Contents
What is cleidocranial dysplasia
Cleidocranial dysplasia also known as cleidocranial dysostosis, Marie-Sainton syndrome or mutational dysostosis, is a genetic condition that primarily affects development of the bones and teeth. Signs and symptoms of cleidocranial dysplasia can vary widely in severity, even within the same family. Clinical continuum ranging from classic cleidocranial dysplasia (triad of delayed closure of the cranial sutures, hypoplastic or aplastic clavicles, and dental abnormalities) to mild cleidocranial dysplasia to isolated dental anomalies without the skeletal features 1. Most individuals come to diagnosis because they have classic features. At birth, affected individuals typically have abnormally large, wide-open fontanels that may remain open throughout life. Delayed maturation of the skull (cranium) which is also characteristic of cleidocranial dysplasia, including delayed closing of the growth lines where the bones of the skull meet (sutures) and larger than normal spaces (fontanelles) between the skull bones that are noticeable as “soft spots” on the heads of infants. The fontanelles normally close in early childhood, but they may remain open throughout life in people with cleidocranial dysplasia. Some individuals with cleidocranial dysplasia have extra pieces of bone called Wormian bones within the sutures.
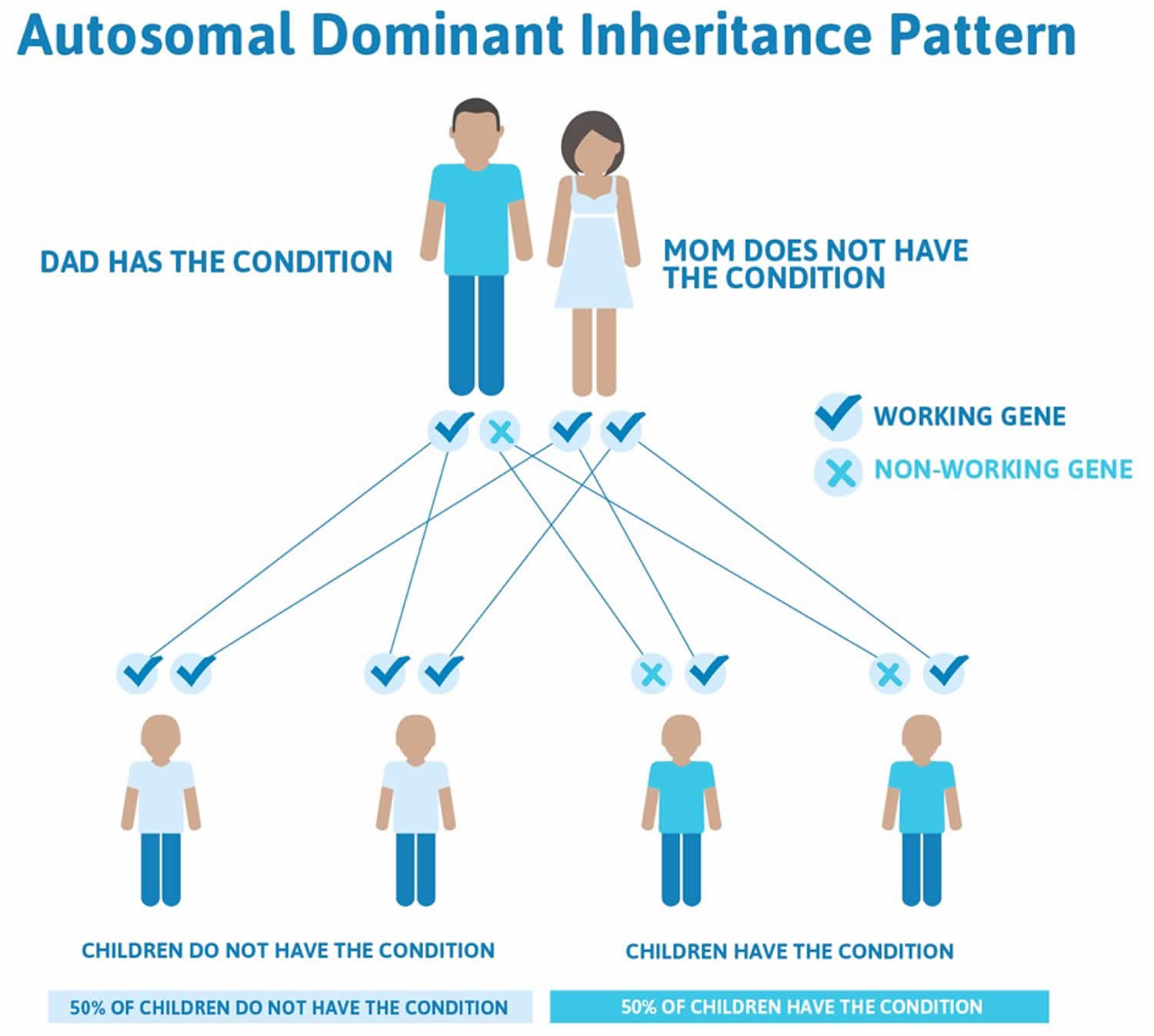
Cleidocranial dysplasia is usually caused by mutations in the RUNX2 gene and cleidocranial dysplasia is inherited in an autosomal dominant manner 1. The proportion of cases caused by a de novo RUNX2 pathogenic variant is high. Each child of an individual with cleidocranial dysplasia spectrum disorder has a 50% chance of inheriting the pathogenic variant. Prenatal diagnosis for pregnancies at increased risk is possible if the pathogenic variant in the family is known.
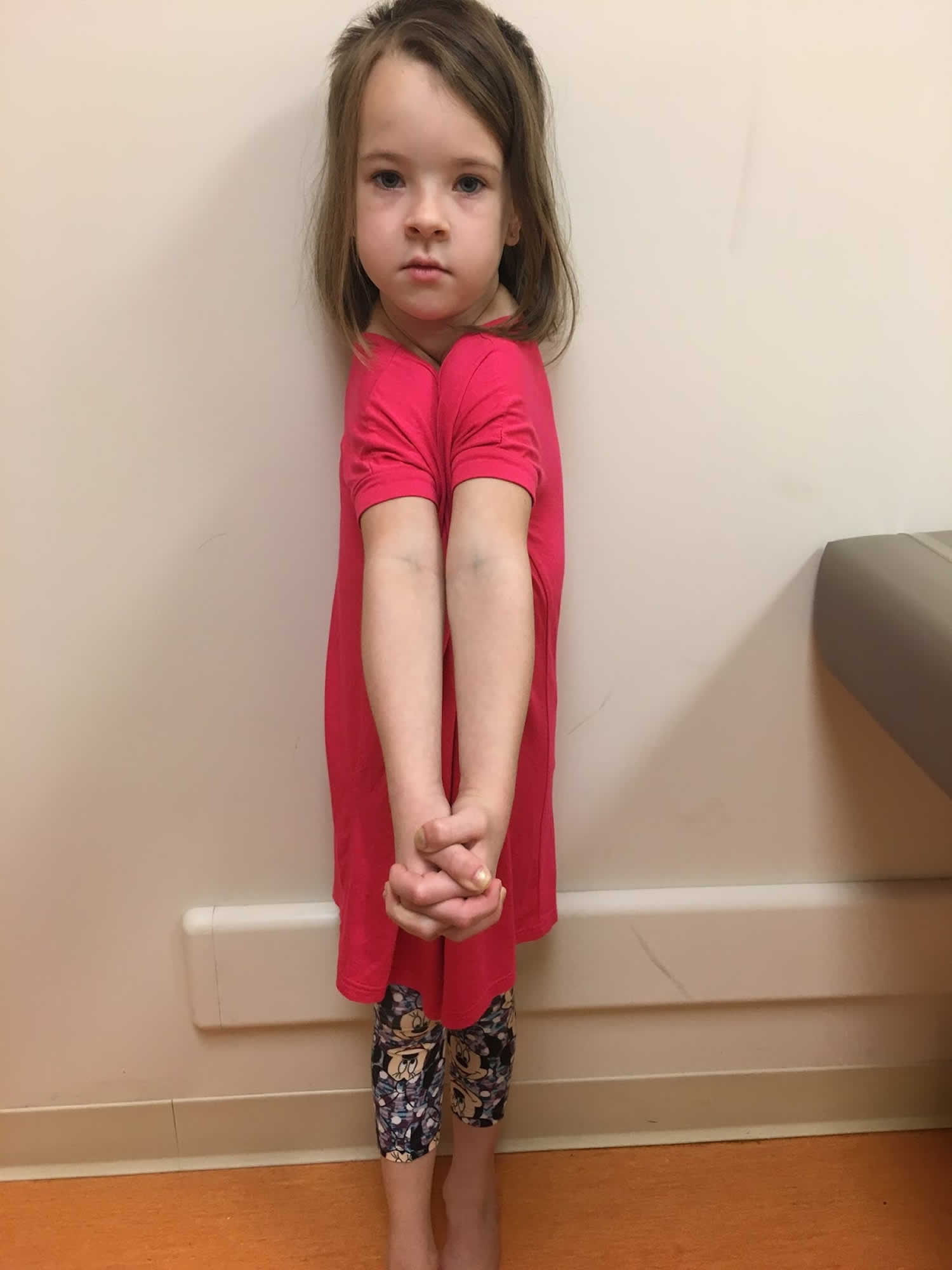
Individuals with clavicular hypoplasia usually have underdeveloped or absent collarbones, also called clavicles (“cleido-” in the condition name refers to these bones). As a result, their shoulders are narrow and sloping, can be brought unusually close together in front of the body, and in some cases can be made to meet in the middle of the body.
Moderate short stature may be observed, with most affected individuals being shorter than other members of their family at the same age. Many also have short, tapered fingers and broad thumbs; flat feet; bowed legs or knock knees; short shoulder blades (scapulae); and an abnormal curvature of the spine (scoliosis). Typical facial features include a wide, short skull (brachycephaly); a prominent forehead; wide-set eyes (hypertelorism); a flat nose; and a small upper jaw.
Dental abnormalities are very common in cleidocranial dysplasia and can include delayed loss of the primary (baby) teeth; delayed appearance of the secondary (adult) teeth; unusually shaped, peg-like teeth; misalignment of the teeth and jaws (malocclusion); and presence of the second permanent molar with the primary dentition, sometimes accompanied by cysts in the gums 2. Individuals with cleidocranial dysplasia spectrum disorder are at increased risk of developing recurrent sinus infections, recurrent ear infections leading to conductive hearing loss, and upper-airway obstruction. Intelligence is typically normal 1.
Individuals with cleidocranial dysplasia often have decreased bone density (osteopenia) and may develop osteoporosis, a condition that makes bones progressively more brittle and prone to fracture, at a relatively early age 2. Women with cleidocranial dysplasia have an increased risk of requiring a cesarean section when delivering a baby, due to a narrow pelvis preventing passage of the infant’s head.
In addition to skeletal and dental abnormalities, some young children with this condition are mildly delayed in the development of motor skills such as crawling and walking, but intelligence is unaffected.
Cleidocranial dysplasia occurs in approximately 1 per million individuals worldwide. Cleidocranial dysplasia is likely underdiagnosed because many affected individuals have mild signs and symptoms 2. Cleidocranial dysplasia affects all ethnic groups. Stevenson et al 3 found the frequency to be 0.12 per 10,000 individuals in the Utah (USA) population, suggesting that the frequency may be higher than previously recognized.
Figure 1. Cleidocranial dysplasia teeth
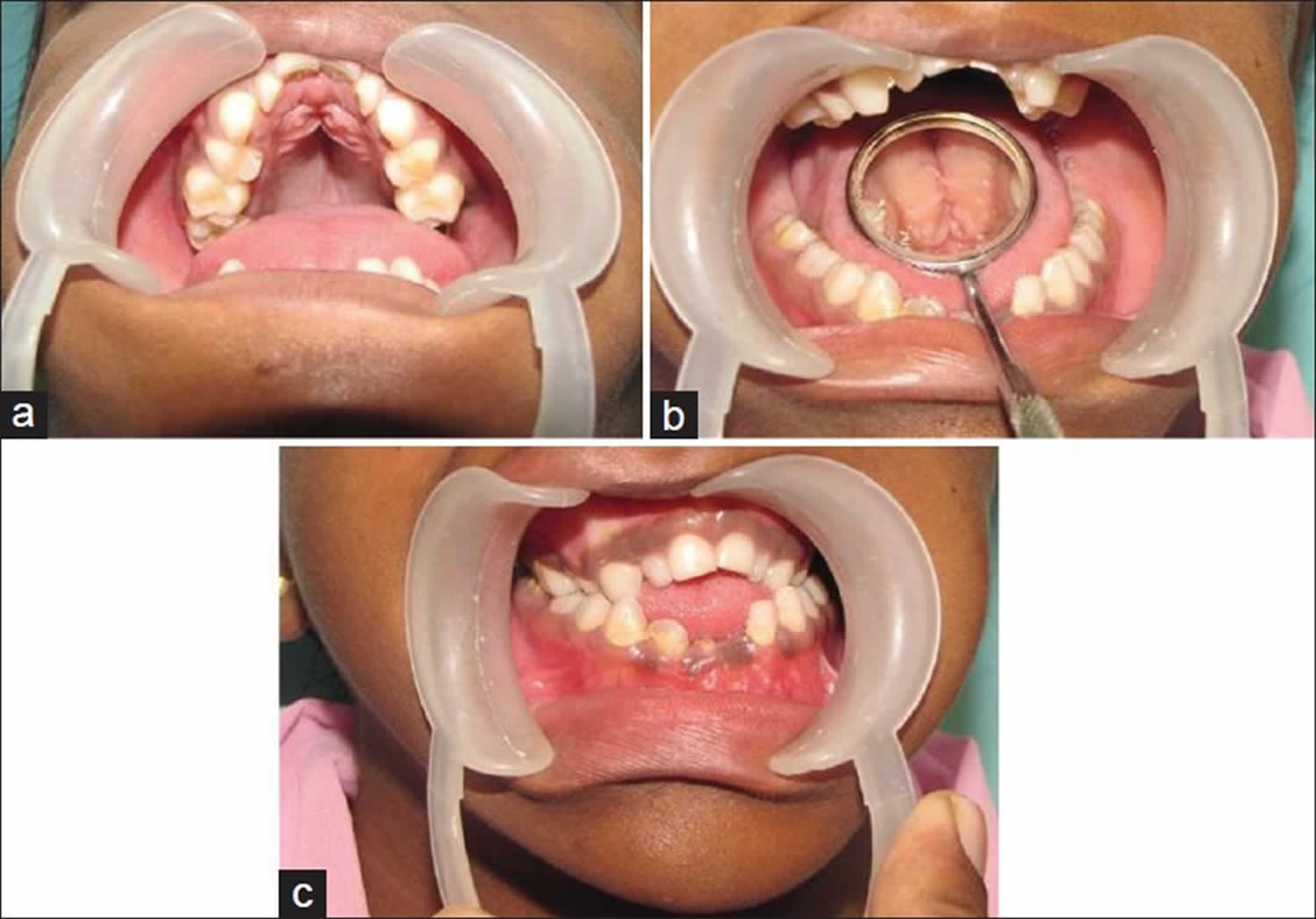
Figure 2. Orthopentamogram of the 9 years old male with cleido-cranial dysplasia showing deciduous retained teeth, numerous supernumerary teeth and dental age lagging behind chronological age

Figure 3. Cleidocranial dysplasia with clavicular hypoplasia – (a) Cone-shaped narrow thorax; (b) Approximation of shoulders to midline
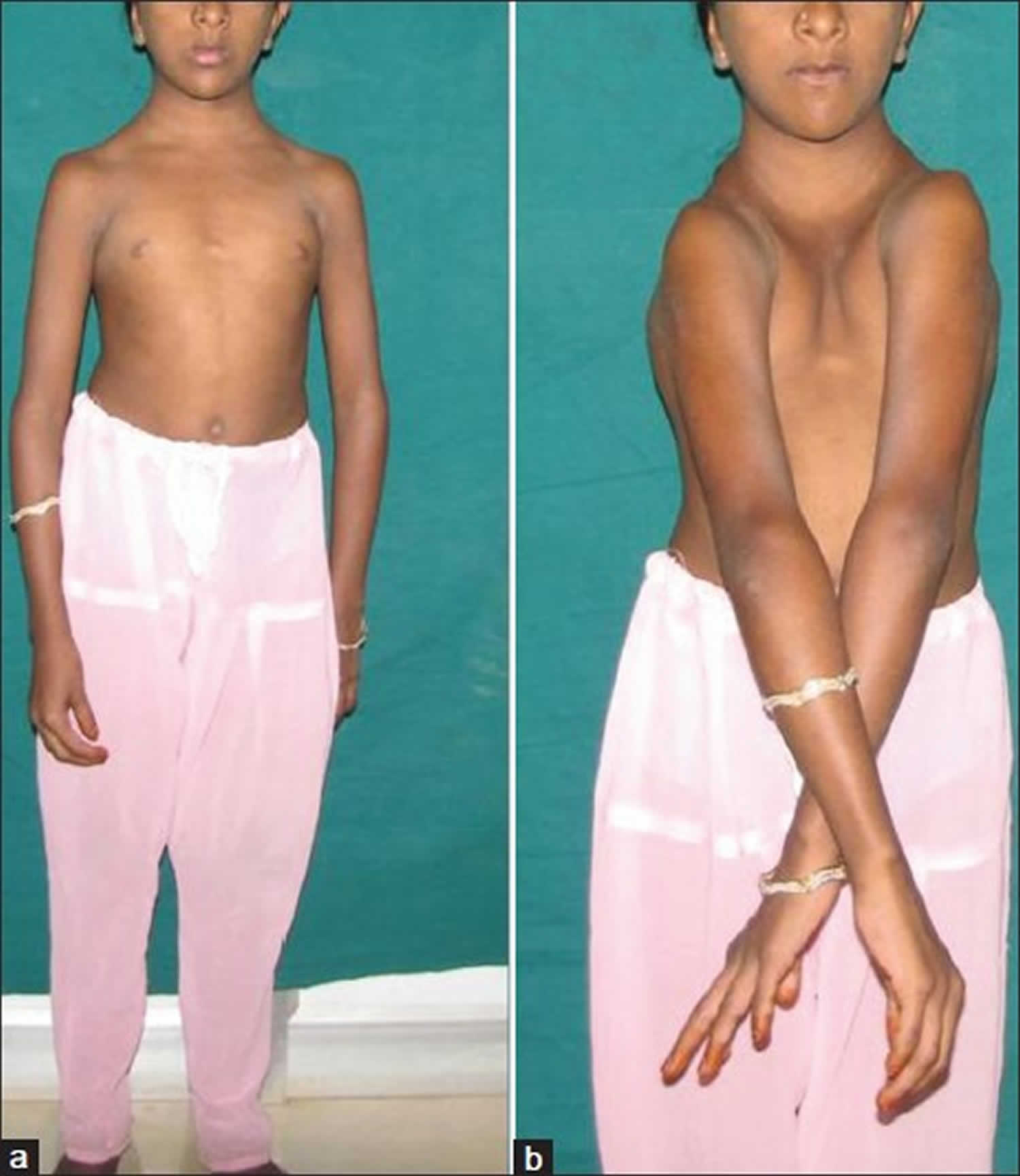
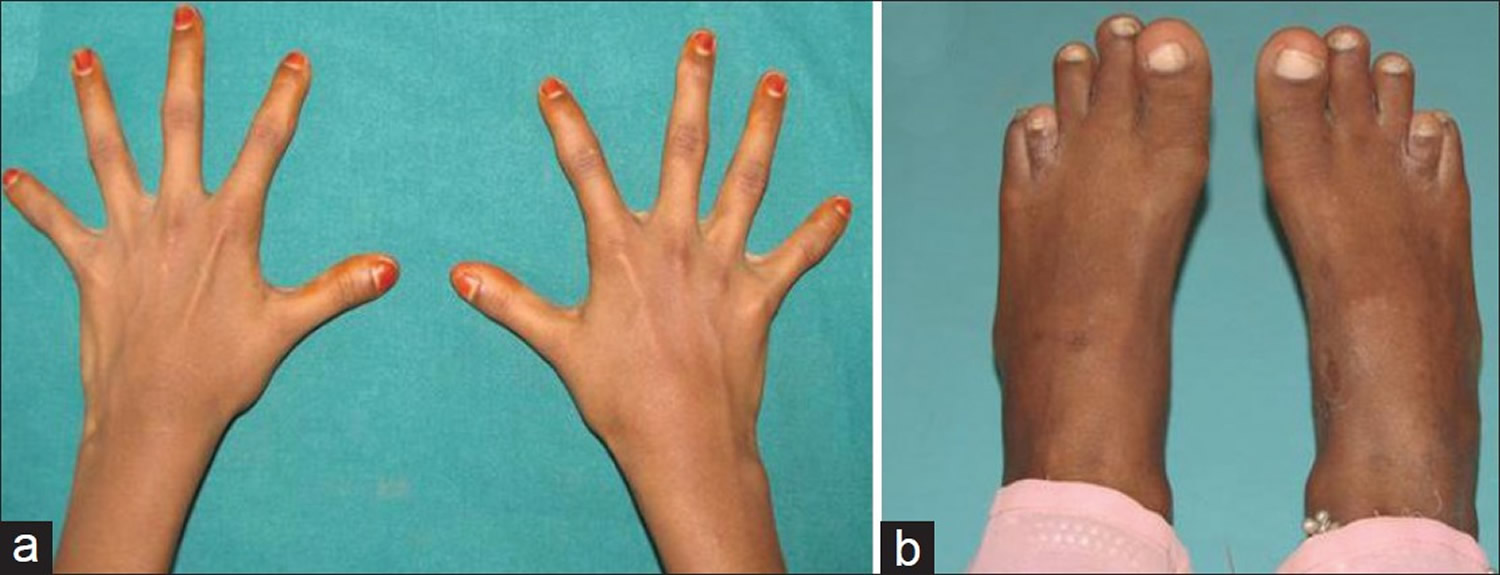
Figure 4. Cleidocranial dysplasia – (a) Fingers showing broad thumb and hypoplastic distal phalanges; (b) Brachydactyly of fourth toe
Cleidocranial dysplasia prognosis
The bone symptoms cause few problems in most cases. Appropriate dental care is important.
Cleidocranial dysplasia possible complications
Complications include dental problems and shoulder dislocations.
Cleidocranial dysplasia symptoms
People with cleidocranial dysostosis have a jaw and brow area that sticks out. The middle of their nose (nasal bridge) is wide.
The collar bones may be missing or abnormally developed. This pushes the shoulders together in front of the body.
Primary teeth do not fall out at the expected time. Adult teeth may develop later than normal, and an extra set of adult teeth grow in. This causes the normal teeth to become crooked.
The condition does not affect a person’s intelligence.
Other symptoms can include:
- Ability to touch shoulders together in front of body
- Delayed closure of fontanelles (“soft spots”)
- Loose joints
- Prominent forehead (frontal bossing)
- Short forearms
- Short fingers
There is often a family history of cleidocranial dysostosis. X-rays are most often taken and may show:
- Undergrowth of the collarbone
- Undergrowth of the shoulder blade
- Failure of the area in the front of the pelvis bone to close
Cleidocranial dysplasia causes
Cleidocranial dysplasia is usually caused by mutations in the RUNX2 gene. The RUNX2 gene provides instructions for making a protein that is involved in the development and maintenance of teeth, bones, and cartilage 6. Cartilage is a tough, flexible tissue that makes up much of the skeleton during early development. Most cartilage is later converted to bone (a process called ossification), except for the cartilage that continues to cover and protect the ends of bones and is present in the nose, airways, and external ears.
The RUNX2 protein is a transcription factor, which means it attaches (binds) to specific regions of DNA and helps control the activity of particular genes. Researchers believe that the RUNX2 protein acts as a “master switch,” regulating a number of other genes involved in the development of cells that build bones (osteoblasts) and in the development of teeth 2.
The RUNX2 gene mutations that cause cleidocranial dysplasia reduce or eliminate the activity of the protein produced from one copy of the RUNX2 gene in each cell, decreasing the total amount of functional RUNX2 protein. This shortage of functional RUNX2 protein interferes with the normal development of bones, cartilage, and teeth, resulting in the signs and symptoms of cleidocranial dysplasia. In rare cases, individuals with a deletion of genetic material that includes RUNX2 and other nearby genes may experience additional features, such as developmental delay, resulting from the loss of these genes.
In about 30 percent of individuals with cleidocranial dysplasia, no mutation in the RUNX2 gene has been found. The cause of the condition in these individuals is unknown 2.
Figure 5. Cleidocranial dysplasia autosomal dominant inheritance pattern
Risk to family members
Parents of a proband
- Some individuals diagnosed with cleidocranial dysplasia spectrum disorder have an affected parent.
- A proband with cleidocranial dysplasia spectrum disorder may have the disorder as the result of a de novo heterozygous RUNX2 pathogenic variant. The proportion of cases caused by a de novo pathogenic variant is high.
- If the pathogenic variant found in the proband cannot be detected in leukocyte DNA of either parent, possible explanations include a de novo pathogenic variant in the proband or germline mosaicism in a parent. Germline mosaicism has been reported [Pal et al 2007].
- Recommendations for the evaluation of parents of a proband with an apparent de novo pathogenic variant include careful clinical examination and consideration of craniofacial and skeletal x-rays if there are signs suggestive of dental or bone abnormalities. (Note: The phenotype may vary between parent and child even though they have the same pathogenic variant.) Molecular genetic testing for the parents of a proband with an apparent de novo pathogenic variant may also be considered.
- The family history of some individuals diagnosed with cleidocranial dysplasia spectrum disorder may appear to be negative because of failure to recognize the disorder in family members. Therefore, an apparently negative family history cannot be confirmed unless a clinical examination with skeletal x-rays and/or molecular genetic testing has been performed on the parents of the proband.
Note: If the parent is the individual in whom the pathogenic variant first occurred, s/he may have somatic mosaicism for the pathogenic variant and may be mildly/minimally affected.
Siblings of a proband
The risk to the siblings of the proband depends on the genetic status of the proband’s parents:
- If a parent of the proband is affected, the risk to the siblings is 50%. (Note: The phenotype may vary among sibs who inherit the RUNX2 pathogenic variant.)
- When the parents are clinically unaffected, the risk to the siblings of a proband appears to be low.
- If the pathogenic variant cannot be detected in the leukocyte DNA of either parent, the empiric recurrence risk to siblings is approximately 1% because of the possibility of parental germline mosaicism. Germline mosaicism has been demonstrated in a family with three affected siblings and an apparently unaffected mother 7.
Offspring of a proband
Each child of an individual with cleidocranial dysplasia spectrum disorder has a 50% chance of inheriting the RUNX2 pathogenic variant.
Other family members
The risk to other family members depends on the status of the proband’s parents: if a parent has features of cleidocranial dysplasia spectrum disorder and/or the RUNX2 pathogenic variant, his or her family members are at risk.
Cleidocranial dysplasia prevention
Genetic counseling is appropriate if a person with a family or personal history of cleidocranial dysostosis is planning to have children.
Genetic Counseling Issues
Considerations in families with an apparent de novo pathogenic variant. When neither parent of a proband with cleidocranial dysplasia spectrum disorder has the RUNX2 pathogenic variant identified in the proband or clinical evidence of the disorder, the RUNX2 pathogenic variant is likely de novo. However, possible non-medical explanations including alternate paternity or maternity (e.g., with assisted reproduction) and undisclosed adoption could also be explored.
Family planning
- The optimal time for determination of genetic risk and discussion of the availability of prenatal testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are affected.
DNA banking is the storage of DNA (typically extracted from white blood cells) for possible future use. Because it is likely that testing methodology and our understanding of genes, allelic variants, and diseases will improve in the future, consideration should be given to banking DNA of affected individuals.
Prenatal Testing and Preimplantation Genetic Diagnosis
Once the RUNX2 pathogenic variant has been identified in an affected family member, prenatal testing for a pregnancy at increased risk and preimplantation genetic diagnosis for cleidocranial dysplasia spectrum disorder are possible.
Ultrasound examination. Classic cleidocranial dysplasia can be diagnosed by ultrasound examination in the offspring of an affected parent as early as 14 weeks’ gestation. The most consistent features are abnormal clavicles, which are either short (<5th centile for gestational age) or partially or totally absent. Other less specific findings include brachycephalic skull with undermineralization, frontal bossing, and generalized immature ossification 8.
Note: Gestational age is expressed as menstrual weeks calculated either from the first day of the last normal menstrual period or by ultrasound measurements.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing, particularly if the testing is being considered for the purpose of pregnancy termination rather than early diagnosis. While most centers would consider decisions regarding prenatal testing to be the choice of the parents, discussion of these issues is appropriate.
Cleidocranial dysplasia treatment
Evaluations following initial diagnosis
To establish the extent of disease and needs in an individual diagnosed with cleidocranial dysplasia spectrum disorder, the following evaluations are recommended if they have not already been completed 1:
- Full skeletal survey including the hands and feet
- DXA scan for those in early adolescence and older
- Dental evaluation by a dentist familiar with cleidocranial dysplasia and its management
- Audiologic evaluation
- Consultation with a clinical geneticist and/or genetic counselor
Treatment of manifestations
If the cranial vault defect is significant, the head needs protection from blunt trauma; helmets may be used for high-risk activities. Surgical cosmesis for depressed forehead or lengthening of hypoplastic clavicles can be considered. If bone density is below normal, treatment with calcium and vitamin D supplementation is considered. Dental procedures to address retention of deciduous dentition, presence of supernumerary teeth, and non-eruption of the permanent dentition. Such procedures may include prosthetic replacements, removal of the supernumerary teeth followed by surgical repositioning of the permanent teeth, and a combination of surgical and orthodontic measures for actively erupting and aligning the impacted permanent teeth. Speech therapy may be required during periods of dental treatment. Aggressive treatment of sinus and middle ear infections; consideration of tympanostomy tubes for recurrent middle ear infections.
Craniofacial. The fontanels close with time in the majority of individuals and cranial remodeling is usually not necessary.
- If the cranial vault defect is significant, the head should be protected from blunt trauma; helmets may be advised for high-risk activities. In these cases, evaluation by a craniofacial surgeon and rehabilitation services are indicated.
- Affected individuals may consider having correction of the depressed forehead or lengthening of the hypoplastic clavicles for cosmetic reasons. There have been reports of successful surgical interventions in a very small number of affected individuals 9.
Skeletal. If bone density is below normal on DXA, treatment with calcium and vitamin D supplementation should be considered.
Dental. Early referral to a dental clinic familiar with leidocranial dysplasia allows for timely planning of necessary procedures.
- The dental problems that need to be addressed include the retention of deciduous dentition, the presence of supernumerary teeth, and the non-eruption of the permanent dentition.
- The goal of treatment is to improve appearance and to provide a functioning masticatory mechanism. The goals may be achieved with prosthetic replacements, with or without prior extractions; by removal of the supernumerary teeth followed by surgical repositioning of the permanent teeth; and by a combination of surgical and orthodontic measures for actively erupting and aligning the impacted permanent teeth.
- Generally, an aggressive approach to coordination of multiple oral surgeries for removal of primary dentition and exposure of permanent dentition is recommended, as watchful waiting for spontaneous eruption after initial delay is not effective.
Speech therapy may be required during periods of dental treatment.
Upper airway obstruction. When symptoms are suggestive, a sleep study is indicated and surgical intervention may be required.
Sinus and middle ear infections require aggressive and timely treatment; tympanostomy tubes should be considered when middle ear infections are recurrent 10.
Endocrinology. The effectiveness of growth hormone (GH) therapy for short stature in this condition has not been proven. Possible adverse effects of growth hormone (GH) therapy on the primary chondrodysplastic growth plate are theoretically possible, as RUNX2 is directly involved in chondrocyte differentiation and growth plate maintenance 11.
Prevention of primary manifestations
Preventive treatment for osteoporosis should be initiated at a young age since peak bone mineral density is achieved in the second and third decade. Early screening for low bone mineral density and appropriate supplementation with vitamin D and calcium are recommended.
Prevention of secondary complications
Anesthetic management of those with cleidocranial dysplasia spectrum disorder needs to be carefully planned since affected individuals may present with a large brachycephalic head with mandibular prognathism and maxillary underdevelopment. In addition, the depressed nasal bridge and hypoplastic sinuses disturb nasal breathing. The dental and craniofacial abnormalities result in predictably difficult airway management. If this is anticipated, an otolaryngologist should be consulted to assist in securing the airway. Alternative anesthetic approaches, including neuraxial block, should be considered, taking into account possible spine abnormalities 12.
Surveillance
Monitoring of children for orthopedic complications, dental abnormalities, upper-airway obstruction, sinus and ear infections, and hearing loss. Monitoring for osteoporosis beginning in early adolescence and every five to ten years thereafter.
Children with cleidocranial dysplasia should be monitored for the following:
- Orthopedic complications
- Dental abnormalities
- Signs and symptoms of upper-airway obstruction
- Sinus and ear infections
- Hearing loss. Regular audiometry in individuals with repeated ear infections allows the identification and early management of hearing loss if it develops.
- Osteoporosis. DXA to measure bone mineral density should be done early in adolescence and every five to ten years thereafter. If there are clinical signs of osteopenia (i.e., increased number of fractures), evaluation and treatment should be started earlier.
All affected individuals should by followed by their primary care physician and receive regular immunizations and anticipatory guidance as recommended.
Agents/circumstances to avoid
Helmets and protective devices should be worn when participating in high-risk sports and activities.
Pregnancy management
Monitoring of affected women during pregnancy for cephalopelvic disproportion. Pregnant women with cleidocranial dysplasia spectrum disorder should be monitored closely for cephalopelvic disproportion, which may require delivery by cesarean section. The primary cesarean section rate among women with a cleidocranial dysplasia spectrum disorder is 69%, which is higher than in controls 13.
- Machol K, Mendoza-Londono R, Lee B. Cleidocranial Dysplasia Spectrum Disorder. 2006 Jan 3 [Updated 2017 Nov 16]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2018. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1513[↩][↩][↩][↩]
- Cleidocranial dysplasia. https://ghr.nlm.nih.gov/condition/cleidocranial-dysplasia[↩][↩][↩][↩][↩]
- Stevenson DA, Carey JC, Byrne JL, Srisukhumbowornchai S, Feldkamp ML. Analysis of skeletal dysplasias in the Utah population. Am J Med Genet A. 2012;158A:1046–54.[↩]
- Cleidocranial dysplasia with hearing loss. Journal of Natural Science, Biology and Medicine 2013, Volume : 4, Issue : 1, Page:245-249 http://www.jnsbm.org/temp/JNatScBiolMed41245-3351314_091833.pdf[↩][↩][↩]
- Verma R, Jindal M, Maheshwari S. Familial Cleidocranial Dysplasia. International Journal of Clinical Pediatric Dentistry. 2010;3(1):57-61. doi:10.5005/jp-journals-10005-1055. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4955046/[↩]
- Cleidocranial dysplasia: a case report. Hemalatha R, Balasubramaniam MR. J Indian Soc Pedod Prev Dent. 2008 Mar; 26(1):40-3. https://www.ncbi.nlm.nih.gov/pubmed/18408272/[↩]
- Pal T, Napierala D, Becker TA, Loscalzo M, Baldridge D, Lee B, Sutphen R. The presence of germ line mosaicism in cleidocranial dysplasia. Clin Genet. 2007;71:589–91.[↩]
- Hermann NV, Hove HD, Jørgensen C, Larsen P, Darvann TA, Kreiborg S, Sundberg K. Prenatal 3D ultrasound diagnostics in cleidocranial dysplasia. Fetal Diagn Ther. 2009;25:36–9.[↩]
- Sewell MD, Higgs DS, Lambert SM. Clavicle lengthening by distraction osteogenesis for congenital clavicular hypoplasia: case series and description of technique. J Pediatr Orthop. 2013;33:314–20.[↩]
- Visosky AM, Johnson J, Bingea B, Gurney T, Lalwani AK. Otolaryngological manifestations of cleidocranial dysplasia, concentrating on audiological findings. Laryngoscope. 2003;113:1508–14.[↩]
- Zheng Q, Sebald E, Zhou G, Chen Y, Wilcox W, Lee B, Krakow D. Dysregulation of chondrogenesis in human cleidocranial dysplasia. Am J Hum Genet. 2005;77:305–12.[↩]
- Ioscovich A, Barth D, Samueloff A, Grisaru-Granovsky S, Halpern S. Anesthetic management of a patient with cleidocranial dysplasia undergoing various obstetric procedures. Int J Obstet Anesth. 2010;19:106–8.[↩]
- Cooper SC, Flaitz CM, Johnston DA, Lee B, Hecht JT. A natural history of cleidocranial dysplasia. Am J Med Genet. 2001;104:1–6.[↩]

{kind=link}