Contents
- What is Ewing’s sarcoma
- Ewing’s sarcoma causes
- Ewing’s sarcoma signs and symptoms
- Ewing’s sarcoma complications
- Ewing’s sarcoma diagnosis
- Ewing’s sarcoma differential diagnosis
- Ewing’s sarcoma stage
- Ewing’s sarcoma treatment
- Ewing’s sarcoma treatment by stage
- Ewing’s sarcoma prognosis
- Ewing’s sarcoma survival rate
What is Ewing’s sarcoma
Ewing sarcoma also called Ewing’s sarcoma or Ewing’s tumor is a type of rare cancer that forms in your bone or soft tissue, most often affecting children and adolescents (between the ages of 10 and 20 years), though it can occur at any age 1, 2. Ewing’s sarcoma primarily develops in the long bones of the legs, the pelvis, the scapula (shoulder blade), the ribs and the vertebrae (bones that protect your spinal cord and make up your spinal column). Usually, Ewing sarcoma originates in a bone, but occasionally, the tumor begins in your muscles, soft tissues, or other organs where it is called extraosseous Ewing sarcoma. Ewing’s sarcoma symptoms include pain and swelling at the tumor site, and potentially fever. In some cases, the affected bone may fracture. Ewing sarcoma diagnosis requires a biopsy where a tissue sample is taken and tested in a laboratory to confirm the presence of cancer cells. Ewing’s sarcoma treatment typically involves a multidisciplinary approach with a team of medical specialists such as medical and orthopedic oncologists, working together to create a treatment plan. Ewing sarcoma treatment may include a combination of surgery to remove the cancer, chemotherapy and radiation therapy. Your treatment depends on the stage of your cancer, your age and sex, and results of the tests on the biopsy sample. In the past, Ewing sarcoma is associated with high mortality rates despite treatments; however, improvements and major advances in local therapy and multiagent adjuvant (add-on) chemotherapy have substantially improved the 5-year survival rate from less than 20% to over 70% 1. Young people diagnosed with Ewing sarcoma are living longer. They sometimes face late effects from the strong cancer treatments. Doctors often suggest long-term monitoring for side effects after treatment. Nevertheless, challenges persist, particularly concerning Ewing sarcoma recurrence rate.
Ewing sarcoma is a very rare form of cancer, accounting for approximately 10% to 15% of primary bone cancers and less than 1% of all childhood cancers 3, 4. Approximately 3 per 1 million children each year are diagnosed with a Ewing sarcoma 5. There are only about 200 to 300 new cases each year in the United States. Although it is rare compared to other types of cancer, Ewing sarcoma is the second most common bone cancer in children and young adults, the most common type of bone cancer is called osteosarcoma. According to data on children younger than 15 years old, approximately 1.7 children out of 1 million develop Ewing sarcoma.
Ewing sarcoma belongs to a family of small blue cell (Round cell) tumors due to its appearance under a microscope. Recently, doctors have defined Ewing’s sarcoma to include 4 types of cancer, referred to as the Ewing family of tumors (EFTs):
- Ewing sarcoma of bone
- Extraosseous Ewing sarcoma. You can also get Ewing sarcoma in the soft tissues of the body. Soft tissues are the connective and supporting tissues in the body. These include:
- fat
- muscle
- blood vessels
- deep skin tissues
- nerves
- tendons and ligaments
- the tissues around the joints
- Peripheral primitive neuroectodermal tumor (pPNET), which may occur in both bone and soft tissue
- Askin’s tumor, a peripheral primitive neuroectodermal tumor (pPNET) that occurs in the bones of the chest
Approximately 87 percent of Ewing sarcomas are Ewing sarcoma of bone, which is a bone tumor that usually occurs in the thigh bones (femurs), pelvis, ribs, or shoulder blades (scapulae). Extraosseous (or extraskeletal) Ewing sarcoma describes tumors in the soft tissues around bones, such as cartilage. Peripheral primitive neuroectodermal tumor (pPNET) occur in nerve tissue and can be found in many parts of the body. A type of peripheral primitive neuroectodermal tumor (pPNET) found in the bones of the chest is called Askin tumor.
Ewing sarcomas most often occur in children and young adults. Affected individuals usually feel stiffness, pain, swelling, or tenderness of the bone or surrounding tissue. Sometimes, there is a lump near the surface of the skin that feels warm and soft to the touch. Often, children have a fever that does not go away. Ewing sarcoma of bone can cause weakening of the involved bone, and affected individuals may have a broken bone with no obvious cause.
It is common for Ewing sarcoma to spread to other parts of the body (metastasize), usually to the lungs, to other bones, or to the bone marrow.
Researchers do not know what causes Ewing sarcoma. Ewing sarcoma happens when cells develop changes in their DNA. A cell’s DNA holds the instructions that tell a cell what to do. In healthy cells, the DNA gives instructions to grow and multiply at a set rate. The instructions tell the cells to die at a set time. In cancer cells, the DNA changes give different instructions. The changes tell the cancer cells to make many more cells quickly. Cancer cells can keep living when healthy cells would die. Nearly all Ewing tumor cells have changes that involve the EWSR1 gene, which is found on chromosome 22. Most often, the change is a swapping of pieces of DNA called a translocation between chromosomes 22 and 11, written as t(11;22), which fuses part of the EWSR1 gene with part of the FLI1 gene, creating the EWSR1/FLI1 fusion gene. Less often, the swap is between chromosomes 22 and 21, or rarely, between 22 and another chromosome. The translocation moves a certain piece of chromosome 11 (or another chromosome) just next to the EWSR1 gene on chromosome 22, causing the EWSR1 gene to be turned on all the time. Activation of the EWSR1 gene leads to overgrowth of the cells and to the development of Ewing sarcomas, but the exact way in which this happens is not yet clear.
In a very small portion of Ewing sarcomas, the cells have translocations that involve the FUS gene (on chromosome 16) instead of the EWSR1 gene.
The gene changes that lead to Ewing tumors are now fairly well known, but it’s still not clear what causes these changes. They might just be random events that sometimes happen inside a cell, without having an outside cause. There are no known lifestyle-related or environmental causes of Ewing tumors, so it’s important to remember that at this time, nothing could have been done to prevent these cancers.
Ewing sarcoma treatment most often includes chemotherapy and surgery. Which treatment you have first will depend on your situation. Other treatment options might include radiation therapy and targeted therapy.
Figure 1. Ewing sarcoma
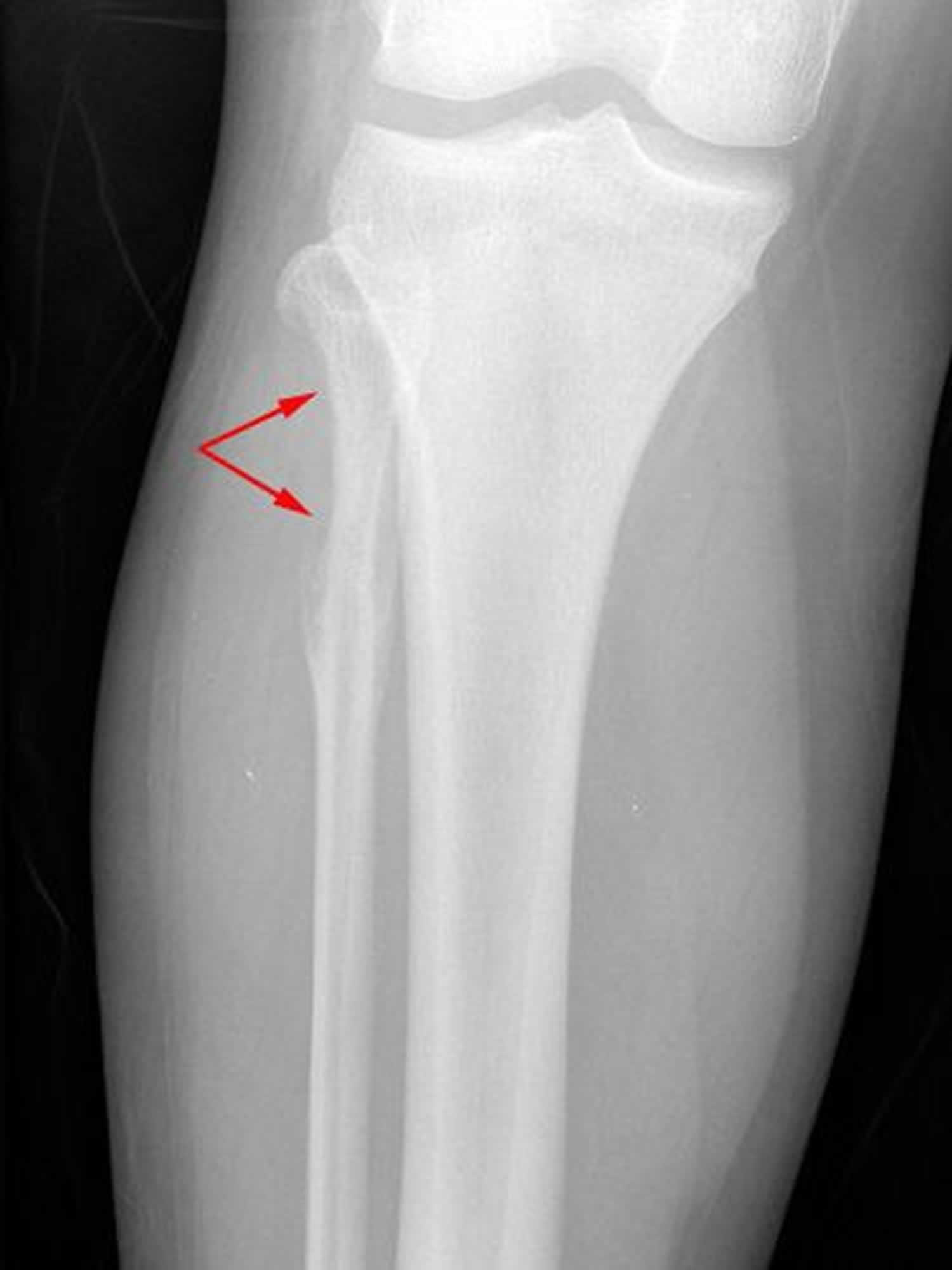
Footnotes: This X-ray shows the knee of a 14-year-old girl with Ewing sarcoma. The tumor is toward the top of the fibula and has destroyed a portion of the bone.
[Source 6 ]Figure 2. Extraskeletal Ewing sarcoma
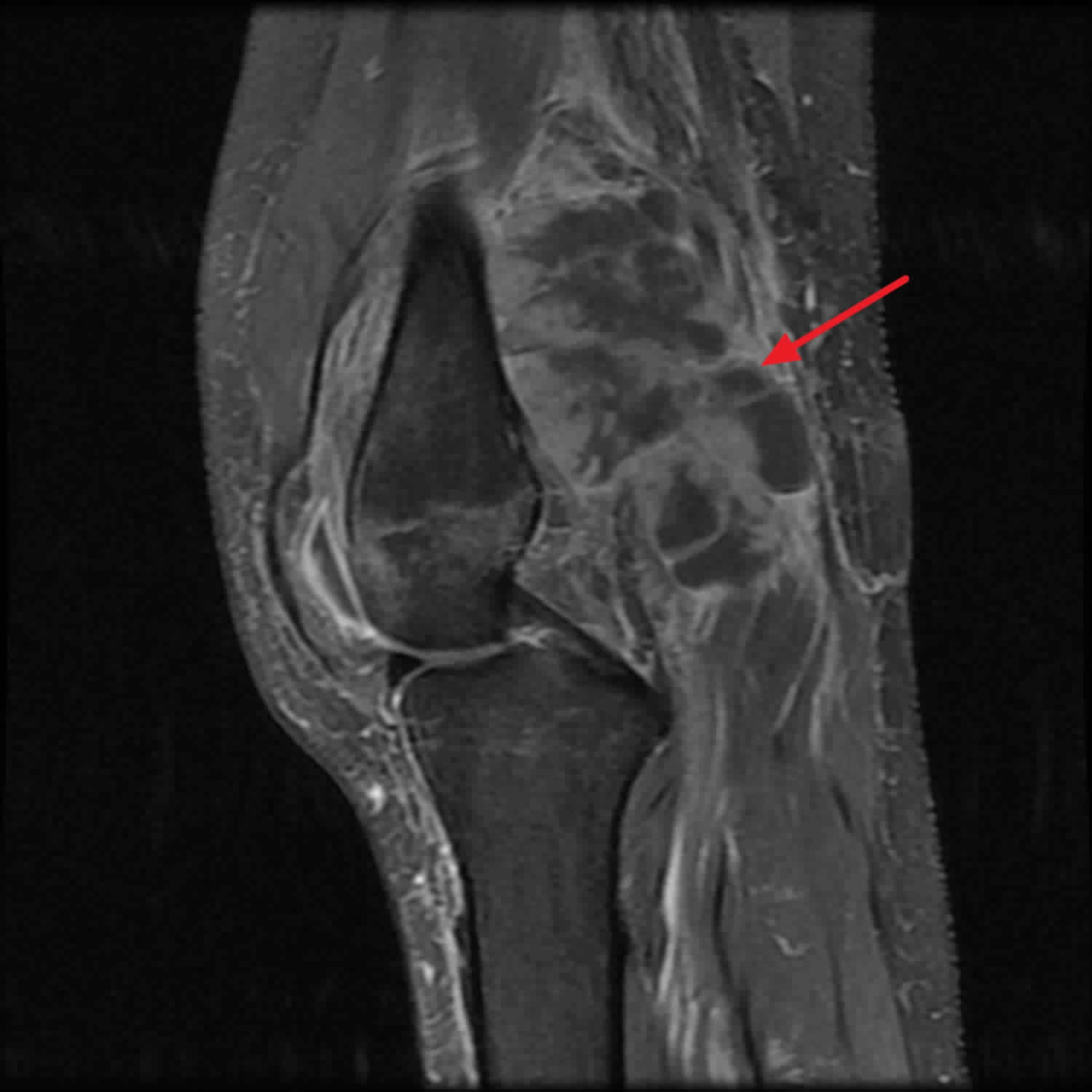
Footnotes: Popliteal fossa-centered mass with extension medially at the level of the medial femoral condyle. Medial aspect involves portions of the medial retinaculum as well as the vastus medialis muscle. The proximal medial collateral ligament complex as well as the medial head of the gastrocnemius are involved with the lesion. Part of the mass is inseparable from underlying periosteum in the posterior medial femoral epicondylar region. Patchy osteopenia of the osseous structures, likely secondary to disuse. Small knee effusion is present.
[Source 7 ]Figure 3. Ewing sarcoma of the chest wall (Askin’s tumor)
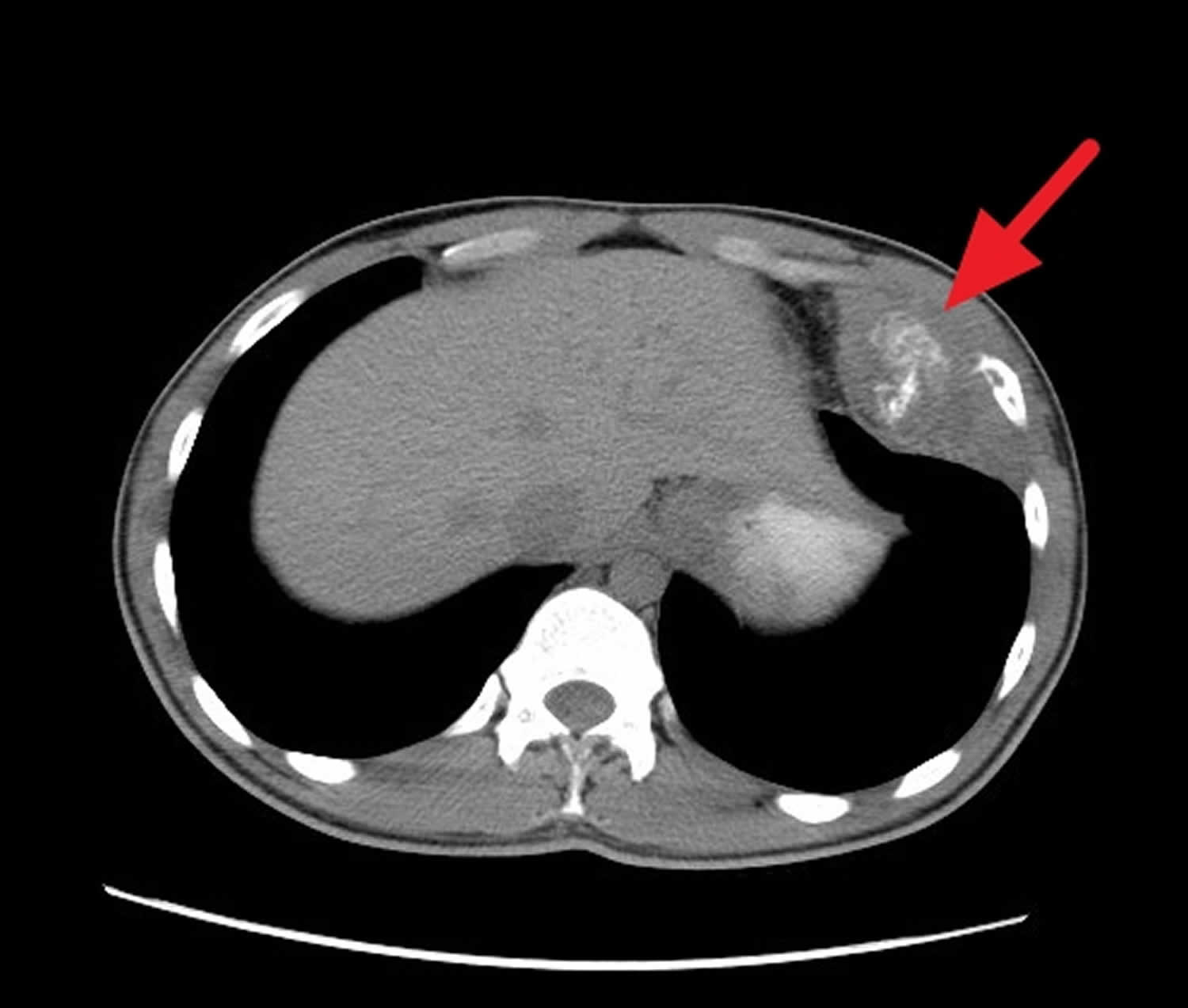
Footnotes: A 20 year old man presenting to his physician with growing chest wall mass. Further evaluation by chest CT with contrast was requested showing lesion arising from the left eleventh rib with oval-shaped ill-defined heterogeneous enhancing soft tissue mass lesion with areas of cystic degeneration and containing calcification is seen arising from the 11th rib antero-medially. It measures around eight by seven by three centimeters in dimensions. Periosteal reaction to the adjacent rib is also seen. Both lung fields appear clear without evidence of metastasis. Hila and mediastinum appear normal without evidence of lymphadenopathy or mass lesions. The liver and spleen appear normal in size and homogenous without evidence of focal lesion. Kidneys, adrenal glands, and pancreas appear normal without evidence of focal lesions. No evidence of significant retroperitoneal, abdominal or pelvic lymphadenopathy or fluid collection. Biopsy of this left chest wall mass was performed and confirmed Ewing’s sarcoma of the chest wall.
[Source 8 ]Ewing’s sarcoma causes
Researchers do not know what causes Ewing sarcoma. Ewing sarcoma happens when cells develop changes in their DNA. A cell’s DNA holds the instructions that tell a cell what to do. In healthy cells, the DNA gives instructions to grow and multiply at a set rate. The instructions tell the cells to die at a set time. In cancer cells, the DNA changes give different instructions. The changes tell the cancer cells to make many more cells quickly. Cancer cells can keep living when healthy cells would die. In Ewing sarcoma, the DNA changes most often affect two genes, the EWSR1 gene on chromosome 22 and the FLI1 gene on chromosome 11 5. A rearrangement (translocation) of genetic material between chromosomes 22 and 11, written as t(11;22), fuses part of the EWSR1 gene with part of the FLI1 gene, creating the EWSR1/FLI1 fusion gene 5. This mutation is acquired during a person’s lifetime and is present only in tumor cells. This type of genetic change, called a somatic mutation, is not inherited 5. The EWSR1/FLI1 fusion gene occurs in approximately 85 percent of Ewing sarcomas. Translocations that fuse the EWSR1 gene with other genes that are related to the FLI1 gene can also cause these types of tumors, although these alternative translocations are relatively uncommon. The fusion proteins produced from the less common gene translocations have the same function as the EWS/FLI protein.
The protein produced from the EWSR1/FLI1 fusion gene called EWS/FLI protein has functions of the protein products of both genes 5. The FLI protein, produced from the FLI1 gene, attaches (binds) to DNA and regulates an activity called transcription, which is the first step in the production of proteins from genes. The FLI protein controls the growth and development of some cell types by regulating the transcription of certain genes. The EWS protein, produced from the EWSR1 gene, also regulates transcription. The EWS/FLI protein has the DNA-binding function of the FLI protein as well as the transcription regulation function of the EWS protein. It is thought that the EWS/FLI protein turns the transcription of a variety of genes on and off abnormally 5. This dysregulation of transcription leads to uncontrolled growth and division (proliferation) and abnormal maturation and survival of cells, causing tumor development 5.
In a very small portion of Ewing sarcomas, the cells have translocations that involve the FUS gene (on chromosome 16) instead of the EWSR1 gene.
If your doctor suspects that you or your child has Ewing sarcoma, the cancer cells may be tested to look for changes in the EWSR1 gene or EWSR1/FLI1 fusion gene.
The gene changes that lead to Ewing tumors are now fairly well known, but it’s still not clear what causes these changes. Doctors have not identified any risk factors that make one person more at risk than another. Ewing sarcoma does not develop as a result of any dietary, social, or behavioral habits. There are no known ways to prevent Ewing sarcoma, and patients or their parents should know that there is nothing they could have done differently to prevent these tumors.
Risk factors for developing Ewing sarcoma
Risk factors for Ewing sarcoma may include:
- Young age. Ewing’s sarcoma can happen at any age. But it is more likely to happen in children and young adults.
- European ancestry. Ewing’s sarcoma is more common in people of European ancestry. It’s much less common in people of African and East Asian ancestry.
- Having Fanconi anemia. Fanconi anemia is a rare inherited bone marrow failure characterized by progressive bone marrow failure, which leads to leads to a decrease in the production of all blood cells (aplastic anemia), increased cancer risk, and physical abnormalities 9, 10. Fanconi anemia is caused mutations in more than 20 genes, with 80 to 90 percent of cases of Fanconi anemia are due to mutations in one of three genes: FANCA, FANCC, and FANCG genes. These genes provide instructions for producing components of the FA core complex. Mutations in any of the genes associated with the FA core complex prevent the complex from functioning properly and disrupt the FA pathway. As a result, DNA damage is not repaired efficiently and interstrand cross-links (ICLs) build up over time. The interstrand cross-links (ICLs) impair DNA replication, which leads to either abnormal cell death due to an inability to make new DNA molecules or uncontrolled cell growth due to a lack of DNA repair. Cells that divide quickly, such as bone marrow cells and cells of the developing fetus, are particularly sensitive to problems with DNA replication. The death of these cells results in the decrease in blood cells and the physical differences that are seen in people with Fanconi anemia. When the buildup of errors in DNA leads to uncontrolled cell growth, affected individuals can develop acute myeloid leukemia or other cancers. Fanconi anemia typically presents in childhood with symptoms like extreme tiredness, frequent infections, and easy bruising, alongside physical signs such as skin pigmentation changes and skeletal malformations. Treatment includes supportive therapies, and a stem cell transplant is the only potentially curative option.
Ewing’s sarcoma signs and symptoms
Ewing’s sarcoma signs and symptoms typically start in and around a bone. Ewing’s sarcoma most often affects bones in your legs and pelvis.
When symptoms happen in and around a bone, they might include:
- A lump in your arm, leg, chest or pelvis that may feel soft or warm.
- Bone pain not caused by an injury that does not go away.
- Break in a bone (pathologic fracture). For example, a bone weakened by a tumor may break after a minor injury. This is called a pathologic fracture.
- Pain, swelling or tenderness at the site of the tumor.
The tumor may be present for many months before it becomes large enough to cause pain and swelling. In some cases, the first symptom of Ewing sarcoma is the presence of a mass.
Sometimes Ewing sarcoma causes symptoms that affect the whole body. These can include:
- Fever.
- Losing weight without trying.
- Tiredness (fatigue).
- Body aches.
Rarely, tumors near the spine can affect nearby nerves, which can lead to back pain, as well as weakness, numbness, or paralysis in the arms or legs.
Tumors that have spread to the lungs can cause shortness of breath.
If you are under 24 years age and have unexplained bone swelling or pain, you should have an urgent X-ray within 2 days. Your primary care physician should refer you to a orthopedic specialist within 2 days if the results of your x-ray suggest you might have a bone cancer.
Ewing’s sarcoma complications
Complications of Ewing sarcoma and its treatment include the following.
- Cancer that spreads (metastasis). Ewing sarcoma can spread from where it started to other areas. Ewing sarcoma most often spreads to your lungs and to other bones. At the time of diagnosis, spread is seen in about one third of children with Ewing sarcoma.
- Long-term side effects of treatments. The strong treatments needed to control Ewing sarcoma can cause major side effects, both in the short and long term. Your doctors (medical and orthopedic oncologists) can help you manage the side effects that happen during treatment. Your doctors also can give you a list of side effects to watch for in the years after treatment.
Ewing’s sarcoma diagnosis
Ewing sarcomas are usually found because of signs or symptoms a person is having. If the doctor suspects a tumor, exams and tests will be needed to find out for sure.
Ewing sarcoma diagnosis usually begins with a physical exam. Based on the findings of the exam, there might other tests and procedures.
If a Ewing tumor is found, other tests will then be needed to learn more about it.
Imaging tests
Imaging tests make pictures of the body. They can show the location and size of a Ewing sarcoma. Tests might include:
- X-ray of the tumor. If a lump in or near a bone doesn’t go away or if the doctor suspects a bone tumor for some other reason, an x-ray of the area will probably be the first test done. Doctors can usually spot a bone tumor on an x-ray and can often tell if it is likely to be a Ewing sarcoma. But other imaging tests might be needed as well. Even if an x-ray image strongly suggests a Ewing tumor, a biopsy (described below) will still be needed to confirm that it is cancer rather than some other problem, such as an infection.
- MRI of the tumor. MRI (magnetic resonance imaging) uses a magnet, radio waves, and a computer to make a series of detailed pictures of areas inside the body, such as the area where the tumor formed. A contrast material called gadolinium is often injected into a vein before the scan to help see details better. An MRI is often done to get a better look at an abnormal area seen on an x-ray. An MRI usually can show if it is likely to be a tumor, an infection, or some type of bone damage from another cause. MRIs can also help determine the extent of a tumor, as they show the marrow inside bones as well as the muscle, fat, and connective tissue around the tumor. Knowing the extent of the tumor is very important when planning surgery or radiation therapy. MRI scans might also be done to see if the cancer has spread to other areas, such as the spine or pelvis (hip area).
- CT scan of the chest. A CT scan combines many x-ray pictures to make detailed cross-sectional images of parts of the body, including soft tissues such as muscles. A contrast material may be injected into a vein before the scan to help see details better. CT scans of the chest are often used to see if a Ewing sarcoma has spread to your lungs. MRI scans are usually a bit better at showing the extent of the main tumor itself, but a CT scan of the tumor may be done as well.
- Bone scan. For a bone scan, a small amount of low-level radioactive material is injected into the blood and travels to the bones. A special camera that can detect the radioactivity then creates a picture of the skeleton. Areas of active bone changes attract the radioactivity and appear as “hot spots” on the skeleton. These areas may suggest the presence of cancer, but other bone diseases can also cause the same pattern. To be sure, other tests such as plain x-rays or MRI scans, or even a bone biopsy, might be needed. A bone scan can help show if a cancer has spread to bones in other parts of your body, so it might be part of the workup for a Ewing sarcoma. This test is useful because it can show the entire skeleton at once. A positron emission tomography (PET) scan can often provide similar information, so a bone scan might not be needed if a PET scan is done.
- Positron emission tomography scan (PET scan). For a PET scan, a form of radioactive sugar (known as fluorodeoxyglucose [FDG]) is injected into the blood. Because cancer cells are growing quickly, they absorb large amounts of the sugar. A special camera can then create a picture of areas of radioactivity in the body. The picture is not detailed like a CT or MRI scan, but it provides helpful information about the whole body. PET scans can be very helpful in showing the spread of Ewing sarcomas and in finding out whether abnormal areas seen on other imaging tests (such as a CT scan) are tumors. PET scans can also be repeated during treatment to see how well it is working. Many machines can do a PET and CT scan at the same time (PET/CT scan). This lets the doctor compare areas of higher radioactivity on the PET scan with the more detailed appearance of that area on the CT scan.
Biopsy
A biopsy of the tumor will be done for testing. A biopsy is a procedure to remove a sample of tissue for testing in a lab. The tissue might be removed using a needle that is put through the skin and into the cancer. Sometimes surgery is needed to get the tissue sample. The sample is tested in a lab to see if it is cancer. Other special tests give more details about the cancer cells to help determine how aggressive the cancer is and what treatment may be best.
Testing the cancer cells for DNA changes
A sample of the cancer cells will be tested in the lab to find which DNA changes are in the cells. Ewing sarcoma cells mostly have changes in the EWSR1 gene. Most often the EWSR1 gene joins with another gene called FLI1. This creates a new gene called EWSR1/FLI1 fusion gene.
CD99 is a surface membrane protein that is expressed in most cases of Ewing sarcoma and is useful in diagnosing these tumors when the results are interpreted in the context of clinical and pathological parameters 11. CD99 positivity is not unique to Ewing sarcoma, and positivity by immunochemistry is found in several other tumors, including synovial sarcoma, non-Hodgkin lymphoma, and gastrointestinal stromal tumors. NKX2.2 is a nuclear antigen that is also commonly assessed by immunohistochemistry to support a diagnosis of Ewing sarcoma, although it is also not 100% specific for this diagnosis 12.
Testing the cancer cells for these gene changes and tumor markers can help confirm your diagnosis 13.
Blood tests
No blood test can be used to diagnose Ewing sarcomas. But certain blood tests may be helpful once a diagnosis has been made.
A complete blood count (CBC) measures the levels of white blood cells, red blood cells, and platelets in the blood. An abnormal complete blood count (CBC) result (that is, low blood cell counts) might suggest the cancer has spread to the bone marrow, where these blood cells are made.
A blood test for levels of an enzyme called lactate dehydrogenase (LDH) is typically done at diagnosis. A high LDH level is often a sign that there is more cancer in the body.
Standard blood tests are done often to check a patient’s general health both before treatment (especially before surgery) and during treatment (such as chemotherapy) to look for possible problems or side effects. These tests often include a CBC to check blood cell levels and blood chemistry tests to measure how well the liver and kidneys are working.
Ewing’s sarcoma differential diagnosis
Symptoms of the following disorders can be similar to those of Ewing sarcoma. Comparisons may be useful for a differential diagnosis. Differential diagnosis of Ewing sarcoma includes other small round cell tumors such as neuroblastoma, rhabdomyosarcoma, lymphoma, neuroectodermal tumors, desmoplastic small round cell tumor, and synovial sarcoma. Additional tumors must also be differentiated from Ewing sarcoma including chondrosarcomas, osteochondromas, medulloblastomas, neuroblastoma, rhabdomyosarcoma, and lymphoma of bone.
Osteosarcoma is a tumor affecting the bones. Osteosarcoma is the most common form of bone cancer. Approximately 60 percent of cases occur in children and adolescents during the second decade of life. Osteosarcomas affect males twice as often as females. The bones most commonly affected are the long bones of the arms and legs. Symptoms may vary depending upon the location and extent of the disease. Pain, swelling, tenderness and eventually the formation of a lump may occur in the affected area. General symptoms may include fever, weight loss, anemia, and lack of energy. Osteosarcomas may weaken the surrounding bone resulting in fractures. Osteosarcomas may spread (metastasize) to other areas of the body. The exact cause of osteosarcoma is unknown.
Osteomyelitis is a bone infection, usually caused by bacteria. Osteomyelitis can be either acute or chronic. Osteomyelitis usually occurs as a result of an infection in one part of the body that is transported through the bloodstream to a bone in a distant location. Among children and teens, the long bones of the legs and arms are most frequently affected. In adults, osteomyelitis most often affects the vertebrae of the spine and/or the hips. Initially there may be several days of fever and a generalized feeling of ill health (malaise). This may be followed by an increase in fever, deep localized bone pain, chills, sweating, swelling and painful or limited movement of the nearby joints. The skin near the affected bone may be red (erythema) and there may be pus, destruction of the surrounding tissue (necrosis) and bone deterioration or deformity.
Eosinophilic granuloma is a subdivision of a rare spectrum of disorders known as Langerhans cell histiocytosis (LCH). Langerhans cell histiocytosis (LCH) is a rare, cancer-like disorder or a myeloid-derived dendritic cell disorder where the body produces too many Langerhans cells (histiocytes), a type of immune cell that can build up various tissues and organs of the body and cause damage to tissues, bones, and organs 14. Langerhans cell histiocytosis (LCH) can affect many parts of the body, but is most common in children and often involves the skin and bones. It is caused by gene mutations and is not inherited. Symptoms and severity vary widely depending on whether the disease affects a single system or multiple systems. It can manifest as a single lesion, such as a bone lesion (eosinophilic granuloma), or as a serious, multi-organ systemic disease affecting the skin, bones, liver, spleen, lungs, lymph nodes, and the hypothalamic-pituitary axis. Prognosis varies greatly depending on the number of involved organs and the presence of dysfunction in high-risk organs like the liver, spleen, and bone marrow. Treatment approaches vary from observation to systemic chemotherapy, corticosteroids, and local therapy.
Ewing’s sarcoma stage
Once a Ewing sarcoma has been diagnosed, tests are done to determine the stage (extent) of the cancer. Staging means how big the cancer is and whether it has spread. The stage is based on results of your imaging tests and biopsies of the main tumor and any other body tissues. Grading means how abnormal the cancer cells look under a microscope. Doctors use the stage and grade of bone cancer to help them decide which treatment you need. Doctors also use a cancer’s stage when talking about survival statistics.
The tests and scans you have to diagnose your cancer give some information about the stage. In bone cancer, staging also takes into account how abnormal the cells look under the microscope (the grade).
Your doctors and surgeons might use one of 2 different systems to stage your bone cancer. These are:
- American Joint Committee on Cancer (AJCC) TNM (Tumor, Node and Metastasis) staging system
- Localized vs. metastatic Ewing tumors
Your doctors and surgeons will explain which one they are using and what this means for you.
Staging and grading for bone cancer is complicated. The most common system doctors use is the American Joint Committee on Cancer (AJCC) TNM system.
Ewing tumor stages can be confusing, so be sure to ask someone on the health care team if you have any questions about the stage of the cancer.
TNM (Tumor, Node and Metastasis) staging system for bone cancers
The American Joint Committee on Cancer (AJCC) uses the TNM system to describe all bone cancers, including Ewing tumors that start in bone.
The AJCC TNM staging system for bone cancers is based on 4 key pieces of information:
- T describes the size of the main (primary) tumor and whether it appears in different areas of the bone.
- N describes the extent of spread to nearby (regional) lymph nodes (small bean-sized collections of immune system cells). Bone tumors rarely spread to the lymph nodes.
- M indicates whether the cancer has metastasized (spread) to other organs of the body. (The most common sites of spread are to the lungs or other bones.)
- G stands for the grade of the tumor, which describes how the cells from biopsy samples look. Low-grade tumor cells look more like normal cells and are less likely to grow and spread quickly, while high-grade tumor cells look more abnormal. All Ewing sarcomas are considered high-grade [G3] tumors.
Once the T, N, M, and G categories have been determined, the information is combined and expressed as an overall stage. The process of assigning a stage number is called stage grouping. The stages are described in Roman numerals from I to IV (1 to 4), and are sometimes divided further.
The following is a summary of the staging for bone cancer. There is no number staging for bone cancer that starts in the back (spine) and pelvis.
- Stage 1A. The cancer is 8 cm or less across. It has not spread to the lymph nodes or other parts of the body. The cancer is low grade or the grade can’t be assessed.
- Stage 1B. The cancer is more than 8 cm across, or is in more than one place in the same bone. It has not spread to the lymph nodes or other parts of the body. The cancer is low grade or the grade can’t be assessed.
- Stage 2A. The cancer is 8 cm or less across. It has not spread to the lymph nodes or other parts of the body. The cancer is high grade.
- Stage 2B. The cancer is more than 8 cm across. It has not spread to the lymph nodes or other parts of the body. The cancer is high grade.
- Stage 3. The cancer is in more than one place in the same bone. It has not spread to the lymph nodes or other parts of the body. The cancer is high grade.
- Stage 4A. The cancer is any grade and is either:
- any size
- in more than one place in the same bone
- There are no cancer cells in the lymph nodes and it has only spread to the lung.
- Stage 4B. This means the cancer has spread to the lymph nodes or to parts of the body such as the liver, other bones or the brain.
Musculoskeletal Tumor Society (MSTS) staging system for bone cancers
A system commonly used to stage bone cancer is the Musculoskeletal Tumor Society (MSTS) system, also known as the Enneking system. It is based on 3 key pieces of information:
- The grade (G) of the cancer, which is a measure of how likely it is to grow and spread, based on how it looks under the microscope. In this system, cancers are either low grade (G1) or high grade (G2). Low-grade cancer cells look more like normal cells and are less likely to grow and spread quickly, while high-grade cancer cells look more abnormal.
- The extent of the primary tumor (T), which is classified as either intracompartmental (T1), meaning it has basically remained within the bone, or extracompartmental (T2), meaning it has grown beyond the bone into other nearby structures.
- If the tumor has metastasized (M), which means it has spread to other areas, either to nearby lymph nodes (bean-sized collections of immune system cells) or other organs. Tumors that have not spread to the lymph nodes or other organs are considered M0, while those that have spread are M1.
These factors are combined to give an overall stage, using Roman numerals from I to III. Stages I and II are divided into A for intracompartmental tumors or B for extracompartmental tumors.
In summary:
- Low-grade, localized tumors are stage I.
- High-grade, localized tumors are stage II.
- Metastatic tumors (regardless of grade) are stage III.
| Stage | Grade | Tumor | Metastasis |
|---|---|---|---|
| IA | G1 | T1 | M0 |
| IB | G1 | T2 | M0 |
| IIA | G2 | T1 | M0 |
| IIB | G2 | T2 | M0 |
| III | G1 or G2 | T1 or T2 | M1 |
AJCC staging system for soft tissue sarcomas
Extraosseous Ewing sarcomas (Ewing tumors that don’t start in bones) are staged like soft tissue sarcomas. The American Joint Committee on Cancer (AJCC) staging system for soft tissue sarcomas is based on 4 key pieces of information:
- T describes the size of the main (primary) tumor.
- N describes the extent of spread to nearby (regional) lymph nodes (small bean-sized collections of immune system cells).
- M indicates whether the cancer has metastasized (spread) to other organs of the body.
- G stands for the grade of the tumor, which describes how the cells from biopsy samples look. Low-grade tumor cells look more like normal cells and are less likely to grow and spread quickly, while high-grade tumor cells look more abnormal. All Ewing sarcomas are considered high-grade [G3] tumors.
Numbers or letters after T, N, M, and G provide more details about each of these factors.
Once the T, N, M, and G categories have been determined, the information is combined and expressed as an overall stage. The process of assigning a stage number is called stage grouping. The stages are described in Roman numerals from I to IV (1 to 4), and are sometimes divided further.
The grade is partly used to determine the stage of a sarcoma. The staging system divides sarcomas into 3 grades (1 to 3). The grade of a sarcoma helps predict how rapidly it will grow and spread. It’s useful in predicting a patient’s outlook and helps determine treatment options.
The grade of a sarcoma is determined using a system known as the French or FNCLCC system, and is based on 3 factors 16:
- Differentiation: Cancer cells are given a score of 1 to 3, with 1 being assigned when they look a lot like normal cells and 3 being used when the cancer cells look very abnormal. Certain types of sarcoma are given a higher score automatically.
- Mitotic count: How many cancer cells are seen dividing under the microscope; given a score from 1 to 3 (a lower score means fewer cells were seen dividing)
- Tumor necrosis: How much of the tumor is made up of dying tissue; given a score from 0 to 2 (a lower score means there was less dying tissue present).
Each factor is given a score, and the scores are added to determine the grade of the tumor. Sarcomas that have cells that look more normal and have fewer cells dividing are generally placed in a low-grade category. Low-grade tumors tend to be slow growing, slower to spread, and often have a better outlook (prognosis) than higher-grade tumors. Certain types of sarcoma are automatically given higher differentiation scores. This affects the overall score so much that they are never considered low grade. Here’s what the grade numbers mean:
- GX: The grade cannot be assessed (because of incomplete information).
- Grade 1 (G1): Total score of 2 or 3
- Grade 2 (G2): Total score of 4 or 5
- Grade 3 (G3): Total score of 6, 7 or 8
All Ewing sarcomas are considered high-grade [G3] tumors.
Table 1. Trunk and Extremities Sarcoma Stages
| American Joint Committee on Cancer (AJCC) stage | Stage grouping | Trunk and Extremities Sarcoma Stage description* |
|---|---|---|
| IA | T1 N0 M0 G1 or GX | The cancer is 5 cm (2 inches) or smaller (T1). It has not spread to nearby lymph nodes (N0) or to distant sites (M0). The cancer is grade 1 (G1) or the grade cannot be assessed (GX). |
| IB | T2, T3, T4 N0 M0 G1 or GX | The cancer is: Larger than 5 cm but not more than 10 cm (T2) OR Larger than 10cm but not more than 15 cm (T3) OR Larger than 15 cm (T4). It has not spread to nearby lymph nodes (N0) or to distant sites (M0). The cancer is grade 1 (G1) or the grade cannot be assessed (GX). |
| II | T1 N0 M0 G2 or G3 | The cancer is 5 cm (2 inches) or smaller (T1). It has not spread to nearby lymph nodes (N0) or to distant sites (M0). The cancer is grade 2 (G2) or grade 3 (G3). |
| IIIA | T2 N0 M0 G2 or G3 | The cancer is larger than 5 cm (2 inches) but not more than 10 cm (T2). It has not spread to nearby lymph nodes (N0) or to distant sites (M0). The cancer is grade 2 (G2) or grade 3 (G3). |
| IIIB | T3 or T4 N0 M0 G2 or G3 | The cancer is: Larger than 10 cm but not more than 15 cm (T3) OR Larger than 15 cm (T4). It has not spread to nearby lymph nodes (N0) or to distant sites (M0). The cancer is grade 2 (G2) or grade 3 (G3). |
| IV | Any T N1 M0 Any G | The cancer is any size (Any T) AND it has spread to nearby lymph nodes (N1). It has not spread to distant sites (M0). It can be any grade. |
| OR | ||
| Any T Any N M1 Any G | The cancer is any size (Any T) AND it has spread to nearby lymph nodes (N1). It has spread to distant sites such as the lungs (M1). It can be any grade. | |
Footnotes: * The following categories are not listed in the table above:
- TX: Main tumor cannot be assessed due to lack of information.
- T0: No evidence of a primary tumor.
- NX: Regional lymph nodes cannot be assessed due to lack of information.
Table 2. Retroperitoneum Sarcoma Stages
| American Joint Committee on Cancer (AJCC) stage | Stage grouping | Retroperitoneum Sarcoma Stage description* |
|---|---|---|
| IA | T1 N0 M0 G1 or GX | The cancer is 5 cm (2 inches) or smaller (T1). It has not spread to nearby lymph nodes (N0) or to distant sites (M0). The cancer is grade 1 (G1) or the grade cannot be assessed (GX). |
| IB | T2, T3, T4 N0 M0 G1 or GX | The cancer is: Larger than 5 cm but not more than 10 cm OR Larger than 10 cm but not more than 15 cm (T3) OR Larger than 15 cm (T4). It has not spread to nearby lymph nodes (N0) or to distant sites (M0). The cancer is grade 1 (G1) or the grade cannot be assessed (GX). |
| II | T1 N0 M0 G2 or G3 | The cancer is 5 cm (2 inches) or smaller (T1). It has not spread to nearby lymph nodes (N0) or to distant sites (M0). The cancer is grade 2 (G2) or grade 3 (G3). |
| IIIA | T2 N0 M0 G2 or G3 | The cancer is larger than 5 cm (2 inches) but not more than 10 cm (T2). It has not spread to nearby lymph nodes (N0) or to distant sites (M0). The cancer is grade 2 (G2) or grade 3 (G3). |
| IIIB | T3 or T4 N0 M0 G2 or G3 | The cancer is: Larger than 10 cm but not more than 15 cm (T3) OR Larger than 15 cm (T4). It has not spread to nearby lymph nodes (N0) or to distant sites (M0). The cancer is grade 2 (G2) or grade 3 (G3). |
| OR | ||
| Any T N1 M0 Any G | The cancer is any size (Any T) AND it has spread to nearby lymph nodes (N1). It has not spread to distant sites (M0). It can be any grade. | |
| IV | Any T Any N M1 Any G | The cancer is any size (Any T) AND it has spread to nearby lymph nodes (N1). It has spread to distant sites such as the lungs (M1). It can be any grade. |
Footnotes: * The following categories are not listed in the table above:
- TX: Main tumor cannot be assessed due to lack of information.
- T0: No evidence of a primary tumor.
- NX: Regional lymph nodes cannot be assessed due to lack of information.
Localized vs. metastatic Ewing sarcomas
When determining how best to treat Ewing sarcomas, doctors typically classify them as either localized or metastatic.
Localized Ewing tumors
Doctors call a Ewing sarcoma localized if it can only be detected in the area where it started or in nearby tissues such as muscle or tendons. A Ewing tumor is considered localized only after all tests have been done including imaging tests such as x-rays, CT or MRI scans, and PET or bone scans, and possibly a bone marrow biopsy, and they don’t show the cancer has spread to distant parts of the body.
Even when imaging tests don’t show that the cancer has spread to distant areas, most patients are likely to have micrometastases (very small areas of cancer spread that can’t be detected with tests). This is why chemotherapy, which can reach all parts of your body, is an important part of treatment for all Ewing sarcomas.
Metastatic Ewing tumors
A metastatic Ewing sarcoma has clearly spread from where it started to distant parts of the body. Most of the time, it spreads to your lungs or to other bones or your bone marrow. Less commonly, it spreads to your liver or lymph nodes.
About 1 in 5 patients will have obvious spread that is found by imaging tests. Many other patients are likely to have small amounts of cancer spread to other parts of the body that can’t be seen on imaging tests.
Grading bone cancer
The grade of bone cancer gives your doctor a guide to how the cancer may behave.
Low grade cancers have cells that look very much like normal cells. These cancers usually grow slowly and are less likely to spread.
High grade cancers have cells that look very abnormal. These cancers generally grow more quickly and are more likely to spread.
In the American Joint Committee on Cancer (AJCC) TNM system, all Ewing tumors are considered high-grade [G3] tumors.
Ewing’s sarcoma treatment
Ewing sarcoma treatment most often includes chemotherapy and surgery. Which treatment you have first will depend on your situation. Other treatment options might include radiation therapy and targeted therapy.
Surgery
The goal of surgery is to remove all the cancer cells. Surgery for Ewing sarcoma might mean removing a small portion of bone and some surrounding tissue. Rarely, it might mean removing the affected arm or leg.
Surgery on an arm or leg might affect the way you can use that limb. Surgeons carefully plan the surgery to minimize this risk, when possible.
Whether surgeons can remove all the cancer without removing the arm or leg depends on several factors. These include the size of the cancer, where it is and whether chemotherapy helps shrink it.
Whenever possible, it’s very important that the biopsy and the surgery to remove the tumor be planned together, and that an experienced orthopedic surgeon at a cancer center does both operations. The biopsy needs to be done in a certain way so that there is a better chance less extensive surgery will be needed later on.
Many types of surgery can be used for Ewing sarcomas. The choice depends on several factors, including:
- The tumor’s size and location
- Your age and overall health
- How likely it is that surgery can remove the tumor with clean margins
- How surgery would change the function of the affected part of the body
Tumors in some soft tissues and certain bones can be removed without causing major disability or deformity. Other tumors might not be able to be removed completely without affecting the function or appearance of that part of the body.
Although all operations to remove Ewing sarcomas are complex, tumors in the arms or legs are generally not as hard to remove as those in other parts of the body, such as the base of the skull, the chest wall, the spine, or the pelvis (hip bones).
Possible side effects of surgery for Ewing sarcomas
- Short-term risks and side effects: Surgery to remove a Ewing tumor is often a long and complex operation. Serious short-term side effects are not common, but they can include reactions to anesthesia, excess bleeding, blood clots, and infections. Pain is common after the operation, and strong pain medicines might be needed for a while after surgery as the site heals.
- Long-term side effects: The long-term side effects of surgery depend mainly on where the tumor is and what type of operation is done.
Complications of limb-sparing surgery can include possible breaking or loosening of bone grafts or prostheses. This is more likely than with bone surgery done for other reasons because the chemo used before and after surgery can increase the risk of infection and affect wound healing. Infections in the area can be very serious because they can be hard to treat, and might require further surgery. Infections are also a concern in people who have had amputations, especially of part of a leg, because the pressure placed on the skin at the site of the amputation can cause the skin to break down over time.
Ewing sarcomas in the arms or legs
For most Ewing sarcomas in an arm or leg, surgery can remove part or all of the affected bone while leaving the arm or leg basically intact. This is known as limb-sparing surgery or limb-salvage surgery. The bone that is removed is replaced either with a bone graft (a piece of bone from another part of the body or from another person) or with an internal prosthesis (a rod-shaped device made of metal and other materials that replaces part or all of a bone). Some newer devices combine a graft and a prosthesis.
If the Ewing sarcoma is in the upper part of the leg, part of the femur (upper leg bone), including the knee, can be removed and replaced with a prosthesis for the bone and knee, which is connected to the lower leg. Tumors in the lower part of the leg are harder to treat this way, because it is harder to remove and reconstruct parts of the lower leg. The humerus (upper arm bone) is also suitable for limb-sparing surgery.
Limb-sparing surgery is a very complex operation. The surgeons who do this type of operation must have special skills and experience. The challenge for the surgeon is to be sure to remove the entire tumor while still saving the nearby tendons, nerves, and blood vessels to keep as much of the limb’s function and appearance as possible. If the tumor has grown into these structures, they will need to be removed as well.
Using an internal prosthesis in a growing child is especially challenging. In the past, it often required several operations over time to replace the prosthesis with a longer one as the child grew. Newer prostheses have become very sophisticated and can often be made longer without any extra surgery. They have tiny devices in them that can lengthen the prosthesis when needed to make room for a child’s growth. But even these prostheses may need to be replaced with a stronger adult prosthesis once the child’s body stops growing.
Some patients may not be able to have limb-sparing surgery because their tumors are in parts of bones that are hard to replace or because the tumors also extend into vital nerves or blood vessels that can’t be removed without severely damaging the limb. These patients usually get radiation therapy instead of surgery.
In rare cases, amputation (removal of part or all of the limb) may be the best option, especially if the cancer comes back in the place where it started, and radiation therapy has already been used.
For an amputation, surgery is usually planned so that muscles and the skin will form a cuff around the remaining bone. This cuff will fit into the end of a prosthetic (artificial) limb. Another option might be to implant a prosthesis into the remaining bone, with the end of the prosthesis remaining outside the skin. This can then be attached to an external prosthesis.
Ewing sarcomas in the chest wall
For a Ewing sarcoma in the chest wall, the surgeon often must remove the diseased area and also remove nearby ribs, which might then be replaced with a man-made material. If the tumor has spread to the lungs, the chest can be opened and the lung tumors removed during an operation called a thoracotomy. Radiation therapy is often given to the chest as well.
Ewing sarcomas in the pelvis (hip bones)
Pelvic tumors can be hard to treat with surgery, and in many cases radiation therapy may be the preferred treatment. But if the tumor responds well to initial chemotherapy, surgery (sometimes followed by radiation therapy) may be an option. Pelvic bones can sometimes be reconstructed after surgery, but in some cases pelvic bones and the leg they are attached to might need to be removed.
Ewing sarcomas in the spine
Ewing sarcomas in or right next to the spine can often be hard to remove completely, so radiation therapy is sometimes a better option. If surgery is done, radiation is often given afterward to try to kill any remaining tumor cells.
Rehabilitation after surgery
This might be the hardest part of treatment, and it can’t be described here completely because it will be different for each patient. Whenever possible, patients and parents should meet with a rehabilitation specialist before surgery to learn about their options and what might be required after surgery.
Physical therapy and rehabilitation are very important for patients who have had surgery for Ewing tumors. Following the recommended rehab program offers the best chance for good long-term limb function. Even with proper rehab, people might still have to adjust to long-term issues such as changes in how they walk or do other tasks, and changes in appearance. Physical, occupational, and other therapies can often help people adjust and cope with these challenges.
Rehab after limb-sparing surgery
Even when only the tumor and part of the bone is removed in a limb-sparing operation, the situation can still be complicated, especially in growing children. Children who have had limb-sparing surgery may need more surgery in the coming years to replace the internal prosthesis with one more suited to their growing body size, and some may eventually need an amputation.
It takes about a year, on average, for patients to learn to walk after limb-sparing surgery on a leg. Physical rehabilitation after limb-sparing surgery is extremely important. If the patient doesn’t actively take part in the rehabilitation program, the salvaged arm or leg might become useless.
Rehab after amputation
If a limb is amputated, the patient must learn to adjust to new ways of doing some things, often with the use of a prosthetic limb. This can be particularly hard for growing children if the prosthetic limb needs changing to keep up with their growth. With proper physical therapy, patients are often able to walk on their own about 3 to 6 months after a leg amputation.
Chemotherapy
Chemotherapy (chemo) is the type of cancer treatment that uses one or more anti-cancer drugs in a standard regimen.
Chemotherapy is sometimes used as the first treatment for Ewing sarcoma, followed by surgery and/or radiation therapy. The anti-cancer drugs may shrink the cancer. That makes it easier to remove the cancer with surgery or target with radiation therapy.
After surgery or radiation therapy, chemotherapy treatments might be used to kill any cancer cells that might remain. As noted in Ewing Tumor Stages, even patients with localized Ewing tumors, who have no obvious cancer spread shown on imaging tests or in bone marrow biopsy samples, are likely to have very small areas of cancer spread (micrometastases). If chemotherapy isn’t given, these small metastases eventually would develop into larger tumors.
For advanced cancer that spreads to other areas of the body, chemotherapy might help relieve pain and slow the growth of the cancer.
Doctors give chemo in cycles, with a period of treatment (often a few days in a row) followed by a rest period to give the body time to recover. A combination of several chemo drugs is used to treat Ewing sarcomas.
In the United States, the most common chemo regimen is known as VDC/IE (or VAC/IE). It alternates between 2 combinations of drugs, given every 2 to 3 weeks 17:
- The first drug combination includes vincristine, doxorubicin (Adriamycin), and cyclophosphamide.
- The second set of drugs includes a combination of ifosfamide and etoposide.
Some doctors may use slightly different drug combinations. These often include many of the same chemo drugs listed above, although other drugs might be used as well especially if the cancer comes back after treatment 17.
Most patients will get chemo for at least 9 weeks before surgery or radiation, and then will get more chemo afterward as well 17. Usually a total of about 14 to 15 cycles of chemo are given, which can take from about 6 months to close to a year to complete, depending on the schedule 17. If the tumor has spread to other parts of the body, these same drugs may be given at higher doses 17.
Soon after the Ewing sarcoma is diagnosed but before starting chemo, your doctor may suggest surgery to put a catheter (a thin, soft tube) into a large vein in your chest. This is sometimes called a venous access device (VAD) or central venous catheter (CVC). One end of the catheter stays in the vein, while the other end lies just under or outside the skin. The catheter usually stays in place for several months, letting the health care team give drugs and draw blood samples without having to stick needles into the veins each time. If you or your child gets such a device, the health care team will teach you how to care for it to reduce the risk of problems such as infections.
Side effects of chemo
When chemo drugs affect cells in the body other than cancer cells, it can lead to side effects. The side effects depend on the type and doses of drugs, and the length of time they are given.
Children tend to have less severe side effects from chemo than adults and often recover from side effects more quickly. This is why doctors can often give them higher doses of chemo to kill the tumor.
Common side effects of many chemo drugs include:
- Hair loss
- Mouth sores
- Loss of appetite
- Nausea and vomiting
- Diarrhea
Chemo can damage your bone marrow, where new blood cells are made. This can lead to low blood cell counts, which can result in:
- Increased chance of infections (from having too few white blood cells)
- Easy bruising or bleeding (from having too few blood platelets)
- Fatigue (from having too few red blood cells)
Most of these side effects tend to go away after treatment is finished. There are often ways to lessen them. For example, drugs can be given to help prevent or reduce nausea and vomiting, or to help get blood cell counts back to normal levels. Be sure to discuss any questions you have about side effects with the cancer care team.
Side effects of certain chemo drugs
Along with the effects listed above, certain chemo drugs can have specific side effects:
- Cyclophosphamide and ifosfamide can damage your urinary bladder, which can cause blood in the urine. The risk of this happening can be lowered by giving the drugs with plenty of fluids and with a drug called mesna, which helps protect the bladder.
- Doxorubicin (Adriamycin) can damage your heart. This risk goes up with the total dose of the drug, so doctors are careful to limit the total dose. The doctor might order a heart function test (such as an echocardiogram) before and during treatment to see if this drug is affecting the heart. A drug called dexrazoxane may be given along with the chemo to help lessen the possible damage.
- Vincristine can cause nerve damage (neuropathy). Some patients may notice tingling and numbness, particularly in the hands and feet. This often goes away or gets better once treatment is stopped, but it may last a long time in some people.
- Etoposide can increase the risk of developing leukemia later on, although this is not common.
Some chemo drugs can affect your (child’s) ability to have children (fertility) later in life. Talk to your doctor about the possible effects of treatment on fertility, and ask if there are options for preserving fertility, such as sperm banking or ovarian tissue banking.
Tests to check for chemo side effects
Before each treatment, lab tests will be done to be sure the liver, kidney, and bone marrow are working well. If not, chemo may need to be delayed or the doses reduced.
- The complete blood count (CBC) checks levels of white blood cells, red blood cells, and blood platelets. These will be watched closely during and after chemo. The white blood cells and platelets usually reach their lowest point about 2 weeks after chemo is given, though this can occur sooner with high-dose regimens.
- Blood chemistry tests measure certain chemicals in the blood that tell doctors how well the liver and the kidneys are working. Some chemo drugs can damage these organs.
- If doxorubicin (Adriamycin) is to be given, tests such as an echocardiogram (an ultrasound of the heart) may be done to check heart function before and during treatment.
Radiation therapy
Radiation therapy treats cancer with powerful energy beams. The energy can come from X-rays, protons or other sources. During radiation therapy, you lie on a table while a machine moves around you. The machine directs radiation to precise points on your body.
Radiation therapy might be suggested after surgery to kill cancer cells that remain. Radiation therapy might be used instead of surgery if an operation is not possible or if it is likely to hurt nearby organs. For example, if the surgery might cause loss of bowel or bladder control, radiation might be used instead.
For advanced Ewing sarcoma, radiation therapy can slow the growth of the cancer and help relieve pain.
Types of radiation therapy
Modern radiation therapy techniques let doctors focus the radiation more precisely than in the past. These techniques include:
- Three-dimensional conformal radiation therapy (3D-CRT): Three-dimensional conformal radiation therapy (3D-CRT) uses the results of imaging tests (such as MRI) and special computers to precisely map the location of the tumor. Several radiation beams are then shaped and aimed at the tumor from different directions. Each beam alone is fairly weak, which makes it less likely to damage normal body tissues, but the beams converge at the tumor to give a higher dose of radiation there.
- Intensity modulated radiation therapy (IMRT): Intensity modulated radiation therapy (IMRT) is an advanced form of 3D therapy that can be especially useful for tumors with complex shapes or tumors near important structures, such as the spine. Along with shaping the beams and aiming them at the tumor from several angles, the intensity (strength) of the beams is adjusted to limit the dose reaching the most sensitive normal tissues. This lets the doctor deliver a higher dose to the tumor. Many major hospitals and cancer centers now use IMRT, especially for tumors in hard-to-treat areas such as the spine or pelvis (hip bones).
- Conformal proton beam radiation therapy: Proton beam therapy is another type of 3D therapy. But instead of using x-rays, it focuses proton beams on the tumor. Unlike x-rays, which release energy both before and after they hit their target, protons cause little damage to tissues they pass through and then release their energy after traveling a certain distance. Doctors can use this property to deliver more radiation to the tumor and do less damage to nearby normal tissues. This approach may be helpful for hard-to-treat tumors, such as those on the spine, skull, or pelvic bones. However, the machines needed to make protons are expensive, and there are not many of them in the United States at this time.
How radiation therapy is done
Radiation therapy is given by a doctor called a radiation oncologist. Before treatments start, your radiation team takes careful measurements with imaging tests such as MRI scans to determine the correct angles for aiming the beams and the right dose of radiation. This planning session is called simulation. Patients may also be fitted with a plastic mold resembling a body cast to keep them in the same position each time so that the radiation can be aimed more accurately.
Most often, radiation treatments are given 5 days a week for several weeks. Each treatment is much like getting an x-ray, but the dose of radiation is much higher. The treatment is not painful. For each session, the patient lies on a special table while a machine delivers the radiation from precise angles.
Each treatment lasts only a few minutes, but the setup time – getting the patient into place for treatment – usually takes longer. Some younger children may be given medicine before each treatment to make them sleep so they won’t move during treatment.
Possible side effects of radiation therapy
Because of the possible side effects of radiation therapy especially in growing children, surgery is often preferred if it is possible. But improvements in the way radiation therapy is given now allow children with Ewing sarcomas to be treated with lower doses than were used in the past, helping to reduce some of the side effects.
The side effects of radiation therapy depend mainly on the dose of radiation and where it is aimed. Some effects may last only a short time, while others might last longer.
Short-term side effects
Radiation might cause some short-term side effects, which usually go away within a week or two after treatment. For example:
- Radiation can affect the skin in areas that are in the path of the radiation. Effects can range from mild sunburn-like changes and hair loss to more severe skin reactions.
- Radiation might lower blood cell counts. This can be worse if radiation is being given at the same time as chemo, which can also lower blood cell counts.
- Radiation to the head and mouth area might cause mouth sores or dry mouth.
- Radiation to the abdomen or pelvis might cause nausea, diarrhea, or urinary problems.
Late and long-term side effects
Some side effects might last longer, or they might not show up until some time after treatment is finished. These types of side effects can often be more serious, especially in growing children, so doctors try to limit them as much as possible.
A serious effect of radiation therapy in children is slowed bone growth, especially in younger children. For example, radiation to the bones in one leg may result in it being much shorter than the other. Radiation of facial bones may cause uneven growth, which might affect how a child looks. But if a child is fully or almost fully grown, this is less likely to be an issue.
Depending on where the radiation is aimed, it can also damage other organs:
- Radiation to your chest wall or lungs can affect your lung and heart function.
- Radiation to your pelvis can damage the bladder or intestines, which can lead to problems with urination, digestion, or bowel movements. It can also damage reproductive organs, which could affect fertility later in life, so doctors do their best to protect these organs by shielding them from the radiation or moving them out of the path of radiation whenever possible.
- Side effects of radiation therapy to the spinal cord or skull may include nerve damage, headaches, and trouble thinking, which usually become most serious a year or two after treatment. Fortunately, Ewing tumors rarely spread to the brain, but they can sometimes extend into the brain from nearby bones of the skull.
Another major concern with radiation therapy is that it might cause a new cancer to form in the part of the body that was treated with the radiation. This is most often a different type of bone cancer called an osteosarcoma. This small risk should not keep children who need radiation from getting it. Still, it’s important to continue follow-up visits with your child’s doctor so that if problems come up they can be found and treated as early as possible.
Targeted therapy
Targeted therapy is a type of drug treatment for cancer that works by attacking specific molecular features, or “targets” of cancer cells that are involved in their growth and survival, such as abnormal genes or proteins. Unlike traditional chemotherapy, targeted therapy aims to minimize damage to healthy cells by selectively blocking cancer growth pathways. By blocking these specific “targets” in the cells, targeted treatments can cause cancer cells to die. To determine if a patient’s cancer has the necessary molecular target, tumor tissue is tested. The specific drug is then chosen based on the test results, and treatment may involve blocking growth signals, inhibiting blood flow to the tumor, delivering toxins to the cells, or working in combination with other treatments. For Ewing sarcoma, researchers are looking at using targeted therapy when the cancer comes back or does not respond to other treatments.
Clinical trials
Clinical trials are studies of new treatments. These studies provide a chance to try the latest treatments. The risk of side effects might not be known. Ask your doctor if you or your child might be able to join a clinical trial.
High-dose chemotherapy and Stem Cell Transplantation
High-dose chemotherapy and stem cell transplantation (often called a bone marrow transplant) is being studied for Ewing sarcomas that are hard to cure with other treatments, such as tumors that have spread (metastasized) to other parts of the body or that have come back after standard treatment. So far, it’s not clear if a stem cell transplant is better than other treatments (such as standard chemotherapy), so most doctors recommend it be done only as part of a clinical trial.
How a stem cell transplant is done
The first step in a stem cell transplant is done before the treatment with high-dose chemo. The patient’s own blood-producing stem cells are collected (harvested) to use later. This type of transplant, where the stem cells are taken from the patient, is known as an autologous transplant. In another type of stem cell transplant, called an allogeneic transplant, the stem cells come from a donor. This type is not used often for treating Ewing sarcomas.
The stem cells are usually collected from the blood using a procedure similar to a blood donation. But instead of going into a collecting bag, the blood goes into a special machine that filters out the stem cells and returns the other parts of the blood to the person’s body. The stem cells are then frozen until the transplant. Stem cells might need to be collected more than once.
Once the stem cells have been frozen and stored, the person gets high-dose chemo, sometimes along with radiation therapy. When the treatment is finished, the patient’s stem cells are thawed and returned to the body in a blood transfusion. The stem cells travel through the bloodstream and settle in the bone marrow. Over the next few weeks, they start to make new, healthy blood cells.
Side effects of stem cell transplants
A stem cell transplant (bone marrow transplant) is a complex treatment that can cause serious or even life-threatening side effects. If your doctor think you might benefit from a bone marrow transplant, it should be done at a cancer center where the staff has experience in doing the procedure and managing the recovery period.
The main side effects from a stem cell transplant are from the chemotherapy. Because high doses of chemo are used, some of these side effects might be more severe than with standard doses of chemo.
Some side effects of a stem cell transplant (bone marrow transplant) might last a long time, or they might not show up until years after the transplant, which is a special concern in children and teens. If a stem cell transplant is recommended for your child, be sure to talk to the cancer care team before the transplant to learn about possible long-term effects your child might have.
Follow-up visits and tests
Once treatment is finished, your doctor will discuss a follow-up schedule with you, including which tests should be done and how often. It’s very important to go to all follow-up appointments. Follow-up visits are needed to check for cancer recurrence, as well as possible side effects of treatment.
Physical exams, along with imaging tests (such as x-rays and CT, MRI, PET, and/or bone scans) are often done every 2 to 3 months for the first couple of years following treatment, and then less often over time if there are no issues. If Ewing tumors come back, it is usually within the first couple of years after treatment. But they can sometimes come back even many years later, so continued follow-up visits are important.
Physical therapy and rehabilitation are typically a very important part of recovery after treatment, and your doctors and other health providers will continue to monitor your (child’s) progress as time goes on.
Blood tests and other tests might be done as well. For example, the chemotherapy drug doxorubicin (Adriamycin) can affect the heart, so tests to measure heart function (such as echocardiograms) will probably be done as well.
During this time, it is very important to report any new symptoms to the doctor right away so that any problems can be found early, when they can be treated most effectively.
Ewing’s sarcoma treatment by stage
Ewing sarcoma treatment is based mainly on where it is in the body and how far it has spread when it’s first found.
Localized Ewing sarcomas
A localized Ewing sarcoma is one that still appears to be confined to the area where it started and maybe also to nearby tissues such as muscle or tendons, based on imaging test and biopsy results. But even people with localized Ewing sarcomas often still have small areas of cancer in other parts of the body that can’t be seen with imaging tests. If these people don’t get chemotherapy (chemo) as part of their treatment, these small areas of cancer cells would eventually become larger tumors. This is why chemo, which can reach all parts of the body, is an important part of treatment for localized Ewing sarcomas.
Once the Ewing sarcoma has been diagnosed and staged, the first treatment is chemotherapy. It’s called neoadjuvant chemotherapy because it’s given before any surgery or radiation therapy. In the United States, patients are given a chemo regimen known as VDC/IE (or VAC/IE), which is a combination of vincristine, doxorubicin (Adriamycin), and cyclophosphamide, alternated with ifosfamide and etoposide, although other combinations of the same drugs are also effective.
After at least 9 weeks of chemo, imaging tests such as CT, MRI, PET, or bone scans are done to see if the tumor is shrinking (or at least isn’t growing) and if it can be surgically removed. If so, surgery is done at this point. The surgery specimen is then sent to a lab to be looked at by a doctor called a pathologist.
If cancer cells are found at or near the edges of the surgery specimen (meaning cancer cells may have been left behind), radiation therapy and chemotherapy (for several months) are used.
If there are no cancer cells at or near the edges of the surgery specimen, chemotherapy can be used without radiation therapy.
If surgery is not an option after the initial chemotherapy (because of the tumor location or some other reason), but the tumor is not growing, radiation therapy (along with chemotherapy) is usually the next treatment given. In some cases this might shrink the tumor enough so that surgery can then be done. This would then be followed by more chemotherapy, possibly with more radiation as well. In other cases where surgery is still not an option, radiation therapy and chemotherapy are the main treatments.
If the Ewing sarcoma continues to grow despite the initial chemotherapy, a second type of chemotherapy (using different drugs) may be tried. Surgery or radiation therapy may also be tried to help keep the tumor under control. This may be followed by more chemotherapy.
Metastatic Ewing sarcomas
Patients who clearly have metastatic disease when they are first diagnosed are harder to treat than patients with localized disease. The outlook tends to be better when the cancer has only spread to your lungs, as opposed to when the cancer has spread to other bones or to your bone marrow.
Treating metastatic disease is similar in many ways to treating localized disease. Chemotherapy is the first treatment, but it often requires a more intense regimen than would be used if the cancer was localized. After a few months, tests such as CT or MRI scans, bone or PET scans, and/or bone marrow biopsies are done to see how the cancer has responded to treatment.
If the cancer remains in only a few small areas after chemo, the main (primary) tumor and all known areas of metastases may be removed with surgery at this point. Other approaches, such as surgery plus radiation therapy (before and/or after surgery) or just radiation therapy to all known metastatic sites, might also be options. During and after these treatments, chemotherapy is given for several months as well.
Doctors at several cancer centers are now studying giving very intensive chemotherapy followed by a stem cell transplant to try to improve the outcome for these patients.
Because these tumors can be hard to treat, clinical trials of newer treatments may be a good option in many cases.
Ewing sarcomas that recur (come back) after treatment
Recurrence of Ewing sarcomas after treatment is less likely now than in the past, but it can happen. If the tumor does come back, treatment depends on a number of factors, including:
- The size and location of the tumor
- If it has spread to different parts of the body
- What types of treatment were used before
- How long it has been since treatment
Chemotherapy, surgery, radiation therapy, or some combination of these may be used to treat recurrent tumors, depending on the situation. Doctors are also studying the usefulness of high-dose chemotherapy followed by a stem cell transplant, as well as targeted drugs and immune therapies. These tumors can be hard to treat, so clinical trials of newer treatments may be a good option.
Ewing’s sarcoma prognosis
Ewing’s sarcoma prognosis varies from patient to patient but, in general, 77% of patients without any spread of the disease will survive at least 5 years after diagnosis with standard treatment 6.
Several factors can improve the likelihood of survival include:
- No known spread of the cancer
- An excellent response to chemotherapy
- Primary tumors being located in the arms and legs, instead of the pelvis
- Complete removal of the tumor
Other factors that can affect prognosis (outlook), survival rates and linked with a better prognosis include:
- Smaller tumor size.
- Main tumor is on an arm or leg as opposed to chest wall or pelvis.
- Normal blood lactate dehydrogenase (LDH) level.
- Good tumor response to chemotherapy.
- Age younger than 10 years.
Distal extremity involvement in Ewing sarcoma is associated with a more favorable prognosis (outlook), than patients with lesions in proximal extremities 18.
After treatment is given, prognosis is affected by:
- whether the tumor was completely removed by surgery
- whether the tumor responded to chemotherapy or radiation therapy
Survivors of Ewing sarcoma require continued follow-up care to monitor any late side effects from treatment, and any recurrence of the tumor. When tumors come back, it usually happens within the first few years after treatment.
If the cancer recurs after initial treatment, prognosis depends on:
- whether the cancer came back more than two years after the initial treatment
- whether the cancer came back where it first formed and in other parts of the body, or whether the cancer came back in only one site
Even when taking these other factors into account, survival rates are at best rough estimates. Your cancer care team is your best source of information on this topic.
Patients with solitary lung metastases are more likely to have a better prognosis than patients with extrapulmonary metastatic sites, and unilateral lung involvement correlates with a better prognosis than bilateral lung involvement 19. The development of both bone and lung metastases and extensive tumors are associated with poor prognosis. Tumor size is an important prognostic factor in studies. Data show that being under 15 years is another significant clinical prognostic factor. Clinical studies have shown that patients with minimal or no viable residual tumor at surgery following neoadjuvant chemotherapy did seem to have a better outcome than the patients with more significant amounts of viable tumors 19. Additionally, inadequate response to neoadjuvant chemotherapy (chemotherapy that is administered before surgery to reduce tumor size and potentially destroy cancer cells that may have spread) is associated with an increased risk of recurrence. EWSR1-ETS translocation is no longer considered an adverse prognostic factor 1.
Ewing’s sarcoma survival rate
Survival rates can give you an idea of what percentage of people with the same type and stage of cancer are still alive a certain amount of time (usually 5 years) after they were diagnosed. They can’t tell you how long you will live, but they may help give you a better understanding of how likely it is that treatment will be successful. Keep in mind that survival rates are estimates and are often based on previous outcomes of large numbers of people who had a specific cancer, but they can’t predict what will happen in any particular person’s case. These statistics can be confusing and may lead you to have more questions. Ask your doctor how these numbers might apply to you or your (child’s) situation.
A relative survival rate compares people with the same type and stage of cancer to people in the overall population. For example, if the 5-year relative survival rate for a specific stage of Ewing tumor is 81%, it means that people who have that cancer are, on average, about 81% as likely as people who don’t have that cancer to live for at least 5 years after being diagnosed.
The SEER (Surveillance, Epidemiology, and End Results) database maintained by the National Cancer Institute tracks 5-year relative survival rates for Ewing sarcomas in the United States, based on how far the cancer has spread. The SEER database, however, does not group cancers by American Joint Committee on Cancer (AJCC) TNM (Tumor, Node, and Metastasis) stages. Instead, it groups cancers into localized, regional, and distant stages:
- Localized: There is no sign that the cancer has spread outside the bone (or other area) where it started.
- Regional: The cancer has spread outside the bone (or other area) and into nearby structures, or it has reached nearby lymph nodes.
- Distant: The cancer has spread to distant parts of the body, such as to the lungs or to bones in other parts of the body.
Table 3. Ewing sarcomas 5-year relative survival rates
| SEER (Surveillance, Epidemiology, and End Results)* stage | 5-year relative survival rate |
| Localized | 81% |
| Regional | 77% |
| Distant | 41% |
| All SEER stages combined | 65% |
Footnotes:
- These numbers are based on people diagnosed with Ewing tumors between 2015 and 2021.
- These numbers apply only to the stage of the cancer when it is first diagnosed. They do not apply later on if the cancer grows, spreads, or comes back after treatment.
- These numbers don’t take everything into account. Survival rates are grouped based on how far the cancer has spread. But other factors, such as those listed below, can also affect a person’s outlook.
- People now being diagnosed with Ewing sarcomas may have a better outlook than these numbers show. Treatments improve over time, and these numbers are based on people who were diagnosed and treated at least 5 years earlier.
- Durer S, Gasalberti DP, Shaikh H. Ewing Sarcoma. [Updated 2024 Jan 8]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK559183[↩][↩][↩]
- PDQ Pediatric Treatment Editorial Board. Ewing Sarcoma and Undifferentiated Small Round Cell Sarcomas of Bone and Soft Tissue Treatment (PDQ®): Health Professional Version. 2024 Nov 27. In: PDQ Cancer Information Summaries [Internet]. Bethesda (MD): National Cancer Institute (US); 2002-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK66045[↩]
- Kim SY, Tsokos M, Helman LJ. Dilemmas associated with congenital ewing sarcoma family tumors. J Pediatr Hematol Oncol. 2008 Jan;30(1):4-7. doi: 10.1097/MPH.0b013e31815cf71f[↩]
- van den Berg H, Dirksen U, Ranft A, Jürgens H. Ewing tumors in infants. Pediatr Blood Cancer. 2008 Apr;50(4):761-4. doi: 10.1002/pbc.21292[↩]
- Ewing sarcoma. https://medlineplus.gov/genetics/condition/ewing-sarcoma[↩][↩][↩][↩][↩][↩][↩]
- Ewing Sarcoma. https://orthoinfo.aaos.org/en/diseases–conditions/ewings-sarcoma[↩][↩]
- Extraskeletal Ewing sarcoma. https://radiopaedia.org/cases/extraskeletal-ewing-sarcoma-1[↩]
- Ewing sarcoma of the rib. https://radiopaedia.org/cases/ewing-sarcoma-of-the-rib[↩]
- Bhandari J, Thada PK, Killeen RB, et al. Fanconi Anemia. [Updated 2024 Jun 19]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK559133[↩]
- Fanconi anemia. https://medlineplus.gov/genetics/condition/fanconi-anemia[↩]
- Parham DM, Hijazi Y, Steinberg SM, Meyer WH, Horowitz M, Tzen CY, Wexler LH, Tsokos M. Neuroectodermal differentiation in Ewing’s sarcoma family of tumors does not predict tumor behavior. Hum Pathol. 1999 Aug;30(8):911-8. doi: 10.1016/s0046-8177(99)90244-7[↩]
- Yoshida A, Sekine S, Tsuta K, Fukayama M, Furuta K, Tsuda H. NKX2.2 is a useful immunohistochemical marker for Ewing sarcoma. Am J Surg Pathol. 2012 Jul;36(7):993-9. doi: 10.1097/PAS.0b013e31824ee43c[↩]
- WHO Classification of Tumours Editorial Board: WHO Classification of Tumours. Volume 3: Soft Tissue and Bone Tumours. 5th ed., IARC Press, 2020.[↩]
- Langerhans Cell Histiocytosis Treatment (PDQ®)–Health Professional Version. https://www.cancer.gov/types/langerhans/hp/langerhans-treatment-pdq[↩]
- Bone Cancer Stages. https://www.cancer.org/cancer/types/bone-cancer/detection-diagnosis-staging/staging.html[↩]
- Soft Tissue Sarcoma Stages. https://www.cancer.org/cancer/types/soft-tissue-sarcoma/detection-diagnosis-staging/staging.html[↩][↩][↩]
- Chemotherapy for Ewing Tumors. https://www.cancer.org/cancer/types/ewing-tumor/treating/chemotherapy.html[↩][↩][↩][↩][↩]
- Moore DD, Haydon RC. Ewing’s sarcoma of bone. Cancer Treat Res. 2014;162:93-115. doi: 10.1007/978-3-319-07323-1_5[↩]
- Bernstein M, Kovar H, Paulussen M, Randall RL, Schuck A, Teot LA, Juergens H. Ewing’s sarcoma family of tumors: current management. Oncologist. 2006 May;11(5):503-19. doi: 10.1634/theoncologist.11-5-503[↩][↩]
- Survival Rates for Ewing Tumors. https://www.cancer.org/cancer/types/ewing-tumor/detection-diagnosis-staging/survival-rates.html[↩]





{kind=link}