What is aplasia
Aplasia means failure of an organ or tissue to develop or to function normally during embryonic life.
Aplasia cutis congenita
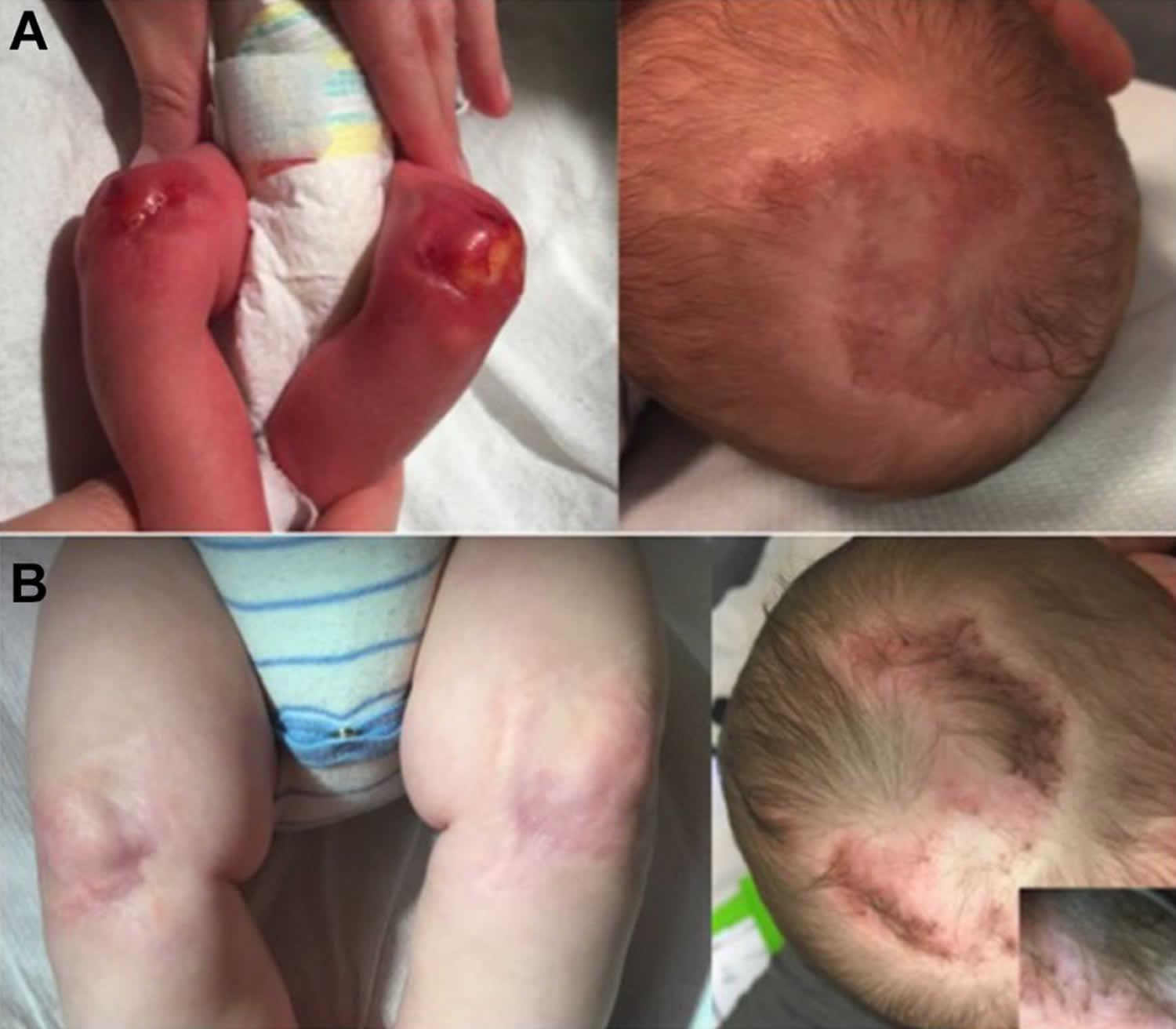
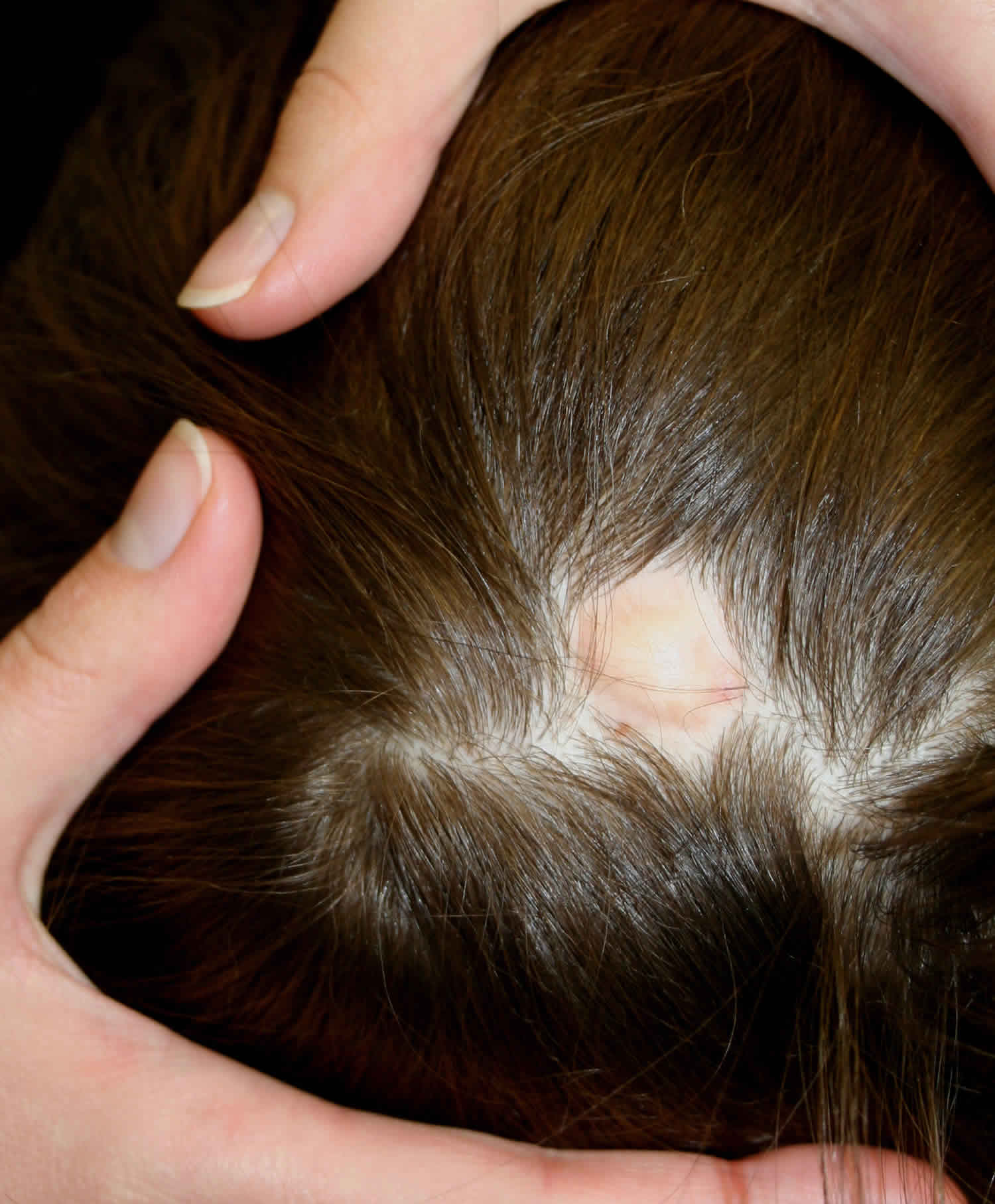
Aplasia cutis congenita is a rare disorder where babies are born with the absence of certain layer(s) of skin, most often on the scalp, but also on the trunk, and/or arms and legs, with or without the absence of underlying structures such as bone 1. The affected area is typically covered with a thin, transparent membrane. The skull and/or underlying areas may be visible and be abnormally developed. Aplasia cutis congenita may be the primary disorder or it may occur in association with other underlying disorders.
Aplasia cutis congenita is a rare congenital condition with an incidence of approximately 1 to 3 out of 10,000 births 2. There is no significant gender or cultural predilection that has been reported in the literature 3. Lesions will typically be noticed at birth, although patients may not present to be evaluated for several months as lesions are often asymptomatic.
While most people with aplasia cutis congenita have no other abnormalities, some people have congenital malformations involving the cardiovascular (heart), gastrointestinal, genitourinary, and central nervous systems 1. The cause of aplasia cutis congenita is unclear and appears to be multifactorial (many different factors appear to play a role); contributing factors may include teratogens, genes, trauma, and compromised blood flow to the skin 1.
Aplasia cutis congenita has been classified into 6 subtypes, some of which are associated with congenital dermatologic syndromes 4. Although most lesions are self-healing, certain locations and clinical characteristics should prompt a more thorough workup to screen for underlying soft tissue anomalies that can potentially be life-threatening 5.
A classification for aplasia cutis congenita was proposed in 1986, which is still accepted today, and presented below 6.
- Group 1: Scalp aplasia cutis congenita without multiple anomalies
- Group 2: Scalp aplasia cutis congenita with limb abnormalities
- Group 3: Scalp aplasia cutis congenita with epidermal and organoid nevi
- Group 4: Aplasia cutis congenita overlying congenital malformations
- Group 5: Aplasia cutis congenita with associated fetus papyraceus or placental infarct
- Group 6: Aplasia cutis congenita with epidermolysis bullosa
- Group 7: Aplasia cutis congenita localized to extremities without blistering
- Group 8: Aplasia cutis congenita due to specific teratogens
- Group 9: Aplasia cutis congenita associated with malformation syndromes
Figure 1. Aplasia cutis congenita
Aplasia cutis congenita cause
There is no one cause for all cases of aplasia cutis congenita 7. Aplasia cutis congenita is thought to be multifactorial, which means that several factors likely interact to cause the condition 8. Factors that may contribute include genetic factors; teratogens (exposures during pregnancy that can harm a developing fetus) such as methimazole, carbimazole, misoprostol, and valproic acid; compromised vasculature to the skin; and trauma 1. Some cases may represent an incomplete or unusual form of a neural tube defect 9. Familial cases of aplasia cutis congenita have been reported 1. Cases that appear to be genetic may be inherited in an autosomal dominant or autosomal recessive manner 9.
Until recently, no specific genetic target had been identified, but a recent study showed BMS1 gene to play a possible role 10. Aplasia cutis congenita can also be associated with several genetic syndromes including Adams-Oliver syndrome, Bart syndrome, and Setleis syndrome as described below 11.
- Adams-Oliver syndrome: aplasia cutis congenita on the scalp plus skull defect plus Cutis marmorata telangiectatica congenital plus limb defects plus cardiac anomalies 12.
- Bart syndrome: aplasia cutis congenita of lower extremities plus epidermolysis bullosa 11.
- Setleis syndrome: Bilateral temporal aplasia cutis congenita plus “leonine” facies 13.
Aplasia cutis congenita signs and symptoms
Individuals born with aplasia cutis congenita lack skin (and therefore hair), in localized areas of the body, usually, but not always, on the scalp (70 percent of cases). In some cases, the trunk, arms, and/or legs may also be involved. Sometimes, the underlying bone may be missing as well as the skin. The affected area(s) are usually replaced with a thin transparent membrane. In some cases, these affected structures and other organs may be seen through the transparent membrane.
Most individuals with aplasia cutis congenita exhibit no other abnormalities. However, in some rare cases, they may experience other physical characteristics including abnormalities of the ears, a form of paralysis (palsy) affecting one side of the face, an abnormally large head (macrocephaly), and/or congenital heart anomalies.
Aplasia cutis congenita may also occur as a physical condition characteristic of several other disorders, including Adams-Oliver syndrome, aplasia cutis congenita-gastrointestinal, and Johanson-Blizzard syndrome.
Aplasia cutis congenita can be associated with underlying morphologic abnormalities in approximately 37% of cases according to Mesrati et al. 14 including underlying bony defects, vascular anomalies, or neurologic malformations, so it is prudent for clinicians to evaluate the disease involvement with imaging. A midline vertex scalp lesion, hair collar sign, and vascular stains have all been shown to be strong indicators for cranial or central nervous system (CNS) involvement 15. Small, scalp lesions are less likely associated with underlying defects and typically heal on their own within a couple of months; therefore, monitoring these lesions without further imaging is acceptable 5. For larger, ulcerative lesions, ultrasound provides a relatively inexpensive evaluation while not putting the child through a great deal of discomfort. If there is any concern for underlying defects on ultrasound, further workup with MRI is warranted. MRI is more sensitive and specific for identifying underlying lesions according to a 2017 retrospective multicenter study 15; however, it is more costly than ultrasound and typically requires the child to be sedated for the duration of the procedure, making this a poor choice for initial screening. If the lesion is purulent or surrounded by erythema, a lab workup including complete blood count, blood cultures and wound cultures would be advised 5.
Aplasia cutis congenita treatment
Treatment of aplasia cutis congenita varies depending on the condition of the infant. Conservative treatment is preferred 16. Small areas (less than 4 cm) without additional findings, usually heal on their own over time 17. Gentle cleansing and application of bland ointments or silver sulfadiazine can help prevent infection. Lesions will typically heal within a few weeks to a few months with an atrophic, hairless scar 17. Larger lesions (greater than 4 cm) are more commonly associated with underlying defects and are at increased risk of complications including hemorrhage, venous thrombosis, and infection 18. Early surgical repair is recommended to avoid these complications. Skin grafting or flap techniques are commonly utilized as some lesions can be several centimeters in size 19. If infection occurs, antibiotics can be used 20. Recently, a variety of specialized dressing materials have been developed and used 7. Ultimately, the decision to use medical, surgical, or both forms of therapy depends primarily on the size, depth, and location of the skin defect 7.
Aplasia cutis congenita prognosis
The long-term outlook (prognosis) for people with aplasia cutis congenita is usually excellent. If the condition is associated with other abnormalities or malformations, the prognosis then depends on the nature and severity of the other condition(s) 7.
Major complications of aplasia cutis congenita are rare, but can include hemorrhage, secondary local infection, meningitis, or sagittal sinus thrombosis. Larger affected areas associated with underlying bony defects can cause death due to central nervous system infection, or hemorrhage from the sagittal sinus. Complications can also result from associated abnormalities or malformations, when present 7.
The estimated mortality rate ranges from 20-55% as a result of serious complications 21. The most common life-threatening complication of aplasia cutis congenita is sagittal sinus bleeding, seen with lesions nearby on the scalp 22. Another potential complication of aplasia cutis congenita includes secondary infection of the lesion. Patients are at an increased risk of cutaneous infections given the fact that the skin’s barrier against environmental microbes is absent or impaired. Severe infections can progress to meningitis if not treated appropriately 23. Prompt management of large scalp lesions, commonly with surgery, can help prevent these complications 15.
Pure red cell aplasia
Pure red cell aplasia is an uncommon disorder in which maturation arrest occurs in the formation of red blood cells resulting in the bone marrow making a reduced number of red blood cells (called anemia). As a result, affected people may experience fatigue, lethargy, and pale skin 24. Erythroblasts are virtually absent in bone marrow; however, white blood cell and platelet production are normal 25. The anemia due to pure red cell aplasia is usually normocytic but can be macrocytic. In 1922, Kaznelson recognized that this condition was a different entity from aplastic anemia, which presents as pancytopenia.
The characteristics of pure red cell aplasia include the following:
- Severe anemia
- Reticulocyte count <1%
- The presence of less than 0.5% mature erythroblasts in the bone marrow
- Normocellular bone marrow in most cases
The cause of pure red cell aplasia is heterogeneous. A rare congenital form of pure red cell aplasia was initially described by Joseph in 1936 and by Diamond and Blackfan in 1938 called Diamond Blackfan syndrome, is an inherited condition that is also associated with other physical abnormalities 25. Congenital pure red cell aplasia is a lifelong disorder and is associated with physical abnormalities.
Pure red cell aplasia can be transient and reversible. Transient erythroblastopenia of childhood can occur after viral infections. Pure red cell aplasia can also be due to medications, infections, pregnancy, renal failure, and conditions such as thymomas, autoimmune disease (such as systemic lupus erythematosus), cancers of the blood, and solid tumors. In many cases, the cause of the condition is unknown (idiopathic) 25. In adults, most cases of chronic pure red cell aplasia are idiopathic.
Secondary pure red cell aplasia occurs in patients with conditions such as the following:
- Autoimmune disorders
- Thymomas
- Systemic lupus erythematosus
- Hematologic malignancies
- Solid tumors
Pure red cell aplasia is an uncommon disorder. The idiopathic form is the most common type of pure red cell aplasia. The incidence of transient and reversible pure red cell aplasia that occurs in childhood and in adults secondary to medications and infections is probably underestimated. The reason for this underestimation is the anemia is self-limiting. Acquired secondary pure red cell aplasia is not common. Diamond-Blackfan syndrome is rare.
No racial, age, or sex predilection is reported in pure red cell aplasia. However, females are more likely to have autoimmune disorders.
Therapeutic approaches include the following:
- Transfusions for severe anemia with cardiorespiratory failure
- Discontinuation of medications that could cause Pure red cell aplasia
- Observation of children with pure red cell aplasia, with treatment if indicated
- Treatment of infections
- Treatment of underlying conditions
- Corticosteroids and immunosuppressive agents
- Plasmapheresis or lymphocytapheresis
The life expectancy of patients with idiopathic pure red cell aplasia is about 1-2 decades 25. The survival of patients with congenital pure red cell aplasia is limited. The lifespan of patients with secondary pure red cell aplasia depends on the course of the underlying disorder.
Pure red cell aplasia pathophysiology
In general, pure red cell aplasia is due to a selective injury, often immunological, that affects the early phase of erythrocyte maturation.
Childhood
Diamond-Blackfan syndrome is a rare congenital pure red cell aplasia that is usually detected at birth, or later during the first 18 months of childhood. Affected individuals usually have a macrocytic anemia. The expression of hemoglobin F and surface “I” antigen in erythrocytes is increased, indicating erythrocyte immaturity.
About one third of these patients have developmental defects, including cleft palates, macroglossia, craniofacial defects, thumb or upper limb abnormalities, cardiac defects, and urogenital malformations. Growth is often retarded 24. A modest increased risk for leukemia and neoplasms is noted.
Diamond-Blackfan syndrome is caused by the deletion of genes for ribosomal protein RPS19 in 25% of patients, leading to defects in ribosome biogenesis. This ribosomopathy and haploinsufficiency may be responsible for impaired mRNA translation and the activation of the tumor suppressor gene TP53 in this disorder 26.
Germ-line mutations in genes encoding components of both the small (RPS24, RPS17, RPS7, RPS10, and RPS26) and large (RPL35A, RPL5, RPL11, and RPL26) ribosomal subunits have also been described in Diamond-Blackfan anemia patients 27. Mutations in the GATA1 gene has been found to cause Diamond-Blackfan anemia in a minority of patients 28. Because GATA1 has been implicated in Diamond-Blackfan anemia, it is possible that non-RP genes may also lead to the characteristic erythroid hypoplasia 27.
De novo cases of Diamond-Blackfan syndrome are believed to be caused by intrauterine damage to early erythroid stem cells 29. A familial history of pure red cell aplasia is evident in approximately 10% of patients.
Transient erythroblastopenia of childhood is a self–limiting, benign disorder. A history of a recent viral infection is usually noted 30. Parvovirus 19 infection should be ruled out.
Adults
Acquired primary (idiopathic) pure red cell aplasia is the most common form of red cell aplasia in adults.
However, pure red cell aplasia can be secondary to underlying disorders. For example, autoimmune disorders (eg, type 1 diabetes, thyroiditis, rheumatoid arthritis, Sjögren syndrome) can be responsible. pure red cell aplasia has been shown to be secondary to T-cell inhibition of marrow erythroid cells. pure red cell aplasia can also be secondary to and is associated with the following:
- Thymoma (1-15%)
- Hematological malignancies (eg, B- and T-cell chronic lymphocytic leukemia)
- T-cell large granular lymphocyte leukemia and solid tumors
- Infections
- Drugs
- Pregnancy 31
- Systemic lupus erythematosus (SLE)
- Renal failure
- Good syndrome (thymoma with combined B- and T-cell deficiency)
Pure red cell aplasia can occur following ABO-mismatched marrow transplantation 32.
The incidence of pure red cell aplasia has increased in patients with chronic renal disease who have received epoetin therapy. This has been ascribed to the generation of antiepoetin antibodies, which occurs more often with epoetin-alpha than with epoetin-beta. This complication may be avoided by using an erythropoietin-mimicking human antibody, which stimulates erythropoiesis but does not appear to induce antiepoetin antibodies and pure red cell aplasia 33.
Pure red cell aplasia causes
Infections such as the following can cause pure red cell aplasia 34:
- HIV infection
- Respiratory tract infections
- Gastroenteritis
- Primary atypical pneumonia
- Infectious mononucleosis
- Mumps
- Viral hepatitis
Most cases of acute transient pure red cell aplasia are caused by parvovirus B19 infection 35. Parvovirus B19 can cross the placenta in infected women and can destroy erythroid cells in the fetus and induce spontaneous abortions. Parvovirus 19 infections can persist longer in immunocompromised patients.
A partial list of medications thought to cause pure red cell aplasia is as follows 36:
- Antiepileptic medications (eg, phenytoin, carbamazepine, sodium valproate)
- Mycophenolate
- Azathioprine
- Chloramphenicol
- Thiamphenicol
- Sulfonamides
- Isoniazid
- Procainamide
- Clopidogrel 37
Originally, thymoma was cited as the primary cause of acquired pure red cell aplasia. However, subsequent studies have revealed that only a small percent of all cases of pure red cell aplasia result from thymomas. Conversely, only 7% of patients with thymomas had pure red cell aplasia.
Pure red cell aplasia signs and symptoms
Presenting symptoms depend on the severity of the anemia. Some patients are virtually asymptomatic, whereas others have an uncompensated anemia, have cardiopulmonary distress, and are transfusion dependent 24.
Patients with aplastic anemia, as opposed to pure red cell aplasia, may have a history of bruising due to thrombocytopenia 38.
Obtaining the history of medications that patients are taking is important. A history of recent infections, such as infectious mononucleosis or viral hepatitis, is important.
Patients who have an underlying hemolytic anemia can become markedly anemic if they develop pure red cell aplasia. This is known as an aplastic crises and is caused by hemolysis that is ongoing while erythrocyte production is impaired. The possibility of an aplastic crisis should be considered in patients with a hemolytic anemia if reticulocyte counts are low and if they have had recent infections. In contrast, the development of anemia in pure red cell aplasia in patients without hemolysis is often gradual and self-limited and, hence, not noticed.
To determine whether the patient has a secondary pure red cell aplasia, ask about the possibility of pregnancy, signs of systemic lupus erythematosus (SLE), signs of a hematological malignancy, and signs of other possible disorders that can cause pure red cell aplasia. A history of miscarriages might suggest SLE.
A history of autoimmune disorders such as type 1 diabetes, thyroiditis, and rheumatoid arthritis should be elicited. Dryness of eyes and mouth occurs in Sjögren syndrome.
Recognize that chronic renal failure and erythropoietin therapy, AB0-incompatible transfusion, and stem cell transplantation are associated with pure red cell aplasia.
Diamond-Blackfan syndrome should be considered in a child with pure red cell aplasia, retarded growth, and developmental defects.
Pure red cell aplasia diagnosis
The classical presentation of pure red cell aplasia is with a normocytic anemia and a reticulocyte count of less than 1%. Bone marrow studies reveal a normocellular marrow with an absence of erythroblasts. Maturation arrest is evidenced by the presence of more immature erythrocyte progenitors.
When the results of those laboratory studies are not consistent with classical pure red cell aplasia, a workup to identify other anemias should be done. If macrocytosis or microcytosis is evident, appropriate diagnostic tests should be indicated. Examination of peripheral smears and bone marrow is important.
Laboratory Studies
The following blood tests should be obtained in suspected pure red cell aplasia:
- Complete blood cell count (CBC) count
- Red blood cell (RBC) indices
- Reticulocyte count
- White blood cell (WBC) differential analysis of white blood cells
Other studies to consider include the following:
- Iron studies, especially iron saturation and serum ferritin levels, are used to diagnose hemosiderosis; this possibility should be considered in patients who have received multiple transfusions.
- Serum vitamin B-12 and folate levels might be indicated in patients with macrocytosis.
- Lactate dehydrogenase (LDH), indirect bilirubin, and serum haptoglobin levels are used to detect hemolysis.
- Hemoglobin A 2 and hemoglobin F are used to rule out thalassemia.
- Flow cytometry is used to diagnose hematological malignancies and T-cell disorders.
Tests to identify infection, including the following, are indicated:
- Parvovirus B19 infection 39
- Hepatitis
- Infectious mononucleosis
Tests to detect autoimmune disorders should include the following:
- Antinuclear antibody test
- C-reactive protein (CRP) level
- Erythrocyte sedimentation rate (ESR)
- Quantitative immunoglobulin analysis
- Direct Coombs test to detect an autoimmune hemolytic anemia
- Tests for thyroiditis, diabetes mellitus, rheumatoid arthritis, Sjögren syndrome, and systemic lupus erythematosus (SLE) might be indicated.
In addition, the following tests are helpful in diagnosing Diamond-Blackfan syndrome:
- Hemoglobin F assay
- “I” antigen on the surface of erythrocytes
- Adenosine deaminase determination
Peripheral smears demonstrate a normocytic anemia in most cases of pure red cell aplasia. However, macrocytic anemia occurs in Diamond-Blackfan and Good syndromes and in HIV infections. Peripheral smears can be used to screen for infectious mononucleosis, megaloblastosis, and hematological malignancies.
Bone marrow histology
Bone marrow aspiration smears in pure red cell aplasia usually reveal a normocellular marrow. An absence of erythroblasts is noted, whereas more immature erythrocyte progenitors are present (maturation arrest). White blood cells and platelet maturation are normal. Bone marrow can be used to evaluate iron stores and help diagnose megaloblastosis and hematological malignancies.
Imaging studies
Uses of imaging studies include the following:
- Positron emission tomography (PET) and computed tomography (CT) scans are used to detect thymomas.
- Spleen size can be determined by ultrasound imaging.
- Appropriate imaging studies are used to help diagnose and evaluate hematological malignancies.
- Duel-energy x-ray absorptiometry scans are used to assess osteopenia and osteoporosis due to corticosteroid therapy.
Pure red cell aplasia treatment
The initial treatment plan should include blood transfusions for patients who are severely anemic and have cardiorespiratory failure. Anemia is more severe in patients with pure red cell aplasia who have ongoing hemolysis (aplastic crises).
Medications that could cause pure red cell aplasia should be discontinued.
Children with pure red cell aplasia should be observed and not aggressively treated to avoid corticosteroid-related growth retardation. This caution is feasible since pure red cell aplasia in children is often transient and reversible. However, transfusion should be administered if indicated.
Infections should be treated. High-dose intravenous immunoglobin therapy should be considered for parvovirus B19 infections 40. Pure red cell aplasia due to medication or infections is usually reversible within a few months, if not earlier. However, immunotherapy may be needed to reverse erythropoiesis-stimulating agent (ESA)–related pure red cell aplasia.
Underlying conditions should be treated. These conditions include a thymoma, hematological malignancies such as T-cell large granular lymphocyte leukemia 41, solid tumors, and systemic lupus erythematosus (SLE). Surgery or gamma irradiation of the thymus should be considered in a patient with a thymoma.
pure red cell aplasia considered to be idiopathic and due to autoimmunity should be initially treated with corticosteroids 24. A response is expected within 4-6 weeks in about 45% of patients. Corticosteroids should be judiciously given to children to avoid growth retardation. Immunosuppressive agents have an important role. Immunosuppressive agents used in pure red cell aplasia include cyclophosphamide, 6-mercaptopurine, azathioprine, and cyclosporine A. Rituximab has been reported to be effective in managing pure red cell aplasia 42. Antithymic globulin (ATG) is another therapeutic option. Danazol has been helpful in some cases but is contraindicated in children. Plasmapheresis has been used to remove autoantibodies.
Autologous and nonmyeloablative allogeneic peripheral stem cell transplantation have been used, especially in patients whose disease is refractory to therapy 43.
Several patients have responded to plasmapheresis or lymphocytapheresis 44.
Iron chelation should be considered in patients who have had multiple transfusions and have evidence of iron overload.
Surgical care
Thymectomy might be indicated in patients with a thymoma. However, the procedure should not be performed in patients with a normal-sized thymus. About 30% of patients with thymomas respond to thymectomy.
Although not effective in most cases, splenectomy might be helpful in refractory cases. Splenectomy is indicated to manage pure red cell aplasia complicated by hypersplenism.
Pure red cell aplasia prognosis
Prognosis varies among the different types of pure red cell aplasia.
Transient erythroblastopenia and other pure red cell aplasia disorders in children and adults are benign with an excellent prognosis.
The prognosis of secondary pure red cell aplasia depends on the course of the underlying condition, such as a thymoma or a hematological malignancy. About 30% of pure red cell aplasia cases due to thymomas are reversed by thymectomy.
Most cases of pure red cell aplasia are idiopathic. About 68% respond to intervention. However, relapses are common. The lifespan of these patients is about 1-2 decades.
Most patients with Diamond-Blackfan syndrome respond to corticosteroid therapy but are prone to relapses. Estimating the lifespan of patients with this disorder is difficult because it is rare.
Prognosis is also influenced by the complications of therapy. Hemosiderosis can develop in multitransfused patients. Corticosteroid therapy can lead to osteopenia and osteoporosis and infections. Pure red cell aplasia can evolve into aplastic anemia and acute myelogenous leukemia, which have high morbidity and mortality rates. Infections acquired during blood product transfusions can also affect prognosis.
- Tamara Buchel, Wendy Devaul, Keith Frey. Photo Quiz: Newborn with a Scalp Lesion. American Family Physician. 2005 Oct 15; 72(8):1589-1571. http://www.aafp.org/afp/2005/1015/p1569.html[↩][↩][↩][↩][↩]
- E Rogvi R, Sommerlund M, Vestergaard ET. [Aplasia cutis congenita is a rare and possibly overlooked congenital anomaly]. Ugeskr. Laeg. 2014 Nov 24;176, 48[↩]
- Martinez-Regueira S, Vazquez-Lopez ME, Somoza-Rubio C, Morales-Redondo R, Gonzalez-Gay MA. Aplasia cutis congenita in a defined population from northwest Spain. Pediatr Dermatol. 2006 Nov-Dec;23(6):528-32[↩]
- Saeidi M, Ehsanipoor F. A Case of Adams-Oliver Syndrome. Adv Biomed Res. 2017;6:167.[↩]
- Humphrey SR, Hu X, Adamson K, Schaus A, Jensen JN, Drolet B. A practical approach to the evaluation and treatment of an infant with aplasia cutis congenita. J Perinatol. 2018 Feb;38(2):110-117[↩][↩][↩]
- Frieden IJ. Aplasia cutis congenita: a clinical review and proposal for classification. J. Am. Acad. Dermatol. 1986 Apr;14(4):646-60[↩]
- Aplasia Cutis Congenita. https://emedicine.medscape.com/article/1110134-overview[↩][↩][↩][↩][↩]
- Alexandros B, Dimitrios G, Elias A, Evangelos D, Andreas M, Sotirios P, Georgios S, Marios TS. Aplasia cutis congenita: Two case reports and discussion of the literature. Surg Neurol Int. 2017;8:273[↩]
- Mary Wu Chang, Seth J. Orlow. Neonatal, Pediatric, and Adolescent Dermatology. In: Irwin M. Freedberg, Arthur Z. Eisen, Klaus Wolff, K. Frank Austen, Lowell A. Goldsmith, and Stephen I. Katz. Fitzpatrick’s Dermatology in General Medicine, 6th edition. New York: McGraw-Hill; 2003[↩][↩]
- Marneros AG. BMS1 is mutated in aplasia cutis congenita. PLoS Genet. 2013 Jun;9(6):e1003573[↩]
- Alfayez Y, Alsharif S, Santli A. A Case of Aplasia Cutis Congenita Type VI: Bart Syndrome. Case Rep Dermatol. 2017 May-Aug;9(2):112-118[↩][↩]
- Saeidi M, Ehsanipoor F. A Case of Adams-Oliver Syndrome. Adv Biomed Res. 2017;6:167[↩]
- Graul-Neumann LM, Stieler KM, Blume-Peytavi U, Tzschach A. Autosomal dominant inheritance in a large family with focal facial dermal dysplasia (Brauer-Setleis syndrome). Am. J. Med. Genet. A. 2009 Feb 15;149A(4):746-50[↩]
- Mesrati H, Amouri M, Chaaben H, Masmoudi A, Boudaya S, Turki H. Aplasia cutis congenita: report of 22 cases. Int. J. Dermatol. 2015 Dec;54(12):1370-5[↩]
- Bessis D, Bigorre M, Malissen N, Captier G, Chiaverini C, Abasq C, Barbarot S, Boccara O, Bourrat E, El Fertit H, Eschard C, Hubiche T, Lacour JP, Leboucq N, Mahé E, Mallet S, Marque M, Martin L, Mazereeuw-Hautier J, Milla N, Phan A, Plantin P, Picot MC, Puzenat E, Rigau V, Vabres P, Fraitag S, Boralevi F., Groupe de Recherche Clinique en Dermatologie Pédiatrique. The scalp hair collar and tuft signs: A retrospective multicenter study of 78 patients with a systematic review of the literature. J. Am. Acad. Dermatol. 2017 Mar;76(3):478-487[↩][↩][↩]
- Duan X, Yang G, Yu D, Yu C, Wang B, Guo Y. Aplasia cutis congenita: A case report and literature review. Exp Ther Med. 2015 Nov; 10(5):1893-1895. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4665749[↩]
- Belkhou A, François C, Bennis Y, Duquennoy-Martinot V, Guerreschi P. [Aplasia cutis congenita: Update and management]. Ann Chir Plast Esthet. 2016 Oct;61(5):450-461[↩][↩]
- Patel DP, Castelo-Soccio L, Yan AC. Aplasia cutis congenita: Evaluation of signs suggesting extracutaneous involvement. Pediatr Dermatol. 2018 Jan;35(1):e59-e61[↩]
- Betancourth-Alvarenga JE, Vázquez-Rueda F, Vargas-Cruz V, Paredes-Esteban RM, Ayala-Montoro J. [Surgical management of aplasia cutis congenita]. An Pediatr (Barc). 2015 Nov;83(5):341-5[↩]
- Aplasia cutis congenita. https://www.dermnetnz.org/topics/aplasia-cutis[↩]
- Ribuffo D, Costantini M, Gullo P, Houseman ND, Taylor GI. Aplasia cutis congenita of the scalp, the skull, and the dura. Scand J Plast Reconstr Surg Hand Surg. 2003;37(3):176-80[↩]
- Johnson R, Offiah A, Cohen MC. Fatal superior sagittal sinus hemorrhage as a complication of aplasia cutis congenita: a case report and literature review. Forensic Sci Med Pathol. 2015 Jun;11(2):243-8[↩]
- Suara RO, Dermody TS. Enterococcal meningitis in an infant complicating congenital cutis aplasia. Pediatr. Infect. Dis. J. 2000 Jul;19(7):668-9[↩]
- Young NS. Pure Red Cell Aplasia. In: Kaushansky K, Lichtman MA, Prchal JT, Levi MM, Press OW, Burns LJ, Caligiuri MA, eds. Williams Hematology. 9th ed. New York, NY: McGraw-Hill Education; 2016. 539-48.[↩][↩][↩][↩]
- Pure red cell aplasia. https://emedicine.medscape.com/article/205695-overview[↩][↩][↩][↩]
- Garçon L, Ge J, Manjunath SH, Mills JA, Apicella M, Parikh S. Ribosomal and hematopoietic defects in induced pluripotent stem cells derived from Diamond Blackfan anemia patients. Blood. 2013 Aug 8. 122(6):912-21[↩]
- Mirabello L, Macari ER, Jessop L, Ellis SR, Myers T, Giri N, et al. Whole-exome sequencing and functional studies identify RPS29 as a novel gene mutated in multicase Diamond-Blackfan anemia families. Blood. 2014 Jul 3. 124 (1):24-32.[↩][↩]
- Gagne KE, Ghazvinian R, Yuan D, Zon RL, Storm K, Mazur-Popinska M, et al. Pearson marrow pancreas syndrome in patients suspected to have Diamond-Blackfan anemia. Blood. 2014 Jul 17. 124 (3):437-40 [↩]
- Van Hook JW, Gill P, Cyr D, Kapur RP. Diamond-Blackfan anemia as an unusual cause of nonimmune hydrops fetalis: a case report. Reprod Med. 40:850-854[↩]
- Cherrick I, Karayalcin G, Lanzkowsky P. Transient erythroblastopenia of childhood. Prospective study of fifty patients. Am J Pediatr Hematol Oncol. 1994 Nov. 16(4):320-4[↩]
- Baker RI, Manoharan A, de Luca E, Begley CG. Pure red cell aplasia of pregnancy: a distinct clinical entity. Br J Haematol. 1993 Nov. 85(3):619-22[↩]
- Bierman PJ, Warkentin P, Hutchins MR, Klassen LW. Pure red cell aplasia following ABO mismatched marrow transplantation for chronic lymphocytic leukemia: response to antithymocyte globulin. Leuk Lymphoma. 1993 Jan. 9(1-2):169-71[↩]
- Liu Z, Stoll VS, Devries PJ, Jakob CG, Xie N, Simmer RL, et al. A potent erythropoietin-mimicking human antibody interacts through a novel binding site. Blood. 2007 Oct 1. 110(7):2408-13[↩]
- Lee TH, Oh SJ, Hong S, Lee KB, Park H, Woo HY. Pure red cell aplasia caused by acute hepatitis a. Chonnam Med J. 2011 Apr. 47(1):51-3[↩]
- Herbert KE, Prince HM, Westerman DA. Pure red-cell aplasia due to parvovirus B19 infection in a patient treated with alemtuzumab. Blood. 2003. 101:1654[↩]
- Smalling R, Foote M, Molineux G, Swanson SJ, Elliott S. Drug-induced and antibody-mediated pure red cell aplasia: a review of literature and current knowledge. Biotechnol Annu Rev. 2004. 10:237-50.[↩]
- Li G, Li ZQ, Yang QY, Yang JD. Acquired pure red cell aplasia due to treatment with clopidogrel: first case report. J Thromb Thrombolysis. 2013 Dec 3.[↩]
- Gupta R, Ezeonyeji A, Thomas A, Scully M, Ehrenstein M, Isenberg D. A case of pure red cell aplasia and immune thrombocytopenia complicating systemic lupus erythematosus: Responseto rituximab and cyclophosphamide. Lupus. 2011. 20(14):1547-1550.[↩]
- Wong S, Brown KE. Development of an improved method of detection of infectious parvovirus B19. J Clin Virol. 2006 Apr. 35(4):407-13[↩]
- Björkholm M. Intravenous immunoglobulin treatment in cytopenic haematological disorders. J Intern Med. 1993 Aug. 234(2):119-26.[↩]
- Dhodapkar MV, Lust JA, Phyliky RL. T-cell large granular lymphocytic leukemia and pure red cell aplasia in a patient with type I autoimmune polyendocrinopathy: response to immunosuppressive therapy. Mayo Clin Proc. 1994 Nov. 69(11):1085-8[↩]
- Gupta R, Ezeonyeji A, Thomas A, Scully M, Ehrenstein M, Isenberg D. A case of pure red cell aplasia and immune thrombocytopenia complicating systemic lupus erythematosus: Responseto rituximab and cyclophosphamide. Lupus. 2011. 20(14):1547-1550[↩]
- Passweg JR, Rabusin M, Musso M, Beguin Y, Cesaro S, Ehninger G, et al. Haematopoetic stem cell transplantation for refractory autoimmune cytopenia. Br J Haematol. 2004 Jun. 125(6):749-55[↩]
- Musso M, Porretto F, Crescimanno A, Polizzi V, Scalone R. Donor lymphocyte infusions for refractory pure red cell aplasia relapsing after both autologous and nonmyeloablative allogeneic peripheral stem cell transplantation. Bone Marrow Transplant. 2004 Apr. 33(7):769-71.[↩]

{kind=link}