Contents
- What is antibiotic resistance
- Why is antibiotic resistance a problem
- How does antibiotic resistance occur?
- What causes antibiotic resistance
- Why is antibiotic resistance dangerous?
- How antibiotic resistant bacteria spread?
- What are ‘superbugs’?
- Is it true that superbugs will kill 10 million people by year 2050?
- How can we stop antibiotic resistance?
- How does antibiotic resistance develop?
- Antibiotic resistance gene
- Antibiotic resistance mechanisms
- How to prevent antibiotic resistance
- Are antibacterial agents, such as antibacterial soaps, a solution?
- Can the effectiveness of existing antibiotics be preserved?
- Can new antibiotics be developed?
- Can antibiotic resistance be overcome?
- Can antibiotic resistance develop from using acne medication?
- Do probiotics have a role in helping to reduce antibiotic resistance?
- Symptoms of antibiotic resistance
- Antibiotic resistance solutions
What is antibiotic resistance
Antibiotic resistance occurs when antibiotics are no longer effective in treating bacterial and fungal infections as well as they used to be. Antibiotic resistance occurs because bacteria or fungi change to protect themselves against the antibiotic. That means the germs are not killed and continue to grow. Using antibiotics can lead to resistance. Each time you take antibiotics, sensitive bacteria are killed. But resistant germs may be left to grow and multiply. They can spread to other people. They can also cause infections that certain antibiotics cannot cure. Methicillin-resistant Staphylococcus aureus (MRSA) is one example. It causes infections that are resistant to several common antibiotics. Methicillin-resistant Staphylococcus aureus (MRSA), also known as ‘golden staph’, it can be picked up in hospitals and unfortunately, is increasingly being picked up in the community. Gonorrhea, a sexually transmitted infection, picked up through unprotected sex, increasing becoming resistant to antibiotics.
Antibiotic resistance is accelerated by the misuse and overuse of antibiotics. We are all part of the problem, and that means we are all part of the solution too. We can all take steps to change the way we use antibiotics.
Infections caused by antibiotic-resistant germs are difficult, and sometimes impossible, to treat. In most cases, antibiotic-resistant infections require extended hospital stays, additional follow-up doctor visits, and costly and toxic alternatives.
Antibiotic resistance does not mean the body is becoming resistant to antibiotics; it is that bacteria have become resistant to the antibiotics designed to kill them.
Antibiotic resistance has the potential to affect people at any stage of life, as well as the healthcare, veterinary, and agriculture industries, making it one of the world’s most urgent public health problems.
Each year in the U.S., at least 2 million people are infected with antibiotic-resistant bacteria, and at least 23,000 people die as a result 1.
No one can completely avoid the risk of resistant infections, but some people are at greater risk than others (for example, people with chronic illnesses). If antibiotics lose their effectiveness, then we lose the ability to treat infections and control public health threats.
Many medical advances are dependent on the ability to fight infections using antibiotics, including joint replacements, organ transplants, cancer therapy, and treatment of chronic diseases like diabetes, asthma, and rheumatoid arthritis.
To help prevent antibiotic resistance:
- Don’t use antibiotics for viruses like colds or flu. Antibiotics don’t work on viruses.
- Don’t pressure your doctor to give you an antibiotic.
- When you take antibiotics, follow the directions carefully. Finish your medicine even if you feel better. If you stop treatment too soon, some bacteria may survive and re-infect you.
- Don’t save antibiotics for later or use someone else’s prescription.
What is an antibiotic?
Antibiotics, also known as antimicrobial drugs, are drugs that fight infections caused by bacteria in both humans and animals. Antibiotics fight these infections either by killing the bacteria or making it difficult for the bacteria to grow and multiply. Antibiotics only treat certain bacterial infections. Antibiotics do not have any effect on viruses.
Any time antibiotics are used, they can cause side effects and lead to antibiotic resistance.
Why is antibiotic resistance a problem
Antibiotic resistance happens when bacteria survive and continue causing infection despite treatment with an antibiotic – the bacteria are no longer sensitive to that antibiotic.
Because the antibiotic no longer works against the resistant bacteria:
- infections take longer to heal
- infections can get worse and lead to more serious problems
- infections are more likely to spread to other people and because bacteria are resistant, the antibiotic may not work for other people, further spreading the problem
You might think it best to use another antibiotic to which bacteria aren’t resistant. However, it may not work as well, and it could have side effects. Also, bacteria may eventually become resistant to this antibiotic too.
For these reasons, antibiotic resistance is a major threat to human health. There is concern that in time, there’ll be bacterial infections that just can’t be treated.
How does antibiotic resistance occur?
Bacteria can change their genes after being exposed to an antibiotic. This allows them to survive antibiotic treatment. Then, when they multiply, they make more resistant bacteria. This is how antibiotic resistance develops.
Table 1. Examples of Defense Strategies for Germs
| Resistance Mechanisms (Defense Strategies) | Description |
|---|---|
| Restrict access of the antibiotic | By limiting the number or changing the size of the openings in the cell wall, resistant bacteria can keep antibiotic drugs from entering the cell altogether. Example: Gram-negative bacteria have an outer layer (membrane) that protects them from their environment. These bacteria can use this membrane to selectively keep antibiotic drugs from entering. |
| Get rid of the antibiotic | Resistant bacteria can use pumps in their cell walls to remove antibiotic drugs that enter the cell. Example: Some Pseudomonas aeruginosa bacteria can produce pumps to get rid of several different important antibiotic drugs, including fluoroquinolones, beta-lactams, chloramphenicol, and trimethoprim. |
| Destroy the antibiotic | Some resistant bacteria use enzymes to break down the antibiotic drug and make it ineffective. Example: Klebsiella pneumoniae bacteria produce enzymes called carbapenemases, which break down carbapenem drugs and most other beta-lactam drugs |
| Change the antibiotic | Other resistant bacteria use enzymes to alter the antibiotic drug so that it loses its effectiveness. Example: Staphylococcus aureus bacteria add compounds to aminoglycoside drugs to change its function. |
| Bypass the effects of the antibiotic | Some antibiotic drugs are designed to disrupt important processes critical to a bacteria’s survival, like the process of making nutrients. If successful, the antibiotic drug will keep the bacterium from performing all the steps needed in the process. Some resistant bacteria, however, have developed different and new processes to get around these drug disruptions. The new process may be slower but they can still bypass the effects of the drug. Example: Some Staphylococcus aureus bacteria can bypass the drug effects of trimethoprim. |
| Change the targets for the antibiotic | Many antibiotic drugs are designed to single out and destroy specific parts (or targets) of a bacterium. Resistant bacteria can change the look of their targets so that the antibiotic does not recognize and destroy them, allowing the bacteria to survive. Example: E. coli bacteria with the mcr-1 gene can add a compound to the outside of the cell wall so that the drug colistin cannot latch onto it. |
What causes antibiotic resistance
Antibiotic overuse is a main cause of antibiotic resistance. This occurs in both humans and animals.
Major causes of antibiotic resistance include:
- Using antibiotics when not needed. Most colds, sore throats, and ear and sinus infections are caused by viruses. Antibiotics don’t work against viruses. Many people don’t understand this and often ask for antibiotics when not needed. This leads to an overuse of antibiotics. The CDC estimates that 1 in 3 antibiotic prescriptions are not needed.
- Not taking antibiotics as prescribed by your doctor — this allows time for the bacteria in your system to become resistant. This includes not taking all of your antibiotics, missing doses, or using leftover antibiotics. Doing so helps the bacteria learn how to grow in spite of the antibiotic. As a result, the infection may not fully respond to treatment the next time the antibiotic is used.
- Misuse of antibiotics. You should never buy antibiotics online without a prescription or take someone else’s antibiotics.
- Exposure from food sources. Antibiotics are widely used in agriculture. This can lead to resistant bacteria in the food supply.
The more bacteria are exposed to antibiotics, the more chances they have to change and become resistant. When you use antibiotics when not needed or don’t take antibiotics properly – such as missing doses or not completing the course – you give bacteria opportunities to become resistant.
A recent prediction from the UK stated that antibiotic resistance could lead to an extra 10 million deaths a year worldwide by 2050, with a financial cost to the world of up to US $100 trillion.
Why is antibiotic resistance dangerous?
It’s dangerous now because it’s leading to fewer options to treat serious bacterial infections. This means people can be sicker for longer, they might need to stay in hospital for longer. In some cases it even can be a life-threatening situation.
Antibiotic resistance causes a number of problems:
- The need for stronger antibiotics with possibly severe side effects
- More expensive treatment
- Harder-to-treat illness spread from person to person
- More hospitalizations and longer stays
- Serious health problems, and even death
How antibiotic resistant bacteria spread?
Antibiotic resistance can spread from person to person or from animals to humans.
In people, it may spread from:
- One patient to other patients or staff in a nursing home, urgent care center, or hospital
- Health care staff to other staff or to patients
- Patients to other people who come in contact with the patient
All animals have bacteria in their intestines. Antibiotics that are given to food-producing animals treat infection and kill some bacteria, but resistant bacteria will often survive and multiply.
Antibiotic resistant bacteria may spread from animals to humans through:
- Food sprayed with water that contains antibiotic resistant bacteria from animal feces
- Other animals that are raised in the same environment
- Animal products that people eat, such as chicken and steak
- Produce, through contaminated water or soil
- The environment, through animal feces
- Prepared food, through contaminated surfaces, such as a cutting board used to cut chicken then chop vegetables without being washed between uses.
People can become sick with antibiotic-resistant infections when they eat food that’s been contaminated with antibiotic-resistant bacteria and not properly prepared, or from handling contaminated animals or surfaces and not practicing proper hand-washing or environmental cleaning afterward.
Antibiotic-resistant infections can lead to mild illnesses, or in some cases, severe illnesses that may lead to death.
The Food and Drug Administration (FDA) regulates antibiotics that are used in humans and animals. In animals raised for food, FDA-approved uses of antibiotics include:
- Treating sick animals
- Controlling illness in a group of animals when some of the animals are sick
- Preventing illness in a group of animals at risk of becoming sick
- Promoting growth and weight gain
The FDA and Centers for Disease Control and Prevention recommend antibiotics that are important for human health be used in food-producing animals only when it’s necessary to ensure the health of the animal, not to promote growth. A doctor who treats animals (veterinarian) should oversee or advise these conditions.
What are ‘superbugs’?
‘Superbugs’ are bacteria that are difficult to treat because they are resistant to several different antibiotics, and especially those that treat other resistant bacteria.
Superbugs such as methicillin-resistant Staphylococcus aureus (MRSA) and multi-drug-resistant strains of Escherichia coli (E. coli) are becoming more common, and can be a real problem in hospitals.
Is it true that superbugs will kill 10 million people by year 2050?
Yes, if things don’t change, we have a really big worldwide public health problem here. The World Health Organization says this is one of the greatest threats facing human health, worldwide currently.
The World Health Organization (WHO) has become quite concerned about the rising levels of resistant bacteria in all areas of the world. To provide some global coordination, WHO issued its Global Strategy for Containment of Antimicrobial Resistance 3, a document aimed at policy-makers that urges governments to take action to help contain antibiotic resistance.
How can we stop antibiotic resistance?
Firstly, only take antibiotics as prescribed and directed by your doctor. Secondly, don’t ask for antibiotics for cold and flu. They don’t work for viruses. And thirdly, practice good hygiene including handwashing to prevent the spread of bacteria.
How does antibiotic resistance develop?
Bacteria can become resistant to antibiotics through several ways. Some bacteria can “neutralize” an antibiotic by changing it in a way that makes it harmless. Others have learned how to pump an antibiotic back outside of the bacteria before it can do any harm. Some bacteria can change their outer structure so the antibiotic has no way to attach to the bacteria it is designed to kill.
After being exposed to antibiotics, sometimes one of the bacteria can survive because it found a way to resist the antibiotic. If even one bacterium becomes resistant to antibiotics, it can then multiply and replace all the bacteria that were killed off. That means that exposure to antibiotics provides selective pressure making the surviving bacteria more likely to be resistant. Bacteria can also become resistant through mutation of their genetic material.
Antibiotic resistance gene
Bacteria have a remarkable genetic plasticity that allows them to respond to a wide array of environmental threats, including the presence of antibiotic molecules that may jeopardize their existence. Bacteria sharing the same ecological niche with antimicrobial-producing organisms have evolved ancient mechanisms to withstand the effect of the harmful antibiotic molecule and consequently, their intrinsic resistance permits them to thrive in its presence. From an evolutionary perspective, bacteria use two major genetic strategies to adapt to the antibiotic “attack”, i) mutations in gene(s) often associated with the mechanism of action of the compound, and ii) acquisition of foreign DNA coding for resistance determinants through horizontal gene transfer (HGT).
Mutational Resistance
In this scenario, a subset of bacterial cells derived from a susceptible population develop mutations in genes that affect the activity of the drug, resulting in preserved cell survival in the presence of the antimicrobial molecule. Once a resistant mutant emerges, the antibiotic eliminates the susceptible population and the resistant bacteria predominate. In many instances, mutational changes leading to resistance are costly to cell homeostasis (i.e., decreased fitness) and are only maintained if needed in the presence of the antibiotic. In general, mutations resulting in antimicrobial resistance alter the antibiotic action via one of the following mechanisms, i) modifications of the antimicrobial target (decreasing the affinity for the drug, see below), i) a decrease in the drug uptake, ii) activation of efflux mechanisms to extrude the harmful molecule, or iv) global changes in important metabolic pathways via modulation of regulatory networks. Thus, resistance arising due to acquired mutational changes is diverse and varies in complexity.
Horizontal Gene Transfer
Acquisition of foreign DNA material through Horizontal Gene Transfer is one of the most important drivers of bacterial evolution and it is frequently responsible for the development of antimicrobial resistance. Most antimicrobial agents used in clinical practice are (or derive from) products naturally found in the environment (mostly soil). As mentioned before, bacteria sharing the environment with these molecules harbor intrinsic genetic determinants of resistance and there is robust evidence suggesting that such “environmental resistome” is a prolific source for the acquisition of antibiotic resistance genes in clinically relevant bacteria. Furthermore, this genetic exchange has been implicated in the dissemination of resistance to many frequently used antibiotics.
Classically, bacteria acquire external genetic material through three main strategies, i) transformation (incorporation of naked DNA), ii) transduction (phage mediated) and, iii) conjugation (bacterial “sex”). Transformation is perhaps the simplest type of Horizontal Gene Transfer, but only a handful of clinically relevant bacterial species are able to “naturally” incorporate naked DNA to develop resistance. Emergence of resistance in the hospital environment often involves conjugation, a very efficient method of gene transfer that involves cell-to-cell contact and is likely to occur at high rates in the gastrointestinal tract of humans under antibiotic treatment. As a general rule, conjugation uses mobile genetic elements (MGEs) as vehicles to share valuable genetic information, although direct transfer from chromosome to chromosome has also been well characterized 4. The most important mobile genetic elements are plasmids and transposons, both of which play a crucial role in the development and dissemination of antimicrobial resistance among clinically relevant organisms.
Finally, one of the most efficient mechanisms for accumulating antimicrobial resistance genes is represented by integrons, which are site-specific recombination systems capable of recruiting open reading frames in the form of mobile gene cassettes. Integrons provide an efficient and rather simple mechanism for the addition of new genes into bacterial chromosomes, along with the necessary machinery to ensure their expression; a robust strategy of genetic interchange and one of the main drivers of bacterial evolution. For details on the mechanisms of Horizontal Gene Transfer the readers are directed to a recent state-of-the-art review 5.
Antibiotic resistance mechanisms
Bacteria have evolved sophisticated mechanisms of drug resistance to avoid killing by antimicrobial molecules, a process that has likely occurred over millions of years of evolution 6. Of note, resistance to one antimicrobial class can usually be achieved through multiple biochemical pathways, and one bacterial cell may be capable of using a cadre of mechanisms of resistance to survive the effect of an antibiotic. As an example, fluoroquinolone (FQ) resistance can occur due to three different biochemical routes, all of which may coexist in the same bacteria at a given time (producing an additive effect and, often, increasing the levels of resistance), i) mutations in genes encoding the target site of FQs (DNA gyrase and topoisomerase IV), ii) over-expression of efflux pumps that extrude the drug from the cell, and iii) protection of the FQ target site by a protein designated Qnr (see below for details on each of these mechanisms). On the other hand, bacterial species seem to have evolved a preference for some mechanisms of resistance over others. For example, the predominant mechanism of resistance to β-lactams in gram-negative bacteria is the production of β-lactamases, whereas resistance to these compounds in gram-positive organisms is mostly achieved by modifications of their target site, the penicillin-binding proteins (PBPs). It has been argued that this phenomenon is likely due to major differences in the cell envelope between gram-negatives and gram-positives. In the former, the presence of an outer membrane permits to “control” the entry of molecules to the periplasmic space. Indeed, most β-lactams require specific porins to reach the penicillin-binding proteins, which are located in the inner membrane. Therefore, the bacterial cell controls the access of these molecules to the periplasmic space allowing the production of β-lactamases in sufficient concentrations to tip the kinetics in favor of the destruction of the antibiotic molecule. Conversely, this “compartmentalization” advantage is absent in gram-positive organisms, although production of β-lactamases also seems to be successful in certain scenarios (e.g., staphylococcal penicillinase).
In order to provide a comprehensive classification of the antibiotic resistance mechanisms, we will categorize them according to the biochemical route involved in resistance, as follows: i) modifications of the antimicrobial molecule, ii) prevention to reach the antibiotic target (by decreasing penetration or actively extruding the antimicrobial compound), iii) changes and/or bypass of target sites, and iv) resistance due to global cell adaptive processes. Each of these mechanistic strategies encompasses specific biochemical pathways that will be described in detail in the reminder of the chapter. Of note, we will focus the discussion on the most relevant mechanisms giving examples that have relevant clinical impact.
Modifications of the Antibiotic Molecule
One of the most successful bacterial strategies to cope with the presence of antibiotics is to produce enzymes that inactivate the drug by adding specific chemical moieties to the compound or that destroy the molecule itself, rendering the antibiotic unable to interact with its target.
Chemical alterations of the antibiotic
The production of enzymes capable of introducing chemical changes to the antimicrobial molecule is a well-known mechanism of acquired antibiotic resistance in both gram-negative and gram-positive bacteria. Interestingly, most of the antibiotics affected by these enzymatic modifications exert their mechanism of action by inhibiting protein synthesis at the ribosome level 7. Many types of modifying enzymes have been described, and the most frequent biochemical reactions they catalyze include i) acetylation (aminoglycosides, chloramphenicol, streptogramins), ii) phosphorylation (aminoglycosides, chloramphenicol), and iii) adenylation (aminoglycosides, lincosamides). Regardless of the biochemical reaction, the resulting effect is often related to steric hindrance that decreases the avidity of the drug for its target, which, in turn, is reflected in higher bacterial MICs.
One of the best examples of resistance via modification of the drug is the presence of aminoglycoside modifying enzymes (AMEs) that covalently modify the hydroxyl or amino groups of the aminoglycoside molecule. Multiple AMEs have been described to date and they have become the predominant mechanism of aminoglycoside resistance worldwide. These enzymes are usually harbored in MGEs, but genes coding for AMEs have also been found as part of the chromosome in certain bacterial species, as seen with some aminoglycoside acetyltransferases in Providencia stuartii, E. faecium and S. marcescens 8. The nomenclature to classify the multiple AMEs considers their biochemical activity (acetyltransferase [ACC], adenyltransferase [ANT] or phosphotransferase [APH]), the site of the modification, which is depicted by a number from 1 to 6 corresponding to the particular carbon on the sugar ring and a single or double apostrophe to symbolize that the reaction occurs in the first or in the second sugar moiety, respectively. In addition, whenever there is more than one enzyme catalyzing the exact same reaction, a roman numeral is used to differentiate them (Figure 1).
Figure 1. Chemical alterations of the antibiotic – different types of aminoglycoside-modifying enzymes and their nomenclature
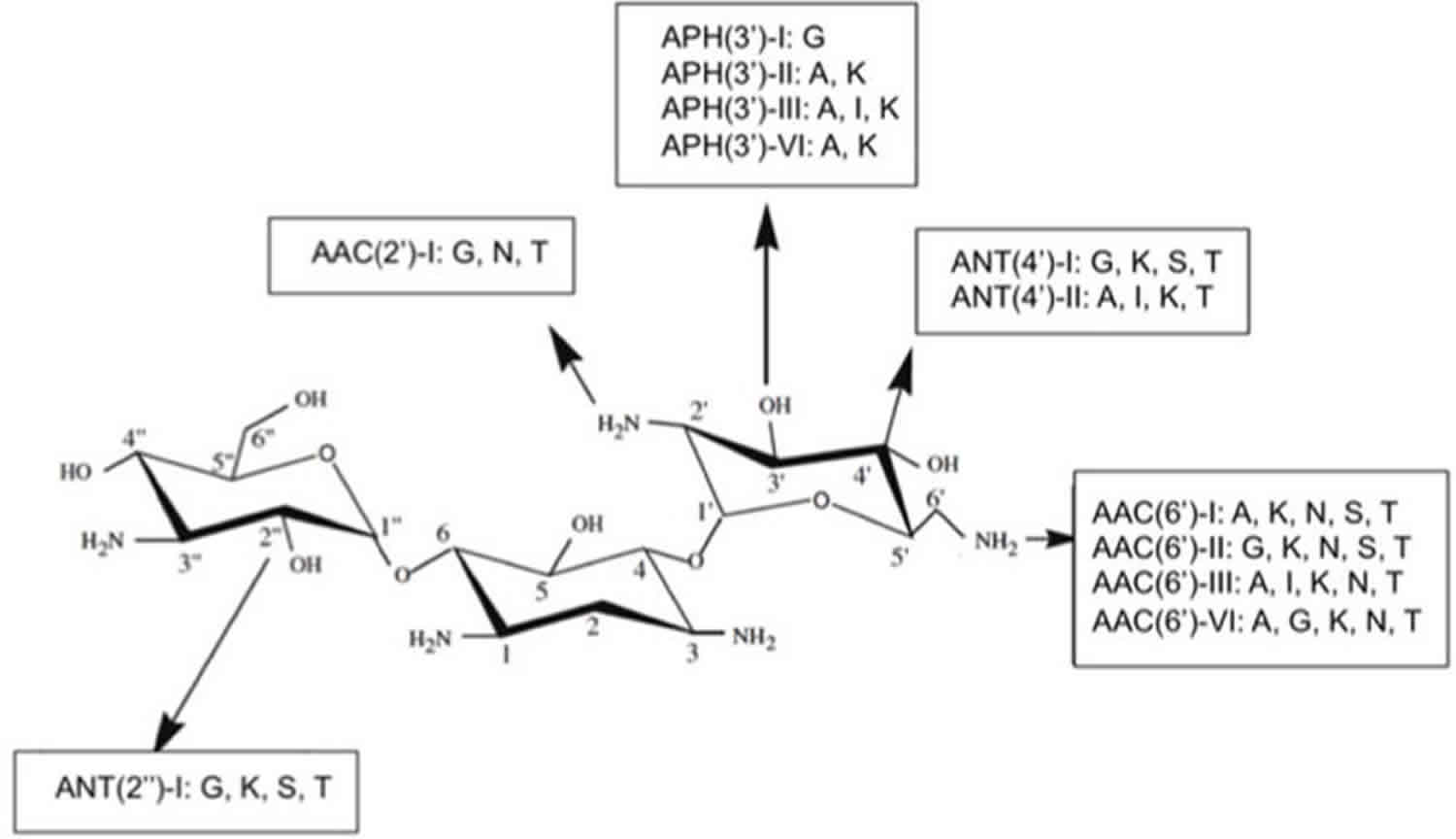
Footnote: Representation of different types of aminoglycoside-modifying enzymes and their nomenclature
Each group of enzymes is identified by their biochemical activity as follows: acetyltransferase (ACC), adenyltransferase (ANT) and phosphotransferase (APH). Next in the enzyme name, an algebraic number in parenthesis indicates the number of the carbon that is inactivated. The ring of the sugar in which the reaction takes place is symbolized by one (first sugar moiety) or two apostrophes (second sugar moiety). Roman numerals are used to differentiate distinct isoenzymes acting in the same site. Not all existing enzymes are shown.
[Source 6 ]There are important differences in the geographical distribution, bacterial species in which these enzymes disseminate and in the specific aminoglycosides they affect. For instance, the APH 9 family is widely distributed in gram-positive and gram-negative bacteria and alters kanamycin and streptomycin, but spares gentamicin and tobramycin. On the other hand, AAC(6′)-I is mainly found in gram-negative clinical isolates including Enterobacteriaceae, Pseudomonas and Acinetobacter and affects most aminoglycosides including amikacin and gentamicin 8. In addition, the activity and distribution of AMEs from a same family also varies. For instance, among the adenyltransferases, which classically affect both gentamicin and tobramycin, the genes encoding ANT(4′), ANT(6′) and ANT(9′) are usually harbored in MGEs of gram-positive bacteria, and ANT(2″) and ANT(3″) are more prevalent in gram-negative organisms 8.
Finally, it is worth mentioning that some of these enzymes have evolved more than a single biochemical activity. Indeed, AAC(6′)APH(2″), which is mainly found in gram-positive organisms, is a bifunctional enzyme (with acetylation and phosphotransferase activities) that likely arose from the fusion of two AMEs encoding genes. This protein confers high-level resistance to all aminoglycosides except for streptomycin and is located on a Tn4001-like transposon widely distributed among enterococci and staphylococci. Furthermore, the presence of this bifunctional enzyme accounts for most of high-level gentamicin resistance detected in enterococci (including in vancomycin-resistant strains) and methicillin-resistant S. aureus worldwide 10.
Another classical example of enzymatic alteration of an antibiotic involves the modification of chloramphenicol, an antibiotic that inhibits protein synthesis by interacting with the peptidyl-transfer center of the 50S ribosomal subunit. The chemical modification of chloramphenicol is mainly driven by the expression of acetyltransferases known as CATs (chloramphenicol acetyltransferases). Multiple cat genes have been described in both gram-positives and gram-negatives and they have been classified in two main types. Type A, which usually result in high-level resistance, and type B that confers low-level chloramphenicol resistance 11. Although these determinants are usually harbored in MGEs such us plasmids and transposons, they have also been reported as being part of the core genome (chromosome) of certain bacteria.
Destruction of the antibiotic molecule
The main mechanism of β-lactam resistance relies on the destruction of these compounds by the action of β- lactamases. These enzymes destroy the amide bond of the β-lactam ring, rendering the antimicrobial ineffective. β-lactamases were first described in the early 1940s, one year before penicillin was introduced to the market, however, there is evidence of their existence for millions of years 12. Infections caused by penicillin-resistant S. aureus became clinically relevant after penicillin became widely available and the mechanism of resistance was found to be a plasmid-encoded penicillinase that was readily transmitted between S. aureus strains, resulting in rapid dissemination of the resistance trait 13. In order to overcome this problem, new β-lactam compounds with wider spectrum of activity and less susceptibility to penicillinases (such as ampicillin) were manufactured. However, during the 1960s a new plasmid-encoded β-lactamase capable of hydrolyzing ampicillin was found among gram-negatives (termed TEM-1 after the name of the patient in which it was originally found [Temoneira]) 14. From then on, the development of newer generations of β-lactams has systematically been followed by the rapid appearance of enzymes capable of destroying any novel compound that reach the market, in a process that is a prime example of antibiotic-driven adaptive bacterial evolution.
Genes encoding for β-lactamases are generally termed bla, followed by the name of the specific enzyme (e.g. blaKPC) and they have been found in the chromosome or localized in MGEs as part of the accessory genome. These genes can also be found forming part of integrons, a situation that facilitates their dissemination. In terms of their expression, transcription of these genes can be constitutive or it may require an external signal to induce their production.
To date more than 1,000 different β-lactamases have been described (http://www.lahey.org/studies/) and many more are likely to continue to be reported, as part of the normal process of bacterial evolution. Two main classification schemes have been proposed in an attempt to group this large number of enzymes. First, the Ambler classification relies on amino acid sequence identity and separates β-lactamases into 4 groups (A, B, C, and D). On the other hand, the Bush-Jacoby classification divides β-lactamases into 4 categories (each with several subgroups) according to their biochemical function, mainly based on substrate specificity 15. A summary of the most important enzymes and their classification is presented in Figure 2.
Figure 2. Mechanism of beta-lactam antibiotic resistance
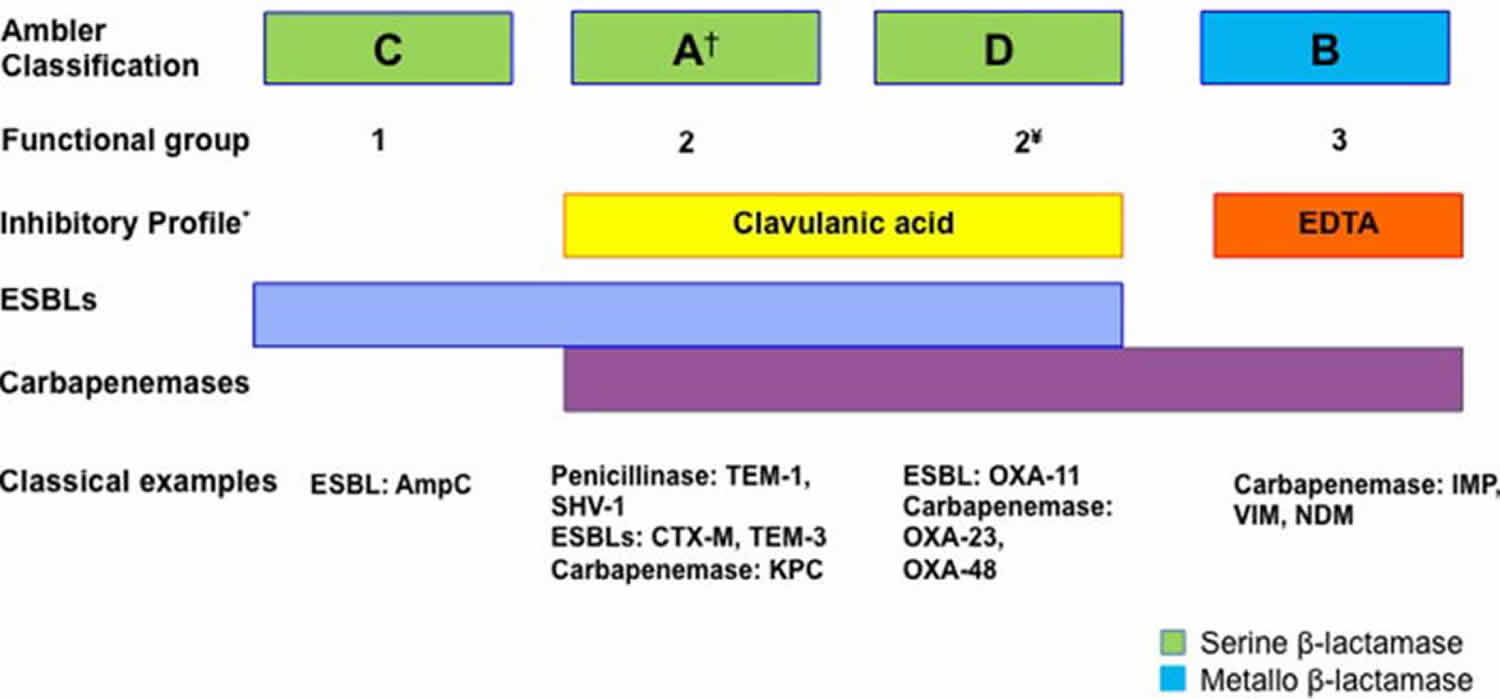
Footnote: Schematic representation of β-lactamases
Molecular classification of B-lactamases follows the Ambler classification. Correlation with the main functional group of the Bush and Jacobi classification is also shown. Of note, the latter classification has several sub-groups that are not shown. Representative examples of each group of enzymes are provided.
† Class A enzymes are the most diverse and include penicillinases, ESBLs and carbapenemases.
¥ Ambler class D enzymes belong to the functional group/subgroup 2d.
* Class A enzymes belonging to the subgroup 2br are resistant to clavulanic acid inhibition.
Abbreviations: EDTA = ehtylenediaminetetraacetic acid; ESBLs = extended-spectrum β-lactamases
[Source 6 ]It is important to note that both classifications mentioned above have caveats and they do not fully overlap. For instance, Ambler classes A and D enzymes are all considered within the group 2 in the Jacoby-Bush system. In addition, while the Ambler classification seems to be easier to follow, the lack of correlation with functional characteristics of the enzymes may lead to confusion. As an example, the Ambler class A group encompasses enzymes with a wide range of biochemical activities, from narrow spectrum β-lactamases to enzymes capable of destroying almost all available β-lactams, including carbapenems. Moreover, enzymes originally classified within a group harboring a particular biochemical profile can evolve into novel enzymes with different substrate specificities usually due to mutations in the active site. A good example of this process is TEM-3, an enzyme that evolved from the original TEM-1 penicillinase after acquiring the ability to hydrolyze third generation cephalosporins and aztreonam (a functional profile that defines it as an “Extended Spectrum β-Lactamase” [ESBL]) due to the development of two amino acid substitutions that altered its function 14.
Deciphering the role of the different types of enzymes and their characteristics is a complex task that requires understanding some of the terminology frequently used in the literature. As mentioned above, an ESBL enzyme has the ability to hydrolyze penicillins, 3rd generation cephalosporins (the hallmark characteristic) and monobactams, but harbor modest (or no) activity against cephamycins and carbapenems. Most of the ESBLs belong to Ambler class A and, as such, they are generally inhibited by clavulanic acid or tazobactam. Importantly, this property distinguishes them from AmpC enzymes, which are class C β-lactamases that also hydrolyze 3rd generation cephalosporins, but are not inhibited by clavulanic acid or tazobactam. Of note, a subgroup of class D OXA enzymes capable of destroying 3rd generation cephalosporins are also considered within the ESBL group (see below and Figure 2). Other clinically relevant group of enzymes is the carbapenemases (a diverse group of β-lactamases with the ability to hydrolyze carbapenems), the most potent β-lactams available in clinical practice. These enzymes can be divided into serine carbapenemases (Ambler class A or D) and metallo-carbapenemases (Ambler class B enzymes). Thus, in the remaining of this section, we will provide examples of the different types of β-lactamases using the Ambler classification as the backbone for discussion.
Class A β-lactamases have a serine residue in the catalytic site, a property that they share with class C and D enzymes. Most class A enzymes are inhibited by clavulanic acid and their spectrum of activity include monobactams but not cephamycins (cefoxitin and cefotetan). Class A enzymes include a wide range of proteins with very different catalytic activities, spanning from penicillinases (TEM-1 and SHV-1 that only hydrolyze penicillin), ESBLs (such as CTX-M) to carbapenemases like KPC (Klebsiella pneumoniae carbapenemase), an enzyme that is currently prevalent in several gram-negative species. We will discuss details on CTX-M (ESBL) and KPC carbapenemases, both class A enzymes with high clinical impact.
CTX-M is a plasmid-encoded ESBL commonly found in K. pneumoniae, E. coli, and other Enterobacteriaceae around the world. In contrast to other Ambler class A ESBLs like TEM-3, this enzyme did not derive from TEM or SHV, rather, the current evidence suggests that it was likely acquired from Kluyvera spp. (an environmental bacterium with no major human pathogenic significance) through HGT 16. Genes encoding CTX-M enzymes have been found in association with insertion sequences (ISEcp1) and with transposable elements such as Tn402-like transposons. These mobile elements can be captured by a broad range of conjugative plasmids or phage-like sequences that can serve as vehicles for dissemination 17. Consequently, CTX-M enzymes have become the most prevalent ESBL worldwide and are responsible for a large proportion of cephalosporin resistance in E. coli and K. pneumoniae.
To date, five different families of class A carbapenemases have been described, of which three are typically chromosomally encoded (IMI [imipenem-hydrolyzing enzyme], SME [Serratia marcescens enzyme] and NMC [not-metallo-enzyme carbapenemase]), and the remaining two (KPC and GES) are classically harbored in plasmids or other MGEs 18. As for other class A enzymes, they are all inhibited by clavulanic acid and tazobactam, and hydrolyze aztreonam but not cephamycins. KPC was first reported in 1996 from a K. pneumoniae recovered from a patient in North Carolina, USA 19. Although these enzymes are predominantly found in Klebsiella spp. (therefore its name, Klebsiella pneumoniae carbapenemase), they have been reported in several other gram-negatives, including Enterobacter spp., E. coli, Proteus mirabilis, and Salmonella spp., among others. Furthermore, they have also been found in non-lactose fermenter organisms such as P. aeruginosa. A total of 22 variants of the blaKPC gene have been described to date, most of them located in plasmids harboring transposable elements (e.g. Tn4401) or in association with insertion sequences like ISKpn6 and ISKpn7 20.
Class B enzymes are also known as metallo- β-lactamases due to the fact that they utilize a metal ion (most usually Zinc) as cofactor (instead of a serine residue) for the nucleophilic attack of the β-lactam ring. They are inhibited by the presence of ion-chelating agents such as EDTA and, similar to class A carbapenemases, they are active against a wide range of β-lactams, including carbapenems. Metallo-β-lactamases are not inhibited by clavulanic acid or tazobactam and while they efficiently hydrolyze cephamycins, aztreonam is typically a poor substrate. These enzymes were discovered over 50 years ago encoded by genes usually located in the chromosome of non-pathogenic bacteria. However, the situation dramatically changed during the 1990’s, when enzymes like IMP and VIM were increasingly reported in clinical strains of Enterobacteriaceae, Pseudomonas spp. and Acinetobacter spp 18. Indeed, genes encoding these enzymes have been found as part of the accessory genome of pathogenic bacteria suggesting HGT. There are ca. 10 types of metallo-carbapenemases, but most of the clinically important ones belong to 4 families, IMP, VIM, SPM and NDM. Considering their high frequency and worldwide spread, we will briefly discuss IMP, VIM and NDM.
The first IMP-type enzymes were described in Japan in the early 1990s in S. marcescens, and since then, more than 20 different subtypes have been described worldwide in Enterobacteriaceae, Pseudomonas spp., and Acinetobacter spp, among other organisms. The blaIMP genes have been found on large-size plasmids and forming part of class 1 integrons 21. Regarding the VIM-type enzymes, they were first described in the late 1990s in Verona, Italy (Verona integron-encoded metallo β-lactamase) and have since spread throughout the globe. These enzymes were initially found in P. aeruginosa, but their association with class 1 integrons, along with reports locating them in different types of MGEs, has likely contributed to their dissemination to many different bacterial species becoming a major concern around the globe. Among the many different variants of VIM described to date, VIM-2 is the most widely distributed enzyme, with reports from Europe, Asia, Africa, and the Americas 22.
More recently (2008), a new carbapenemase was identified in a K. pneumoniae isolate recovered from a Swedish patient who had been previously admitted to a hospital in New Delhi, India. The enzyme was designated NDM-1, in reference to its origin (New Delhi Metallo β-lactamase) 23. NDM-1 shares little amino acid identity with other members of the Ambler class B enzymes (e.g. 32% with VIM-1), but its hydrolytic profile is very similar to all of them. The blaNDM gene has been found in several types of plasmids readily transferable among different species of gram-negatives, and it has also been associated with the presence of insertion sequences such as the ISAba125. In contrast to other genes encoding metallo-enzymes, blaNDM is not usually related to integron-like structures 24. Nevertheless, NDM-1 rapidly spread around the globe, becoming a prime example of how a resistance determinant can readily disseminate worldwide despite many efforts to avoid its transmission. Moreover, MGEs-containing genes coding for NDM enzymes generally carry multiple other resistance determinants such as genes encoding other carbapenemases (e.g. VIM-type and OXA-type enzymes), ESBL, AMEs, methylases conferring resistance to macrolides, the quinolone resistance Qnr protein, enzymes that modify rifampin and proteins involved in resistance to sulfamethoxazole, among others. Thus, the presence of NDM-1 is frequently accompanied by a multidrug-resistant phenotype.
The emergence of NDM-1 is particularly concerning because the blaNDM gene has shown to be readily transmissible among different types of gram-negative organisms, spreading to many countries in a short span of time and becoming one of the most feared resistance determinants in several parts of the world 25. In addition, in the Indian subcontinent (i.e. India and Pakistan), the blaNDM gene is not only extensively disseminated among nosocomial pathogens, but it is frequently found in community-associated isolates. Furthermore, several reports have found NDM-1 producing gram-negative bacteria in the soil and drinking water for human consumption, suggesting that these genes may be disseminating through the human microbiota 22.
Class C β-lactamases confer resistance to all penicillins and cephalosporins (although cefepime is usually a poor substrate), including cephamycins. They do not reliably hydrolyze aztreonam and are not inhibited by clavulanic acid. The most clinically relevant class C enzyme is AmpC, which is a cephalosporinase that is generally encoded on the chromosome (although the blaAMPc gene has also been found in plasmids). Production of chromosomal AmpC is a hallmark of E. cloacae, E. aerogenes, C. freundii, S. marcescens, Providencia sp., Morganella morganii, and P. aeruginosa, among others. In contrast, P. mirabilis, P. vulgaris, Klebsiella spp. and Stenotrophomonas spp. are classical examples of species in which the blaampC gene is absent from the core genome 26.
The expression of ampC is generally inducible and is under strict control of a complex regulatory mechanism that has been best studied in Enterobacter spp. AmpR is a transcriptional regulator of the LysR family that acts as a repressor of the transcription of blaAMPc. Under non-inducing conditions (absence of β-lactams), AmpR is bound to peptidoglycan precursors (UDP-MurNAc pentapeptides) and interaction of AmpR with its cognate promoter does not occur (resulting in absence of blaAMPc transcription). In contrast, in the presence of β-lactams, the alterations in cell wall homeostasis result in accumulation of peptidoglycan byproducts such as anhydro-muropeptides that compete for the same AmpR binding site with the UDP-MurNAc pentapeptides. As result of this competition, AmpR is released and is able to interact with the blaAMPc promoter, activating transcription of the gene 27.
Another mechanism by which ampC is overexpressed is through AmpD, a cytosolic amidase that recycles muropeptides. AmpD effectively reduces the concentration of anhydro-UDP-MurNAc tri-, tetra- and pentapeptides preventing displacement of UDP-MurNAc pentapeptide from AmpR and, therefore, ampC overexpression. Mutations in ampD are often seen in isolates that constitutively overproduce AmpC, affecting the clinical efficacy of cephalosporins. As mentioned, cefepime is not a good substrate for AmpC enzymes; however, high-level production of AmpC may markedly increase cefepime MICs 28.
Class D β-lactamases include a wide range of enzymes that were initially differentiated from the class A penicillinases due to their ability to hydrolyze oxacillin (hence their name) and because they were poorly inhibited by clavulanic acid. Many OXA variants have been described, including enzymes with the ability to degrade third generation cephalosporins (ESBLs) (e.g., OXA-11 from P. aeruginosa) and carbapenems (e.g., OXA-23 from A. baumanii). For example, OXA-48 is a widely disseminated class D carbapenemase which was originally described in 2001 in Turkey from a multidrug resistant isolate of K. pneumoniae. OXA-48 and its variants are now widely spread in clinical isolates of K. pneumoniae and other Enterobacteriaceae, and have also been found in A. baumanii 29. Many other types of OXA enzymes have been described to date possessing a variety of hydrolytic profiles and encoded by genes that are often found in a wide range of MGEs. In certain instances, the OXA-containing MGE inserts in the chromosome, resulting in core-genome genes encoding OXA enzymes. This phenomenon has been often described in Acinetobacter with both OXA-51 and OXA-69 encoded by genes located in the chromosome 29.
Although Class D enzymes are particularly prevalent in A. baumanii, they have been reported in many other clinically relevant organisms, such as E. coli, Enterobacter spp., K. pneumoniae and P. aeruginosa, among others. Furthermore, intra- and interspecies transmission of some of these genes has been particularly successful, with enzymes like OXA-23 and OXA-58 currently being spread around the globe.
Decreased Antibiotic Penetration and Efflux
Decreased permeability
Many of the antibiotics used in clinical practice have intracellular bacterial targets or, in case of gram-negative bacteria, located in the cytoplasmic membrane (the inner membrane). Therefore, the compound must penetrate the outer and/or cytoplasmic membrane in order to exert its antimicrobial effect. Bacteria have developed mechanisms to prevent the antibiotic from reaching its intracellular or periplasmic target by decreasing the uptake of the antimicrobial molecule. This mechanism is particularly important in gram-negative bacteria (for the reason specified above), limiting the influx of substances from the external milieu. In fact, the outer membrane acts as the first-line of defense against the penetration of multiple toxic compounds, including several antimicrobial agents. Hydrophilic molecules such as β-lactams, tetracyclines and some fluoroquinolones are particularly affected by changes in permeability of the outer membrane since they often use water-filled diffusion channels known as porins to cross this barrier 30. The prime example of the efficiency of this natural barrier is the fact that vancomycin, a glycopeptide antibiotic, is not active against gram-negative organisms due to the lack of penetration through the outer membrane. Likewise, the innate low susceptibility of Pseudomonas and Acinetobacter baumanii to β-lactams (compared to Enterobacteriaceae) can be explained, at least in part, to a reduced number and/or differential expression of porins 31.
Several types of porins have been described, and they can be classified according to their structure (trimeric vs. monomeric), their selectivity and the regulation of their expression. Among the best-characterized porins, the three major proteins produced by E. coli (known as OmpF, OmpC and PhoE) and the P. aeruginosa OprD (also known as protein D2) are classical examples of porin-mediated antibiotic resistance. Alterations of porins could be achieved by 3 general processes, i) a shift in the type of porins expressed, ii) a change in the level of porin expression, and iii) impairment of the porin function. Importantly, changes in permeability through any of these mechanisms frequently result in low-level resistance and are often associated with other mechanisms of resistance, such as increased expression of efflux pumps (see below) 32.
One classic example of porin-mediated resistance is the aberrant production of OprD in P. aeruginosa, which is normally used for the uptake of basic amino acids and antibiotics (i.e., imipenem, a potent anti-pseudomonal antibiotic from the carbapenem class). Mutations in the oprD gene have been shown to arise in clinical isolates of P. aeruginosa during therapy 33. Furthermore, clinical and in vitro studies have shown that these changes can produce resistance alone or in conjunction with over expression of an efflux pump and/or the production of a carbapenem-hydrolyzing enzyme, resulting in high levels of resistance to carbapenems.
Another example relates to clinical isolates of K. pneumoniae recovered before and after antimicrobial therapy, The post-therapy isolates were found to exhibit a shift in porin expression from OmpK35 to OmpK36 (the latter possessing a smaller channel size). This alteration in the type of porin expressed correlated with a 4 – 8 fold decrease in susceptibility for a wide range of β-lactam antimicrobials 34. Similar examples are found in other bacterial species of clinical importance such as E. cloacae, Salmonella spp., Neisseria gonorrhoeae, and A. baumanii.
Efflux Pumps
The production of complex bacterial machineries capable to extrude a toxic compound out of the cell can also result in antimicrobial resistance. The description of an efflux system able to pump tetracycline out of the cytoplasm of E. coli dates from the early 1980s and was among the first to be described 35. Since then, many classes of efflux pumps have been characterized in both gram-negative and gram-positive pathogens. These systems may be substrate-specific (for a particular antibiotic such as tet determinants for tetracycline and mef genes for macrolides in pneumococci) or with broad substrate specificity, which are usually found in MDR bacteria 36. This mechanism of resistance affects a wide range of antimicrobial classes including protein synthesis inhibitors, fluoroquinolones, β-lactams, carbapenems and polymyxins. The genes encoding efflux pumps can be located in MGEs (as initially described for the tet gene) or in the chromosome. Importantly, chromosomally encoded pumps can explain the inherent resistance of some bacterial species to a particular antibiotic (e.g. E. faecalis intrinsic resistance to streptogramin A, see below) 37.
There are 5 major families of efflux pumps, including i) the major facilitator superfamily (MFS), ii) the small multidrug resistance family (SMR), iii) the resistance-nodulation-cell-division family (RND), iv) the ATP-binding cassette family (ABC), and v) the multidrug and toxic compound extrusion family (MATE). These families differ in terms of structural conformation, energy source, range of substrates they are able to extrude and in the type of bacterial organisms in which they are distributed 38 (Figure 3).
Figure 3. Mechanism of antibiotic resistance – decreased antibiotic penetration
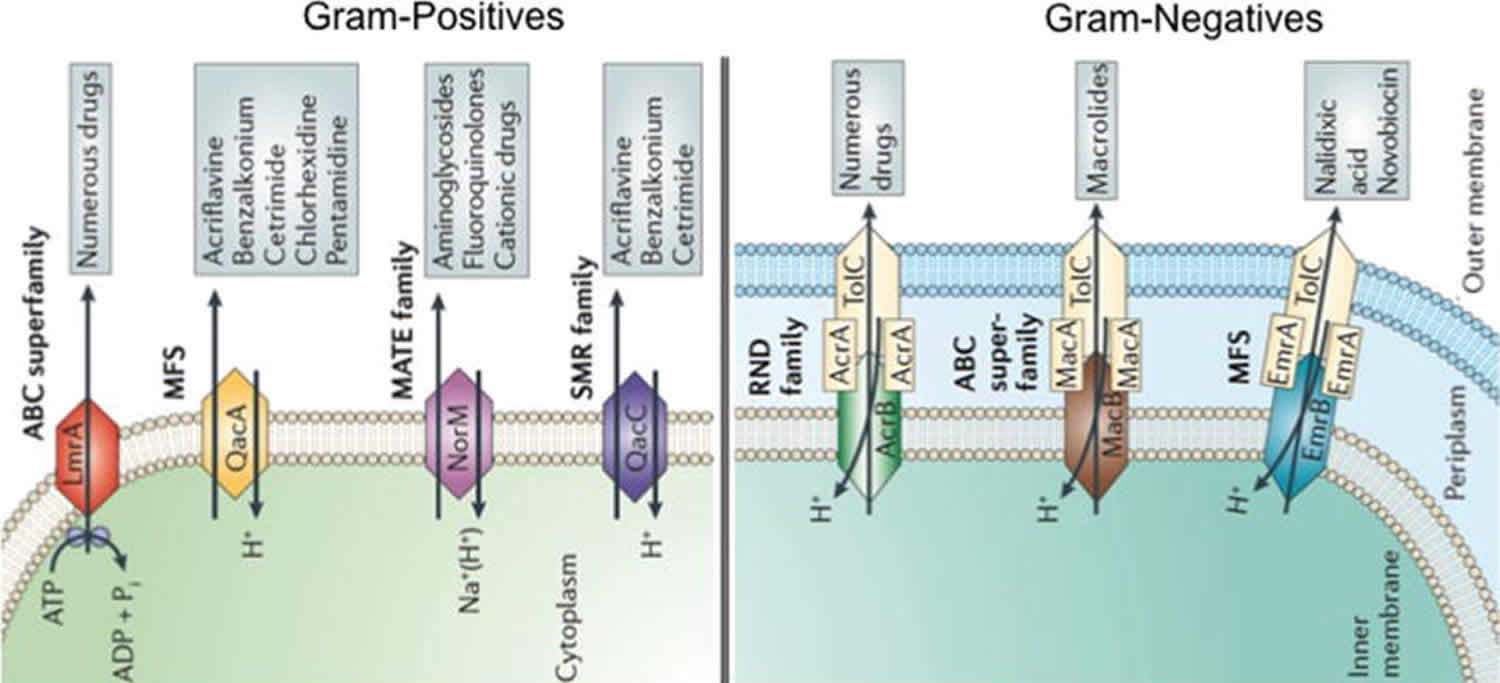
Footnote: Representation of different types of efflux pumps in Gram-positive and Gram-negative bacteria.
The five major families of efflux pumps are shown, ATP-binding cassette (ABC) superfamily, the major facilitator superfamily (MFS), the multidrug and toxic-compound extrusion (MATE) family, the small multidrug resistance (SMR) family and the resistance nodulation division (RND) family. A diagrammatic comparison of all the families showing their source of energy and examples of drugs and compounds that serve as a substrate are shown.
[Source 6 ]Tetracycline resistance is one of the classic examples of efflux-mediated resistance, where the Tet efflux pumps (belonging to the MFS family) extrude tetracyclines using proton exchange as the source of energy. Currently, more than 20 different tet genes have been described, most of which are harbored in MGEs. The majority of these genes are preferentially found in gram-negatives, with Tet(K) and Tet(L) being among the few exceptions that predominate in gram-positive organisms. Importantly, many of these pumps affect tetracycline and doxycycline, but do not decrease minocycline or tigecycline susceptibility, as they are not able to use these compounds as substrates 39. In addition to the tetracycline-specific transport systems, several MDR efflux pumps like AcrAB-TolC in Enterobacteriaceae and MexAB-OprM in P. aeruginosa (both belonging to the RND family) are able to extrude tetracyclines (including tigecycline) as part of their contribution to multidrug resistance 40. Of note, MDR pumps belonging to the RND family are frequently found in the chromosome of clinically relevant gram-negative bacteria and determine varying degrees of intrinsic resistance to several antimicrobials. Efflux pumps that belong to this family are organized as tripartite structures spanning the width of the gram-negative cell envelope and selectively communicating the cytoplasm with the external environment. Among them, one of the best studied is the AcrAB-TolC system (classically found in E. coli), which is composed of a transporter protein located in the inner membrane (AcrB), a linker protein located in the periplasmic space (AcrA), and a protein channel located in the outer membrane (TolC) (Dijun Du, Cell2015). RND pumps function as proton antiporters and are able to transport a wide array of substrates, conferring resistance to tetracyclines, chloramphenicol, some β-lactams, novobiocin, fusidic acid and fluoroquinolones. In addition, they are capable of extruding several toxic compounds like bile salts, cationic dyes and disinfectants, among many others. Crystalographic studies have provided insight on the structure and function of these pumps, improving our understanding of how these systems operate. Indeed, they have shown that AcrB has two binding pockets with different substrate preferences and that compounds are moved out of the cell through a series of conformational changes in a functionally rotating mechanism that finishes with the substrate being extruded via TolC (a process that requires an interaction with the periplasmic accessory protein AcrA). Of note, recent investigations have described a small protein named ArcZ, which has been shown to modulate and enhance the affinity of AcrB for certain molecules such as chloramphenicol and tetracycline through a mechanism that is yet to be determined.
Another important phenotype of clinical relevance mediated by the efflux mechanism is that of resistance to macrolides. The best characterized efflux pumps are encoded by the mef genes (mefA and mefE) that extrudes the macrolide class of antibiotics (e.g., erythromycin). The Mef pumps are mainly found in S. pyogenes and S. pneumoniae, along with other streptococci and gram-positive organisms. MefA is usually carried in a transposon (Tn1207) located in the chromosome and MefE is harbored in the so called “MEGA-element”, a fragment of DNA known as the macrolide efflux genetic assembly element that has been found inserted in different regions of the bacterial chromosome. Importantly, macrolide resistance due to these pumps does not result in cross-resistance to lincosamides and streptogramins (the so called MLSB group) 41.
Other efflux pumps resulting in macrolide resistance in gram-positives include MsrA and MsrC, which belong to the ABC transporter family. MsrA is a plasmid-borne determinant that was initially described in Staphylococcus epidermidis. MsrC is a chromosomally encoded protein described in E. faecalis that produces low-level resistance to macrolides and streptogramin B. Finally, another predicted efflux pump is Lsa (encoded by the chromosomal gene lsa), which is responsible for the intrinsic resistance of E. faecalis to lincosamides and streptogramin A (LSA phenotype) 36.
Changes in Target Sites
A common strategy for bacteria to develop antimicrobial resistance is to avoid the action of the antibiotic by interfering with their target site. To achieve this, bacteria have evolved different tactics, including protection of the target (avoiding the antibiotic to reach its binding site) and modifications of the target site that result in decreased affinity for the antibiotic molecule.
Target protection
Although some of the genetic determinants coding for proteins that mediate target protection have been found in the bacterial chromosome, most of the clinically relevant genes involved in this mechanism of resistance are carried by MGEs. Examples of drugs affected by this mechanism include tetracycline (Tet[M] and Tet[O]), fluoroquinolones (Qnr) and fusidic acid (FusB and FusC).
One of the classic and best-studied examples of the target protection mechanism is the tetracycline resistance determinants Tet(M) and Tet(O). Tet(M) was initially described in Streptococcus spp. and Tet(O) in Campylobacter jejuni, but they are now both widely distributed among different bacterial species, likely because they have been found in several plasmids and in broad-range conjugative transposons 42. These proteins belong to the translation factor superfamily of GTPases and act as homologues of elongation factors (EF-G and EF-Tu) used in protein synthesis. TetO and TetM interact with the ribosome and dislodge the tetracycline from its binding site in a GTP-dependent manner. Dönhöfer et al. recently showed that TetM directly dislodges and releases tetracycline from the ribosome by an interaction between the domain IV of the 16S rRNA and the tetracycline binding site. Furthermore, this interaction alters the ribosomal conformation, preventing rebinding of the antibiotic 43. Similarly, TetO has also been shown to compete with tetracycline for the same ribosomal space and to alter the geometry of the binding site of the antibiotic, displacing the molecule from the ribosome and allowing protein synthesis to resume 44.
Another example of target protection is the quinolone resistance protein Qnr, which is a plasmid-mediated fluoroquinolone resistance determinant frequently found in clinical isolates. Initially described in a clinical isolate of K. pneumoniae in the mid-1990s 45, Qnr belongs to the pentapeptide repeat protein family and it acts as a DNA homologue that competes for the DNA binding site of the DNA gyrase and topoisomerase IV. It is thought that this reduction in the DNA gyrase-DNA interaction decreases the opportunities of the quinolone molecule to form and stabilize the gyrase-cleaved DNA-quinolone complex that is lethal for the cell 46. Several different qnr alleles have been described to date, namely qnrA, qnrB, qnrC, qnrD, qnrS and qnrVC, all of which have a similar mechanism of action. Importantly, the presence of Qnr confers low-level quinolone resistance. However, harboring Qnr-encoding genes has been shown to promote the emergence of highly resistant isolates by facilitating the selection of mutants with point mutations in genes encoding the DNA gyrase and/or topoisomerase IV 47 (the predominant target of the fluoroquinolone class of antibiotics, see below).
Modification of the target site
Introducing modifications to the target site is one of the most common mechanisms of antibiotic resistance in bacterial pathogens affecting almost all families of antimicrobial compounds. These target changes may consist of i) point mutations in the genes encoding the target site, ii) enzymatic alterations of the binding site (e.g. addition of methyl groups), and/or iii) replacement or bypass of the original target. As mentioned, regardless of the type of change, the final effect is always the same, a decrease in the affinity of the antibiotic for the target site. Classical examples of each of these strategies will be detailed below.
Mutations of the target site
One of the most classical examples of mutational resistance is the development of rifampin (RIF) resistance. RIF is a rifamycin that blocks bacterial transcription by inhibiting the DNA-dependent RNA polymerase, which is a complex enzyme with a α2ββ’σ subunit structure. RIF binding pocket is a highly conserved structure located in the β subunit of the RNA polymerase (encoded by rpoB), and after binding, the antibiotic molecule interrupts transcription by directly blocking the path of the nascent RNA 48. High-level RIF resistance has been shown to occur by single-step point mutations resulting in amino acid substitutions in the rpoB gene and many different genetic changes have been reported. Of note, while these mutations result in decreased affinity of the drug for its target, they usually spare the catalytic activity of the polymerase, permitting transcription to continue 49.
Another well-characterized example of mutational resistance involves the mechanism of FQ resistance (as briefly mentioned above). FQs kill bacteria by altering DNA replication through the inhibition of two crucial enzymes, DNA gyrase and topoisomerase IV. Development of chromosomal mutations in the genes encoding subunits of the above-mentioned enzymes (gyrA-gyrB and parC-parE for DNA gyrase and topoisomerase IV, respectively) is the most frequent mechanism of acquired resistance to these compounds. Importantly, since FQs interact with two enzymes (DNA gyrase and topoisomerase), and both of them are essential for bacterial survival, the level of resistance achieved by developing changes in one of the enzymes will depend on the potency with which the antimicrobial inhibits the unaltered target. Thus, in contrast to the case of RIF, clinically relevant FQ resistance frequently requires an accumulation of genetic changes over time, with the first mutation producing minor increases in the MIC 50.
Finally, another good example of antibiotic resistance arising due to mutational changes is resistance to oxazolidinones (linezolid and tedizolid). These drugs are synthetic bacteriostatic antibiotics with broad gram-positive activity that exert their mechanism through an interaction with the A site of bacterial ribosomes. Such interaction inhibits protein synthesis by interfering with the positioning of the aminoacyl-tRNA. Linezolid is the most widely used antibiotic of this class, as tedizolid was only recently approved for clinical use. Although linezolid resistance remains an uncommon phenomenon, it has been well described in most clinically relevant gram-positives. The most commonly characterized mechanisms of linezolid resistance include mutations in genes encoding the domain V of the 23S rRNA and/or the ribosomal proteins L3 and L4 (rplC and rplD, respectively), and methylation of A2503 (E. coli numbering) in the 23S rRNA mediated by the Cfr enzyme (see below) (Figure 4) 51.
Mutations in genes encoding the central loop of the domain V of the 23S rRNA in the 50S ribosomal subunit are the most frequent determinants of linezolid resistance. A number of mutations have been described to date, and the most frequent change found in clinical isolates appears to be the transition G2576T (Escherichia coli numbering). Regardless of the position and type of genetic change, these mutations result in decreased affinity of the drug for its ribosomal target. Importantly, since bacteria carry multiple copies of the 23S rRNA genes, mutations need to accumulate in multiple alleles to yield a clinically relevant phenotype (gene-dose effect) 52. In addition, substitutions in the L3 and L4 ribosomal proteins have also been associated with development of linezolid resistance in vivo and in vitro, both alone and in combination with other resistance determinants 51.
Figure 4. Mechanism of antibiotic resistance – mutations of the target site
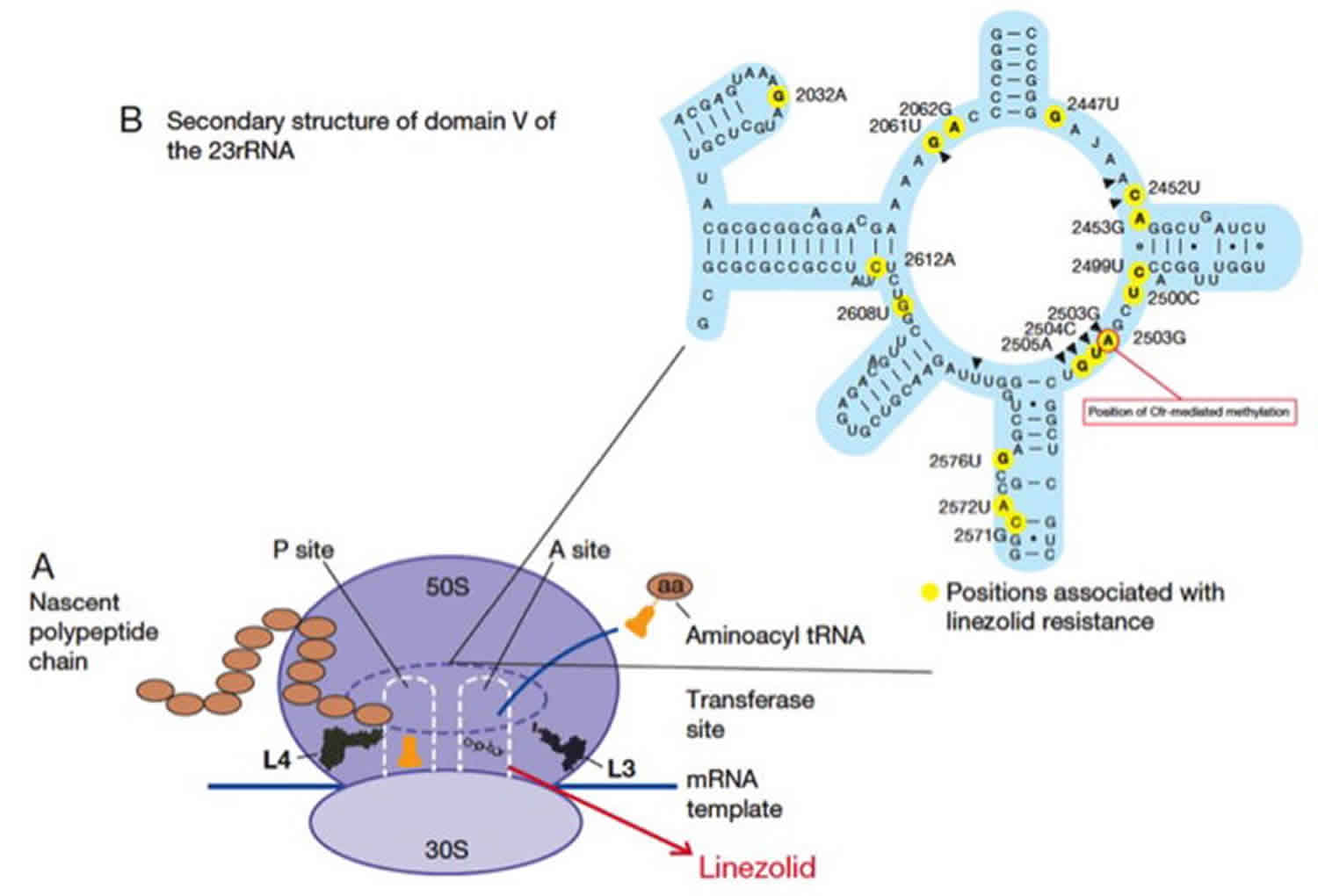
Footnote: Schematic representation of the mechanism of action and resistance to linezolid.
Panel A. Linezolid interferes with the positioning of aminoacyl-tRNA by interactions with the peptidyl-transferase center (PTC). Ribosomal proteins L3 and L4 associated with resistance are shown.
Panel B. Representation of domain V of 23S rRNA showing mutations associated with linezolid resistance. Position A2503, which is the target of Cfr methylation, is highlighted.
[Source 6 ]Enzymatic alteration of the target site
One of the best characterized examples of resistance through enzymatic modification of the target site is the methylation of the ribosome catalyzed by an enzyme encoded by the erm genes (erythromycin ribosomal methylation), which results in macrolide resistance. These enzymes are capable of mono- or dimethylating an adenine residue in position A2058 of the domain V of the 23rRNA of the 50S ribosomal subunit. Due to this biochemical change, the binding of the antimicrobial molecule to its target is impaired. Importantly, since macrolides, lincosamides, and streptogramin B antibiotics have overlapping binding sites in the 23S rRNA, expression of the erm genes confers cross-resistance to all members of the MLSB group 53. More than 30 different erm genes have been described, many of them located in MGEs, which may account for their ample distribution among different genera, including aerobic and anaerobic gram-positive and gram-negative bacteria. In staphylococci, the most important erm genes are ermA (mostly distributed in a transposon in MRSA) and erm(C) (found in plasmids in methicillin-susceptible S. aureus). On the other hand, erm(B) has been more frequently reported in enterococci and pneumococci (where it was first described), located in plasmids and conjugative and non-conjugative transposons such as Tn917 and Tn551. Importantly, these genes are widely distributed and have now been found in over 30 different bacterial genera 54. Erm-mediated resistance carries an important bacterial fitness cost due to less efficient translation by the methylated ribosome. Hence, although the MLSB phenotype can be constitutively expressed, in most cases it is subject to strict control via a complex posttranscriptional gene regulation. Through this mechanism, bacteria growing in the absence of antibiotics produce an inactive mRNA transcript that cannot be translated into the desired protein (in this case a methylase). Conversely, in the presence of antibiotic, the transcript becomes active and the system is primed to confer rapid resistance. This is best characterized by the inducible MLSB phenotype of the erm(C) operon in S. aureus, which is conformed by the erm(C) gene, an upstream gene encoding a leader peptide and an intergenic region (Figure 5). In the absence of an inducer, transcription of the operon generates an mRNA with a secondary structure that conceals the ribosomal binding site upstream of erm. Translation proceeds through the leader peptide, then terminates, preventing the production of ErmC. In the presence erythromycin (but also other macrolides), the ribosome stalls due to inhibition by the antibiotic during translation of the leader peptide allowing a conformational change in the ermC mRNA that unmasks its ribosomal binding site, resulting in efficient translation of erm(C) 55. Thus, bacteria have evolved a sophisticated mRNA-based control mechanism to tightly regulate the expression of these methylases, ensuring a high efficiency of action in the presence of the antibiotic while minimizing the fitness costs for the bacterial population. The array of compounds capable of inducing the MLSB phenotype varies among different erm genes, but as a general rule the best inducer is erythromycin while the inducing ability of other macrolides varies. Similarly, the system is usually not induced by lincosamides or streptogramins. However, the use of these agents against isolates carrying inducible erm genes may result in the selection of constitutive mutants in vivo (particularly in severe infections) leading to therapeutic failures.
Another relevant example of enzymatic alteration of the target is Cfr-mediated linezolid resistance. The cfr gene is a plasmid-borne determinant initially described in 2000 in a bovine isolate of Staphylococcus sciuri and first reported in humans in 2005 in an S. aureus isolated from a patient in Colombia 56. Since then, it has been found in several species of human pathogens, including S. aureus, E. faecalis, E. faecium and some Gram-negative bacteria. This gene encodes the Cfr enzyme, which is a member of the S-adenosyl-L-methionine (SAM) methylase family that also confers resistance to phenicols, lincosamides, pleuromutilins, and streptogramin A. Moreover, cfr has been associated with various MGEs suggesting that it has an enhanced potential of spread and to cause transferable linezolid resistance in the future. Importantly, carriage of cfr does not appear to confer resistance to the recently FDA-approved oxazolidinone tedizolid 57.
Figure 5. Mechanism of antibiotic resistance – enzymatic alteration of the target site
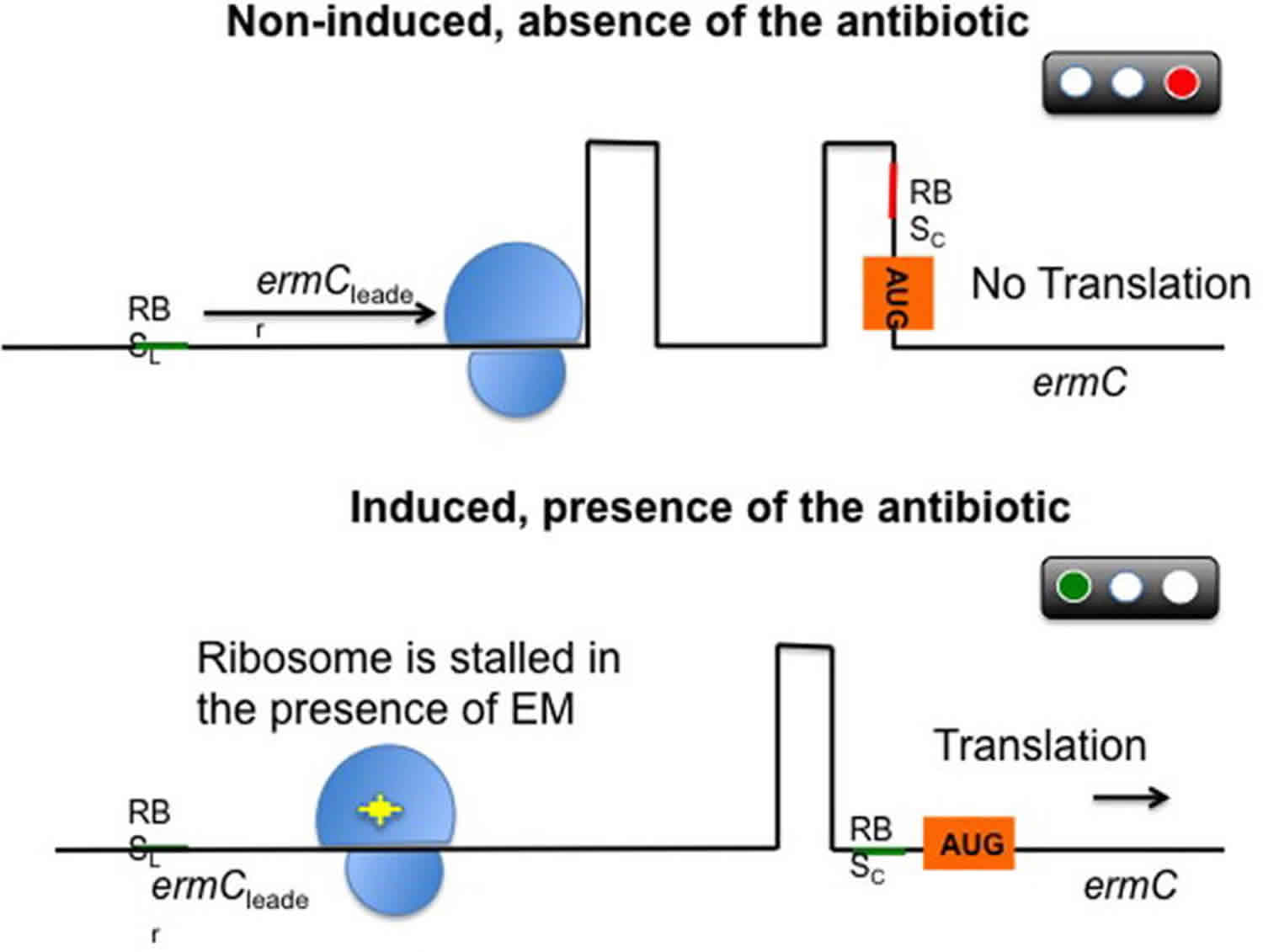
Footnote: Schematic representation of the post-transcriptional control of the ermC gene.
Under non-inducing conditions, the ErmC leader peptide is produced and the ermC mRNA forms two hairpins, preventing the ribosome to recognize the ribosomal binding site (RBS) of ermC. As a result, translation is inhibited. After exposure to erythromycin (EM, yellow star), the antibiotic interacts with the ribosome and binds tightly to the leader peptide, stalling progression of translation. This phenomenon releases the ermC RBS and permits translation.
RBSL, ribosomal binding site of the leader; RBSC, ribosomal binding site of ermC; AUG, initiation codon. Ribosome represented in blue and erythromycin in yellow.
[Source 6 ]Complete replacement or bypass of the target site
Using this strategy, bacteria are capable of evolving new targets that accomplish similar biochemical functions of the original target but are not inhibited by the antimicrobial molecule. The most relevant clinical examples include methicillin resistance in S. aureus due to the acquisition of an exogenous PBP (PBP2a) and vancomycin resistance in enterococci through modifications of the peptidoglycan structure mediated by the van gene clusters. Finally, another route to avoid the antimicrobial action is to “bypass” the metabolic pathway they inhibit by overproducing the antibiotic target. A relevant example of this mechanism is resistance to trimethoprim-sulfamethoxazole (TMP-SMX). In the remainder of the section we will provide further details of the examples mentioned above.
The antibacterial activity of β-lactams relies on their ability to disrupt cell wall synthesis through inhibition of PBPs which are important enzymes responsible for the transpeptidation and transglycosylation of peptidoglycan units emerging from the cytoplasm. Resistance to methicillin (a semisynthetic penicillin stable against the staphylococcal penicillinase) in S. aureus results from the acquisition of a foreign gene (likely from Staphylococcus sciuri) designated mecA often located in a large DNA fragment designated staphylococcal chromosomal cassette mec (SCCmec). The mecA gene encodes PBP2a, a PBP that has low affinity for all β-lactams, including penicillins, cephalosporins (except for last generation compounds) and carbapenems. Acquisition of mecA renders most β-lactams useless against MRSA and alternative therapies need to be used in serious infections. Of note, PBP2a carries a transpeptidase domain, but it does not function as a transglycosylase (class B PBP), therefore, it requires the activity of other native PBPs to perform the latter function and fully crosslink peptidoglycan. Specifically, the penicillin-insensitive transglycosylase domain of PBP2 (a class A PBP) is particularly important to achieve transglycosylation of peptidoglycan in the presence of β-lactams in mecA-carrying MRSA isolates.
As mentioned above, the mecA gene is usually found as part of a gene cassette inserted into a larger MGE (SCCmec), whose basic components include mecA, mecR1 (encoding the signal transducer protein MecR1), mecI (encoding the repressor protein Mecl), and ccr (encoding a recombinase; cassette chromosome recombinase). To date, 11 different SCCmec allotypes have been described with varying degrees of genetic homology and different sizes, insertion sequences and accompanying resistance genes 58. Importantly, SCCmec types seem to differ between different MRSA clones. Indeed, community-associated MRSA strains appear to harbor shorter SCCmec cassettes (e.g SCCmec type IV) and carry less antibiotic resistance determinants, whereas hospital-associated (HA) isolates possess longer elements (e.g. SCCmec type II) and are usually multidrug resistant 59.
A two-component regulatory system that includes the repressor protein Mecl and the signal transducer MecR1 regulates the expression of mecA. Once MecR1 senses the presence of β-lactams in the environment, it triggers a signal transduction cascade that removes the MecI repressor from its DNA binding site resulting in transcription of mecA and its regulatory genes. These events culminate with the production of PBP2a, which is the hallmark of methicillin resistance in S. aureus 59.
Another important example of the replacement and bypass strategy to achieve resistance is related to vancomycin resistance. Similar to β-lactams, glycopeptides (i.e., vancomycin and teicoplanin) kill bacteria by inhibiting cell wall synthesis. However, unlike β-lactams, glycopeptides do not directly interact with PBPs. Instead, they bind to the terminal D-alanine-D-alanine (D-Ala-D-Ala) of the pentapeptide moiety of the nascent peptidoglycan precursors (lipid II), preventing PBP-mediated cross-linking and resulting in inhibition of cell wall synthesis. It has been postulated that the main effect of the binding of vancomycin to D-Ala-D-Ala-ending precursors emerging from the cytoplasm is alteration of transglycosylation (presumably due to steric hindrance) preventing further processing of the cell wall and leading to bacterial death 60.
Vancomycin resistance is especially relevant in enterococci (particularly E. faecium) and it is usually accompanied by the presence of other resistance determinants, making the treatment of infections caused by these organisms an important clinical challenge 61. Vancomycin resistance in enterococci involves the acquisition of a group of genes (designated van gene clusters) that code for a biochemical machinery that remodels the synthesis of peptidoglycan by, i) changing the last D-Ala for either D-lactate (high-level resistance) or D-serine (low-level resistance), and ii) destroying the “normal” D-Ala-D-Ala ending precursors to prevent vancomycin binding to the cell wall precursors. The change of D-Ala for D-lactate removes a single hydrogen bond between the vancomycin molecule and its target (D-Ala-D-Ala moiety) decreasing the antibiotic affinity for the precursor ca. 1,000 fold. Although the change of D-Ala for D-Ser does not remove any of the 5 hydrogen bonds between vancomycin and its target, the presence of the hydroxyl group of serine affects the interaction of the antibiotic with the precursors reducing its affinity, albeit less markedly than with the D-Lac replacement 62.
The origin of the van genes has been a topic of intense investigation. Genes nearly identical to those of the vanA gene cluster (the most prevalent in clinical enterococcal strains) have been found in soil organisms such as Paenibacillus thiaminoluticus and P. apiaries 63. To date, nine distinct enterococcal van clusters have been described (vanA, vanB, vanC, vanD, vanE, vanG, vanL, vanM and vanN). The vanADLM clusters synthesize precursors ending in D-Lac whereas the vanCEGN produce D-Ser-ending peptidoglycan. Most clinical VRE isolates carry the vanA or vanB gene clusters, which are usually found in MGEs either associated with plasmids or inserted in the chromosome. We will provide further detail of the biochemical mechanism of VanA-mediated resistance (involving both vancomycin and teicoplanin). The reader is referred to other comprehensive reviews for additional details on glycopeptide resistance 62.
The vanA gene cluster is usually located on a Tn3-family transposon designated Tn1546, which has been found on both conjugative and non-conjugative plasmids. This gene cluster consists of 7 genes coding for three groups of proteins, i) a classical two-component regulatory system that regulates the expression of resistance (VanS is the histidine kinase and VanR the response regulator of the system), ii) enzymes necessary for the synthesis of new peptidoglycan precursors, namely a dehydrogenase (VanH) and an amino acid ligase with altered substrate specificity (VanA) capable of producing D-Ala-D-Lac, and iii) enzymes that destroy the normal D-Ala-D-Ala-ending precursors (VanX and VanY). Of note, an additional gene, vanZ, is present in Tn1546, but its function remains unknown.
Induction of the vanA gene cluster appears to involve initial sensing by VanS of the accumulation of substrates resulting from inhibition of glycosyltransferase activity 64. This initial step results in an ATP-dependent phosphorylation of the response regulator VanR, which subsequently binds to two promoters, one of them located upstream of its own gene (vanR) and the other upstream of vanH in Tn1546 65. The vanH gene encodes a dehydrogenase enzyme necessary for the production of D-lactate using pyruvate as substrate. D-Lac is then bound to a molecule of D-Ala by the VanA ligase and the D-Ala-D-Lac dipeptide is subsequently added to the nascent tripeptide (MurNAc-L-Ala1-γ-D-Glu2-L-Lys3) to form the altered peptidoglycan unit (UDP-MurNAc-pentadepsipeptide; Mur-NAc-L-Ala1-γ-D-Glu2-L-Lys3-D-Ala4-D-Lac5) (Figure 6).
Figure 6. Mechanism of antibiotic resistance – complete replacement or bypass of the target site
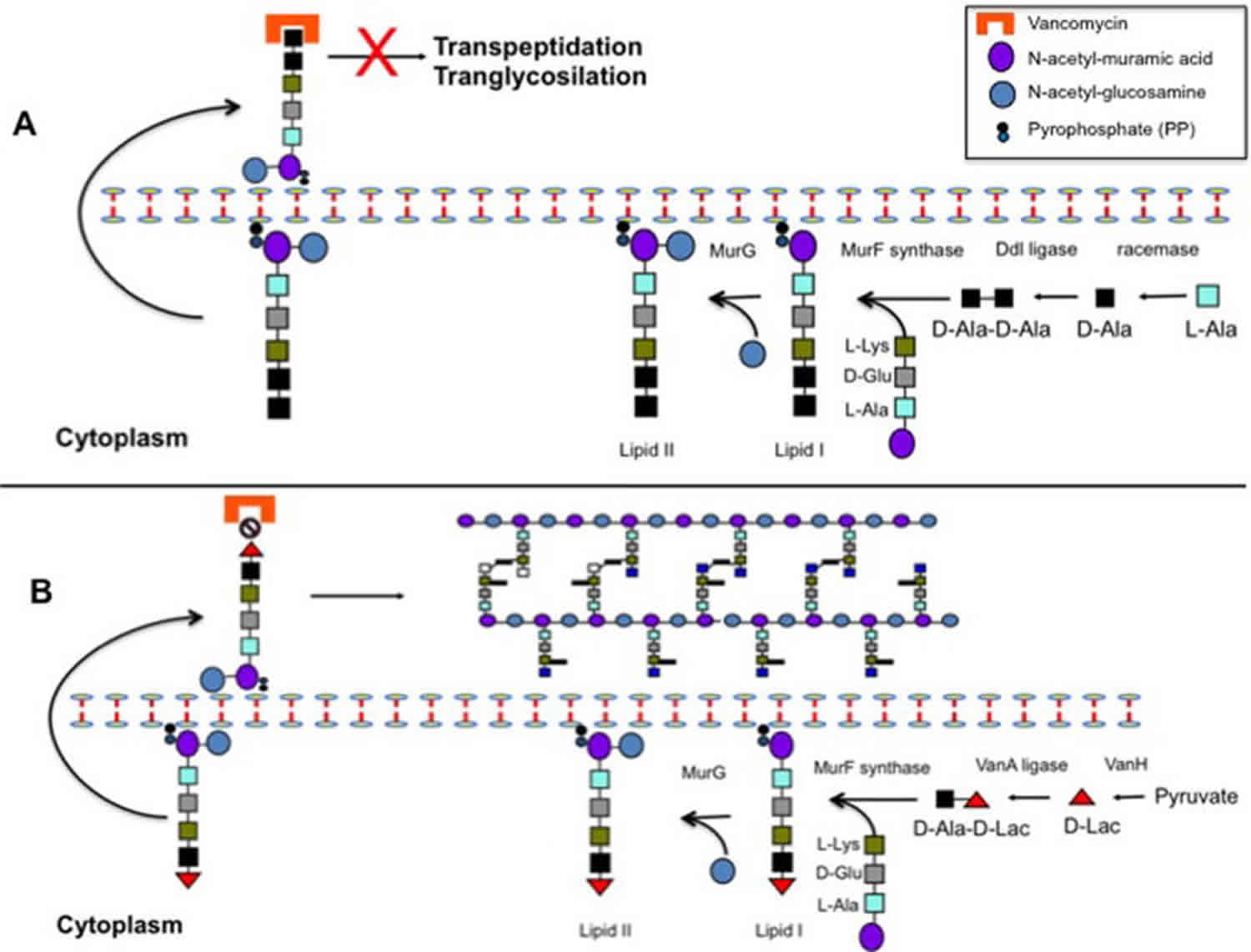
Footnote: Schematic representation of peptidoglycan biosynthesis and mechanisms of vancomycin action (A) and resistance (B)
Panel A depicts normal peptidoglycan production and shows that binding of the antibiotic to the terminal D-Ala-D-Ala of the peptidoglycan precursors prevents transpeptidation and transglycosylation, interrupting cell wall synthesis and resulting in bacterial death.
Panel B shows the change in peptidoglycan synthesis produced by the expression of the vanA gene cluster. Change of the terminal dipeptide from D-Ala-D-Ala to D-Ala-D-Lac markedly reduces the binding of vancomycin to the peptidoglycan target permitting cell wall synthesis to continue.
[Source 6 ]As mentioned above, the other genes of Tn1546 code for enzymes that destroy D-Ala ending precursors. The vanX gene encodes a D,D-dipeptidase that hydrolyzes any D-Ala-D-Ala produced in the “normal” peptidoglycan synthesis pathway and vanY codes for a membrane-bound D,D-carboxypeptidase that removes the last D-Ala of normal ending precursors, ensuring that no D-Ala-D-Ala-ending pentapeptides (that could potentially bind vancomycin) are exposed on the cell surface. Finally, Tn1546 harbors another gene (designated vanZ) that appears to be involved in teicoplanin resistance (but not vancomycin) whose function is unknown 66.
Development of high-level vancomycin resistance in S. aureus (vancomycin-resistant S. aureus, VRSA) was first described in 2002 and was the result of acquisition by an MRSA strain of the vanA gene cluster from a VRE (E. faecalis) isolate 67. However, occurrence of this phenomenon continues to be rare. Although transfer of an enterococcal plasmid containing the vanA gene cluster in Tn1546 to S. aureus has been shown to occur in vitro, the efficiency of this mechanism is low since replication of enterococcal plasmids in staphylococci is frequently suboptimal. However, a potentially more worrisome scenario is the acquisition of the vanA gene cluster by community-associated strains using native staphylococcal plasmids. Indeed, a recent report described such phenomenon where the vanA gene cluster was harbored in a highly transferable staphylococcal plasmid originally identified in community-associated S. aureus isolates. The isolate was found in a bloodstream isolate of MRSA recovered from a Brazilian patient 68 and transfer to a methicillin-susceptible isolate within the same patient was also documented.
The vanB gene cluster harbor similar genes to those carried by the vanA cluster with the difference that the VanSB sensor kinase does not appear to be activated by the presence of teicoplanin. Thus, isolates harboring the vanB cluster remain susceptible to this glycopeptide. The vanB gene cluster is also carried by mobile elements in Tn1547 or related conjugative transposons and has been identified in pheromone-responsive plasmids. In addition, the vanB cluster lacks the vanZ gene and carries an additional gene (designated vanW) whose function remains to be established.
The prototypical gene cluster responsible for low-level vancomycin resistance and the production of D-Ala-D-Ser peptidoglycan precursors is vanC. Of note, enterococci carrying the vanEGLN clusters also produce D-Ala-D-Ser and exhibit low level of vancomycin resistance and carry similar genes to those described in vanC. The main differences in terms of gene content and biochemical activities of VanC-type mediated resistance compared with VanA and VanB are, i) they encode a unique racemase (VanT) capable of producing D-serine using L-serine as substrate, ii) they possess a ligase (vanCEGLN) with the ability to synthesize a D-Ala-D-Ser dipeptides, and iii) they often harbor a single gene (vanXY) encoding both D-Ala-D-Ala dipeptidase and carboxypeptidase activities that are normally coded for two distinct genes in the other clusters (vanX and vanY, see above)
On rare occasions, VRE strains can develop null mutations in the native D-Ala-D-Ala ligase (ddl) that abolish the normal production of D-Ala-D-Ala for peptidoglycan synthesis. Thus, strains harboring such mutations rely on the production of altered peptidoglycan precursors for cell wall synthesis by the inducible van clusters (e.g., vanA). Therefore, cell survival depends on the permanent presence of the antibiotic to induce the system (hence, these isolates are designated vancomycin-dependent enterococci). This phenotype seems to be unstable since mutations in the VanS sensor or promoter regions frequently revert the phenotype 69.
As mentioned above, another well-described strategy of “target bypass” is by increasing the production of the antimicrobial target with the objective of overwhelming the antibiotic by increasing the amount of targets available. One of the best examples of this mechanism is development of resistance to TMP-SMX. This drug impairs bacterial synthesis of purines and some important amino acids by altering the production of folate, exploiting the fact that most bacteria are unable to incorporate folate from external sources. Therefore, bacteria rely on their own biochemical machinery for folate synthesis. The synthetic pathway of folate involves two major enzymes, namely i) dihydropteroic acid synthase (DHPS), which forms dihydrofolate from para-aminobenzoic acid (inhibited by SMX), and ii) dihydrofolate reductase (DHFR), which catalyzes the formation of tetrahydrofolate from dihydrofolate (inhibited by TMP).
Although development of resistance to TMP-SMX can be achieved through several strategies including amino acid changes in the above enzymes (decreasing their affinity for the antibiotic molecules, target modification) and acquisition of external genes encoding DHPS or DHFR that are less sensitive to inhibition by TMP/SMX (target bypass), a “clever” bypass strategy is the overproduction of DHFR or DHPS through mutations in the promoter region of the DNA encoding these enzymes. These mutations result in the production of increased quantities of the above enzymes, “overwhelming” the ability of TMP-SMX to inhibit folate production and permitting bacterial survival 70. Interestingly, enterococci use another “bypass” strategy by incorporating exogenous tetrahydrofolic acid and folinic acid when added to the media. This ability to use folate from different sources is correlated with up to 25-fold increase in the MICs of TMP-SMX and are thought to impair the antimicrobial activity in vivo 71.
Antibiotic resistance due to global cell adaptations
Through years of evolution, bacteria have developed sophisticated mechanisms to cope with environmental stressors and pressures in order to survive the most hostile environments, including the human body. Bacteria need to compete for nutrients and avoid the attack of molecules produced by other rival organisms in order to gain the “upper hand”. Inside a particular host, bacterial organisms are constantly attacked by the host’s immune system and in order establish themselves in particular biological niches, it is crucial that they adapt and cope with these stressful situations. Thus, bacterial pathogens have devised very complex mechanisms to avoid the disruption of pivotal cellular process such as cell wall synthesis and membrane homeostasis. Development of resistance to daptomycin (DAP) and vancomycin (low-level in S. aureus) are the most clinically relevant examples of resistance phenotypes that are the result of a global cell adaptive response to the antibacterial attack.
DAP is a lipopeptide antibiotic related to cationic antimicrobial peptides (CAMPs) produced by the innate immune system that exerts its bactericidal effect by altering cell envelope homeostasis. The bactericidal activity of DAP requires four important steps (Figure 7). First, DAP is complexed with calcium (rendering the molecule positively charged) and, subsequently, is directed to the CM target by electrostatic interactions with the usually negatively charged cell membrane (CM). Of note, recent evidence suggests that DAP mainly targets the CM at the level of the division septum 72. Second, once the antibiotic molecules reach the CM, it initially oligomerizes at the outer leaflet of the CM and, subsequently, these DAP oligomers reach the inner CM leaflet. DAP’s oligomerization at the outer leaflet of the CM appears to be dependent on the presence of the phospholipid phosphatidylglycerol (PG) 73. Additionally, another phospholipid (cardiolipin, CL), seems to play an important role in the translocation of DAP oligomers from the outer of the inner leaflet, but its contribution is not completely understood. In fact, there is evidence to suggest that the presence of CL in high concentrations may prevent translocation of DAP oligomers into the inner leaflet of the phospholipid bilayer 73. Third, once the DAP oligomers reach the inner leaflet of the CM, they organize and form transmembrane pore-like structures that are likely to alter the physicochemical properties of the CM and promote leakage of ions (e.g, potassium) from the cytoplasm, causing important electrochemical alterations. Finally, these structural and functional CM alterations lead to bacterial death in the absence of cell lysis by mechanisms that are not fully understood.
Figure 7. Mechanism of antibiotic resistance – global cell adaptations
Footnote: Diagrammatic representation of the mechanism of action of daptomycin.
[Source 6 ]Bacteria have developed ancient systems of defense to withstand CAMP action and possess a cadre of regulatory systems that are involved in protecting the cell envelope when under attack by CAMPs. In enterococci, work using whole genome sequencing of a clinical strain-pair of E. faecalis that developed DAP resistance (DAP-R) over the course of therapy, revealed that changes in a three-component regulatory system designated LiaFSR (which orchestrates the cell-envelope stress response in gram-positives) are paramount in the development of DAP-R 74. In B. subtilis, where the system was first characterized, and other Gram-positive bacteria, LiaFSR (and the homolog system VraTSR in S. aureus) is composed of three proteins, i) LiaF (VraT), a transmembrane protein that appears to negatively regulate the system, ii) LiaS (VraS), a classical sensor-histidine kinase protein that phosphorylates the response regulator, and iii) LiaR (VraR), the response regulator of the system. Indeed, a single deletion of an isoleucine at position 177 of LiaF increased the DAP MIC from 1 to 4 μg/mL (established clinical breakpoint is 4 μg/ml) and, more importantly, was sufficient to abolish the bactericidal activity of DAP 75. Moreover, in a recent genomic analyses of 19 DAP non-susceptible E. faecium (DAP MICs from 3 to 48 μg/mL, clinical breakpoint is 4 μg/mL), the most frequently identified mutations were in liaFSR, supporting the hypothesis that changes in this system are a pivotal step towards DAP-R in enterococci 76. Furthermore, the majority (75%) of DAP-susceptible E. faecium isolates recovered from bacteremic patients whose MIC was in the higher range of susceptibility (i.e. between 3 and 4 μg/mL) harbored mutations in LiaFSR. Conversely, none of the isolates of the same collection with DAP MIC ≤ 2 μg/mL exhibited changes in this system 77. More importantly, these changes were sufficient to abolish the in vitro bactericidal activity of DAP and were associated with a clinical failure in a neutropenic patient with VRE bacteremia 78.
The mechanism by which LiaFSR results in DAP-R is not fully understood. Furthermore, the specific mechanism through which this system orchestrates the cell-envelope response to stress is still a matter of active research. In B. subtilis, the lia locus consists of six genes, liaIH-liaGFSR, of which liaGFSR are constitutively expressed at a low basal level due to the presence of a weak constitutive promoter upstream of liaG. In contrast, expression of liaIH is completely LiaR-dependent. Although LiaR regulates other genes, Wolf et al. provided evidence indicating that liaIH is the only relevant target of LiaR-dependent gene expression in wild-type cells. The physiological role of LiaI and LiaH are not completely understood. LiaI is a small hydrophobic protein of unknown function with two putative transmembrane helices and LiaH is a member of the phage-shock protein family that forms large oligomeric rings-like structures (resembling what has been reported with PspA in E. coli). Importantly, the LiaFSR system constitutes a cell envelope stress-sensing/response system that is highly conserved in Firmicutes bacteria 79.
In enterococci, recent evidence suggests that LiaR mediates a reorganization of anionic phospholipids (i.e., cardiolipin) in the CM associated with DAP-R. In a clinical strain of DAP-R, development of resistance was clearly associated with redistribution of CL microdomains from the septum to other CM areas 80. This mislocalization seems to “divert” DAP from its principle CM septal target, resulting in bacterial survival to the DAP “attack”. Furthermore, deletion of LiaR completely reverted DAP susceptibility and restored the organization of CL domains 81.
Other regulatory systems involved in cell envelope homeostasis have also been associated with DAP-R. For example, YycFG (WalKR), an essential two-component regulatory system that has been implicated in cell-wall synthesis and homeostasis, has been found to be important to DAP-R both in enterococci and S. aureus 82. Although the exact mechanism mediating this phenomenon has not been fully elucidated, it appears to involve alteration in cell wall metabolism that results in changes in surface charge producing electrostatic ‘repulsion” of the positively charged calcium-DAP complex from the cell envelope.
A second group of genes that have been shown to contribute to the development of DAP-R correspond to enzymes involved in the metabolism of CM phospholipids. For example, two enzymes, a glycerol-phosphodiester phosphodiesterase (GdpD) and cardiolipin synthase (Cls) were found to enhance the DAP-R phenotype in the background of liaFSR mutations in E. faecalis 74. These changes seem to alter the CM phospholipid composition mainly by decreasing the amount of PG. Other enzymes such as MprF, PG synthase (PgsA), cyclic fatty acid synthase (Cfa) and geranyltransferase (98) involved in CM phospholipid homeostasis have also been linked to DAP-R. In S. aureus, MprF (a lysyl-PG [LPG] synthase) has been one of the most studied enzymes, and inactivation of this protein reversed DAP-R. This enzyme harbors two domains, i) a lysyl-transferase domain that transfers the positively-charged amino acid lysine from its tRNA carrier to PG (LPG) in the inner leaflet of the CM, and ii) a flipase domain, through which newly synthesized LPG is translocated from the inner to the outer leaflet of the CM (99). Mutations in mprF appear to produce a gain of function of the enzyme and, as a result, the cell surface becomes more positively charged, repelling the DAP-calcium complex (also with positive charge). Of interest, a homolog of LiaFSR (VraTSR) seems to also contribute to the DAP-R phenotype mediated by changes in MprF in S. aureus 83.
As discussed above, the development of high-level vancomycin resistance mediated by the acquisition of the van gene cluster is a rare event in S. aureus. However, a much more common problem is the finding of S. aureus isolates with intermediate susceptibility to vancomycin (known as VISA isolates), exhibiting MICs between 4 – 8 μg/mL. This phenomenon was first reported in Japan in 1997 and led to therapeutic failure 84. The isolate, designated Mu50, was derived from a vancomycin susceptible strain known as Mu3 (≤ 2 μg/mL). Population analyses of Mu3 later confirmed that this strain contained a subpopulation of bacterial cells capable of surviving at concentrations of vancomycin above 2 μg/mL (clinical breakpoint for susceptibility), a phenomenon that has now been designated the heterogenous-VISA (hVISA) phenotype. As the “resistant” subpopulation is difficult to identify, detection of hVISA becomes very challenging and some S. aureus strains reported as “susceptible” to vancomycin by standard susceptibility testing may still exhibit this phenotype 85. Indeed, S. aureus within the range of susceptibility, with vancomycin MICs > 1 and ≤ 2 μg/ml have been more frequently associated with the hVISA phenotype. Due to the difficulties on detecting these strains, vancomycin failures have been increasingly reported in deep-seated infections. Published studies estimate that the overall prevalence of MRSA strains with hVISA/VISA profile range between 0 and 8.24%, but it can be as high as 30% in selected populations (e.g. patients with MRSA infective endocarditis) 86.
The hVISA/VISA isolates usually emerge in vivo in patients with a history of an MRSA infection that failed to a prolonged course of vancomycin therapy. From a mechanistic point of view, the development of the hVISA/VISA does not occur by the acquisition of foreign DNA material (as seen in VRSA), rather, the phenotype appears to be the result of sequential and ordered genetic changes that usually involve genes forming part of regulatory systems controlling cell envelope homeostasis (similar to that described above for DAP). The specific mechanisms that lead to the hVISA/VISA phenotype remain to be completely understood. However, the available evidence shows that the regulatory systems most consistently implicated in this mechanism of resistance are YycFG (WalKR), VraSR (homolog of LiaFSR), and GraRS 87. Interestingly, these two- and three component regulatory systems are involved in cell wall homeostasis, supporting the notion that selection of the hVISA/VISA phenotype involves important remodeling of the cell wall in order to survive the antimicrobial attack.
Apart from the above regulatory systems, another change that has been frequently associated with the VISA phenotype is mutations in rpoB (encoding the B subunit of the RNA polymerase). Indeed, Watanabe et al. analyzed 38 VISA isolates from 10 different countries and demonstrated that mutations in the rpoB gene were present in the majority (71%) of the isolates 88. However, the mechanisms by which mutation if rpoB lead to reduced vancomycin and DAP susceptibility are unclear.
Phenotypically, hVISA/VISA strains exhibit distinct metabolic characteristics that may include i) increase in fructose utilization, ii) increased fatty acid metabolism, iii) impaired acetate metabolism and tricarboxylic acid cycle, iv) decrease in glutamate availability, and iv) increase expression of cell wall synthesis genes. These global homeostatic changes appear to lead to a reduced autolytic activity with thickened cell wall and an increase amount of free D-Ala-D-Ala dipeptides with less peptidoglycan cross-linking 85. In addition, VISA strains bind vancomycin more avidly than their non –VISA counterparts, however, diffusion of the antibiotic molecule into the inner part of the cell wall appears to be impaired. Hence, it has been postulated that these changes result in “trapping” of vancomycin in outer layers of the peptidoglycan preventing the antibiotic molecule from reaching its target of precursors emerging from the cytoplasmic membrane. As a result, cell wall synthesis and peptidoglycan cross-linking continues to be uninterrupted.
Finally, a striking feature of many hVISA/VISA strains is the ability to revert from one phenotype to another (or even to a fully vancomycin susceptible phenotype) in the absence of vancomycin exposure. Therefore, there seems to be a “price” to pay for developing resistance and this is yet another example of the ability of bacteria to adapt to the environment by means of their remarkable genetic plasticity.
How to prevent antibiotic resistance
Antibiotic resistance can’t be totally stopped, but it can be slowed down by sensibly using antibiotics. You can help by:
- Not taking antibiotics for a cold or the flu, including cough and sore throat; viruses cause most colds, and antibiotics don’t work against viruses
- Telling your doctor you only want antibiotics when necessary – such as for serious bacterial infections such as pneumonia
- Taking your antibiotic as prescribed, and completing the full course
- Never saving antibiotics for the next time you’re sick
- Never taking antibiotics prescribed for someone else
- Get vaccinated. Vaccination is one of the best ways to prevent illnesses. Every year, thousands of Americans get sick from diseases that could be prevented by vaccines. Talk to your child’s healthcare provider about recommended vaccines, and learn more about vaccines recommended for all ages.
- Having good hygiene practices to avoid spreading infections. By washing your hands often and thoroughly with soap and water, you are helping to prevent disease – and therefore the need for antibiotics.
- Prepare Food Safely. Cooking meat thoroughly and handling food hygienically will help to prevent food-borne illnesses.
- Protect yourself from sexually transmitted disease (STD). Gonorrhea, a common sexually transmitted disease (STD), is becoming harder to treat due to increasing drug resistance. If you are diagnosed with gonorrhea and your symptoms continue for more than a few days after receiving treatment, then return to a healthcare provider to be checked again.
- Stay healthy when traveling abroad.
Are antibacterial agents, such as antibacterial soaps, a solution?
In institutions such as hospitals and nursing homes, these agents are useful and appropriate when used under strict guidelines for specific purposes. However, there is some concern that antibacterials could promote antibiotic resistance and their usefulness by the general public is unproven.
Can the effectiveness of existing antibiotics be preserved?
To preserve the potency of existing antibiotics, overall antibiotic use must be decreased. Physicians, pharmacists, and the general public must avoid careless use of these valuable drugs. Antibiotics must be prescribed only for bacterial infections and in the proper dose for the correct amount of time. Narrow spectrum drugs should be chosen by doctors whenever possible to avoid destroying populations of beneficial bacteria along with the disease-causing bacteria. In addition, non-therapeutic uses of antibiotics in farm animals and agriculture should be eliminated.
Can new antibiotics be developed?
The epidemic of resistant bacteria has spurred renewed interest in finding novel antibiotics. The process of producing a new antibiotic, however, is long and expensive, requiring approximately ten years and $300 million to bring a new antibiotic to market. Many efforts to find novel drugs in fungi and soil result in compounds that are the same or very similar to previously discovered antibiotics. Thus, resistance eventually develops to these new antibiotics. Heavy use of the latest antibiotic can lead to the emergence of resistance in as little as two years. Nonetheless, scientists are still searching for new antibiotics by looking in unusual places such as in bacteria living deep below the earth’s surface, in the skin of frogs and in certain insects.
Can antibiotic resistance be overcome?
One approach taken by scientists to combat antibiotic resistance is to strengthen the action of existing antibiotics by modifying them so the bacterial enzymes that cause resistance cannot attack them. Alternately, “decoy” molecules can be used along with the antibiotic, so that the bacterium’s resistance enzyme attacks the decoy molecule rather than the antibiotic. Decoy molecules such as clavulanic acid or sulbactam are already in use for blocking the beta-lactamase enzymes that destroy the penicillin family of drugs.
An alternative approach to the antibiotic resistance problem is to interfere with the mechanisms that promote resistance, rather than to attempt to kill the bacteria. For example, interfering with the duplication or movement of a bacterium’s genetic material would eliminate the transfer of resistance genes between bacteria.
Can antibiotic resistance develop from using acne medication?
Yes. Antibiotic use, appropriate or not, contributes to the development of antibiotic resistance. This is true for acne medications that contain antibiotics. Short- and long-term use of antibiotics for treatment or prevention of bacterial infections should be under the direction of a healthcare professional to ensure appropriate use and detection of resistance.
Do probiotics have a role in helping to reduce antibiotic resistance?
Probiotics are defined as microorganisms that when administered in sufficient quantities may improve health. There are a variety of probiotics that have been studied for various health benefits. Their role in preventing drug-resistant infections in humans has not been established. The Centers for Disease Control and Prevention (CDC) is actively researching the subject. Although some studies have shown benefit, the data are not conclusive enough for CDC to issue specific recommendations at this time.
Symptoms of antibiotic resistance
Some infections, like skin infections, appear as redness, pain, or drainage at an IV catheter site or surgery site. Symptoms of a C. difficile infection include severe diarrhea, loss of appetite, abdominal pain/tenderness, and nausea. Often these symptoms come with a fever.
Antibiotic resistance solutions
- Prevent Infections and their Spread
- Improve Antibiotic Prescribing
- Be Alert and Take Action
Healthcare professional can prevent the spread of antibiotic resistance by:
- Prescribing an antibiotic only when it is likely to benefit the patient.
- Prescribing an antibiotic that targets the bacteria that is most likely causing their patient’s illness when an antibiotic is likely to provide benefit.
- Encouraging patients to use the antibiotic as instructed.
- Collaborating with each other, office staff, and patients to promote appropriate antibiotic use.
World Health Organization
For World Health Day 2011, the World Health Organization (WHO) introduced a 6-point policy package to combat the spread of antimicrobial resistance (https://www.who.int/topics/antimicrobial_resistance/en/). As part of its core functions, WHO has called together a Strategic and Technical Advisory Group to review and help shape a global strategy to tackle the growing challenge of antimicrobial resistance, and to advise WHO on the coordination role it should be playing in the fight against antimicrobial resistance.
European Centre for Disease Prevention and Control
The European Centre for Disease Prevention and Control (ECDC) conducts surveillance for antibiotic-resistant infections in the European Union and European Economic Areas. In addition to playing a key role with TATFAR, ECDC hosts an annual European Antibiotic Awareness Day (https://ecdc.europa.eu/en/antimicrobial-resistance).
Transatlantic Taskforce on Antimicrobial Resistance
The Transatlantic Taskforce on Antimicrobial Resistance (https://www.cdc.gov/drugresistance/tatfar/index.html) was created in 2009 with the goal of improving cooperation between the United States and the European Union in 3 key areas:
- Appropriate therapeutic use of antimicrobial drugs in medical and veterinary communities
- Prevention of healthcare and community-associated drug-resistant infections
- Strategies for improving the pipeline of new antimicrobial drugs.
- About Antimicrobial Resistance. https://www.cdc.gov/drugresistance/about.html[↩]
- How Antibiotic Resistance Happens. https://www.cdc.gov/drugresistance/about/how-resistance-happens.html[↩]
- http://apps.who.int/iris/bitstream/handle/10665/66860/WHO_CDS_CSR_DRS_2001.2.pdf;jsessionid=C12055DED7AEA88072DBF31B62BEDBC4?sequence=1[↩]
- Manson JM, Hancock LE, Gilmore MS. Mechanism of chromosomal transfer of Enterococcus faecalis pathogenicity island, capsule, antimicrobial resistance, and other traits. Proc Natl Acad Sci U S A. 2010 Jul 6;107(27):12269–74[↩]
- Thomas CM, Nielsen KM. Mechanisms of, and barriers to, horizontal gene transfer between bacteria. Nat Rev Microbiol. 2005 Sep;3(9):711–21.[↩]
- Munita JM, Arias CA. Mechanisms of Antibiotic Resistance. Microbiol Spectr. 2016;4(2):10.1128/microbiolspec.VMBF-0016-2015. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4888801[↩][↩][↩][↩][↩][↩][↩][↩]
- Wilson DN. Ribosome-targeting antibiotics and mechanisms of bacterial resistance. Nat Rev Microbiol. 2014 Jan;12(1):35–48[↩]
- Ramirez MS, Tolmasky ME. Aminoglycoside modifying enzymes. Drug Resist Updat. 2010 Dec;13(6):151–71.[↩][↩][↩]
- DiazGranados CA, Zimmer SM, Klein M, Jernigan JA. Comparison of mortality associated with vancomycin-resistant and vancomycin-susceptible enterococcal bloodstream infections: a meta-analysis. Clin Infect Dis. 2005 Aug 1;41(3):327–33.[↩]
- Hollenbeck BL, Rice LB. Intrinsic and acquired resistance mechanisms in enterococcus. Virulence. 2012 Aug 15;3(5):421–33[↩]
- Schwarz S, Kehrenberg C, Doublet B, Cloeckaert A. Molecular basis of bacterial resistance to chloramphenicol and florfenicol. FEMS Microbiol Rev. 2004 Nov;28(5):519–42.[↩]
- D’Costa VM, King CE, Kalan L, Morar M, Sung WW, Schwarz C, Froese D, Zazula G, Calmels F, Debruyne R, Golding GB, Poinar HN, Wright GD. Antibiotic resistance is ancient. Nature. 2011 Aug 31;477(7365):457–61.[↩]
- Bush K. Proliferation and significance of clinically relevant β-lactamases. Ann N Y Acad Sci. 2013 Jan;1277:84–90.[↩]
- Paterson DL, Bonomo RA. Extended-spectrum beta-lactamases: a clinical update. Clin Microbiol Rev. 2005 Oct;18(4):657–86.[↩][↩]
- Bush K. The ABCD’s of β-lactamase nomenclature. J Infect Chemother. 2013 Aug;19(4):549–59.[↩]
- Bonnet R. Growing group of extended-spectrum beta lactamases: the CTX-M enzymes. Antimicrob Agents Chemother. 2004 Jan;48(1):1–14.[↩]
- Poirel L, Lartigue MF, Decousser JW, Nordmann P. ISEcp1B-mediated transposition of blaCTX-M in Escherichia coli. Antimicrob Agents Chemother. 2005;49:447–450.[↩]
- Queenan AM, Bush K. Carbapenemases: the versatile beta-lactamases. Clin Microbiol Rev. 2007 Jul;20(3):440–58.[↩][↩]
- Yigit H, Queenan AM, Anderson GJ, Domenech-Sanchez A, Biddle JW, Steward CD, Alberti S, Bush K, Tenover FC. Novel carbapenem-hydrolyzing beta-lactamase, KPC-1, from a carbapenem-resistant strain of Klebsiella pneumoniae. Antimicrob Agents Chemother. 2001 Apr;45(4):1151–61.[↩]
- Nordmann P, Cuzon G, Naas T. The real threat of Klebsiella pneumoniae carbapenemase-producing bacteria. Lancet Infect Dis. 2009 Apr;9(4):228–36.[↩]
- Poirel L, Pitout JD, Nordmann P. Carbapenemases: molecular diversity and clinical consequences. Future Microbiol. 2007 Oct;2(5):501–12.[↩]
- Cornaglia G, Giamarellou H, Rossolini GM. Metallo-β-lactamases: a last frontier for β-lactams? Lancet Infect Dis. 2011 May;11(5):381–93.[↩][↩]
- Kumarasamy KK, Toleman MA, Walsh TR, Bagaria J, Butt F, Balakrishnan R, Chaudhary U, Doumith M, Giske CG, Irfan S, Krishnan P, Kumar AV, Maharjan S, Mushtaq S, Noorie T, Paterson DL, Pearson A, Perry C, Pike R, Rao B, Ray U, Sarma JB, Sharma M, Sheridan E, Thirunarayan MA, Turton J, Upadhyay S, Warner M, Welfare W, Livermore DM, Woodford N. Emergence of a new antibiotic resistance mechanism in India, Pakistan, and the UK: a molecular, biological, and epidemiological study. Lancet Infect Dis. 2010 Sep;10(9):597–602.[↩]
- Nordmann P, Poirel L, Walsh TR, Livermore DM. The emerging NDM carbapenemases. Trends Microbiol. 2011 Dec;19(12):588–95.[↩]
- Walsh TR, Weeks J, Livermore DM, Toleman MA. Dissemination of NDM-1 positive bacteria in the New Delhi environment and its implications for human health: an environmental point prevalence study. Lancet Infect Dis. 2011 May;11(5):355–62.[↩]
- Jacoby GA. AmpC beta-lactamases. Clin Microbiol Rev. 2009 Jan;22(1):161–82.[↩]
- Johnson JW, Fisher JF, Mobashery S. Bacterial cell wall recycling. Ann N Y Acad Sci. 2013 Jan;1277:54–75.[↩]
- Schmidtke AJ, Hanson ND. Model system to evaluate the effect of ampD mutations on AmpC-mediated beta-lactam resistance. Antimicrob Agents Chemother. 2006 Jun;50(6):2030–7.[↩]
- Evans BA, Amyes SG. OXA β-lactamases. Clin Microbiol Rev. 2014 Apr;27(2):241–63.[↩][↩]
- Pagès JM, James CE, Winterhalter M. The porin and the permeating antibiotic: a selective diffusion barrier in Gram-negative bacteria. Nat Rev Microbiol. 2008 Dec;6(12):893–903.[↩]
- Hancock RE, Brinkman FS. Function of pseudomonas porins in uptake and efflux. Annu Rev Microbiol. 2002;56:17–38.[↩]
- Nikaido H. Molecular basis of bacterial outer membrane permeability revisited. Microbiol Mol Biol Rev. 2003 Dec;67(4):593–656.[↩]
- Quinn JP, Dudek EJ, DiVincenzo CA, Lucks DA, Lerner SA. Emergence of resistance to imipenem during therapy for Pseudomonas aeruginosa infections. J Infect Dis. 1986 Aug;154(2):289–94.[↩]
- Hasdemir UO, Chevalier J, Nordmann P, Pagès JM. Detection and prevalence of active drug efflux mechanism in various multidrugresistant Klebsiella pneumoniae strains from Turkey. J Clin Microbiol. 2004;42:2701–2706.[↩]
- McMurry LM, Petrucci RE, Jr, Levy SB. Active efflux of tetracycline encoded by four genetically different tetracycline resistance determinants in Escherichia coli. Proc Natl Acad Sci USA. 1980;77:3974–7.[↩]
- Poole K. Efflux-mediated antimicrobial resistance. J Antimicrob Chemother. 2005 Jul;56(1):20–51.[↩][↩]
- Singh KV, Weinstock GM, Murray BE. An Enterococcus faecalis ABC homologue (Lsa) is required for the resistance of this species to clindamycin and quinupristin–dalfopristin. Antimicrob Agents Chemother. 2002;46:1845–50.[↩]
- Piddock LJ. Clinically relevant chromosomally encoded multidrug resistance efflux pumps in bacteria. Clin Microbiol Rev. 2006 Apr;19(2):382–402.[↩]
- Roberts MC. Update on acquired tetracycline resistance genes. FEMS Microbiol Lett. 2005 Apr 15;245(2):195–203.[↩]
- Visalli MA, Murphy E, Projan SJ, Bradford PA. AcrAB multidrug efflux pump is associated with reduced levels of susceptibility to tigecycline (GAR-936) in Proteus mirabilis. Antimicrob Agents Chemother. 2003 Feb;47(2):665–9.[↩]
- Ross JI, Eady EA, Cove JH, Cunliffe WJ, Baumberg S, Wootton JC. Inducible erythromycin resistance in staphylococci is encoded by a member of the ATP-binding transport super-gene family. Mol Microbiol. 1990;4:1207–14.[↩]
- Connell SR, Tracz DM, Nierhaus KH, Taylor DE. Ribosomal protection proteins and their mechanism of tetracycline resistance. Antimicrob Agents Chemother. 2003 Dec;47(12):3675–81.[↩]
- Dönhöfer A, Franckenberg S, Wickles S, Berninghausen O, Beckmann R, Wilson DN. Structural basis for TetM-mediated tetracycline resistance. Proc Natl Acad Sci USA. 2012 Oct 16;109(42):16900–5.[↩]
- Li W, Atkinson GC, Thakor NS, Allas U, Lu CC, Chan KY, Tenson T, Schulten K, Wilson KS, Hauryliuk V, Frank J. Mechanism of tetracycline resistance by ribosomal protection protein Tet(O) Nat Commun. 2013;4:1477.[↩]
- Martinez-Martinez L, Pascual A, Jacoby GA. Quinolone resistance from a transferable plasmid. Lancet. 1998;351:797–9.[↩]
- Rodríguez-Martínez JM, Cano ME, Velasco C, Martínez-Martínez L, Pascual A. Plasmid-mediated quinolone resistance: an update. J Infect Chemother. 2011 Apr;17(2):149–82.[↩]
- Aldred KJ, Kerns RJ, Osheroff N. Mechanism of quinolone action and resistance. Biochemistry. 2014 Mar 18;53(10):1565–74.[↩]
- Campbell EA, Korzheva N, Mustaev A, Murakami K, Nair S, Goldfarb A, Darst SA. Structural mechanism for rifampicin inhibition of bacterial rna polymerase. Cell. 2001 Mar 23;104(6):901–12.[↩]
- Floss HG, Yu TW. Rifamycin-mode of action, resistance, and biosynthesis. Chem Rev. 2005 Feb;105(2):621–32.[↩]
- Hooper DC. Fluoroquinolone resistance among Gram-positive cocci. Lancet Infect Dis. 2002 Sep;2(9):530–8.[↩]
- Mendes RE, Deshpande LM, Jones RN. Linezolid update: stable in vitro activity following more than a decade of clinical use and summary of associated resistance mechanisms. Drug Resist Updat. 2014;17(1–2):1–12.[↩][↩]
- Marshall SH, Donskey CJ, Hutton-Thomas R, Salata RA, Rice LB. Gene dosage and linezolid resistance in Enterococcus faecium and Enterococcus faecalis. Antimicrob Agents Chemother. 2002;46(10):3334–6.[↩]
- Leclercq R. Mechanisms of resistance to macrolides and lincosamides: nature of the resistance elements and their clinical implications. Clin Infect Dis. 2002 Feb 15;34(4):482–92.[↩]
- Roberts MC. Update on macrolide-lincosamide-streptogramin, ketolide, and oxazolidinone resistance genes. FEMS Microbiol Lett. 2008 May;282(2):147–59.[↩]
- Katz L, Ashley GW. Translation and protein synthesis: macrolides. Chem Rev. 2005;105:499–528.[↩]
- Toh SM, Xiong L, Arias CA, Villegas MV, Lolans K, Quinn J, Mankin AS. Acquisition of a natural resistance gene renders a clinical strain of methicillin-resistant Staphylococcus aureus resistant to the synthetic antibiotic linezolid. Mol Microbiol. 2007;64(6):1506–14.[↩]
- Locke JB, Zurenko GE, Shaw KJ, Bartizal K. Tedizolid for the management of human infections: in vitro characteristics. Clin Infect Dis. 2014;58(Suppl 1):S35–42.[↩]
- Hiramatsu K, Ito T, Tsubakishita S, Sasaki T, Takeuchi F, Morimoto Y, Katayama Y, Matsuo M, Kuwahara-Arai K, Hishinuma T, Baba T. Genomic basis for methicillin resistance in Staphylococcus aureus. Infect Chemother. 2013;45:117.[↩]
- Chambers HF, Deleo FR. Waves of resistance: Staphylococcus aureus in the antibiotic era. Nat Rev Microbiol. 2009;7(9):629–41.[↩][↩]
- Reynolds PE. Structure, biochemistry and mechanism of action of glycopeptide antibiotics. Eur J Clin Microbiol Infect Dis. 1989 Nov;8(11):943–50.[↩]
- Arias CA, Murray BE. The rise of the Enterococcus: beyond vancomycin resistance. Nat Rev Microbiol. 2012;10:266–78.[↩]
- Miller WR, Munita JM, Arias CA. Mechanisms of antibiotic resistance in enterococci. Expert Rev Anti Infect Ther. 2014 Oct;12(10):1221–36.[↩][↩]
- Guardabassi L, Agersø Y. Genes homologous to glycopeptide resistance vanA are widespread in soil microbial communities. FEMS Microbiol Lett. 2006;259:221–5.[↩]
- Arthur M. Antibiotics: vancomycin sensing. Nat Chem Biol. 2010 May;6(5):313–5.[↩]
- Arthur M, Molinas C, Courvalin P. The VanS-VanR two-component regulatory system controls synthesis of depsipeptide peptidoglycan precursors in Enterococcus faecium BM4147. J Bacteriol. 1992 Apr;174(8):2582–91[↩]
- Courvalin P. Vancomycin resistance in gram-positive cocci. Clin Infect Dis. 2006;42:S25–34.[↩]
- Sievert DM, Rudrik JT, Patel JB, McDonald LC, Wilkins MJ, Hageman JC. Vancomycin-resistant Staphylococcus aureus in the United States, 2002–2006. Clin Infect Dis. 2008;46(5):668–74.[↩]
- Rossi F, Diaz L, Wollam A, Panesso D, Zhou Y, Rincon S, Narechania A, Xing G, Di Gioia TS, Doi A, Tran TT, Reyes J, Munita JM, Carvajal LP, Hernandez-Roldan A, Brandão D, van der Heijden IM, Murray BE, Planet PJ, Weinstock GM, Arias CA. Transferable vancomycin resistance in a community-associated MRSA lineage. N Engl J Med. 2014;370(16):1524–31.[↩]
- Van Bambeke F, Chauvel M, Reynolds PE, Fraimow HS, Courvalin P. Vancomycin-dependent Enterococcus faecalis clinical isolates and revertant mutants. Antimicrob Agents Chemother. 1999;43:41–47.[↩]
- Huovinen P. Resistance to trimethoprim sulfamethoxazole. Clin Infect Dis. 2001 Jun 1;32(11):1608–14.[↩]
- Hamilton-Miller JM. Reversal of activity of trimethoprim against Gram-positive cocci by thymidine, thymine and ‘folates’ J Antimicrob Chemother. 1988;22:35–9.[↩]
- Pogliano J, Pogliano N, Silverman JA. Daptomycin-mediated reorganization of membrane architecture causes mislocalization of essential cell division proteins. J Bacteriol. 2012;194(17):4494–504.[↩]
- Zhang T, Muraih JK, Tishbi N, Herskowitz J, Victor RL, Silverman J, Uwumarenogie S, Taylor SD, Palmer M, Mintzer E. Cardiolipin prevents membrane translocation and permeabilization by daptomycin. J Biol Chem. 2014 Apr 25;289(17):11584–91.[↩][↩]
- Arias CA, Panesso D, McGrath DM, Qin X, Mojica MF, Miller C, Diaz L, Tran TT, Rincon S, Barbu EM, Reyes J, Roh JH, Lobos E, Sodergren E, Pasqualini R, Arap W, Quinn JP, Shamoo Y, Murray BE, Weinstock GM. Genetic basis for in vivo daptomycin resistance in enterococci. N Engl J Med. 2011;365(10):892–900.[↩][↩]
- Munita JM, Tran TT, Diaz L, Panesso D, Reyes J, Murray BE, Arias CA. A liaF codon deletion abolishes daptomycin bactericidal activity against vancomycin-resistant Enterococcus faecalis. Antimicrob Agents Chemother. 2013 Jun;57(6):2831–3[↩]
- Diaz L, Tran TT, Munita JM, Miller WR, Rincon S, Carvajal LP, Wollam A, Reyes J, Panesso D, Rojas NL, Shamoo Y, Murray BE, Weinstock GM, Arias CA. Whole-genome analyses of Enterococcus faecium isolates with diverse daptomycin MICs. Antimicrob Agents Chemother. 2014;58(8):4527–34.[↩]
- Munita JM, Panesso D, Diaz L, Tran TT, Reyes J, Wanger A, Murray BE, Arias CA. Correlation between mutations in liaFSR of Enterococcus faecium and MIC of daptomycin: revisiting daptomycin breakpoints. Antimicrob Agents Chemother. 2012;56(8):4354–9.[↩]
- Munita JM, Mishra NN, Alvarez D, Tran TT, Diaz L, Panesso D, Reyes J, Murray BE, Adachi JA, Bayer AS, Arias CA. Failure of high-dose daptomycin for bacteremia caused by daptomycin-susceptible Enterococcus faecium harboring LiaSR substitutions. Clin Infect Dis. 2014;59(9):1277–80.[↩]
- Wolf D, Kalamorz F, Wecke T, Juszczak A, Mäder U, Homuth G, Jordan S, Kirstein J, Hoppert M, Voigt B, Hecker M, Mascher T. In-depth profiling of the LiaR response of Bacillus subtilis. J Bacteriol. 2010 Sep;192(18):4680–93.[↩]
- Tran TT, Panesso D, Mishra NN, Mileykovskaya E, Guan Z, Munita JM, Reyes J, Diaz L, Weinstock GM, Murray BE, Shamoo Y, Dowhan W, Bayer AS, Arias CA. Daptomycin-resistant Enterococcus faecalis diverts the antibiotic molecule from the division septum and remodels cell membrane phospholipids. MBio. 2013;4(4) pii: e00281-13.[↩]
- Reyes J, Panesso D, Tran TT, Mishra NN, Cruz MR, Munita JM, Singh KV, Yeaman MR, Murray BE, Shamoo Y, Garsin D, Bayer AS, Arias CA. A liaR Deletion Restores Susceptibility to Daptomycin and Antimicrobial Peptides in Multidrug-Resistant. Enterococcus faecalis J Infect Dis. 2014 Oct 31; pii: jiu602[↩]
- Tran TT, Panesso D, Gao H, Roh JH, Munita JM, Reyes J, Diaz L, Lobos EA, Shamoo Y, Mishra NN, Bayer AS, Murray BE, Weinstock GM, Arias CA. Whole-genome analysis of a daptomycin-susceptible enterococcus faecium strain and its daptomycin-resistant variant arising during therapy. Antimicrob Agents Chemother. 2013 Jan;57(1):261–8.[↩]
- Bayer AS, Schneider T, Sahl HG. Mechanisms of daptomycin resistance in Staphylococcus aureus: role of the cell membrane and cell wall. Ann N Y Acad Sci. 2013;1277:139–58.[↩]
- Hiramatsu K, Hanaki H, Ino T, Yabuta K, Oguri T, Tenover FC. Methicillin-resistant Staphylococcus aureus clinical strain with reduced vancomycin susceptibility. J Antimicrob Chemother. 1997 Jul;40(1):135–6.[↩]
- Howden BP, Davies JK, Johnson PD, Stinear TP, Grayson ML. Reduced vancomycin susceptibility in Staphylococcus aureus, including vancomycin-intermediate and heterogeneous vancomycin-intermediate strains: resistance mechanisms, laboratory detection, and clinical implications. Clin Microbiol Rev. 2010 Jan;23(1):99–139.[↩][↩]
- Stryjewski ME, Corey GR. Methicillin-resistant Staphylococcus aureus: an evolving pathogen. Clin Infect Dis. 2014 Jan;58(Suppl 1):S10–9[↩]
- Gardete S, Tomasz A. Mechanisms of vancomycin resistance in Staphylococcus aureus. J Clin Invest. 2014;124(7):2836–40.[↩]
- Watanabe Y, Cui L, Katayama Y, Kozue K, Hiramatsu K. Impact of rpoB mutations on reduced vancomycin susceptibility in Staphylococcus aureus. J Clin Microbiol. 2011;49(7):2680–2684.[↩]

{kind=link}