Contents
What is aniridia
Aniridia is a rare sight-threatening genetic eye disorder in which there is complete or partial absence of the colored part of the eye (the iris) that is associated with other ocular features, some present from birth (congenital aniridia) and some arising progressively over time 1. These iris abnormalities may cause the pupils to be abnormal or misshapen. Aniridia can cause reduction in the sharpness of vision (visual acuity) and increased sensitivity to light (photophobia).
People with aniridia can also have other eye problems. Increased pressure in the eye (glaucoma) typically appears in late childhood or early adolescence. Clouding of the lens of the eye (cataracts), occur in 50 percent to 85 percent of people with aniridia. In about 10 percent of affected people, the structures that carry information from the eyes to the brain (optic nerves) are underdeveloped. Individuals with aniridia may also have involuntary eye movements (nystagmus) or underdevelopment of the region at the back of the eye responsible for sharp central vision (foveal hypoplasia). Many of these eye problems contribute to progressive vision loss in affected individuals. The severity of symptoms is typically the same in both eyes. The phenotype may vary between and within families; however, affected individuals usually show little inter-ocular difference.
Rarely, people with aniridia have behavioral problems, developmental delay, and problems detecting odors.
Most cases are diagnosed at birth with an obvious iris/pupillary abnormality or in infancy with nystagmus (usually apparent by six weeks of age). Photophobia may also be present. Slit-lamp examination commonly discloses small anterior polar cataracts, sometimes with persistent pupillary membrane strands attached.
The major diagnostic feature of aniridia is congenital partial or complete hypoplasia of the iris; foveal hypoplasia with reduced visual acuity is almost always present and associates with early onset nystagmus. Other frequently associated ocular abnormalities, generally with later onset, include cataract, glaucoma and corneal opacification and vascularization secondary to limbal stem cell deficiency 2.
Despite their many ocular problems, most individuals with aniridia retain useful vision with appropriate ophthalmologic management.
Congenital aniridia is a rare disease which affects both eyes. Aniridia occurs in 1 in 50,000 to 100,000 newborns worldwide. In the vast majority of congenital aniridia cases is autosomal dominant inherited mutations or deletions of the paired box gene-6 (PAX6). Most of the other cases of congenital aniridia are sporadic. Sporadic aniridia may correlate with WAGR syndrome (Wilm’s tumor, aniridia, genitourinary anomalies, and mental retardation) in which the adjacent PAX6 and Wilms tumor (WT1) genes are both deleted 3. A minority of congenital aniridia may be transmitted autosomal recessively called as the Gillespie syndrome (aniridia, cerebellar ataxia, and mental retardation) 3.
Acquired aniridia occurs after trauma or ocular surgery.
Figure 1. Aniridia (Congenital aniridia, left eye. Initial examination shows a large, dark, and dilated pupil that does not react to light)
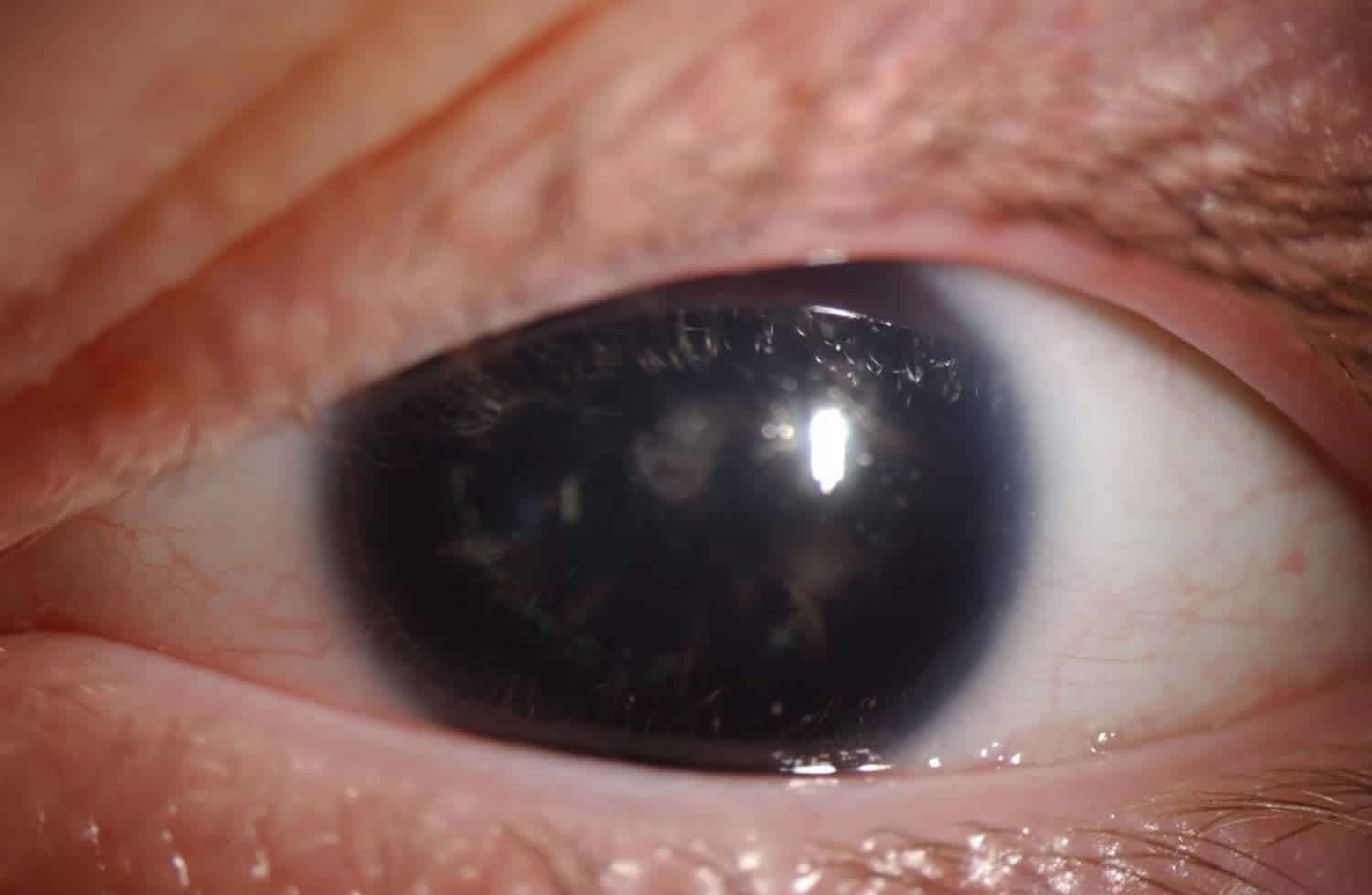
Figure 2. Partial aniridia
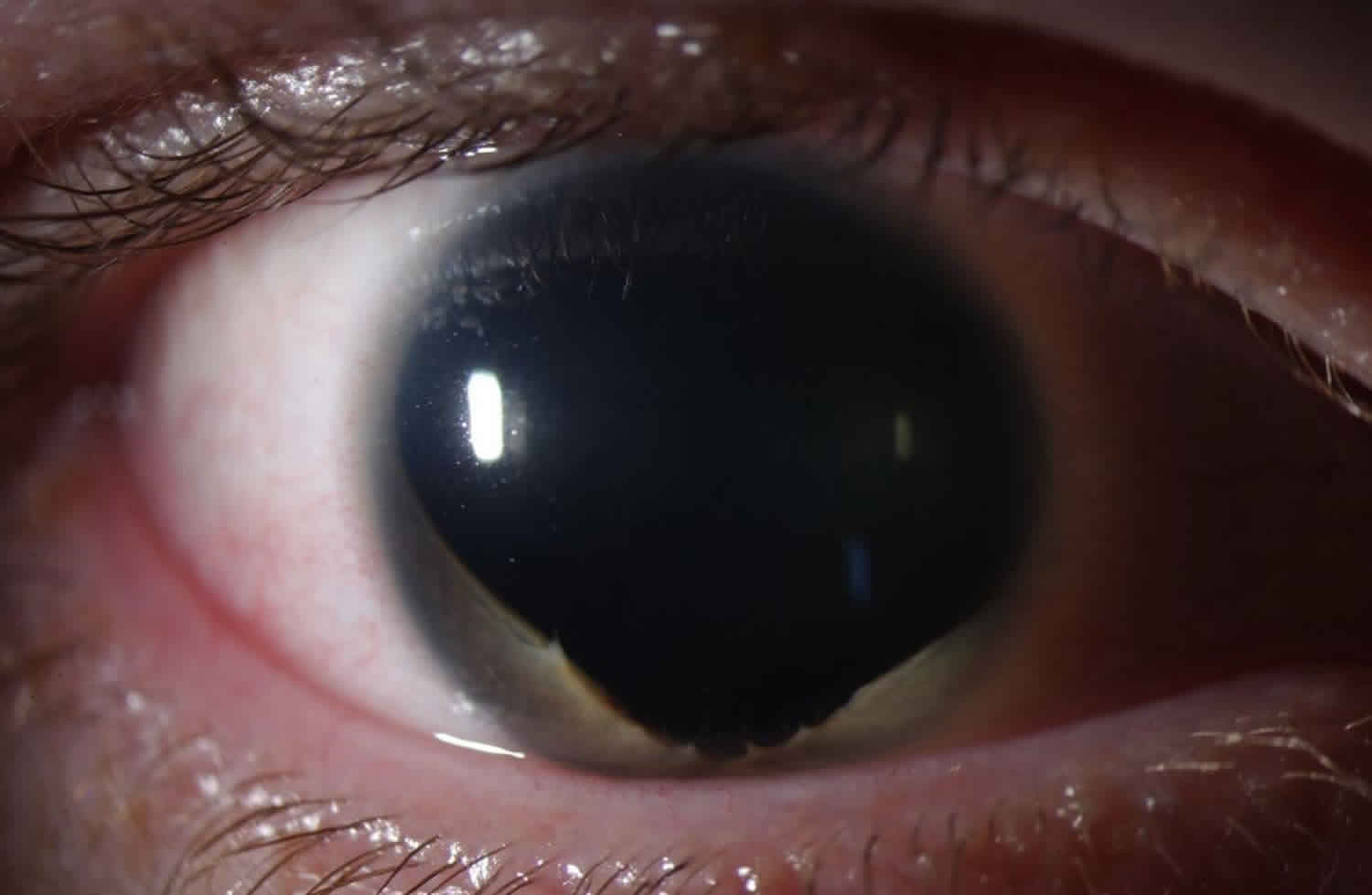
Footnote: Aniridia can also present as partial absence of the iris as demonstrated here, with residual iris on the inferior margin. Of note, there is almost always residual iris in all cases of aniridia, but the iris might be so small as only to be visible with careful exam or gonioscopy.
Aniridia causes
The exact cause of congenital aniridia is unknown. However, the paired box gene (PAX6) gene mutations at chromosome 11p13 is a crucial factor in pathogenesis. The PAX6 gene provides instructions for making a protein that is involved in the early development of the eyes, brain and spinal cord (central nervous system), and the pancreas. Within the brain, the PAX6 protein is involved in the development of a specialized group of brain cells that process smell (the olfactory bulb). The PAX6 protein attaches (binds) to specific regions of DNA and regulates the activity of other genes. On the basis of this role, the PAX6 protein is called a transcription factor. Following birth, the PAX6 protein regulates several genes that likely contribute to the maintenance of different eye structures.
Mutations in the PAX6 gene result in the production of a nonfunctional PAX6 protein that is unable to bind to DNA and regulate the activity of other genes. A lack of functional PAX6 protein disrupts the formation of the eyes during embryonic development.
Three hundred PAX6 mutations have been identified; 286 are associated with congenital eye malformations; 257 of them cause aniridia and the reminder 29 cause related ocular phenotypes such as Peters anomaly, foveal hypoplasia, and optic nerve anomalies 1:
The Aniridia phenotype may be the result of:
- Nonsense mutations (39%)
- Splice mutations (13%)
- Frameshift deletions and insertions (25%)
- Inframe insertions and deletions (6%)
- Missense mutations (12%)
- Run-on mutations (5%)
- Most lead to loss of protein function.
- In the non-aniridia eye disorders, 69% are missense mutations.
In the Aniridic phenotype, 94% of all intragenic point mutations lead to the introduction of a premature termination codon (PTC), or to C-terminal extensions (CTE), or aminoacid substitutions (missense mutations).
Premature termination codon (PTC) mutations comprise nonsense mutations, frame-shifting insertions and deletions and most splice mutations. C-terminal extensions (CTE) mutations cause the open reading frame to continue into the 3’ untranslated region. Missense mutations produce a reduced-function protein, resulting in the variant ocular phenotypes or (if protein function is greatly reduced) in aniridia.
Various genes implicated in the pathogenesis of congenital aniridia include:
- Aniridia 1 (AN1) – This results from heterozygous mutation of the PAX6 gene at chromosome 11p13. It is inherited autosomal dominantly, and also includes late-onset corneal dystrophy and congenital cataracts. Other associations include Peters anomaly, reduced smell sensation, hypoplasia of corpus callosum and/or anterior commissure, and the absence of the pineal gland.
- Aniridia 2 (AN2) (autosomal dominant) results from mutation of a cis-regulatory element of the PAX6 gene. This regulatory region is at the intron of the adjacent ELP4 gene (11p13).
- Aniridia 3 (AN3) (autosomal dominant) is due to a mutation in the TRIM44 gene (11p13).
- Gillespie syndrome occurs due to a mutation of the ITPR1 gene at chromosome 3p26.1.
Research has suggested a probable linkage of autosomal dominant aniridia and acid phosphatase 1 (ACP1) at chromosome 2p (short arm of chromosome 2) 4.
Aniridia inheritance pattern
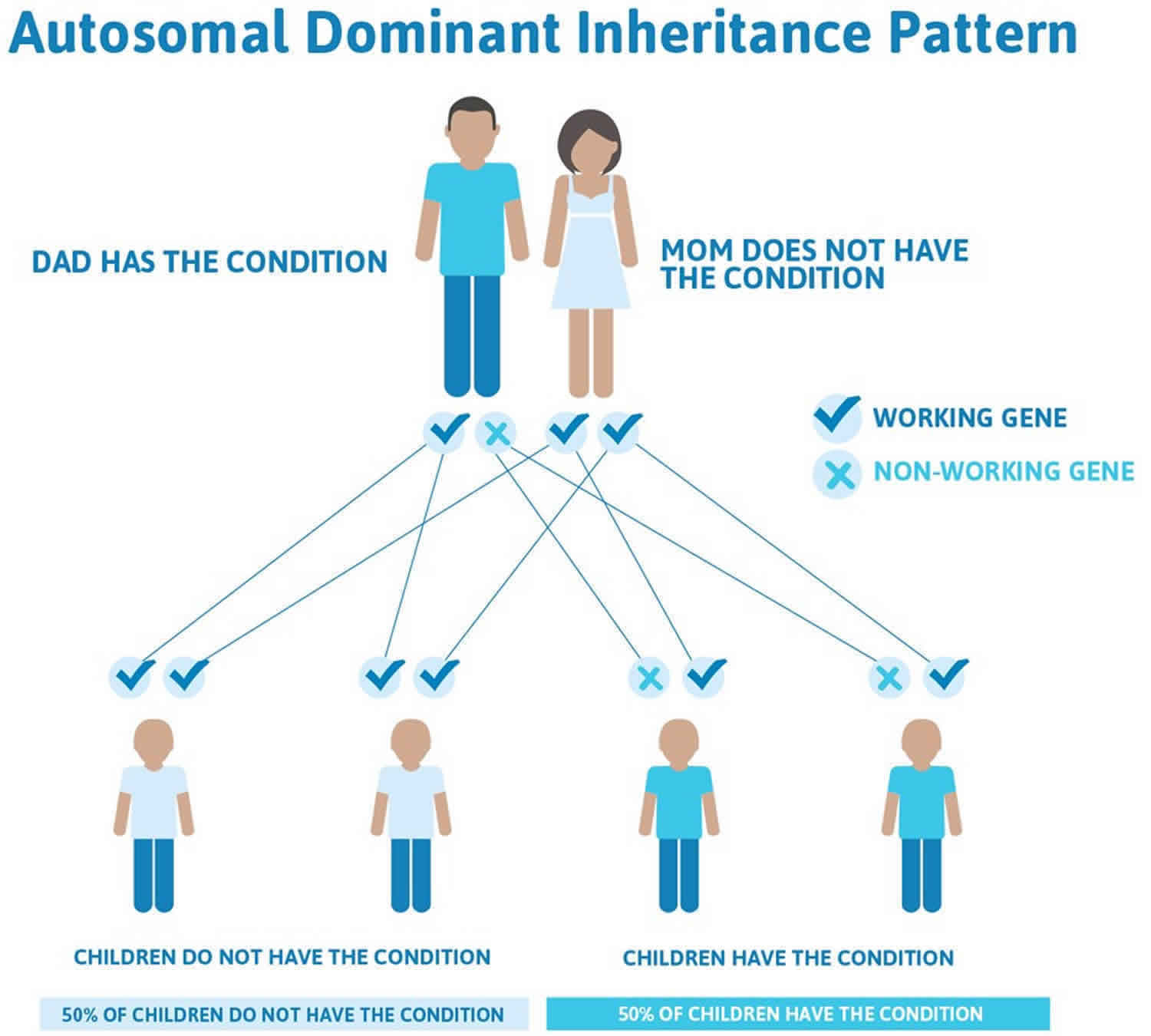
The autosomal dominant mode of inheritance, which means one copy of the altered gene in each cell is sufficient to cause the disorder, accounts for two-thirds of aniridia cases and has no systemic implications. Penetrance is complete but expressivity is variable 1. It is caused by mutation in the PAX6 gene on chromosome 11p13 or deletion of the regulatory regions that control its expression.
In cases where the autosomal dominant condition does run in the family, the chance for an affected person to have a child with the same condition is 50% regardless of whether it is a boy or a girl. These possible outcomes occur randomly. The chance remains the same in every pregnancy and is the same for boys and girls.
- When one parent has the abnormal gene, they will pass on either their normal gene or their abnormal gene to their child. Each of their children therefore has a 50% (1 in 2) chance of inheriting the changed gene and being affected by the condition.
- There is also a 50% (1 in 2) chance that a child will inherit the normal copy of the gene. If this happens the child will not be affected by the disorder and cannot pass it on to any of his or her children.
The remaining one-third of cases result from new mutations (sporadic aniridia) in the gene and occur in people with no history of the disorder in their family 5. The sporadic form occurs due to de novo deletions on chromosome 11p13 involving the PAX6 gene. Larger deletions affecting the adjacent WT1 (Wilms tumor) gene are the underlying cause of the WAGR syndrome (Wilms tumor, Aniridia, Genitourinary anomalies, and mental Retardation) 6.
Twenty five to thirty percent of patients with sporadic aniridia develop Wilms’ tumor. According to Gronskov et al 7 patients with sporadic aniridia had a relative risk of 67 (CI, 8.1-241) of developing Wilms tumor and if there is contiguous gene deletion of PAX6 and WT1 patients have up to 50% risk of developing this tumor.
Autosomal recessive aniridia accounts for approximately 2% of all cases. It is associated with cerebellar ataxia and mental retardation (Gillespie syndrome) 7. Nelson et al 8 considered that the specific iris abnormality characterized by circumpupillary aplasia leading to a fixed dilated pupil is pathognomonic for Gillespie syndrome. In fact, it may help distinguish from other forms of aniridia and a presumptive diagnosis of Gillespie syndrome can be made during the first months of life. Owing to its rarity, the inheritance pattern and molecular basis of this syndrome are still unclear. However, at least one form may be caused by a heterozygous PAX6 mutation.
Figure 3. Aniridia autosomal dominant inheritance pattern (accounts for two-thirds of aniridia cases)
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Congenital aniridia classification
There are three recognized clinical phenotypes of aniridia:
- Isolated aniridia without systemic involvement: 85% of cases are not associated with overt systemic abnormalities, although deep phenotyping can reveal reduced olfaction and brain MRI abnormalities.
- WAGR Syndrome: 13% of cases have the WAGR association of Wilms tumor (nephroblastoma), Aniridia, Genitourinary abnormalities, and mental Retardation. Some of these patients have other malformations, such as hemihypertrophy. Ambiguous genitalia, mental and growth retardation, and multiple minor anomalies (anomalies of the pinna, inguinal and umbilical hernias, microcephaly) are part of the WAGR association. 38 % of WAGR patients develop nephropathy and renal failure in their lifetime.
- Gilliespie Syndrome: 2% of cases have Gillespie syndrome of aniridia, developmental delay, and cerebellar ataxia.
There are congenital ocular findings, including bilateral, total, or partial absence of the iris.- Aniridia is a misnomer, because careful gonioscopic examination of all patients, even those in whom no iris is visible, results in finding some iris tissue. More often than not, some iris tissue is visible. It is important to distinguish aniridia from the anomalous iris that results from mutations in ACTA2. Patients with the mutations in the latter gene have other congenital malformations that involve the heart, aorta, other blood vessels, and smooth muscles.
- Aniridia is a malformation of the whole eyeball, with abnormalities in most ocular tissues. Patients may have microcornea, keratopathy with corneal pannus and corneal opacification resulting from absence of limbal stem cells.
- Cataracts are found in 85% of cases, and dislocated lenses in 35%.
- Secondary glaucoma occurs in up to 50% of patients; glaucoma usually develops after the second decade of life as the peripheral iris stump adheres to the angle, occluding it. Goniodysgenesis is noted in some cases with rare cases of infantile glaucoma.
- Foveal and optic nerve hypoplasia are frequently present.
- Nystagmus is observed in the majority of cases, and photophobia is common.
- Bilateral ptosis has been reported.
- Visual acuity is less than 20/60 in almost all patients and less than 20/200 in more than 60%.
- Rare families, with a very mild phenotype, have been reported in whom the incidence of cataracts and glaucoma is less than the classical, more common form of the disease, and in whom visual acuity is better preserved. It is more likely that missense mutations in PAX6 cause mild phenotypes than nonsense mutations or deletions.
- Some PAX6 mutations can result in partial phenotypic features of classic aniridia, such as keratitis, foveal hypoplasia, or cataract, without evident or prominent absence of iris tissue.
- Some patients with PAX6 mutations may present with Peters anomaly or with isolated cataracts or foveal hypoplasia.
- Mutations in other genes can cause overlapping phenotypes (eg, FOXC1, CYP1B1) that can sometimes be differentiated by the absence of foveal hypoplasia.
Autosomal dominant congenital aniridia
Autosomal dominant congenital aniridia consists of at least 2/3rd cases of congenital aniridia and may be present in up to 85% cases with aniridia. This variant is autosomal dominant and the most common form of aniridia; it has complete penetrance but variable expressivity. Thus family members may have different severity of aniridia, ocular involvement, and visual acuity. Some cases may have subtle iris hypoplasia or iris coloboma.
Sporadic congenital aniridia
There is a de novo mutation of the PAX6 gene and may consist of 13% to 33% of cases with aniridia. An important fact is that this variant of aniridia correlates with nephroblastoma (Wilm’s tumor) as a part of WAGR syndrome (Wilms tumor, aniridia, genitourinary anomalies, and mental retardation), also known as Miller’s syndrome. The genitourinary anomalies include undescended testes in males and streak gonads (nonfunctional ovaries) and/or bicornuate uterus in females. Intellectual disability includes difficulty in learning and processing information. There might be other manifestations including attention deficit hyperactivity disorder, obsessive-compulsive disorder, depression, and anxiety. There may be craniofacial dysmorphism.
WAGR syndrome occurs as the chromosomal location of the PAX6 gene at 11p13 is very close to WT1 (Wilm’s tumor) gene. There is a partial deletion of the short arm of chromosome 11 involving both PAX6 and WT1 in this syndrome. Patients with WAGR syndrome have around 45 to 60% chance of developing nephroblastoma. Approximately 25% to 33% of patients with sporadic congenital aniridia may develop nephroblastoma before 3 years of age 9.
Autosomal recessive congenital aniridia
Autosomal recessive congenital aniridiaconsists of 1% to 3% of the all congenital aniridia cases and is the least common variant of congenital aniridia. It has associations with cerebellar ataxia and mental retardation (Gillespie syndrome).
Aniridia symptoms
The patients usually complain of photophobia related to the severity of aniridia and poor vision (due to many reasons including foveal hypoplasia and optic nerve head hypoplasia) since early childhood. Parents may bring the child for the abnormal appearance of the eyes.
Nystagmus and strabismus are common due to poor vision since childhood.
Other ocular findings include:
- Aniridia– The term aniridia is a misnomer, since a small portion of iris tissue is almost always found on gonioscopic or ultrasound biomicroscopy. The iris may be partially absent (detectable with retroillumination) or there may be an apparent complete absence of iris on external observation. However, most cases have small hypoplastic iris tissue visible on gonioscopy. All cases have some hypoplastic iris tissue on histopathological examination 9. Thus, the term aniridia is a misnomer and ‘iridemia’ may be a better term to describe the disease. Variations range from almost total absence to only mild hypoplasia of the iris. In the less severe cases, a round, normal-appearing pupil may be found. The pupil size may be normal, but there may be loss of the iris surface architecture or the presence of iris transillumination 1. Other iris changes include partial iris defects (resembling a coloboma) or eccentric pupil/misshapen pupils (corectopia) and iris ectropion 8. The iris may be partially absent (detectable with retroillumination) or there may be an apparent complete absence of iris on external observation. However, most cases have small hypoplastic iris tissue visible on gonioscopy. All cases have some hypoplastic iris tissue on histopathological examination 9.
- Bilateral ptosis – Up to 10% pf patients may have bilateral, usually symmetrical ptosis 2.
- Meibomian gland dysfunction
- Aniridia associated keratopathy -Aniridia-associated keratopathy (AAK) is a late and progressive manifestation. There is dry eye and tear film instability. The ocular surface may be very poor and unstable predisposing to epithelial defect and/or infective keratitis. A classification of aniridia associated keratopathy into three stages (mild, moderate, and severe limbal insufficiency) has been suggested 10. It is a significant threat to vision and is thought to have an incidence of 20% 11. Aniridia-associated keratopathy is caused mainly by limbal stem-cell deficiency but also by a combination of other factors such as an abnormal differentiated epithelium, corneal pannus, corneal epithelial irregularity, abnormal cell adhesion, impaired healing response and infiltration of conjunctival cells (conjunctivalization of the cornea). Corneal opacity, corneal scarring (subepithelial fibrosis), and vascularization are seen peripherally and may encroach the central cornea. Corneal portends a bad prognosis after penetrating keratoplasty and predisposes to graft rejection and failure. The trigger event for corneal deterioration may be surgical intervention with excessive manipulation of the limbus, or after the application of topical antimetabolites in order to treat the aniridia-associated glaucoma. Inadequate tear production is common, exacerbating the ocular surface disease. The first signs of aniridia-associated keratopathy appear in the first decade of life, with thickening and vascularization of the peripheral cornea, which gradually advances into the central cornea, ending in pancorneal vascularization, opacification and keratinisation. Central corneal thickness is often increased. Aniridia-associated keratopathy may be caused by a deficiency in matrix metalloproteinase 9 (MMP-9), which is also regulated by PAX6. In PAX6 mutation animal models, MMP-9 deficiency results in the accumulation of fibrin and the infiltration of inflammatory cells. There is also a significant increase in stromal cell apoptosis, which disrupts the orderly arrangement of the collagen fibers of the cornea, results in subsequent loss of transparency.
- Microcornea
- Glaucoma – Ocular Hypertension and Glaucoma are common. The exact prevalence is unknown. Congenital Glaucoma with or without buphthalmos is rare in infants with aniridia; it usually develops in later childhood or adulthood. In the former, the reported incidence is 6% to 75% according to Nelson et al. 8. Glaucoma develops due to angle abnormalities which obstruct the outflow of the aqueous humor through Schlemm’s canal. Margo et al studied the histopathology of seven enucleated eyes from children with aniridia and glaucoma. They reported an abnormal angle development. Grant and Walton analyzed a series of gonioscopic examinations in aniridic patients with glaucoma versus aniridic patients without glaucoma and found that initially the stroma of the iris extends forward onto the trabecular meshwork forming synechiae-like attachments, followed by a more homogeneous sheet, eventually resulting in angle closure. Less frequently, they found that the angle remained open until adolescence and then was closed by the anterior rotation of the entire iris leaflet. The corneal thickness is usually high which might influence the measurement of the intraocular pressure. In the early years of life, the angle of the anterior chamber is generally open on gonioscopy, and congenital glaucoma/buphthalmos is rare in aniridia. However, within 2nd-decade angle closure due to peripheral anterior synechia is noted which may lead to an increase in the intraocular pressure.
- Persistent pupillary membrane
- Rudimentary iris stump on gonioscopy – Gonioscopy may show iris fibers which lie at the space between the rudimentary small hypoplastic iris and the trabecular meshwork. These fibers contract and pull the rudimentary iris forward thereby causing synechial angle closure glaucoma
- Superior subluxation of the lens (clear or cataractous), ectopia lentis (noted in up to 56% of cases), dislocation of the lens 9
- Cataract or opacities of the crystalline lens or its capsule may be present in 50% to 85% of patients 9. Congenital lens opacities (especially polar) are common. Occasionally there is persistent vascularization of the anterior lens capsule (tunica vasculosa lentis) or remnants of pupillary membrane 1. The lens opacities are rarely dense enough to require lens extraction in infancy, but visually significant lens opacities eventually develop in 50%-85% of patients, usually during the first two decades of life 1. Lens subluxation or dislocation occur but are infrequent. Scheider et al found that the anterior capsule of aniridic cataracts is very fragile.
- Microspherophakia
- Optic nerve head hypoplasia is present in 75% of cases in a series of 12 patients with aniridia 12. A reduced disc diameter may confirm the hypoplasia: width of the retinal arteriole at the disc margin of 6.1 to less than 14.6 (normal value 14.6 ± 2.4) 13
- Foveal hypoplasia – Foveal hypoplasia, characterized by the absence of a defined foveal avascular zone, the absence of pigmentation at the fovea, and blood vessels may cross through the area where fovea is presumed to be present. There is an absence of foveal depression, and inner retinal layers are present all over the macular region. In a normal fovea, inner retinal layers are absent, and there is a depression corresponding to the foveola. Pendular horizontal nystagmus is usually present by 6 weeks of age. ERG testing discloses variable retinal dysfunction from nearly normal to severely abnormal. Rods and cones are equally affected. The etiology of this retinal dysfunction remains unclear. Perhaps it may be due to foveal aplasia or hypoplasia, secondary to PAX6 mutation or phototoxicity as a result of iris maldevelopment.
- Fundal coloboma
- Refractive error strabismus -Both are frequent in aniridic patients. According to Nelson et al 8 esotropia is the most frequent deviation encountered. High refractive errors are not uncommon.
- Aniridic fibrosis syndrome – ‘A progressive anterior fibrosis syndrome in patients with postsurgical congenital aniridia’ has been described 14. Usually, there is a history of multiple surgeries including cataract surgery, glaucoma surgery, and keratoplasty. This condition is characterized by retrocorneal and retrolenticular membranes arising from the rudimentary iris which may involve ciliary processes and anterior retina also. This progressive membrane results in anterior displacement of the intraocular lens and corneal decompensation.
Other associations of aniridia include:
- Sensory deficiency including:
- Abnormal olfaction
- Hearing loss 15
- AGR triad (aniridia, genitourinary abnormalities, mental retardation)
- Absent or hypoplastic patella (aniridia and absent patella) in family
Central Nervous System
Patients with isolated aniridia may show reduced olfaction (probably the most common functional deficit) and cognition , behavioral problems, or developmental delay. MRI studies have demonstrated abnormalities of the anterior comissure, anterior cingulated cortex, cerebellum, temporal and occipital lobes, corpus callosum, pineal gland and olfactory bulb 1.
Hearing
Individuals with aniridia may show central auditory processing difficulties (from abnormal interhemispheric transfer), which may cause hearing difficulties.
WAGR syndrome
Children with sporadic aniridia must be investigated to determine if it is caused by a chromosomal rearrangement affecting both the PAX6 and WT1 genes (Wilms tumour, pediatric nephroblatoma predisposition gene). However, chances of Wilms tumor is very very rare in familial aniridia. Classical WAGR syndrome includes Wilms tumor with Aniridia, Genitourinary abnormalities and mental Retardation, but the phenotype is highly variable. The term ‘WAGR syndrome’ is used even in the absence of all four classical features.
- Wilms tumor risk for individuals with a cytogenetically visible deletion of 11p13 or a submicroscopic deletion that involves PAX6 and WT1 is up to 50%. Patients with WAGR, Wilms tumors are more likely to be bilateral, present earlier and to have more favorable tumor histology with better prognosis than those with isolated Wilms tumor.
- Aniridia is the most consistent symptom and is typically severe. Although cases without classical aniridia have been described.
- Genitourinary abnormalities include cryptorchidism (in 60% of males), uterine abnormalities, hypospadias, ambiguous genitalia, streak ovaries, urethral strictures, ureteric abnormalities, and gonadoblastoma.
- Intellectual disability and behavioral abnormalities in WAGR syndrome are highly variable and both may not be present in some patients:
- Seventy percent of individuals with WAGR syndrome have intellectual disability (defined as IQ <74);
- Behavioral abnormalities include attention deficit hyperactivity disorder (ADHD), autism spectrum disorders (anxiety, depression, and obsessive compulsive disorder).
- Neurologic abnormalities occur in up to one-third of individuals with WAGR syndrome. Findings include epilepsy, hypertonia or hypotonia, microcephaly, enlarged ventricles and corpus callosum agenesis.
- Development kidney abnormalities, focal segmental glomerulosclerosis and end-stage renal disease (ESRD) are more common than Wilms tumor. The rate of ESRD is 36% with unilateral Wilms tumor and 90% with bilateral Wilms tumor. Approximately 25% of individuals with WAGR syndrome have proteinuria ranging from minimal to overt nephrotic syndrome.
- A variety of dysmorphic features and metabolic abnormalities, including obesity, craniofacial dysmorphism, hemihypertrophy, growth retardation, scoliosis, and kyphosis.
Gillespies’ Syndrome
It is a rare variant form of aniridia associated with cerebellar ataxia, ptosis and mental retardation. The molecular basis of classical Gillespie syndrome remains unidentified. Owing to its rarity the inheritance pattern is unclear. PAX6 intragenic mutations have been reported in two individuals with phenotypes overlapping this syndrome, however these cases probably have atypical aniridia.
Aniridia complications
Aniridia may be complicated by:
- Glaucoma – Around 50% to 75% of patient with aniridia develop glaucoma, which however is usually not seen at birth. It usually occurs after 10 to 20 years of age likely due to blockage of the trabecular meshwork by the contracting iris strands of the rudimentary iris tissue or maldeveloped angle of the anterior chamber (goniodysgenesis). Glaucoma is the predominant cause of blindness in aniridia. The treatment of glaucoma is complicated and the glaucoma is often resistant to both medical and surgical treatment. The prognosis of glaucoma associated with aniridia is guarded.
- Aniridic fibrosis syndrome
- Painful or painless blind eye
- Systemic associations including Wilm’s tumor
Aniridia diagnosis
The fundus fluorescein angiogram reveals the lack of a well-defined foveal avascular zone.
Optical coherence tomography in foveal hypoplasia shows the absence of foveal depression and the presence of the inner retinal layers.
In sporadic congenital aniridia, Wilm’s tumor must be ruled out by regular ultrasonogram of the abdomen. Also, genetic evaluation for PAX6 and WT1 gene is possible.
Ophthalmic examination of the family members is of utmost importance.
Aniridia treatment
In early childhood, the management includes optimal refractive correction, amblyopia therapy, and correction of squint.
Painted contact lenses may reduce photophobia, improve cosmesis, and improve vision. Tinted contact lenses may also be an option. Such contact lenses also reduce nystagmus. Photochromatic or tinted glasses may be helpful.
The food and drug administration has approved an artificial iris implant. It may improve the disabling glare, light sensitivity during day or night, improve cosmesis and vision-related quality of life. However, there is a risk of development or worsening of glaucoma, and limbal stem cell deficiency. Customflex artificial iris may be used to treat
- Aniridia (congenital or traumatic)
- Iris defects due to various causes including albinism, or after excision of iris mass.
Glaucoma – The medications usually fail to control glaucoma, and surgical intervention is needed. Glaucoma presenting before 1 year of age is very difficult to treat. Surgery is often necessary for glaucoma by 20 years of age in patients with congenital aniridia. The options for surgical therapy include:
- Goniotomy
- Trabeculotomy
- Trabeculectomy with mitomycin C
- Combined trabeculectomy and trabeculotomy
- Glaucoma drainage devices may be implanted as a primary modality in aniridic glaucoma 16.
The success of surgical intervention may not be significant. In a study, success rates for various interventions were:
- 9% for trabeculectomy
- 25% for cyclocryotherapy; however side effects of phthisis and cataract was noted
- 83% for Molteno implant 17
Okada and colleagues 18 noted that ‘mean good intraocular pressure control period after the filtering surgery was 14.6 months’. The success rate of trabeculectomy is better in older patients and Nelson and colleagues reported successful trabeculectomies in 11 of 14 cases with the minimum follow-up of 1 year 9. Ultimately, if other modalities fail to control intraocular pressure, cycloablative therapies may be needed which include diode laser cyclophotocoagulation and cyclocryotherapy. Prophylactic goniotomy may prevent the development of glaucoma in congenital aniridia 19.
The cataract in aniridia may need extraction. Surgery is difficult due to zonular weakness and fragility of the lens capsule. The eye may be left aphakic, and postoperatively contact lens or aphakic glasses may be prescribed. Special intraocular lens (IOL) for aniridia is available which have a peripheral colored region like iris of different pigmentation. Tinted IOLs are also available which can reduce photophobia. In extreme subluxation, scleral-fixated IOL may be necessary. Surgical trauma may increase the chances of glaucoma, corneal decompensation, and limbal stem cell deficiency.
Aniridia-associated keratopathy:
- Phase 0 (subclinical limbal stem cell deficiency and phase 1 (slight limbal stem cell deficiency, less than 2 corneal epithelial erosion or ulcer in last 6months, corneal pannus less than 1mm from limbus, mild watering and photophobia and fluorescein staining abnormality) 20. The dry eye and keratopathy need frequent artificial tear drops (free of preservatives). Antiglaucoma drops with preservatives may worsen the corneal surface.
- Phase 2 (Moderate limbal insufficiency, constant redness, watering, and photophobia, more than 3 corneal erosions or ulcers in last 6 months, constant tear film instability, vascular pannus) 20 – amniotic membrane transplant or autologous serum is used.
- Phase 3 (severe limba insufficiency, corneal vascularization, loss of vision)- Autologous transplant of limbal epithelial cell is not possible due to bilateral involvement. Management options include:
- 2 stage surgery involving initial replenishment of the limbal stem cells followed by surgery for visual rehabilitation
- Options for replenishing limbal stem cells include:
- Keratolimbal allografts
- Cultured limbal stem cell transplant
- Cultured oral mucosal epithelial transplant 21
- Options for visual rehabilitation include
- Deep anterior lamellar keratoplasty
- Penetrating keratoplasty
- Options for replenishing limbal stem cells include:
- Simultaneous surgeries for both replenishment of the limbal stem cells and restoration of media clarity by keratoplasty have also been successful 22.
- Boston keratoprosthesis may be helpful in the visual rehabilitation of patients with severe aniridia-associated keratopathy 23.
- 2 stage surgery involving initial replenishment of the limbal stem cells followed by surgery for visual rehabilitation
However, the survival of corneal graft is often poor due to multiple causes including corneal vascularization, limbal stem cell deficiency, severe dry eye, and glaucoma.
The associated squint and ptosis may need correction.
Aniridic fibrosis syndrome needs early surgical intervention to remove the membranes.
Systemic diseases need a collaborative approach of specialists of different subspecialties.
Aniridia prognosis
Visual prognosis is usually poor. Up to 86% of patients with aniridia may have a visual acuity of 20/100 or worse in the better eye. The cause of visual decline include
- Foveal hypoplasia
- Optic nerve head hypoplasia
- Glaucoma
- Cataract
- Keratopathy
However, members of some families with familial aniridia may have good vision of at least 20/30 in up to 61% of cases 24.
- Melanie Hingorani, Isabel Hanson and Veronica van Heyningen, Aniridia, European Journal of Human Genetics. (2012) 20, 1011–1017; doi:10.1038/ ejhg.2012.100; published online 13 June 2012[↩][↩][↩][↩][↩][↩][↩]
- Melanie Hingorani, Anthony Moore, Aniridia, Includes: Isolated Aniridia, Wilms Tumor-Aniridia-Genital Anomalies-Retardation (WAGR) Syndrome, Bookshelf ID: NBK1360, 2008[↩][↩]
- Tripathy K, Salini UB B. Aniridia. [Updated 2019 Apr 28]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2019 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK538133[↩][↩]
- Ferrell RE, Chakravarti A, Hittner HM, Riccardi VM. Autosomal dominant aniridia: probable linkage to acid phosphatase-1 locus on chromosome 2. Proc. Natl. Acad. Sci. U.S.A. 1980 Mar;77(3):1580-2.[↩]
- Lim HT, Seo EJ, Kin GH et al, Comparison between Aniridia with and without PAX 6 mutations, clinical and molecular analysis of 14 Korean patients, Ophthalmology. 2012; 119:1258-1264[↩]
- Ton, C. C. T., Hirvonen, H., Miwa, H., et al Positional cloning and characterization of a paired box- and homeobox-containing gene from the aniridia region. Cell. 1991; 67: 1059-1074, 1991[↩]
- Gronskov K, Olsen JH, Sand A, et al.T. Population-based risk estimates of Wilms tumor in sporadic aniridia. A comprehensive mutation screening procedure of PAX6 identifies 80% of mutations in aniridia. Hum Genet. 2001;109:11–8.[↩][↩]
- Nelson LB, Spaeth GL, Nowinski TS, et al. Aniridia. A review. Surv Ophthalmol.1984; 28:621–642[↩][↩][↩][↩]
- Nelson LB, Spaeth GL, Nowinski TS, Margo CE, Jackson L. Aniridia. A review. Surv Ophthalmol. 1984 May-Jun;28(6):621-42.[↩][↩][↩][↩][↩][↩]
- Lee H, Khan R, O’Keefe M. Aniridia: current pathology and management. Acta Ophthalmol. 2008 Nov;86(7):708-15[↩]
- Lee H, Khan R, O’Keefe M: Aniridia: current pathology and management. Acta Ophthalmol. 2008; 86: 708–715[↩]
- Layman PR, Anderson DR, Flynn JT. Frequent occurrence of hypoplastic optic disks in patients with aniridia. Am. J. Ophthalmol. 1974 Apr;77(4):513-6.[↩]
- Berlin HS, Ritch R. The treatment of glaucoma secondary aniridia. Mt. Sinai J. Med. 1981 Mar-Apr;48(2):111-5[↩]
- Tsai JH, Freeman JM, Chan CC, Schwartz GS, Derby EA, Petersen MR, Holland EJ. A progressive anterior fibrosis syndrome in patients with postsurgical congenital aniridia. Am. J. Ophthalmol. 2005 Dec;140(6):1075-9[↩]
- Hingorani M, Hanson I, van Heyningen V. Aniridia. Eur. J. Hum. Genet. 2012 Oct;20(10):1011-7[↩]
- Arroyave CP, Scott IU, Gedde SJ, Parrish RK, Feuer WJ. Use of glaucoma drainage devices in the management of glaucoma associated with aniridia. Am. J. Ophthalmol. 2003 Feb;135(2):155-9[↩]
- Wiggins RE, Tomey KF. The results of glaucoma surgery in aniridia. Arch. Ophthalmol. 1992 Apr;110(4):503-5[↩]
- Okada K, Mishima HK, Masumoto M, Tsumamoto Y, Tsukamoto H, Takamatsu M. Results of filtering surgery in young patients with aniridia. Hiroshima J. Med. Sci. 2000 Sep;49(3):135-8.[↩]
- Chen TC, Walton DS. Goniosurgery for prevention of aniridic glaucoma. Trans Am Ophthalmol Soc. 1998;96:155-65; discussion 165-9[↩]
- Calvão-Pires P, Santos-Silva R, Falcão-Reis F, Rocha-Sousa A. Congenital Aniridia: Clinic, Genetics, Therapeutics, and Prognosis. Int Sch Res Notices. 2014;2014:305350[↩][↩]
- Dobrowolski D, Orzechowska-Wylegala B, Wowra B, Wroblewska-Czajka E, Grolik M, Szczubialka K, Nowakowska M, Puzzolo D, Wylegala EA, Micali A, Aragona P. Cultivated Oral Mucosa Epithelium in Ocular Surface Reconstruction in Aniridia Patients. Biomed Res Int. 2015;2015:281870[↩]
- Omoto M, Shimmura S, Hatou S, Ichihashi Y, Kawakita T, Tsubota K. Simultaneous deep anterior lamellar keratoplasty and limbal allograft in bilateral limbal stem cell deficiency. Jpn. J. Ophthalmol. 2010 Nov;54(6):537-43[↩]
- Rixen JJ, Cohen AW, Kitzmann AS, Wagoner MD, Goins KM. Treatment of aniridia with Boston type I keratoprosthesis. Cornea. 2013 Jul;32(7):947-50[↩]
- Elsas FJ, Maumenee IH, Kenyon KR, Yoder F. Familial aniridia with preserved ocular function. Am. J. Ophthalmol. 1977 May;83(5):718-24.[↩]


{kind=link}