Contents
- X-Linked Protoporphyria
X-Linked Protoporphyria
X-Linked Protoporphyria (XLP) also called X-linked dominant protoporphyria or X-linked dominant erythropoietic protoporphyria is an extremely rare genetic disorder of the heme-biosynthetic pathway known as the porphyrias that usually present in early life with nonblistering painful and abnormal sensitivity to the sun (photosensitivity) that can cause severe pain, burning, and itching of sun-exposed skin 1, 2, 3, 4, 5, 6. X-linked protoporphyria (XLP) symptoms may occur immediately or shortly after exposure to the sun, including direct exposure or indirect exposure such as sunlight that passes through window glass or that is reflected off water or sand. Redness and swelling of affected areas can also occur. Blistering and severe scarring occur infrequently. Chronic episodes of photosensitivity may lead to changes in the skin of sun-exposed areas. Some individuals with X-linked protoporphyria (XLP) eventually develop potentially severe liver disease.
X-linked protoporphyria (XLP) is caused by gain-of-function mutations to the 5-aminolevulinate synthase 2 (ALAS2) gene located on the X chromosome and is inherited as an X-linked dominant trait 2. Males often develop a severe form of the disorder while females may not develop any symptoms (asymptomatic) or can develop a form as severe as that seen in males. The ALAS2 gene is located on the short arm (p) of the X chromosome (Xp11.21)*. The ALAS2 gene provides instructions for making an enzyme called 5′-aminolevulinate synthase 2 or erythroid ALA-synthase 6. The 5′-aminolevulinate synthase 2 or erythroid ALA-synthase (ALAS2) enzyme is found only in developing red blood cells found in bone marrow that develop into red blood cells (erythrocytes) called erythroblasts. 5′-aminolevulinate synthase 2 or erythroid ALA-synthase (ALAS2) enzyme also plays an important role in the production of heme. Heme is a component of iron-containing proteins called hemoproteins, including hemoglobin (the protein that carries oxygen in the blood). Heme is vital for all of the body’s organs, although it is most abundant in the blood, bone marrow, and liver. The production of heme is a multi-step process that requires 8 different enzymes. ALA-synthase (ALAS2) enzyme is responsible for the first step in this process, the formation of a compound called delta-aminolevulinic acid (δ-ALA or ALA) (see Figures 4 and 5 below). Mutations of the ALAS2 gene lead to the overproduction of ALA-synthase or 5′-aminolevulinate synthase 2 enzyme, which, in turn, results in the rate of delta-aminolevulinic acid (δ-ALA or ALA) formation being increased, and the insertion of iron into protoporphyrin IX (PPIX) by ferrochelatase (FECH) the last step in heme synthesis becomes the rate-limiting for heme synthesis in erythroid tissues (the tissues where red blood cells or erythrocytes are produced and develop, in adults, the primary erythroid tissue is the bone marrow) resulting in accumulation of protoporphyrin IX (PPIX) in the the bone marrow 7, 8. If protoporphyrin IX (PPIX) compounds build up in erythroblasts (a precursor to red blood cells in the bone marrow), they can leak out and be transported through the bloodstream to your skin and other tissues such as your liver 7, 8. High levels of protoporphyrin IX (PPIX) in the skin cause the oversensitivity to sunlight that is characteristic of X-linked protoporphyria (XLP) 9, 10. For example, when protoporphyrins absorb energy from sunlight, they enter an excited state (photoactivation) and this abnormal activation results in the characteristic damage to the skin. Accumulation of protoporphyrins in the liver causes toxic damage to the liver and may contribute to the formation of gallstones. Protoporphyrin is formed within red blood cells in the bone marrow and then enters the blood plasma, which carries it to the skin where it can be photoactivated by sunlight and cause damage. The liver removes protoporphyrin from the blood plasma and secretes it into the bile.
Protoporphyrin IX (PPIX) is released from the bone marrow into the circulating red blood cells and plasma where it is taken up by the liver and vascular endothelium including the superficial skin vasculature. The protoporphyrin IX (PPIX) molecules are photodynamic (light sensitive chemical that is activated by light) and absorb light radiation in visible blue-violet light in the Soret band and to a lesser degree in the long-wave UV region 11, 12. When protoporphyrin IX (PPIX) molecules absorb light they enter an excited energy state. This energy is presumably released as fluorescence and by formation of singlet oxygen and other oxygen radicals that can produce tissue and vessel damage secondary to activation of the complement system 2. The release of histamines, kinins, and chemotactic factors may bring about skin damage 13. Accumulated liver (hepatic) protoporphyrin IX (PPIX) can precipitate in liver cells (hepatocytes) and small bile ducts (bile canaliculi) that collect bile secreted by these liver cells (hepatocytes), causing liver toxicity, decreased bile formation and flow, and cholestatic liver failure in some patients 14, 15. Large amounts of porphyrins in the gallbladder can also cause gallstones. Less commonly, a buildup of porphyrins in the liver can result in liver damage that leads to cirrhosis and liver failure.
X-linked protoporphyria (XLP) accounts for 2 to 10% of protoporphyria cases with about 2% of cases in Europe and approximately 10% of cases in the United States 16, 17, 18.
The diagnosis of X-linked protoporphyria (XLP) is established in a male index case (the affected individual through whom a family with a genetic disorder is ascertained) with markedly increased free red blood cell (erythrocyte) protoporphyrin IX (PPIX) and zinc-chelated erythrocyte protoporphyrin IX (PPIX) by identification of a hemizygous pathogenic gain-of-function variant in ALAS2 on molecular genetic testing.
The diagnosis of X-linked protoporphyria (XLP) is established in a female index case with increased free erythrocyte protoporphyrin IX (PPIX) and zinc-chelated erythrocyte protoporphyrin IX (PPIX) by identification of a heterozygous pathogenic gain-of-function variant in ALAS2 on molecular genetic testing.
The treatment of X-linked protoporphyria is directed toward the specific symptoms that are apparent in each individual. Treatment may require the coordinated efforts of a team of specialists. Pediatricians, hematologists, dermatologists, hepatologists, and other healthcare professionals may need to systematically and comprehensively plan an affected child’s treatment. Genetic counseling may benefit affected individuals and their families.
There is no specific, FDA-approved therapy for individuals with X-linked protoporphyria (XLP). Because the disorder is so rare, most treatment information is based on erythropoietic protoporphyria (EPP), which is clinically similar to X-linked protoporphyria (XLP).
Avoidance of sunlight will benefit affected individuals and can include the use of clothing styles with long sleeves and pant legs, made with double layers of fabric or of light-exclusive fabrics, wide brimmed hats, gloves, and sunglasses. Topical sunscreens are generally ineffective, unless they contain light-reflective ingredients (e.g., zinc oxide). Some tanning products with ingredients that increase pigmentation may be helpful. Affected individuals may also benefit from window tinting and the use of vinyl or films to cover the windows of their homes and cars.
Avoidance of sunlight can potentially cause vitamin D deficiency and some individuals may require supplemental vitamin D.
A high potency form of oral beta-carotene (Lumitene) may be given to improve an affected individual’s tolerance of sunlight. This drug causes skin discoloration and may improve tolerance to sunlight. Oral Lumitene (beta-carotene) (120–180 mg/dL) has been used to improve tolerance to sunlight if the dose is adjusted to maintain serum carotene levels in the range of 10-15 μmol/L (600–800 μg/dL), causing mild skin discoloration due to carotenemia. The beneficial effects of beta-carotene (Lumitene) may involve quenching of singlet oxygen or free radicals. However, a systematic review of about 25 studies showed that the available data are unable to prove efficacy of treatments including beta-carotene, N-acetyl cysteine, and vitamin C 19. For more information on oral beta-carotene (Lumitene) treatment, contact the American Porphyria Foundation and the Prophyria Consortium of the Rare Diseases Clinical Research Network.
Another drug sometimes used to improve tolerance to sunlight is cysteine.
In 2019, the Food and Drug Administration (FDA) approved Afamelanotide (Scenesse®) for the treatment of adult patients with erythropoietic protoporphyria (EPP). Afamelanotide (Scenesse) is a controlled-release, long-acting injectable implant, alpha-melanocyte-stimulating hormone (α-MSH) analogue, that increases eumelanin by binding to the melanocortin-1 receptor and provides sun protection and improves sun tolerance by increasing skin pigmentation and antioxidant properties 20, 21. Afamelanotide (Scenesse) was available in Europe for a period of time before its approval in the United States. Afamelanotide (Scenesse) was approved for patients with the erythropoietic protoporphyria (EPP) by the European Medicines Agency in 2014, and by the FDA in October 2019. Afamelanotide showed positive results in Phase III clinical trials in the US and Europe 22. Long-term studies in Europe show good compliance, clinical effectiveness, and improved quality of life 23.
Other treatment is symptomatic and supportive. Individuals with high levels of protoporphyrin in the plasma and red blood cells should be observed closely by a physician for possible liver malfunction that could eventually lead to liver failure.
When iron deficiency is present, iron supplements may be given. A drug called Prevalite (cholestyramine) or activated charcoal maybe prescribed to interrupt the circulation of protoporphyrin through the liver and intestines in patients with liver disease.
Liver transplantation has been performed as a life-saving measure in patients with erythropoietic protoporphyria (EPP) and X-linked protoporphyria (XLP) related liver failure. Bone marrow transplant can also be performed after liver transplant to prevent further damage to the liver.
X-linked protoporphyria (XLP) patients should also receive vaccination against hepatitis A and B to prevent other causes of liver damage.
X-linked protoporphyria (XLP) patients should be seen at least yearly to monitor protoporphyrin levels, anemia, liver enzymes, iron and vitamin D levels.
Figure 1. Porphyrin molecular structure
Footnote: Molecular structure of porphyrin (M represent metal ions, such as Mg, Cu, Fe, Zn, etc.).
[Source 24 ]Figure 2. Hemoglobin molecular structure
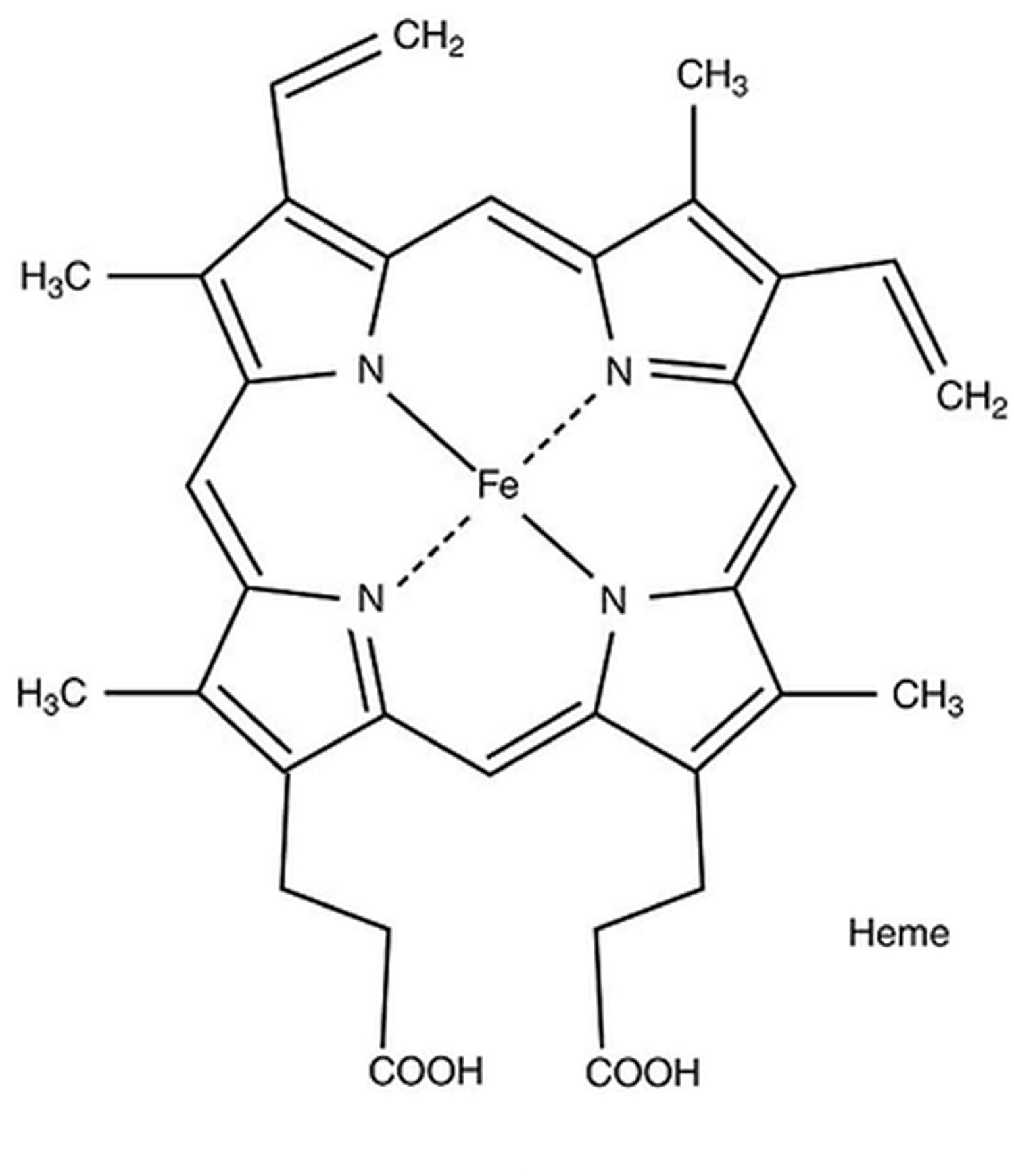
Figure 3. Heme (haem) molecular structure
Footnote: Heme A and heme B molecular structures
[Source 26 ]Figure 4. Heme biosynthesis pathway
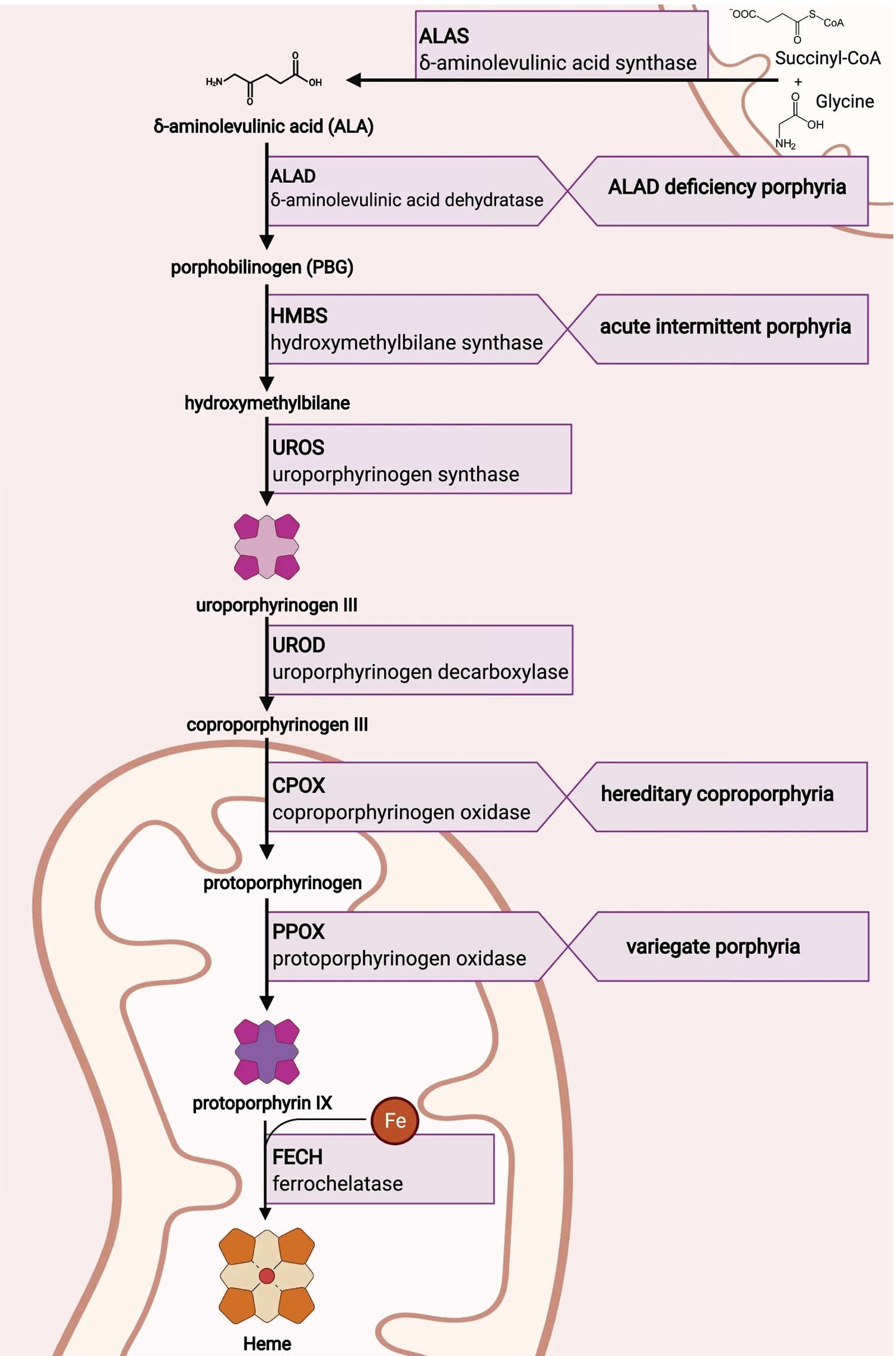
Figure 5. Heme synthesis pathway and porphyrias
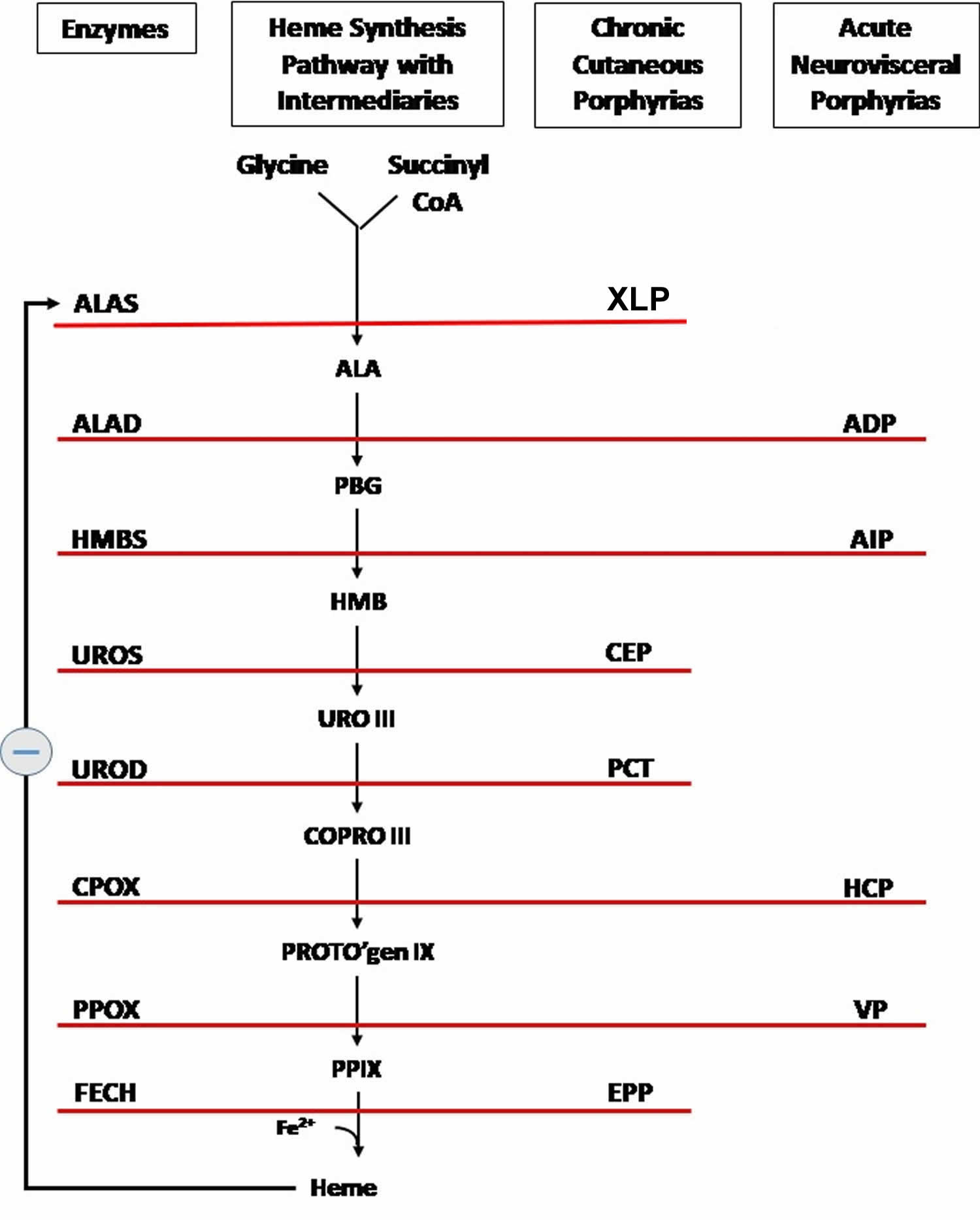
Footnotes: The heme biosynthetic pathway requires 8 enzymatic steps. Heme synthesis pathway showing the enzymes involved in the heme synthesis pathway and the associated porphyrias with the disruption of each specific enzyme. Gain-of-function variants in ALAS2 result in X-linked protoporphyria (XLP), and loss-of-functions variants in FECH result in erythropoietic protoporphyria (EPP). In both X-linked protoporphyria (XLP) and erythropoietic protoporphyria (EPP), metal-free protoporphyrin IX (PPIX) accumulates in erythroblasts, erythrocytes, the plasma, and the biliary system. Metal-free protoporphyrin IX (PPIX) is photosensitive, particularly to visible light in the blue range, and the light-mediated activation of metal-free protoporphyrin IX (PPIX) produces free radicals that damage the surrounding tissues.
Enzymes, encoded by genes, catalyze each of the steps. Gene mutations cause deficient enzyme production. Disruptions are indicated by red lines connecting enzymes with the resultant porphyrias. ALAS (ALAS2) = aminolevulinate synthase (aminolevulinate synthase 2); ALAD = delta-aminolevulinic acid dehydratase; PBGD = porphobilinogen dehydratase; HMBS = hydroxymethylbilane synthase; UROS = uroporphyrinogen-III synthase; UROD = uroporphyrinogen III decarboxylase; CPOX = coproporphyrinogen-III oxidase; PPOX = protoporphyrinogen oxidase; FECH = ferrochelatase.
Porphyrias resulting from disruption of enzyme production. XLP (X-linked protoporphyria); ADP (aminolevulinic acid dehydratase porphyria); AIP (acute intermittent porphyria); CEP (congenital erythropoietic porphyria); PCT (porphyria cutanea tarda); HCP (hereditary coproporphyria); VP (variegate porphyria); EPP (erythropoietic protoporphyria).
Abbreviations: ALA = aminolevulinic acid; PBG = porphobilinogen; HMB = hydroxymethylbilane; URO III = uroporphyrinogen III; COPRO III = coproporphyrinogen III; PROTO’gen IX protoporphyrinogen IX; PPIX = protoporphyrin IX; Fe2+ = iron.
[Source 27 ]X-Linked Protoporphyria cause
X-linked protoporphyria (XLP) is caused by gain-of-function mutations to the 5-aminolevulinate synthase 2 (ALAS2) gene located on the X chromosome and is inherited as an X-linked dominant trait 2. Males often develop a severe form of the disorder while females may not develop any symptoms (asymptomatic) or can develop a form as severe as that seen in males. The ALAS2 gene is located on the short arm (p) of the X chromosome (Xp11.21)*. The ALAS2 gene provides instructions for making an enzyme called 5′-aminolevulinate synthase 2 or erythroid ALA-synthase 6. The 5′-aminolevulinate synthase 2 or erythroid ALA-synthase (ALAS2) enzyme is found only in developing red blood cells found in bone marrow that develop into red blood cells (erythrocytes) called erythroblasts. 5′-aminolevulinate synthase 2 or erythroid ALA-synthase (ALAS2) enzyme also plays an important role in the production of heme. Heme is a component of iron-containing proteins called hemoproteins, including hemoglobin (the protein that carries oxygen in the blood). Heme is vital for all of the body’s organs, although it is most abundant in the blood, bone marrow, and liver. The production of heme is a multi-step process that requires 8 different enzymes. ALA-synthase (ALAS2) enzyme is responsible for the first step in this process, the formation of a compound called delta-aminolevulinic acid (δ-ALA or ALA). Mutations of the ALAS2 gene lead to the overproduction of ALA-synthase or 5′-aminolevulinate synthase 2 enzyme, which, in turn, results in the rate of delta-aminolevulinic acid (δ-ALA or ALA) formation being increased, and the insertion of iron into protoporphyrin IX (PPIX) by ferrochelatase (FECH) the last step in heme synthesis becomes the rate-limiting for heme synthesis in erythroid tissues (the tissues where red blood cells or erythrocytes are produced and develop, in adults, the primary erythroid tissue is the bone marrow) resulting in accumulation of protoporphyrin IX (PPIX) in the the bone marrow 7, 8. If protoporphyrin IX (PPIX) compounds build up in erythroblasts (a precursor to red blood cells in the bone marrow), they can leak out and be transported through the bloodstream to your skin and other tissues such as your liver 7, 8. High levels of protoporphyrin IX (PPIX) in the skin cause the oversensitivity to sunlight that is characteristic of X-linked protoporphyria (XLP) 9, 10. For example, when protoporphyrins absorb energy from sunlight, they enter an excited state (photoactivation) and this abnormal activation results in the characteristic damage to the skin. Accumulation of protoporphyrins in the liver causes toxic damage to the liver and may contribute to the formation of gallstones. Protoporphyrin is formed within red blood cells in the bone marrow and then enters the blood plasma, which carries it to the skin where it can be photoactivated by sunlight and cause damage. The liver removes protoporphyrin from the blood plasma and secretes it into the bile.
Chromosomes are found in the nucleus of all body cells. They carry the genetic characteristics of each individual. Pairs of human chromosomes are numbered from 1 through 22, with an unequal 23rd pair of X and Y chromosomes for males and two X chromosomes for females. Each chromosome has a short arm designated as “p” and a long arm identified by the letter “q”. Chromosomes are further subdivided into bands that are numbered. For example, “chromosome Xp22.2-22.1” refers to bands 22.2 through 22.1 on the short arm of chromosome X.
X-linked protoporphyria (XLP) results from gain-of-function mutations in erythroid-specific ALAS2 28. Mutations associated with X-linked protoporphyria (XLP) have only been observed in exon 11, which encodes the C-terminus, and result in a gain-of-function of ALAS2. These mutations result in stop or frameshift lesions that prematurely truncate or abnormally elongate the wild-type enzyme, leading to increased ALAS2 activity 28, 18. In X-linked protoporphyria (XLP), all males are affected 1. In heterozygous females (females with one normal copy and one mutated copy of ALAS2 gene on their X chromosomes) with X-linked protoporphyria (XLP), the random X-inactivation pattern directly influences the penetrance and the severity of the phenotype. X-linked protoporphyria (XLP) females can be asymptomatic clinically with normal protoporphyrins, be asymptomatic clinically with slightly elevated protoporphyrin levels or have significant symptoms based on the pattern of X-inactivation 29, 1.
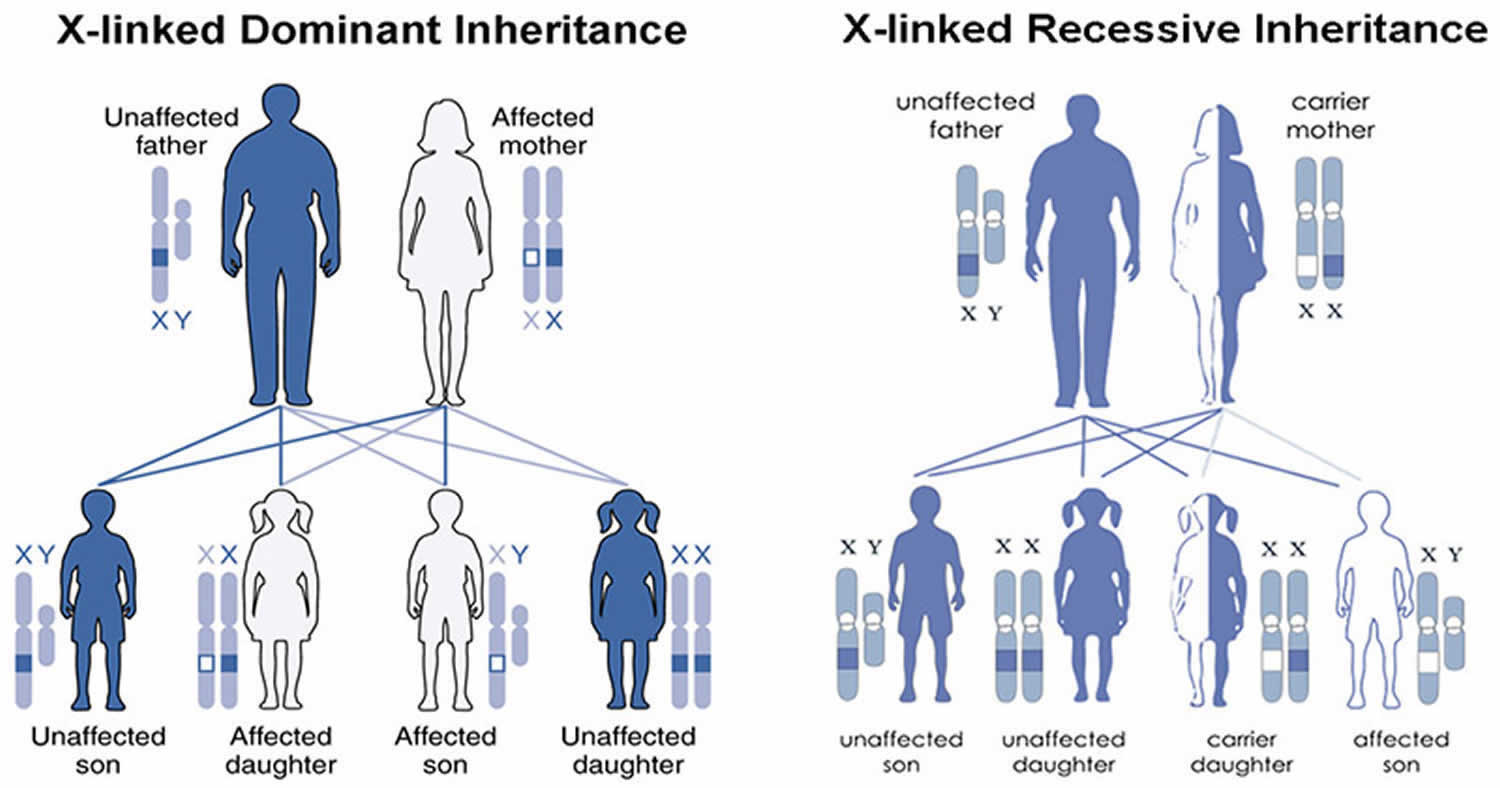
X-linked protoporphyria inheritance pattern
X-linked protoporphyria (XLP) is passed down through families in an X-linked manner 30. Males have one X chromosome and one Y chromosome, while females have two X chromosomes.This means that males have only one copy of the ALAS2 gene and females have two copies of the ALAS2 gene. When a male has a mutation is his single copy of ALAS2 gene, he is expected to have symptoms of X-linked protoporphyria (XLP). In a woman with a mutation in one of her ALAS2 genes, the second functioning copy of the ALAS2 gene can help compensate and may lead to less severe symptoms or no symptoms at all. It is not possible to predict or control the severity of X-linked protoporphyria (XLP) in females. Men with XLP pass on their X chromosome to their daughters and their Y chromosome to their sons. Therefore, a man with X-linked protoporphyria (XLP) with pass on his genetic change to all his daughters, and none of his sons. For a female with XLP, she will pass on the X chromosome with the genetic change 50% of the time. Thus, in each pregnancy, there is a 50% chance of having a child with a mutation in ALAS2 gene.
Genetic counseling is recommended for affected individuals and their families.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://abgc.learningbuilder.com/Search/Public/MemberRole/Verification) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (https://clinics.acmg.net/) has a searchable database of medical genetics clinic services in the United States.
Figure 6. X-linked protoporphyria (XLP) inheritance pattern
X-Linked Protoporphyria Pathophysiology
Bone marrow reticulocytes are thought to be the primary source of the accumulated protoporphyrin IX (PPIX) that is excreted in bile and feces 1. Most of the excess protoporphyrin IX (PPIX) in circulating erythrocytes is found in a small percentage of cells, and the rate of protoporphyrin IX (PPIX) leakage from these cells is proportional to their protoporphyrin content.
The skin of persons with X-linked protoporphyria (XLP) is maximally sensitive to visible blue-violet light near 400 nm, which corresponds to the so-called “Soret band” (the narrow peak absorption maximum that is characteristic for protoporphyrin and other porphyrins) 1. When porphyrins absorb light they enter an excited energy state. This energy is presumably released as fluorescence and by formation of singlet oxygen and other oxygen radicals that can produce tissue and vessel damage. This may involve lipid peroxidation, oxidation of amino acids, and cross-linking of proteins in cell membranes 1.
Photoactivation of the complement system and release of histamine, kinins, and chemotactic factors may mediate skin damage 1. Histologic changes occur predominantly in the upper dermis and include deposition of amorphous material containing immunoglobulin, complement components, glycoproteins, glycosaminoglycans, and lipids around blood vessels. Damage to capillary endothelial cells in the upper dermis has been demonstrated immediately after light exposure in this disease 31.
Long-term observations of individuals with protoporphyria generally show little change in protoporphyrin levels in erythrocytes, plasma, and feces 32. In contrast, severe liver complications, when they occur, often follow increasing accumulation of protoporphyrin in erythrocytes, plasma, and liver. Iron deficiency and factors that impair liver function sometimes contribute. Enterohepatic circulation of protoporphyrin may favor its return and retention in the liver, especially when liver function is impaired. Liver damage probably results at least in part from protoporphyrin accumulation itself 1. As this porphyrin is insoluble, it tends to form crystalline structures in liver cells, can impair mitochondrial functions in liver cells, and can decrease hepatic bile formation and flow 33.
X-Linked Protoporphyria signs and symptoms
Hypersensitivity of the skin to sunlight beginning in infancy or childhood is the characteristic finding of X-linked protoporphyria (XLP) 9, 1. Most patients develop symptoms within 30 minutes of sun exposure. Affected individuals develop tingling, burning, pain, and itching of the skin after exposure to sunlight. Sometimes these symptoms are accompanied by swelling and redness (erythema) of the affected areas 9, 1. Large blisters and severe scarring, which are common to other forms of cutaneous porphyria, usually do not occur in individuals with X-linked protoporphyria 1. The back of the hands and face are most commonly affected but any sun exposed area can be affected. Symptoms may be noticed as quickly as a few minutes after exposure to the sun 34. Although most symptoms usually subside within 24-48 hours (may take up to 4–7 days), pain and a red or purple discoloration of the skin may persist for several days after the initial incident 34, 35. Pain is disproportionately severe in relation to the visible skin lesions. Pain associated with X-linked protoporphyria can be excruciating and is often resistant to pain medications, even narcotics.
Photosensitivity is lifelong. Repeated episodes of photosensitivity may eventually causes changes in the skin of affected individuals. Such changes include thickening and hardening of the sun-exposed skin (lichenification), development of a rough or leathery texture, small facial pock-like pits, and grooving around the lips and loss of lunulae of the nails 13, 1.
Some individuals with X-linked protoporphyria develop liver disease, which can range from mild liver abnormalities to liver failure 1. Information on liver disease is limited, but the risk of liver disease is believed to be higher in X-linked protoporphyria than in erythropoietic protoporphyria (EPP). Affected individuals may experience back pain and severe abdominal pain especially in the upper right area of the abdomen. In some affected individuals, the flow of bile through the gallbladder and bile ducts may be interrupted (cholestasis) leading to gallstones. These stones can cause obstruction and inflammation of the gallbladder (cholecystitis). Scarring of the liver (cirrhosis) may also develop and some individuals may eventually develop end stage liver failure.
Additional symptoms have been reported in individuals with X-linked protoporphyria including mild anemia (low levels of circulating red blood cells) and iron deficiency.
The signs and symptoms in heterozygous females (females with one normal copy and one mutated copy of ALAS2 gene on their X chromosomes) ranges from asymptomatic to as severe as in affected males 1.
Figure 7. X-linked protoporphyria
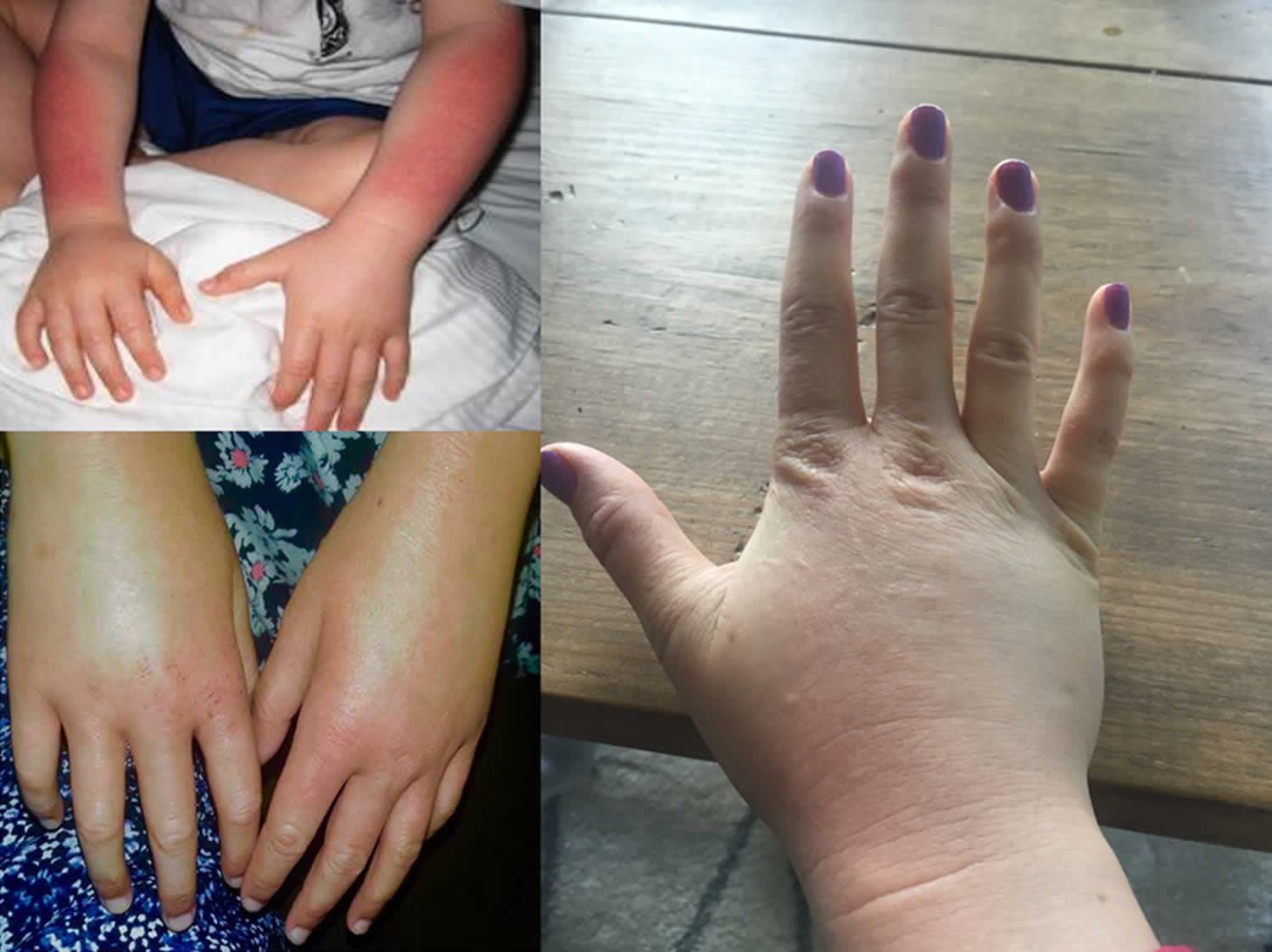
Footnote: Skin redness (erythema) and swelling (edema) on the back of hands and forearm seen after acute phototoxic episode.
[Source 2 ]Liver disease
The excess protoporphyrin in erythropoietic protoporphyria (EPP) and X-linked protoporphyria (XLP) is excreted by the liver into the bile where it enters the enterohepatic circulation 36. Progressive accumulation of protoporphyrin IX (PPIX) may occur in the liver when the biliary excretion does not keep pace with the load being presented to the liver. When hepatocellular damage reaches a critical stage, protoporphyrin IX (PPIX) accumulation will rapidly accelerate due to marked impairment of biliary excretion 15, 37, 1. Concomitant conditions such as viral hepatitis, excessive alcohol consumption, and use of drugs which induce cholestasis may contribute to worsening liver disease. End-stage liver disease is typically preceded by an elevation in plasma and erythrocyte protoporphyrin IX (PPIX) levels. Patients may also develop a motor neuropathy in the setting of liver failure 38, 39.
The excess amounts of free protoporphyrin IX (PPIX) may become insoluble and aggregate in the hepatocytes and small biliary canaliculi leading to obstruction to bile flow and cholestasis. About 20–30% of patients with erythropoietic protoporphyria (EPP) will have elevations in serum aminotransferases20. Protoporphyrin in bile may also crystallize forming gallstones. In one series, cholelithiasis were seen in 23.5% of patients 35.
Anemia
Mild anemia, typically microcytic anemia can be seen in erythropoietic protoporphyria (EPP) patients 40. Patients with erythropoietic protoporphyria (EPP) appear to have an abnormal iron metabolism but the mechanism of iron deficiency is unclear 41, 42. The cause of microcytic anemia and low iron and ferritin levels in erythropoietic protoporphyria (EPP) patients is unknown 43, 44. Previous studies suggest that erythropoietic protoporphyria (EPP) and X-linked protoporphyria (XLP) patients have normal iron absorption and an appropriate hepcidin response 45. The iron deficiency in erythropoietic protoporphyria (EPP) and X-linked protoporphyria (XLP) patients does not appear to be related to chronic inflammation or iron loss. The cause and mechanism of iron deficiency in these patients remains to be resolved.
Vitamin D deficiency
X-linked protoporphyria (XLP) and erythropoietic protoporphyria (EPP) patients can develop vitamin D deficiency secondary to sun avoidance 46, 47. A recent report showed that the prevalence of osteopenia (low bone mineral density) and osteoporosis is increased in patients in erythropoietic protoporphyria (EPP) 48.
X-Linked Protoporphyria diagnosis
A diagnosis of X-linked protoporphyria is based upon identification of characteristic symptoms (e.g., non-blistering photosensitivity), a detailed patient history, a thorough clinical evaluation, and a variety of specialized tests 9, 49, 50, 1.
X-linked protoporphyria (XLP) should be suspected in individuals with the following clinical findings and initial laboratory findings 1:
- Clinical findings
- Skin photosensitivity, usually beginning in childhood
- Burning, tingling, pain, and itching of the skin (the most common findings); may occur within minutes of sun/light exposure, followed later by erythema and swelling
- Painful symptoms; may occur without obvious skin damage
- Absent or sparse blisters and bullae
- Note: The absence of skin damage (e.g., scarring), vesicles, and bullae often make it difficult to suspect the diagnosis.
- Liver complications, particularly cholestatic liver disease, may develop in fewer than 5% of affected individuals.
A diagnosis of X-linked protoporphyria may be made through blood tests that can detect markedly increased levels of metal-free and zinc-bound protoporphyrins within red blood cells (erythrocytes). A higher ratio of zinc-bound protoporphyrin to metal-free protoporphyrin can differentiate X-linked protoporphyria from erythropoietic protoporphyria.
Note: It is essential to use an assay for erythrocyte protoporphyrin that distinguishes between free protoporphyrin and zinc-chelated protoporphyrin to differentiate X-linked protoporphyria (XLP) from erythropoietic protoporphyria (EPP) and several other conditions that may lead to elevation of erythrocyte protoporphyrins.
- Initial laboratory findings. Detection of markedly increased free erythrocyte protoporphyrin IX (PPIX) and zinc-chelated erythrocyte protoporphyrin IX (PPIX) is the most sensitive biochemical diagnostic test for X-linked protoporphyria (XLP).
- Erythroid-specific 5-aminolevulinate synthase 2 (ALAS2) enzyme activity >100% of normal
- Free protoporphyrin/zinc-chelated protoporphyrin ratio 90:10 to 50:50
- Urine Protoporphyrins not detectable
- Stool Protoporphyrin normal or increased
- Plasma porphyrins increased
Molecular genetic testing can confirm a diagnosis of X-linked protoporphyria (XLP) by detecting mutations in the ALAS2 gene (the only gene known to cause this disorder).
Additional tests may be performed such as blood tests to evaluate anemia and iron stores in the body and vitamin D levels, or an abdominal sonogram to detect and evaluate liver disease potentially associated with X-linked protoporphyria.
Newer imaging modalities such as Fibroscan® may be useful in evaluating liver fibrosis; however, this has not been validated in erythropoietic protoporphyria (EPP) or X-linked protoporphyria (XLP) 1.
X-linked protoporphyria differential diagnosis
X-linked protoporphyria differential diagnosis include 51:
- Phototoxic drug reaction: Phototoxic drug reaction is a non-immunologic skin reaction that appears acutely within minutes to hours on sun-exposed skin after taking photosensitising medications. There must be a history of the introduction of any new drug. Phototoxic skin damage begins when the drug or its metabolite within the skin absorbs ultraviolet radiation (UVR) or visible light. Patients experience painful reddish skin immediately after sun exposure 52, 53.
- Hydroa vacciniforme: Hydroa vacciniforme is one of the rarest forms of photosensitivity dermatoses. Hydroa vacciniforme affects sun-exposed skin and is characterized by recurrent fluid-filled blisters (‘hydroa’) over sun-exposed sites that heal with pox-like (‘vacciniform’) scars 54.
- Solar urticaria: Solar urticaria is a condition in which exposure to sunlight or an artificial light source emitting ultraviolet radiation causes urticaria 55. Like EPP, symptoms often develop within minutes. Symptoms of solar urticaria are often itchy rather than painful. The reaction may subside within a few minutes or it may persist for up to an hour or more where it can become very disabling. The cause of solar urticaria is not clearly defined but may be due to an antigen-antibody reaction 55. It seems that a chemical created in the body (a photoallergen) reacts with UV radiation to cause an allergic reaction that manifests as urticaria.
- Polymorphic light eruption (PMLE): Polymorphic light eruption also called a sun allergy or sun poisoning. Polymorphic light eruption is a common seasonal, acquired, idiopathic photodermatosis occurring in spring and early summer that typically occurs during the first three decades of life 56. Symptoms occur in sun-exposed areas. Patients present with discrete lesions such as pruritic papules, vesicles, or plaques on sun-exposed areas.
- Discoid lupus erythematosus: It presents as scaly erythematous plaques on sun-exposed areas.
- Sunburn: It is a transient inflammatory skin response to ultraviolet radiation from sunlight or artificial sources. Sunburn can occur in individuals without an underlying dermatologic condition, with sensitivity depending on the degree of skin pigmentation and hair and eye color 57.
X-Linked Protoporphyria treatment
The treatment of X-linked protoporphyria is directed toward the specific symptoms that are apparent in each individual. Treatment may require the coordinated efforts of a team of specialists. Pediatricians, hematologists, dermatologists, hepatologists, and other healthcare professionals may need to systematically and comprehensively plan an affected child’s treatment. Genetic counseling may benefit affected individuals and their families.
There is no specific, FDA-approved therapy for individuals with X-linked protoporphyria (XLP). Because the disorder is so rare, most treatment information is based on erythropoietic protoporphyria (EPP), which is clinically similar to X-linked protoporphyria (XLP).
Avoidance of sunlight will benefit affected individuals and can include the use of clothing styles with long sleeves and pant legs, made with double layers of fabric or of light-exclusive fabrics, wide brimmed hats, gloves, and sunglasses. Topical sunscreens are generally ineffective, unless they contain light-reflective ingredients (e.g., zinc oxide). Some tanning products with ingredients that increase pigmentation may be helpful. Affected individuals may also benefit from window tinting and the use of vinyl or films to cover the windows of their homes and cars.
Avoidance of sunlight can potentially cause vitamin D deficiency and some individuals may require supplemental vitamin D.
A high potency form of oral beta-carotene (Lumitene) may be given to improve an affected individual’s tolerance of sunlight. This drug causes skin discoloration and may improve tolerance to sunlight. Oral Lumitene (beta-carotene) (120–180 mg/dL) has been used to improve tolerance to sunlight if the dose is adjusted to maintain serum carotene levels in the range of 10-15 μmol/L (600–800 μg/dL), causing mild skin discoloration due to carotenemia. The beneficial effects of beta-carotene (Lumitene) may involve quenching of singlet oxygen or free radicals. However, a systematic review of about 25 studies showed that the available data are unable to prove efficacy of treatments including beta-carotene, N-acetyl cysteine, and vitamin C 19. For more information on oral beta-carotene (Lumitene) treatment, contact the American Porphyria Foundation and the Prophyria Consortium of the Rare Diseases Clinical Research Network.
Another drug sometimes used to improve tolerance to sunlight is cysteine.
In 2019, the Food and Drug Administration (FDA) approved Afamelanotide (Scenesse®) for the treatment of adult patients with erythropoietic protoporphyria (EPP). Afamelanotide (Scenesse) is a controlled-release, long-acting injectable implant, alpha-melanocyte-stimulating hormone (α-MSH) analogue, that increases eumelanin by binding to the melanocortin-1 receptor and provides sun protection and improves sun tolerance by increasing skin pigmentation and antioxidant properties 20, 21. Afamelanotide (Scenesse) was available in Europe for a period of time before its approval in the United States. Afamelanotide (Scenesse) was approved for patients with the erythropoietic protoporphyria (EPP) by the European Medicines Agency in 2014, and by the FDA in October 2019. Afamelanotide showed positive results in Phase III clinical trials in the US and Europe 22. Long-term studies in Europe show good compliance, clinical effectiveness, and improved quality of life 23.
Other treatment is symptomatic and supportive.
X-linked protoporphyria (XLP) patients should be seen at least yearly to monitor protoporphyrin levels, anemia, liver enzymes, iron and vitamin D levels.
Liver disease treatment
Treatment of liver disease and complications, which may be accompanied by motor neuropathy, is difficult.
- Cholestyramine and other porphyrin absorbents, such as activated charcoal, may interrupt the enterohepatic circulation of protoporphyrin and promote its fecal excretion, leading to some improvement 58.
- Cholestyramine absorbs porphyrin. Cholestyramine may interrupt the recirculation of protoporphyrin secreted into the bile back into the liver and promote its excretion through the feces. Other drugs that absorb porphyrins such as activated charcoal have also been used to treat affected individuals.
- Cholestyramine and other porphyrin absorbents, such as activated charcoal may lead to improvement of liver disease.
- Plasmapheresis and intravenous hemin have been used to treat people with erythropoietic protoporphyria (EPP) 59.
- Liver transplantation has been performed as a lifesaving measure in individuals with severe protoporphyric liver disease 38, 60. However, many liver transplant recipients experience a recurrence of the protoporphyric liver disease in the transplanted liver. Combined bone marrow and liver transplantation is indicated in patients with liver failure to prevent future damage to the allografts 61, and sequential liver and bone marrow transplantation has been successful in curing protoporphyric liver disease 62.
- Bone marrow transplantation has also been attempted without liver transplantation in some instances. A child age two years with X-linked protoporphyria (XLP) and stage 4 liver fibrosis was treated with a hematopoietic progenitor cell transplantation that stabilized his liver disease, thus avoiding liver transplantation 63.
Individuals with any form of protoporphyria should avoid substances associated with cholestasis including alcohol and certain drugs such as estrogens.
In patients with cholestatic liver failure, use of protective filters for artificial lights in the operating room to prevent phototoxic damage during procedures such as endoscopy and surgery 64.
Additional treatment
- Vitamin D supplementation is advised as patients are predisposed to vitamin D deficiency resulting from sun avoidance.
- Immunizations for hepatitis A and B are recommended as well.
- Iron supplementation may be attempted in persons with X-linked protoporphyria (XLP) who have anemia and low ferritin levels. Iron supplementation therapy requires strict monitoring by physicians. Whatley et al 7 reported some evidence of diminished iron stores in males with X-linked protoporphyria (XLP); in one patient with iron deficiency, iron repletion decreased protoporphyrin accumulation and corrected the anemia. Subsequent reports indicate that iron supplementation can improve protoporphyrin levels, liver damage, and anemia in X-linked protoporphyria (XLP) 65. A pilot study using oral iron supplementation in persons with X-linked protoporphyria (XLP) showed a reduction in protoporphyrin levels, but also carries a risk of increased photosensitivity 66.
Pregnancy Management
There is no information on pregnancy management in X-linked protoporphyria (XLP). Based on experience with erythropoietic protoporphyria (EPP), pregnancy is unlikely to be complicated by X-linked protoporphyria (XLP) 67.
Investigational Therapies
A Phase 2 clinical trial with MT-7117, an oral small molecule that works as a melanocortin 1 receptor agonist and increases skin pigmentation in subjects with erythropoietic protoporphyria (EPP) has been completed 68. A Phase 3 clinical trial for adults and children is planned for MT-7117.
X-Linked Protoporphyria prognosis
The natural history of X-linked protoporphyria (XLP) is not as well characterized as that of the autosomal recessive type of erythropoietic protoporphyria (EPP) 1. A natural history study from the US described 22 individuals with X-linked protoporphyria (XLP) from seven unrelated families 35.
X-linked protoporphyria (XLP) in Males
While the skin signs and symptoms in males with X-linked protoporphyria (XLP) are similar to those of erythropoietic erythropoietic protoporphyria (EPP), Balwani et al 35 suggest that males with X-linked protoporphyria (XLP) have significantly higher protoporphyrin levels and increased risk of liver dysfunction.
Photosensitivity
Onset of photosensitivity is typically in infancy or childhood with the first exposure to sun; in most individuals with X-linked protoporphyria (XLP) the photosensitivity is lifelong.
Most males with X-linked protoporphyria (XLP) develop acute cutaneous photosensitivity within five to 30 minutes following exposure to sun or ultraviolet light. Photosensitivity symptoms are provoked mainly by visible blue-violet light in the Soret band, to a lesser degree in the long-wave UV region.
The initial symptoms reported are tingling, burning, and/or itching that may be accompanied by swelling and redness. Symptoms vary based on the intensity and duration of sun exposure; pain may be severe and refractory to narcotic analgesics, persisting for hours or days after the initial phototoxic reaction. Symptoms may seem out of proportion to the visible skin lesions. Blistering lesions are uncommon.
Affected males are also sensitive to sunlight that passes through window glass, which does not block long-wave UVA or visible light.
Skin signs and symptoms
Multiple episodes of acute photosensitivity may lead to chronic changes of sun-exposed skin (lichenification, leathery pseudovesicles, grooving around the lips) and loss of lunulae of the nails. The back of the hands is most notably affected.
Severe scarring is rare, as are hyper- or hypopigmentation, skin friability, and hirsutism.
Unlike in other cutaneous porphyrias, blistering and scarring rarely occur.
Liver problems
Protoporphyrin is not excreted in the urine by the kidneys, but is taken up by the liver and excreted in the bile. Accumulated protoporphyrin in the bile can form stones, reduce bile flow, and damage the liver. Protoporphyric liver disease may cause back pain and severe abdominal pain (especially in the right upper quadrant).
The information on X-linked protoporphyria (XLP) and liver disease is limited 1. The risk for liver dysfunction in X-linked protoporphyria (XLP) (observed in 5/31 affected individuals) is higher than the risk in erythropoietic protoporphyria (EPP) 7. A natural history study in the US showed that 40% of males with X-linked protoporphyria (XLP) had a history of abnormal liver enzymes compared to 33% of persons with erythropoietic protoporphyria (EPP). Gallstones were seen in 40% of males with X-linked protoporphyria (XLP) and 33.3% of females with X-linked protoporphyria (XLP) compared to 22.1% of individuals with erythropoietic protoporphyria (EPP).
Note that the information on liver involvement presented below is based on experience with liver disease in autosomal recessive erythropoietic protoporphyria (EPP). Gallstones composed in part of protoporphyrin may be symptomatic in individuals with X-linked protoporphyria (XLP) and need to be excluded as a cause of biliary obstruction in persons with hepatic decompensation.
Life-threatening liver complications are preceded by increased levels of plasma and erythrocyte protoporphyrins, worsening liver function tests, increased photosensitivity, and increased deposition of protoporphyrins in liver cells and bile canaliculi. End-stage liver disease may be accompanied by motor neuropathy, similar to that seen in acute porphyrias. Comorbid conditions, such as viral hepatitis, alcohol abuse, and use of oral contraceptives, which may impair hepatic function or protoporphyrin metabolism, may contribute to hepatic disease in some 38.
Blood problems
Anemia and abnormal iron metabolism can occur in X-linked protoporphyria (XLP). Mild anemia with microcytosis and hypochromia or occasionally reticulocytosis can be seen 1. However, hemolysis is absent or mild 1. In a recent series, 30% of males with X-linked protoporphyria (XLP) and 75% of females with X-linked protoporphyria (XLP) were anemic 35.
Vitamin D deficiency
Individuals with X-linked protoporphyria (XLP) who avoid sun/light are at risk for vitamin D deficiency 46, 47, 40.
Triggering factors
Unlike the triggering factors for acute hepatic porphyrias, the only known precipitating factor for X-linked protoporphyria (XLP) is sunlight.
X-linked protoporphyria (XLP) in Females
The signs and symptoms of X-linked protoporphyria (XLP) in heterozygous females, the consequence of random X-chromosome inactivation, ranges from as severe as in affected males to asymptomatic. The median age of symptom onset for females with X-linked protoporphyria (XLP) was 11 years. Following sun exposure, symptom onset ranged from within ten minutes to none 35.
X-Linked Protoporphyria life expectancy
People with X-linked protoporphyria (XLP) generally have a life expectancy similar to those without porphyria, except for those with advanced liver disease 1.
- Balwani M, Desnick R; Porphyrias Consortium of the NIH-Sponsored Rare Diseases Clinical Research Network. X-Linked Protoporphyria. 2013 Feb 14 [Updated 2019 Nov 27]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK121284[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Balwani M. Erythropoietic Protoporphyria and X-Linked Protoporphyria: pathophysiology, genetics, clinical manifestations, and management. Mol Genet Metab. 2019 Nov;128(3):298-303. doi: 10.1016/j.ymgme.2019.01.020[↩][↩][↩][↩][↩]
- Levy C, Naik H, Overbey J, Hedstrom K, et al.; Porphyrias Consortium of the Rare Diseases Clinical Research Network. Liver involvement in a large cohort of patients with erythropoietic protoporphyria or X-linked protoporphyria. Hepatol Commun. 2025 Feb 19;9(3):e0657. doi: 10.1097/HC9.0000000000000657[↩]
- Balwani M, Doheny D, Bishop DF, et al. Loss-of-function ferrochelatase and gain-of-function erythroid-specific 5-aminolevulinate synthase mutations causing erythropoietic protoporphyria and x-linked protoporphyria in North American patients reveal novel mutations and a high prevalence of X-linked protoporphyria. Mol Med. 2013;19(1):26–35. doi: 10.2119/molmed.2012.00340[↩]
- Whatley SD, Ducamp S, Gouya L, et al. C-terminal deletions in the ALAS2 gene lead to gain of function and cause X-linked dominant protoporphyria without anemia or iron overload. Am J Hum Genet. 2008;83(3):408–414. doi: 10.1016/j.ajhg.2008.08.003[↩]
- ALAS2 gene. https://medlineplus.gov/genetics/gene/alas2[↩][↩][↩]
- Whatley SD, Ducamp S, Gouya L, Grandchamp B, Beaumont C, Badminton MN, Elder GH, Holme SA, Anstey AV, Parker M, Corrigall AV, Meissner PN, Hift RJ, Marsden JT, Ma Y, Mieli-Vergani G, Deybach JC, Puy H. C-terminal deletions in the ALAS2 gene lead to gain of function and cause X-linked dominant protoporphyria without anemia or iron overload. Am J Hum Genet. 2008 Sep;83(3):408-14. doi: 10.1016/j.ajhg.2008.08.003[↩][↩][↩][↩][↩][↩]
- Manceau H, Gouya L, Puy H. Acute hepatic and erythropoietic porphyrias: from ALA synthases 1 and 2 to new molecular bases and treatments. Curr Opin Hematol. 2017 May;24(3):198-207. doi: 10.1097/MOH.0000000000000330[↩][↩][↩][↩]
- Anderson KE, Sassa S, Bishop DF, and Desnick RJ (2001). Disorders of Heme Biosynthesis: X-Linked Sideroblastic Anemia and the Porphyrias In The Metabolic and Molecurlar Bases of Inherited Disease, Scriver CR, Beaudet AL, Sly WS, and Valle D, eds. (New York, McGraw-Hill: ), pp 2961–3062.[↩][↩][↩][↩][↩]
- Whatley SD, Ducamp S, Gouya L, et al. C-terminal deletions in the ALAS2 gene lead to gain of function and cause X-linked dominant protoporphyria without anemia or iron overload. Am J Hum Genet. 2008 Sep;83(3):408-14. doi: 10.1016/j.ajhg.2008.08.003[↩][↩]
- Poh-Fitzpatrick MB. Erythropoietic protoporphyria. Int J Dermatol. 1978 Jun;17(5):359-69. doi: 10.1111/ijd.1978.17.5.359[↩]
- Lecha M, Puy H, Deybach JC. Erythropoietic protoporphyria. Orphanet J Rare Dis. 2009 Sep 10;4:19. doi: 10.1186/1750-1172-4-19[↩]
- Poh-Fitzpatrick MB. Porphyrias: photosensitivity and phototherapy. Methods Enzymol. 2000;319:485-93. doi: 10.1016/s0076-6879(00)19045-7[↩][↩]
- Bloomer JR. The liver in protoporphyria. Hepatology. 1988 Mar-Apr;8(2):402-7. doi: 10.1002/hep.1840080235[↩]
- Bloomer JR. Hepatic protoporphyrin metabolism in patients with advanced protoporphyric liver disease. Yale J Biol Med. 1997 Jul-Aug;70(4):323-30. https://pmc.ncbi.nlm.nih.gov/articles/instance/2589331/pdf/yjbm00029-0044.pdf[↩][↩]
- Dickey AK, Naik H, Keel SB, et al. Porphyrias Consortium of the Rare Diseases Clinical Research Network. Evidence-based consensus guidelines for the diagnosis and management of erythropoietic protoporphyria and X-linked protoporphyria. J Am Acad Dermatol. 2023 Dec;89(6):1227-1237. doi: 10.1016/j.jaad.2022.08.036[↩]
- Whatley SD, Mason NG, Holme SA, Anstey AV, Elder GH, Badminton MN. Molecular epidemiology of erythropoietic protoporphyria in the U.K. Br J Dermatol. 2010 Mar;162(3):642-6. doi: 10.1111/j.1365-2133.2010.09631.x[↩]
- Balwani M, Doheny D, Bishop DF, Nazarenko I, Yasuda M, Dailey HA, Anderson KE, Bissell DM, Bloomer J, Bonkovsky HL, Phillips JD, Liu L, Desnick RJ; Porphyrias Consortium of the National Institutes of Health Rare Diseases Clinical Research Network. Loss-of-function ferrochelatase and gain-of-function erythroid-specific 5-aminolevulinate synthase mutations causing erythropoietic protoporphyria and x-linked protoporphyria in North American patients reveal novel mutations and a high prevalence of X-linked protoporphyria. Mol Med. 2013 Apr 30;19(1):26-35. doi: 10.2119/molmed.2012.00340[↩][↩]
- Minder EI, Schneider-Yin X, Steurer J, Bachmann LM. A systematic review of treatment options for dermal photosensitivity in erythropoietic protoporphyria. Cell Mol Biol (Noisy-le-grand). 2009 Feb 16;55(1):84-97.[↩][↩]
- Harms JH, Lautenschlager S, Minder CE, Minder EI. Mitigating photosensitivity of erythropoietic protoporphyria patients by an agonistic analog of alpha-melanocyte stimulating hormone. Photochem Photobiol. 2009 Nov-Dec;85(6):1434-9. doi: 10.1111/j.1751-1097.2009.00595.x[↩][↩]
- Minder EI. Afamelanotide, an agonistic analog of α-melanocyte-stimulating hormone, in dermal phototoxicity of erythropoietic protoporphyria. Expert Opin Investig Drugs. 2010 Dec;19(12):1591-602. doi: 10.1517/13543784.2010.535515[↩][↩]
- Langendonk JG, Balwani M, Anderson KE, Bonkovsky HL, Anstey AV, Bissell DM, Bloomer J, Edwards C, Neumann NJ, Parker C, Phillips JD, Lim HW, Hamzavi I, Deybach JC, Kauppinen R, Rhodes LE, Frank J, Murphy GM, Karstens FPJ, Sijbrands EJG, de Rooij FWM, Lebwohl M, Naik H, Goding CR, Wilson JHP, Desnick RJ. Afamelanotide for Erythropoietic Protoporphyria. N Engl J Med. 2015 Jul 2;373(1):48-59. doi: 10.1056/NEJMoa1411481[↩][↩]
- Biolcati G, Marchesini E, Sorge F, Barbieri L, Schneider-Yin X, Minder EI. Long-term observational study of afamelanotide in 115 patients with erythropoietic protoporphyria. Br J Dermatol. 2015 Jun;172(6):1601-1612. doi: 10.1111/bjd.13598[↩][↩]
- Lin, Jou & Shi, Donglu. (2021). Photothermal and photovoltaic properties of transparent thin films of porphyrin compounds for energy applications. Applied Physics Reviews. 8. 011302. https://doi.org/10.1063/5.0036961[↩]
- Panawala, Lakna. (2017). What is the Function of Hemoglobin in the Human Body. https://www.researchgate.net/publication/313841668_What_is_the_Function_of_Hemoglobin_in_the_Human_Body[↩]
- Heme and Bilirubin Metabolism. https://themedicalbiochemistrypage.org/heme-and-bilirubin-metabolism/[↩]
- Edel Y, Mamet R. Porphyria: What Is It and Who Should Be Evaluated? Rambam Maimonides Med J. 2018 Apr 19;9(2):e0013. doi: 10.5041/RMMJ.10333[↩]
- Whatley SD, Ducamp S, Gouya L, et al. C-terminal deletions in the ALAS2 gene lead to gain of function and cause X-linked dominant protoporphyria without anemia or iron overload. Am J Hum Genet 2008;83(3):408–414. doi: 10.1016/j.ajhg.2008.08.003[↩][↩]
- Brancaleoni V, Balwani M, Granata F, Graziadei G, Missineo P, Fiorentino V, Fustinoni S, Cappellini MD, Naik H, Desnick RJ, Di Pierro E. X-chromosomal inactivation directly influences the phenotypic manifestation of X-linked protoporphyria. Clin Genet. 2016 Jan;89(1):20-6. doi: 10.1111/cge.12562[↩]
- Erythropoietic Protoporphyria and X-Linked Protoporphyria. https://rarediseases.org/rare-diseases/erythropoietic-protoporphyria[↩]
- Schneider-Yin X, Gouya L, Meier-Weinand A, Deybach JC, Minder EI. New insights into the pathogenesis of erythropoietic protoporphyria and their impact on patient care. Eur J Pediatr. 2000 Oct;159(10):719-25. doi: 10.1007/s004310000494[↩]
- Gou E, Weng C, Greene T, Anderson KE, Phillips JD. Longitudinal Analysis of Erythrocyte and Plasma Protoporphyrin Levels in Patients with Protoporphyria. J Appl Lab Med. 2018 Sep 1;3(2):213-221. doi: 10.1373/jalm.2017.025874[↩]
- Anderson KE, Sassa S, Bishop DF, Desnick RJ. Disorders of heme biosynthesis: X-linked sideroblastic anemias and the porphyrias. In: Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Vogelstein B, eds. The Metabolic and Molecular Bases of Inherited Disease. 8 ed. New York, NY: McGraw-Hill; 2001:2991-3062.[↩]
- Poh-Fitzpatrick MB. The “priming phenomenon” in the acute phototoxicity of erythropoietic protoporphyria. J Am Acad Dermatol. 1989 Aug;21(2 Pt 1):311. doi: 10.1016/s0190-9622(89)70187-0[↩][↩]
- Balwani M, Naik H, Anderson KE, Bissell DM, Bloomer J, Bonkovsky HL, Phillips JD, Overbey JR, Wang B, Singal AK, Liu LU, Desnick RJ. Clinical, Biochemical, and Genetic Characterization of North American Patients With Erythropoietic Protoporphyria and X-linked Protoporphyria. JAMA Dermatol. 2017 Aug 1;153(8):789-796. doi: 10.1001/jamadermatol.2017.1557[↩][↩][↩][↩][↩][↩]
- Anstey AV, Hift RJ. Liver disease in erythropoietic protoporphyria: insights and implications for management. Postgrad Med J. 2007 Dec;83(986):739-48. doi: 10.1136/gut.2006.097576[↩]
- Balwani M, Bloomer J, Desnick R; Porphyrias Consortium of the NIH-Sponsored Rare Diseases Clinical Research Network. Erythropoietic Protoporphyria, Autosomal Recessive. 2012 Sep 27 [Updated 2017 Sep 7]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK100826[↩]
- McGuire BM, Bonkovsky HL, Carithers RL Jr, Chung RT, Goldstein LI, Lake JR, Lok AS, Potter CJ, Rand E, Voigt MD, Davis PR, Bloomer JR. Liver transplantation for erythropoietic protoporphyria liver disease. Liver Transpl. 2005 Dec;11(12):1590-6. doi: 10.1002/lt.20620[↩][↩][↩]
- Singal AK, Parker C, Bowden C, Thapar M, Liu L, McGuire BM. Liver transplantation in the management of porphyria. Hepatology. 2014 Sep;60(3):1082-9. doi: 10.1002/hep.27086[↩]
- Wahlin S, Floderus Y, Stål P, Harper P. Erythropoietic protoporphyria in Sweden: demographic, clinical, biochemical and genetic characteristics. J Intern Med. 2011 Mar;269(3):278-88. doi: 10.1111/j.1365-2796.2010.02236.x[↩][↩]
- Holme SA, Worwood M, Anstey AV, Elder GH, Badminton MN. Erythropoiesis and iron metabolism in dominant erythropoietic protoporphyria. Blood. 2007 Dec 1;110(12):4108-10. doi: 10.1182/blood-2007-04-088120[↩]
- Lyoumi S, Abitbol M, Andrieu V, Henin D, Robert E, Schmitt C, Gouya L, de Verneuil H, Deybach JC, Montagutelli X, Beaumont C, Puy H. Increased plasma transferrin, altered body iron distribution, and microcytic hypochromic anemia in ferrochelatase-deficient mice. Blood. 2007 Jan 15;109(2):811-8. doi: 10.1182/blood-2006-04-014142[↩]
- Holme SA, Thomas CL, Whatley SD, Bentley DP, Anstey AV, Badminton MN. Symptomatic response of erythropoietic protoporphyria to iron supplementation. J Am Acad Dermatol. 2007 Jun;56(6):1070-2. doi: 10.1016/j.jaad.2006.11.030[↩]
- Barman-Aksoezen J, Girelli D, Aurizi C, Schneider-Yin X, Campostrini N, Barbieri L, Minder EI, Biolcati G. Disturbed iron metabolism in erythropoietic protoporphyria and association of GDF15 and gender with disease severity. J Inherit Metab Dis. 2017 May;40(3):433-441. doi: 10.1007/s10545-017-0017-7[↩]
- Bossi K, Lee J, Schmeltzer P, Holburton E, Groseclose G, Besur S, Hwang S, Bonkovsky HL. Homeostasis of iron and hepcidin in erythropoietic protoporphyria. Eur J Clin Invest. 2015 Oct;45(10):1032-41. doi: 10.1111/eci.12503[↩]
- Holme SA, Anstey AV, Badminton MN, Elder GH. Serum 25-hydroxyvitamin D in erythropoietic protoporphyria. Br J Dermatol. 2008 Jul;159(1):211-3. doi: 10.1111/j.1365-2133.2008.08616.x[↩][↩]
- Spelt JM, de Rooij FW, Wilson JH, Zandbergen AA. Vitamin D deficiency in patients with erythropoietic protoporphyria. J Inherit Metab Dis. 2010 Dec;33 Suppl 3:S1-4. doi: 10.1007/s10545-008-1037-0[↩][↩]
- Biewenga M, Matawlie RHS, Friesema ECH, Koole-Lesuis H, Langeveld M, Wilson JHP, Langendonk JG. Osteoporosis in patients with erythropoietic protoporphyria. Br J Dermatol. 2017 Dec;177(6):1693-1698. doi: 10.1111/bjd.15893[↩]
- Gouya L, Puy H, Lamoril J, Da Silva V, Grandchamp B, Nordmann Y, Deybach JC. Inheritance in erythropoietic protoporphyria: a common wild-type ferrochelatase allelic variant with low expression accounts for clinical manifestation. Blood. 1999 Mar 15;93(6):2105-10. https://doi.org/10.1182/blood.V93.6.2105.406k28_2105_2110[↩]
- Gou EW, Balwani M, Bissell DM, Bloomer JR, Bonkovsky HL, Desnick RJ, Naik H, Phillips JD, Singal AK, Wang B, Keel S, Anderson KE. Pitfalls in Erythrocyte Protoporphyrin Measurement for Diagnosis and Monitoring of Protoporphyrias. Clin Chem. 2015 Dec;61(12):1453-6. doi: 10.1373/clinchem.2015.245456[↩]
- Ahmed jan N, Masood S. Erythropoietic Protoporphyria. [Updated 2023 Feb 16]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK563141[↩]
- Coffey A, Leung DH, Quintanilla NM. Erythropoietic Protoporphyria: Initial Diagnosis With Cholestatic Liver Disease. Pediatrics. 2018 Apr;141(Suppl 5):S445-S450. doi: 10.1542/peds.2016-1625[↩]
- Drug-induced photosensitivity. https://dermnetnz.org/topics/drug-induced-photosensitivity[↩]
- Hydroa vacciniforme. https://dermnetnz.org/topics/hydroa-vacciniforme[↩]
- Solar urticaria. https://dermnetnz.org/topics/solar-urticaria[↩][↩]
- Polymorphic light eruption. https://dermnetnz.org/topics/polymorphic-light-eruption[↩]
- de Castro Maqueda G, Gutiérrez-Manzanedo JV, González-Montesinos JL, Vaz Pardal C, Rivas Ruiz F, de Troya Martín M. Sun Exposure and Photoprotection: Habits, Knowledge and Attitudes Among Elite Kitesurfers. J Cancer Educ. 2022 Jun;37(3):517-523. doi: 10.1007/s13187-020-01838-7[↩]
- McCullough AJ, Barron D, Mullen KD, Petrelli M, Park MC, Mukhtar H, Bickers DR. Fecal protoporphyrin excretion in erythropoietic protoporphyria: effect of cholestyramine and bile acid feeding. Gastroenterology. 1988 Jan;94(1):177-81. doi: 10.1016/0016-5085(88)90627-0[↩]
- Do KD, Banner BF, Katz E, Szymanski IO, Bonkovsky HL. Benefits of chronic plasmapheresis and intravenous heme-albumin in erythropoietic protoporphyria after orthotopic liver transplantation. Transplantation. 2002 Feb 15;73(3):469-72. doi: 10.1097/00007890-200202150-00024[↩]
- Wahlin S, Stal P, Adam R, Karam V, Porte R, Seehofer D, Gunson BK, Hillingsø J, Klempnauer JL, Schmidt J, Alexander G, O’Grady J, Clavien PA, Salizzoni M, Paul A, Rolles K, Ericzon BG, Harper P; European Liver and Intestine Transplant Association. Liver transplantation for erythropoietic protoporphyria in Europe. Liver Transpl. 2011 Sep;17(9):1021-6. doi: 10.1002/lt.22341[↩]
- Rand EB, Bunin N, Cochran W, Ruchelli E, Olthoff KM, Bloomer JR. Sequential liver and bone marrow transplantation for treatment of erythropoietic protoporphyria. Pediatrics. 2006 Dec;118(6):e1896-9. doi: 10.1542/peds.2006-0833[↩]
- Wahlin S, Harper P. The role for BMT in erythropoietic protoporphyria. Bone Marrow Transplant. 2010 Feb;45(2):393-4. doi: 10.1038/bmt.2009.132[↩]
- Butler DF, Ginn KF, Daniel JF, Bloomer JR, Kats A, Shreve N, Myers GD. Bone marrow transplant for X-linked protoporphyria with severe hepatic fibrosis. Pediatr Transplant. 2015 Jun;19(4):E106-10. doi: 10.1111/petr.12472[↩]
- Wahlin S, Srikanthan N, Hamre B, Harper P, Brun A. Protection from phototoxic injury during surgery and endoscopy in erythropoietic protoporphyria. Liver Transpl. 2008 Sep;14(9):1340-6. doi: 10.1002/lt.21527[↩]
- Landefeld C, Kentouche K, Gruhn B, Stauch T, Rößler S, Schuppan D, Whatley SD, Beck JF, Stölzel U. X-linked protoporphyria: Iron supplementation improves protoporphyrin overload, liver damage and anaemia. Br J Haematol. 2016 May;173(3):482-4. doi: 10.1111/bjh.13612[↩]
- Balwani M. Effects of iron supplementation in EPP and XLP. Milan, Italy: International Congress on Porphyrins and Porphyria: From Bench to Care. 2019.[↩]
- Poh-Fitzpatrick MB. Human protoporphyria: reduced cutaneous photosensitivity and lower erythrocyte porphyrin levels during pregnancy. J Am Acad Dermatol. 1997 Jan;36(1):40-3. doi: 10.1016/s0190-9622(97)70323-2[↩]
- Study to Evaluate Efficacy, Safety, and Tolerability of MT-7117 in Subjects With Erythropoietic Protoporphyria. https://clinicaltrials.gov/study/NCT03520036[↩]




{kind=link}