Contents
What is ubiquitin
Ubiquitin is a small 76 amino acid protein modifier of approximately 8.5 kDa that covalently attaches to the lysine residues of target proteins via its carboxy-terminal glycine residue, forming an iso-peptide linkage, in an ATP-dependent fashion 1. Four genes in the human genome code for ubiquitin: UBB, UBC, UBA52 and RPS27A 1. Ubiquitin through its chemical conjugation onto proteins (ubiquitination) plays major roles in regulating almost all cellular processes from tagging them for proteasomal degradation (protein degradation), protein trafficking, cell-cycle regulation, DNA repair, apoptosis and signal transduction 2 and protein-protein interaction 3. The ubiquitination process is catalyzed by three enzymes, including ubiquitin-activating enzyme (E1), ubiquitin-conjugating enzyme (E2) and ubiquitin ligase enzyme (E3). In the first step, ubiquitin is activated and binds to C-terminus of an E1 enzyme with the use of ATP, followed by the conjugation of the activated ubiquitin to an E2 enzyme. Next, E2 interacts with an E3 ubiquitin ligase that binds to a substrate protein, and transfers the ubiquitin to the lysine residues in the substrates. All the enzymes are then dissociated form the substrate protein to complete the ubiquitination process. These steps can be repeated to form polyubiquitinated chains 4.
Since ubiquitin itself contains seven lysines, it can attach repeatedly to other ubiquitins, allowing the formation of polyubiquitin chains. Owing to diverse lysine residues in proteins, proteins can be either ubiquitinated on a single lysine residue or on multiple lysine residues, resulting in monoubiquitination and multiubiquitination, respectively. Moreover, proteins can also be polyubiquitinated by homotypically attaching a number of ubiquitin molecules on the same lysine residue sequentially 5. In addition, when the attachments of additional ubiquitin to the lysine residues of the previous ubiquitin molecule at different positions, it leads to the formation of branched polyubiquitination chains 6. Ubiquitination can also occur on cysteine, threonine and serine residues in some cases, which make protein ubiquitination more diverse 7. Therefore, ubiquitin exists intracellularly either as a monomer, a substrate-conjugated polyubiquitin or monoubiquitin, or free (or unanchored) ubiquitin chains, and there is a dynamic equilibrium among the three forms in the cell. The ubiquitination process can be reversed by deubiquitinating enzymes (DUBs), which are ubiquitin-specific proteases. Up to date, it is estimated that there are more than 30 ubiquitin-conjugating enzymes (E2s), 1000 ubiquitin ligase enzymes (E3s) and 100 deubiquitinases (DUBs) discovered in mammalian cells 8. Nevertheless, the mechanism of the distinct selection of E2s and E3s for different types of ubiquitination remains largely elusive 9.
Ubiquitin itself can also itself become ubiquitinated, resulting in a poly-ubiquitin chain defined by the specific modified lysine residue or in the case of M1/linear chains, the methionine residue of ubiquitin 10. The type of chain created has a distinct effect on the fate of its substrate protein; for instance,K48 chains are known for their involvement in targeting proteins for proteasomal degradation 11. Ubiquitiniquitination is a reversible process; deubiquitinases (DUBs) remove ubiquitin from a protein or edit the length and type of a ubiquitinchain 3.
Unanchored ubiquitin chains – that is, poly-ubiquitin that is not tethered onto a substrate protein – also exist in the cell. Unanchored ubiquitin chains can arise when a deubiquitinase removes an intact chain from a protein, or they can be generated anew through E1/E2/E3 cycles. Although unanchored poly-ubiquitin is not well understood, it has been implicated as a participant in several cellular processes, including NF-κB signaling and autophagy 12. The prevailing view is that unanchored ubiquitin chains are quickly disassembled by deubiquitinases and recycled as mono-ubiquitin 13. Studies in yeast and in cultured mammalian cells have suggested that the buildup of free poly-ubiquitin might become toxic by perturbing ubiquitin-dependent proteasomal degradation 14.
The diversity of protein ubiquitination is of great significance for protein functions. Different ubiquitination chains display variant structures that can be recognized by different ubiquitin binding domains in a specific manner, leading to different fates of proteins through different pathways. Monoubiquitination was reported to be important for regulation of histone functions, modulation of DNA repair, receptor endocytosis and gene expression 15. The polyubiquitination chains through K63 is thought to be involved in the regulation of kinase activity, signal transduction and DNA damage response 16. The K48-and K11-linked polyubiquitination, on the other hand, mainly guides proteins for proteasome-mediated degradation 17. The monoubiquitination of histone H2A is mediated by the branched ubiquitination chains on RING1B E3 ubiquitin ligase through ubiquitinating lysine residues at position 6, 27 and 48 18. In a word, the structural diversity of ubiquitination chains serves as important signal for distinct functions of proteins. The generation of various types of ubiquitination chains arises from the distinct combination of E2 and E3 enzymes used in the ubiquitination processes.
Ubiquitin is an abundant protein in eukaryotic cells constituting ∼0.1–5% of total proteins, therefore it is assumed that it is redundantly expressed 19. However, due to its pervasive use and large number of substrates to be ubiquitinated in a cell, ubiquitin does not seem to be produced in excess, rather the free pool of ubiquitin is maintained at an adequate level depending on the cell conditions.
In yeast as well as in most higher eukaryotes, ubiquitin is initially expressed in the form of different precursors: polyubiquitin, a linear fusion protein consisting of four or more ubiquitin copies in a head-to-tail configuration, and fusion proteins between ubiquitin and usually ubiquitinL40 and ubiquitinS27, that are large and small essential ribosomal polypeptides, L40 and S27, respectively 20. These ubiquitin precursors are cleaved by deubiquitinases to release identical functional monomeric ubiquitin units. Mammals have four ubiquitin genes, two of which encode polyubiquitin and the other two encode fusions with ribosomal proteins 20. The two polyubiquitin-encoding genes, Ubb and Ubc, express usually four and nine tandem repeats of ubiquitin, respectively. Thus, the polyubiquitin genes seem redundant; however, the importance of the polyubiquitin-encoding genes was highlighted in the knockout mouse of either Ubb or Ubc 21. In the case of Ubc, disruption of Ubc in mice is embryonically lethal, possibly due to the lack of fetal liver proliferation at midgestation 19. Analysis of Ubc−/− mouse embryo fibroblasts showed 40% reduction in ubiquitin level compared with the control. On the other hand, mice lacking Ubb are born normally at the expected Mendelian frequency 21. However, they are infertile due to the failure of progression of meiosis in germ cells 21. Consistently, significantly low ubiquitin levels were found in the testis and germinal vesicle oocytes in 5-month-old mice whereas other organs were not significantly affected. Furthermore, the ubiquitinb null mice develop adult-onset obesity due to the degeneration of hypothalamic neurons involved in the control of energy balance and feeding. Although the ubiquitin level was not reduced in the whole brain, it was reduced in the hypothalamus by 30%. Therefore, a modest reduction in ubiquitin level seems to cause infertility and neurodegeneration in mice.
Other than the mutations in ubiquitin-encoding genes, mutations in several Dubs cause reduction of ubiquitin and various defects. In yeast, deletion of Dub encoding genes, including DOA4 and UBP6, can reduce the amount of monomeric ubiquitin 22. These mutants are sensitive to canavanine, and the defects are compensated by expression of excess ubiquitin. In mice, UCH-L1 is an abundant brain-specific deubiquitinase, constituting ∼1–5% of the total proteins in the brain 23. The mutation in UCH-Ll is responsible for gracile axonal dystrophy (gad) in mice 24. The mice develop synaptic dysfunction and degeneration of neurons. In the brain of gad mice, monomeric ubiquitin level is reduced by 20–30% compared with control mice, suggesting that a low level of ubiquitin is a possible cause of the disease (16). Since UCH-L1 binds ubiquitin, it is suggested that UCH-L1 stabilizes ubiquitin or prevent ubiquitin from degradation 23. Similarly, ataxia (axJ) mutation, a spontaneous recessive mutation, is caused by reduced expression of Usp14, a homolog of yeast Ubp6 25. The axJ mice develop neurological dysfunctions including progressive motor system abnormalities, ataxia, loss of movement and premature death. In the axJ mice, monomeric ubiquitin level is reduced by 30–40%.
Curiously, not only a small amount of ubiquitin but also ubiquitin surplus is not beneficial to cells. In yeast, overexpression of ubiquitin renders cells sensitive to certain kinds of stresses such as treatment with cadmium, arsenite and paromycin 26. In addition, overexpression of ubiquitin worsens cell growth when it is introduced in mutants of ubiquitin-proteasome system (UPS)-related genes, such as cdc48 temperature-sensitive mutant 27. Such mutants exhibit accumulation of ubiquitinated proteins, which could consequently lead to further accumulation of cytotoxic ubiquitinated proteins 27.
Ubiquitin proteasome pathway
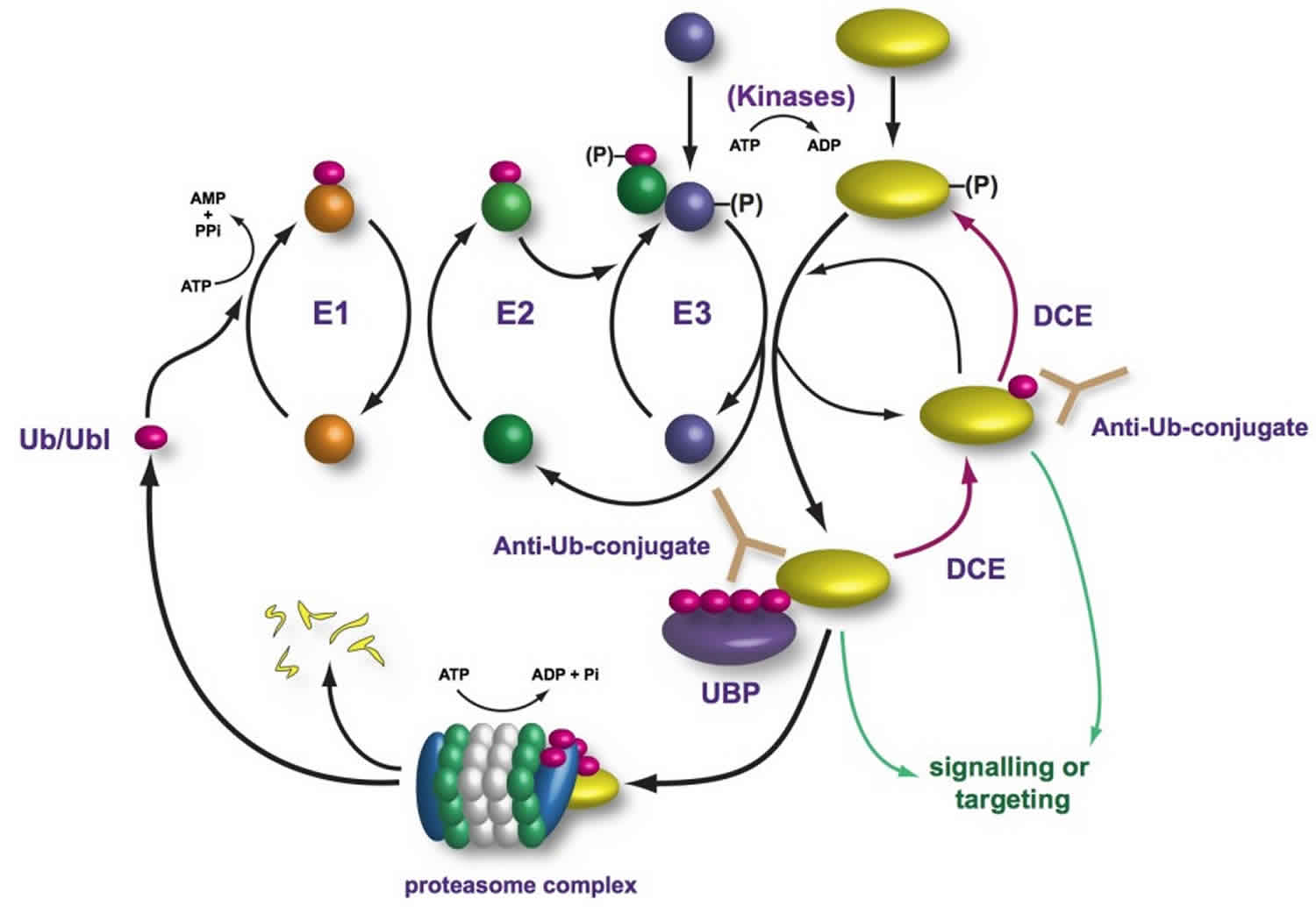
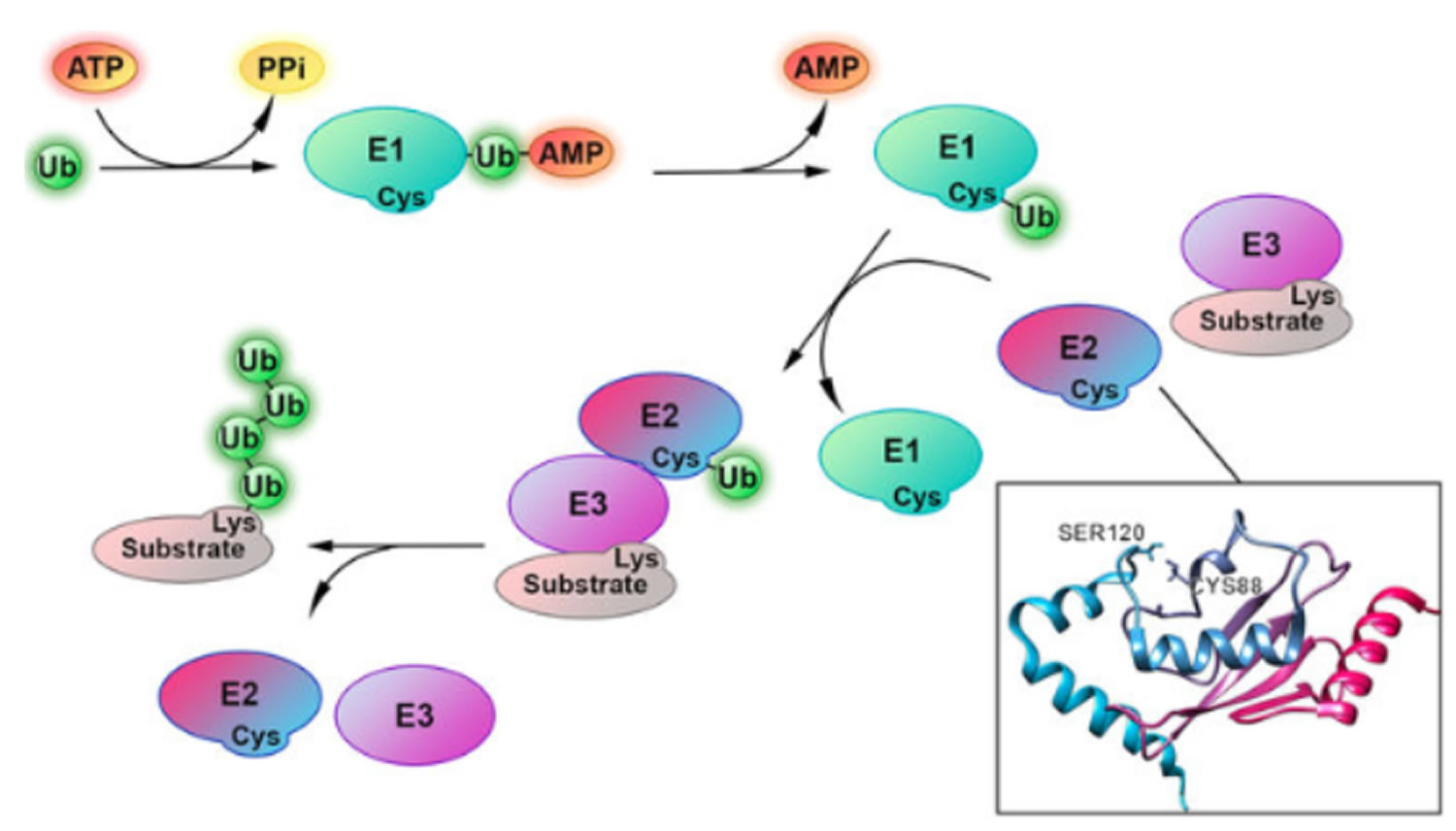
In the first step of the ubiquitin conjugation cascade, the carboxyl group of Gly-76 of ubiquitin is activated by ubiquitin-activating enzyme (E1). This step involves the hydrolysis of ATP to PPi to generate an ubiquitinyl adenylate intermediate bound to an E1 enzyme. Subsequently, an active site Cys residue of E1 covalently links to ubiquitin via a high-energy thioester linkage, with the concomitant release of AMP. Following activation, activated ubiquitin is then transferred by transacylation reaction to a thiol group of an active site Cys residue of E2 (ubiquitin carrier protein or ubiquitin-conjugating enzyme). Finally, E2 shuttles ubiquitin either directly to a protein substrate by itself or in cooperation with ubiquitin-protein ligase (E3), to form an amide isopeptide bond between the carboxyl group of Gly-76 of ubiquitin and an ε-amino group of the protein substrate’s internal Lys residue. The last step occurs by first transferring ubiquitin from E2 to E3, which accepts ubiquitin in a similar thiol linkage and then to the protein substrate. In some cases, however, covalent linkage between E3 and ubiquitin is not observed and it appears that ubiquitin is directly transferred from E2 to the protein substrate in a ternary E2-E3-substrate complex.
Once the protein substrate is mono-ubiquitinated, a polyubiquitin chain is formed through the same ubiquitination conjugation cascade, as described above, in which the carboxyl group of the carboxy terminal Gly-76 of ubiquitin is covalently linked to an internal Lys residue of ubiquitin that is already conjugated to the protein substrate.
It should be emphasized that the specificity of ubiquitination is largely determined by a series of E3 enzymes and E3 multiprotein complexes, each of which is specific to one or a few corresponding protein substrate(s) and of E2 enzymes, each of which is dedicated to their cognate E3 enzyme(s). As a result, different combinations of E2 and E3 enzymes allow selective tagging and degradation of specific intracellular proteins. In contrast, there is a single family of E1 that is highly conserved.
Figure 1. Ubiquitin proteasome pathway
Abbreviations: Ub = ubiquitin; E1 = ubiquitin-activating enzyme; E2 = ubiquitin-conjugating enzyme; E3 = ubiquitin ligase enzyme; Lys = lysine; Cys = cystine
Enzymes of the ubiquitination cascade system
Ubiquitin-activating enzyme (E1)
Ubiquitin-activating enzymes (E1) initiate the activation and conjugation of ubiquitin and ubiquitin-like modifiers (UBLs) in a cascade of events that modifies substrate proteins resulting in proteasomal or lysosomal degradation, changes in intracellular localization, activity or other signalling events. To date eight ubiquitin-activating enzymes (E1) enzymes have been identified, two characterized from the human genome comprise three domains; an adenylation domain which binds ATP and ubiquitin, a catalytic cysteine domain (CCD) which binds activated ubiquitin, and a C-terminal ubiquitin-fold domain (Ufd) which recruits specific E2 conjugating enzymes. E1 enzymes utilise ATP to activate the terminal glycine residue of ubiquitin/UBL generating a covalent thioester linkage between ‘activated’ ubiquitin/UBL and the E1 enzyme itself. Ubiquitin/UBL is then transferred to the sulphydryl group of the active-site cysteine on an E2 conjugating enzyme in a transthiolation reaction.
Ubiquitin-conjugating enzyme or ubiquitin-carrier protein (E2)
Ubiquitin conjugating enzyme (E2) form a ubiquitin-thioester intermediate in the ubiquitylation pathway. There are predicted to be 38 ubiquitin-conjugating enzyme (E2) conjugating enzymes (of which Ubiquigent offer 32) and four classes (Class I-IV) of the enzyme exist, all of which contain a catalytic (Ubc) domain with an active site cysteine. After transfer of ubiquitin/UBL (ubiquitin-like modifier) from the ubiquitin-activating enzyme (E1) activating enzyme to the ubiquitin-conjugating enzyme (E2) via a transthiolation reaction, the loaded ubiquitin-conjugating enzyme (E2) interacts with E3 ligases to transfer the activated ubiquitin/UBL to the substrate lysine acceptor residue; either directly from the E2 or via a lysine residue on the E3 ligase itself.
All ubiquitin-conjugating enzymes (E2s) contain a conserved ~130 amino acid-long catalytic core sequence i.e., UBC domain. An active-site Cys residue within the UBC domain is essential since it is required to form an ubiquitin-thioester intermediate. As a result, mutation of an active-site Cys residue abolishes UBC activity. Ubiquitin-conjugating enzymes (E2s) are subdivided on the basis of their distinct primary sequences, presumably reflecting their different specificities for their cognate E3s. For example, class I E2s, such as UBC4, UBC5, UBC7, and UBC9–13 consist exclusively of UBC domain and may require their cognate ubiquitin ligase enzymes (E3s) for recognition of substrates, since they cannot alone transfer ubiquitin to substrates. Class II (UBC1, UBC2/RAD6, UBC3/CDC34, UBC6, and UBC8), class III (UBC6), and class IV ubiquitin-conjugating enzymes (E2s) contain unique carboxy or amino terminal extensions or both that may mediate substrate specificity as well as intracellular localization.
Among the 13 known yeast ubiquitin-conjugating enzymes (E2s), only UBC3 and UBC9 are encoded by essential genes and both are involved in regulating cell cycle progression. UBC3/CDC34, for example, is involved in the degradation of SIC1 CDK inhibitor, which is necessary for the G1 to S phase transition, in conjunction with CDC53, SKP1, and CDC4. In addition, UBC2/RAD6 is involved in DNA repair in cooperation with its cognate E3α15 to degrade so-called N-end rule protein substrates. It is generally believed that E2s together with their cognate E3s recognize specific protein substrates. Such examples and specific E3s will be described below in more detail.
Ubiquitin ligase enzymes (E3s)
E3 ligases or ligase complexes recognize specific motifs in their substrate(s) and catalyze the transfer of ubiquitin directly or indirectly from a thioester intermediate from their cognate ubiquitin-conjugating enzyme (E2) to form an amide isopeptide bond between protein substrate and ubiquitin. In general, there are a few possible mechanisms for recognition of protein substrates by the different ubiquitin ligase enzymes (E3s). For example, some substrates carry degradation signals that are constitutively active, whereas other substrates require a covalent modification of the sequence motif, such as phosphorylation. Conversely, ubiquitin ligase enzymes (E3s) themselves can be altered or bound with an ancillary protein to recognize a specific subset of substrates.
E3 ligases confer specificity to the ubiquitylation pathway by recognising target substrates and mediating transfer of ubiquitin or ubiquitin like modifiers (UBLs) from an E2 conjugating enzyme to a substrate. There are estimated to be >700 E3 ligases forming several classes of enzyme; N-end rule ubiquitin E3 ligases, Homology to E6AP C-Terminus (HECT) domain E3s, Really Interesting New Gene (RING) domain E3s, U-Box containing E3s, Inhibitor of Apoptosis (IAP) E3s, and Cullin-RING E3 ligases (CRLs). The classes of E3 ligases are continually expanding. HECT E3 ligases form a thioester intermediate with the C-terminus of activated ubiquitin via a catalytic cysteine residue on the E3. In this case, ubiquitin is transferred from an E2 via the E3 to a substrate protein lysine side chain. RING and U-Box E3 ligases do not possess a catalytic cysteine residue and bring the E2-ubiquitin complex and substrate into close proximity to mediate the transfer of the ubiquitin directly from the E2 to the substrate.
Ubiquitin function
Ubiquitin through its chemical conjugation onto proteins (ubiquitination) plays major roles in regulating almost all cellular processes from tagging them for proteasomal degradation (protein degradation), protein trafficking, cell-cycle regulation, DNA repair, apoptosis and signal transduction 2 and protein-protein interaction 3. Among the various functions of ubiquitin, the most characterized function is serving as a tag for selective proteolysis by the 26S proteasome. Multiple ubiquitins are covalently added to a substrate successively by ubiquitin-activating (E1), ubiquitin-conjugating (E2) and ubiquitin ligase (E3) enzymes, producing a substrate conjugated with polyubiquitin. The ubiquitinated substrates are recognized and degraded by the 26S proteasome after the polyubiquitin chain is processed off and recovered by deubiquitinases.
The ubiquitination system functions in a wide variety of cellular processes, including:
- Antigen processing
- Apoptosis
- Biogenesis of organelles
- Cell cycle and division
- DNA transcription and repair
- Differentiation and development
- Immune response and inflammation
- Neural and muscular degeneration
- Morphogenesis of neural networks
- Modulation of cell surface receptors, ion channels and the secretory pathway
- Response to stress and extracellular modulators
- Ribosome biogenesis
- Viral infection
In addition, ubiquitination is also critical in the vacuolar sorting process of both endocytic and biosynthetic membrane proteins 2. At the plasma membrane, ubiquitin serves as a signal for endocytosis, and at the endosome, ubiquitin serves as a signal to sort cargo proteins into the multivesicular body, which is a critical step to their transport to lysosomes. ubiquitin is removed from the cargo by deubiquitinases before its entry into the multivesicular body.
Human diseases due to defects of the ubiquitin proteasome pathway
Reflecting the fact that the regulation of diverse short-lived proteins and regulatory proteins is mediated by the ubiquitin-proteasome pathway, it is not surprising that defects of various components of this complex biochemical machinery result in a range of human diseases. As a result, these components would provide an attractive novel target for therapeutic intervention. A few examples of inherited and acquired human diseases due to the defects of the ubiquitin-proteasome pathway are described below, emphasizing the molecular mechanism of pathogenesis.
Inherited Disorders
Angelman’s syndrome
Angelman’s syndrome is a complex neurological disorder 28 due to various genetic mechanisms that map to human chromosome 15q11–q13 29. Clinical characteristics of this disorder include developmental delay, speech impairment, movement or balance disorder, and behavioral uniqueness 30. Further genetic analysis revealed that truncated mutants of the UBE3A gene on chromosome 15q were identified in some patients with Angelman’s syndrome 31. UBE3A gene encodes a protein called E6-AP protein ligase (also known as ubiquitin-protein ligase or E3). Although the true substrate(s) of this E6-AP in Angelman’s syndrome patients has not yet been identified, the ubiquitin-proteasome pathway plays an important role during brain development and perturbation of this pathway causes a neurolopathological disorder.
Liddle syndrome
Liddle et al. 32 recognized this rare hereditary disorder in a family who were unable to maintain the proper balance of salt and water in the body, resulting in abnormally high blood pressure. The molecular mechanism of Liddle syndrome was later understood by Yale researchers 33 who observed mutations in the carboxy terminus of β and γ subunit of the epithelial sodium channel in the kidney. Among many different mutations, deletion or mutation of the proline-rich (PPxY) region leads to constitutive activation of the kidney sodium channel, resulting in retention of excessive amounts of salt and water. Using a yeast two-hybrid screen and in vitro binding studies 34, WW domain of an ubiquitin ligase, Nedd4, is found to bind specifically to the PPxY sequences of the above-mentioned kidney sodium channel. Taken together, these findings suggests a novel mechanism regulating sodium reabsorption, in which failure of proper ubiquitination of the sodium channel results in increased retention of the sodium channel at the cell surface and thus excessive reabsorption of sodium and water leading to hypertension. It should be noted, however, that this short-lived sodium channel, after ubiquitination, is targeted and degraded in the lysosome, but not by the 26S proteasome machinery.
Acquired Disorders
Cervical cancer
The most important risk factor for cervical cancer is human papillomavirus (HPV) infection. In cases of cervical cancer caused by the high-risk strains of HPV, levels of the tumor suppressor protein p53 were found to be unusually low due to the presence of the E6 proteins encoded by the oncogenic HPV 35. This E6-promoted degradation of p53 involves the ubiquitin-proteasome pathway and represents a major mechanism, by which virus transforms the host cells. The E6 oncoprotein was later found to modify the functions of E6-AP 36 whose absence prevents ubiquitination of p53 by E6-AP 37. Specifically, E6 appears to function as an adapter protein to maintain a ternary complex of E6-AP ubiquitin-protein ligase (E3), its cognate UbcH8 ubiquitin-conjugating enzyme (E2), and a target substrate to ensure specific ubiquitination of substrate molecules 38. Interestingly, E6 also promotes the ubiquitination and degradation of E6-AP 39.
Colorectal cancer
Colorectal cancer, which arises in the epithelium of the lumen of the colon and rectum, is the second most common cause of cancer death in the United States. Generally speaking, the transformation of the normal epithelial cells into cancer cells occurs through a multistage process as a result of many different defects. One of most well-characterized genetic defects is found in the APC (adenomatous polyposis coli) gene located on chromosome 5q21. Mutations of the APC gene have been documented in familial adenomatous polyposis and in more than 80% of the sporadic colorectal cancer patients 40.
Among its many functions, APC is known to control the cellular level of β-catenin by forming a complex with Axin, which recruits and facilitates ubiquitination of β-catenin for subsequent degradation by the 26S proteasome 41. This process appears to be dependent on the phosphorylation of both APC and β-catenin by glycogen synthase kinase-3 (GSK-3) 42. In cancer cells expressing mutant APC, β-catenin accumulates 43 and interacts with T-cell factor as well as activates transcription of the proto-oncogenes, such as cyclin D1 and c-myc 44. Mutations of Ser to amino acid residues that cannot be phosphorylated stabilize β-catenin 45. It appears that ubiquitination of β-catenin is catalyzed by the SCF complex and its cognate E2. A receptor component of the SCF E3 ligase complex, i.e., F box- and WD domain-containing β-TrCP binds to the phosphorylated form of β-catenin, and over-production of a defective β-TrCP blocks β-catenin degradation 46.
Alzheimer’s disease
Alzheimer’s disease is a chronic brain disorder due to a progressive degeneration of brain tissue, leading to loss of mental functions, such as memory and learning. Alzheimer’s disease is marked by abnormal aggregates (senile plaques) and irregular knots (neurofibrillary tangles) of brain matter. By immunohistochemical analysis of protein contents in intracellular inclusion bodies, ubiquitin is found in both neurofibrillary tangles and senile plaques. Although it is not yet clear whether accumulation of ubiquitin conjugates is primary or secondary, a growing number of evidence indicate the involvement of the ubiquitin-proteasome pathway in Alzheimer’s disease. For example, findings of a simultaneous frameshift mutation of both β amyloid precursor protein (βAPP) and ubiquitin have implicated the ubiquitin-dependent pathway in the pathogenesis of Alzheimer’s disease 47. In addition, treatment of cells expressing presenilins 1 and 2 (PS1 and PS2) with proteasome inhibitors was shown to increase the PS1/PS2-mediated production of both APPα and Aβ40, which are βAPP maturation products 48. Senile plaques are thought to derive from the abnormal level of Aβ40 production 49. PS1 and PS2 are also found to be the proteasome substrates 50. Taken altogether, the ubiquitin-proteasome pathway appears to control the level of PS1 and PS2, thereby regulating the βAPP maturation pathway.
- Yoko Kimura, Keiji Tanaka, Regulatory mechanisms involved in the control of ubiquitin homeostasis, The Journal of Biochemistry, Volume 147, Issue 6, June 2010, Pages 793–798, https://doi.org/10.1093/jb/mvq044[↩][↩]
- Mukhopadhyay D, Riezman H. Proteasome-independent functions of ubiquitin in endocytosis and signaling, Science , 2007, vol. 315, pg. 201-205[↩][↩][↩]
- SWATEK, K. N. & KOMANDER, D. 2016. Ubiquitin modifications. Cell Res, 26, 399-422.[↩][↩][↩]
- Scheffner, M., Nuber, U., and Huibregtse, J. M. (1995) Protein Ubiquitination Involving an E1-E2-E3 Enzyme Ubiquitin Thioester Cascade. Nature373, 81-83[↩]
- Spence, J., Sadis, S., Haas, A. L., and Finley, D. (1995) A Ubiquitin Mutant with Specific Defects in DNA-Repair and Multiubiquitination. Molecular and Cellular Biology15, 1265-1273[↩]
- Meyer, H. J., and Rape, M. (2014) Enhanced Protein Degradation by Branched Ubiquitin Chains. Cell157, 910-921[↩]
- Tokarev, A. A., Munguia, J., and Guatelli, J. C. (2011) Serine-Threonine Ubiquitination Mediates Downregulation of BST-2/Tetherin and Reliefof Restricted Virion Release by HIV-1 Vpu. J Virol85, 51-63[↩]
- Reyes-Turcu FE, Ventii KH, Wilkinson KD. Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes, Annu. Rev. Biochem. , 2009, vol. 78, pg. 363-397[↩]
- Metzger, M. B., Hristova, V. A., and Weissman, A. M. (2012) HECT and RING finger families of E3 ubiquitin ligases at a glance. J Cell Sci125, 531-537[↩]
- YAU, R. G., DOERNER, K., CASTELLANOS, E. R., HAAKONSEN, D. L., WERNER, A., WANG, N., YANG, X. W., MARTINEZ-MARTIN, N., MATSUMOTO, M. L., DIXIT, V. M. & RAPE, M. 2017. Assembly and Function of Heterotypic Ubiquitin Chains in Cell-Cycle and Protein Quality Control. Cell, 171, 918-933.e20.[↩]
- THROWER, J. S., HOFFMAN, L., RECHSTEINER, M. & PICKART, C. M. 2000. Recognition of the polyubiquitin proteolytic signal. Embo j, 19, 94-102.[↩]
- LEE, B. H., LU, Y., PRADO, M. A., SHI, Y., TIAN, G., SUN, S., ELSASSER, S., GYGI, S. P., KING, R. W. & FINLEY, D. 2016. USP14 deubiquitinates proteasome-bound substrates that are ubiquitinated at multiple sites. Nature, 532, 398-401[↩]
- RISTIC, G., TSOU, W. L. & TODI, S. V. 2014. An optimal ubiquitin-proteasome pathway in the nervous system: the role of deubiquitinating enzymes. Front Mol Neurosci, 7, 72.[↩]
- WANG, C. H., CHEN, G. C. & CHIEN, C. T. 2014. The deubiquitinase Leon/USP5 regulates ubiquitin homeostasis during Drosophila development. Biochem Biophys Res Commun, 452, 369-75.[↩]
- Zhang, Y. F., Li, D. Y., Zhang, H. J., Hong, Y. B., Huang, L., Liu, S. X., Li, X. H., Ouyang, Z. G., and Song, F. M. (2015) Tomato histone H2B monoubiquitination enzymes SlHUB1 and SlHUB2 contribute to disease resistance against Botrytis cinerea through modulating the balance between SA-and JA/ET-mediated signaling pathways. Bmc Plant Biol15[↩]
- Silva, G. M., Finley, D., and Vogel, C. (2015) K63 polyubiquitination is a new modulator of the oxidative stress response. Nat Struct Mol Biol22, 116-123[↩]
- Wickliffe, K. E., Williamson, A., Meyer, H. J., Kelly, A., and Rape, M. (2011) K11-linked ubiquitin chains as novel regulators of cell division. Trends Cell Biol21, 656-663[↩]
- Fang, J., Chen, T. P., Chadwick, B., Li, E., and Zhang, Y. (2004) Ring1b-mediated H2A ubiquitination associates with inactive X chromosomes and is involved in initiation of X inactivation. J Biol Chem279, 52812-52815[↩]
- Ryu KY, Maehr R, Gilchrist CA, Long MA, Bouley DM, Mueller B, Ploegh HL, Kopito RR. The mouse polyubiquitin gene UbC is essential for fetal liver development, cell-cycle progression and stress tolerance, EMBO J. , 2007, vol. 26, pg. 2693-2706[↩][↩]
- Redman KL, Rechsteiner M. Identification of the long ubiquitin extension as ribosomal protein S27a, Nature , 1989, vol. 338, pg. 438-440[↩][↩]
- Ryu KY, Sinnar SA, Reinholdt LG, Vaccari S, Hall S, Garcia MA, Zaitseva TS, Bouley DM, Boekelheide K, Handel MA, Conti M, Kopito RR. The mouse polyubiquitin gene Ubb is essential for meiotic progression, Mol. Cell. Biol. , 2008, vol. 28, pg. 1136-1146[↩][↩][↩]
- Chernova TA, Allen KD, Wesoloski LM, Shanks JR, Chernoff YO, Wilkinson KD. Pleiotropic effects of Ubp6 loss on drug sensitivities and yeast prion are due to depletion of the free ubiquitin pool, J. Biol. Chem. , 2003, vol. 278, pg. 52102-52115[↩]
- Osaka H, Wang YL, Takada K, Takizawa S, Setsuie R, Li H, Sato Y, Nishikawa K, Sun YJ, Sakurai M, Harada T, Hara Y, Kimura I, Chiba S, Namikawa K, Kiyama H, Noda M, Aoki S, Wada K. Ubiquitin carboxy-terminal hydrolase L1 binds to and stabilizes monoubiquitin in neuron, Hum. Mol. Genet. , 2003, vol. 12, pg. 1945-1958[↩][↩]
- Saigoh K, Wang YL, Suh JG, Yamanishi T, Sakai Y, Kiyosawa H, Harada T, Ichihara N, Wakana S, Kikuchi T, Wada K. Intragenic deletion in the gene encoding ubiquitin carboxy-terminal hydrolase in gad mice, Nat. Genet. , 1999, vol. 23, pg. 47-51[↩]
- Anderson C, Crimmins S, Wilson JA, Korbel GA, Ploegh HL, Wilson SM. Loss of Usp14 results in reduced levels of ubiquitin in ataxia mice, J. Neurochem. , 2005, vol. 95, pg. 724-731[↩]
- Chen Y, Piper PW. Consequences of the overexpression of ubiquitin in yeast: elevated tolerances of osmostress, ethanol and canavanine, yet reduced tolerances of cadmium, arsenite and paromomycin, Biochim. Biophys. Acta , 1995, vol. 1268, pg. 59-64[↩]
- Kimura Y, Yashiroda H, Kudo T, Koitabashi S, Murata S, Kakizuka A, Tanaka K. An inhibitor of a deubiquitinating enzyme regulates ubiquitin homeostasis, Cell , 2009, vol. 137, pg. 549-559[↩][↩]
- Angelman H. Dev Med Child Neurol. 1965;7:681–688[↩]
- Mann MR, Bartolomei MS. Hum Mol Genet. 1999;8:1867–1873[↩]
- Williams CA, Angelman H, Clayton-Smith J, Driscoll DJ, Hendrickson JE, Knoll JH, Magenis RE, Schinzel A, Wagstaff J, Whidden EM, et al. Am J Med Genet. 1995;56:237–238[↩]
- Matsuura T, Sutcliffe JS, Fang P, Galjaard RJ, Jiang YH, Benton CS, Rommens JM, Beaudet AL. Nat Genet. 1997;15:74–77.[↩]
- Liddle G, Bledsoe T, Coppage W. Trans Assoc Am Phys. 1963;76:199–213[↩]
- Shimkets RA, Warnock DG, Bositis CM, Nelson-Williams C, Hansson JH, Schambelan M, Gill JR, Jr, Ulick S, Milora RV, Findling JW, et al. Cell. 1994;79:407–414[↩]
- WW domains of Nedd4 bind to the proline-rich PY motifs in the epithelial Na+ channel deleted in Liddle’s syndrome. Staub O, Dho S, Henry P, Correa J, Ishikawa T, McGlade J, Rotin D. EMBO J. 1996 May 15; 15(10):2371-80.[↩]
- The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Scheffner M, Werness BA, Huibregtse JM, Levine AJ, Howley PM. Cell. 1990 Dec 21; 63(6):1129-36.[↩]
- The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Scheffner M, Huibregtse JM, Vierstra RD, Howley PM. Cell. 1993 Nov 5; 75(3):495-505.[↩]
- The role of E6AP in the regulation of p53 protein levels in human papillomavirus (HPV)-positive and HPV-negative cells. Talis AL, Huibregtse JM, Howley PM. J Biol Chem. 1998 Mar 13; 273(11):6439-45.[↩]
- Basal and human papillomavirus E6 oncoprotein-induced degradation of Myc proteins by the ubiquitin pathway. Gross-Mesilaty S, Reinstein E, Bercovich B, Tobias KE, Schwartz AL, Kahana C, Ciechanover A. Proc Natl Acad Sci U S A. 1998 Jul 7; 95(14):8058-63.[↩]
- Human papillomavirus type 16 E6 induces self-ubiquitination of the E6AP ubiquitin-protein ligase. Kao WH, Beaudenon SL, Talis AL, Huibregtse JM, Howley PM. J Virol. 2000 Jul; 74(14):6408-17.[↩]
- Lessons from hereditary colorectal cancer. Kinzler KW, Vogelstein B. Cell. 1996 Oct 18; 87(2):159-70.[↩]
- beta-catenin is a target for the ubiquitin-proteasome pathway. Aberle H, Bauer A, Stappert J, Kispert A, Kemler R. EMBO J. 1997 Jul 1; 16(13):3797-804.[↩]
- GSK-3beta-dependent phosphorylation of adenomatous polyposis coli gene product can be modulated by beta-catenin and protein phosphatase 2A complexed with Axin. Ikeda S, Kishida M, Matsuura Y, Usui H, Kikuchi A. Oncogene. 2000 Jan 27; 19(4):537-45.[↩]
- Regulation of intracellular beta-catenin levels by the adenomatous polyposis coli (APC) tumor-suppressor protein. Munemitsu S, Albert I, Souza B, Rubinfeld B, Polakis P. Proc Natl Acad Sci U S A. 1995 Mar 28; 92(7):3046-50.[↩]
- Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Tetsu O, McCormick F. Nature. 1999 Apr 1; 398(6726):422-6.[↩]
- Stabilization of beta-catenin by genetic defects in melanoma cell lines. Rubinfeld B, Robbins P, El-Gamil M, Albert I, Porfiri E, Polakis P. Science. 1997 Mar 21; 275(5307):1790-2.[↩]
- The human F box protein beta-Trcp associates with the Cul1/Skp1 complex and regulates the stability of beta-catenin. Latres E, Chiaur DS, Pagano M. Oncogene. 1999 Jan 28; 18(4):849-54.[↩]
- Frameshift mutants of beta amyloid precursor protein and ubiquitin-B in Alzheimer’s and Down patients. van Leeuwen FW, de Kleijn DP, van den Hurk HH, Neubauer A, Sonnemans MA, Sluijs JA, Köycü S, Ramdjielal RD, Salehi A, Martens GJ, Grosveld FG, Peter J, Burbach H, Hol EM. Science. 1998 Jan 9; 279(5348):242-7.[↩]
- Role of the proteasome in Alzheimer’s disease. Checler F, da Costa CA, Ancolio K, Chevallier N, Lopez-Perez E, Marambaud P. Biochim Biophys Acta. 2000 Jul 26; 1502(1):133-8.[↩]
- Processing of the beta-amyloid precursor protein and its regulation in Alzheimer’s disease. Checler F. J Neurochem. 1995 Oct; 65(4):1431-44.[↩]
- Alzheimer’s disease-linked mutation of presenilin 2 (N141I-PS2) drastically lowers APPalpha secretion: control by the proteasome. Marambaud P, Alves da Costa C, Ancolio K, Checler F. Biochem Biophys Res Commun. 1998 Nov 9; 252(1):134-8.[↩]





{kind=link}