Contents
What is episcleritis
Episcleritis (episcleritis periodica fugax) is an inflammatory condition affecting the episclera or episcleral tissue between the conjunctiva (the clear mucous membrane lining the inner eyelids and sclera) and the sclera (the white part of the eye) that occurs in the absence of an infection. Episcleritis is not an infection. The red appearance caused by this condition looks similar to conjunctivitis, but there is no discharge. There is no apparent cause, but it can be associated with an underlying systemic inflammatory or rheumatologic condition such as rosacea, lupus or rheumatoid arthritis. Typical symptoms include generalized or local redness of the eyes that may be accompanied by mild soreness or discomfort but no visual problems. Swelling usually goes away without treatment in a couple of weeks. However, the swelling can go and come back more than once over time. Although treatment is often unnecessary, corticosteroid eyedrops can be used. Episcleritis most often goes away on its own in 1 to 2 weeks. Using corticosteroid eye drops may help ease the symptoms faster.
Episcleritis is a relatively common, benign, self-limited cause of red eye, due to inflammation of the episcleral tissues. There are two forms of this condition, nodular and simple 1:
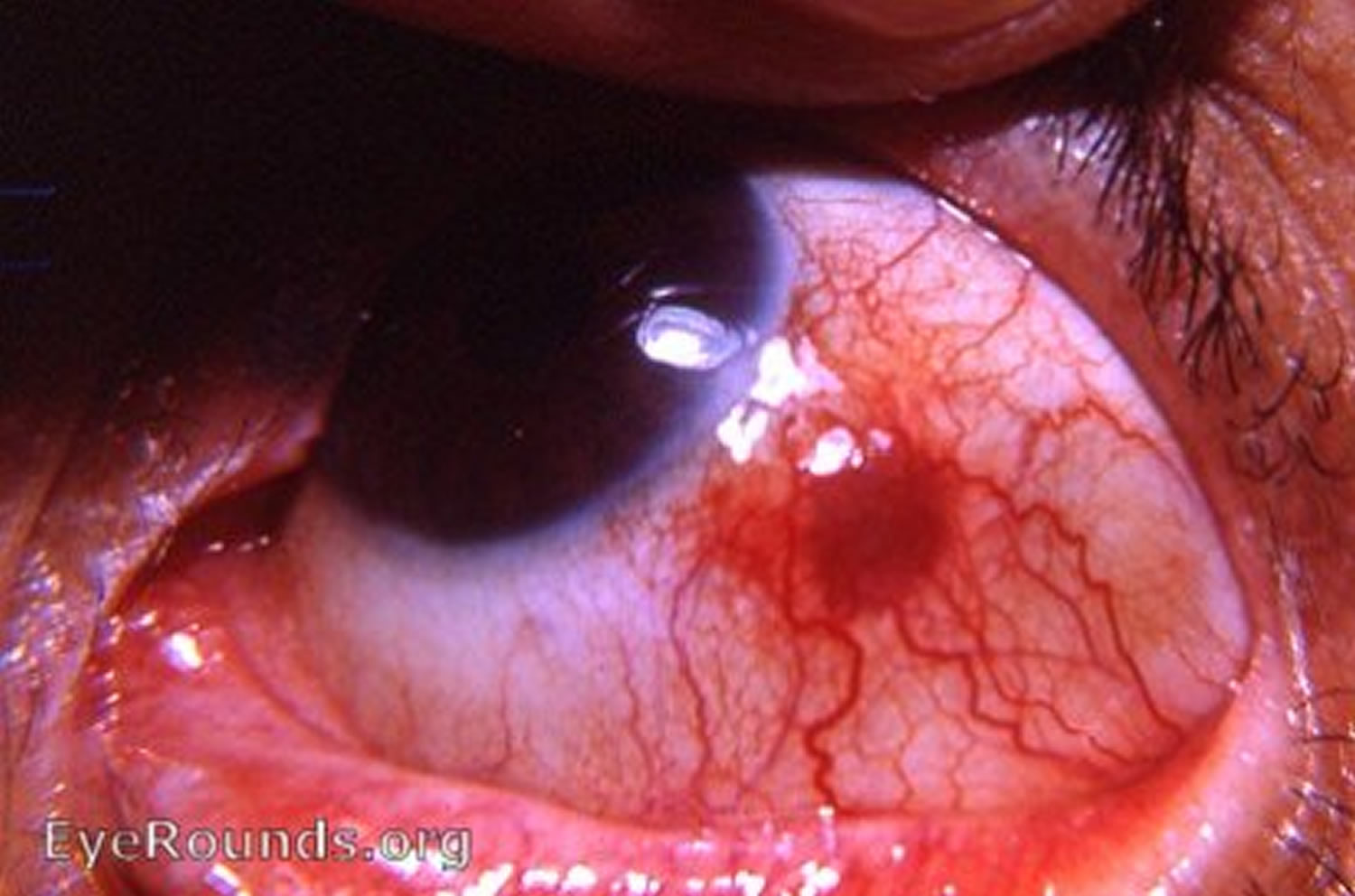
- Nodular episcleritis is characterized by a discrete, elevated area of inflamed episcleral tissue.
- Simple episcleritis, vascular congestion is present in the absence of an obvious nodule.
A 2013 study estimated incidence of episcleritis as 41.0 per 100,000 per year and prevalence at 52.6. The simple variety is more common than nodular. According to one study, approximately sixty-seven percent of simple episcleritis is “sectoral” (involving only one sector or area of the episclera) and thirty-three percent is diffuse (involving the entire episclera).
Episclera anatomy
The episclera is a fibroelastic structure consisting of two layers loosely joined together. The outer parietal layer, with the vessels of the superficial episcleral capillary plexus, is the more superficial layer. The superficial vessels appear straight and are arranged in a radial fashion. The deeper visceral layer contains a highly anastomotic network of vessels. Both of the vessel networks originate from the anterior ciliary arteries, which stem from the muscular branches of the ophthalmic artery. The episclera lies between the superficial scleral stroma and Tenon’s capsule. In episcleritis, vascular congestion occurs in the superficial episcleral plexus. The episclera as well as Tenon’s capsule become infiltrated with inflammatory cells. The sclera is spared.
Figure 1. Episclera and sclera anatomy
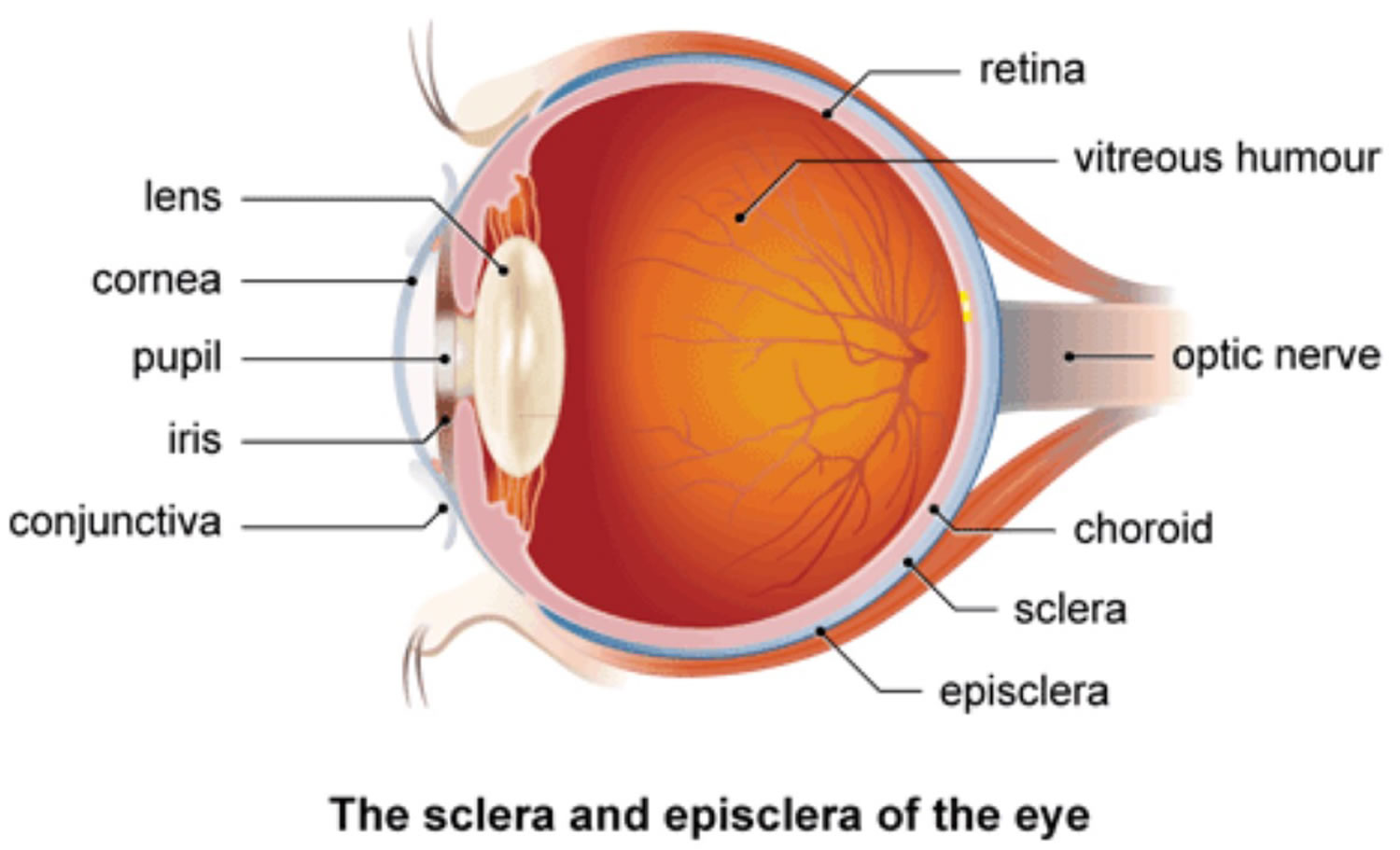 Figure 2. Episcleritis
Figure 2. Episcleritis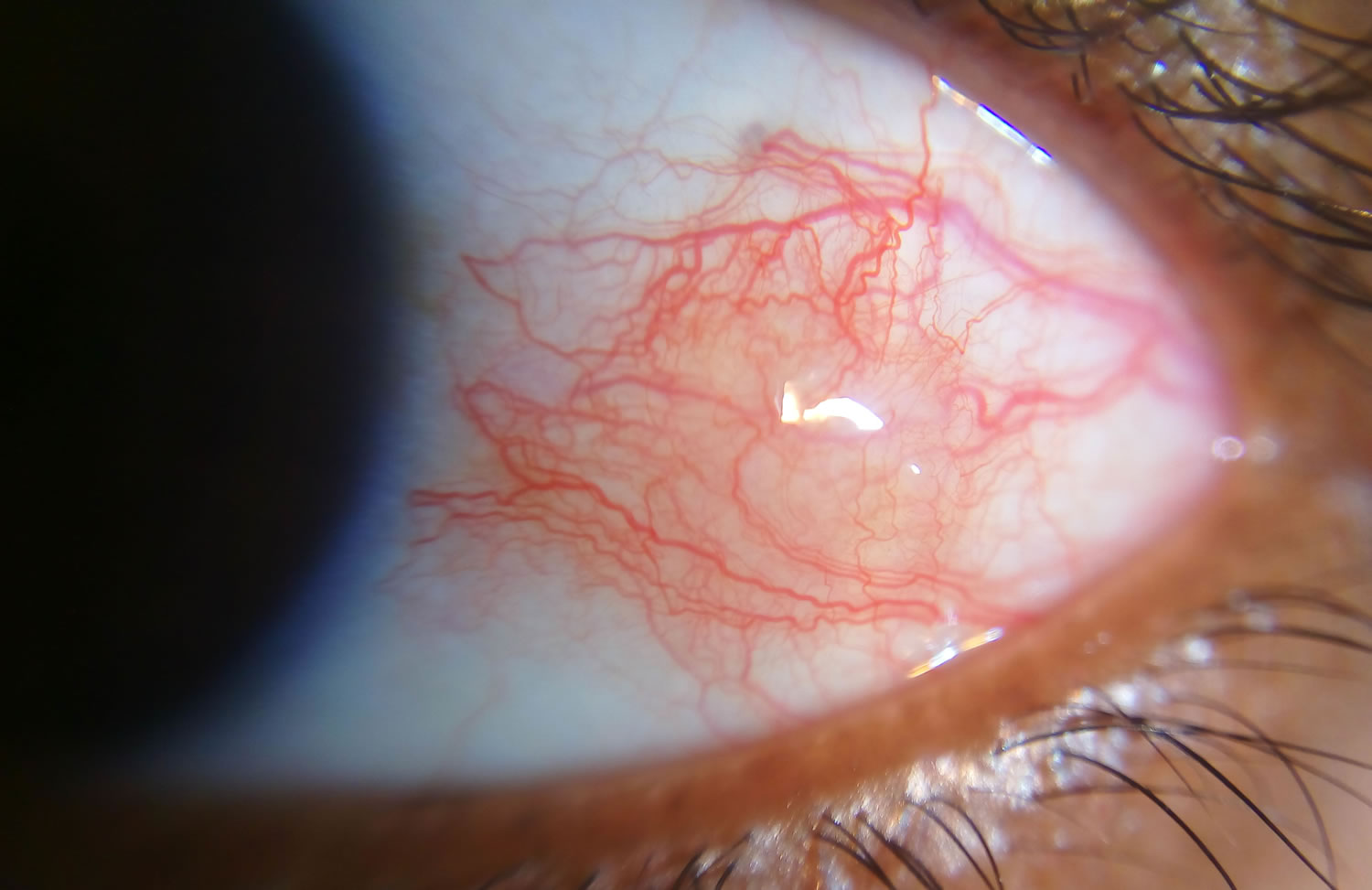
Figure 3. Nodular episcleritis
Episcleritis vs Scleritis
Scleritis or inflammation of the sclera, is the inflammation in the episcleral and scleral tissues with injection in both superficial and deep episcleral vessels. It may involve the cornea, adjacent episclera and the uvea and thus can be vision-threatening. Scleritis is often associated with an underlying systemic disease in up to 50% of patients. Scleritis can present as a painful red eye with or without vision loss.
The most common form of scleritis is anterior scleritis, is defined as scleral inflammation anterior to the extraocular recti muscles. Posterior scleritis is defined as involvement of the sclera posterior to the insertion of the rectus muscles.
- Anterior scleritis, the most common form, can be subdivided into diffuse, nodular, or necrotizing forms. In the diffuse form, anterior scleral edema is present along with dilation of the deep episcleral vessels. The entire anterior sclera or just a portion may be involved. In nodular disease, a distinct nodule of scleral edema is present. The nodules may be single or multiple in appearance and are often tender to palpation. Necrotizing anterior scleritis is the most severe form of scleritis. It is characterized by severe pain and extreme scleral tenderness. Severe vasculitis as well as infarction and necrosis with exposure of the choroid may result. A rare form of necrotizing anterior scleritis without pain can be called scleromalacia perforans. The sclera is notably white, avascular and thin. Both choroidal exposure and staphyloma formation may occur.
- Posterior scleritis, although rare, can manifest as serous retinal detachment, choroidal folds, or both. There is often loss of vision as well as pain upon eye movement.
Histologically, the appearance of episcleritis and scleritis differs in that the sclera is not involved in the former. In episcleritis, hyperemia, edema and infiltration of the superficial tissue is noted along with dilated and congested vascular networks. There is chronic, non-granulomatous infiltrate consisting of lymphocytes and plasma cells.
In scleritis, scleral edema and inflammation are present in all forms of disease. There is often a zonal granulomatous reaction that may be localized or diffuse. If localized, it may result in near total loss of scleral tissue in that region. Most commonly, the inflammation begins in one area and spreads circumferentially until the entire anterior segment is involved.
Inflammation of the sclera can involve a non-granulomatous process (lymphocytes, plasma cells, macrophages) or a granulomatous process (epitheliod cells, multinucleated giant cells) with or without associated scleral necrosis.
Scleritis causes
There are many connective tissue disorders that are associated with scleral disease. Scleritis is often linked to autoimmune diseases. These diseases occur when the body’s immune system attacks and destroys healthy body tissue by mistake. Diseases such as rheumatoid arthritis and systemic lupus erythematosus can cause this problem. Rheumatoid arthritis is the most common. Sometimes the cause is unknown.
Scleritis may also be infectious or surgically/trauma-induced. There is no known HLA association.
Scleritis occurs most often in people between the ages of 30 and 60. It is rare in children.
As there are different forms of scleritis, the pathophysiology is also varied. Scleritis associated with autoimmune disease is characterized by zonal necrosis of the sclera surrounded by granulomatous inflammation and vasculitis. Eosinophilic fibrinoid material may be found at the center of the granuloma. These eyes may exhibit vasculitis with fibrinoid necrosis and neutrophil invasion of the vessel wall.
There is an increase in inflammatory cells including T-cells of all types and macrophages. T-cells and macrophages tend to infiltrate the deep episcleral tissue with clusters of B-cells in perivascular areas. There may be cell-mediated immune response as there is increased HLA-DR expression as well as increased IL-2 receptor expression on the T-cells. Plasma cells may be involved in the production of matrix metalloproteinases and TNF-alpha. In idiopathic necrotizing scleritis, there may be small foci of scleral necrosis and mainly nongranulomatous inflammation with mainly mononuclear cells (lymphocytes, plasma cells and macrophages). Microabscesses may be found in addition to necrotizing inflammation in infectious scleritis.
Vasculitis is not prominent in non-necrotizing scleritis.
Treatments for scleritis may include:
- Corticosteroid eye drops to help reduce the inflammation
- Corticosteroid pills
- Newer, nonsteroid anti-inflammatory drugs (NSAIDs) in some cases
- Certain anticancer drugs (immune-suppressants) for severe cases
If scleritis is caused by an underlying disease, treatment of that disease may be needed.
Risk factors for scleritis
As scleritis is associated with systemic autoimmune diseases, it is more common in women. It usually occurs in the fourth to sixth decades of life. Men are more likely to have infectious scleritis than women. Patients with a history of pterygium surgery with adjunctive mitomycin C administration or beta irradiation are at higher risk of infectious scleritis due to defects in the overlying conjunctiva from calcific plaque formation and scleral necrosis. Bilateral scleritis is more often seen in patients with rheumatic disease. Two or more surgical procedures may be associated with the onset of surgically induced scleritis.
Scleritis prevention
Most scleritis cases cannot be prevented.
People with autoimmune diseases, may need to have regular check-ups with an ophthalmologist familiar with the condition.
Scleritis signs and symptoms
The onset of scleritis is gradual. Most patients develop severe boring or piercing eye pain over several days. Globe tenderness and redness may involve the whole eye or a small localized area. A severe pain that may involve the eye and orbit is usually present. This pain is characteristically dull and boring in nature and exacerbated by eye movements. Worsening of the pain during eye movement is due to the extraocular muscle insertions into the sclera. It may be worse at night and awakens the patient while sleeping. This pain may radiate to involve the ear, scalp, face and jaw.
Symptoms of scleritis include:
- Blurred vision
- Eye pain and tenderness — severe
- Red patches on the normally white part of the eye
- Sensitivity to light — very painful
- Tearing of the eye
A rare form of this disease causes no eye pain or redness.
Scleritis signs
Scleritis presents with a characteristic violet-bluish hue with scleral edema and dilatation. Other signs vary depending on the location of the scleritis and degree of involvement. In the anterior segment there may be associated keratitis with corneal infiltrates or thinning, uveitis, and trabeculitis. With posterior scleritis, there may be chorioretinal granulomas, retinal vasculitis, serous retinal detachment and optic nerve edema with or without cotton-wool spots.
Non-ocular signs are important in the evaluation of the many systemic associations of scleritis. Epistaxis, sinusitis and hemoptysis are present in granulomatosis with polyangiitis (formerly known as Wegener’s). Arthritis with skin nodules, pericarditis, and anemia are features of rheumatoid arthritis. Systemic lupus erythematous may present with a malar rash, photosensitivity, pleuritis, pericarditis and seizures. In addition to scleritis, myalgias, weight loss, fever, purpura, nephropathy and hypertension may be signs of polyarteritis nodosa.
Scleritis complications
Complications may include:
- Return of scleritis
- Side effects of long-term corticosteroid therapy
- Perforation of the eyeball, leading to vision loss if the condition is left untreated
Complications are frequent and include peripheral keratitis, uveitis, cataract and glaucoma. Central stromal keratitis may also occur in the absence of treatment. Sclerokeratitis in which peripheral cornea is opacified by fibrosis and lipid deposition with neighboring scleritis may occur particularly with herpes zoster scleritis. Sclerosing keratitis may present with crystalline deposits in the posterior corneal lamellae. Sclerokeratitis may move centrally gradually and thus opacify a large segment of the cornea. Vitritis (cells and debris in vitreous) and exudative detachments occur in posterior scleritis.
Scleritis diagnosis
The diagnosis of scleritis is clinical. However, laboratory testing is often necessary to discover any associated connective tissue and autoimmune disease.
Your health care provider will perform the following tests:
- Eye exam
- Physical exam and blood tests to look for conditions that may be causing the problem
Scleritis presents with a characteristic violet-bluish hue with scleral edema and dilatation. Examination in natural light is useful in differentiating the subtle color differences between scleritis and episcleritis. On slit-lamp biomicroscopy, inflamed scleral vessels often have a criss-crossed pattern and are adherent to the sclera. They cannot be moved with a cotton-tipped applicator, which differentiates inflamed scleral vessels from more superficial episcleral vessels. Red-free light with the slit lamp also accentuates the visibility of the blood vessels and areas of capillary nonperfusion. Finally, the conjunctival and superficial vessels may blanch with 2.5-10% phenylephrine but deep vessels are not affected. The globe is also often tender to touch.
It is important for your doctor to determine if you have scleritis. The symptoms can also be a less severe form of inflammation, such as episcleritis.
Diagnostic tests
B-scan ultrasonography and orbital magnetic resonance imaging (MRI) may be used for the detection of posterior scleritis. Ultrasonographic changes include scleral and choroidal thickening, scleral nodules, distended optic nerve sheath, fluid in Tenons capsule, or retinal detachment.
Laboratory test
As scleritis may occur in association with many systemic diseases, laboratory workup may be extensive. As mentioned earlier, the autoimmune connective tissue diseases of rheumatoid arthritis, lupus, sero-negative spondylarthropathies and vasculitides such as granulomatosis with polyangiitis and polyarteritis nodosa are most frequently seen. In addition to complete physical examination, laboratory studies should include assessment of blood pressure, renal function, and acute phase response. Laboratory tests include complete blood count (CBC) with differential, erythrocye sedimentation rate (ESR) or C-reactive protein (CRP), serum autoantibody screen (including antinuclear antibodies, anti-DNA antibodies, rheumatoid factor, antineutrophil cytoplasmic antibodies), urinalysis, syphilis serology, serum uric acid and sarcoidosis screen.
Scleritis treatment
The primary goal of treatment of scleritis is to minimize inflammation and thus reduce damage to ocular structures.
Medical therapy
NSAIDS
Oral non-steroidal anti-inflammatory drugs (NSAIDs) are the first-line agent for mild-to-moderate scleritis. These consist of non-selective or selective cyclo-oxygenase inhibitors (COX inhibitors). Non-selective COX-inhibitors such as flurbiprofen, indomethacin and ibuprofen may be used. Indomethacin 50mg three times a day or 600mg of ibuprofen three times a day may be used. Patients using oral NSAIDS should be warned of the side effects of gastrointestinal (GI) side effects including gastric bleeding. Patients with renal compromise must be warned of renal toxicity. NSAIDS that are selective COX-2 inhibitors may have fewer GI side effects but may have more cardiovascular side effects.
Corticosteroids
Topical corticosteroids may reduce ocular inflammation but treatment is generally systemic. Corticosteroids may be used in patients unresponsive to COX-inhibitors or those with posterior or necrotizing disease. A typical starting dose may be 1mg/kg/day of prednisone. This dose should be tapered to the best-tolerated dose. Pulsed intravenous methylprednisolone at 0.5-1g may be required initially for severe scleritis. Side effects of steroids that patients should be made aware of include elevated intraocular pressure, decreased resistance to infection, gastric irritation, osteoporosis, weight gain, hyperglycemia, and mood changes.
Immunomodulatory agents
If the disease is inadequately controlled on corticosteroids, immunomodulatory therapy may be necessary. Likewise, immunomodulatory agents should be considered in those who might otherwise be on chronic steroid use. Consultation with a rheumatologist or other internist is recommended. Patients with rheumatoid arthritis may be placed on methotrexate. Patients with granulomatosis with polyangiitis may require cyclosphosphamide or mycophenolate. Cyclosporine is nephrotoxic and thus may be used as adjunct therapy allowing for lower corticosteroid dosing. Mycophenolate mofetil may eliminate the need for corticosteroids. However, there is a risk of hematologic and hepatic toxicity.
More recently, tumor necrosis factor (TNF) alpha inhibitors such as infliximab have shown promise in the treatment of non-infectious scleritis refractory to other treatment. Treatment consists of repeated infusions as the treatment effect is short-lived. TNF-alpha inhibitors may also result in a drug-induced lupus-like syndrome as well as increased risk of lymphoproliferative disease. All patients on immunomodulatory therapy must be closely monitored for development of systemic complications with these medications.
Medical follow up
Adjustment of medications and dosages is based on the level of clinical response. Laboratory testing may be ordered regularly to follow the therapeutic levels of the medication, to monitor for systemic toxicity, or to determine treatment efficacy.
Surgery
Clinical examination is usually sufficient for diagnosis. Formal biopsy may be performed to exclude a neoplastic or infective cause. However, one must be prepared to place a scleral reinforcement graft or other patch graft as severe thinning may result in the presentation of intraocular contents. Small corneal perforations may be treated with bandage contact lens or corneal glue until inflammation is adequately controlled, allowing for surgery.
Primary indications for surgical intervention include scleral perforation or the presence of excessive scleral thinning with a high risk of rupture. High-grade astigmatism caused by staphyloma formation may also be treated. Reinforcement of the sclera may be achieved with preserved donor sclera, periosteum or fascia lata. A lamellar or perforating keratoplasty may be necessary. Cataract surgery should only be performed when the scleritis has been in remission for 2-3 months. Small incision clear corneal surgery is preferred, and one must anticipate a return of inflammation in the postsurgical period.
Surgical follow up
Surgical biopsy of the sclera should be avoided in active disease, though if absolutely necessary, the surgeon should be prepared to bolster the affeted tissue with either fresh or banked tissue (i.e., preserved pericardium, banked sclera or fascia lata). Areas with imminent scleral perforation warrant surgical intervention, though the majority of patients often have scleral thinning or staphyloma formation that do not require scleral reinforcement.
Scleritis prognosis
Visual loss is related to the severity of the scleritis. Patients with mild or moderate scleritis usually maintain excellent vision. Scleritis may be active for several months or years before going into long-term remission. Patients with necrotizing scleritis have a high incidence of visual loss and an increased mortality rate.
Episcleritis causes
Most cases of episcleritis are idiopathic or unknown 1.
Approximately 26-36% of patients have an associated systemic disorder 1. These include:
- Collagen-vascular diseases (rheumatoid arthritis, Crohn’s disease, ulcerative colitis, psoriatic arthritis, systemic lupus erythematosus, reactive arthritis (formerly Reiter’s syndrome), relapsing polychondritis, ankylosing spondylitis, and pustulotic arthro-osteitis), vasculitides (polyarteritis nodosa, temporal arteritis, Cogan’s syndrome, Churg-Strauss syndrome, granulomatosis with polyangiitis, Behcet’s disease). The most common collagen vascular disease association is with rheumatoid arthritis.
- Dermatologic disease (rosacea, pyoderma gangrenosum, Sweet’s syndrome),
- Metabolic disease (gout), and atopy.
- Malignancies, usually T-cell leukemia and Hodgkin’s lymphoma, can be associated with episcleritis.
- Foreign bodies and chemical injuries can also serve as inciting factors.
- Infectious agents do exist and include bacteria, mycobacteria, spirochetes (Treponema, Borrelia), Chlamydia, Actinomyces, fungi, herpes zoster & simplex, mumps, and chikungunya. Protozoa such as Acanthamoeba and Toxoplasmosis should be considered. Toxocara is another, albeit rare, cause.
- Medications such as topiramate and pamidronate can cause episcleritis.
Risk factors for episcleritis
Most studies have shown that female adults are affected more commonly than male adults. However, one study of a pediatric population, revealed that boys were affected more commonly than girls. There are no specific risk factors; however, as mentioned above, a subset of patients will have an associated systemic disease. One study 2 found that 51% of patients have some concurrent eye disease.
Episcleritis prevention
There is no primary prevention for episcleritis.
Episcleritis signs and symptoms
Patients with episcleritis will report the acute or gradual onset of diffuse or localized eye redness, usually unilateral. Some may not report any other symptoms, while others may report discomfort, photophobia, or tenderness. Complaints of severe pain or ocular discharge should prompt reconsideration of the diagnosis of episcleritis.
Episcleritis symptoms include:
- A pink or purple color to the normally white part of the eye
- Eye pain
- Eye tenderness
- Sensitivity to light
- Tearing of the eye
Episcleritis signs
Episcleritis is characterized by an area of diffuse or sectoral, bright red or pink bulbar injection. This is in contrast to the violaceous hue of scleritis. Eyelid edema and conjunctival chemosis may be present.
Episcleritis complications
Episcleritis is largely benign; however, there have been reports of a few complications in patients with recurrent disease. These include anterior and intermediate uveitis, as well as corneal dellen (adjacent an episcleral nodule) and peripheral corneal infiltrates (adjacent to episcleral inflammation). Declining vision, in the setting of episcleritis, is typically attributed to advancing cataracts. Glaucoma has also been noted in a minority of patients. Both cataracts and glaucoma could be related to steroid use as part of the management of episcleritis.
Episcleritis diagnosis
Episcleritis is a clinical diagnosis, based primarily on history as well as external and slit lamp examination.
History
Classically, patients 20-50 years old will present with either acute (simple episcleritis) or gradual (nodular episcleritis) onset of redness in the eye, possibly associated with pain. In simple episcleritis, the episode usually peaks in about 12 hours and then slowly resolves over the next 2-3 days. It tends to recur in the same eye or both eyes at the same time; over time, the attacks become less frequent and, over years, disappear completely. The attacks may move between eyes. In nodular episcleritis, the redness is typically noted when the patient wakes up in the morning. Over the next few days, the redness enlarges, usually causing more discomfort, and takes on a nodular appearance. Episodes of nodular episcleritis are self-limited but tend to last longer than attacks of simple episcleritis.
Physical examination
The area of injection should be examined with the slit lamp. If the examiner uses a narrow, bright slit beam, nodular episcleritis can be distinguished from scleritis. In nodular scleritis, the inner reflection, which rests on the sclera and visceral layer, will remain undisturbed while the outer reflection will be displaced forward by the episcleral nodule. In scleritis, both of light beams will be displaced forward. Also important to note is that the nodule in episcleritis is freely mobile over the scleral tissue that lies underneath.
Diagnostic tests
In practice, the differentiation of episcleritis and scleritis is often aided by the instillation of phenylephrine 2.5%. The phenylephrine blanches the conjunctival and episcleral vessels but leaves the scleral vessels undisturbed. If a patient’s eye redness improves after phenylephrine instillation, the diagnosis of episcleritis can be made. According to Krachmer et al 3, phenylephrine 2.5% eye drops blanch conjunctival vessels, allowing the differentiation of conjunctivitis and episcleritis. Instillation of phenylephrine 10% will result in blanching of the superficial episcleral vascular network but not the deep plexus, thus distinguishing between episcleritis and scleritis.
Episcleritis treatment
Management is generally supportive alone. Episcleritis typically clears on its own without treatment and reassurance is the primary step in management. Some patients, however, may experience significant pain or discomfort or may dislike the appearance of the condition. In such cases, supportive measures, such as cool compresses and iced artificial tears, or medical therapy can be initiated.
There are no surgical therapies for episcleritis.
Medical therapy
Oral NSAIDs (nonsteroidal anti-inflammatory drugs), typically 800 mg ibuprofen three times daily, are the mainstay of treatment for episcleritis. Alternative medications include indomethacin 75 mg twice daily or flurbiprofen 100 mg three times daily. Studies comparing topical flurbiprofen and ketorolac to placebo found no difference in effectiveness in resolving the injection. The use of weak topical steroids (administered 1-4 times daily until symptoms resolve) is sometimes employed, but this is controversial. Although they bring about a timely control of the condition, steroids may increase the risk of recurrence and cause ‘rebound’ redness followed by a more intense attack. In patients with collagen vascular disease, measures targeted at the underlying disease itself can achieve control of the episcleritis.
Episcleritis frequently occurs in the setting of dry eye syndrome and blepharitis and attention to these two underlying issues is likely to be of benefit.
Medical follow-up
Regular follow-up is not required unless a patient does not notice any improvement in his or her symptoms. Most isolated episodes of episcleritis resolve completely over 2-3 weeks. Those cases that are associated with systemic disease can take on a more prolonged course with multiple recurrences.
Episcleritis prognosis
Episcleritis is a benign, self-limited condition that usually resolves completely over the course of a few weeks. However, treatment with corticosteroid eye drops may make symptoms go away sooner.

{kind=link}