Contents
What is osteogenesis imperfecta
Osteogenesis imperfecta also known as ‘brittle bone disease’, is a group of rare genetic disorders that mainly affects the protein collagen, which is found in bone, teeth, skin, tendons, and parts of the eye. The term osteogenesis imperfecta means “imperfect bone formation”. People with osteogenesis imperfecta have bones that break easily, often from mild trauma or with no apparent cause 1. Multiple fractures are common, and in severe cases, can occur even before birth. Milder cases may involve only a few fractures over a person’s lifetime.
Osteogenesis imperfecta can also cause weak muscles, brittle teeth, a curved spine, and hearing loss. Osteogenesis imperfecta is caused by one of several genes (COL1A1, COL1A2, CRTAP, and P3H1 genes) that aren’t working properly. When these genes don’t work, it affects how you make collagen, a protein that helps make bones strong.
Osteogenesis imperfecta can range from mild to severe, and symptoms vary from person to person. A person may have just a few or as many as several hundred fractures in a lifetime.
There are at least eight recognized forms of osteogenesis imperfecta, designated type I through type VIII. The types can be distinguished by their signs and symptoms, although their characteristic features overlap. Type I is the mildest form of osteogenesis imperfecta and type II is the most severe; other types of this condition have signs and symptoms that fall somewhere between these two extremes. Increasingly, genetic factors are used to define the different forms of osteogenesis imperfecta.
Osteogenesis imperfecta affects an estimated 6 to 7 per 100,000 people worldwide. Types I and IV are the most common forms of osteogenesis imperfecta, affecting 4 to 5 per 100,000 people. This translates to approximately 50,000 people in the United States. Osteogenesis imperfecta occurs equally in both males and females and in all races and ethnic groups.
The milder forms of osteogenesis imperfecta, including type I, are characterized by bone fractures during childhood and adolescence that often result from minor trauma. Fractures occur less frequently in adulthood. People with mild forms of the condition typically have a blue or grey tint to the part of the eye that is usually white (the sclera), and may develop hearing loss in adulthood. Affected individuals are usually of normal or near normal height.
Other types of osteogenesis imperfecta are more severe, causing frequent bone fractures that may begin before birth and result from little or no trauma. Additional features of these conditions can include blue sclerae, short stature, hearing loss, respiratory problems, and a disorder of tooth development called dentinogenesis imperfecta. The most severe forms of osteogenesis imperfecta, particularly type II, can include an abnormally small, fragile rib cage and underdeveloped lungs. Infants with these abnormalities have life-threatening problems with breathing and often die shortly after birth.
No single test can identify osteogenesis imperfecta. Your doctor uses your medical and family history, physical exam, and imaging and lab tests to diagnose osteogenesis imperfecta. Your doctor may also test your collagen (from skin) or genes (from blood). There is no cure for osteogenesis imperfecta, but you can manage symptoms. Treatments include exercise, pain medicine, physical therapy, wheelchairs, braces, and surgery.
Although there is no cure for osteogenesis imperfecta, symptoms can be managed. Treatments for osteogenesis imperfecta may include:
- Care for broken bones
- Care for brittle teeth
- Pain medication
- Physical therapy
- Use of wheelchairs, braces, and other aids
- Surgery.
One type of surgery is called “rodding.” Metal rods are put inside the long bones to:
- Strengthen them
- Fix bone malformations
- Prevent bone malformations.
A healthy lifestyle also helps people with osteogenesis imperfecta. You can help prevent broken bones and maintain your health if you:
- Exercise (swimming, water therapy, walking)
- Keep a healthy weight
- Eat a balanced diet
- Do not smoke
- Do not drink a lot of alcohol and caffeine
- Do not take steroid medicines.
Proper care helps children and adults who have osteogenesis imperfecta to:
- Stay active
- Make bones more dense
- Keep muscles strong.
Osteogenesis imperfecta types
There are 8 main types of osteogenesis imperfecta. People with types 2, 3, 7, and 8 tend to have severe symptoms. People with types 4, 5, and 6 tend to have more moderate symptoms. People with type 1 tend to have mild symptoms.
Osteogenesis imperfecta type 1
Osteogenesis type I is the most common and usually the mildest form of osteogenesis imperfecta. In most cases, it is characterized by multiple bone fractures usually occurring during childhood through puberty. Fractures usually begin when an affected child begins to walk; fractures during the newborn (neonatal) period are rare. The frequency of fractures usually declines after puberty. Repeated fractures may result in slight malformation of the bones of the arms and legs (e.g., bowing of the tibia and femur).
A distinguishing feature associated with osteogenesis imperfecta type I is bluish discoloration of the whites of the eyes (blue sclera). In some cases, individuals with osteogenesis imperfecta type I may develop abnormalities affecting the middle and/or inner ears contributing to, or resulting in, hearing impairment (i.e., conductive and/or sensorineural hearing loss). Hearing loss occurs most often in the third decade of life; however, it can occur as early as the second decade or as late as the seventh.
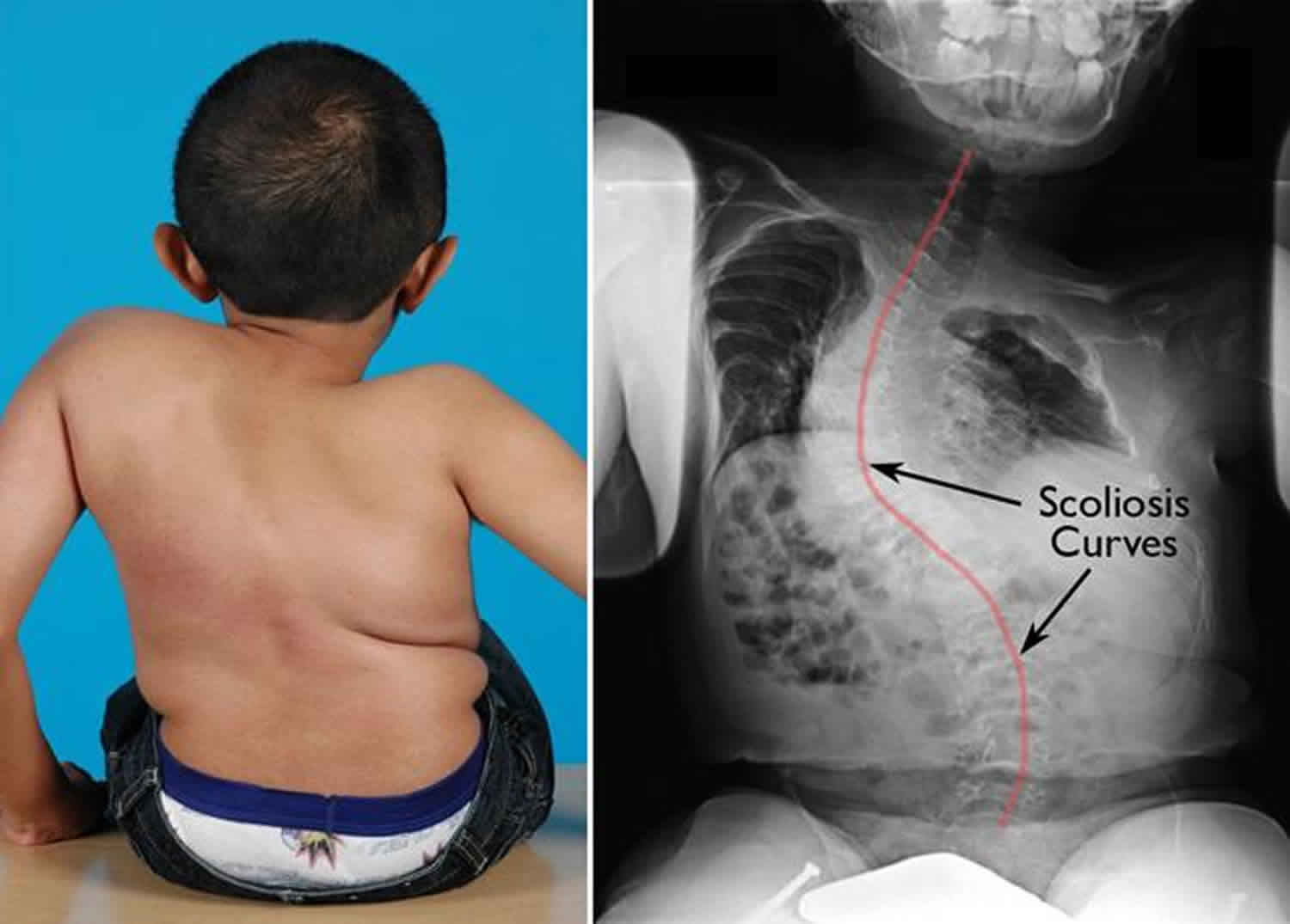
Individuals with osteogenesis imperfecta type I may have a triangular facial appearance and an abnormally large head (macrocephaly). In approximately 50 percent of cases, individuals with osteogenesis imperfecta type I will experience growth deficiencies after birth (postnatal), resulting in mild short stature (e.g., affected individuals will be shorter than unaffected family members). Approximately 20 percent of adults with osteogenesis imperfecta type I develop abnormal sideways or front-to-back curvature of the spine (scoliosis or kyphosis).
Additional symptoms associated with osteogenesis imperfecta type I include loose (hyperextensible) joints, low muscle tone (hypotonia), and thin skin that bruises easily.
Some researchers believe that a subgroup of osteogenesis imperfecta type I exists in which affected individuals experience dental abnormalities in addition to the abovementioned features.
Key clinical features
Most common and mildest form of osteogenesis imperfecta. It can be so mild that health care providers do not diagnose it in some people until they are adults.
- Bone fractures occur mostly in years before puberty and decrease in frequency after puberty.
- Normal height; a few inches shorter than same gender relatives
- Bone deformity absent or minimal.
- Brittle teeth in rare cases
- Hearing loss in some cases, often beginning in early 20s or 30s.
- Sclera (whites of the eyes) usually have a blue, purple, or gray tint.
- Easy bruising
- Mild delay in motor skills
- Normal or near-normal height.
- Loose joints and muscle weakness.
- Triangular face.
- Tendency toward spinal curvature.
- Collagen structure is normal, but the amount is less than normal.
Osteogenesis imperfecta type 2
Osteogenesis imperfecta type II is the most severe type of osteogenesis imperfecta. Affected infants often experience life-threatening complications at, or shortly after, birth. Infants with osteogenesis imperfecta type II have low birth weight, abnormally short arms and legs (limbs), and bluish discoloration of the whites of the eyes (blue sclera). In addition, affected infants may have extremely fragile bones and numerous fractures present at birth. The ribs and long bones of the legs of affected infants are often malformed.
Infants with osteogenesis imperfecta type II often have underdeveloped lungs and an abnormally small upper chest (thorax) that may result in life-threatening respiratory insufficiency. In some cases, affected infants may experience congestive heart failure.
Infants with osteogenesis imperfecta type II may also have a small, narrow nose; a small jaw (micrognathia); and an abnormally soft top of the skull (calvaria) with abnormally large soft spots (large fonatanelle). Affected infants may also have abnormally thin, fragile skin and low muscle tone (hypotonia).
osteogenesis imperfecta type II has been subdivided into three subgroups (A, B, and C) based upon small differences in bone formation seen only on x-rays (radiographic features).
Key clinical features
- Most severe form; usually results in stillborn birth or death in the first months of life
- Severe bone deformity
- Frequently lethal at or shortly after birth, often due to respiratory problems.
- Numerous fractures and severe bone deformity.
- Small stature with underdeveloped lungs.
- Tinted sclera.
- Collagen improperly formed.
Osteogenesis imperfecta type 3
Osteogenesis imperfecta type III is characterized by extremely fragile bones, multiple fractures, and malformed bones. Multiple fractures are often present at birth. Fractures and malformation of various bones (most often the ribs and long bones) may become worse (progressive malformation) as affected infants and children age.
Progressive malformation of various bones may result in short stature, sideways and front-to-back curvature of the spine (scoliosis and kyphosis), and malformation of the area where the bone in the back of the skull (occipital bone) and the top of the spine meet (basilar impression). In some cases, affected individuals may develop pulmonary insufficiency and respiratory problems. In severe cases, progressive bone malformation may result in affected individuals requiring wheelchairs.
Infants with osteogenesis imperfecta type III may have a slight blue discoloration to the whites of the eyes (blue sclera) at birth. In most cases, the bluish tinge fades during the first year of life. Affected infants may have a triangular facial appearance due to an abnormally prominent forehead (frontal bossing) and an abnormally small jaw (micrognathia). In some cases, hearing impairment and brittle, discolored teeth (dentinogenesis imperfecta) may also be present.
Key clinical features
- Most severe, nonlethal form
- Bones fracture easily. Fractures often present at birth, and x-rays may reveal healed fractures that occurred before birth.
- Hundreds of fractures starting very early in life
- Severe bone deformities and physical disability that worsen over time
- Sclera may be blue, purple, or gray tint.
- Triangular face and prominent forehead.
- Scoliosis (abnormal curving of the spine).
- Respiratory problems possible.
- Sunken or protruding chest wall.
- Barrel-shaped rib cage.
- Triangular face.
- Brittle teeth
- Hearing loss
- Very short height
- Motor skill delays
- Loose joints and poor muscle development in arms and legs.
- Collagen improperly formed.
- Usually need wheelchairs
Osteogenesis imperfecta type 4
Individuals with osteogenesis imperfecta type IV have fragile bones that often fracture easily. Fractures are more common before puberty. Affected individuals experience mild to moderate bone malformation and are usually shorter than average. Affected individuals may develop sideways and front-to-back curvature of the spine (scoliosis and kyphosis).
Individuals with osteogenesis imperfecta type IV may have a triangular facial appearance. In most cases, the whites of the eyes (sclera) are normal or pale blue during infancy. As an affected infant ages, the pale blue discoloration of the sclera fades. Affected individuals may also experience hearing impairment and brittle, discolored teeth (dentinogenesis imperfecta).
Key clinical features
- Similar to type I but with mild to moderate bone deformity
- Bones fracture easily. Dozens of fractures on average, most of which occur before puberty or after middle age
- Motor skill delays
- People with type IV often need braces or crutches to walk
- Shorter than average height.
- Sclera are white or near-white (i.e. normal in color).
- Mild to moderate bone deformity.
- Tendency toward spinal curvature (i.e. scoliosis).
- Barrel-shaped rib cage.
- Triangular face.
- Large head.
- Hearing loss in some cases
- Brittle teeth
- Collagen improperly formed.
- Easy bruising
Osteogenesis imperfecta type 5
By studying the appearance of osteogenesis imperfecta bone under the microscope, investigators noticed that some people who are clinically within the Type IV group had a distinct pattern to their bone. When they reviewed the full medical history of these people, they found that groups had other features in common. They named these groups Types V and VI osteogenesis imperfecta. The mutations causing these forms of osteogenesis imperfecta have not been identified, but people in these two groups do not have mutations in the type I collagen genes.
Key clinical features
- Clinically similar to Type IV in appearance and symptoms of osteogenesis imperfecta.
- Severely limited ability to twist forearms clockwise or counterclockwise.
- A dense band seen on x-rays adjacent to the growth plate of the long bones.
- Unusually large calluses (hypertrophic calluses) at the sites of fractures or surgical procedures. (A callus is an area of new bone that is laid down at the fracture site as part of the healing process.)
- Calcification of the membrane between the radius and ulna (the bones of the forearm). This leads to restriction of forearm rotation.
- Normal sclera.
- Normal teeth.
- Distinguished from Type IV by differing bone features at microscopic level. Bone has a “mesh-like” appearance when viewed under the microscope.
- Dominant inheritance pattern
Osteogenesis imperfecta type 6
Key clinical features
- Clinically similar to Type IV in appearance and symptoms of osteogenesis imperfecta.
- The alkaline phosphatase (an enzyme linked to bone formation) activity level is slightly elevated in osteogenesis imperfecta Type VI. This can be determined by a blood test.
- Bone has a distinctive “fish-scale” appearance when viewed under the microscope.
- Diagnosed by bone biopsy.
- Whether this form is inherited in a dominant or recessive manner is unknown, but researchers believe the mode of inheritance is most likely recessive.
- Eight people with this type of osteogenesis imperfecta have been identified.
Recessive Forms of osteogenesis imperfecta
After years of research, two forms of osteogenesis imperfecta that are inherited in a recessive manner were discovered in 2006. Both types are caused by genes that affect collagen formation. These forms provide information for people who have severe or moderately severe osteogenesis imperfecta but who do not have a primary collagen mutation.
Osteogenesis imperfecta type 7
Key clinical features
- The first described cases resemble Type IV osteogenesis imperfecta in many aspects of appearance and symptoms.
- In other instances the appearance and symptoms are similar to Type II lethal osteogenesis imperfecta, except infants had white sclera, a small head and a round face.
- Short stature.
- Short humerus (arm bone) and short femur (upper leg bone)
- Coxa vera is common (the acutely angled femur head affects the hip socket).
- Results from recessive inheritance of a mutation to the CRTAP (cartilage-associated protein) gene. Partial function of CRTAP leads to moderate symptoms while total absence of CRTAP was lethal in all 4 identified cases.
Osteogenesis imperfecta type 8
Key clinical features
- Resembles lethal Type II or Type III osteogenesis imperfecta in appearance and symptoms except that infants have white sclera.
- Severe growth deficiency.
- Extreme skeletal under mineralization.
- Caused by a deficiency of P3H1 (Prolyl 3-hydroxylase 1) due to a mutation to the LEPRE1 gene.
Osteogenesis imperfecta life expectancy
Osteogenesis imperfecta is a lifelong condition. Respiratory failure is the most frequent cause of death for people with osteogenesis imperfecta, followed by accidental trauma. The most severe forms may cause death in infancy. With good medical management and supportive care, the majority of people who have osteogenesis imperfecta will lead healthy, productive lives and can expect an average life span.
Osteogenesis imperfecta symptoms
All people with osteogenesis imperfecta have brittle bones. osteogenesis imperfecta can range from mild to severe and symptoms vary from person to person. Some of the symptoms that people with osteogenesis imperfecta may have are:
- Malformed bones
- Short, small body
- Loose joints
- Muscle weakness
- Sclera (whites of the eyes) that look blue, purple, or gray
- Triangular face
- Barrel-shaped rib cage
- Curved spine
- Brittle teeth
- Hearing loss (often starting in 20s or 30s)
- Breathing problems
- Type 1 collagen that does not work well
- Not enough collagen.
Non-skeletal Issues of Osteogenesis Imperfecta
Early definitions of osteogenesis imperfecta (osteogenesis imperfecta) refer to a syndrome of fragile bones plus hearing loss. This suggests that the problems seen in people with osteogenesis imperfecta include more than just brittle bones. In dominant osteogenesis imperfecta, the mutation in type 1 collagen affects not only the skeleton but also the collagen-rich tissues in other organ systems. Like the skeletal manifestations, the non-skeletal clinical features vary in the degree of severity. In some instances, age is also a factor in severity. Research is needed to fully understand the prevalence and best approach to management of non-skeletal issues.
Dental Oral-Facial
osteogenesis imperfecta affects the growth of both jaws and tooth development. In addition, about 50% of people with osteogenesis imperfecta also have dentinogenesis imperfecta. Regular dental care is recommended for all people with osteogenesis imperfecta beginning within 6 months after the primary teeth erupt and continuing throughout life. Other common oral cavity problems related to osteogenesis imperfecta include impacted teeth, anterior and posterior open and cross bites and skeletal Class III malocclusion.
Lung Problems
In people with severe forms of osteogenesis imperfecta respiratory complications are a leading cause of death. Although respiratory problems are usually more severe in those with severe osteogenesis imperfecta, the primary collagen defect affects lung tissue in all people with osteogenesis imperfecta, including those with a mild phenotype. Altered lung tissue predisposes the person to respiratory infections.
Rates of asthma and pneumonia are higher in children and adults who have osteogenesis imperfecta than in the unaffected population. Chest wall deformities, rib fractures, scoliosis, kyphoscoliosis, chest wall muscle weakness, limited mobility and the effects of gastrointestinal problems such as constipation and reflux all contribute to poor pulmonary function. Fatigue, breathlessness and wheezing are frequent symptoms. Manifestations can include asthma, recurrent pneumonia, exercise intolerance, and sleep apnea.
Hearing Issues
Approximately 50% of adults with osteogenesis imperfecta have a measurable hearing loss, which typically arises in the second or third decade of life. Bone quality and structural abnormalities of the ear bones — including visible deformities in the ossicles and inner ear– contribute to the loss. Environmental factors affecting hearing may cause a loss sooner than in the unaffected population. Most hearing loss in osteogenesis imperfecta is mixed but conductive, and sensorineural types of loss are seen. Severity ranges from mild to profound. In addition, some people report tinnitus and vertigo. Treatments include hearing aids and/or surgery such as stapedectomy or cochlear implant. Physicians should be alert to dangers of ototoxins in this population.
Cardiovascular
The prevalence of cardiovascular problems among patients with osteogenesis imperfecta is unknown. The most frequently reported cardiovascular issues are hypertension, and mitral valve prolapse. Heart valve problems are believed to develop over time. They are more of a concern for adults who have osteogenesis imperfecta, but are infrequently seen in children. Cardiac surgery, including valve replacement, has been successful.
Neurological
Intelligence is typically normal in osteogenesis imperfecta. People with osteogenesis imperfecta often have enlarged heads, called macrocephaly. They can also have a condition called hydrocephalus, in which fluid builds up inside the skull, causing the brain to swell. Enlarged head circumference is seen in infants and children who have osteogenesis imperfecta and may or may not be caused by hydrocephalus. Evaluation is required; shunting is possible.
People with severe osteogenesis imperfecta often have basilar invagination, a malformation of the spinal column that puts pressure on the spinal cord and brain stem and those with severe osteogenesis imperfecta (Type III) have a greater risk. Basilar invagination worsens over time and can cause severe headaches, changes in facial sensation, lack of control over muscle movements, and difficulty swallowing. If untreated, basilar invagination can lead to rapid neurological decline and inability to breathe.
Vision
People with osteogenesis imperfecta seem to experience the common refractive errors such as myopia, hyperopia or astigmatism at about the same rate as people in the unaffected population. More serious conditions like glaucoma and retinal detachment are seen in adults, but the incidence is unknown. Blue sclerae, a frequently described finding, may be associated with corneal thinning. Scleral thickness is normal in osteogenesis imperfecta Type I but may be thin in other types of osteogenesis imperfecta.
Connective Tissue
Blood Vessels, Skin, Tendons and Ligaments
- Thin blood vessels and thin skin may cause people to bruise easily.
- Skin may be stiffer and less elastic, increasing the risk for scarring.
- Reduced muscle strength may be significant in those with moderate and severe forms of osteogenesis imperfecta.
- Joint laxity is common and contributes to frequent sprains and dislocations particularly of the ankles, hips, shoulders, thumbs and elbows.
- Flat feet are common, particularly in Type I (the mildest type).
- Hernias may be present at birth and occur more frequently in children with osteogenesis imperfecta than in the general population.
Endocrine
- People with more severe forms of osteogenesis imperfecta are often short however growth hormones and other hormones are typically normal.
- Excessive diaphoresis has been reported in individuals of all types of osteogenesis imperfecta.
- Young women with osteogenesis imperfecta may start menstruating later than other women.
Gastrointestinal
- Constipation is common in children and adults with osteogenesis imperfecta.
- Spine, hip and pelvic deformities contribute to constipation in severely affected children.
- Treatments include diet, hydration, physical activity and medication.
- Celiac disease, gluten sensitivity, and colitis are reported in children, teens and adults with all types of osteogenesis imperfecta, but it is not clear if these are more common than in the general population.
Pain
People who have osteogenesis imperfecta experience acute pain from an injury; surgical pain; and varying degrees of chronic pain. Back pain may be due to compression fractures of the spine or spine curves such as scoliosis or kyphosis. Some people may have pain without clear evidence of a fracture. Adequate pain management is essential to maintaining mobility and improving quality of life. People with osteogenesis imperfecta feel the same level of pain as others but may complain less since they experience pain more often than others.
Osteogenesis imperfecta causes
Mutations in the COL1A1, COL1A2, CRTAP, and P3H1 genes cause osteogenesis imperfecta.
Mutations in the COL1A1 and COL1A2 genes are responsible for more than 90 percent of all cases of osteogenesis imperfecta. These genes provide instructions for making proteins that are used to assemble type I collagen. This type of collagen is the most abundant protein in bone, skin, and other connective tissues that provide structure and strength to the body.
Most of the mutations that cause osteogenesis imperfecta type I occur in the COL1A1 gene. These genetic changes reduce the amount of type I collagen produced in the body, which causes bones to be brittle and to fracture easily. The mutations responsible for most cases of osteogenesis imperfecta types II, III, and IV occur in either the COL1A1 or COL1A2 gene. These mutations typically alter the structure of type I collagen molecules. A defect in the structure of type I collagen weakens connective tissues, particularly bone, resulting in the characteristic features of osteogenesis imperfecta.
Mutations in the CRTAP and P3H1 genes are responsible for rare, often severe cases of osteogenesis imperfecta. Cases caused by CRTAP mutations are usually classified as type VII; when P3H1 mutations underlie the condition, it is classified as type VIII. The proteins produced from these genes work together to process collagen into its mature form. Mutations in either gene disrupt the normal folding, assembly, and secretion of collagen molecules. These defects weaken connective tissues, leading to severe bone abnormalities and problems with growth.
In cases of osteogenesis imperfecta without identified mutations in one of the genes described above, the cause of the disorder is unknown. These cases include osteogenesis imperfecta types V and VI. Researchers are working to identify additional genes that may be responsible for these conditions.
Osteogenesis imperfecta inheritance pattern
Most cases (85-90%) of osteogenesis imperfecta have an autosomal dominant pattern of inheritance, which means one copy of the altered gene in each cell is sufficient to cause the condition. Many people with type I or type IV osteogenesis imperfecta inherit a mutation from a parent who has the disorder. Most infants with more severe forms of osteogenesis imperfecta (such as type II and type III) have no history of the condition in their family. In these infants, the condition is caused by new (sporadic) mutations in the COL1A1 or COL1A2 gene.
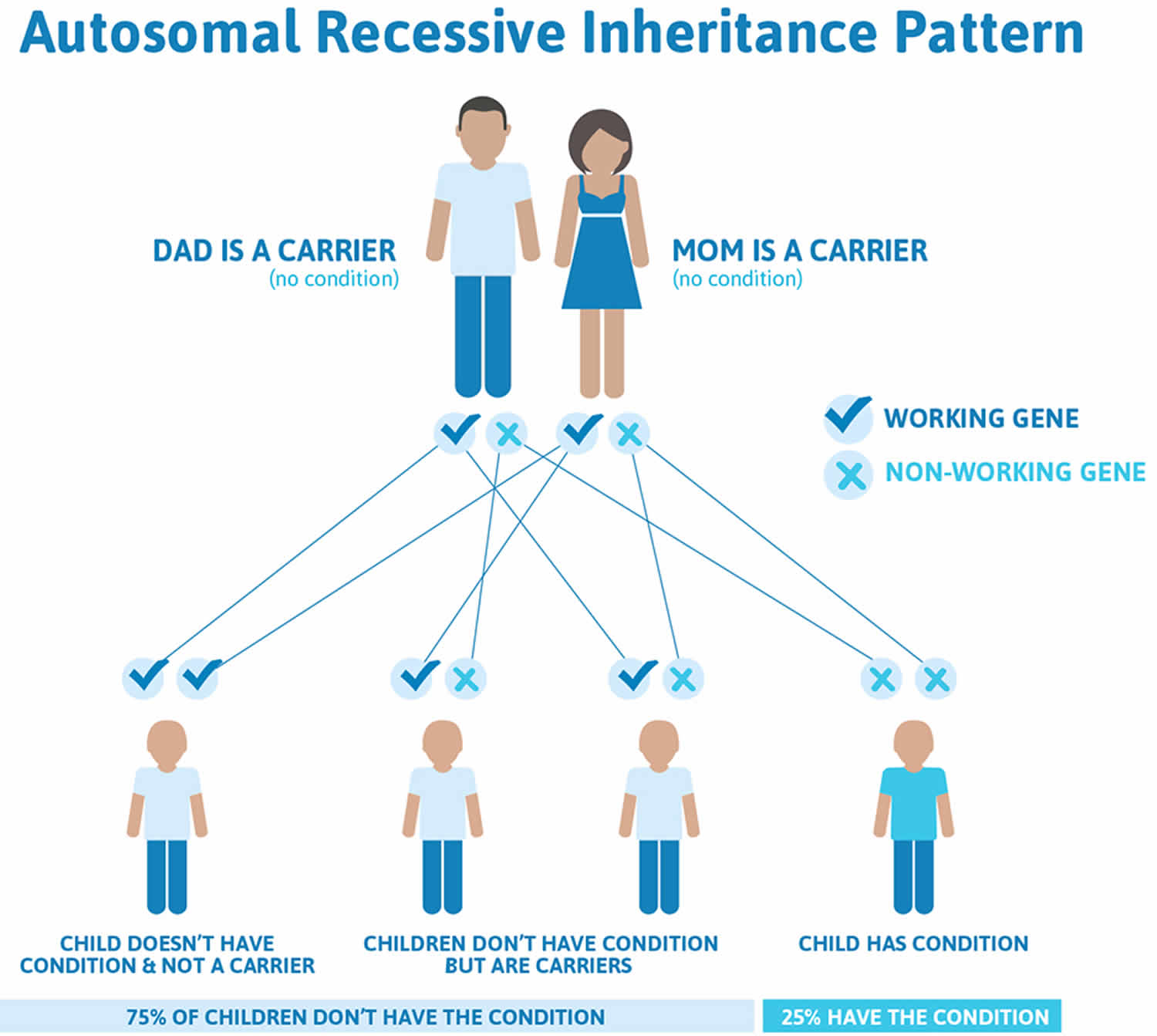
Less commonly (10-15 percent) of osteogenesis imperfecta (type VII and type VIII) has an autosomal recessive pattern of inheritance. Autosomal recessive inheritance means two copies of the gene in each cell are altered. The parents of a child with an autosomal recessive disorder typically are not affected, but each carry one copy of the altered gene. Some cases of osteogenesis imperfecta type III are autosomal recessive; these cases usually result from mutations in genes other than COL1A1 and COL1A2. When osteogenesis imperfecta is caused by mutations in the CRTAP or P3H1 gene, the condition also has an autosomal recessive pattern of inheritance.
Figure 1. Autosomal dominant pattern of inheritance
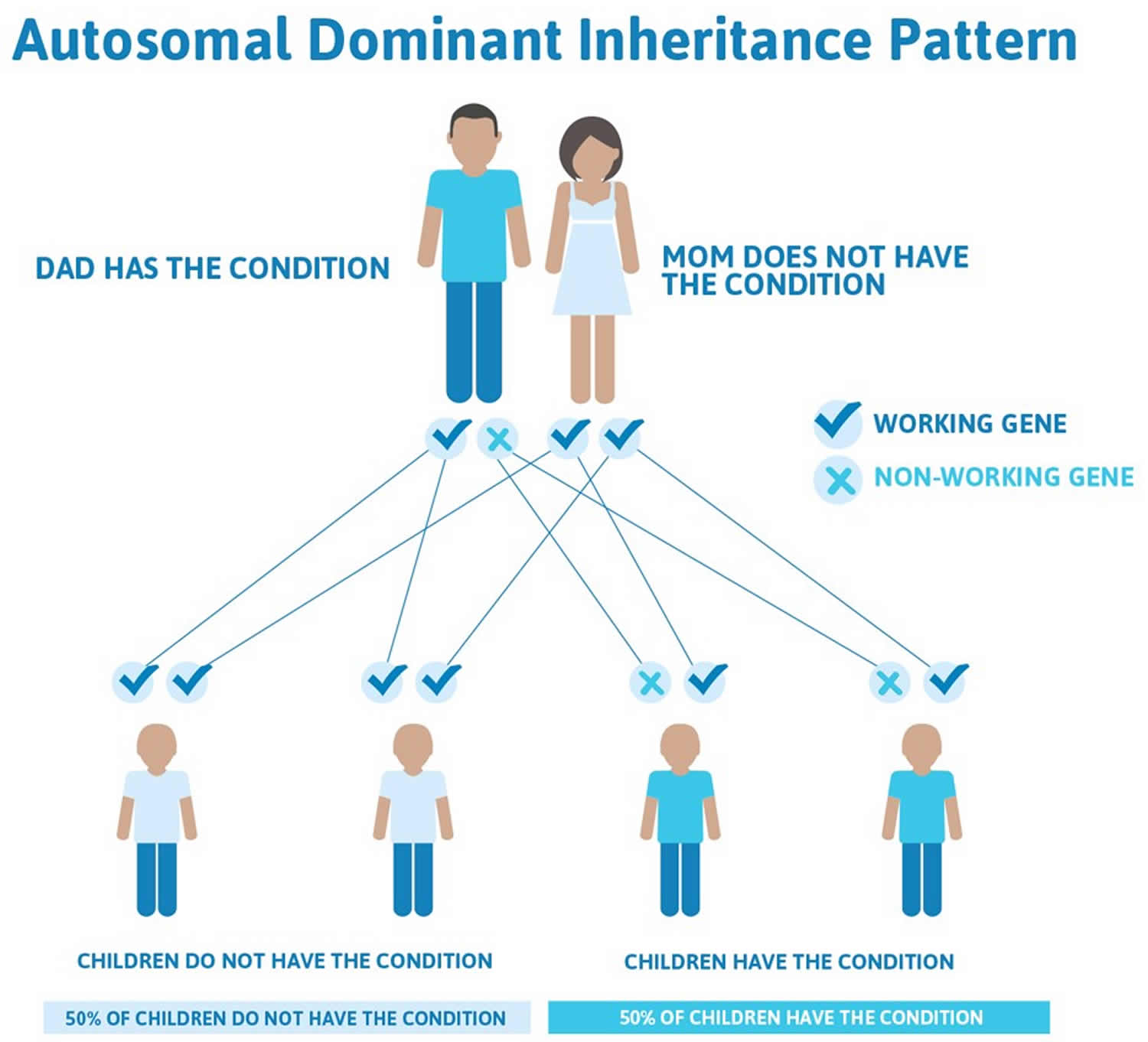
Figure 2. Autosomal recessive pattern of inheritance
Osteogenesis Imperfecta diagnosis
No single test can identify osteogenesis imperfecta. To diagnose osteogenesis imperfecta, doctors look at:
- Family history
- Medical history
- Results from a physical exam
- X rays.
Your doctor may also test your collagen (from skin) or genes (from blood). It may take a few weeks to learn the results of the tests. These tests spot osteogenesis imperfecta in 9 out of 10 people who have it.
Osteogenesis imperfecta treatment
There is not yet a cure for osteogenesis imperfecta. Treatment is directed toward preventing or controlling the symptoms, maximizing independent mobility, and developing optimal bone mass and muscle strength. Care of fractures, extensive surgical and dental procedures, and physical therapy are often recommended for people with osteogenesis imperfecta. Use of wheelchairs, braces, and other mobility aids is common, particularly (although not exclusively) among people with more severe types of osteogenesis imperfecta.
People with osteogenesis imperfecta are encouraged to exercise as much as possible to promote muscle and bone strength, which can help prevent fractures. Swimming and water therapy are common exercise choices for people with osteogenesis imperfecta, as water allows independent movement with little risk of fracture. For those who are able, walking (with or without mobility aids) is excellent exercise. People with osteogenesis imperfecta should consult their physician and/or physical therapist to discuss appropriate and safe exercise.
Children and adults with osteogenesis imperfecta will also benefit from maintaining a healthy weight, eating a nutritious diet, and avoiding activities such as smoking, excessive alcohol and caffeine consumption, and taking steroid medications — all of which may deplete bone and make bones more fragile.
A surgical procedure called “rodding” is frequently considered for people with osteogenesis imperfecta. This treatment involves inserting metal rods through the length of the long bones to strengthen them and prevent and/or correct deformities. Plastic braces are replacing plaster casts as protective devices because they permit greater freedom of movement and can be used in water. Inflatable suits can provide added protection, especially to very young children.
Surgery may prove necessary for individuals with severe malformation of the bones of the spine or basilar impression. Dental procedures may be necessary to correct various dental abnormalities. Affected individuals, especially adults, should be monitored for hearing impairment often associated with osteogenesis imperfecta. Genetic counseling may be of benefit for affected individuals and their families. Other treatment is symptomatic and supportive.
Several medications and other treatments are being explored for their potential use to treat osteogenesis imperfecta. These include growth hormone treatment, treatment with intravenous and oral drugs called bisphosphonates, an injected drug called teriparatide (for adults only) and gene therapies. It is not clear if people with recessive osteogenesis imperfecta will respond in the same manner as people with dominant osteogenesis imperfecta to these treatments.
Pediatric Treatment Summary
Medications
- IV bisphosphonates (pamidronate (Aredia®) and zoledronate (Reclast®/Zometa®) to increase bone mass; may be started in neonatal period if infant can breathe on own.
- Supplement with vitamin D and calcium as needed.
Physical and occupational therapy beginning in infancy
- Both land and aquatic based programs.
- To improve function, address joint laxity, and improve cardiopulmonary status.
- To aid development of fine motor skills.
- To identify appropriate mobility aids.
Surgery
- To correct bowing of long bones; may involve osteotomies.
- To insert rods (intramedullary rodding surgery) to support long bones.
- To stabilize the spine.
Diet and Nutrition
Children with osteogenesis imperfecta need a balanced diet containing adequate water, fiber, calcium and vitamin D calibrated to their age and size. Nutrition counseling for parent and child may be beneficial.
- Slow weight gain is seen in infants and may not be failure to thrive.
- Swallowing difficulties, reported in some toddlers, may require referral to an occupational or speech therapist, or a nutritionist who treats feeding disorders.
- Small appetite is seen in children of all ages. Inactivity, pain, medications and depression are potential causes.
- Constipation is seen in children of all ages and with all types of osteogenesis imperfecta and can be recurrent. Short stature, inactivity, pelvic deformity and difficulty with hydration are contributing factors.
- Weight control is important, as obesity places a strain on the fragile skeleton and can lead to loss of mobility. Hormonal changes related to puberty can contribute to unhealthy weight gain, especially in girls who have osteogenesis imperfecta.
Mental Health
Living with osteogenesis imperfecta presents emotional as well as physical challenges for the child, parents and siblings. Health care providers are encouraged to note signs of depression, substance abuse, and fearfulness, and to make referrals to mental health professionals. Children may experience low self-esteem and anxiety. Children with the milder forms of osteogenesis imperfecta may struggle to cope with having a hidden disorder that can be misunderstood by their peers. Older children may become discouraged by the repeated need to re-learn mobility skills, receive painful therapies and miss out on activities with their peers due to fractures and reduced mobility. Relationships between siblings can be strained.
Development
Osteogenesis imperfecta does not affect a child’s ability to think and learn, but children with osteogenesis imperfecta often demonstrate delays in meeting developmental milestones. These delays can be the result of repeated immobilizations after fractures, misalignment of the long bones and joints and the general hypotonia and ligamentous laxity common in osteogenesis imperfecta. Interventions can include physical and occupational therapy, braces, use of adaptive equipment and mobility aides. Small-muscle development, especially in hands and fingers, is likewise affected. To the best of our current knowledge, the incidence of autism, hyperactivity and childhood cancers is believed to be similar to that seen in the unaffected population.
Growth
Mild to significant short stature and a slow growth rate occurs in osteogenesis imperfecta. Hip and back pain due to poor alignment and leg length discrepancies occur in all types of osteogenesis imperfecta and should be evaluated by an orthopedist and/or a gait specialist. Height and weight charts for the child with osteogenesis imperfecta are available.
Adult Treatment Summary
Medications
- IV and oral bisphosphonates such as alendronate (Fosamax®), teriparatide (Forteo®) or other osteoporosis drugs to maintain bone mass.
- Supplement with vitamin D and calcium as needed
Physical and Occupational Therapy
- To maintain mobility.
- To adjust to physical changes due to injury and/or aging.
Surgery
- Joint replacement,
- Fracture Repair,
- Rod repair.
Monitoring for Children and Adults
In addition to monitoring for general health and when a patient is receiving drug therapy, regularly evaluate:
- Lax joints.
- Muscle strength.
- Pain level changes; assess for a non-osteogenesis imperfecta problem.
- Spine curve progression.
Fracture Care and Repair
The effects of immobility are cumulative. Long periods of immobility further weaken bones and lead to muscle loss, weakness, and more fractures. Orthopedists who are familiar with osteogenesis imperfecta recommend:
- Cast or splint with lightweight materials for shortest term possible.
- Limit use of the spica cast.
- Avoid the use of plates or pins for surgical fracture repair.
- Plan for rehabilitation services after fracture or surgery for people with all types of osteogenesis imperfecta including Type I.
- Acute Care for fractures
- Immobilize for comfort;
- Fracture may not be visible in first set of x-rays particularly in child with severe osteogenesis imperfecta;
- Address pain control;
- Address muscle spasms.
Rehabilitation and Safe Exercise
Goals for physical and occupational therapy include expanding and maintain function, promoting independence and moving into community based recreational physical activity programs. A typical program includes muscle strengthening and aerobic conditioning that focuses on the individual’s needs. Physical therapy often begins in infancy to counteract delays in motor skill development. It may be needed across the life span in response to injury, surgery or aging. Older children and teens benefit from continued physical activity such as adapted physical education and appropriate recreational activities. Swimming and water therapy are particularly well-suited to people with osteogenesis imperfecta of all ages as it allows independent movement with little fracture risk. Walking is also excellent exercise for those who are able with or without mobility aids.
Orthopedic Surgery
Surgery may be needed to repair a broken bone, correct bone deformities such as bowing, or stabilize the spine. Many children with osteogenesis imperfecta undergo rodding surgery to insert metal rods into the long bones. Issues related to orthopedic surgery for children and adults include positioning, thermal instability, anesthesia, and potential for excessive bleeding.
Spine curves and fractures are seen in all types of osteogenesis imperfecta including the milder forms. Onset of scoliosis and other curves is often earlier than in most children. Curves may continue to change during the adult years. Vertebral fractures are also seen across the life span. Recommendations include:
- Monitor for scoliosis, kyphosis, and vertebral compression fractures.
- Evaluate for curve starting at age 2-3 years in Type III osteogenesis imperfecta or age 6-8 years in mild osteogenesis imperfecta.
- X-rays should be taken every 1-2 years, as observation alone may not be effective.
- For people who cannot stand independently, a standard scoliosis exam should consist of a seated AP and lateral x-ray with a level pelvis, except in cases of pelvic obliquity.
- If the person is able to stand independently, a standing x-ray with a level pelvis should be taken.
- Surgery is generally recommended following progression to 60 degrees.
- Factors to consider include age, balanced curve, leg length discrepancy, trunk imbalance, lung function and bone quality.
- Use of an intermittent soft brace can be effective in mild Type I osteogenesis imperfecta and may work for mild Type IV osteogenesis imperfecta. It is not recommended for other Types of osteogenesis imperfecta due to stresses placed on sternum.
- Research into the role of exercise to improve or prevent scoliosis is ongoing.
Medications
At this time, there are no drug therapies specifically developed to treat osteogenesis imperfecta in either children or adults. The drug therapies currently in use are based on medications developed to treat age-related osteoporosis or cancer-related bone loss in adults in the general population. Bone strengthening in an effort to reduce the frequency and seriousness of fractures is a key treatment issue for children and adults who have osteogenesis imperfecta.
Bisphosphonates: Bisphosphonate drugs which are currently approved by the US Food and Drug Administration to prevent and treat osteoporosis and bone complications of cancer have become a standard treatment to increase bone density in children with moderated and severe osteogenesis imperfecta. There are many different types of bisphosphonates. The two that are most often prescribed for people with osteogenesis imperfecta are pamidronate (Aredia®) and zolendronic acid (Reclast®). Pamidronate has been studied the longest. Bisphosphonate treatment has been shown to reduce pain, and increase bone mineral density in children. The effects of such treatment on fracture frequency and bone deformity are less clear, but there are studies that have shown reduction in fracture rates in treated children. Side effects include a drop in blood levels of calcium especially at the start of the treatment and delayed bone healing after osteotomies. The longer-term side effects of pamidronate treatment are not well known. Several different protocols are currently in use. When to start treatment, most effective dose, and how long to treat are issues that continue to be studied. In children, once bisphosphonate treatment is initiated, it is important to maintain it (often at a reduced maintenance dose) until growth plates are fused in order to protect the new bone formed during the growing process. Bisphosphonates do not build new bone, or improve the quality of existing bone. They do not address any of the non-skeletal features of osteogenesis imperfecta.
Zoledronic acid is a newer bisphosphonate that is given intravenously like pamidronate but only requires one 30-60 minute infusion every 6 months. It is as effective as pamidronate when the treatment is started; however the best dose for long-term treatment is not yet known. Alendronate (Fosamax®) is a bisphosphonate given orally. It has been shown to increase bone density but does not appear to have a significant effect on fracture rates or pain in children with osteogenesis imperfecta. There is no evidence at this time of osteonecrosis of the jaw in children who have osteogenesis imperfecta who are treated with the appropriate dose of a bisphosphonate. This problem has not been studied in adults with osteogenesis imperfecta. See the supporting document Bisphosphonate Protocols for information on administering these drugs.
Use of Bone Building Drugs for Adults with osteogenesis imperfecta: Adults who have osteogenesis imperfecta frequently are treated with bisphosphonates and other drugs developed to treat age-related osteoporosis. Adults may receive oral or intravenous bisphosphonates. Unlike the experience with children, treatment with bisphosphonates has not been shown to significantly decrease fractures in the osteogenesis imperfecta adult. In addition in studies conducted on adults with osteoporosis, it has been noted that some individuals experience musculoskeletal pain due to bisphosphonate treatment. This is now mentioned as a warning on the product insert for most types of bisphosphonates. Bone building drugs are typically not prescribed to young adults.
Teriparatide (Forteo®) is a medication that builds bone rather than preserving existing amount of bone like bisphosphonates. Teriparatide has been shown to increase bone mineral density and increase bone strength, but the effects on fracture are not clear. It appears that this form of treatment may be more beneficial to those with milder forms for osteogenesis imperfecta. It is given as a daily injection. It has been approved by the US FDA as a treatment for osteoporosis in adults but it is not approved for use in children who are still growing.
Growth Hormone Therapy has been studied as a treatment for children with osteogenesis imperfecta. Although people who have osteogenesis imperfecta are sometimes short in stature, this does not appear to be due to a lack of growth hormone. Studies indicate the some children with osteogenesis imperfecta (moderate osteogenesis imperfecta Type IV) respond to a daily injection with an initial increase in growth. Responders show changes in bone architecture and small increased in BMD (bone mineral density); bone strength and growth are also positively affected. There is no data at this time regarding any effect on fracture rate
Mobility Aids
Correct sizing of aids – walkers, canes, crutches and wheelchairs—is important to mobility and to reducing use-related injuries. It is not unusual for children and adults to use different mobility aids depending on whether they are at home, or out in the community.
Other Treatment
Additional treatments include treatment for hearing loss, cardiac, vision, respiratory and other problems associated with osteogenesis imperfecta.
- Osteogenesis imperfecta. https://ghr.nlm.nih.gov/condition/osteogenesis-imperfecta[↩]

{kind=link}