Contents
- Erdheim–Chester disease
- Erdheim–Chester disease cause
- Erdheim–Chester disease pathology outlines
- Erdheim–Chester disease symptoms
- Erdheim–Chester disease diagnosis
- Erdheim–Chester disease treatment
- Immunotherapy (Interferon-alpha)
- BRAF-inhibitors (vemurafenib, dabrafenib)
- MEK-inhibitors (trametinib, cobimetinib)
- Cladribine (2-CdA)
- Anakinra (Kineret)
- Imatinib (Gleevec)
- Infliximab (Remicade)
- Tocilizumab (Actemra)
- Methotrexate
- Sirolimus (Rapamune)
- Systemic corticosteroids
- Surgical debulking
- Radiation treatment
- Other treatments
- Monitoring on treatment
- Relapsed Erdheim–Chester disease treatment
- Erdheim–Chester disease prognosis
- Erdheim–Chester disease life expectancy
Erdheim–Chester disease
Erdheim-Chester disease (ECD) also known as lipid granulomatosis or polyostotic sclerosing histiocytosis, is a rare slow-growing blood cancer, a non-Langerhans cell histiocytosis neoplasm, which is characterized by the infiltration of histiocytes that contain large amounts of fatty (lipid) material (xanthomatous histiocytes); certain lymphocytes; and distinctive, large cells with multiple nuclei (Touton giant cells) and scattered plasma cells with surrounding fibrosis in multiple tissues and organs 1, 2, 3, 4, 5, 6, 7, 8. The histiocytes are positive by immunohistochemistry for CD68, and CD163, negative for CD1a and Langerin, and have a variable expression of S-100 protein 9. Histiocytes are large phagocytic cells (macrophages) that normally play a role in responding to infection and injury. A phagocytic cell is any “scavenger cell” that engulfs and destroys invading microorganisms or cellular debris. Histiocytes normally function to destroy foreign substances and protect the body from infection. In Erdheim-Chester disease, the excess production of histiocytes (histiocytosis) leads to inflammation that can damage organs and tissues throughout the body, causing them to become thickened, dense, and scarred (fibrotic); this tissue damage may lead to organ failure 5.
Individuals affected by Erdheim-Chester disease are typically middle-aged adults between their 4th and 7th decades of life 10, 11, 12, with patients have been diagnosed between the ages of 7 to 84 years with a mean age of 46 years at diagnosis in the United States (range, 20-74 years) and 56 years in the French cohort (range, 29-86 years) 13, 14, , 15 and cases involving children have been documented in the medical literature 16, 17, 18, 19.
Only about 1500 Erdheim–Chester disease cases have been reported worldwide 20. Since Erdheim–Chester disease is so rare and it was not discussed in the common textbooks of medicine, many doctors have never heard of it 12. The rarity of Erdheim–Chester disease, coupled with its heterogenous manifestations, can make the diagnosis extremely challenging with many patients go on years before they get a correct diagnosis 12. Erdheim–Chester disease patients may see multiple healthcare providers and undergo several biopsies that have, historically, led to delayed diagnosis and institution of therapy in most patients, with the average time from symptom onset to diagnosis being a few months to several years 13, 21. One scenario leading to delayed diagnosis occurs when histopathological features of the biopsy material are suggestive of, but not considered diagnostic for Erdheim–Chester disease because of the absence of classical Erdheim–Chester disease histopathologic features 9. Erdheim–Chester disease is not exclusively a pathologic diagnosis, and it is necessary to interpret histopathologic features in conjunction with clinical, radiographic, and, as of recently, molecular findings 9.
Erdheim-Chester disease is a rare disorder; its exact prevalence is unknown 5. More than 500 affected individuals worldwide have been described in the medical literature 5. For unknown reasons, men are slightly more likely to develop Erdheim-Chester disease, accounting for about 60 percent of cases 11, 22 and some studies suggest that male patients are diagnosed at a more advanced age than female patients 23.
The clinical presentation of Erdheim–Chester disease is highly heterogeneous, ranging from smouldering unifocal forms to multiorgan life-threatening disease due to histiocytes infiltration of critical organ systems 24. Almost any organ can be involved, but the most common sites of histiocytes infiltration include the long-bones, skin, tissues behind the eyeballs (retro-orbital), pericardial and myocardial infiltration, lungs with interstitial lung disease, large-blood vessels involvement, kidneys, brain, pituitary gland and/or additional tissues and organs. Associated symptoms and findings and disease course depend on the specific location and extent of such involvement. In many affected individuals, the most common presenting symptom of Erdheim–Chester disease is bone pain, usually affecting the knees and legs, that is similar on both sides of the body (symmetrical). In some cases, more generalized symptoms may also develop, including weight loss, fever, muscle and joint aches; and a general feeling of discomfort, weakness, and fatigue (malaise) 11, 25. Furthermore, people with Erdheim-Chester disease may present with a variety of symptoms, ranging from focal neurological deficits to multiorgan failure 26. Erdheim-Chester disease patients may also present with focal neurological signs, bulging eyeballs (exophthalmos), scarring behind the peritoneal cavity (retroperitoneal fibrosis), diabetes insipidus, and shortness of breath (dyspnea) due to extraskeletal involvement of these organs.
Erdheim–Chester disease involvement of more than one organ is not uncommon. While the long bones are the most frequently affected sites, more than 50% of patients have involvement in other parts of the body. Lung symptoms are due to progressive scarring and thickening of the lung tissue and can affect the ability of the heart to pump enough blood to the lungs and the rest of the body. Diabetes insipidus is believed to occur in 30% of patients. Erdheim–Chester disease of the heart causes problems when histiocytes accumulate in the valves or heart muscle and affect the heart’s ability to pump blood through the arteries.
Erdheim–Chester disease characteristically affects certain regions of the long bones of the legs, including the shafts (diaphyses) and the areas (i.e., metaphyses) where the shafts converge with the ends (epiphyses) 25. The ends of the long bones are usually spared or may have mild changes. Infiltration by histiocytes typically leads to widespread or patchy increases in bone density as well as hardening (osteosclerosis) and thickening of bone. In some rare cases, there may also be involvement of other bones, such as the lower jawbone (mandible) or certain bones of the spinal column (vertebrae).
Erdheim–Chester disease may also be characterized by involvement of the skin, tissues behind the eyeballs (retrobulbar region); the lungs; the brain; the pituitary gland, the region containing organs at the back of the abdominal cavity (retroperitoneum) and/or other sites. Associated symptoms and disease course may vary from case to case, depending on the site and degree of involvement 25.
Some individuals with Erdheim–Chester disease may develop soft, yellowish, fatty plaques or nodules on the eyelids (xanthelasma) or skin (cutaneous xanthomas). In addition, involvement of the retrobulbar region may lead to marked protrusion of the eyeballs (exophthalmos) and other symptoms and findings 25.
In those with lung (pulmonary) involvement, progressive scarring and thickening of lung tissue (pulmonary fibrosis) may lead to a dry cough, increasingly labored breathing (dyspnea) with exertion, insufficient oxygenation of the blood, impaired ability of the heart to pump enough blood to the lungs and the rest of the body (heart failure), and potentially life-threatening complications.
In some affected individuals, there may also be infiltration of the pituitary gland, leading to diabetes insipidus. Diabetes insipidus is a metabolic condition in which insufficient secretion of antidiuretic hormone (ADH also known as vasopressin) by the pituitary gland leads to the passing of large amounts of dilute urine (polyuria) and excessive thirst (polydipsia). Antidiuretic hormone (ADH also known as vasopressin) normally reduces the amount of water lost in urine. The pituitary gland produces several hormones, including ADH; it is controlled by and connected to a region of the brain called the hypothalamus.)
In some rare cases, Erdheim–Chester disease may also be characterized by involvement of other brain regions, such as part of the lowest region of the brain (brainstem) and the cerebellum, which is involved in coordinating voluntary movement, balance and posture. Associated neurologic symptoms may be variable from person to person. However, such abnormalities often include impaired muscular coordination (ataxia); an abnormal staggering manner of walking (gait); slurred speech (dysarthria) and/or involuntary, rhythmic, rapid eye movements (nystagmus).
Erdheim–Chester disease may also be characterized by infiltration and associated scarring of tissues within the retroperitoneal region (retroperitoneal fibrosis). In some cases, such changes may result in obstruction of the tubes (i.e., ureters) that carry urine from the kidneys into the bladder, causing abnormal swelling of the kidneys with urine (hydronephrosis), impaired kidney (renal) function, and possible renal failure. A few cases have also been described in which retroperitoneal fibrosis has involved the major artery of the body (aorta) and its branching blood vessels (periaortic fibrosis).
The course of Erdheim-Chester disease is variable, depending on the extent of involvement occurring outside of bone (extraosseous involvement) and affecting internal organs (visceral involvement). In some cases, Erdheim-Chester disease progression and associated organ system dysfunction may lead to potentially life-threatening complications, such as due to pulmonary fibrosis, heart failure, and/or renal failure.
Erdheim–Chester disease is named for the two scientists who originally described the disease. The first two cases of Erdheim-Chester disease was first described in 1930 as “lipid granulomatosis” by Jakob Erdheim, an Austrian pathologist, and William Chester, an American pathologist 27, 28. In 1972, Dr. Ronald Jaffe reported a third case and coined the name Erdheim-Chester disease (ECD).
Erdheim-Chester disease was previously considered an inflammatory or autoimmune disease, but was recognized as a blood cancer in 2016 by the World Health Organization. This was due to the discovery of cancer-causing DNA changes (mutations) in BRAF-V600E and other genes in biopsy samples from most patients 29.
The underlying cause of Erdheim-Chester disease is thought to be a malignancy of the myeloid progenitor cells. Erdheim–Chester disease is driven by somatic mutations in proto-oncogenes such as BRAF-V600E gene and MAPK (RAS-RAF-MEK-ERK) or phosphatidylinositol 3-kinase (PI3K)-AKT pathway, while immune-mediated mechanisms contribute to disease development and progression 30. Detection of the characteristic BRAF mutation in subsets of dendritic cells, mature monocytes, committed myeloid progenitors, and CD34+ cells is helpful for the diagnosis. For symptomatic patients with the BRAF mutation, a BRAF inhibitor like vemurafenib is available as targeted therapy. Mutations affecting other signalling molecules (e.g. NRAS, KRAS and ALK) may also be found. These may be treated with MEK inhibitors 31, 32.
A definitive diagnosis of Erdheim-Chester disease is usually based on the constellation of clinical symptoms, biopsy, and bone and/or PET-CT scan 33. Increasingly, molecular testing of biopsies is playing a part in an Erdheim–Chester disease diagnosis, close to 90% or more patients are seen to have a genetic mutation in the MAPK (RAS-RAF-MEK-ERK) or PI3K-AKT pathway 34, 35, 29, 13, 36, 37, 38, 39, 40, 41, 33. These mutations are thought to be acquired rather than inherited genetic alterations, and therefore, these mutations will not be passed through generations. Therefore, if there is a suspicion of Erdheim–Chester disease, it is important for patients to be seen at specialized centers which have expertise in hematology, pathology, radiology, neurology, and other subspecialties to accurately diagnose Erdheim–Chester disease and implement treatment in a timely manner.
There is no cure for Erdheim–Chester disease 9, 13. Asymptomatic patients will generally be monitored until they develop specific symptoms. Treatment of symptomatic Erdheim–Chester disease patients will be guided by the type of somatic mutations and by organ specific involvement such as the central nervous system (brain and spinal cord). Reports indicate that various treatments have been used with variable success. Further research is needed to determine optimal treatments for Erdheim–Chester disease. The scientific field is evolving day by day and more effective treatment strategies and guidelines to monitor patients effectively would be available in near future.
Zelboraf (vemurafenib) is the only therapy approved by the U.S. Food and Drug Administration (FDA) to treat certain adult patients with Erdheim–Chester disease who have the BRAF-V600 gene mutation. Targeted therapies are being tried including BRAF-inhibitors, MEK-inhibitors, interferon alfa, glucocorticoids, mTOR inhibitors or systemic therapies such as cytotoxic chemotherapy or cytokine-directed therapy. Radiation therapy is not used since Erdheim–Chester disease is not radiosensitive. Surgery has a limited role due to extensive disease.
Patients are encouraged to enroll in clinical trials for Erdheim–Chester disease as several trials are underway. Additional treatment for individuals with Erdheim–Chester disease is symptomatic and supportive.
Figure 1. Erdheim–Chester disease

Footnotes: Diverse manifestations of Erdheim–Chester disease. (A) Coronal postcontrast chest computed tomography (CT) demonstrates extensive soft tissue sheathing of the thoracic aorta. (B) Enhancing lesions in the hypothalamic pituitary axis (HPA), brainstem, and cerebellar peduncle is shown in sagittal gadolinium enhanced T1 magnetic resonance imaging (MRI). (C) Three-dimensional fast imaging using steady-state acquisition (3D-FIESTA) MRI of the heart showing right atrial mass from Erdheim–Chester disease. (D) Maximal intensity projection (MIP) of 18F-fluoro-deoxyglucose (FDG)–positron emission tomography (PET) demonstrates typical hypermetabolic Erdheim–Chester disease lesions throughout the appendicular skeleton with greatest activity of the disease in the legs. (E) Irregular bilateral enhancing of Erdheim–Chester disease lesions in the middle cerebellar peduncles are demonstrated by postgadolinium axial T1 MRI. (F) Expansile irregularly enhancing Erdheim–Chester disease lesions in the pons seen on postgadolinium axial T1 MRI. (G) Maximal intensity projection (MIP) of FDG-PET demonstrating Erdheim–Chester disease lesions with increased uptake in distal femur, orbit, multilevel thoracolumbar spine roots, and right atrium. (H) Periorbital xanthelasmas from Erdheim–Chester disease. (I-J) “Hairy kidney” hypermetabolic and contrast-enhancing perinephric infiltrates are shown on axial-fused FDG PET-CT and contrast-enhanced axial CT scan. (K) High-resolution axial CT scan image of the chest demonstrating reticulonodular opacities from Erdheim–Chester disease. (L) Atrophic or neurodegenerative changes in the brainstem and cerebellum are shown by axial T2-FLAIR MRI.
[Source 9 ]Figure 2. Erdheim–Chester disease bone lesions

Footnotes: The red arrows show: (A) Whole-body bone scan with technetium-99 showing avid uptake in the knees and left hip of an Erdheim–Chester disease patient. Less intense uptake occurs bilaterally in the humeri and distal tibias. (B) FDG PET-CT scan showing increased FDG uptake in the knees and proximal and distal tibias. (C) Bone radiograph showing cortical osteosclerosis in the right humeri. (D) Bone radiograph showing cortical osteosclerosis in the distal right radius. (E) CT scan of the right knee of an Erdheim–Chester disease patient showing cortical osteosclerosis and mottled appearance of the bone. (F) MRI scan of the right knee of an Erdheim–Chester disease patient showing serpiginous areas of T1 signal hypointensity, indicative of osteosclerosis. (G) Panoramic radiograph showing bilateral osteosclerosis of the mandible and maxillary sinus disease.
[Source 13 ]Figure 3. Erdheim–Chester disease patients cardiovascular, retroperitoneal, and lung images

Footnotes: (A) Sagittal reconstruction postcontrast CT demonstrates encasement of the thoracic aorta down to the bifurcation. (B) Coronal FDG PET-CT scan showing increased FDG uptake in the thoracic and abdominal aorta. Symmetrically encased kidneys “hairy kidney” showing increased FDG uptake. (C) Axial CT with contrast demonstrates mass-like enhancement encasing the kidneys symmetrically (“hairy kidney”). In addition, there is circumferential encasement and narrowing of the abdominal aorta (arrow). (D) Cardiac CT showing partial encasement of the right coronary artery. (E) Cardiac CT showing partial encasement of the right coronary artery. (F) Axial postcontrast CT image of the upper abdomen demonstrating mass-like perinephric stranding surrounding the kidneys with bilateral hydronephrosis. Hyperdense material within the right collecting system is a ureteral stent. (G) Postcontrast coronal MRI image of the kidneys demonstrating extension of the perinephric mass into the adrenal bed and encasement of the adrenals (arrows). (H) High-resolution CT of the chest showing interstitial fibrosis.
[Source 13 ]Figure 4. Erdheim–Chester disease central nervous system lesions

Footnotes: (A) Axial post contrast brain MRI showing suprasellar and cerebellar involvement in a patient with Erdheim–Chester disease. (B) Sagittal post contrast brain MRI (from panel A) showing suprasellar and cerebellar tumors in a patient with Erdheim–Chester disease. (C) Axial fluid attenuated inversion recovery brain MRI showing Erdheim–Chester disease tumors in cerebral hemispheres. (D) Axial fluid attenuated inversion recovery brain MRI showing Erdheim–Chester disease tumors in cerebral hemisphere. (E) Sagittal post contrast brain MRI showing an Erdheim–Chester disease tumor with cystic components in the midbrain-pons of a patient with Erdheim–Chester disease. (F) T2 fluid attenuated inversion recovery MRI image showing increased symmetrical signal intensity in the cerebellum. (G) Neurodegeneration and atrophy of the cerebellum in a patient with Erdheim–Chester disease seen on brain MRI. (H) Orbital involvement with tissue accumulation in the intraconal space secondary to histiocytes accumulation in Erdheim–Chester disease. (I) Orbital involvement showing increased thickening of the lateral rectus muscle. (J) Pituitary stalk is thickened secondary to macrophage accumulation in Erdheim–Chester disease and deviated to the right. (K) Hematoxylin and eosin stain for brain lesion showing foamy macrophages and inflammation in brain mass biopsy specimen (original magnification ×40). (L) CD68 KP-1 stain of panel K highlighting the foamy macrophages (original magnification ×40).
[Source 13 ]Figure 5. Erdheim–Chester disease xanthelasma-like skin lesions

Footnotes: Clinical features of xanthelasma-like lesions. Multiple yellowish papules/plaques or nodules most commonly located on the inner canthus of the eyelids (A) but also on the outer canthus (B). Color variations from yellow to brown-gray (C) and extensive lesions of the forehead (D) and temporal region.
[Source 42 ]Figure 6. Erdheim–Chester disease skin lesions

Footnote: (A) Periorbital xanthelasmas in a patient with Erdheim–Chester disease. Note mild exophthalmos secondary to retro-orbital mass. (B) Upper lid xanthelasmas are apparent when the patient’s eyes are closed. (C) Skin lesions containing foamy macrophages negative for S-100 protein but with activating ALK gene fusion. (D) Skin lesion positive for BRAF-V600E mutation. (E) Foamy macrophages in skin lesion. Inset shows lipid-laden macrophages interspersed with inflammatory cells (original magnification ×20; hematoxylin and eosin stain).
[Source 13 ]Figure 7. Erdheim-Chester disease skin

Footnote: A 45-year-old woman with childhood Langerhans cell histiocytosis (LCH) presented with complaints of a new widespread eruption across her face, arms, and legs. The patient described an occasionally pruritic, nonpainful, papular rash that would erupt sporadically without a known trigger and resolve to leave hyperpigmented macules. On physical examination, erythema was noted on her face, arms, chest, and back. Upon closer inspection, the erythema was composed of numerous, pinpoint, pink-to-red papules. In addition, examination found yellow papules coalescing into thin plaques along her bilateral temples and periorbital regions and scattered 2- to 4-mm red brown papules across her arms and legs. Biopsies performed on the arm, leg, and face, found foamy cells and multinucleated cells in the dermis without 2-toned cytoplasm. Immunohistochemistry found positive CD68 and negative CD1a and S-100 staining, consistent with non-Langerhans cell histiocytosis histiocytosis. The tissue was tested for this mutation and found to be BRAF V600E positive. A positron emission tomography–computerized tomography (PET-CT) scan was performed to evaluate for multiorgan involvement. The PET-CT scan was significant for multifocal 18F flurodeoxyglucose avid sclerotic changes in the sternum, sacrum, and bilateral symmetric uptake in the femoral and tibial bones, and the diagnosis of Erdheim–Chester disease was made based on these pathognomonic radiologic findings.
[Source 43 ]Figure 8. Erdheim-Chester disease skin lesions

Footnote: Clinical features of nonxanthelasma cutaneous Erdheim-Chester disease lesions. (A) Papulonodular erythematous lesions on the face. (B) Tumoral lesion on the flank. (C) Brown infiltrated patches on the trunk. (D) Hyperkeratotic patches on the legs.
[Source 42 ]Figure 9. Erdheim–Chester disease signs and symptoms

What is histiocytosis?
Histiocytosis is a general name for a group of disorders or “syndromes” that involve an abnormal increase in the number of specialized white blood cells that are called histiocytes 44. The term histiocyte refers to a bone marrow derived progenitor cell that differentiates, depending on the cytokine and growth factor milieu, into a dendritic antigen presenting cell (Langerhans cell, dermal dendrocyte) or a phagocytic cell (tissue macrophage) 45.
Recently, new knowledge about this family of diseases has led experts to develop a new classification. Five categories have been proposed 46, 47:
- L group — includes Langerhans cell histiocytosis (LCH), Erdheim-Chester disease, Indeterminate-cell histiocytosis and mixed Langerhans cell histiocytosis/Erdheim-Chester disease
- C group — includes non-Langerhans cell histiocytosis that involves the skin
- Cutaneous non-Langerhans cell histiocytosis
- Xanthomatous granuloma (XG) family: juvenile xanthogranuloma, adult xanthogranuloma, solitary reticulohistiocytoma, benign cephalic histiocytosis, generalized eruptive histiocytosis, progressive nodular histiocytosis
- Non-Xanthomatous granuloma (non-XG) family: Cutaneous Rosai-Dorfman disease, necrobiotic xanthogranuloma, other
- Cutaneous non-Langerhans cell histiocytosis with a major systemic component
- Xanthomatous granuloma (XG) family: xanthoma disseminatum
- Non-Xanthomatous granuloma (non-XG) family: multicentric reticulohistiocytosis
- Cutaneous non-Langerhans cell histiocytosis
- M group — includes Primary malignant histiocytoses and Secondary malignant histiocytoses
- R group — includes Rosai-Dorfman disease
- Familial Rosai-Dorfman disease
- Sporadic Rosai-Dorfman disease
- Classical Rosai-Dorfman disease
Extranodal Rosai-Dorfman disease - Rosai-Dorfman disease with neoplasia or immune disease
- Unclassified
- Classical Rosai-Dorfman disease
- H Group — includes hemophagocytic lymphohistiocytosis
- Primary hemophagocytic lymphohistiocytosis : Monogenic inherited conditions leading to hemophagocytic lymphohistiocytosis
- Secondary hemophagocytic lymphohistiocytosis (non-Mendelian hemophagocytic lymphohistiocytosis)
- Hemophagocytic lymphohistiocytosis of unknown/uncertain origin.
Erdheim–Chester disease cause
Erdheim–Chester disease is thought to represent cancer of myeloid progenitor cells that is characterized by excessive proliferation and accumulation of CD63+ CD1a- histiocytes that contain large amounts of fatty (lipid) material (xanthomatous histiocytes); certain lymphocytes; and distinctive, large cells with multiple nuclei (Touton giant cells), with associated scarring or overgrowth of fibrous connective tissue (fibrosis). Lymphocytes are an immune system cell type that originates in the bone marrow. Histiocytes are large phagocytic cells (macrophages) that normally play a role in responding to infection and injury. A phagocytic cell is any “scavenger cell” that engulfs and destroys invading microorganisms or cellular debris. Histiocytes normally function to destroy foreign substances and protect the body from infection. In Erdheim-Chester disease, the excess production of histiocytes (histiocytosis) and fatty nodular (xanthogranulomatous) cell deposits leads to inflammation that can damage organs and tissues throughout the body, causing them to become thickened, dense, and scarred (fibrotic); this tissue damage may lead to organ failure 5.
More than half of people with Erdheim-Chester disease have acquired somatic mutation in the BRAF-V600E gene. Mutations in other genes such as mitogen-activated protein kinase (MAPK) pathway (MAP2K1, NRAS, KRAS, and ARAF) and phosphatidylinositol 3-kinase (PI3K)-AKT pathways have been discovered in a large proportion of Erdheim-Chester disease patients 40, 34, 35, 29, 13, 36, 37, 38, 39, 40, 41, , 29, 40. Chronic, uncontrolled inflammation is also a significant component of Erdheim–Chester disease process 12. It is believed that the inflammation is the result of the body’s immune response to a more underlying process in the disease, as opposed to being the primary cause of the disease.
The BRAF-V600E gene provides instructions for making a protein that helps transmit chemical signals from outside the cell to the cell’s nucleus. This protein is part of a signaling pathway known as the RAS/RAF/MEK/MAPK pathway, which controls several important cell functions. Mutant BRAF increases cell proliferation and drives the malignant process in Erdheim–Chester disease. Specifically, the RAS/MAPK pathway regulates the growth and division (proliferation) of cells, the process by which cells mature to carry out specific functions (differentiation), cell movement (migration), and the self-destruction of cells (apoptosis).
The BRAF gene mutation that causes Erdheim-Chester disease is somatic, which means that it occurs during a person’s lifetime and is present only in certain cells. The mutation occurs in histiocytes or in immature precursor cells that will develop into histiocytes. This mutation leads to production of a BRAF protein that is abnormally active, which disrupts regulation of cell growth and division. The unregulated overproduction of histiocytes results in their accumulation in the body’s tissues and organs, leading to the signs and symptoms of Erdheim-Chester disease.
In 2015, whole-exome sequencing on 14 Erdheim–Chester disease fresh-frozen specimens identified point mutations in the ARAF, MAP2K1, NRAS, and PI3KCA genes 29. This study also found novel MAP2K1 mutations in 14% of cases (2 of 14). An additional 9 activating MAP2K1 mutations were discovered in 50% of BRAF–wild-type, archived Erdheim–Chester disease cases (9 of 18) evaluated by targeted sequencing in a validation cohort in this study 29. These MAP2K1 mutations have been demonstrated in vitro to cause constitutive activation of MEK1 29. Although MAP2K1 mutations can be seen in Erdheim–Chester disease, they are not exclusive to histiocytosis and can be seen in other blood neoplasms as well. Other mutations in Erdheim–Chester disease include activating mutations in NRAS and KRAS, which have been described in 4 independent studies 37, 48, 49, 50. PI3K-AKT pathway alterations (PI3KCA mutations) have been reported in 2 studies 29, 48. Mutations in ARAF were found in 21% of Erdheim–Chester disease specimens (3 of 14) with 2 of them being mutually exclusive of BRAF-V600E 29. Functionally activating gene fusions involving the ALK gene have also been reported in Erdheim–Chester disease 29, 51. Interestingly, similar MAPK–extracellular signal-regulated kinase (ERK) pathway mutations have been recently reported in 57% to 100% of histiocytic sarcoma specimens, suggesting a potential correlation between the 2 neoplasms 52, 53.
Figure 10. MAPK pathway

Footnotes: Overview of MAPK (mitogen-activated protein kinase) and PI3K-AKT signaling and the diverse kinase alterations discovered in select histiocytic neoplasms. (A) Diagram of the MAPK and PI3K-AKT signaling pathways with a description of the activation of the RAS proteins (HRAS, KRAS, and NRAS) with annotation of the signaling proteins affected by genetic alterations in the histiocytic neoplasms. (B) Pie chart illustrating a composite of the known kinase alterations in Langerhans cell histiocytosis. (C) Pie chart showing a composite of the known kinase alterations in Erdheim-Chester disease. (D) Pie chart demonstrating the published kinase alterations in Rosai-Dorfman-Destombes disease.
[Source 4 ]Figure 11. Molecular alterations in Erdheim–Chester disease

Footnotes: (A) Image depicting MAPK pathway signaling in Erdheim–Chester disease with therapeutic targets. The lightning bolts indicate the most common genes that are altered in Erdheim–Chester disease. (B) Composite of all somatic alterations (mutations and fusions) that have been reported in Erdheim–Chester disease to date. *Mutations identified in plasma cell-free DNA analysis only; therefore, these could reflect somatic mutations from another diagnostic entity (eg, clonal hematopoiesis).
[Source 9 ]Erdheim–Chester disease pathology outlines
Although classic histopathologic findings include foamy histiocytes with small nuclei and surrounding fibrosis, multinucleated giant cells, and Touton giant cells, Erdheim–Chester disease has a spectrum of histopathological features within the xanthogranuloma family, often in a milieu of reactive lymphocytes, plasma cells, and rarely neutrophils.45 Erdheim–Chester disease may also display atypical features such as florid lymphohistiocytic infiltrates or fibrotic lamellae with only scattered foamy histiocytes and rare/absent Touton giant cells.46 On immunohistochemistry (IHC), histiocytes are positive for CD68, CD163, factor XIIIa, and fascin, and negative for CD1a and CD207 (langerin). Although Erdheim–Chester disease was classically thought to be negative for S100 by immunohistochemistry, weak or focally positive staining has been observed in 20% to 30% cases (Table 1).6, 46 Although it is important to differentiate Erdheim–Chester disease from other histiocytic disorders for appropriate diagnosis, as well as to identify overlapping entities (Table 1), absence of the classical description of foamy histiocytes and Touton cells does not preclude a diagnosis of Erdheim–Chester disease.
Table 1. Summary of the pathological, molecular, and radiological features of the histiocytic disorders
| Disease | Erdheim–Chester disease | Juvenile xanthogranuloma / adult xanthogranuloma | ALK+ histiocytosis | Rosai-Dorfman-Destombes disease | Langerhans cell histiocytosis |
|---|---|---|---|---|---|
| Pathologic features | |||||
| Xanthomatous histiocytes | Yes | Yes | Variable | No | No |
| Touton giant cells | Yes (mainly dermal sites) | Yes (mainly dermal sites) | Rarely | No | No |
| Emperipolesis (intracytoplasmic inflammatory cells including plasma cells and lymphocytes) | Rare | Rare | Rare | Abundant | No |
| Nuclear features | Bland; round-to-oval; small; no grooves | Bland; round-to-oval; small; no grooves | Bland; round-to-oval; small; typically no grooves | Large round; hypochromatic | Oval; retiform irregular nuclear contours |
| Nucleoli | Inconspicuous | Inconspicuous | Inconspicuous | Variable inconspicuous to distinct | Inconspicuous |
| Cytoplasm | Classically abundant, xanthomatous but often overlap with JXG/AXG | Compact; pink; glassy; progressively xanthomatous | Abundant; eosinophilic; typically not xanthomatous | Abundant foamy, clear without xanthomatous features; frequent emperipolesis | Abundant; eosinophilic |
| Immunophenotype | |||||
| CD68 (cytoplasmic) | ++ | ++ | ++ | ++ | + (paranuclear cytoplasmic dot) |
| CD163 (surface) | ++ | ++ | ++ | ++ | — |
| CD14 (surface) | ++ | ++ | ++ | ++ | — |
| CD1a (surface) | − | − | − | − | ++ |
| CD207 (Langerin) (cytoplasmic) | − | − | − | − | ++ |
| S100 (cytoplasmic/nuclear) | −/+ (light) | −/+ (light) | −/++ (in some cases dark staining) | + | + |
| Factor XIIIa (cytoplasmic) | + | + | + | + | − |
| Fascin (cytoplasmic) | + | + | + | + | − |
| CD45 (light surface) | + | + | + | + | + |
| BRAF VE1 (cytoplasmic) | ++* | − (Positive cases should be strongly favored to be in ECD family) | − | − (Rare case reports ++) | ++* |
| ALK (cytoplasmic) | ++* | ++* | ++* | − | − |
| NTRK1(cytoplasmic) | ++* | ++* | − | − | − |
| Molecular features | |||||
| BRAF V600E | Frequent (50%) | Reported (3%) | No | Reported (3%) | Frequent (55%) |
| MAP2K1 | Common (18%) | Common (12%) | No | Common (15%) | Common (15%) |
| RAS isoforms (KRAS, NRAS) | Common (8%) | Common (10%) | No | Common (30%) | Rare (2%) |
| BRAF deletions | Rare (2%) | No | No | No | Common (6%) |
| PI3K isoforms (PIK3CA, PIK3CD) | Reported (3%) | Rare (1%) | No | No | Rare (1%) |
| ARAF | Reported (4%) | Rare (1%) | No | Reported (3%) | Rare (1%) |
| Other BRAF missense | No | No | No | No | Reported (3%) |
| RAF1 | Rare (1%) | No | No | No | No |
| MAP2K2 | Rare (1%) | No | No | No | No |
| MAP3K1 | Reported (1 case) (Amplification) | No | No | No | Reported |
| CSF1R | Rare (1%) | Common (10%) | No | Rare (1%) | Rare (1%) |
| BRAF fusions | Rare (2%) | Common (6%) | No | No | Reported (3%) |
| ALK fusions | Reported (3%) | Reported (3%) | Frequent (100%) | No | No |
| NTRK1 fusions | Rare (1%) | Common (10%) | No | No | No |
| RET fusions | No | Reported (3%) | No | No | No |
| ETV3-NCOA2 fusion | No | No | No | No | Rare (1%) |
Footnote: * Moderate to strong positivity should correlate with molecular alteration; BRAF VE1, ALK and pTRK are mutually exclusive.
Immunophenotype key: − = negative; + = weak positive; ++ = moderate to strong positive.
[Source 9 ]Erdheim–Chester disease symptoms
The signs and symptoms of Erdheim–Chester disease depend on the number and degree of organ involvement. The signs and symptoms of Erdheim-Chester disease usually appear between the ages of 40 and 70, although Erdheim-Chester disease can occur at any age including children. The severity of the condition varies widely; some affected individuals have few or no associated health problems, while others have severe complications that can be life-threatening. People with Erdheim-Chester disease often have bone pain, especially in the lower legs and upper arms, due to an abnormal increase in bone density (osteosclerosis). Damage to the pituitary gland (a structure at the base of the brain that produces several hormones, including a hormone that controls the amount of water released in the urine) may result in hormonal problems such as a condition called diabetes insipidus that leads to excessive urination. Abnormally high pressure of the cerebrospinal fluid within the skull (intracranial hypertension) caused by accumulation of histiocytes in the brain may result in headaches, seizures, cognitive impairment, or problems with movement or sensation. People with this condition can also have shortness of breath, heart or kidney disease, protruding eyes (exophthalmos), skin growths, or inability to conceive a child (infertility). Affected individuals may also experience fever, night sweats, fatigue, weakness, and weight loss.
It is important to note that not all organ involvements may result in symptoms. The signs and symptoms listed below may indicate Erdheim–Chester disease involvement but are not necessarily diagnostic.
Erdheim–Chester disease characteristically affects certain regions of the long bones of the legs, including the shafts (diaphyses) and the areas (i.e., metaphyses) where the shafts converge with the ends (epiphyses) 25. The ends of the long bones are usually spared or may have mild changes. Infiltration by histiocytes typically leads to widespread or patchy increases in bone density as well as hardening (osteosclerosis) and thickening of bone. In some rare cases, there may also be involvement of other bones, such as the lower jawbone (mandible) or certain bones of the spinal column (vertebrae).
Erdheim–Chester disease may also be characterized by involvement of the skin, tissues behind the eyeballs (retrobulbar region); the lungs; the brain; the pituitary gland, the region containing organs at the back of the abdominal cavity (retroperitoneum) and/or other sites. Associated symptoms and disease course may vary from case to case, depending on the site and degree of involvement 25.
Some individuals with Erdheim–Chester disease may develop soft, yellowish, fatty plaques or nodules on the eyelids (xanthelasma) or skin (cutaneous xanthomas). In addition, involvement of the retrobulbar region may lead to marked protrusion of the eyeballs (exophthalmos) and other symptoms and findings 25.
In those with lung (pulmonary) involvement, progressive scarring and thickening of lung tissue (pulmonary fibrosis) may lead to a dry cough, increasingly labored breathing (dyspnea) with exertion, insufficient oxygenation of the blood, impaired ability of the heart to pump enough blood to the lungs and the rest of the body (heart failure), and potentially life-threatening complications.
In some affected individuals, there may also be infiltration of the pituitary gland, leading to diabetes insipidus. Diabetes insipidus is a metabolic condition in which insufficient secretion of antidiuretic hormone (ADH also known as vasopressin) by the pituitary gland leads to the passing of large amounts of dilute urine (polyuria) and excessive thirst (polydipsia). Antidiuretic hormone (ADH also known as vasopressin) normally reduces the amount of water lost in urine. The pituitary gland produces several hormones, including ADH; it is controlled by and connected to a region of the brain called the hypothalamus.)
In some rare cases, Erdheim–Chester disease may also be characterized by involvement of other brain regions, such as part of the lowest region of the brain (brainstem) and the cerebellum, which is involved in coordinating voluntary movement, balance and posture. Associated neurologic symptoms may be variable from person to person. However, such abnormalities often include impaired muscular coordination (ataxia); an abnormal staggering manner of walking (gait); slurred speech (dysarthria) and/or involuntary, rhythmic, rapid eye movements (nystagmus).
Erdheim–Chester disease may also be characterized by infiltration and associated scarring of tissues within the retroperitoneal region (retroperitoneal fibrosis). In some cases, such changes may result in obstruction of the tubes (i.e., ureters) that carry urine from the kidneys into the bladder, causing abnormal swelling of the kidneys with urine (hydronephrosis), impaired kidney (renal) function, and possible renal failure. A few cases have also been described in which retroperitoneal fibrosis has involved the major artery of the body (aorta) and its branching blood vessels (periaortic fibrosis).
Erdheim–Chester disease varies greatly from patient to patient, and some but not all of these symptoms may be present 54:
- Bone (>90%): Erdheim–Chester disease involves the long bones around the knees in most cases, but can affect any other bone of the body. Most common symptom is bone pain in legs or knees (50%), usually on both sides (bilateral) 13. Full-body (skull-to-toes) FDG-PET-CT scan or 99mTc bone scintigraphy shows bilateral symmetric osteosclerosis of metadiaphysis of femur, tibia, and fibula in >95% cases and is pathognomonic; skull and axial skeleton less commonly involved than Langerhans cell histiocytosis, which typically shows lytic punched-out lesions rather than sclerotic lesions that are seen in Erdheim–Chester disease 55.
- Retroperitoneum (tissue surrounding kidneys and large blood vessels like aorta) and kidneys (50-60%): Can manifest as infiltrative perinephric soft tissue thickening, or hairy growth around the kidneys affecting the kidney function or around the large blood vessels like the aorta 14. On CT scan of the body, this can appear as “hairy kidney” or “coated aorta”. Perinephric infiltrates can rarely extend to involve the renal pelvis and/or renal ureters causing hydronephrosis and renal failure requiring dialysis and nephrostomy with stent placement 13; in some cases, it may also extend to involve the adrenal glands 56; longstanding perinephric Erdheim–Chester disease may lead to atrophy of kidneys. The most common symptoms include abdominal or lower back pain, painful or difficult urination and kidney failure.
- Nervous system including the brain (25-50%): Erdheim–Chester disease can involve the parts of the brain that control balance and coordination (brainstem and cerebellum), although infiltrations can occur throughout the central nervous system (brain and spinal cord) affecting the spinal cord and nerves arising from it 57. Retinal involvement has been reported as well 58. Rarely, Erdheim–Chester disease may present with nontumorous neurodegenerative-like (atrophic) changes in the brainstem and cerebellum 9. Special imaging of the brain (MRI) is required to assess the involvement of nervous system by Erdheim–Chester disease. Clinical manifestations depend on the site of involvement and symptoms include difficulty with coordination, difficulty difficulty maintaining balance (ataxia), cognitive impairment, headaches, recurrent falls, slurred speech (dysarthria), behavior disorders, muscle weakness, peripheral neuropathy and some patients may present with mood lability 13, 3.
- Mood and memory difficulties (50-70%): Erdheim–Chester disease can cause difficulty with memory and mood in a large proportion of patients despite a completely normal MRI scan of the brain. Symptoms include mood lability, uncontrollable crying, anxiety, depression, difficulty remembering things
- Pituitary gland and other hormone deficiencies (40-70%): Anterior and posterior pituitary involvement most commonly presents as diabetes insipidus (25% to 50%) and is often the first manifestation of the disease. Diabetes insipidus may precede the diagnosis of Erdheim–Chester disease by several years, sometimes decades 13, 59, 57, 60. Other pituitary hormone deficiencies that are commonly seen are growth hormone, gonadotropin, thyrotropin, and corticotropin; hyperprolactinemia can be seen in 15% to 30% patients 59, 60. The pituitary hormone deficiencies may or may not be associated with abnormal pituitary MRI scan (i.e., normal pituitary imaging does not exclude hormonal deficiency). An abnormal pituitary stalk MRI is commonly associated with hypopituitarism; primary hypothyroidism (20%), hypogonadism (19%), and adrenal insufficiency (6%) can be seen as well 59. Other hormones that can be deficient are thyroid, adrenal, and sex hormones (testosterone in men and estrogen in women). Symptoms include excessive thirst and urination, fatigue, low libido, cold intolerance and weight gain.
- Heart (50-60%): Cardiovascular involvement may be asymptomatic but can be seen in 50% to 70% patients at imaging evaluation by CT and/or MRI 61, 62, 63; most common findings include pericardial infiltration with pericardial effusion (which may be complicated by cardiac tamponade) or heart muscle infiltration in the form of right atrioventricular pseudotumor (40%) based on appearance on MRI scan of the heart 64, 65, 66, 13, 67, 63; circumferential soft tissue sheathing of the thoracic/abdominal aorta and its branches may be seen as “coated” aorta may be seen on CT scan (50% to 60%) 13, 68; periarterial involvement of kidney blood vessels may lead to renovascular hypertension (20%), is responsive to stenting 66 and can be monitored with renal artery dopplers. Involvement of other visceral blood vessels has been reported as well; coronary arteries (blood vessels supplying the heart) may be involved in 30% to 50% of patients 61, 69. Symptoms include shortness of breath, fatigue, and swelling of feet, ankles, and lower legs.
- Lung and respiratory system (40-50%): Lung involvement in Erdheim–Chester disease is mostly asymptomatic and seen radiologically in ∼50% of cases, involving either the lung tissue or pleura and no association with cigarette smoking has been reported 13, 14. Erdheim–Chester disease may also involve facial sinuses, with maxillary sinus thickening in ∼50% patients 13. Symptoms include shortness of breath, dry cough and sinus congestion. Plain chest radiographs can be normal, findings on CT of the chest may include mediastinal infiltration, pleural thickening or effusions, interlobular septal thickening, ground-glass opacities, or centrilobular opacities 70, 69, 71; lung function tests commonly reveal a more restrictive as compared with an obstructive pattern 13, 70.
- Eyes (25-30%): Erdheim–Chester disease can cause accumulation of cells in the bony cavity of the skull that houses the eyeball (orbit), causing pressure and visual disturbances. Symptoms include retro-orbital pain and redness, bulging eyes (exophthalmos), difficulty with vision including double vision, oculomotor nerve palsy or vision loss 13, 72.
- Skin and Eyelids (20-30%): Seen in 20% to 30% patients, with one-half of the patients manifesting skin lesions as initial Erdheim–Chester disease presentation 13, 14, 73. Most commonly seen as yellowish-orange plaques around the eyes called xanthelasmas. Rarely can occur as yellowish-brown papules or plaques on the face, neck, armpits, groin, or back 73. It may also present as subcutaneous nodules or granuloma annulare-like lesions 73. Symptoms include rash or soft, fatty, yellowish-orange bumps around the eyes.
- Liver, spleen, lymph nodes, and bone marrow (10%): Mostly asymptomatic involvement. In one study, 11% of Erdheim–Chester disease patients had liver and spleen involvement, respectively, although the prevalence has been lower in other series 13, 74. Erdheim–Chester disease rarely involves the lymph nodes, but may involve bone marrow in 8% of cases 75. In 10% of cases, Erdheim–Chester disease can be associated with other blood cancers such as myelodysplastic syndrome, myeloproliferative neoplasm and mixed myelodysplastic/myeloproliferative overlap syndrome including chronic myelomonocytic leukemia 76. Usually asymptomatic, however can manifest as abnormally low or high blood cells on routine testing (CBC).
- Additional symptoms, regardless of organ involvement: fatigue, weight loss, low-grade fevers, night sweats, muscle and joint aches
Erdheim–Chester disease diagnosis
A diagnosis of Erdheim–Chester disease is made based upon a thorough clinical evaluation, a detailed patient history, identification of characteristic symptoms, and findings of a biopsy of the affected tissue along with imaging studies. Imaging studies may include plain x-rays; advanced imaging techniques, including computed tomography (CT) scanning, magnetic resonance imaging (MRI), FDG-PET-CT and/or a bone scan (bone scintigraphy); and/or other tests. Plain x-rays of involved bones typically reveal symmetrical increased hardening and thickening, mainly in the metaphyses and diaphyses with sparing of the epiphyses, a finding that is considered distinctive of Erdheim–Chester disease. In addition, the diagnosis may be confirmed by removal (biopsy) and microscopic evaluation of tissue samples that demonstrate infiltration by fatty (lipid)-laden, foamy histiocytes with certain non-Langerhans cellular features and distinctive, large cells with multiple nuclei (Touton giant cells). The tissue samples should be reviewed by a trained pathologist and imaging by a radiologist who has knowledge of Erdheim–Chester disease. Molecular testing of biopsy specimens can identify specific somatic mutations that can direct specific treatments.
Figure 12. Erdheim–Chester disease diagnostic algorithm

Abbreviations: ACTH = adrenocorticotropic hormone; ECG = electrocardiogram; FSH/LH = follicle-stimulating hormone/luteinizing hormone; HEENT = head, eyes, ears, nose, and throat; IGF-1 = insulin-like growth factor 1; TSH = thyroid-stimulating hormone.
[Source 9 ]Figure 13. Erdheim–Chester disease tissue biopsy diagnostic algorithm

Baseline evaluation
When a doctor learns of suggestive symptoms, they will often order tests. It is important to make a systematic study of the organs possibly affected: skeleton, lungs, heart, central nervous system, kidneys, eyes, pituitary, skin, and/or teeth.
The goal of the evaluation in newly diagnosed patients is to define the extent of disease involvement, assess subsequent risk of end-organ compromise, and define a plan of treatment and surveillance. Regardless of symptoms, experts recommend FDG PET-CT imaging including the brain and distal extremities, MRI of the brain with gadolinium, and cardiac MRI in all newly diagnosed patients 9. Even in cases in which 99mTc bone scanning has been performed initially for diagnostic purposes, FDG PET-CT is recommended for initial evaluation to assess organ involvement and as a tool for guiding biopsy targets 78, 79, 80. Dedicated CT of the chest, abdomen, and pelvis is recommended to demonstrate pulmonary, periaortic, and perinephric infiltrates. In some cases, further organ-specific imaging may be necessary based on clinical and radiologic findings to better characterize the involvement of certain sites. Additionally, laboratory studies are needed to assess endocrinopathies, peripheral blood count abnormalities, renal/hepatic function, immunological assessment, and the degree of inflammation.
X-rays and electrocardiograms (EKG or ECG) are often the initial tests. Non-invasive scans such as CT scan, CT/PET scan, MRI, bone scan, or echocardiogram may be warranted depending upon the symptoms, clinical signs, and/or results from previous tests.
If a mass or lesion is found within the body, a tissue biopsy might be performed as part of the diagnostic workup. A pathologist will study the tissue sample that is obtained via the biopsy. Erdheim–Chester disease affected tissue will usually contain clusters of lipid-laden, foamy histiocytes with signs of chronic inflammation, often with Touton-type giant cells, fibrosis, and possible fat necrosis. The pathologist will study the tissue sample based on the above-mentioned criteria.
A bone biopsy might be warranted. If Erdheim–Chester disease has affected the bones, the bone biopsy will typically show sclerotic bone.
Although brain lesions are sometimes seen with Erdheim–Chester disease, it is not always possible to perform a biopsy on these lesions depending on where they appear in the brain. Doctors will normally see these lesions when an MRI of the brain is performed. These lesions will show T2 hyperintensity and often intense gadolinium enhancement. The lesions most often occur in the pons and cerebellum, but they can also occur in the tissue overlying the brain (the meninges) or anywhere else in the brain. These findings might be confused with multiple sclerosis as they mimic a demyelinating process normally seen in multiple sclerosis.
If there are multiple lesions involving bone and soft tissue which are suspected to be involved by Erdheim–Chester disease and could be biopsied, preference should be given to biopsying the soft tissue lesion. When biopsying a bone lesion, tissue processing is required with decalcification. This process could alter the proteins and cellular DNA in the sample which could impair genetic sequencing studies, which are important for accurate diagnosis and making treatment recommendations.
Typical skeletal findings:
- X-rays showing bilateral and symmetric cortical osteosclerosis of the diaphyseal and metaphyseal (middle and the ends of a bone) regions in the long bones and/or symmetric and abnormally increased labeling of the distal ends of the long bones of the lower limbs, and sometimes the upper limbs, on 99Tc bone scintigraphy.
- In PET scan, 18F-fluoro-deoxyglucose (FDG) avid (increased glucose uptake and appears to have an uptake of blue to yellow to red color) around the mid and the ends of the long bones especially in the femur and the tibia.
- These bone findings are evident in around 95% of patients with Erdheim–Chester disease. However, 5% of patients could have Erdheim–Chester disease without the classic aforementioned bone findings.
- If there is a suspicion of brain involvement, an MRI of the brain should be commenced as the PET scan would not be able to evaluate brain involvement.
- If there is suspicion of cardiac involvement, an MRI of the heart should be undertaken for disease evaluation.
- The usual recommendation is to conduct an MRI of the brain and an MRI of the heart along with the PET/CT scan to better stage the disease at the time of diagnosis.
A defining feature of Erdheim–Chester disease is symmetric osteosclerosis of the metadiaphysis of the lower-extremity bones on studies such as plain radiographs, 99mTc bone scintigraphy, 18F-fluoro-deoxyglucose (FDG)–positron emission tomography (PET), computed tomography (CT), or magnetic resonance imaging (MRI) (Figure 1). Although bone scintigraphy is the most sensitive of these for detecting osseous lesions, FDG PET-CT is preferred as a diagnostic test by virtue of its ability to assess other organ involvement 81, 82. It must be noted that it is crucial to obtain the PET-CT scan as a full-body (skull-to-toes) test, as compared with skull-to-mid-thigh, as the latter may not capture these characteristic osseous lesions. Previously proposed diagnostic criteria required presence of both osteosclerotic lesions in the legs 83, 15. However, a small proportion of Erdheim–Chester disease (∼5%) may not demonstrate long-bone involvement6 and in such cases the diagnosis hinges on other features.
Tissue biopsy and histopathologic assessment
Tissue biopsy is required in all Erdheim–Chester disease cases, not only for confirmation of diagnosis, but also to allow identification of associated mutations for therapeutic purposes. The selection of the biopsy site for specimen acquisition can be challenging in Erdheim–Chester disease due to low tumor cellularity and heterogeneity of lesions. The diagnosis is often made by biopsy of one of the skin, osseous, or soft tissue perinephric infiltrates/lesions using a percutaneous CT-guided approach. If an FDG PET-CT has been performed prior to biopsy, we recommend a biopsy of the most FDG-avid sites that are accessible and safe, especially in cases of bony lesions. Because of the variable components of histiocytic infiltrate and surrounding stroma, multiple core biopsies are recommended to optimize the yield of tissue for histopathologic review and molecular testing. Biopsy of xanthelasmas or other skin lesions, if present, offers a less-invasive alternative 84. If DNA-based testing is planned, it is important to coordinate tissue handling of bone biopsies because standard decalcification of bone samples will lead to the destruction of informative DNA. Alternatively, an EDTA-based decalcification method can be used, which can help preserve DNA integrity.
Molecular assessment of tissue for alterations in MAPK-ERK and other pathways
As most Erdheim–Chester disease patients harbor activating somatic mutations or fusions in the genes of the MAPK-ERK or the PI3K-AKT pathway, molecular profiling of biopsy material can increase confidence in an Erdheim–Chester disease diagnosis in cases with ambiguous histopathological findings and/or absence of osseous lesions. It is notable that tissue genotyping may not uncover a driver alteration in a small proportion of patients (10% to 15%) or there may be insufficient cellularity in the specimen to conduct molecular analysis 85. In such cases, a properly validated BRAF-VE1 or phosphorylated ERK stain may help if there is moderate to strong cytoplasmic staining in the lesional cells.
BRAF-V600E mutation testing should be pursued for all patients. There are several methods to test for this mutation, including immunohistochemistry (IHC), polymerase chain reaction (PCR), pyrosequencing, droplet digital PCR (ddPCR), and targeted-capture next-generation sequencing (NGS). Although immunohistochemistry (IHC) is a cost-efficient and reliable method of testing for the BRAF-V600E protein in other cancers, some experts suggested that this method is not as sensitive for the evaluation of Erdheim–Chester disease material, similar to previous reports in Langerhans cell histiocytosis 86. Hence, all negative or equivocal immunohistochemistry (IHC) tests should be confirmed by a sensitive sequencing technique on the same or alternative tissue specimens. Although not available at most clinical laboratories, droplet digital PCR (ddPCR) is the most sensitive method, and, in many cases, BRAF-V600E is present at remarkably low allele fractions (<5%) 87. Clinical presentations with cerebral, cardiac, and orbital disease may prompt more exhaustive testing for BRAF-V600E because the incidence of this mutation is highest in these phenotypes 14.
Typical histologic findings:
- On tissue biopsies, typically there is an infiltrate of bland appearing histiocytes characterized by abundant foamy (xanthomatous) cytoplasm with surrounding fibrosis. Touton giant cells are frequently present. In a subset of cases, these typical morphologic features may not be present.
- The lesional histiocytes by immunohistochemistry (IHC) show positive staining for CD68, CD163, Factor13a, and S100 (variable).
- BRAF V600E is positive by immunohistochemistry in approximately 50-60% of cases. They lack expression of CD1a and langerin. However, the absence of BRAF V600E by immunohistochemistry does not necessarily rule out the presence of this mutation. Therefore, genetic sequencing on the tumor tissue should be undertaken through publically available platforms (TEMPUS, FoundationOne, etc.).
In cases without the BRAF-V600E mutation, some experts recommend targeted-capture next-generation sequencing with a commercially available assay to test alterations in other genes of the MAPK-ERK and PI3K-AKT pathways (KRAS, NRAS, ARAF, RAF1, MAP2K1, MAP2K2, BRAF indels, and PI3KCA). Of note, ∼40% of BRAF-V600–wild-type patients will harbor a mutation in MAP2K1. Most of these panels also include RNA sequencing that tests known oncogenic kinase fusions that have been reported previously in Erdheim–Chester disease. Data regarding concordance between tumor-based sequencing and cell-free DNA (cfDNA)-based sequencing have varied, with high concordance in BRAF-V600E mutant cases and low in others 49, 34, 88. In cases for which a tissue specimen is insufficient for molecular analysis, cell-free DNA testing is a reasonable alternative.
Erdheim–Chester disease treatment
A “cure” for Erdheim–Chester disease has yet to be discovered. Most patients with Erdheim–Chester disease require systemic treatment at diagnosis, with the exception of asymptomatic nonvital single-organ (eg, bone) or minimally symptomatic disease that may be monitored 9, 13. The best treatments available today control and sometimes shrink the growths associated with Erdheim–Chester disease. The limited evidence available suggests that when some treatments are discontinued, the disease will progress again, usually quickly. Consequently, some successful treatments may be continued indefinitely.
It is also important to keep in mind that not all patients need treatment. Some patients could be followed without treatment for many months and sometimes for many years. Therefore, patients and their doctors should discuss the indications for therapy and treatment options in detail before selecting an appropriate treatment strategy for patients. Most targeted therapy that is used to treat Erdheim–Chester disease carry side effects and therefore, these agents should be selected after an extensive discussion about the risks and benefits of therapy.
Because of the rarity of Erdheim–Chester disease, clinical trials have not been historically conducted. However, this is changing. Today there are several studies and clinical trials open to Erdheim–Chester disease patients. When Erdheim–Chester disease patients enroll in a clinical trial, they can have access to some of the newest and best treatments available to Erdheim–Chester disease patients. All patients are encouraged to talk with their medical team about whether a clinical trial would be appropriate for their situation. Clinical trials not only allow patients access to the newest treatment options but also pave the way for treatments to be scientifically proven effective in the treatment of Erdheim–Chester disease. This can lead to the approval of Erdheim–Chester disease treatments which would help all Erdheim–Chester disease patients gain access to the treatment.
For these reasons, there is no treatment plan that is accepted by the medical community as the “best available.” However, there are a number of doctors who have documented their findings with a particular treatment plan. Many of these treatments are based on anecdotal experience, but over time are showing they do help patients.
For patients with multisystem BRAF-V600-mutant Erdheim–Chester disease with life-threatening cardiac or neurologic involvement, some experts first-line recommendation is BRAF-inhibitors such as vemurafenib or dabrafenib 9. The choice of BRAF-inhibitor may be guided by the toxicity profile in light of the particular patient’s clinical status, and the experience of the treating clinician. For BRAF-V600–mutated Erdheim–Chester disease without end-organ dysfunction, it is appropriate to consider either BRAF-inhibitor or immunosuppressive/cytotoxic therapy, balancing Erdheim–Chester disease symptom management and side effects of treatment 9. For patients without BRAF-V600–Erdheim–Chester disease, some experts recommend pursuing next-generation sequencing to evaluate other MAPK-ERK pathway alterations that can be treated with a MEK-inhibitor. “Empiric” treatment with MEK-inhibitors for BRAF-V600–wild-type Erdheim–Chester disease without an identified MAPK pathway mutation is a reasonable approach for an acutely ill patient with heart or central nervous system (brain and spinal cord) involvement without viable alternative therapies 9. In patients with central nervous system (brain and spinal cord) involvement, higher doses of targeted (BRAF- or MEK-inhibitor) therapies or dual therapy for BRAF-V600–mutated Erdheim–Chester disease may be considered to attain robust response and may be tailored subsequently based on tolerance. It is to be noted that the outcomes of associated myeloid neoplasms under targeted therapies are not currently known. For patients without access to targeted therapies, interferon-alpha (IFN-α) or PEGylated interferon-alpha (PEG-IFN-α) present efficacious treatment options, although the latter may be slightly better tolerated 9. A retrospective cohort review of 165 Erdheim–Chester disease patients from the French registry reported an overall survival (OS) benefit with the use of interferon-alpha (IFN-α), PEGylated interferon-alpha (PEG-IFN-α), or targeted therapies 14. In patients with mixed histiocytosis (Erdheim–Chester disease/Langerhans cell histiocytosis (LCH) overlap), however, IFN-α therapy may be suboptimal (given the non-Erdheim–Chester disease component of disease) and targeted therapies are favored 89. One of the challenges with targeted therapies or interferon (IFN) treatment is the risk of disease relapse at discontinuation, necessitating a prolonged duration of treatment. Hence, in patients who are clinically fit to receive systemic chemotherapy and/or are unable to access targeted agents or tolerate them, cladribine may be considered as a limited duration treatment to offer sustained response. For patients with low-burden disease involving bones and retroperitoneum, a biologic agent, especially anakinra can be used.
In addition, early initiation of rehab services is needed for Erdheim–Chester disease patients. Depending upon the patient’s symptoms, this may include physical therapy, occupational therapy, and/or speech/swallowing therapy.
Figure 14. Erdheim–Chester disease with nervous system involvement treatment plan

Immunotherapy (Interferon-alpha)
Interferon-alpha (IFN-α), normally considered the “first-line” of treatment, with a number of papers available reporting on the effectiveness of this treatment for patients who can tolerate it 90, 91, 92, 27, 93, 94. Interferon (IFN) is a protein the body creates when it is trying to fight off a foreign agent such as a virus. If a doctor believes this is the best course of action for a patient he will prescribe the medicine to the patient and continue monitoring the patient for any adverse side effects. With interferon treatment, a patient is given the medicine by injection (shot). There are two forms of interferon, one requiring an injection 3 times a week, and another PEGylated interferon-alpha (PEG-IFN-α) requiring injections only once a week. This treatment is normally given for extended periods of time. It is a treatment that can be given in the home (much like an insulin shot); the patient usually does not need to go into a doctor’s office or hospital to obtain the injection. The patient may experience some side effects such as fatigue (flu-like feelings). However, for some patients, these side effects diminish with time 93. It was recently reported that a typical dosage for the treatment of Erdheim–Chester disease with interferon-alpha is an injection under the skin of 3mIU, three times per week. For patients with the central nervous system (CNS) or cardiac involvement, an injection under the skin of a high dosage of 6-9 mIU, three times per week may be considered. A reported typical dosage for treating Erdheim–Chester disease with PEGylated interferon-alpha is an injection under the skin of 135ug once a week.
BRAF-inhibitors (vemurafenib, dabrafenib)
The FDA has approved vemurafenib as a treatment for Erdheim–Chester disease patients testing positive for the BRAF-V600E mutation 95. The approval was based on data gathered during a successful clinical trial at Memorial Sloan Kettering. Zelboraf (vemurafenib) is currently the only FDA-approved treatment for Erdheim–Chester disease. A little over 50% of Erdheim–Chester disease patients test positive for the BRAF-V600E mutation. These findings have led some treating physicians to begin using BRAF-inhibitors to treat Erdheim–Chester disease should the patient test positive for this mutation. Emerging data suggests that Erdheim–Chester disease patients should be tested for the BRAF-mutation using ultra-sensitive methods as there may be a high rate of false-negative results occurring.
Recent reports indicate a typical dose of vemurafenib for the treatment of Erdheim–Chester disease is 480 – 960 mg daily 95. Experience to date shows that Erdheim–Chester disease patients do not develop a resistance to vemurafenib as seen in some other cancers. However, research is showing that patients may need to remain on this treatment indefinitely 95. A phase 2 clinical trial to study the effects of dabrafenib and trametinib in the treatment of BRAF-V600E positive Erdheim–Chester disease patients was performed. For updates, see https://www.clinicaltrials.gov/study/NCT02281760. Patients enrolled in this trial are reporting good results. Finally, recent research is suggesting that those patients who test negative for the BRAF mutation may have other mutations that would allow focused treatment. All Erdheim–Chester disease patients and their medical teams are urged to stay up-to-date with the latest research in this area.
MEK-inhibitors (trametinib, cobimetinib)
Some of the newest research in the treatment of Erdheim–Chester disease includes the use of MEK inhibitors. These treatments are being studied at some of the Erdheim–Chester disease Referral Care Centers (https://erdheim-chester.org/care-centers) and are for both BRAF-positive and BRAF-negative patients. The treatments are being used alone in some cases and sometimes with BRAF-inhibitors when the patient is BRAF-positive. Patients and/or their medical teams are encouraged to contact the lead physicians at an Erdheim–Chester disease Referral Care Center for more information about this emerging treatment.
Cladribine (2-CdA)
Sometimes a doctor will make the decision that chemotherapy is the best way to treat Erdheim–Chester disease in a particular patient. Chemotherapy or chemo means that a chemical is used to kill certain cells in the body in an attempt to fight off disease. The specific chemotherapy treatment that has shown favorable results for some Erdheim–Chester disease patients is cladribine (2-CdA). 3 published cases and a retrospective series of 21 patients treated with cladribine was published demonstrating ∼50% clinical or radiologic response rate 96, 97, 98. Cladribine (2-CdA) is usually used for a specific period of time. All chemotherapy treatments must be given with close monitoring by a doctor. Cladribine has been shown to negatively impact Erdheim–Chester disease patients’ immune systems and therefore most specialists refrain from administering more than 2 to 4 courses of this treatment. Prophylactic antimicrobials against Pneumocystis jirovecii (cotrimoxazole) and viruses (acyclovir, valacyclovir) should be added during the duration of the treatment and until the lymphocyte count normalize 9.
Anakinra (Kineret)
Kineret is approved for the reduction in signs and symptoms and slowing the progression of structural damage in moderately to severely active rheumatoid arthritis (RA). Kineret can lower the ability of the immune system to fight infections. It has been reported that Kineret has shown good results for some patients with mild forms of Erdheim–Chester disease, without the central nervous system (CNS) or cardiovascular involvement. A paper was published in August 2010 with the results of two patients who were treated with anakinra with positive results 99. Since that time additional case studies have been published showing positive results 100, 101, 102. Yet other reports have shown poor efficacy of anakinra in complicated cases that include central nervous system or cardiovascular involvement 100, 103, 104, 101, 105. The reported typical dosage of anakinra in the treatment of Erdheim–Chester disease is an injection of 100 mg daily. Similar to interferon-alpha (IFN-α), subcutaneous injection and intolerable adverse effects in one-third of patients (injection-site rash, headache, nasopharyngitis) may be a barrier to the use of anakinra 9.
Imatinib (Gleevec)
Imatinib is the first approved drug to directly turn off the signal of a protein known to cause certain cancers 106. Little has been published regarding this treatment for Erdheim–Chester disease. However, one article is available that describes the use of Imatinib with six patients having multi-system involvement, who were not responding well to other treatments 107. Two were stabilized and four continued to worsen on this treatment. Most experienced stabilized cardiovascular involvement. Another article was published in November 2010 which provides data on 3 patients with histiocytic diseases given Imatinib, one of which was an Erdheim–Chester disease patient 108. The conclusion of this article is, “Imatinib may be an effective treatment option for some patients with these diseases” 108. Currently, there are a handful of Erdheim–Chester disease Global Alliance patients who are on this treatment. The typically reported dosage of imatinib for the treatment of Erdheim–Chester disease is 400 mg by mouth daily 9.
Infliximab (Remicade)
Infliximab is a biologic drug normally used to treat autoimmune diseases. 2 patients with cardiac Erdheim–Chester disease refractory to treatment with IFN-α had clinical improvement when treated infliximab 109 and another case series of 12 patients showed ∼40% response rates mainly cardiac and cerebellar Erdheim–Chester disease 110. In general, Infliximab has shown little efficacy in treating Erdheim–Chester disease. However, it may have limited use for the treatment of patients with mild Erdheim–Chester disease or for those patients who show intolerance to other treatments. Some suggest that Infliximab might also be considered as a treatment for those patients whose targeted therapy, such as a BRAF- and/or MEK-inhibitor, has been interrupted 95.
Tocilizumab (Actemra)
Tocilizumab (Actemra) is approved for the treatment of moderately to severely active rheumatoid arthritis (RA) and adults with giant cell arteritis 111. Tocilizumab is classified as an interleukin-6 (IL-6) receptor inhibitor. Emerging data on the use of Tocilizumab as an Erdheim–Chester disease treatment shows it may decrease c-reactive protein (CRP) levels as well as decrease fluorodeoxyglucose (FDG) intake. Tocilizumab may be effective patients with bone only or cardiac Erdheim–Chester disease but seems to have poor effects on central nervous system (CNS) involvement. 2 of 3 patients in a pilot phase 2 trial had a favorable response, both without central nervous system (CNS) involvement 112. Although there are no published papers on this treatment, patients have experienced some bone pain relief while on this treatment.
Methotrexate
Methotrexate is usually used to treat cancer or autoimmune diseases. Methotrexate has been used to treat some Erdheim–Chester disease patients. Weekly oral methotrexate was not shown to be efficacious in a case series of 13 patients, except for prolonged disease improvement and stabilization in 2 patients with ocular (subconjunctival and choroidal) Erdheim–Chester disease 113. In some cases, Methotrexate has been used alone and in at least one case it was used with Infliximab and Mycophenolic Acid. Some patients have reported receiving high doses of methotrexate when the rapid progression of Erdheim–Chester disease has been identified 114, whereas other patients have received lower maintenance doses of Methotrexate.
Sirolimus (Rapamune)
The data shows that Sirolimus (an mTOR-inhibitor) has induced stabilization and is somewhat well tolerated in some patients with multisystemic Erdheim–Chester disease, especially for those with kidney involvement. A phase 2 trial used Sirolimus with prednisone in 10 Erdheim–Chester disease patients and resulted in an objective response rate (percentage of people in a study or treatment group who have a partial response or complete response to the treatment within a certain period of time) of 80% in at least 1 disease site 63. Responses were seen in 50% of retroperitoneal and 75% of cardiovascular Erdheim–Chester disease, but no patients achieved complete remission (disappearance of all signs of cancer in response to treatment). None of the 5 patients who were tested showed the presence of PI3KCA mutations, but mTOR pathway activation was demonstrated in tissues using immunohistochemistry. mTOR inhibitors are not recommended as first-line therapy for Erdheim–Chester disease treatment but may be a therapeutic option in refractory Erdheim–Chester disease patients. Sirolimus is not recommended for people with pleural, pericardial, or ascitic untreated effusions. Immunosuppressants reduce the effectiveness of the immune system and require patients with this therapy to be extremely careful in reducing the risk of contracting infections.
Systemic corticosteroids
Corticosteroids are hormones that are produced in the body as a response to stress, foreign bodies, inflammation, etc. Corticosteroids (e.g., prednisone) are not considered effective as monotherapy for Erdheim–Chester disease, although they may be used as adjuncts to improve acute symptoms related to tissue swelling such as in the case of orbital disease with impending vision loss. In a French cohort study, treatment with corticosteroids and immunosuppressive agents was not associated with improved survival 27. If a doctor believes corticosteroids are a good course of action for a patient, he will prescribe the medicine to the patient and continue monitoring the patient for any adverse side effects. Corticosteroids are often taken orally (by mouth) daily. The patient may experience some side effects that should be closely monitored by a doctor.
Surgical debulking
Due to the multifocal nature of Erdheim–Chester disease requiring systemic therapies, surgical resection is generally not curative. If a mass has formed as the result of Erdheim–Chester disease, a doctor may recommend removing as much of the mass as practical using surgical techniques. This is done with the intent to allow other treatments to work more effectively and/or to improve the quality of life where a reduction in mass size may help to reduce symptoms. The decision regarding surgical debulking is done by a doctor on a case-by-case basis.
Radiation treatment
Erdheim–Chester disease is not a radiosensitive disease, and experts do not recommend radiation therapy 115. The exceptions to this include situations in which immediate palliation of symptoms is needed such as for large tumors causing central nervous system, eye, or internal organ compromise. Sometimes a doctor may try to treat a mass using radiation techniques, typically to palliate bone pain associated with Erdheim–Chester disease lesions. Radiation treatment is done in a hospital or clinical setting.
Other treatments
Other treatments that some patients have found helpful in managing symptoms of Erdheim–Chester disease include:
- Bisphosphonates. Bisphosphonates are a class of drugs that prevent the loss of bone mass, used to treat osteoporosis and similar diseases. High-potency intravenous bisphosphonates have been shown to modify progression of skeletal metastasis in several forms of cancer. Bisphosphonates may contribute to the reduction of bone pain and to the management of the skeletal involvement in Erdheim–Chester disease.
- Modafinil (Provigil). Some patients have been treated with provigil to help relieve some of the fatigue/sleepiness symptoms they experience with Erdheim–Chester disease. Patients report varying levels of success regarding the relief provided. This medication will not treat the disease, but has simply been tried as a way to help patients cope with severe fatigue.
- Long-term Antibiotics. Many Erdheim–Chester disease patients report an increased tendency to suffer from infections and have a more difficult time recovering from them. Some treatments can reduce a patient’s immune system, putting them at a higher risk for infection. Most patients deal with common infections such as sinus, upper respiratory, urinary tract, etc., but others may have to deal with more opportunistic infections that occur when the immune system is compromised. The following are various methods used by some doctors and patients to help keep infections under control.
- Visit your local doctor as soon as you suspect an infection.
- Some doctors arrange a standing order for antibiotics in the event their patient cannot see him/her in a timely manner. This is done on a case by case basis. Even when using this method, it is very important to see your doctor as soon as possible when you suspect an infection. Choosing the best antibiotic for a particular infection can be very important and this can only be done with proper testing. Many times an infection may be the result of a virus in which case an antibiotic will NOT help in fighting the infection at all.
- Still other doctors place patients on long term courses of certain antibiotics as a prophylactic treatment to prevent an infection. This is done on a case-by-case basis and requires close monitoring by a doctor. There are differing opinions on the benefits versus risks of this approach to treating those with chronic infections.
Monitoring on treatment
Once treatment is initiated, patients will likely need monitoring for assessing the response to therapy and also management of side effects of treatments. It is recommended that those with Erdheim–Chester disease have the following tests performed to understand the areas of involvement:
- Physical exam, including neurological exam
- Skin exams in patients on BRAF- and MEK-inhibitor treatments
- PET scan to check for reduced brightness and size of the tumor sites
- Additional imaging studies (CT, MRI) may be undertaken based on organs involved at diagnosis. Imaging (PET, CT, MRI) is usually repeated every few months and can be variable for each patient
- Blood tests to assess for changes from the disease or the treatments
- A systematic cardiac and aortic evaluation. For patients with cardiac involvement, an additional study should be performed every three months until the progression is stabilized with treatment, at which point studies are often extended to every six months.
- An MRI evaluation of the brain with specific studies on the cerebellum. If CNS involvement is found or suspected, studies should be repeated every three months until progression is stabilized with treatment. At that point, studies are often reduced to once every six months.
- Renal and renovascular evaluation, with additional studies every three months after starting therapy, extending to every six months once the disease is stabilized.
- PET/CT scans to access and monitor the activity and extent of disease and response to therapy; this is typically done every 3 to 6 months until the disease is stabilized.
- A check of vitamin B12 levels (400 pg/ml is the recommended level).
- A check of vitamin E levels that could be involved in neurological manifestations.
- An evaluation of the pituitary function and treatment with the help of an endocrinologist. Hormone levels, including testosterone, ADH, thyroid hormones, insulin, ACTH and PTH should all be considered.
Relapsed Erdheim–Chester disease treatment
If the tumor grows back after treatment, it is called as relapsed disease. In such cases, one of the other targeted or conventional treatments than used previously may be utilized. In some cases, a repeat biopsy of tumor cells may be needed for confirmation of diagnosis and mutation testing. If a clinical trial is available, that may be a consideration as well.
Erdheim–Chester disease prognosis
Erdheim–Chester disease prognosis is variable and depends mainly on the extent and site of disease and the response to therapy 12, 9. Erdheim–Chester disease may range from asymptomatic bone lesions to multi-systemic life-threatening forms. Scientific literature, mainly published before the year 2010, often reports a very poor prognosis but treatment has improved since. In a large French cohort of 165 Erdheim–Chester disease patients, factors associated with worse overall survival included advanced age and disease involving the central nervous system, lungs, and retroperitoneum 14. Advances in understanding the pathology, the genetic makeup, the diagnosis and therapeutics of Erdheim–Chester disease during the last 5 years have made tremendous improvement in 5-year overall survival rates from 43% in a study from 1996 to 83% in a recent study 14, 15. Due to this, mortality and morbidity have considerably decreased. With the advent of targeted therapies, even severe manifestations of Erdheim–Chester disease have become a chronic, rather than fatal, illness. It is also important to note that there are patients who are living high-quality lives with Erdheim–Chester disease for decades and referral for rehabilitation and supportive care can be of benefit 12. On rare occasions, patients still succumb to advanced disease with irreversible organ damage, especially cerebellar dysfunction.
Erdheim–Chester disease life expectancy
Erdheim–Chester disease prognosis is variable and depends mainly on the extent and site of disease and the response to therapy 12, 9. Erdheim–Chester disease may range from asymptomatic bone lesions to multi-systemic life-threatening forms. While some individuals with Erdheim–Chester disease may experience a relatively stable disease course and have a near-normal life expectancy, others may face more aggressive forms of the disease that can significantly impact their lifespan. Early diagnosis of Erdheim–Chester disease with appropriate medical management, and close monitoring could potentially optimize outcomes for individuals with Erdheim–Chester disease 116, 117, 118, 43, 11. Lesions in the central nervous system (brain and spinal cord) are an independent factor for poor prognosis and are responsible for one third of deaths in Erdheim–Chester disease patients 119.
It is important for patients with Erdheim–Chester disease to work closely with a multidisciplinary medical team to develop an individualized treatment plan and receive ongoing care to manage the disease effectively. Advances in understanding the pathology, the genetic makeup, the diagnosis and therapeutics of Erdheim–Chester disease during the last 5 years have made tremendous improvement in 5-year overall survival rates from 43% in a study from 1996 to 83% in a recent study 14, 15. Due to this, mortality and morbidity have considerably decreased. With the advent of targeted therapies, even severe manifestations of Erdheim–Chester disease have become a chronic, rather than fatal, illness. It is also important to note that there are patients who are living high-quality lives with Erdheim–Chester disease for decades and referral for rehabilitation and supportive care can be of benefit 12. On rare occasions, patients still succumb to advanced disease with irreversible organ damage, especially cerebellar dysfunction.
- Aubart FC, Poupel L, Saint-Charles F, Charlotte F, Arsafi Y, Frisdal E, Roos-Weil D, Emile JF, Amoura Z, Guerin M, Lesnik P, Haroche J, Le Goff W. Profound systemic alteration of the immune phenotype and an immunoglobulin switch in Erdheim-Chester disease in 78 patients from a single center. Haematologica. 2022 Jun 1;107(6):1347-1357. doi: 10.3324/haematol.2021.279118[↩]
- Haroche J, Cohen-Aubart F, Amoura Z. Erdheim-Chester disease. Blood. 2020 Apr 16;135(16):1311-1318. doi: 10.1182/blood.2019002766 [↩]
- Diamond EL, Reiner AS, Buthorn JJ, Shuk E, Applebaum AJ, Hyman DM, Abdel-Wahab O, Rampal R, Janku F, Brewer K, Campbell J, Mao JJ, Atkinson TM, Panageas KS. A scale for patient-reported symptom assessment for patients with Erdheim-Chester disease. Blood Adv. 2019 Apr 9;3(7):934-938. doi: 10.1182/bloodadvances.2018030502[↩][↩]
- Cohen Aubart F, Idbaih A, Emile JF, Amoura Z, Abdel-Wahab O, Durham BH, Haroche J, Diamond EL. Histiocytosis and the nervous system: from diagnosis to targeted therapies. Neuro Oncol. 2021 Sep 1;23(9):1433-1446. doi: 10.1093/neuonc/noab107[↩][↩][↩]
- Erdheim-Chester disease. https://medlineplus.gov/genetics/condition/erdheim-chester-disease[↩][↩][↩][↩][↩]
- Qiao, J., Ma, R., Peng, X. et al. Erdheim-Chester disease with bilateral orbital masses and multi-systemic symptoms: two case reports. World J Surg Onc 21, 233 (2023). https://doi.org/10.1186/s12957-023-03123-5[↩]
- Garbino N, Punzo B, Todisco A, Cirillo G, Cavaliere C. Whole body positron emission tomography-MRI of Erdheim-Chester disease: a case report. Quant Imaging Med Surg. 2020 Dec;10(12):2379-2386. doi: 10.21037/qims-19-953[↩]
- Goyal G, Young JR, Koster MJ, Tobin WO, Vassallo R, Ryu JH, Davidge-Pitts CJ, Hurtado MD, Ravindran A, Sartori Valinotti JC, Bennani NN, Shah MV, Rech KL, Go RS; Mayo Clinic Histiocytosis Working Group. The Mayo Clinic Histiocytosis Working Group Consensus Statement for the Diagnosis and Evaluation of Adult Patients With Histiocytic Neoplasms: Erdheim-Chester Disease, Langerhans Cell Histiocytosis, and Rosai-Dorfman Disease. Mayo Clin Proc. 2019 Oct;94(10):2054-2071. doi: 10.1016/j.mayocp.2019.02.023[↩]
- Goyal G, Heaney ML, Collin M, Cohen-Aubart F, Vaglio A, Durham BH, Hershkovitz-Rokah O, Girschikofsky M, Jacobsen ED, Toyama K, Goodman AM, Hendrie P, Cao XX, Estrada-Veras JI, Shpilberg O, Abdo A, Kurokawa M, Dagna L, McClain KL, Mazor RD, Picarsic J, Janku F, Go RS, Haroche J, Diamond EL. Erdheim-Chester disease: consensus recommendations for evaluation, diagnosis, and treatment in the molecular era. Blood. 2020 May 28;135(22):1929-1945. https://doi.org/10.1182/blood.2019003507[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Volpicelli ER, Doyle L, Annes JP, Murray MF, Jacobsen E, Murphy GF, Saavedra AP. Erdheim-Chester disease presenting with cutaneous involvement: a case report and literature review. J Cutan Pathol. 2011 Mar;38(3):280-5. doi: 10.1111/j.1600-0560.2010.01650.x[↩]
- Mazor RD, Manevich-Mazor M, Shoenfeld Y. Erdheim-Chester Disease: a comprehensive review of the literature. Orphanet J Rare Dis. 2013 Sep 8;8:137. doi: 10.1186/1750-1172-8-137[↩][↩][↩][↩]
- What is Erdheim-Chester Disease? https://erdheim-chester.org/about-ecd[↩][↩][↩][↩][↩][↩][↩][↩]
- Estrada-Veras JI, O’Brien KJ, Boyd LC, Dave RH, Durham B, Xi L, Malayeri AA, Chen MY, Gardner PJ, Alvarado-Enriquez JR, Shah N, Abdel-Wahab O, Gochuico BR, Raffeld M, Jaffe ES, Gahl WA. The clinical spectrum of Erdheim-Chester disease: an observational cohort study. Blood Adv. 2017 Feb 14;1(6):357-366. doi: 10.1182/bloodadvances.2016001784[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Cohen-Aubart F, Emile JF, Carrat F, Helias-Rodzewicz Z, Taly V, Charlotte F, Cluzel P, Donadieu J, Idbaih A, Barete S, Amoura Z, Haroche J. Phenotypes and survival in Erdheim-Chester disease: Results from a 165-patient cohort. Am J Hematol. 2018 May;93(5):E114-E117. doi: 10.1002/ajh.25055[↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Veyssier-Belot C, Cacoub P, Caparros-Lefebvre D, Wechsler J, Brun B, Remy M, Wallaert B, Petit H, Grimaldi A, Wechsler B, Godeau P. Erdheim-Chester disease. Clinical and radiologic characteristics of 59 cases. Medicine (Baltimore). 1996 May;75(3):157-69. doi: 10.1097/00005792-199605000-00005[↩][↩][↩][↩]
- Clerico A, Ragni G, Cappelli C, Schiavetti A, Gonfiantini M, Uccini S. Erdheim-Chester disease in a child. Med Pediatr Oncol. 2003 Dec;41(6):575-7. doi: 10.1002/mpo.10402[↩]
- Tran TA, Fabre M, Pariente D, Craiu I, Haroche J, Charlotte F, Eid P, Durrbach A, Taoufik Y, Kone-Paut I. Erdheim-Chester disease in childhood: a challenging diagnosis and treatment. J Pediatr Hematol Oncol. 2009 Oct;31(10):782-6. doi: 10.1097/MPH.0b013e3181b76827[↩]
- Song SY, Lee SW, Ryu KH, Sung SH. Erdheim-Chester disease with multisystem involvement in a 4-year-old. Pediatr Radiol. 2012 May;42(5):632-5. doi: 10.1007/s00247-011-2235-8[↩]
- Tran TA, Pariente D, Lecron JC, Delwail A, Taoufik Y, Meinzer U. Treatment of pediatric Erdheim-Chester disease with interleukin-1-targeting drugs. Arthritis Rheum. 2011 Dec;63(12):4031-2. doi: 10.1002/art.30638[↩]
- Erdheim-Chester disease. https://histio.org/histiocytic-disorders/erdheim-chester-disease[↩]
- Haroche J, Amoura Z, Wechsler B, Veyssier-Belot C, Charlotte F, Piette JC. Maladie d’Erdheim-Chester [Erdheim-Chester disease]. Presse Med. 2007 Nov;36(11 Pt 2):1663-8. French. doi: 10.1016/j.lpm.2007.04.032[↩]
- Drier A, Haroche J, Savatovsky J, Godenèche G, Dormont D, Chiras J, Amoura Z, Bonneville F. Cerebral, facial, and orbital involvement in Erdheim-Chester disease: CT and MR imaging findings. Radiology. 2010 May;255(2):586-94. doi: 10.1148/radiol.10090320[↩]
- Allen TC, Chevez-Barrios P, Shetlar DJ, Cagle PT. Pulmonary and ophthalmic involvement with Erdheim-Chester disease: a case report and review of the literature. Arch Pathol Lab Med. 2004 Dec;128(12):1428-31. doi: 10.5858/2004-128-1428-PAOIWE[↩]
- Carpinteri R, Patelli I, Casanueva FF, Giustina A. Pituitary tumours: inflammatory and granulomatous expansive lesions of the pituitary. Best Pract Res Clin Endocrinol Metab. 2009 Oct;23(5):639-50. doi: 10.1016/j.beem.2009.05.009[↩]
- Erdheim Chester Disease. https://rarediseases.org/rare-diseases/erdheim-chester-disease[↩][↩][↩][↩][↩][↩][↩]
- Fortman BJ, Beall DP. Erdheim-Chester disease of the retroperitoneum: a rare cause of ureteral obstruction. AJR Am J Roentgenol. 2001 May;176(5):1330-1. doi: 10.2214/ajr.176.5.1761330[↩]
- Arnaud L, Hervier B, Néel A, Hamidou MA, Kahn JE, Wechsler B, Pérez-Pastor G, Blomberg B, Fuzibet JG, Dubourguet F, Marinho A, Magnette C, Noel V, Pavic M, Casper J, Beucher AB, Costedoat-Chalumeau N, Aaron L, Salvatierra J, Graux C, Cacoub P, Delcey V, Dechant C, Bindi P, Herbaut C, Graziani G, Amoura Z, Haroche J. CNS involvement and treatment with interferon-α are independent prognostic factors in Erdheim-Chester disease: a multicenter survival analysis of 53 patients. Blood. 2011 Mar 10;117(10):2778-82. doi: 10.1182/blood-2010-06-294108[↩][↩][↩]
- Adawi M, Bisharat B, Bowirrat A. Erdheim-Chester disease (ECD): Case report, clinical and basic investigations, and review of literature. Medicine (Baltimore). 2016 Oct;95(42):e5167. doi: 10.1097/MD.0000000000005167[↩]
- Diamond EL, Durham BH, Haroche J, et al. Diverse and Targetable Kinase Alterations Drive Histiocytic Neoplasms. Cancer Discov. 2016 Feb;6(2):154-65. doi: 10.1158/2159-8290.CD-15-0913[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Pegoraro, F., Papo, M., Maniscalco, V. et al. Erdheim–Chester disease: a rapidly evolving disease model. Leukemia 34, 2840–2857 (2020). https://doi.org/10.1038/s41375-020-0944-4[↩]
- Diamond EL, Durham BH, Haroche J, Yao Z, Ma J, Parikh SA, Wang Z, Choi J, Kim E, Cohen-Aubart F, Lee SC, Gao Y, Micol JB, Campbell P, Walsh MP, Sylvester B, Dolgalev I, Aminova O, Heguy A, Zappile P, Nakitandwe J, Ganzel C, Dalton JD, Ellison DW, Estrada-Veras J, Lacouture M, Gahl WA, Stephens PJ, Miller VA, Ross JS, Ali SM, Briggs SR, Fasan O, Block J, Héritier S, Donadieu J, Solit DB, Hyman DM, Baselga J, Janku F, Taylor BS, Park CY, Amoura Z, Dogan A, Emile JF, Rosen N, Gruber TA, Abdel-Wahab O. Diverse and Targetable Kinase Alterations Drive Histiocytic Neoplasms. Cancer Discov. 2016 Feb;6(2):154-65. doi: 10.1158/2159-8290.CD-15-0913[↩]
- Goyal G, Heaney ML, Collin M, Cohen-Aubart F, Vaglio A, Durham BH, Hershkovitz-Rokah O, Girschikofsky M, Jacobsen ED, Toyama K, Goodman AM, Hendrie P, Cao XX, Estrada-Veras JI, Shpilberg O, Abdo A, Kurokawa M, Dagna L, McClain KL, Mazor RD, Picarsic J, Janku F, Go RS, Haroche J, Diamond EL. Erdheim-Chester disease: consensus recommendations for evaluation, diagnosis, and treatment in the molecular era. Blood. 2020 May 28;135(22):1929-1945. doi: 10.1182/blood.2019003507[↩]
- How is Erdheim-Chester Disease Diagnosed? https://erdheim-chester.org/diagnosis[↩][↩]
- Janku F, Diamond EL, Goodman AM, Raghavan VK, Barnes TG, Kato S, Abdel-Wahab O, Durham BH, Meric-Bernstam F, Kurzrock R. Molecular Profiling of Tumor Tissue and Plasma Cell-Free DNA from Patients with Non-Langerhans Cell Histiocytosis. Mol Cancer Ther. 2019 Jun;18(6):1149-1157. doi: 10.1158/1535-7163.MCT-18-1244[↩][↩][↩]
- Diamond EL, Durham BH, Ulaner GA, Drill E, Buthorn J, Ki M, Bitner L, Cho H, Young RJ, Francis JH, Rampal R, Lacouture M, Brody LA, Ozkaya N, Dogan A, Rosen N, Iasonos A, Abdel-Wahab O, Hyman DM. Efficacy of MEK inhibition in patients with histiocytic neoplasms. Nature. 2019 Mar;567(7749):521-524. doi: 10.1038/s41586-019-1012-y[↩][↩]
- Bentel JM, Thomas MA, Rodgers JJ, Arooj M, Gray E, Allcock R, Fermoyle S, Mancera RL, Cannell P, Parry J. Erdheim-Chester disease associated with a novel, complex BRAF p.Thr599_Val600delinsArgGlu mutation. BMJ Case Rep. 2017 Apr 28;2017:bcr2017219720. doi: 10.1136/bcr-2017-219720[↩][↩]
- Diamond EL, Abdel-Wahab O, Pentsova E, Borsu L, Chiu A, Teruya-Feldstein J, Hyman DM, Rosenblum M. Detection of an NRAS mutation in Erdheim-Chester disease. Blood. 2013 Aug 8;122(6):1089-91. doi: 10.1182/blood-2013-02-482984[↩][↩][↩]
- Emile JF, Charlotte F, Amoura Z, Haroche J. BRAF mutations in Erdheim-Chester disease. J Clin Oncol. 2013 Jan 20;31(3):398. doi: 10.1200/JCO.2012.46.9676[↩][↩]
- Emile JF, Diamond EL, Hélias-Rodzewicz Z, et al. Recurrent RAS and PIK3CA mutations in Erdheim-Chester disease. Blood. 2014 Nov 6;124(19):3016-9. doi: 10.1182/blood-2014-04-570937[↩][↩]
- Haroche J, Charlotte F, Arnaud L, von Deimling A, Hélias-Rodzewicz Z, Hervier B, Cohen-Aubart F, Launay D, Lesot A, Mokhtari K, Canioni D, Galmiche L, Rose C, Schmalzing M, Croockewit S, Kambouchner M, Copin MC, Fraitag S, Sahm F, Brousse N, Amoura Z, Donadieu J, Emile JF. High prevalence of BRAF V600E mutations in Erdheim-Chester disease but not in other non-Langerhans cell histiocytoses. Blood. 2012 Sep 27;120(13):2700-3. doi: 10.1182/blood-2012-05-430140[↩][↩][↩][↩]
- Goyal G, Lau D, Nagle AM, Vassallo R, Rech KL, Ryu JH, Davidge-Pitts CJ, Tobin WO, Koster MJ, Bennani NN, Shah MV, Liu MC, Go RS; Mayo Clinic Histiocytosis Working Group. Tumor mutational burden and other predictive immunotherapy markers in histiocytic neoplasms. Blood. 2019 Apr 4;133(14):1607-1610. doi: 10.1182/blood-2018-12-893917[↩][↩]
- Chasset F, Barete S, Charlotte F, Cohen-Aubart F, Arnaud L, Le Pelletier F, Emile JF, Francès C, Amoura Z, Haroche J. Cutaneous manifestations of Erdheim-Chester disease (ECD): Clinical, pathological, and molecular features in a monocentric series of 40 patients. J Am Acad Dermatol. 2016 Mar;74(3):513-20. doi: 10.1016/j.jaad.2015.11.007[↩][↩]
- Neckman JP, Kim J, Mathur M, Myung P, Girardi M. Diverse cutaneous manifestations of Erdheim-Chester disease in a woman with a history of Langerhans cell histiocytosis. JAAD Case Rep. 2016 Mar 5;2(2):128-31. doi: 10.1016/j.jdcr.2015.10.010[↩][↩]
- Histiocytosis. https://medlineplus.gov/ency/article/000068.htm[↩]
- 15 – Histiocytic Proliferative Disorders. Dermatopathology Foundations in Diagnostic Pathology 2010, Pages 619-630. https://doi.org/10.1016/B978-0-443-06654-2.00015-9[↩]
- Rodriguez-Galindo C, Allen CE. Langerhans cell histiocytosis. Blood. 2020 Apr 16;135(16):1319-1331. https://doi.org/10.1182/blood.2019000934[↩]
- Emile JF, Abla O, Fraitag S, Horne A, Haroche J, Donadieu J, Requena-Caballero L, Jordan MB, Abdel-Wahab O, Allen CE, Charlotte F, Diamond EL, Egeler RM, Fischer A, Herrera JG, Henter JI, Janku F, Merad M, Picarsic J, Rodriguez-Galindo C, Rollins BJ, Tazi A, Vassallo R, Weiss LM; Histiocyte Society. Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood. 2016 Jun 2;127(22):2672-81. doi: 10.1182/blood-2016-01-690636[↩]
- Emile JF, Diamond EL, Hélias-Rodzewicz Z, Cohen-Aubart F, Charlotte F, Hyman DM, Kim E, Rampal R, Patel M, Ganzel C, Aumann S, Faucher G, Le Gall C, Leroy K, Colombat M, Kahn JE, Trad S, Nizard P, Donadieu J, Taly V, Amoura Z, Abdel-Wahab O, Haroche J. Recurrent RAS and PIK3CA mutations in Erdheim-Chester disease. Blood. 2014 Nov 6;124(19):3016-9. doi: 10.1182/blood-2014-04-570937[↩][↩]
- Hyman DM, Diamond EL, Vibat CR, Hassaine L, Poole JC, Patel M, Holley VR, Cabrilo G, Lu TT, Arcila ME, Chung YR, Rampal R, Lacouture ME, Rosen N, Meric-Bernstam F, Baselga J, Kurzrock R, Erlander MG, Janku F, Abdel-Wahab O. Prospective blinded study of BRAFV600E mutation detection in cell-free DNA of patients with systemic histiocytic disorders. Cancer Discov. 2015 Jan;5(1):64-71. doi: 10.1158/2159-8290.CD-14-0742[↩][↩]
- Nordmann TM, Juengling FD, Recher M, Berger CT, Kalbermatten D, Wicki A, Paasinen-Sohns A, Cathomas G, Tzankov A, Daikeler T. Trametinib after disease reactivation under dabrafenib in Erdheim-Chester disease with both BRAF and KRAS mutations. Blood. 2017 Feb 16;129(7):879-882. doi: 10.1182/blood-2016-09-740217[↩]
- Durham BH, Lopez Rodrigo E, Picarsic J, et al. Activating mutations in CSF1R and additional receptor tyrosine kinases in histiocytic neoplasms. Nat Med. 2019 Dec;25(12):1839-1842. doi: 10.1038/s41591-019-0653-6[↩]
- Egan C, Nicolae A, Lack J, Chung HJ, Skarshaug S, Pham TA, Navarro W, Abdullaev Z, Aguilera NS, Xi L, Pack S, Pittaluga S, Jaffe ES, Raffeld M. Genomic profiling of primary histiocytic sarcoma reveals two molecular subgroups. Haematologica. 2020 Apr;105(4):951-960. doi: 10.3324/haematol.2019.230375[↩]
- Shanmugam V, Griffin GK, Jacobsen ED, Fletcher CDM, Sholl LM, Hornick JL. Identification of diverse activating mutations of the RAS-MAPK pathway in histiocytic sarcoma. Mod Pathol. 2019 Jun;32(6):830-843. doi: 10.1038/s41379-018-0200-x[↩]
- Erdheim-Chester Disease. https://histio.org/histiocytic-disorders/erdheim-chester-disease[↩]
- Ambrosini V, Savelli F, Merli E, Zompatori M, Nanni C, Allegri V, Fanti S. F-18 FDG PET/CT detects muscle involvement in Erdheim-Chester disease. Clin Nucl Med. 2012 Feb;37(2):196-7. doi: 10.1097/RLU.0b013e31823e9d54[↩]
- Dion E, Graef C, Haroche J, Renard-Penna R, Cluzel P, Wechsler B, Piette JC, Grenier PA. Imaging of thoracoabdominal involvement in Erdheim-Chester disease. AJR Am J Roentgenol. 2004 Nov;183(5):1253-60. doi: 10.2214/ajr.183.5.1831253[↩]
- Parks NE, Goyal G, Go RS, Mandrekar J, Tobin WO. Neuroradiologic manifestations of Erdheim-Chester disease. Neurol Clin Pract. 2018 Feb;8(1):15-20. doi: 10.1212/CPJ.0000000000000422[↩][↩]
- Mazor RD, Manevich-Mazor M, Kesler A, Aizenstein O, Eshed I, Jaffe R, Pessach Y, Goldberg I, Sprecher E, Yaish I, Gural A, Ganzel C, Shoenfeld Y. Clinical considerations and key issues in the management of patients with Erdheim-Chester Disease: a seven case series. BMC Med. 2014 Dec 1;12:221. doi: 10.1186/s12916-014-0221-3[↩]
- Hurtado M, Cortes T, Goyal G, Maraka S, O’Keeffe D, Erickson D, Shah M, Go R, Davidge-Pitts C. OR32-1 Endocrine Manifestations of Erdheim-Chester Disease: The Mayo Clinic Experience. J Endocr Soc. 2019 Apr 30;3(Suppl 1):OR32-1. doi: 10.1210/js.2019-OR32-1[↩][↩][↩]
- Courtillot C, Laugier Robiolle S, Cohen Aubart F, Leban M, Renard-Penna R, Drier A, Charlotte F, Amoura Z, Touraine P, Haroche J. Endocrine Manifestations in a Monocentric Cohort of 64 Patients With Erdheim-Chester Disease. J Clin Endocrinol Metab. 2016 Jan;101(1):305-13. doi: 10.1210/jc.2015-3357[↩][↩]
- Brun AL, Touitou-Gottenberg D, Haroche J, Toledano D, Cluzel P, Beigelman-Aubry C, Piette JC, Amoura Z, Grenier PA. Erdheim-Chester disease: CT findings of thoracic involvement. Eur Radiol. 2010 Nov;20(11):2579-87. doi: 10.1007/s00330-010-1830-7[↩][↩]
- Haroche J, Cluzel P, Toledano D, Montalescot G, Touitou D, Grenier PA, Piette JC, Amoura Z. Images in cardiovascular medicine. Cardiac involvement in Erdheim-Chester disease: magnetic resonance and computed tomographic scan imaging in a monocentric series of 37 patients. Circulation. 2009 Jun 30;119(25):e597-8. doi: 10.1161/CIRCULATIONAHA.108.825075[↩]
- Gianfreda D, Nicastro M, Galetti M, Alberici F, Corradi D, Becchi G, Baldari G, De Filippo M, Ferretti S, Moroni G, Foti R, Di Gangi M, Jeannin G, Saffroy R, Emile JF, Buzio C, Vaglio A. Sirolimus plus prednisone for Erdheim-Chester disease: an open-label trial. Blood. 2015 Sep 3;126(10):1163-71. doi: 10.1182/blood-2015-01-620377[↩][↩][↩]
- Buono A, Bassi I, Santolamazza C, Moreo A, Pedrotti P, Sacco A, Morici N, Giannattasio C, Oliva F, Ammirati E. Getting to the heart of the matter in a multisystem disorder: Erdheim-Chester disease. Lancet. 2019 Aug 17;394(10198):e19. doi: 10.1016/S0140-6736(19)31787-8[↩]
- Ghotra AS, Thompson K, Lopez-Mattei J, Bawa D, Hernandez R, Banchs J, Palaskas N, Iliescu C, Kim P, Yusuf SW, Hassan SA. Cardiovascular manifestations of Erdheim-Chester disease. Echocardiography. 2019 Feb;36(2):229-236. doi: 10.1111/echo.14231[↩]
- Haroche J, Amoura Z, Dion E, Wechsler B, Costedoat-Chalumeau N, Cacoub P, Isnard R, Généreau T, Wechsler J, Weber N, Graef C, Cluzel P, Grenier P, Piette JC. Cardiovascular involvement, an overlooked feature of Erdheim-Chester disease: report of 6 new cases and a literature review. Medicine (Baltimore). 2004 Nov;83(6):371-392. doi: 10.1097/01.md.0000145368.17934.91[↩][↩]
- Costa IBSDS, Costa FAS, Bittar CS, Rizk SI, Abdo ANR, Siqueira SAC, Rocha V, Pereira J, Ky B, Hajjar LA. Cardiac Tamponade as the First Manifestation of Erdheim-Chester Disease. JACC CardioOncol. 2020 Jun 16;2(2):324-328. doi: 10.1016/j.jaccao.2020.03.004[↩]
- Villatoro-Villar M, Bold MS, Warrington KJ, Crowson CS, Goyal G, Shah M, Go RS, Koster MJ. Arterial involvement in Erdheim-Chester disease: A retrospective cohort study. Medicine (Baltimore). 2018 Dec;97(49):e13452. doi: 10.1097/MD.0000000000013452[↩]
- Mirmomen SM, Sirajuddin A, Nikpanah M, Symons R, Paschall AK, Papageorgiou I, Gahl WA, O’Brien K, Estrada-Veras JI, Malayeri AA. Thoracic involvement in Erdheim-Chester disease: computed tomography imaging findings and their association with the BRAFV600E mutation. Eur Radiol. 2018 Nov;28(11):4635-4642. doi: 10.1007/s00330-018-5421-3[↩][↩]
- Haroutunian SG, O’Brien KJ, Estrada-Veras JI, Yao J, Boyd LC, Mathur K, Gahl WA, Mirmomen SM, Malayeri AA, Kleiner DE, Jaffe ES, Gochuico BR. Clinical and Histopathologic Features of Interstitial Lung Disease in Erdheim⁻Chester Disease. J Clin Med. 2018 Aug 28;7(9):243. doi: 10.3390/jcm7090243[↩][↩]
- Toya T, Ogura M, Toyama K, Yoshimi A, Shinozaki-Ushiku A, Honda A, Honda K, Hosoya N, Murakami Y, Kawashima H, Nannya Y, Arai S, Nakamura F, Shinoda Y, Nangaku M, Miyagawa K, Fukayama M, Moriya-Saito A, Katayama I, Ogura T, Kurokawa M. Prognostic factors of Erdheim-Chester disease: a nationwide survey in Japan. Haematologica. 2018 Nov;103(11):1815-1824. doi: 10.3324/haematol.2018.190728[↩]
- Karcioglu ZA, Sharara N, Boles TL, Nasr AM. Orbital xanthogranuloma: clinical and morphologic features in eight patients. Ophthalmic Plast Reconstr Surg. 2003 Sep;19(5):372-81. doi: 10.1097/01.IOP.0000083642.15174.83[↩]
- Kobic A, Shah KK, Schmitt AR, Goyal G, Go RS, Guo R, Rech KL, Sartori-Valinotti JC; Mayo Clinic Histiocytosis Working Group. Erdheim-Chester disease: expanding the spectrum of cutaneous manifestations. Br J Dermatol. 2020 Feb;182(2):405-409. doi: 10.1111/bjd.18153[↩][↩][↩]
- Shah, M.V., Call, T.G., Hook, C.C., Johnston, P., Letendre, L.L., Colgan, J.P., Pardanani, A.D., Clarke, B.L., Grogg, K.L., & Go, R.S. (2014). Clinical Presentation, Diagnosis, Treatment, and Outcome of Patients with Erdheim-Chester Disease: The Mayo Clinic Experience. Blood, 124, 1405-1405. https://doi.org/10.1182/blood.V124.21.1405.1405[↩]
- Goyal G, Ravindran A, Liu Y, He R, Shah MV, Bennani NN, Patnaik MM, Rech KL, Go RS; Mayo Clinic Histiocytosis Working Group. Bone marrow findings in Erdheim-Chester disease: increased prevalence of chronic myeloid neoplasms. Haematologica. 2020 Jan 31;105(2):e84-e86. doi: 10.3324/haematol.2019.234187[↩]
- Papo M, Diamond EL, Cohen-Aubart F, Emile JF, Roos-Weil D, Gupta N, Durham BH, Ozkaya N, Dogan A, Ulaner GA, Rampal R, Kahn JE, Sené T, Charlotte F, Hervier B, Besnard C, Bernard OA, Settegrana C, Droin N, Hélias-Rodzewicz Z, Amoura Z, Abdel-Wahab O, Haroche J. High prevalence of myeloid neoplasms in adults with non-Langerhans cell histiocytosis. Blood. 2017 Aug 24;130(8):1007-1013. doi: 10.1182/blood-2017-01-761718[↩]
- Erdheim-Chester Disease: An Algorithmic Approach to a Diagnosis. https://erdheim-chester.org/news/erdheim-chester-disease-an-algorithmic-approach-to-a-diagnosis[↩]
- Lin E. FDG PET/CT for biopsy guidance in Erdheim-Chester disease. Clin Nucl Med. 2007 Nov;32(11):860-1. doi: 10.1097/RLU.0b013e318156bc9b[↩]
- Namwongprom S, Núñez R, Kim EE, Macapinlac HA. Tc-99m MDP bone scintigraphy and positron emission tomography/computed tomography (PET/CT) imaging in Erdheim-Chester disease. Clin Nucl Med. 2007 Jan;32(1):35-8. doi: 10.1097/01.rlu.0000249758.49841.fa[↩]
- Steňová E, Steňo B, Povinec P, Ondriaš F, Rampalová J. FDG-PET in the Erdheim-Chester disease: its diagnostic and follow-up role. Rheumatol Int. 2012 Mar;32(3):675-8. doi: 10.1007/s00296-010-1676-y[↩]
- García-Gómez FJ, Acevedo-Báñez I, Martínez-Castillo R, Tirado-Hospital JL, Cuenca-Cuenca JI, Pachón-Garrudo VM, Álvarez-Pérez RM, García-Jiménez R, Rivas-Infante E, García-Morillo JS, Borrego-Dorado I. The role of 18FDG, 18FDOPA PET/CT and 99mTc bone scintigraphy imaging in Erdheim-Chester disease. Eur J Radiol. 2015 Aug;84(8):1586-1592. doi: 10.1016/j.ejrad.2015.04.022[↩]
- Arnaud L, Malek Z, Archambaud F, Kas A, Toledano D, Drier A, Zeitoun D, Cluzel P, Grenier PA, Chiras J, Piette JC, Amoura Z, Haroche J. 18F-fluorodeoxyglucose-positron emission tomography scanning is more useful in followup than in the initial assessment of patients with Erdheim-Chester disease. Arthritis Rheum. 2009 Oct;60(10):3128-38. doi: 10.1002/art.24848[↩]
- Diamond EL, Dagna L, Hyman DM, Cavalli G, Janku F, Estrada-Veras J, Ferrarini M, Abdel-Wahab O, Heaney ML, Scheel PJ, Feeley NK, Ferrero E, McClain KL, Vaglio A, Colby T, Arnaud L, Haroche J. Consensus guidelines for the diagnosis and clinical management of Erdheim-Chester disease. Blood. 2014 Jul 24;124(4):483-92. doi: 10.1182/blood-2014-03-561381[↩]
- Ozkaya N, Rosenblum MK, Durham BH, Pichardo JD, Abdel-Wahab O, Hameed MR, Busam KJ, Travis WD, Diamond EL, Dogan A. The histopathology of Erdheim-Chester disease: a comprehensive review of a molecularly characterized cohort. Mod Pathol. 2018 Apr;31(4):581-597. doi: 10.1038/modpathol.2017.160[↩]
- Durham BH. Molecular characterization of the histiocytoses: Neoplasia of dendritic cells and macrophages. Semin Cell Dev Biol. 2019 Feb;86:62-76. doi: 10.1016/j.semcdb.2018.03.002[↩]
- Ballester LY, Cantu MD, Lim KPH, Sarabia SF, Ferguson LS, Renee Webb C, Allen CE, McClain KL, Mohila CA, Punia JN, Roy A, López-Terrada DH, John Hicks M, Fisher KE. The use of BRAF V600E mutation-specific immunohistochemistry in pediatric Langerhans cell histiocytosis. Hematol Oncol. 2018 Feb;36(1):307-315. doi: 10.1002/hon.2388[↩]
- Melloul S, Hélias-Rodzewicz Z, Cohen-Aubart F, Charlotte F, Fraitag S, Terrones N, Riller Q, Chazal T, Héritier S, Moreau A, Kambouchner M, Copin MC, Donadieu J, Taly V, Amoura Z, Haroche J, Emile JF. Highly sensitive methods are required to detect mutations in histiocytoses. Haematologica. 2019 Mar;104(3):e97-e99. doi: 10.3324/haematol.2018.201194[↩]
- Janku F, Angenendt P, Tsimberidou AM, Fu S, Naing A, Falchook GS, Hong DS, Holley VR, Cabrilo G, Wheler JJ, Piha-Paul SA, Zinner RG, Bedikian AY, Overman MJ, Kee BK, Kim KB, Kopetz ES, Luthra R, Diehl F, Meric-Bernstam F, Kurzrock R. Actionable mutations in plasma cell-free DNA in patients with advanced cancers referred for experimental targeted therapies. Oncotarget. 2015 May 20;6(14):12809-21. doi: 10.18632/oncotarget.3373. Erratum in: Oncotarget. 2015 Sep 15;6(27):24581.[↩]
- Hervier B, Haroche J, Arnaud L, Charlotte F, Donadieu J, Néel A, Lifermann F, Villabona C, Graffin B, Hermine O, Rigolet A, Roubille C, Hachulla E, Carmoi T, Bézier M, Meignin V, Conrad M, Marie L, Kostrzewa E, Michot JM, Barete S, Taly V, Cury K, Emile JF, Amoura Z; French Histiocytoses Study Group. Association of both Langerhans cell histiocytosis and Erdheim-Chester disease linked to the BRAFV600E mutation. Blood. 2014 Aug 14;124(7):1119-26. doi: 10.1182/blood-2013-12-543793[↩]
- Braiteh F, Boxrud C, Esmaeli B, Kurzrock R. Successful treatment of Erdheim-Chester disease, a non-Langerhans-cell histiocytosis, with interferon-alpha. Blood. 2005 Nov 1;106(9):2992-4. doi: 10.1182/blood-2005-06-2238[↩]
- Haroche J, Amoura Z, Trad SG, Wechsler B, Cluzel P, Grenier PA, Piette JC. Variability in the efficacy of interferon-alpha in Erdheim-Chester disease by patient and site of involvement: results in eight patients. Arthritis Rheum. 2006 Oct;54(10):3330-6. doi: 10.1002/art.22165[↩]
- Suzuki HI, Hosoya N, Miyagawa K, Ota S, Nakashima H, Makita N, Kurokawa M. Erdheim-Chester disease: multisystem involvement and management with interferon-alpha. Leuk Res. 2010 Jan;34(1):e21-4. doi: 10.1016/j.leukres.2009.07.026[↩]
- Hervier B, Arnaud L, Charlotte F, Wechsler B, Piette JC, Amoura Z, Haroche J. Treatment of Erdheim-Chester disease with long-term high-dose interferon-α. Semin Arthritis Rheum. 2012 Jun;41(6):907-13. doi: 10.1016/j.semarthrit.2011.11.004[↩][↩]
- Cao XX, Niu N, Sun J, Cai H, Wang FD, Wang YN, Duan MH, Zhou DB, Li J. Clinical and positron emission tomography responses to long-term high-dose interferon-α treatment among patients with Erdheim-Chester disease. Orphanet J Rare Dis. 2019 Jan 10;14(1):11. doi: 10.1186/s13023-018-0988-y[↩]
- How is Erdheim-Chester Disease Treated? https://erdheim-chester.org/treatments[↩][↩][↩][↩]
- Adam Z, Koukalová R, Sprláková A, Rehák Z, Cervinek L, Szturz P, Krejcí M, Pour L, Zahradová L, Moulis M, Prásek J, Chaloupka R, Hájek R, Mayer J. Uspesná lécba Erdheimovy-Chesterovy nemoci chemoterapií obsahující 2-chlorodeoxyadenosin. Popis dvou prípadů a prehled literatury [Successful treatment of Erdheim-Chester disease by 2-chlorodeoxyadenosine-based chemotherapy. Two case studies and a literature review]. Vnitr Lek. 2011 Jun;57(6):576-89. Czech.[↩]
- Azadeh N, Tazelaar HD, Gotway MB, Mookadam F, Fonseca R. Erdheim Chester Disease treated successfully with cladribine. Respir Med Case Rep. 2016 Apr 4;18:37-40. doi: 10.1016/j.rmcr.2016.03.008[↩]
- Goyal G, Shah MV, Call TG, Litzow MR, Hogan WJ, Go RS. Clinical and Radiologic Responses to Cladribine for the Treatment of Erdheim-Chester Disease. JAMA Oncol. 2017 Sep 1;3(9):1253-1256. doi: 10.1001/jamaoncol.2017.0041[↩]
- Aouba A, Georgin-Lavialle S, Pagnoux C, Martin Silva N, Renand A, Galateau-Salle F, Le Toquin S, Bensadoun H, Larousserie F, Silvera S, Provost N, Candon S, Seror R, de Menthon M, Hermine O, Guillevin L, Bienvenu B. Rationale and efficacy of interleukin-1 targeting in Erdheim-Chester disease. Blood. 2010 Nov 18;116(20):4070-6. doi: 10.1182/blood-2010-04-279240[↩]
- Diamond EL, Abdel-Wahab O, Durham BH, Dogan A, Ozkaya N, Brody L, Arcila M, Bowers C, Fluchel M. Anakinra as efficacious therapy for 2 cases of intracranial Erdheim-Chester disease. Blood. 2016 Oct 6;128(14):1896-1898. doi: 10.1182/blood-2016-06-725143[↩][↩]
- Franconieri F, Deshayes S, de Boysson H, Trad S, Martin Silva N, Terrier B, Bienvenu B, Galateau-Sallé F, Emile JF, Johnson AC, Aouba A. Superior efficacy and similar safety of double dose anakinra in Erdheim-Chester disease after single dose treatment. Oncoimmunology. 2018 Apr 9;7(8):e1450712. doi: 10.1080/2162402X.2018.1450712[↩][↩]
- Franconieri F, Martin-Silva N, de Boysson H, Galateau-Salle F, Emile JF, Bienvenu B, Aouba A. Superior efficacy and tolerance of reduced doses of vemurafenib plus anakinra in Erdheim-Chester disease: Towards the paradigm of combined targeting and immune therapies. Acta Oncol. 2016 Jul;55(7):930-2. doi: 10.3109/0284186X.2015.1120885[↩]
- Cohen-Aubart F, Maksud P, Saadoun D, Drier A, Charlotte F, Cluzel P, Amoura Z, Haroche J. Variability in the efficacy of the IL1 receptor antagonist anakinra for treating Erdheim-Chester disease. Blood. 2016 Mar 17;127(11):1509-12. doi: 10.1182/blood-2015-09-672667[↩]
- Goyal G, Shah MV, Call TG, Litzow MR, Wolanskyj-Spinner AP, Koster MJ, Tobin WO, Vassallo R, Ryu JH, Hook CC, Hogan WJ, Go RS. Efficacy of biological agents in the treatment of Erdheim-Chester disease. Br J Haematol. 2018 Nov;183(3):520-524. doi: 10.1111/bjh.14997[↩]
- Jan H. Schirmer, Christoph Thorns, Frank Moosig, Julia U. Holle, Treatment failure by canakinumab in a patient with progressive multisystemic Erdheim–Chester disease refractory to anakinra: successful use of vemurafenib, Rheumatology, Volume 54, Issue 10, October 2015, Pages 1932–1934, https://doi.org/10.1093/rheumatology/kev237[↩]
- Haroche J, Amoura Z, Charlotte F, Salvatierra J, Wechsler B, Graux C, Brousse N, Piette JC. Imatinib mesylate for platelet-derived growth factor receptor-beta-positive Erdheim-Chester histiocytosis. Blood. 2008 Jun 1;111(11):5413-5. doi: 10.1182/blood-2008-03-148304[↩]
- Giorgio Zauli, Paola Secchiero; Role of TRAIL in osteoclastogenesis. Blood 2008; 111 (11): 5413. doi: https://doi.org/10.1182/blood-2008-03-145300[↩]
- Janku F, Amin HM, Yang D, Garrido-Laguna I, Trent JC, Kurzrock R. Response of histiocytoses to imatinib mesylate: fire to ashes. J Clin Oncol. 2010 Nov 1;28(31):e633-6. doi: 10.1200/JCO.2010.29.9073[↩][↩]
- Dagna L, Corti A, Langheim S, Guglielmi B, De Cobelli F, Doglioni C, Fragasso G, Sabbadini MG, Ferrarini M. Tumor necrosis factor α as a master regulator of inflammation in Erdheim-Chester disease: rationale for the treatment of patients with infliximab. J Clin Oncol. 2012 Oct 1;30(28):e286-90. doi: 10.1200/JCO.2012.41.9911[↩]
- Cohen-Aubart F, Maksud P, Emile JF, Benameur N, Charlotte F, Cluzel P, Amoura Z, Haroche J. Efficacy of infliximab in the treatment of Erdheim-Chester disease. Ann Rheum Dis. 2018 Sep;77(9):1387-1390. doi: 10.1136/annrheumdis-2017-212678[↩]
- FDA approves first drug to specifically treat giant cell arteritis. https://www.fda.gov/news-events/press-announcements/fda-approves-first-drug-specifically-treat-giant-cell-arteritis[↩]
- Berti A, Cavalli G, Guglielmi B, Biavasco R, Campochiaro C, Tomelleri A, Nicoletti R, Panzacchi A, Ferrarini M, Dagna L. Tocilizumab in patients with multisystem Erdheim-Chester disease. Oncoimmunology. 2017 Apr 20;6(6):e1318237. doi: 10.1080/2162402X.2017.1318237[↩]
- Goyal G, Shah MV, Call TG, Hook CC, Hogan WJ, Go RS. Clinical and radiological responses to oral methotrexate alone or in combination with other agents in Erdheim-Chester disease. Blood Cancer J. 2017 Dec 15;7(12):647. doi: 10.1038/s41408-017-0034-7[↩]
- Ho P, Smith C. High-dose methotrexate for the treatment of relapsed central nervous system erdheim-chester disease. Case Rep Hematol. 2014;2014:269359. doi: 10.1155/2014/269359[↩]
- Miller RC, Villà S, Kamer S, Pasquier D, Poortmans P, Micke O, Call TG. Palliative treatment of Erdheim-Chester disease with radiotherapy: a Rare Cancer Network study. Radiother Oncol. 2006 Sep;80(3):323-6. doi: 10.1016/j.radonc.2006.07.034[↩]
- Cives M, Simone V, Rizzo FM, et al. Erdheim-Chester disease: a systematic review. Crit Rev Oncol Hematol. 2015;95:1–11. doi: 10.1016/j.critrevonc.2015.02.004[↩]
- Haroche J, Arnaud L, Cohen-Aubart F, et al. Erdheim-Chester disease. Rheum Dis Clin North Am. 2013;39:299–311. doi: 10.1016/j.rdc.2013.02.011[↩]
- Cavalli G, Guglielmi B, Berti A, et al. The multifaceted clinical presentations and manifestations of Erdheim-Chester disease: comprehensive review of the literature and of 10 new cases. Ann Rheum Dis. 2013;72:1691–5. doi: 10.1136/annrheumdis-2012-202542[↩]
- Lachenal F, Cotton F, Desmurs-Clavel H, et al. Neurological manifestations and neuroradiological presentation of Erdheim-Chester disease: report of 6 cases and systematic review of the literature. J Neurol. 2006;253:1267–77. doi: 10.1007/s00415-006-0160-9[↩]
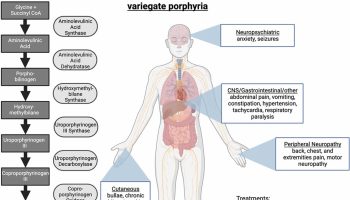
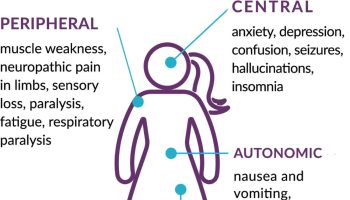

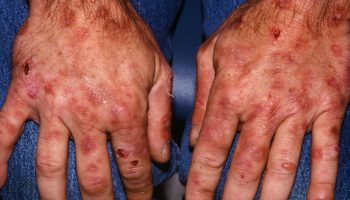
{kind=link}