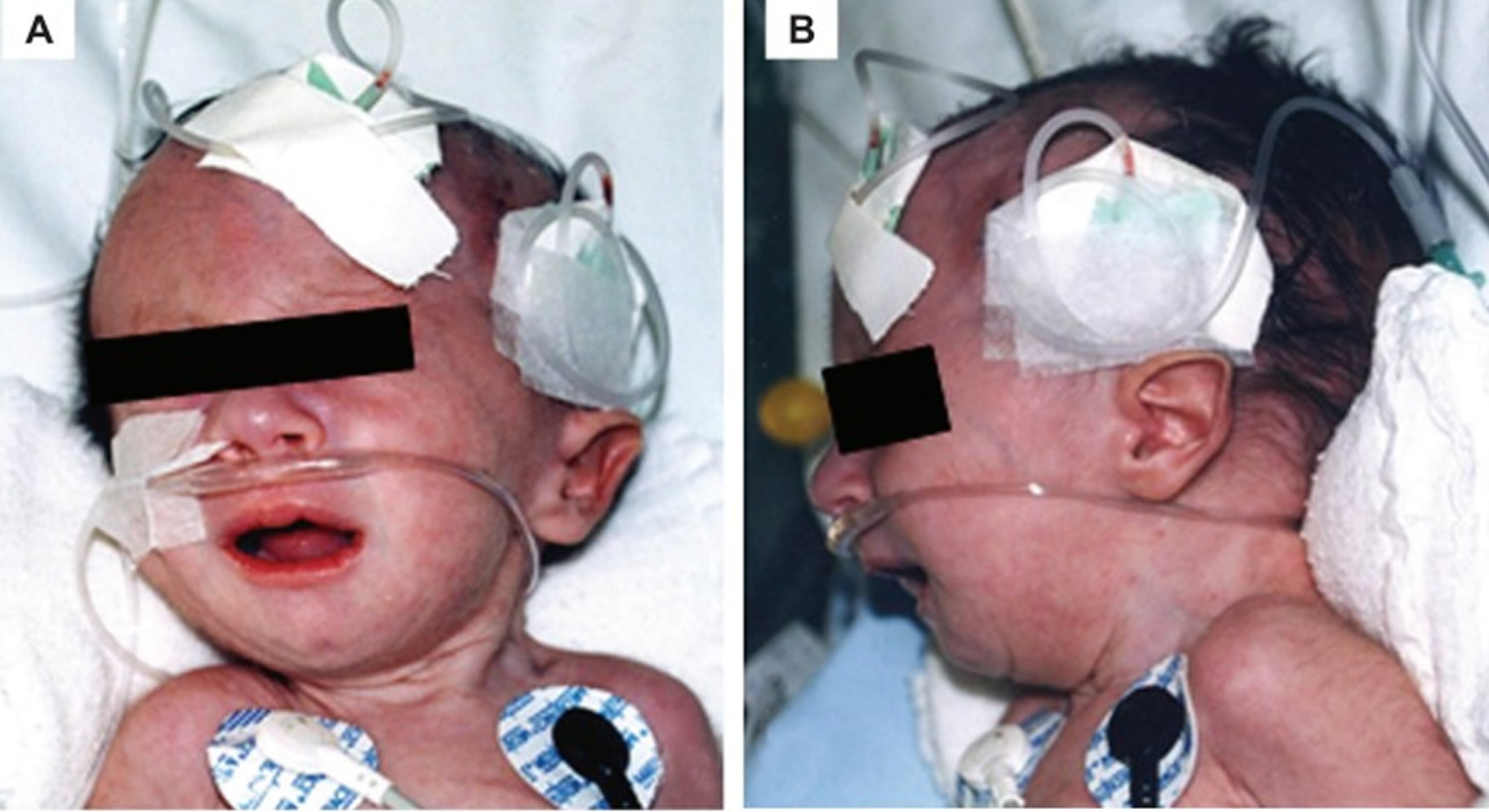
In some cases, the chromosomal abnormality may be present in only a percentage of cells, whereas other cells contain the normal chromosomal pair (mosaicism). Depending on the specific location of the duplicated (trisomic) portion of chromosome 18–as well as the percentage of cells containing the abnormality–symptoms and findings may be extremely variable from case to case. However, in many affected infants, such abnormalities may include growth deficiency, feeding and breathing difficulties, developmental delays, mental retardation, and, in affected males, undescended testes (cryptorchidism). Individuals with trisomy 18 syndrome may also have distinctive malformations of the head and facial (craniofacial) area, such as a prominent back portion of the head; low-set, malformed ears; an abnormally small jaw (micrognathia); a small mouth with an unusually narrow roof (palate); and an upturned nose. Affected infants may also have narrow eyelid folds (palpebral fissures), widely spaced eyes (ocular hypertelorism), and drooping of the upper eyelids (ptosis). Malformations of the hands and feet are also often present, including overlapped, flexed fingers; webbing of the second and third toes; and a deformity in which the heels are turned inward and the soles are flexed (clubfeet [talipes equinovarus]). Infants with trisomy 18 syndrome may also have a small pelvis with limited movements of the hips, a short breastbone (sternum), kidney malformations, and structural heart (cardiac) defects at birth (congenital). Such cardiac defects may include an abnormal opening in the partition dividing the lower chambers of the heart (ventricular septal defect) or persistence of the fetal opening between the two major arteries (aorta, pulmonary artery) emerging from the heart (patent ductus arteriosus). Congenital heart defects and respiratory difficulties may lead to potentially life-threatening complications during infancy or childhood.
Mosaic trisomy 18
Mosaic trisomy 18 can be a less severe form of Edwards’ syndrome, as only some of the cells have the extra copy of chromosome 18, rather than every cell.
How severely affected the baby is depends on the number of and type of cells that have the extra chromosome. Some babies may only be mildly affected, while some can be severely disabled.
Around seven in every 10 babies born with mosaic trisomy 18 will live for at least a year and, in rare cases, may survive into early adulthood.
Partial trisomy 18
In partial trisomy 18 only a section of the additional chromosome 18 is present in the cells, rather than a whole additional chromosome 18.
This type of Edwards’ syndrome is more likely if one the parents has a minor alteration in their chromosomes, so blood samples are often requested from both parents to check for this and to help them understand the risks for future pregnancies.
How severely affected the baby is will depend on which part of chromosome 18 is present in the cells.
Figure 1. Trisomy 18 baby – craniofacial findings of a baby with Edwards’ syndrome micrognathia (B) and low-set ear faun (A and B)
Trisomy 18 life expectancy
Due to the presence of several life-threatening medical problems, many individuals with trisomy 18 die before birth or within their first month. Some babies with less severe types of Edwards’ syndrome, such as mosaic or partial trisomy 18, about five to ten percent of children with this condition live past their first year, and these children often have severe intellectual and physical disabilities. Edwards’ syndrome children very rarely survive into early adulthood.
Trisomy 18 symptoms
Babies with Edwards’ syndrome can have a wide range of different problems. Babies with trisomy 18 (Edwards syndrome) often have slow growth before birth (intrauterine growth retardation) and a low birth weight. Affected individuals may have heart defects and abnormalities of other organs that develop before birth. Other features of trisomy 18 include a small, abnormally shaped head; a small jaw and mouth; and clenched fists with overlapping fingers.
Symptoms may include:
- Clenched hands
- Crossed legs
- Feet with a rounded bottom (rocker-bottom feet)
- Low birth weight
- Low-set ears
- Mental delay
- Poorly developed fingernails
- Small head (microcephaly)
- Small jaw (micrognathia)
- Undescended testicle
- Unusual shaped chest (pectus carinatum)
- Long fingers that overlap, with underdeveloped thumbs and clenched fists
- Smooth feet with rounded soles
- A cleft lip and palate
- An exomphalos (where the intestines are held in a sac outside the tummy)
Babies with Edwards’ syndrome also typically have:
- heart and kidney problems
- feeding problems – leading to poor growth
- breathing problems
- hernias in the wall of their stomach (where internal tissues push through a weakness in the muscle wall)
- bone abnormalities – such as a curved spine
- frequent infections of the lungs and urinary system
- a severe learning disability
Edwards syndrome symptoms
Trisomy 18 fetuses can have multiple anomalies in multiple systems. Over 130 features have been reported. Trisomy 18 has highest incidence of major structural anomalies. Features include:
- congenital heart disease: 90-95%
- atrial septal defect (ASD)
- ventricular septal defect (VSD)
- patent ductus arteriosus (PDA)
- dextrocardia
- central nervous system / spinal abnormalities: 70%
- intrauterine growth retardation (IUGR): 60-90% (tends to occur from early in gestation)
- facial/calvarial abnormalities
- micrognathia
- dolichocephaly: strawberry skull: as a result of frontal lobe hypoplasia
- low set ears
- hypertelorism
- cleft lip +/- palate
- cystic hygroma: ~20%
- skeletal abnormalities
- hand anomalies
- clenched hands with overlap of 2nd and 3rd digits: 80%
- radial ray anomalies
- absent thumb
- feet anomalies
- rockerbottom feet: typical feature
- club feet (talipes equinovarus)
- prominent occiput
- short neck
- hand anomalies
- umbilical cord
- single umbilical artery: 80%
- umbilical cord cysts
- umbilical cord pseudocysts
- gastrointestinal: thoracic anomalies
- bowel containing omphalocoele: 20-25%
- congenital diaphragmatic hernia: ~10%
- renal anomalies
- antenatal hydronephrosis
- urethral duplication
- horseshoe kidney: 20% 5
The symptoms and findings associated with trisomy 18 syndrome (Edwards syndrome) may be extremely variable. In addition, if only a percentage of cells contains the chromosomal abnormality (mosaicism), associated symptoms may tend to be less severe. However, certain findings before birth (prenatally) and during infancy are considered characteristic of trisomy 18 syndrome. In many cases, there is abnormally decreased movement during fetal development. In addition, many affected newborns are born abnormally early or late (altered gestation). Additional characteristic findings typically include poor suckling ability and associated feeding difficulties, failure to grow and gain weight at the expected rate (failure to thrive), mental retardation, and distinctive malformations of the head and facial (craniofacial) area. In many infants with trisomy 18 syndrome, other physical abnormalities may also be present, such as undescended testes in affected males (cryptorchidism), malformations of the hands and feet, additional skeletal defects, and structural abnormalities of the heart (congenital heart defects).
A “full-term” infant is born anywhere from 37 to 42 weeks of gestation. (Gestation refers to the period of development from fertilization of the egg [ovum] to birth.) However, about one third of newborns with trisomy 18 syndrome are born prior to 37 weeks’ gestation (premature infant), and approximately one third are born after 42 weeks (postmature infant). Affected infants also tend to have a low birth weight, growth delays, and severe feeding difficulties. In addition, at birth, skeletal muscles and underlying connective and fatty tissues (subcutaneous and adipose tissue) may be underdeveloped (hypoplastic). Additional characteristic abnormalities during infancy may include diminished muscle tone (hypotonia) followed by unusually increased tone (hypertonia) and muscle stiffness (rigidity); an abnormally weak, feeble cry; a decreased response to environmental sounds; and/or repeated episodes in which there is a temporary cessation of breathing (apneic episodes).
Many infants with trisomy 18 syndrome also have distinctive malformations of the craniofacial region. These may include a small head (microcephaly) that appears unusually long and narrow (dolichocephaly); a prominent back region of the head (occiput); a small mouth (microstomia); incomplete closure of the roof of the mouth (cleft palate) and/or an abnormal groove in the upper lip (cleft lip); a small jaw (micrognathia); or a short, webbed neck. Affected infants may also have an upturned nose; low-set, malformed ears; widely spaced eyes (ocular hypertelorism) with slanted or narrow eyelid folds (palpebral folds); and vertical skin folds covering the eyes’ inner corners (epicanthal folds).
Trisomy 18 syndrome may also be characterized by additional eye (ocular) abnormalities. For example, there may be drooping of the upper eyelids (ptosis) and an inability to completely close the eyes. Some affected infants also have clouding of the normally transparent front regions of the eyes (corneas); loss of transparency of the lenses (cataracts); absence or defects of tissue (colobomas) affecting the colored regions of the eyes (irides); or unusual smallness of the eyes (microphthalmia). Additional ocular abnormalities may include abnormal deviation of one eye in relation to the other (strabismus); inequality in the diameter of the pupils (anisocoria); rapid involuntary eye movements (nystagmus); and/or a decreased response to visual stimuli.
Many infants with trisomy 18 syndrome also have characteristic malformations of the hands and feet. The hands are typically clenched, with overlapping of the index finger (second finger) over the third finger and the “pinky” (fifth finger) over the fourth. Frequent findings also include unusual skin ridge patterns (dermatoglyphics) on the fingers and palms; underdeveloped (hypoplastic) nails, particularly those of the fifth fingers and toes; and abnormal deviation of the great toes (hallux) in an upwardly bent position (dorsiflexion). In some cases, additional malformations may be present, such as the presence of extra fingers or toes (polydactyly); webbing (syndactyly) of certain toes; or a deformity in which the feet appear shaped like the rocker of a rocking chair (“rocker-bottom feet”) with abnormal prominence of the heel bones (calcaneus). Some infants also have a foot deformity in which the heels are turned inward and the soles are flexed (club feet [talipes equinovarus]).
Many infants with trisomy 18 syndrome also have additional skeletal deformities, such as a short breastbone (sternum); a small pelvis with limited outward movements (abduction) of the hips; or abnormalities of the ribs. In addition, in some cases, there may be defects of certain bones of the spinal column (vertebrae), including sideways curvature of the spine (scoliosis); underdevelopment of one half of certain vertebrae (hemivertebrae); or abnormal fusion of vertebrae.
As mentioned above, in males with the disorder, the testes fail to descend into the scrotum (cryptorchidism). Trisomy 18 syndrome may also be associated with additional genital abnormalities. In some affected males, there may be division of the scrotum into two parts (bifid scrotum) and/or abnormal placement of the urinary opening (hypospadias), such as on the underside of the penis. In some females with the disorder, there is underdevelopment (hypoplasia) of the outer skin folds (labia majora) surrounding the vaginal opening and abnormal prominence of the relatively small, sensitive protrusion (clitoris) that forms part of the female external genitalia.
Trisomy 18 syndrome is also often characterized by structural heart (cardiac) defects that are present at birth (congenital heart defects). Many affected infants have an abnormal opening in the fibrous partition (septum) that separates the lower chambers of the heart (ventricular septal defect). Another common finding is abnormal persistence of the fetal opening between the two major arteries (aorta, pulmonary artery) that emerge from the heart (patent ductus arteriosus). In some cases, additional cardiac defects may be present. These may include narrowing (stenosis) of the opening between the right ventricle and the artery that carries oxygen-deficient blood to the lungs (pulmonary artery); narrowing of the major artery that transports oxygen-rich blood from the heart to all parts of the body (coarctation of the aorta); and/or abnormalities of certain heart valves.
In some instances, infants with trisomy 18 syndrome have additional physical defects, such as malformations of the abdominal wall and the kidneys. For example, there may be protrusion of portions of the intestine through an abnormal opening in muscles of the groin (inguinal hernia) or the abdominal wall near the navel (umbilical hernia). Some infants with trisomy 18 syndrome have an omphalocele, a birth defect in which varying amounts of intestines or other abdominal organs (viscera), covered by a membrane-like sac, protrude through an opening in the abdominal wall near the navel. In addition, in some affected infants, the kidneys may be abnormally positioned (ectopic) or joined together (horseshoe kidneys) or contain multiple cysts (polycystic kidneys). There may also be swelling of the kidneys with urine due to narrowing or blockage of the tubes that carry urine from the kidneys to the bladder (hydronephrosis).
Some affected infants may also have malformations of the brain and spinal cord (central nervous system [CNS]). These may include absence (agenesis) or underdevelopment (hypoplasia) of the thick band of nerve fibers connecting the two hemispheres of the brain (corpus callosum); protrusion of part of the spinal cord and its surrounding membranes (meninges) through an abnormal opening in the spinal column (myelomeningocele); or other malformations. Trisomy 18 syndrome is also typically characterized by mental retardation, with severely impaired intellectual, verbal, and motor development.
In many cases, congenital heart defects, respiratory difficulties (e.g., apneic episodes, aspiration), or other abnormalities associated with the disorder may lead to potentially life-threatening complications during infancy or childhood.
Trisomy 18 causes
Most cases of trisomy 18 (Edwards syndrome) result from having three copies of chromosome 18 in each cell in the body instead of the usual two copies. The extra genetic material disrupts the normal course of development, causing the characteristic features of trisomy 18.
Chromosomes are found in the nucleus of all body cells. They carry the genetic characteristics of each individual. Pairs of human chromosomes are numbered from 1 through 22, with an unequal 23rd pair of X and Y chromosomes for males and two X chromosomes for females. Each chromosome has a short arm designated as “p” and a long arm identified by the letter “q.” Chromosomes are further subdivided into bands that are numbered.
Approximately 5 percent of people with trisomy 18 have an extra copy of chromosome 18 in only some of the body’s cells. In these people, the condition is called mosaic trisomy 18. The severity of mosaic trisomy 18 depends on the type and number of cells that have the extra chromosome. The development of individuals with this form of trisomy 18 may range from normal to severely affected.
Very rarely, part of the long (q) arm of chromosome 18 becomes attached (translocated) to another chromosome during the formation of reproductive cells (eggs and sperm) or very early in embryonic development. Affected individuals have two copies of chromosome 18, plus the extra material from chromosome 18 attached to another chromosome. People with this genetic change are said to have partial trisomy 18. If only part of the q arm is present in three copies, the physical signs of partial trisomy 18 may be less severe than those typically seen in trisomy 18. If the entire q arm is present in three copies, individuals may be as severely affected as if they had three full copies of chromosome 18.
Most cases of trisomy 18 are not inherited, but occur as random events during the formation of eggs and sperm. An error in cell division called nondisjunction results in a reproductive cell with an abnormal number of chromosomes. For example, an egg or sperm cell may gain an extra copy of chromosome 18. If one of these atypical reproductive cells contributes to the genetic makeup of a child, the child will have an extra chromosome 18 in each of the body’s cells.
Mosaic trisomy 18 is also not inherited. It occurs as a random event during cell division early in embryonic development. As a result, some of the body’s cells have the usual two copies of chromosome 18, and other cells have three copies of this chromosome.
Partial trisomy 18 can be inherited. An unaffected person can carry a rearrangement of genetic material between chromosome 18 and another chromosome. This rearrangement is called a balanced translocation because there is no extra material from chromosome 18. Although they do not have signs of trisomy 18, people who carry this type of balanced translocation are at an increased risk of having children with the condition.
Trisomy 18 diagnosis
During pregnancy
Pregnant women are offered screening for Edwards’ syndrome between 10 and 14 weeks of pregnancy to assess the chances of their baby having the condition.
This screening test is known as the combined test, and it also screens for Down’s syndrome (trisomy 21) and Patau’s syndrome (trisomy 13).
During the combined test you will have a blood test and a special ultrasound scan where the fluid at the back of the baby’s neck (nuchal translucency) is measured.
If the combined test shows that you have a higher risk of having a baby with Edwards’ syndrome, you will be offered a diagnostic test to find out for certain if your baby has the condition.
This involves analyzing a sample of your baby’s cells to check if they have an extra copy of chromosome 18.
There are two different ways of getting this sample of cells – chorionic villus sampling (CVS), which collects a sample from the placenta, or amniocentesis, which collects a sample of the amniotic fluid from around your baby.
These are invasive tests that do have a risk of causing a miscarriage. Your doctor will discuss these risks with you.
Later in pregnancy, usually when you are 18-21 weeks pregnant, you will also be offered a scan that looks for physical abnormalities, known as congenital anomalies.
A newer test has also been developed that can be performed by taking a sample of blood from the mother, at 10-12 weeks, and testing the baby’s DNA that is found within it. This is known as “non-invasive prenatal testing.”
An exam during pregnancy may show an unusually large uterus and extra amniotic fluid. There may be an unusually small placenta when the baby is born. A physical exam of the infant may show unusual fingerprint patterns. X-rays may show a short breast bone.
During fetal ultrasonography, reflected sound waves create an image of the developing fetus, potentially revealing findings that may suggest a chromosomal disorder or other abnormalities. In addition, screening tests that reveal abnormal levels of certain “markers” in the mother’s blood may suggest an increased risk of trisomy 18 syndrome or other chromosomal abnormalities (e.g., Down syndrome). Such tests measure the levels of specific substances in the blood, including alpha-fetoprotein (AFP); human chorionic gonadotropin (hCG); unconjugated estriol; or other markers. If such screening studies reveal abnormal levels of these markers, additional testing may be recommended, such as amniocentesis with chromosome analysis.
Prenatal tests, such as amniocentesis or CVS, may also be offered to all pregnant women who are aged 35 or older; for known translocation carriers; and/or due to other factors. During amniocentesis, a sample of fluid that surrounds the developing fetus is removed and analyzed, while CVS involves the removal of tissue samples from a portion of the placenta. Chromosomal analysis performed on the fluid or tissue samples may confirm the presence of trisomy 18.
The diagnosis of trisomy 18 syndrome may also be made or confirmed after birth (postnatally) based upon a thorough clinical evaluation, detection of characteristic physical findings, and chromosomal analysis. For infants diagnosed with Edwards syndrome, careful monitoring and specialized testing may be conducted to ensure early detection and appropriate management of certain conditions that may be associated with trisomy 18 syndrome.
Other signs include:
- Hole, split, or cleft in the iris of the eye (coloboma)
- Separation between the left and right side of the abdominal muscle (diastasis recti)
- Umbilical hernia or inguinal hernia
There are often signs of congenital heart disease, such as:
- Atrial septal defect (ASD)
- Patent ductus arteriosus (PDA)
- Ventricular septal defect (VSD)
Tests may also show kidney problems, including:
- Horseshoe kidney
- Hydronephrosis
- Polycystic kidney
Making a decision
If your baby is diagnosed with Edwards’ syndrome during your pregnancy your doctor will talk to you about how you want to move forward. They will discuss the options of either continuing with the pregnancy or ending it with a termination, as it is such a severe condition.
This is a very difficult situation and it is normal to feel a whole range of emotions. It may help to talk to your doctor, partner, family and friends about what you are thinking and how you are feeling.
If your baby is diagnosed with Edwards’ syndrome, either before birth or afterwards, you’ll be offered counseling and support.
You can also contact Support Organization for Trisomy 18, 13 and Related Disorders (https://trisomy.org/) which has information that could help families having children with trisomies.
Edwards syndrome treatment
There is no cure for Edwards’ syndrome and the symptoms can be very difficult to manage. You are likely to need help from a wide range of health professionals.
The treatment of trisomy 18 syndrome is directed toward the specific symptoms that are apparent in each individual. Such treatment may require the coordinated efforts of a multidisciplinary team of medical professionals.
For many affected infants, supportive measures may be required to improve feeding and the intake of necessary nutrients. Such measures may include the delivery of liquid nutrients to the stomach through a tube inserted through the nose (nasogastric tube feeding). In addition, oxygen therapy may be required to ensure sufficient oxygen supply to bodily tissues.
In some cases, recommended treatment may include surgical correction of certain abnormalities associated with the disorder. The surgical procedures performed will depend upon the nature and severity of the anatomical abnormalities, their associated symptoms, and other factors.
A supportive team approach for children with this disorder may be of benefit and may include special education, physical therapy, and/or other medical or social services. Genetic counseling will also be of benefit for families of affected children. Other treatment for this disorder is symptomatic and supportive.
- Cereda A, Carey JC. The trisomy 18 syndrome. Orphanet Journal of Rare Diseases. 2012;7:81. doi:10.1186/1750-1172-7-81. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3520824/[↩][↩][↩]
- Rosa RFM, Rosa RCM, Lorenzen MB, Zen PRG, Graziadio C, Paskulin GA. Craniofacial abnormalities among patients with Edwards Syndrome. Revista Paulista de Pediatria. 2013;31(3):293-298. doi:10.1590/S0103-05822013000300004. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4182981/[↩]
- Epelman M, Daneman A, Blaser SI et-al. Differential diagnosis of intracranial cystic lesions at head US: correlation with CT and MR imaging. Radiographics. 26 (1): 173-96. doi:10.1148/rg.261055033[↩]
- Entezami M, Albig M, Knoll U et-al. Ultrasound Diagnosis of Fetal Anomalies. Thieme. (2003) ISBN:1588902129[↩]
- Kumar P, Burton BK. Congenital malformations, evidence-based evaluation and management. McGraw-Hill Professional. (2007) ISBN:0071471898[↩]





{kind=link}