Contents
Roberts syndrome
Roberts syndrome also known as SC phocomelia syndrome, RBS, Appelt-Gerken-Lenz syndrome, hypomelia hypotrichosis facial hemangioma syndrome, Roberts-SC phocomelia syndrome, tetraphocomelia-cleft palate syndrome or pseudothalidomide syndrome, is an extremely rare genetic autosomal recessive disease characterized by craniofacial deformities, limb abnormalities and intellectual disability 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17. Affected individuals are born with abnormalities of all four limbs and typically have shortened arm and leg bones (hypomelia). They may also have phocomelia (a birth defect in which infants born with phocomelia will have arms and/or legs that are severely shortened or sometimes completely absent); abnormal or missing fingers and toes; joint deformities (contractures); and numerous facial abnormalities including cleft lip with or without opening in the roof of the mouth (cleft palate); micrognathia; ear abnormalities; wide-set eyes (hypertelorism); down-slanting palpebral fissures; small nostrils; and a beaked nose. Microcephaly (baby’s head is significantly smaller than expected), intellectual disability, and heart, kidney or genital abnormalities may also be present. The limb defects are similar to those seen in thalidomide embryopathy; hence, Roberts syndrome is also known as pseudothalidomide syndrome 2.
This disease was first described in 1919 by John Roberts who reported phocomelia, bilateral cleft lip and cleft palate, and protrusion of the intermaxillary region in three children of an Italian couple who were first cousins 18. But earlier reports of tetra phocomelia and facial anomalies are cited since 1670s 10.
Roberts syndrome was previously referred as “pseudothalidomide syndrome”, based on the similarities regarding limb malformations with thalidomide syndrome patients 19. In 1969, J Herrmann and colleagues 20 described a syndrome of intrauterine and postnatal growth retardation, mild symmetric reduction of the limbs, flexion contractures of various joints, multiple minor anomalies (including capillary hemangiomas of the face, cloudy corneas, hypoplastic cartilages of the ears and nose, micrognathia, and scanty, silvery-blond hair), and autosomal recessive inheritance. They named this condition the pseudothalidomide or ‘SC syndrome’. SC phocomelia syndrome was originally thought to be distinct from Roberts syndrome; however, it is now considered to be a mild variant of Roberts syndrome 20. “SC” represents the first letters of the surnames of the two families first diagnosed with this disorder 20.
Roberts syndrome is an extremely rare condition with only about 150 cases have been described in literature 4, 21, 22, 23, 24.
Roberts syndrome is caused by mutation in the “establishment of cohesion 1 homolog 2” (ESCO2) gene on chromosome 8p21.1 (short arm of chromosome 8), resulting in the loss of acetyltransferase activities and manifesting as premature centromere separation in metaphase chromosomes 25, 26, 27, 28, 29, 30, 31. The ESCO2 gene is an essential gene that targets the DNA-binding cohesin complex 32, 33, 34, 35. The ESCO2 gene provides instructions for making a protein that is important for proper chromosome separation during cell division. Before cells divide, they must copy all of their chromosomes. The copied DNA from each chromosome is arranged into two identical structures, called sister chromatids. The ESCO2 protein plays an important role in establishing the glue that holds the sister chromatids together until the chromosomes are ready to separate. Aneuploidy with random chromosome loss and micronuclei and/or nuclear lobulation in the interphase cells are also characteristics of Roberts syndrome 35.
Roberts syndrome occurs equally in both males and females, and is common among closely related parents (parental consanguinity) 2, 24.
The diagnosis of Roberts syndrome can be made clinically in individuals with characteristic intrauterine growth restriction, limb malformation, and craniofacial abnormalities 11, 36. However, the gold standard for diagnosis is cytogenetic testing 37. A high index of suspicion is needed as Roberts syndrome is extremely rare. Regular antenatal ultrasounds play a significant role in preventing Roberts syndrome, especially in consanguineous couples. An experienced sonographer can suspect Roberts syndrome with prenatal ultrasound where structural defects can be well defined 11, 36, 38, 39. Postnatal cytogenetic testing of the biopsied tissue confirms the diagnosis 2, 40. In low-income countries, clinical diagnosis can be made where genetic diagnosis is not feasible 2.
Mildly affected individuals can survive to adulthood, but infants with a severe form of Roberts syndrome result in spontaneous abortion, stillbirth, or die shortly after birth within 1 month 41, 42, 6. The treatment of Roberts syndrome is limited to prevention, surgery to correct physical malformations, prostheses, special education, speech therapy, and treatments for organ dysfunction 42, 6.
Figure 1. Roberts syndrome
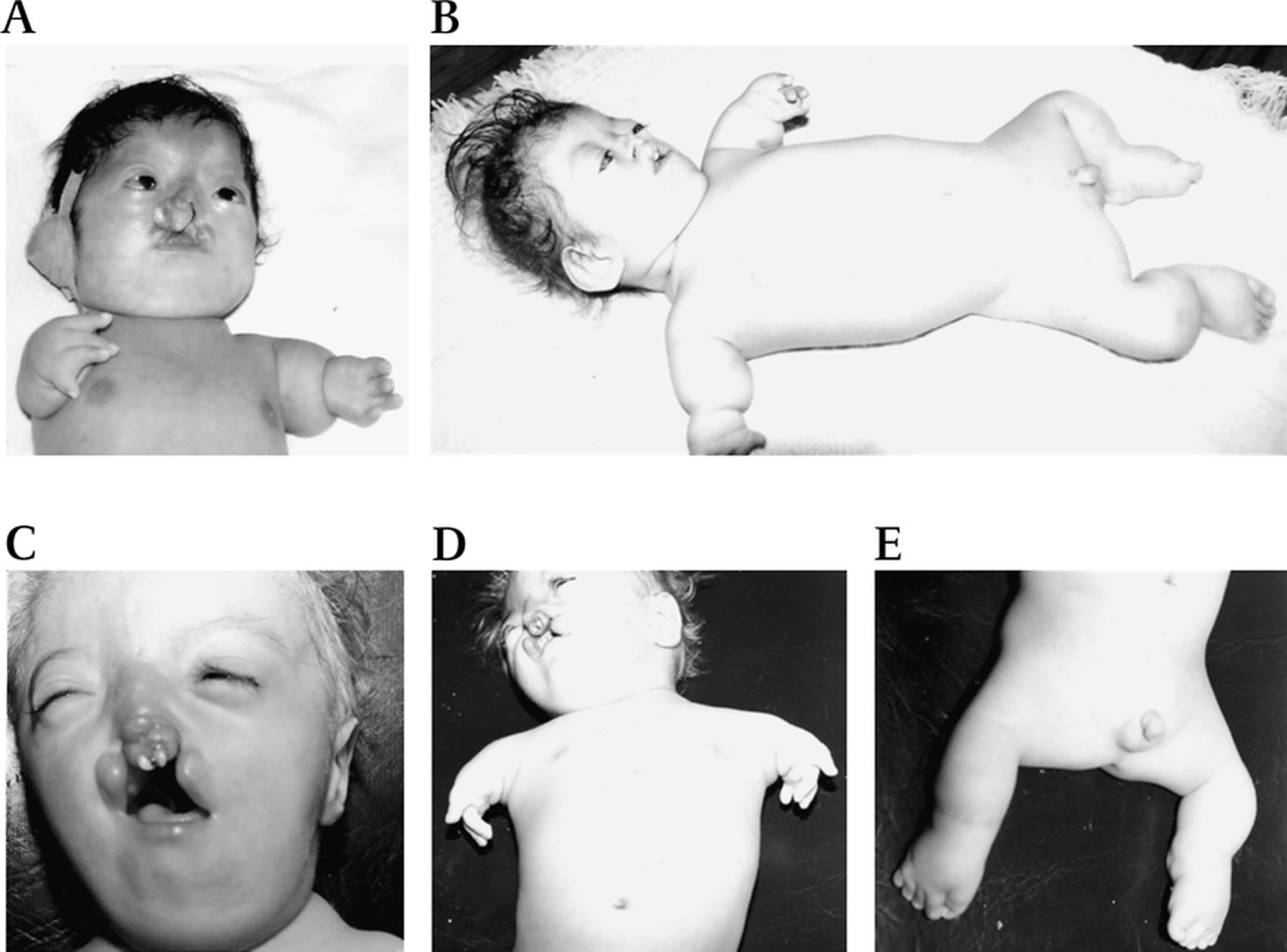
Footnotes: Photographs of patients with Roberts syndrome (RBS). (A,B) Patient with the ESCO2 c.505C>T mutation in the neonatal period (A) and 1 year later (B). (C,D,E) Patient with ESCO2 c.879_880delAG mutation. (A,C) Roberts syndrome facies with microcephaly, hypoplastic nasal alae, malar hypoplasia, hypertelorism, micrognathia, hemangioma, exophthalmos, down-slanting palpebral fissures and cleft lip and palate. (B,D,E) Roberts syndrome limb reduction is mesomelic, symmetric and follows a cephalocaudal pattern in which the arms are more severely affected than the legs.
[Source 43 ]Roberts syndrome causes
Roberts syndrome is caused by mutation in the “establishment of cohesion 1 homolog 2” (ESCO2) gene on chromosome 8p21.1 (short arm of chromosome 8), resulting in the loss of acetyltransferase activities and manifesting as premature centromere separation in metaphase chromosomes 25, 26, 27, 28, 29, 30, 31. The ESCO2 gene is an essential gene that targets the DNA-binding cohesin complex 32, 33, 34, 35. The ESCO2 gene provides instructions for making a protein that is important for proper chromosome separation during cell division. Before cells divide, they must copy all of their chromosomes. The copied DNA from each chromosome is arranged into two identical structures, called sister chromatids. The ESCO2 protein plays an important role in establishing the glue that holds the sister chromatids together until the chromosomes are ready to separate. Aneuploidy with random chromosome loss and micronuclei and/or nuclear lobulation in the interphase cells are also characteristics of Roberts syndrome 35.
All identified mutations in the ESCO2 gene prevent the cell from producing any functional ESCO2 protein, which causes some of the glue between sister chromatids to be missing around the chromosome’s constriction point (centromere). In Roberts syndrome, cells respond to abnormal sister chromatid attachment by delaying cell division. Delayed cell division can be a signal that the cell should undergo self-destruction. The signs and symptoms of Roberts syndrome may result from the loss of cells from various tissues during early development. Because both mildly and severely affected individuals lack any functional ESCO2 protein, the underlying cause of the variation in disease severity remains unknown. Researchers suspect that other genetic and environmental factors may be involved 5, 3.
Preimplantation genetic testing are important in the management of Roberts syndrome 2. This has become imperative because the parents of an affected child are obligate heterozygotes (i.e. presumed to be carriers of one ESCO2 pathogenic variant based on family history). Molecular genetic testing is recommended for the parents of a proband to confirm that each parent is heterozygous for an ESCO2 pathogenic variant and allow reliable recurrence risk assessment 2.
Roberts syndrome inheritance pattern
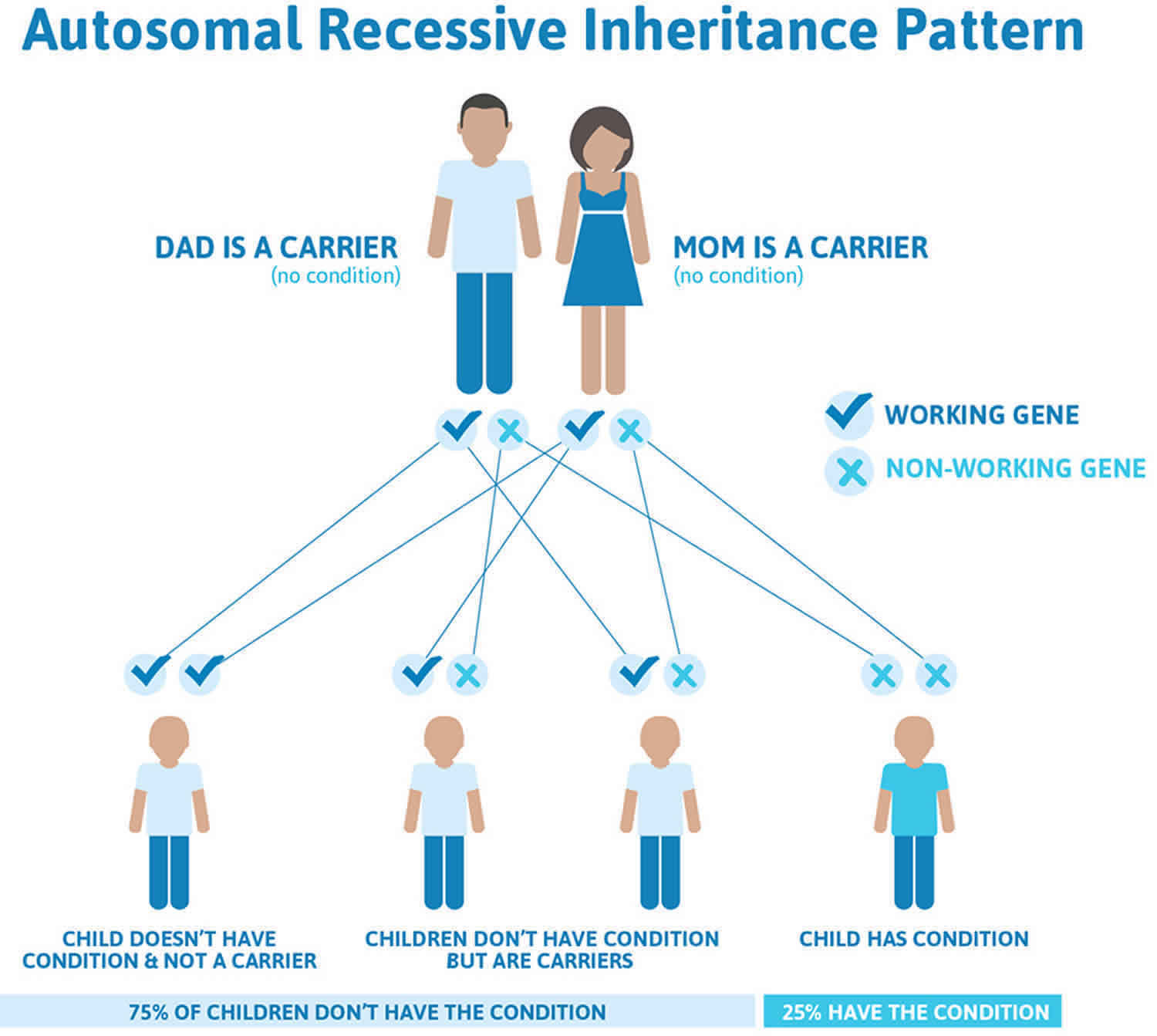
Roberts syndrome is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition. Although a new (de novo) mutations has not been reported in ESCO2 spectrum disorder to date, de novo mutations are known to occur at a low but appreciable rate in autosomal recessive disorders. Heterozygotes (carriers) are asymptomatic and are not at risk of developing the disorder.
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Figure 2. Roberts syndrome autosomal recessive inheritance pattern
Roberts syndrome symptoms
Individuals with Roberts syndrome grow slowly before and after birth. Mild to severe intellectual impairment occurs in about half of all people with Roberts syndrome.
Children with Roberts syndrome are born with abnormalities of all four limbs. They have shortened arm and leg bones (hypomelia), particularly the bones in their forearms and lower legs. In severe cases, the limbs may be so short that the hands and feet are located very close to the body (phocomelia). People with Roberts syndrome may also have abnormal or missing fingers and toes, and joint deformities (contractures) commonly occur at the elbows and knees. The limb abnormalities are very similar on the right and left sides of the body, but arms are usually more severely affected than legs.
Individuals with Roberts syndrome typically have numerous facial abnormalities, including an opening in the lip (a cleft lip) with or without an opening in the roof of the mouth (cleft palate), a small chin (micrognathia), ear abnormalities, wide-set eyes (hypertelorism), outer corners of the eyes that point downward (down-slanting palpebral fissures), small nostrils, and a beaked nose. They may have a small head size (microcephaly) or clouding of the clear front covering of the eyes (corneal opacities). In severe cases affected individuals have a sac-like protrusion of the brain (encephalocele) at the front of their head. In addition, people with Roberts syndrome may have heart, kidney, and genital abnormalities.
Children with Roberts syndrome have sparse hair, silvery blonde scalp hair.
Occasionally, Roberts syndrome patients have third cranial nerve paralysis related to cavernous angioma 44; additional extracranial neoplasms have been reported, 1 case with malignant melanoma, 1 with rhabdomyosarcoma 45, 46, 47.
Moyamoya disease (2%) and stroke secondary to intracranial aneurysms (4%) occurred during late adolescence and young adulthood 48, 43.
Infants with a severe form of Roberts syndrome are often stillborn or die shortly after birth. Mildly affected individuals may live into adulthood.
Table 1. Roberts syndrome clinical features
| Clinical Feature | Proportion of Individuals with Clinical Feature | Comment |
|---|---|---|
| Growth deficiency | 49/49 (100%) | Growth restriction of prenatal onset is the most consistent finding in all affected individuals. Postnatal growth restriction can be moderate to severe and correlates with the severity of the limb and craniofacial malformations. |
| Microcephaly | 38/40 (95%) | |
| Phocomelia | 53/53 (100%) | Upper limbs only (21%) Upper & lower limbs (79%) |
| Bone fusions | 15/23 (65%) | Knees, ankles, wrists, elbows, talipes equinovarus, syndactyly (18%) Flexion contractures (16%) |
| Cleft lip & palate | 29/48 (56%) | Cleft palate only (5%) |
| Developmental delay / intellectual disability | 23/38 (61%) | Intellectual disability is present in the majority of affected individuals; normal intellectual and social development have also been reported 49, 50, 51, 52 |
| Eye abnormalities | (37%) | Corneal opacities (33%) Microphthalmia (8%) Nystagmus (8%) Glaucoma (8%) |
| Urogenital anomalies | 14/30 (48%) | Cryptorchidism (22%) Enlarged phallus (33%) Enlarged clitoris (45%) |
| Kindey anomalies | 1/8 (12%) | Polycystic kidney (2%), horseshoe kidney, hydronephrosis (2%) |
| Heart anomalies | 10/34 (29%) | Atrial septal defect (ASD), ventricular septal defect (VSD), patent ductus arteriosus (PDA) |
Roberts syndrome diagnosis
The diagnosis of Roberts syndrome can be made clinically in individuals with characteristic intrauterine growth restriction, limb malformation, and craniofacial abnormalities 11, 36. However, the gold standard for diagnosis is cytogenetic testing 37.
Roberts syndrome should be suspected in an individual with the following clinical findings and family history.
- Prenatal growth restriction ranging from mild to severe. Mean birth length and weight is below the third centile in most term and prematurely born affected infants.
- Limb malformations including bilateral symmetric tetraphocomelia or hypomelia caused by mesomelic shortening. Upper limbs are more severely affected than lower limbs.
- Hand abnormalities. Most commonly oligodactyly with thumb aplasia or hypoplasia, followed by fifth finger clinodactyly or hypoplasia
- Flexion contractures of the knees, ankles, wrists, and elbows; talipes equinovarus
- Craniofacial abnormalities including bilateral cleft lip and/or palate, micrognathia, widely spaced eyes, exophthalmos, downslanted palpebral fissures, malar flattening, underdeveloped ala nasi, and ear malformation
- Developmental delay / intellectual disability is not always present, segregates by families, and probably correlates with the presence of corneal opacities. There are families with severe phocomelia and bilateral cleft lip and palate with no corneal opacities and no intellectual disability. When present, intellectual disability ranges from mild to severe.
- Urogenital abnormalities: cryptorchidism, enlarged phallus
- Kidney anomalies: horseshoe kidney, polycystic kidney
- Heart defects: ventricular septal defect
- Eye abnormalities: corneal opacities
- Sparse hair
Regular antenatal ultrasounds play a significant role in preventing Roberts syndrome, especially in consanguineous couples. An experienced sonographer can suspect Roberts syndrome with prenatal ultrasound where structural defects can be well defined 11, 36, 38, 39. Postnatal cytogenetic testing of the biopsied tissue confirms the diagnosis 2, 40. In low-income countries, clinical diagnosis can be made where genetic diagnosis is not feasible 2.
Cytogenetic preparations are stained by Giemsa or C-banding techniques, which show premature centromere separation (PCS) and their centromeres separate during metaphase rather than anaphase. The second chromosomal abnormality is heterochromatin repulsion and chromosome with heterochromatin repulsion experience separation of heterochromatin regions during metaphase. Both abnormalities give the chromosome “a railroad track” appearance 23, 33, 34, 35. Other findings which may be found on the chromosome in Roberts syndrome include aneuploidy, micronucleation, and multilobulated nuclei 55.
Family history consistent with autosomal recessive inheritance, including consanguinity (a union between two individuals who are related as second cousins or closer) 40.
Roberts syndrome treatment
Roberts syndrome treatment is based on the affected individual’s symptoms and specific needs with the involvement of a multidisciplinary team of plastic surgeon, pediatrician, orthopedics, physiotherapist, speech therapist, cardiologist, nephrologist, eye specialist, social workers, and genetic counselors 42, 6. The aim of treatment in mild forms is to improve the quality of life of children affected with Roberts syndrome. The possible corrections include correction of cleft lip and palate, surgical correction of limb abnormalities, and management of cognitive disabilities 52, 40.
Roberts syndrome is characterized by growth retardation (mild to severe), craniofacial anomalies (microcephaly, dolichocephaly, cleft palate, micrognathia, premaxillary prominence, microbrachycephaly, downslanted palpebral fissures, widely spaced eyes, exophthalmos, corneal clouding, ear malformations), limb malformations (bi-/tetra-phocomelia, hypomelia, oligodactyly with thumb aplasia, syndactyly, clinodactyly), more severe to the upper limbs 56, 57, 58, 59, 60, 40.
- Gross motor dysfunction. Physical therapy is recommended to maximize mobility and to reduce the risk for later-onset orthopedic complications (e.g., contractures, scoliosis, hip dislocation). Consider use of durable medical equipment and positioning devices as needed (e.g., wheelchairs, walkers, bath chairs, orthotics, adaptive strollers).
- Fine motor dysfunction. Occupational therapy is recommended for difficulty with fine motor skills that affect adaptive function such as feeding, grooming, dressing, and writing.
- Communication issues. Consider evaluation for alternative means of communication (e.g., augmentative and alternative communication [AAC]) for individuals who have expressive language difficulties. An augmentative and alternative communication evaluation can be completed by a speech-language pathologist who has expertise in the area. The evaluation will consider cognitive abilities and sensory impairments to determine the most appropriate form of communication. AAC devices can range from low-tech, such as picture exchange communication, to high-tech, such as voice-generating devices. Contrary to popular belief, AAC devices do not hinder verbal development of speech, but rather support optimal speech and language development.
- Social/Behavioral Concerns. Consultation with a developmental pediatrician may be helpful in guiding parents through appropriate behavior management strategies or providing prescription medications, when necessary. Concerns about serious aggressive or destructive behavior can be addressed by a pediatric psychiatrist.
Table 2. Roberts syndrome treatment
| Symptoms / Concern | Treatment | Considerations/Other |
|---|---|---|
| Poor weight gain / Weight-faltering | Assessment of caloric intake; high-calorie foods/formulas &/or supplementation via nasogastric or enteral feeding may be necessary. | |
| Limb malformations | Multidisciplinary neuromuscular clinic physical medicine, physical therapy or occupational therapy | Maximize gross motor & fine motor skills through physical therapy or occupational therapy & use of adaptive devices; prostheses as indicated. |
| Orthopedics | Consider: physical therapy to improve range of motion; Stretching, night splints, serial casts, surgery. Avoid periods of prolonged immobilization (e.g., following surgery). | |
| Hand malformations | Hand surgeon | Hand surgery as needed for early & proper development of prehensile grasp |
| Cleft palate | Specialized cleft bottles; timing & type of surgical repair determined by craniofacial team 1 | If micrognathia also present, may be evaluated for Pierre Robin sequence |
| Developmental delay / intellectual disability | The following information represents typical management recommendations for individuals with developmental delay / intellectual disability in the United States; standard recommendations may vary from country to country. Ages 0-3 years: Referral to an early intervention program is recommended for access to occupational, physical, speech, and feeding therapy as well as infant mental health services, special educators, and sensory impairment specialists. In the US, early intervention is a federally funded program available in all states that provides in-home services to target individual therapy needs. Ages 3-5 years: In the US, developmental preschool through the local public school district is recommended. Before placement, an evaluation is made to determine needed services and therapies and an individualized education plan (IEP) is developed for those who qualify based on established motor, language, social, or cognitive delay. The early intervention program typically assists with this transition. Developmental preschool is center based; for children too medically unstable to attend, home-based services are provided. All ages: Consultation with a developmental pediatrician is recommended to ensure the involvement of appropriate community, state, and educational agencies (US) and to support parents in maximizing quality of life. | |
| Speech issues | Assessment by speech language pathologist as part of multidisciplinary craniofacial team | Interventions depend on etiology of speech issues & may incl speech therapy, surgery, &/or use of assistive communication devices. |
| Urogenital abnormalities | Per treating nephrologist/urologist | |
| Renal anomalies | ||
| Congenital heart defects | Per treating cardiologist | |
| Eye abnormalities | Per treating ophthalmologist | |
| Stroke | Per treating neurologist | |
| Family/ Community | Ensure appropriate social work involvement to connect families w/local resources, respite, & support. Coordinate care to manage multiple subspecialty appointments, equipment, medications, & supplies. | Ongoing assessment of need for palliative care involvement &/or home nursing Consider involvement in adaptive sports or Special Olympics. |
Table 3. Roberts syndrome recommended surveillance
| Concern | Evaluation | Frequency |
|---|---|---|
| Growth | Primary care provider: monitor growth incl weight, height, & head circumference, blood pressure, risk of infection. | At every visit |
| Development | Monitor developmental progress & educational needs, mobility & self-care needs. Assess for household barriers to mobility/self-help. | At every visit |
| Limb malformations | Physical medicine, occupational therapy / physical therapy assessment of mobility, self-help skills, and need for architectural modifications at home | At every visit |
| Cleft lip/palate | Multidisciplinary craniofacial team: Assess equipment & techniques for feeding infants w/cleft palate. Determine surgical repair timing & type of procedure. Audiologic evaluation as needed | In infants: visit frequency determined by feeding & respiratory issues In children: varies depending on comorbidities; at least annually |
| Speech | Speech assessment by speech language pathologist familiar w/cleft palate & neuromuscular contributors to speech issues Consider speech therapy, surgical interventions, & augmentative communication devices. | At least annually |
| Renal anomalies | Renal function test, blood pressure measurement | Annually |
| Congenital heart defects | Per treating cardiologist / cardiac surgeon | Per treating cardiologist / cardiac surgeon |
| Eye abnormalities | Visual acuity | Per treating ophthalmologist |
| Malignancies | Skin, musculoskeletal, & intracranial | Annually |
| Stroke | Aneurysms, vascular malformation, & intracranial neoplasms | Starting in adolescence |
| Infection risk | Ig serum level | During infancy |
| Family/ Community | Architectural adaptations at home based on level of physical & intellectual disability | According to developmental & physical needs |
Roberts syndrome prognosis
Roberts syndrome prognosis depends on the severity of structural malformations and involvement of other organs 40. Infants with a severe form of Roberts syndrome are often stillborn or die shortly after birth. Mildly affected individuals may live into adulthood 61, 62. Roberts-SC phocomelia represents the milder form of Roberts syndrome and typically survive to adulthood 61. The cause of death has not been reported for most affected individuals. The majority of infants with Roberts syndrome die due to heart, kidney abnormalities and neonatal infective complications 2. Reported causes of death are infection (5 individuals), aneurysm/hemorrhage (3 individuals), and cancer (3 individuals) 8, 43. Perinatal mortality is correlated with severity of the malformations; early death is usually secondary to respiratory complications and infection 10.
- Roberts J. A child with double cleft of lip and palate, protrusion of the intermixillary portion of the upper jaw and imperfect development of the bones of the four extremitis. Ann Surg. 1919;70:252–254.[↩]
- Okpala BC, Echendu ST, Ikechebelu JI, Eleje GU, Joe-Ikechebelu NN, Nwajiaku LA, Nwachukwu CE, Igbodike EP, Nnoruka MC, Okpala AN, Ofojebe CJ, Umeononihu OS. Roberts syndrome with tetraphocomelia: A case report and literature review. SAGE Open Med Case Rep. 2022 Apr 21;10:2050313X221094077. doi: 10.1177/2050313X221094077[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Mfarej MG, Skibbens RV. An ever-changing landscape in Roberts syndrome biology: Implications for macromolecular damage. PLoS Genet. 2020 Dec 31;16(12):e1009219. doi: 10.1371/journal.pgen.1009219[↩][↩]
- Zhou J, Yang X, Jin X, Jia Z, Lu H, Qi Z. Long-term survival after corrective surgeries in two patients with severe deformities due to Roberts syndrome: A Case report and review of the literature. Exp Ther Med. 2018 Feb;15(2):1702-1711. doi: 10.3892/etm.2017.5592[↩][↩]
- Roberts syndrome. https://medlineplus.gov/genetics/condition/roberts-syndrome[↩][↩]
- Gordillo M, Vega H, Jabs EW. Roberts syndrome Gene Reviews, 1993, University of Washington; Seattle WA.[↩][↩][↩][↩]
- Freeman MV, Williams DW, Schimke RN, Temtamy SA, Vachier E, German J. The Roberts syndrome. Clin Genet. 1974 Jan;5(1):1-16. doi: 10.1111/j.1399-0004.1974.tb01652.x[↩]
- Herrmann J, Opitz JM. The SC phocomelia and the Roberts syndrome: nosologic aspects. Eur J Pediatr. 1977 Jun 1;125(2):117-34. doi: 10.1007/BF00489985[↩][↩]
- Waldenmaier C, Aldenhoff P, Klemm T. The Roberts’ syndrome. Hum Genet. 1978 Feb 16;40(3):345-9. doi: 10.1007/BF00272196[↩]
- Van Den Berg DJ, Francke U. Roberts syndrome: a review of 100 cases and a new rating system for severity. Am J Med Genet. 1993 Nov 15;47(7):1104-23. doi: 10.1002/ajmg.1320470735[↩][↩][↩]
- Ayaz R, Göktaş E, Balasar M. A case of Roberts syndrome: its ultrasonographic characteristics and genetic diagnosis. Perinatal J 2020; 28(3): 212–216.[↩][↩][↩][↩][↩]
- Borjas-Lucio CG, Briones-Bernal BL, Gutiérrez HT, et al. Roberts syndrome—an isolated case. Medicina Universitaria 2017; 19(75): 98–99.[↩]
- Urban M, Rogalla P, Tinschert S, Krietsch P. Tetraphocomelia and bilateral cleft lip in a historical case of Roberts syndrome [Virchow, 1898]. Am J Med Genet. 1997 Oct 31;72(3):307-14. doi: 10.1002/(sici)1096-8628(19971031)72:3<307::aid-ajmg11>3.0.co;2-x[↩]
- Bates AW. Autopsy on a case of Roberts syndrome reported in 1672: the earliest description? Am J Med Genet A. 2003 Feb 15;117A(1):92-6. doi: 10.1002/ajmg.a.10864[↩]
- Robins DB, Ladda RL, Thieme GA, Boal DK, Emanuel BS, Zackai EH. Prenatal detection of Roberts-SC phocomelia syndrome: report of 2 sibs with characteristic manifestations. Am J Med Genet. 1989 Mar;32(3):390-4. doi: 10.1002/ajmg.1320320325[↩]
- Maheshwari A, Kumar P, Dutta S, Narang A. Roberts-SC phocomelia syndrome. Indian J Pediatr. 2001 Jun;68(6):557-9. doi: 10.1007/BF02723253[↩]
- Bates AW. A case of Roberts syndrome described in 1737. J Med Genet. 2001 Aug;38(8):565-7. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1734913/pdf/v038p00565.pdf[↩]
- Roberts JB. A child with double cleft of lip and palate, protrusion of the intermaxillary portion of the upper jab and imperfect development of the bones of the four extremeties. Ann Surg. 1919;70:252–3.[↩]
- Jurenka SB. Variations in IQ of two patients with pseudothalidomide syndrome. Dev Med Child Neurol. 1976 Aug;18(4):525-7. doi: 10.1111/j.1469-8749.1976.tb03693.x[↩]
- Herrmann J, Feingold M, Tuffli GA, Opitz JM. A familial dysmorphogenetic syndrome of limb deformities, characteristic facial appearance and associated anomalies: the ‘pseudothalidomide’ or ‘SC-syndrome’. Birth Defects Orig Art Ser. 1969;V:81–89.[↩][↩][↩]
- Ismail S, Essawi M, Sedky N, Hassan H, Fayez A, Helmy N, Shehab M, Farouk D, Elruby M, Otaify G, Eldarsh A, Hosny L, Gaber K, Aboul-Ezz EHA, Ramzy MI, Mehrez MI, Hassib NF, Elhadidi SMA, Aglan MS, Temtamy SA. ROBERTS SYNDROME: CLINICAL AND CYTOGENETIC STUDIES IN 8 EGYPTIAN PATIENTS AND MOLECULAR STUDIES IN 4 PATIENTS WITH GENOTYPE/PHENOTYPE CORRELATION. Genet Couns. 2016;27(3):305-323.[↩]
- Murthy J, Dewan M, Hussain A. Roberts-SC syndrome, a rare syndrome and cleft palate repair. Indian J Plast Surg. 2008 Jul;41(2):222-5. doi: 10.4103/0970-0358.44939[↩]
- Vega H, Waisfisz Q, Gordillo M, Sakai N, Yanagihara I, Yamada M, van Gosliga D, Kayserili H, Xu C, Ozono K, Jabs EW, Inui K, Joenje H. Roberts syndrome is caused by mutations in ESCO2, a human homolog of yeast ECO1 that is essential for the establishment of sister chromatid cohesion. Nat Genet. 2005 May;37(5):468-70. doi: 10.1038/ng1548[↩][↩]
- Olney RS, Hoyme HE, Roche F, Ferguson K, Hintz S, Madan A. Limb/pelvis hypoplasia/aplasia with skull defect (Schinzel phocomelia): distinctive features and prenatal detection. Am J Med Genet. 2001 Nov 1;103(4):295-301.[↩][↩]
- Hou F, Zou H. Two Human Orthologues of Eco1/Ctf7 Acetyltransferases Are Both Required for Proper Sister-Chromatid Cohesion. Mol Biol Cell. 2005;16 (8):3908–18. 10.1091/mbc.e04-12-1063[↩][↩]
- Schüle OA, Johnston K, Pai S, Francke U. Inactivating Mutations in ESCO2 Cause SC Phocomelia and Roberts Syndrome: No Phenotype-Genotype Correlation. Am J Hum Genet. 2005;77:1117–28. 10.1086/498695[↩][↩]
- Gordillo M, Vega H, Trainer AH, Hou F, Sakai N, Luque R, et al. The molecular mechanism underlying Roberts syndrome involves loss of ESCO2 acetyltransferase activity. Hum Mol Genet. 2008;17 (14):2172–80. 10.1093/hmg/ddn116[↩][↩]
- Ivanov D, Schleiffer A, Eisenbacher F, Mechtler K, Haering CH, Nasmyth K. Eco1 Is a Novel Acetyltransferase that Can Acetylate Protein Involved in Cohesion. Curr Biol. 2002;12 (4):323–8. 10.1016/s0960-9822(02)00681-4[↩][↩]
- Ünal E, Heidinger-Pauli JM, Kim W, Guacci V, Onn I, Gygi SP, et al. A Molecular Determinant for the Establishment of Sister Chromatid Cohesion. Science. 2008;321 (5888):566–9. 10.1126/science.1157880[↩][↩]
- Rolef Ben-Shahar T, Heeger S, Lehane C, East P, Flynn H, Skehel M, et al. Eco-dependent cohesin acetylation during establishment of sister chromatid cohesion. Science. 2008;321 (5888):563–6. 10.1126/science.1157774[↩][↩]
- Heindinger-Pauli JM, Ünal E, Koshland D. Distinct Targets of the Eco1 Acetyltransferase Modulate Cohesion in S Phase and in Response to DNA Damage. Mol Cell. 2009;34 (3):311–21. 10.1016/j.molcel.2009.04.008[↩][↩]
- Temtamy SA, Ismail S, Helmy NA. Roberts syndrome: study of 4 new Rgyptian cases with comparison of clinical and cytogenetic findings. Genet Couns. 2006;17(1):1-13.[↩][↩]
- Al Kaissi A, Csepan R, Klaushofer K, Grill F. Femoral-tibial-synostosis in a child with Roberts syndrome (Pseudothalidomide): a case report. Cases J. 2008 Aug 18;1(1):109. doi: 10.1186/1757-1626-1-109[↩][↩][↩]
- Abbas R, Waqar S, Ahmad TM, Irfan Waheed KA, Sultan T, Qureshi AU. A child with Roberts syndrome. J Coll Physicians Surg Pak. 2011 Jul;21(7):431-3.[↩][↩][↩]
- Gordillo M, Vega H, Trainer AH, Hou F, Sakai N, Luque R, Kayserili H, Basaran S, Skovby F, Hennekam RC, Uzielli ML, Schnur RE, Manouvrier S, Chang S, Blair E, Hurst JA, Forzano F, Meins M, Simola KO, Raas-Rothschild A, Schultz RA, McDaniel LD, Ozono K, Inui K, Zou H, Jabs EW. The molecular mechanism underlying Roberts syndrome involves loss of ESCO2 acetyltransferase activity. Hum Mol Genet. 2008 Jul 15;17(14):2172-80. doi: 10.1093/hmg/ddn116[↩][↩][↩][↩][↩]
- Socolov RV, Andreescu NI, Haliciu AM, Gorduza EV, Dumitrache F, Balan RA, Puiu M, Dobrescu MA, Socolov DG. Intrapartum diagnostic of Roberts syndrome – case presentation. Rom J Morphol Embryol. 2015;56(2):585-8. https://rjme.ro/RJME/resources/files/560215585588.pdf[↩][↩][↩][↩]
- Malla TM, Pandith AA, Dar FA, et al. Cytogenetic diagnosis of Roberts SC phocomelia syndrome: first report from Kashmir. Egypt J Med Human Genet 2016; 17: 137–140.[↩][↩]
- Dulnuan DJ, Matsuoka M, Uketa E, Hayashi K, Murotsuki J, Nishimura G, Hata T. Antenatal three-dimensional sonographic features of Roberts syndrome. Arch Gynecol Obstet. 2011 Jul;284(1):241-4. doi: 10.1007/s00404-011-1910-1[↩][↩]
- Paladini D, Palmieri S, Lecora M, Perone L, Di Meglio A, D’Armiento M, Cascioli C, Martinelli P. Prenatal ultrasound diagnosis of Roberts syndrome in a family with negative history. Ultrasound Obstet Gynecol. 1996 Mar;7(3):208-10. doi: 10.1046/j.1469-0705.1996.07030208.x[↩][↩]
- Vega H, Gordillo M, Jabs EW. ESCO2 Spectrum Disorder. 2006 Apr 18 [Updated 2020 Mar 26]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1153[↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Roberts syndrome. https://rarediseases.info.nih.gov/diseases/7387/index[↩]
- Van Den Berg DJ, Francke U. Roberts syndrome: A review of 100 cases and a new rating system for severity. Am J Med Genet. 1993;47:1104–23. 10.1002/ajmg.1320470735[↩][↩][↩]
- Vega H, Trainer AH, Gordillo M, Crosier M, Kayserili H, Skovby F, Uzielli ML, Schnur RE, Manouvrier S, Blair E, Hurst JA, Forzano F, Meins M, Simola KO, Raas-Rothschild A, Hennekam RC, Jabs EW. Phenotypic variability in 49 cases of ESCO2 mutations, including novel missense and codon deletion in the acetyltransferase domain, correlates with ESCO2 expression and establishes the clinical criteria for Roberts syndrome. J Med Genet. 2010 Jan;47(1):30-7. doi: 10.1136/jmg.2009.068395[↩][↩][↩][↩]
- Ogilvy CS, Pakzaban P, Lee JM. Oculomotor nerve cavernous angioma in a patient with Roberts syndrome. Surg Neurol. 1993 Jul;40(1):39-42. doi: 10.1016/0090-3019(93)90168-z[↩]
- Parry DM, Mulvihill JJ, Tsai SE, Kaiser-Kupfer MI, Cowan JM. SC phocomelia syndrome, premature centromere separation, and congenital cranial nerve paralysis in two sisters, one with malignant melanoma. Am J Med Genet. 1986 Aug;24(4):653-72. doi: 10.1002/ajmg.1320240410[↩]
- Wenger SL, Blatt J, Steele MW, Lloyd DA, Bellinger M, Phebus CK, Horn M, Jaffe R. Rhabdomyosarcoma in Roberts syndrome. Cancer Genet Cytogenet. 1988 Apr;31(2):285-9. doi: 10.1016/0165-4608(88)90230-0[↩]
- Feingold M. History of C-patient with SC-Roberts/pseudothalidamide syndrome. Am J Med Genet. 1992 Jul 15;43(5):898-9. doi: 10.1002/ajmg.1320430532[↩]
- Herrmann J, Feingold M, Tuffli GA, Opitz JM. A familial dysmorphogenetic syndrome of limb deformities, characteristic facial appearance and associated anomalies: the ‘pseudothalidomide’ or ‘SC-syndrome’. Birth Defects Orig Art Ser. 1969;5:81–9.[↩]
- Petrinelli P, Antonelli A, Marcucci L, Dallapiccola B. Premature centromere splitting in a presumptive mild form of Roberts syndrome. Hum Genet. 1984;66(1):96-9. doi: 10.1007/BF00275195[↩]
- Stanley WS, Pai GS, Horger EO 3rd, Yan YS, McNeal KS. Incidental detection of premature centromere separation in amniocytes associated with a mild form of Roberts syndrome. Prenat Diagn. 1988 Oct;8(8):565-9. doi: 10.1002/pd.1970080803[↩]
- Maserati E, Pasquali F, Zuffardi O, Buttitta P, Cuoco C, Defant G, Gimelli G, Fraccaro M. Roberts syndrome: phenotypic variation, cytogenetic definition and heterozygote detection. Ann Genet. 1991;34(3-4):239-46.[↩]
- Holden KR, Jabs EW, Sponseller PD. Roberts/pseudothalidomide syndrome and normal intelligence: approaches to diagnosis and management. Dev Med Child Neurol. 1992 Jun;34(6):534-9. doi: 10.1111/j.1469-8749.1992.tb11475.x[↩][↩]
- Hennekam RCM, Krantz ID, Allanson JE. Gorlin’s Syndromes of the Head and Neck. 5 ed. New York, NY: Oxford University Press; 2010:999-1003.[↩]
- Vega H, Gordillo M, Jabs EW. Inborn Errors of Development. 3 ed. New York, NY: Oxford University Press; 2016:1001-4.[↩]
- Jabs EW, Tuck-Muller CM, Cusano R, Rattner JB. Studies of mitotic and centromeric abnormalities in Roberts syndrome: implications for a defect in the mitotic mechanism. Chromosoma. 1991 May;100(4):251-61. doi: 10.1007/BF00344159[↩]
- O’Brien HR, Mustard HS. An adult living case of total phocomelia. J Am Med Assoc. 1921;77:1964–1967. doi: 10.1001/jama.1921.02630510030009[↩]
- Freeman MV, Williams DW, Schimke RN, Temtamy SA, Vachier E, German J. The Roberts syndrome. Clin Genet. 1974;5:1–16. doi: 10.1111/j.1399-0004.1974.tb01652.x[↩]
- Leonard P, Rendle-Short J, Skardoon L. Roberts’-SC phocomelia syndrome with cytogenetic findings. Hum Genet. 1982;60:379–380. doi: 10.1007/BF00569225[↩]
- Stanley WS, Pai GS, Horger EO, III, Yan YS, McNeal KS. Incidental detection of premature centromere separation in amniocytrs associated with a mild form of Roberts syndrome. Prenat Diagn. 1988;8:565–569. doi: 10.1002/pd.1970080803[↩]
- Judge C. A sibship with the pseudothalidomide syndrome and an association with Rh incompatibility. Med J Aust. 1973 Aug 11;2(6):280-1. doi: 10.5694/j.1326-5377.1973.tb128828.x[↩]
- Goh ES, Li C, Horsburgh S, Kasai Y, Kolomietz E, Morel CF. The Roberts syndrome/SC phocomelia spectrum–a case report of an adult with review of the literature. Am J Med Genet A. 2010 Feb;152A(2):472-8. doi: 10.1002/ajmg.a.33261[↩][↩]
- Sezer A, Kayhan G, Zenker M, Percin EF. Hypopigmented patches in Roberts/SC phocomelia syndrome occur via aneuploidy susceptibility. Eur J Med Genet. 2019 Dec;62(12):103608. doi: 10.1016/j.ejmg.2018.12.013[↩]




{kind=link}