Contents
- What is gonadotropin
- Follicle stimulating hormone (FSH)
- Luteinizing hormone (LH)
- Human chorionic gonadotropin (hCG)
- Human chorionic gonadotropin function
- Human chorionic gonadotropin angiogenic actions
- Human chorionic gonadotropin immunological actions
- Human chorionic gonadotropin test
- How is human chorionic gonadotropin used?
- What does human chorionic gonadotropin test result mean?
- How does the test that I do at home myself compare with the results of a test done in a lab?
- When is a blood human chorionic gonadotropin test ordered instead of a urine human chorionic gonadotropin?
- How many days after a miscarriage would it take for a urine pregnancy test to show a negative result?
- What is an ectopic pregnancy?
- Gonadotropin deficiency
- Gonadotropin deficiency diagnosis and testing
- Gonadotropin deficiency treatment
- Males with isolated gonadotropin-releasing hormone deficiency Age ≥18 Years
- Male Infants/Adolescents with Suspicion of isolated gonadotropin-releasing hormone deficiency
- Females with isolated gonadotropin-releasing hormone deficiency
- Fertility Options in Patients with isolated gonadotropin-releasing hormone (GnRH) deficiency if Fertility Induction is Unsuccessful
- Prevention of secondary complications
- Surveillance
- Evaluation of relatives at risk
- Gonadotropin treatment
- Follicle stimulating hormone (FSH) therapeutic uses
- Luteinizing hormone (LH) use Infertility and Assisted Reproductive Technology
- Gonadotropins use in Assisted Reproductive Technology
- Human Chorionic Gonadotropin (hCG) in Assisted Reproductive Technology
- Hypogonadotropic Hypogonadism Treatment in Males
- Human chorionic gonadotropin therapy
What is gonadotropin
In the body, there are two types of gonadotropins, luteinizing hormone (LH) and follicle-stimulating hormone (FSH), that are secreted from the anterior pituitary gland and that act on the gonads (i.e., the ovaries or testes) that are essential for normal sexual maturation and reproductive function in both males and females 1, 2, 3. Another type of gonadotropin found in women is human chorionic gonadotropin (hCG), which is produced by the placenta during pregnancy 3. The detection of human chorionic gonadotropin (hCG) forms the basis of pregnancy tests. Gonadotropins are glycoprotein hormones that regulate ovarian and testicular function and are essential for normal growth, sexual development and reproduction. In men, luteinizing hormone (LH) stimulates the Leydig cells in the testicles to make testosterone whilst the follicle-stimulating hormone (FSH) stimulates the development of sperm (spermatogenesis) by acting on special cells in the testes called Sertoli cells 4, 5, 6, 7. In women, gonadotropins (LH and FSH) cause the ovaries to make estrogen and progesterone. Gonadotrophs, cells that constitute about 10 percent of the pituitary gland, secrete two primary gonadotropins: luteinizing hormone (LH) and follicle-stimulating hormone (FSH) 2. Gonadotropin-releasing hormone (GnRH) causes the pituitary gland in the brain to make and secrete the hormones luteinizing hormone (LH) and follicle-stimulating hormone (FSH). The amount and rate of secretion of these hormones vary widely at different ages and at different times during the menstrual cycle in women. Secretion of luteinizing hormone (LH) and follicle-stimulating hormone (FSH) is low in both males and females prior to puberty. Following puberty, more luteinizing hormone (LH) than follicle-stimulating hormone (FSH) is secreted. During the menstrual cycle there is a dramatic increase in the serum concentrations of both hormones at the time of ovulation, and the secretion of both hormones increases 10- to 15-fold in postmenopausal women.
The gonadotropins, luteinizing hormone (LH) and follicle-stimulating hormone (FSH), are both glycoproteins made up of an alpha (α) and beta (β) subunit 8, 9. The alpha subunits are identical between the two hormones, but the beta subunit of each is different and gives each hormone its biological specificity 10, 11. The alpha (α) subunit of luteinizing hormone (LH) is made up of 92 amino acids, and the beta (β) subunit contains 120 amino acids 12, 11. The secretion of these hormones is regulated by the release of gonadotropin-releasing hormone (GnRH) secreted by the hypothalamus.
Luteinizing hormone (LH) plays an important role in sexual development and is produced by the pea-sized pituitary gland in the brain.
In children, luteinizing hormone (LH) levels are high right after birth, but then fall, remaining low until puberty approaches (usually between ages 10 and 14). At this time the hypothalamus, an almond-sized area of the brain that links the nervous system with the hormone-producing endocrine system, releases gonadotropin-releasing hormone (GnRH) that starts the changes of puberty. Gonadotropin-releasing hormone (GnRH) signals the pituitary gland to release two puberty hormones into the bloodstream: luteinizing hormone (LH) and follicle-stimulating hormone (FSH).
Pulsatile secretion of gonadotropin-releasing hormone (GnRH) into the hypophyseal portal circulation represents the initial neuroendocrine step in the regulation of the hypothalamo-pituitary-gonadal axis in both sexes. Thus, this specialized gonadotropin-releasing hormone (GnRH) neuronal network plays a commanding role in this biologic hierarchy and controls episodic gonadotropin secretion, modulates gonadal steroid feedback, and ultimately determines the initiation or suppression of pubertal development and fertility across the life cycle 13.
Under normal conditions, the gonadotropin-releasing hormone (GnRH) neuronal network undergoes a series of dynamic changes from fetal life to adulthood. The initiation of gonadotropin-releasing hormone (GnRH) secretion is initiated in early fetal life and remains active until the first several months of infancy (representing the “mini-puberty”), and then becomes remarkably dampened during the years of the childhood “quiescence” 14. At puberty, unknown biologic triggers re-ignite gonadotropin-releasing hormone (GnRH) secretion, resulting in full sexual maturation. Therefore, the controls of the reproductive axis are in dynamic flux, turning on and turning off in response to as-yet-unknown biologic signals at various points in the reproductive life cycle.
- In boys, luteinizing hormone (LH) and follicle-stimulating hormone (FSH) work together to get the testes to begin producing testosterone, the hormone responsible for the physical changes of puberty and the production of sperm. Testosterone is the hormone that causes most of the changes in a boy’s body during puberty. Sperm cells must be produced for men to reproduce.
- In girls, luteinizing hormone (LH) and follicle-stimulating hormone (FSH) prompt the ovaries to begin producing the hormone estrogen, which causes a girl’s body to mature and prepares her for menstruation. Estrogen, along with luteinizing hormone (LH) and follicle-stimulating hormone (FSH), causes a girl’s body to mature and prepares her for pregnancy.
- In men, follicle-stimulating hormone (FSH) stimulates the development of spermatozoa, in large part by acting on special cells in the testes called Sertoli cells. Luteinizing hormone (LH) stimulates the secretion of androgen (male) hormones by specialized cells in the testes called Leydig cells.
- In women, follicle-stimulating hormone (FSH) stimulates the synthesis of estrogens and the maturation of cells lining the spherical egg-containing structures known as Graafian follicles. In menstruating women, there is a preovulatory surge in serum follicle-stimulating hormone (FSH) and luteinizing hormone (LH) concentrations. The preovulatory surge of luteinizing hormone (LH) is essential for rupture of the Graafian follicle (ovulation), after which the egg enters the fallopian tube and travels to the uterus. The empty Graafian follicle becomes filled with progesterone-producing cells, transforming it into a corpus luteum. Luteinizing hormone (LH) stimulates the production of progesterone by the corpus luteum.
- Inhibin, a hormone secreted by the Graafian follicles of the ovaries and by the Sertoli cells of the testes, inhibits the secretion of follicle-stimulating hormone (FSH) from the pituitary gonadotrophs.
Patients with diseases involving the anterior pituitary gland often have gonadotropin deficiency. Thus, the disappearance of menstrual periods may be the first sign of a pituitary tumor or other pituitary disease in women. In men the most common symptoms of gonadotropin deficiency are loss of libido and erectile dysfunction. Isolated deficiencies of both luteinizing hormone (LH) and follicle-stimulating hormone (FSH) do occur but only rarely. In men isolated luteinizing hormone (LH) deficiency (“fertile eunuch”) is characterized by symptoms and signs of androgen deficiency; however, there is sufficient secretion of follicle-stimulating hormone (FSH) to permit the maturation of spermatozoa. Some pituitary tumors produce an excess of luteinizing hormone (LH) or follicle-stimulating hormone (FSH), whereas other pituitary tumors produce the hormonally inactive alpha chain subunit of the glycoprotein hormones.
Because luteinizing hormone (LH) and follicle-stimulating hormone (FSH) work so closely with each other, doctors often perform these tests together, as well as tests for testosterone (the major male sex hormone) and estradiol (a form of estrogen, the major female sex hormone). Taken together, the results can often provide a more complete picture of a person’s sexual maturation, and the well-being of the endocrine glands that produce these hormones.
Figure 1. Gonadotropins
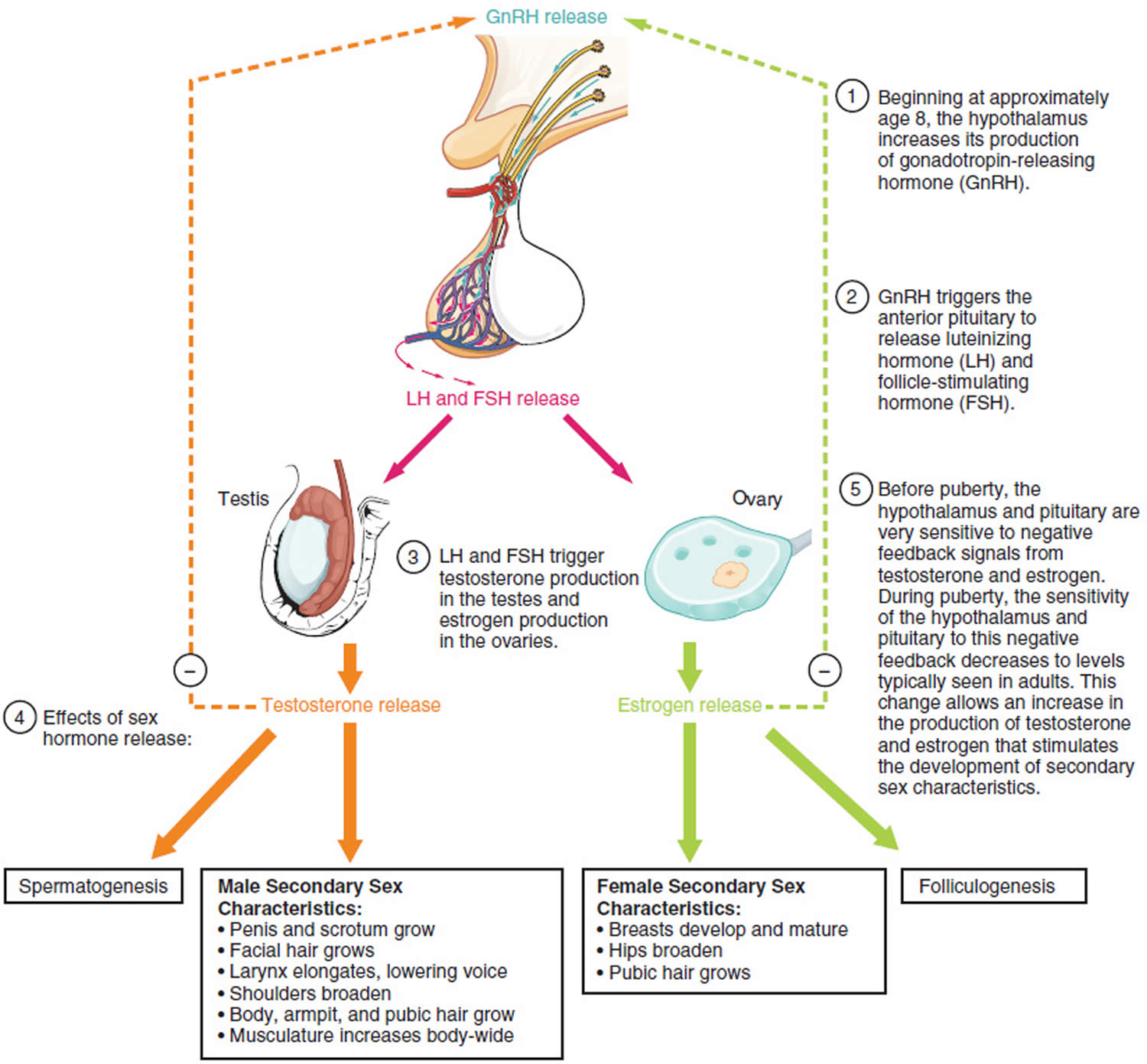
Figure 2. The pituitary gland location
Figure 3. The hypothalamus and pituitary gland (anterior and posterior) endocrine pathways and target organs
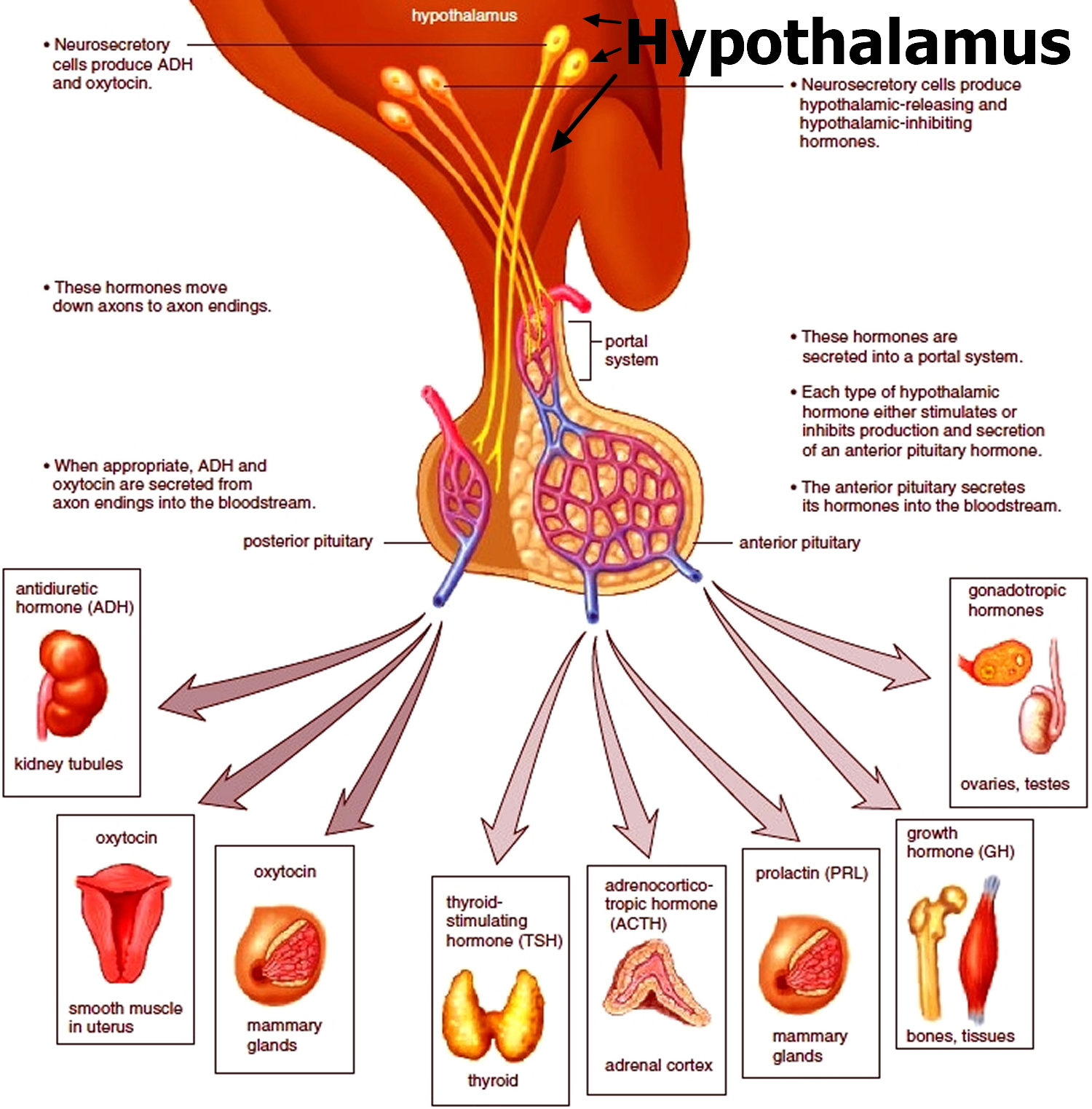
Figure 4. Hypothalamic–pituitary–gonadal axis
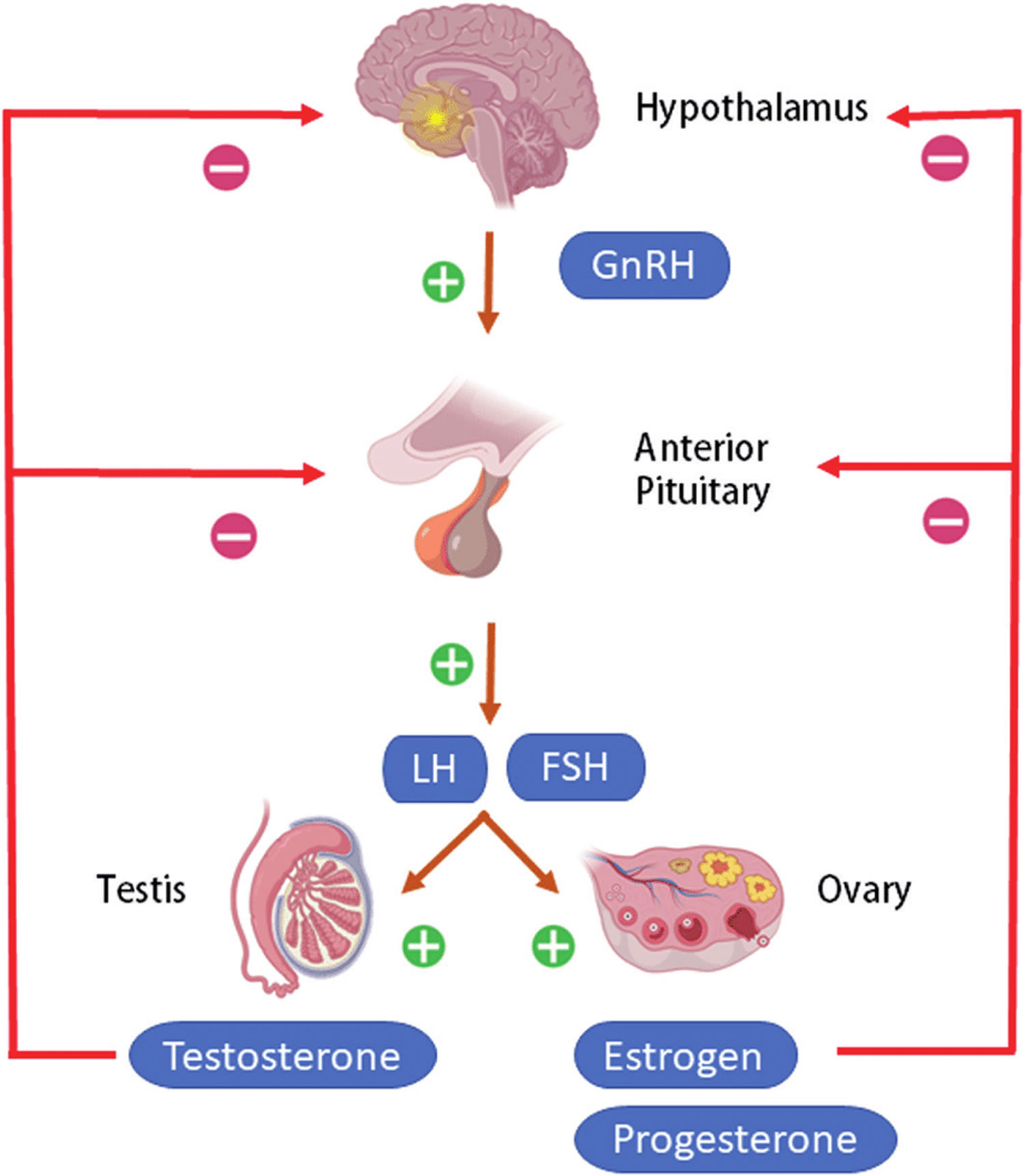
Footnotes: Hypothalamic–pituitary–gonadal (HPG) axis also known as the hypothalamic–pituitary–ovarian/testicular axis refers to the hypothalamus, pituitary gland, and gonadal glands as if these individual endocrine glands were a single entity. The hypothalamic control of reproduction is coordinated through the release of gonadotropin-releasing hormone (GnRH). Pulsatile secretion of gonadotropin-releasing hormone (GnRH) from the hypothalamus is required for both the initiation and maintenance of the hypothalamic–pituitary–ovarian/testicular axis in human. Pulsatile GnRH stimulates secretion of the gonadotropins, luteinizing hormone (LH) and follicle-stimulating hormone (FSH), from the anterior pituitary gonadotropes. Gonadotropins, luteinizing hormone (LH) and follicle-stimulating hormone (FSH) in turn function at the gonads to stimulate the production of gametes and promote gonadal release of sex steroids [i.e., testosterone (T), estradiol (primarily 17β-estradiol, E2), and progesterone (P4)]. In females, ovarian follicles are stimulated by follicle-stimulating hormone (FSH) to grow and mature; luteinizing hormone (LH) stimulates ovulation and corpus luteum formation. In males, LH stimulates testicular Leydig cells to synthesize and secrete testosterone (T), which in turn maintains spermatogenesis in Sertoli cells through its paracrine action and exerts sexual and anabolic actions. While FSH acts on Sertoli cells to produce androgen-binding protein, which is critical for spermatogenesis initiation and ultimately augments sperm production. Along with guiding reproductive function in the peripheral tissues, these gonadal steroids [i.e., testosterone (T), estradiol (primarily 17β-estradiol, E2) and progesterone (P4)] secreted by ovaries and testis can inhibit gonadotropin-releasing hormone (GnRH)’s hypothalamic synthesis via a feedback loop, hence playing a vital role in regulating reproductive function. In both men and women, gonadal failure results in increased LH, because of loss of the negative feedback of estrogen at the hypothalamus and pituitary in women and from decreases in both androgen and estrogen feedback in men. In response to decreased levels of sex steroids as well as the loss of inhibin, FSH levels are also elevated following gonadal damage. Serum gonadotropin [luteinizing hormone (LH) and follicle-stimulating hormone (FSH)] and sex steroid values will differentiate between reproductive failure at the gonadal level or at the hypothalamic/pituitary level. Sex steroid levels will be low in both, but serum gonadotropin levels will be high in primary gonadal failure and low in those with hypothalamic or pituitary disease. The hallmark of primary gonadal failure from any cause is elevation of gonadotropin [luteinizing hormone (LH) and follicle-stimulating hormone (FSH)] levels, and this is the usual state in postpubertal patients receiving substantial doses of chemotherapy agents. Cranial radiation, on the other hand, may result in significant hypothalamic-pituitary dysfunction and secondary gonadal failure with low serum levels of gonadotropins.
[Source 15 ]Figure 5. Testosterone production in men
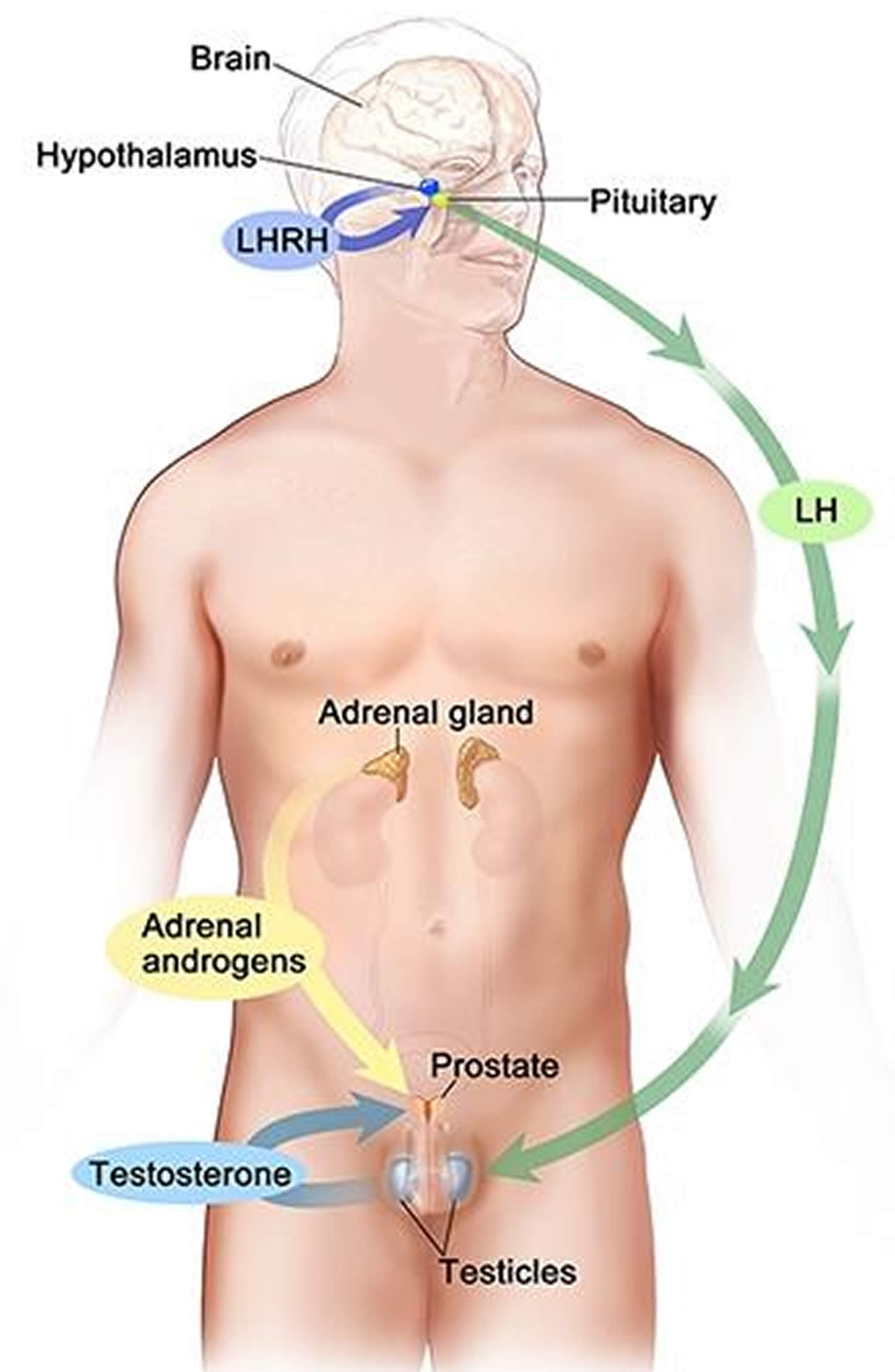
Footnotes: Drawing shows that testosterone (androgen) production is regulated by luteinizing hormone (LH) and luteinizing hormone-releasing hormone (LHRH). The hypothalamus releases luteinizing hormone-releasing hormone (LHRH), which stimulates the release of LH (luteinizing hormone) from the pituitary gland. Luteinizing hormone (LH) acts on Leydig cells in the testes to produce the majority of testosterone in the body. Most of the remaining testosterone are produced by the adrenal glands. Testosterone are taken up by prostate cells, where they either bind to the androgen receptor (AR) directly or are converted to dihydrotestosterone (DHT), which has a greater binding affinity for the androgen receptor than testosterone.
Figure 6. Estrogen and progesterone production in premenopausal women
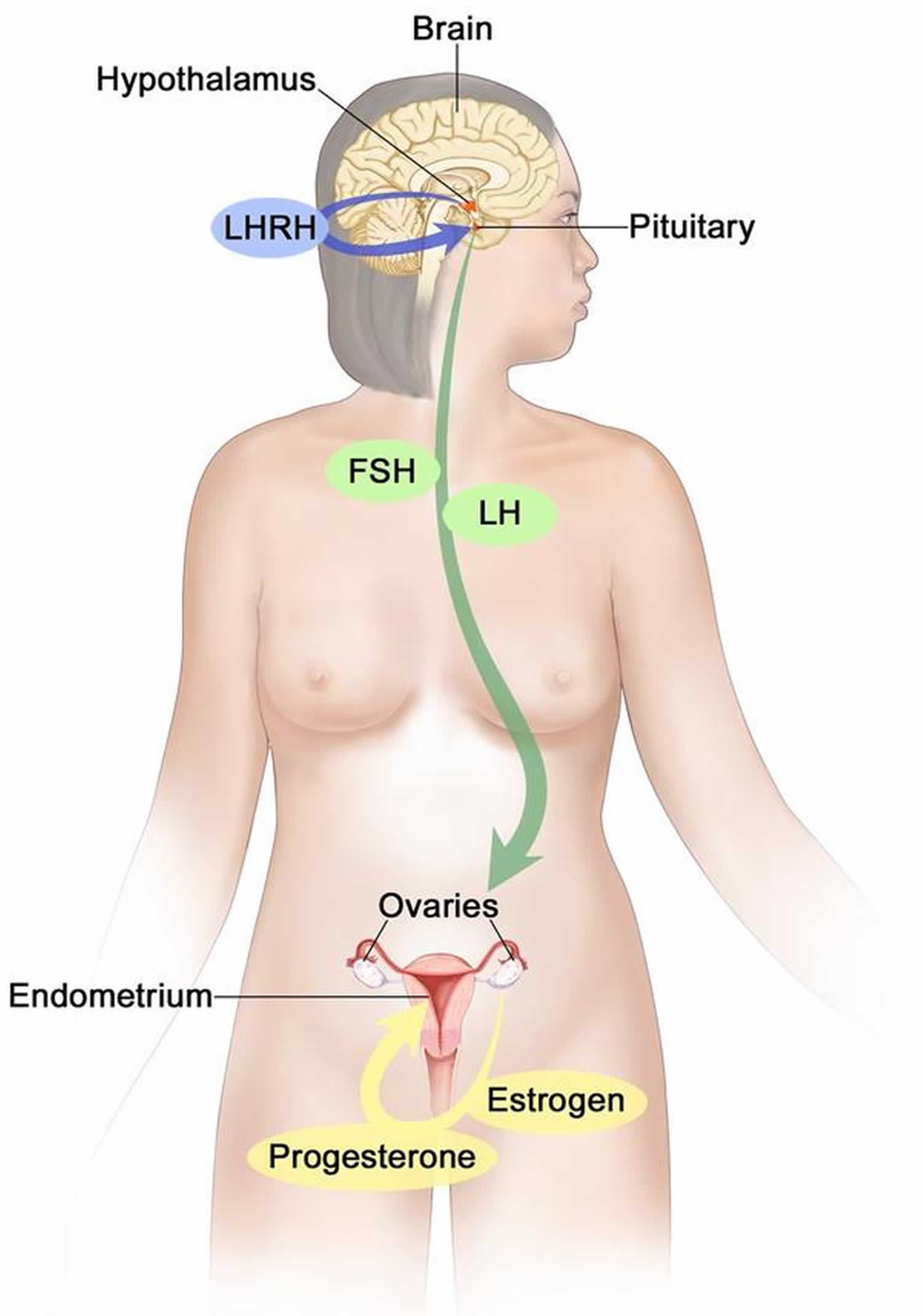
Footnotes: Drawing shows that in premenopausal women, estrogen and progesterone production by the ovaries is regulated by luteinizing hormone (LH) and luteinizing hormone-releasing hormone (LHRH). The hypothalamus releases LHRH, which then causes the pituitary gland to make and secrete LH and follicle-stimulating hormone (FSH). LH and FSH cause the ovaries to make estrogen and progesterone, which act on the endometrium (inner lining of the uterus). When estrogen and progesterone production reaches a certain level during the menstrual cycle, these hormones act on the hypothalamus and pituitary to turn off production of LHRH, LH, and FSH. Estrogen is a steroid hormone that is responsible for the growth and regulation of the female reproductive system and secondary sex characteristics. Estrogen is produced by the granulosa cells of the developing follicle and exerts negative feedback on LH production in the early part of the menstrual cycle. However, once estrogen levels reach a critical level as oocytes mature within the ovary in preparation for ovulation, estrogen begins to exert positive feedback on LH production, leading to the LH surge through its effects on GnRH pulse frequency. Estrogen also has many other effects that are important for bone health and cardiovascular health in premenopausal patients 16.
Follicle stimulating hormone (FSH)
Follicle stimulating hormone (FSH) is a hormone produced by the anterior pituitary in response to gonadotropin-releasing hormone (GnRH) from the hypothalamus 17, 18. Follicle stimulating hormone (FSH) is a glycoprotein hormone with alpha (α) and beta (β) subunits. The beta subunit is unique to FSH, while the alpha subunit is the same as in thyroid stimulating hormone (TSH), human chorionic gonadotropin (hCG), and luteinizing hormone (LH) 19. Follicle stimulating hormone (FSH) regulates growth, sexual development and reproduction, including menstruation, follicular development and ovulation 1, 2.
FSH is regulated, at least in part, by gonadotropin-releasing hormone (GnRH) produced in the hypothalamus in response to multiple signals including circulating levels of sex hormones (i.e, estrogen, progesterone, and testosterone). The hypothalamus produces GnRH, and it is released into the hypophyseal portal circulation to act on G-protein-coupled receptors at gonadotropic cells of the anterior pituitary. Those gonadotropic cells produce FSH and luteinizing hormone (LH) and release them into the peripheral circulation. Gonadal sex hormones, estrogen, progesterone, and testosterone exert negative feedback, thus decreasing the secretion of FSH and LH 20.
Gonadotropin-releasing hormone (GnRH) release occurs in a pulsatile manner, with low pulse frequencies stimulating more FSH production and high pulse frequencies stimulating more luteinizing hormone (LH) production 18. Continuous use of gonadotropin-releasing hormone (GnRH) suppresses the release of FSH and LH from the anterior pituitary which inhibits ovulation and estrogen production in women. Clinically, GnRH agonists like leuprolide work via this mechanism 21. In women, negative feedback from estrogen levels inhibits FSH secretion 22. In men, levels of inhibin B, secreted by the Sertoli cells in response to FSH, inhibit FSH secretion via negative feedback 23.
- In females, FSH receptors are located in the Granulosa cells that surround developing ovarian follicles of the ovaries 24. FSH stimulates the growth and maturation of immature oocytes into mature (Graafian) secondary follicles before ovulation. FSH Receptors are G-protein coupled receptors and are found in the Granulosa cells and Granulosa cells initially produce the estrogen needed to maturate the developing dominant ovarian follicle. After 2 days of sustained elevation of estrogen levels, the LH surge causes luteinization of the Granulosa cells into LH receptive cells. This transition enables Granulosa cells to respond to LH levels and produce progesterone 16. The corpus luteum grows and secretes progesterone and some estrogen, which makes the endometrium more receptive to implantation. If fertilization does not occur, progesterone/estrogen levels fall, and the corpus luteum dies, forming the corpus albicans. These falling hormone levels stimulate FSH to begin recruiting follicles for the next cycle. If fertilization does occur, human chorionic gonadotropin (hCG) produced by the early placenta preserves the corpus luteum, maintaining progesterone levels until the placenta is able to make sufficient progesterone to support the pregnancy 25.
- In males, FSH receptors are found in the Sertoli cells of the testes 19. In men, FSH promotes the production (spermatogenesis) and function of sperm and androgen responsiveness in the testes.
Therefore, FSH is essential for sexual maturation and reproduction in both men and women. Partially and highly purified human menopausal urine derived FSH (Menotropins or human menopausal gonadotropin [hMG] or Menopur which also has LH activity); industrial production of therapeutic grade urinary FSH (urofollitropin, Bravelle) and recombinant DNA (rDNA)-derived human FSH (follitropin alpha, Follistim, Gonal F) are available and approved for use in treatment of infertility and hypogonadism 26. They are generally given by subcutaneous injection daily or several times weekly. The dose and appropriate regimen vary by indication. These agents should be used only by doctors with expertise in management of infertility and hypogonadism.
Follicle stimulating hormone (FSH) function
During fetal development, gonadotropin-releasing hormone (GnRH) producing neurons develop from the epithelium of the medial olfactory pit and then migrate to the hypothalamus 27. The anterior pituitary gland develops from Rathke’s pouch, a portion of the oral cavity 19. In the second and third trimesters of pregnancy, as well during the first 3 to 6 months of infancy, the pituitary gland secretes luteinizing hormone (LH) and follicle stimulating hormone (FSH) 28. LH and FSH levels peak mid-pregnancy as the first ovarian follicle or seminiferous tubule mature 27.
In males, follicle stimulating hormone (FSH) stimulates Sertoli cell proliferation, which is the most significant contributor to testicular volume in children 28. The Sertoli cells produce an anti-mullerian hormone (AMH), which causes the involution of the Mullerian ducts, preventing the formation of female internal genitalia 27.
During puberty, the hypothalamus secretes GnRH in a pulsatile manner, which stimulates the anterior pituitary to increase secretion of LH and FSH 27. In the male, follicle stimulating hormone (FSH) in combination with testosterone, which is under the control of luteinizing hormone (LH), is required for the initiation and maintenance of the quality and quantity of normal production of sperm (spermatogenesis) 29. In transgenic mice it appears FSH to be not essential for male fertility 30, spermatogenesis is not completely normal in the absence of FSH and, furthermore, the requirement for FSH is more critical in primates than in rats.
In the female follicle stimulating hormone (FSH) is necessary for the selection and growth of ovarian follicles and for the production of estrogens from androgen substrates. The gonadotrophic effects of FSH may be subserved by a number of intermediaries 31 that form part of the cellular and tissue response to FSH stimulation culminating in ovulation 32. Such cellular responses illustrate the complex nature of FSH since they indicate that FSH activity has many components, i.e., FSH is a growth factor or tropic hormone, a secretagogue, and a modulator of cellular development 33. It is generally thought that FSH exerts most of its intracellular actions via the cAMP-mediated signaling pathway, although FSH may also utilize other signal transduction pathways such as calcium ion 34.
The biological activity of FSH is the sum of a complex combination of processes: release from the pituitary, survival in the circulation, transport to the site of action (i.e., the gonad), binding to the receptor, and activation of signal transduction pathways.
The lack of the normal infancy peak of luteinizing hormone (LH) and follicle stimulating hormone (FSH) might identify and diagnose infants, especially males, who have hypogonadotropic hypogonadism.
Girls with monosomy Turner syndrome (45, XO) have an elevation of FSH up to 6 years old due to lack of negative feedback from nonfunctional ovaries, while girls with mosaicism (45, X/46, XX) have a much lower FSH elevation due to partial ovarian function 27.
FSH function in Females
- Estrogen production: FSH stimulates granulosa cells in the ovarian follicles to synthesize aromatase, which converts androgens produced by the thecal cells to estradiol.
- Follicular development and the menstrual cycle: During the follicular phase of the menstrual cycle, FSH stimulates the maturation of ovarian follicles. As a dominant follicle takes over and secretes estradiol and inhibin, FSH secretion is suppressed. When the dominant follicle produces enough estradiol to maintain levels of 200 to 300 pg/ml for 48 hours, the hypothalamus responds with a surge of GnRH which stimulates the secretion of gonadotropic hormones instead inhibiting them. FSH peaks at the same time as the LH surge that causes ovulation. FSH then remains low throughout the luteal phase, preventing the development of new follicles 19.
FSH function in Males
Follicle stimulating hormone (FSH), along with testosterone, is necessary for maintaining normal sperm count and function. Studies have shown that FSH deprivation not only lowers sperm count but also affects the quality of the remaining sperm 35.
Follicle stimulating hormone (FSH) diagnostic use
Elevated levels of follicle stimulating hormone (FSH) are associated with unresponsive gonads or hyperfunctioning pituitary adenomas. Low levels of FSH are associated with either hypothalamic or anterior pituitary dysfunction. The measurement of FSH in the blood is widely employed in the diagnosis of disorders of reproduction and development.
Clinical situations where follicle stimulating hormone (FSH) measurements are useful or are commonly requested 36:
- Anovulatory infertility (irregular and inconsistent menstrual blood flow [oligomenorrhea] or absence of monthly menstruation [amenorrhea]): To help determine whether cause is pituitary or gonadal in origin and to aid diagnosis of conditions such as polycystic ovary syndrome (PCOS)
- Suspected premature puberty: In addition to steroid hormone levels
- Delayed puberty: In addition to steroid hormone levels
- Azoospermia or severe low sperm count (oligospermia): To differentiate pituitary and gonadal causes
- Ovarian reserve: As a biological marker for the number of releasable oocytes; may be enhanced by measurements of inhibin and ovarian ultrasound to accurately stage the timing of the sample
- Menopausal status: A frequently requested test; FSH is not a good marker for timing of the menopause or of perimenopausal state
The primary use of follicle stimulating hormone (FSH) measurements is for assessment of gonadal function. Through classical endocrine feedback pathways, an elevated level of FSH indicates reduced gonadal function or gonadal failure, whereas a normal serum concentration of FSH suggests normal gonadal function (see Figures 4 to 6). A low serum FSH may indicate a problem at the level of the hypothalamus or pituitary.
A measurement of serum follicle stimulating hormone (FSH), with measurement of luteinizing hormone (LH) and either estradiol (E2) or testosterone (T), may be helpful in children with suspected premature puberty or in cases of delayed puberty, particularly as the application of sensitive assay methodologies permits detection of hormonal changes before clinical changes of puberty are observed 37. FSH measurement is indicated in men with azoospermia or severe oligospermia (low sperm count) to help determine the degree to which the problem is due to gonadal failure 38.
Ovarian reserve, or the total number of remaining oocytes within the ovary, declines with ovarian age, but this does not always equate with the age of the woman. A baseline measurement of serum FSH concentration, usually on day 3 of the menstrual cycle, is a fairly good predictor of ovarian reserve in women of reproductive years 39. A fluctuating baseline FSH level is indicative of compromised ovarian function. The picture is further enhanced if measurement of FSH is combined with serum estradiol and inhibin 40. In an irregular menstrual cycle it can be difficult to time collection of samples correctly, and therefore more than one sample may have to be taken, often in combination with an ultrasound scan of the ovaries, to help determine the stage in the cycle 41, 42, 43, 44. Measurement of FSH is also helpful in determining the presence of common disorders of reproduction such as polycystic ovary syndrome (PCOS), when classically the serum luteinizing hormone (LH) concentration is elevated, while FSH is usually normal 45. A single measurement of FSH is not predictive of the timing of menopause and is not usually recommended for this purpose, although it may be useful in developing a differential diagnosis to exclude other causes (endocarditis or pheochromocytoma for example) of symptoms such as hot flushes. Although various studies have been performed to characterize the perimenopausal status 46, 47, the practical use of FSH measurement is in the prediction of ovarian response to stimulation in the context of assisted reproduction.
In the United States FSH assays are used for investigations of menopausal status, diagnoses of infertility/amenorrhea, and infertility in men. Essentially, your doctor will wish to detect gross changes in FSH levels from the normal ranges associated with primary gonadal failure and hypogonadotrophic hypogonadism. The ratio of LH to FSH has been proposed as a good predictor of ovarian hyperstimulation syndrome 48. In such cases, particularly where a ratio of two measurements is made, it is important to maintain continuity of unitage between estimates derived from different assays over a period of time and thus from one standard preparation to the next.
FSH levels and Male infertility
If males present with small, firm testes and absence of sperm in the ejaculate (azoospermia) or low sperm count (oligospermia), elevated FSH levels can be used to differentiate Klinefelter syndrome from hypothalamic or pituitary insufficiency. If testicular size is normal and patients present with azoospermia or oligospermia, FSH levels can be used to determine whether the cause is a primary impairment of spermatogenesis or obstructive. In an obstructive cause of infertility, FSH levels remain normal, while a primary impairment of spermatogenesis will present with elevated FSH levels.
Several FSH preparations have been used to treat secondary hypogonadism in males. These preparations have been reasonably successful at inducing spermatogenesis and achieving paternity 35.
Polycystic ovary syndrome (PCOS)
With PCOS, many small sacs of fluid called cysts develop along the outer edge of the ovary. The small fluid-filled cysts contain immature eggs. These are called follicles. The follicles fail to regularly release eggs.
The symptoms of PCOS vary. A diagnosis of PCOS is made when you have at least two of these:
- Irregular periods. Having few menstrual periods or having periods that aren’t regular are common signs of PCOS. So is having periods that last for many days or longer than is typical for a period. For example, you might have fewer than nine periods a year. And those periods may occur more than 35 days apart. You may have trouble getting pregnant.
- Too much androgen (testosterone). High levels of the hormone androgen may result in excess facial and body hair. This is called hirsutism. Sometimes, severe acne and male-pattern baldness can happen, too.
- Polycystic ovaries. Your ovaries might be bigger. Many follicles containing immature eggs may develop around the edge of the ovary. The ovaries might not work the way they should.
PCOS signs and symptoms are typically more severe in people with obesity.
The exact cause of PCOS is unknown. Early diagnosis and treatment along with weight loss may lower the risk of long-term complications such as type 2 diabetes and heart disease.
In PCOS, the LH:FSH ratio is skewed due to persistently rapid GnRH pulses. These GnRH pulses lead to an increased LH: FSH ratio. This skewed ratio leads to the theca cells of the ovaries producing excess androgen while the granulosa cells do not produce enough aromatase to convert the androgens to estradiol 18.
There is no cure for PCOS with treatment focuses on managing the things that are concerning you. This could include infertility, hirsutism, acne or obesity. Specific treatment might involve lifestyle changes or medication. Birth control pills help women have normal periods, reduce male hormone levels, and clear acne. Treatments for infertility caused by PCOS may include medicines, surgery, and in vitro fertilization (IVF).
Hypogonadotropic Hypogonadism
Hypogonadotropic hypogonadism (HH) is a form of hypogonadism that is due to a problem with the pituitary gland or hypothalamus. Hypogonadism is a condition in which the male testes or the female ovaries produce little or no sex hormones.
Hypogonadotropic hypogonadism (HH) is caused by a lack of gonadotropin-releasing hormone (GnRH), follicle stimulating hormone (FSH) and luteinizing hormone (LH) that normally stimulate the ovaries or testes.
There are several causes of hypogonadotropic hypogonadism 50:
- Damage to the pituitary gland or hypothalamus from surgery, injury, tumor, infection, or radiation
- Genetic defects. Kallmann syndrome is an inherited form of hypogonadotropic hypogonadism. Some people with this condition also lose their sense of smell (anosmia).
- High doses or long-term use of opioid or steroid (glucocorticoid) medicines
- High prolactin level (a different hormone released by the pituitary)
- Severe stress
- Nutritional problems (both rapid weight gain or weight loss)
- Long-term (chronic) medical diseases, including chronic inflammation or infections
- Drug use, such as heroin or use or abuse of prescription opioid medicines
- Certain medical conditions, such as iron overload
Hypogonadotropic hypogonadism treatment depends on the source of the problem, but may involve:
- Injections of testosterone (in males)
- Slow-release testosterone skin patch (in males)
- Testosterone gels (in males)
- Estrogen and progesterone pills or skin patches (in females)
- Gonadotropin-releasing hormone (GnRH) injections
- Human chorionic gonadotropin (hCG) injections
Kallman syndrome
Kallmann syndrome also called congenital hypogonadotropic hypogonadism is a rare genetic disorder in humans that is defined by a delayed or absent of signs of puberty along with an absent or impaired sense of smell (anosmia) 51, 52. In Kallmann syndrome, the sense of smell is either diminished (hyposmia) or completely absent (anosmia). This feature distinguishes Kallmann syndrome from most other forms of hypogonadotropic hypogonadism such as normosmic idiopathic hypogonadotropic hypogonadism, which do not affect the sense of smell. Many people with Kallmann syndrome are not aware that they are unable to detect odors until the impairment is discovered through testing.
Kallmann syndrome can have a wide variety of additional signs and symptoms. These include a failure of one kidney to develop (unilateral renal agenesis), abnormalities of bones in the fingers or toes, a cleft lip with or without an opening in the roof of the mouth (a cleft palate), abnormal eye movements, hearing loss, and abnormalities of tooth development 51, 52. Some affected individuals have a feature called bimanual synkinesis, in which the movements of one hand are mirrored by the other hand. Bimanual synkinesis can make it difficult to do tasks that require the hands to move separately, such as playing a musical instrument.
Kallmann syndrome is caused by mutation (changes) in more than 20 genes 51, 52. Among the most common causes of Kallmann syndrome are mutations in the ANOS1, CHD7, FGF8, FGFR1, PROK2, or PROKR2 gene. In some cases, affected individuals have mutations in more than one of these genes. Additionally, researchers have identified mutations in other genes that may contribute to the development and features of Kallmann syndrome, but are unlikely to cause the disease on their own.
The genes associated with Kallmann syndrome play roles in the development of certain areas of the brain before birth. Although some of their specific functions are unclear, these genes appear to be involved in the formation and movement (migration) of a group of nerve cells that are specialized to process the sense of smell (olfactory neurons). These nerve cells originate in the developing nose and then migrate together to a structure in the front of the brain called the olfactory bulb, which is critical for the perception of odors. Studies suggest that the genes associated with Kallmann syndrome are also involved in the migration of neurons that produce a hormone called gonadotropin-releasing hormone (GnRH). Like olfactory neurons, GnRH-producing neurons migrate from the developing nose to the front of the brain. GnRH controls the production of several hormones that direct sexual development before birth and during puberty. These hormones are important for the normal function of the ovaries in women and testes in men.
Studies suggest that mutations in genes associated with Kallmann syndrome disrupt the migration of olfactory nerve cells and GnRH-producing nerve cells in the developing brain. If olfactory nerve cells do not extend to the olfactory bulb, a person’s sense of smell will be impaired or absent. Misplacement of GnRH-producing neurons in the brain prevents the production of other sex hormones, which interferes with normal sexual development and causes the characteristic features of hypogonadotropic hypogonadism. It is unclear how gene mutations lead to the other signs and symptoms that can occur in Kallmann syndrome. Because the features of this condition vary among individuals, additional genetic and environmental factors likely contribute to this disease.
Together, mutations in known genes account for about 30 percent of all cases of Kallmann syndrome. In cases without a mutation in one of the identified genes, the cause of the condition is unknown. Researchers are looking for additional genetic changes that can cause Kallmann syndrome.
Each of these genes have varied pattern of affecting families, i.e. inheritance (the way that the disorder passes from parents to offspring). All forms of Mendelian inheritance (autosomal dominant, autosomal recessive, and X-lined recessive) as well more complex oligogenic inheritance patterns are now recognized. Understanding the genetic basis of the disorder is crucial not only for genetic counseling for determine the risk of transmission to the next generation, but also for fostering new gene discovery as well as bench-to-bedside research.
Most of the time, people with Kallmann syndrome resulting from an ANOS1 gene mutation inherit the mutation from their mothers, who carry a single altered copy of the gene in each cell (and generally do not have any signs or symptoms of the condition). Other people have Kallmann syndrome as a result of a new mutation in the ANOS1 gene.
The diagnosis of Kallmann syndrome is based on the clinical evidence of arrested sexual maturation or hypogonadism and the incomplete sexual maturation by Tanner staging on physical examination. Apart from the physical examination, biochemical testing is also critical for diagnosis of Kallmann syndrome. As GnRH is not measurable, serum concentration of the gonadotropins (LH and FSH) and sex hormones are used for diagnosis.
Kallmann syndrome treatment involve hormone replacement therapies and this is usually tailored the clinical need of the patients. Typically, once the diagnosis is made, in both sexes, treatment is aimed at inducing puberty and maintaining normal hormonal levels. Subsequently, treatment may also be need for inducing fertility for achieving pregnancy.
Stress-Induced Hypogonadotropic Hypogonadism
When calorie intake falls short of energy expenditure, the physiological stress decreases hypothalamic gonadotropin-releasing hormone (GnRH) pulse frequency and amplitude, leading to low FSH and LH levels. This explains the anovulation and amenorrhea that can occur in female athletes and individuals with eating disorders due to lack of adequate caloric intake or excessive exercise 18, 19.
Primary Ovarian Insufficiency
In cases of amenorrhea with elevated levels of FSH, the problem lies in the ovaries. Premature ovarian failure occurs when ovarian failure and menopause occur before age 40. When this happens, FSH levels are elevated due to the lack of negative feedback from the ovaries. Although there may be multiple genetic causes, most cases are idiopathic.
Turner syndrome is the most common genetic disorder causing premature ovarian failure. Turner syndrome is a rare chromosomal disorder that affects only females. Turner syndrome is caused by the loss of an X chromosome where one of the X chromosomes (sex chromosomes) is missing (XO karyotype) or partially missing 53, 54, 55. The reason that this occurs is unknown and is believed to result from a random event (Turner Syndrome. https://rarediseases.org/rare-diseases/turner-syndrome/)). In some people, the chromosomal abnormality appears to arise spontaneously (de novo) due to an error in the division of a parent’s reproductive cells, either in the father’s sperm or the mother’s egg (Turner Syndrome. https://rarediseases.org/rare-diseases/turner-syndrome/)).
The genetic changes of Turner syndrome may be one of the following (Turner Syndrome. https://rarediseases.org/rare-diseases/turner-syndrome/)):
- Monosomy. The complete absence of an X chromosome generally occurs because of an error in the father’s sperm or in the mother’s egg. This results in every cell in the body having only one X chromosome.
- Mosaicism. In many people with Turner syndrome, only a certain percentage of cells may be affected. This is referred to as mosaicism. Specifically, some cells have the normal 46 chromosomes (one cell line) while other cells do not have the normal 46 chromosomes (second cell line). This second cell line may contain various abnormalities such as partial or complete loss of the X chromosome. In these cases, the loss of genetic material from the X chromosome usually occurs because of spontaneous errors very early during fetal development. Theoretically, individuals with Turner syndrome mosaicism may have fewer developmental problems because fewer cells are affected. However, this is difficult to predict. Further research is necessary to completely understand the complicated factors involved in the development of the various symptoms associated with Turner syndrome.
- X chromosome changes. Changed or missing parts of one of the X chromosomes can occur. Cells have one complete and one altered copy. This error can occur in the sperm or egg with all cells having one complete and one altered copy. Or the error can occur in cell division in early fetal development so that only some cells contain the changed or missing parts of one of the X chromosomes (mosaicism).
- Y chromosome material. In a small percentage of Turner syndrome cases, some cells have one copy of the X chromosome and other cells have one copy of the X chromosome and some Y chromosome material. These individuals develop biologically as female, but the presence of Y chromosome material increases the risk of developing a type of cancer called gonadoblastoma.
Turner syndrome can cause many different symptoms. The symptoms may be mild for some people. But for others, Turner syndrome can cause serious health problems. People with Turner syndrome may be born with heart and kidney defects. They usually don’t have typical sexual development and are infertile. They are also at risk for other health problems such as high blood pressure, type 2 diabetes, osteoporosis, and thyroid problems.
Some of the symptoms of Turner syndrome affect a person’s appearance. Most people with Turner syndrome are shorter than average. They may also have physical features such as:
- A neck that is short and has extra skin (a “webbed” neck)
- A low hairline in the back
- Low-set ears
- Swollen hands and feet
Girls with Turner syndrome will present with primary amenorrhea and underdeveloped ovaries (streak ovaries) and elevated FSH levels 56.
Turner syndrome may be diagnosed before birth (prenatally), during infancy or in early childhood. Occasionally, in females with mild signs and symptoms of Turner syndrome, the diagnosis is delayed until the teen or young adult years.
There is no cure for Turner syndrome, but there are treatments for some of the symptoms. Girls and women with Turner syndrome need ongoing medical care from a variety of specialists. Regular checkups and appropriate care can help most girls and women lead healthy, independent lives.
The primary treatments for nearly all girls and women with Turner syndrome include hormone therapies:
- Growth hormone (GH). Growth hormone therapy — usually given daily as an injection of recombinant human growth hormone — is typically recommended to increase height as much as possible at appropriate times during early childhood until the early teen years. Starting treatment early can improve height and bone growth.
- Estrogen replacement therapy (ERT). Most girls with Turner syndrome need to start estrogen and related hormone therapy in order to begin puberty. Often, estrogen therapy is started around age 11 or 12 years. Estrogen helps to promote breast development and improve the size (volume) of the uterus. Estrogen helps with bone mineralization, and when used with growth hormone, may also help with height. Estrogen replacement therapy usually continues throughout life, until the average age of menopause is reached.
Other treatments are tailored to address particular problems as needed. Assisted reproduction technologies can help some women with Turner syndrome get pregnant. Regular checkups have shown substantial improvements in the health and quality of life for girls and women with Turner syndrome.
Pituitary Adenomas
Pituitary adenomas can develop from any of the cell types in the pituitary. Pituitary adenomas derived from gonadotropic cells are most often nonfunctioning or function within normal hormone levels and are diagnosed due to symptoms from mass effect rather than hormone secretions. However, in sporadic, these tumors can secrete excess FSH and/or LH and can cause ovarian hyperstimulation 57.
GnRH Agonists and Antagonists medications
GnRH agonists initially stimulate secretion of LH and FSH, but when given continuously, suppress LH and FSH release. This results in ovarian suppression and decreased estrogen levels. GnRH antagonists can acutely suppress LH and FSH secretion 19.
Both GnRH agonists and antagonists have a role in the treatment of certain breast and prostate cancers, endometriosis, and uterine leiomyomas.
Assisted Reproduction Techniques (ART)
Techniques such as in vitro fertilization (IVF) and intracytoplasmic sperm injection (ICSI) help couples by using FSH to stimulate multiple follicles in the ovaries to harvest multiple eggs for fertilization. FSH is available as urinary FSH with or without LH or recombinant FSH 58. GnRH agonists or antagonists can be used during these cycles to prevent the LH surge and ovulation 19.
Luteinizing hormone (LH)
Luteinizing hormone (LH) is a hormone produced by the anterior pituitary in response to gonadotropin-releasing hormone (GnRH) from the hypothalamus 11, 18. Luteinizing hormone (LH) is essential for sexual development and reproduction in both men and women 1, 2. LH is regulated by gonadotropin-releasing hormone (GnRH) from the hypothalamus which is sensitive to circulating levels of sex hormones (i.e., estrogen, progesterone, and testosterone). Gonadal sex hormones, estrogen, progesterone, and testosterone exert negative feedback, thus decreasing the secretion of LH 20.
LH interacts with receptors on ovarian follicles and promotes their maturation. In the middle of the menstrual cycle, a surge of LH triggers ovulation and production of progesterone by the corpus luteum that is necessary for the maturation of the uterine endometrium for implantation of the fertilized egg. In males, LH stimulates production of testosterone by the testes. Luteinizing hormone (LH) is used clinically in assisted reproduction techniques (ART) and in vitro fertilization (IVF) to stimulate ovarian follicle maturation. Both urinary derived (menotropin, Menopur, which also has FSH activity) and recombinant forms (lutropin alfa: Luveris) of human LH have been developed, but not all are available in the United States 26. LH is generally administered by subcutaneous injection in a cyclic and step-wise fashion. The dosages and regimens of administration vary by indication. These agents should be used only by health care workers with expertise in management of infertility and hypogonadism.
Luteinizing hormone (LH) function
Luteinizing hormone (LH) release is stimulated by gonadotropin-releasing hormone (GnRH) and inhibited by estrogen in females and testosterone in males. Luteinizing hormone (LH) has various functions, which differ between women and men. In both sexes, LH contributes to the maturation of primordial germ cells. In men, LH causes the testes’ Leydig cells to produce testosterone. In women, LH triggers the creation of steroid hormones from the ovaries 59. Additionally, LH helps regulate the length and order of the menstrual cycle in females by playing roles in ovulation and implantation of an egg in the uterus 60.
Luteinizing hormone (LH) function in Fetal Development
Luteinizing hormone (LH) and human chorionic gonadotropin (hCG) are 2 essential hormones in the development of both sexes. Their levels can be seen to fluctuate throughout development. In male fetuses, human chorionic gonadotropin (hCG) begins at a high level in the plasma and quickly decreases between weeks 10 and 20 of gestation and then slowly declines afterward 11. In contrast, LH secretion increases by week 10, peaks before week 20, and decreases gradually 11. Increased plasma levels of human chorionic gonadotropin (hCG) early on in gestation are a more significant contributor to testosterone production by Leydig cells than LH early in the development of a fetus 11. However, as LH levels rise, the regulation of testosterone formation changes to LH, which is driven by weeks 15 to 20 of gestation 11. This change in regulation can be exemplified by anencephalic male fetuses that are deficient in LH. In these fetuses, normal development of the male reproductive tract occurs while hCG levels are high initially. However, due to the lack of LH, the development of the external genitalia is impeded when hCG levels decrease around gestational weeks 15 to 20 61.
In female fetuses, the peak levels of LH are higher than in male fetuses; this has been thought to be due to negative feedback of higher testosterone levels on the hypothalamic-pituitary-gonadal axis in male fetuses. Female fetuses have a lower level of gonadal hormones during gestation because the development of the female reproductive tract is not dependent on circulating levels of LH or human chorionic gonadotropin (hCG). The developing ovary does not express LH/choriogonadotropin receptors until the 16th week of gestation, which is why there is minimal steroidogenesis in the ovary until after delivery of the fetus 61.
Luteinizing hormone (LH) function After Delivery
After delivery, regardless of sex, a sharp increase in luteinizing hormone (LH) levels is seen because the mother withdraws estrogen. After this temporary increase, LH levels begin to decline and stay at low basal levels until prepuberty starts in both sexes 61.
Luteinizing hormone (LH) function At Puberty
In the years leading up to puberty in both sexes, there is a slow increase in the secretion of luteinizing hormone (LH) at night. As puberty progresses, LH begins to be secreted less so in a nighttime pattern followed by a pulsatile pattern throughout the whole day. This increase in gonadotropin secretion helps to stimulate gonadal steroidogenesis, which is important for maturation 61.
Luteinizing hormone (LH) function in Males
In males, luteinizing hormone (LH) stimulates testosterone release by the Leydig cells of the testes.
Luteinizing hormone (LH) function in Females
In females, luteinizing hormone (LH) stimulates steroid hormone release from the ovaries, ovulation, and the release of progesterone after ovulation by the corpus luteum 62. Ovulation is made possible by the combined actions of the hypothalamus, pituitary, and ovary 60. The hypothalamus begins the ovulation process by releasing gonadotropin-releasing hormone (GnRH) in a pulsatile fashion. This pulsatile release causes the anterior pituitary to release luteinizing hormone (LH) and follicle-stimulating hormone (FSH), which then act on the ovarian follicle. The ovarian follicle comprises 3 essential cells: theca, granulosa, and oocyte. Luteinizing hormone (LH) causes the theca cells to make androstenedione. Androstenedione then converts to estradiol via aromatase, which follicle-stimulating hormone (FSH) stimulates. Upon achieving a critical concentration of estradiol, the negative feedback on LH that normally occurs by estrogen is shut off, and it begins to have positive feedback on LH release, which causes an “LH surge”, which initiates ovulation. Once ovulation has occurred, the ovarian follicle becomes the corpus luteum. The corpus luteum secretes progesterone and is stimulated by LH or human chorionic gonadotropin (hCG) if a pregnancy occurs 19.
Progesterone is a steroid hormone that is responsible for preparing the endometrium for the uterine implantation of the fertilized egg and maintenance of pregnancy 60, 63. If a fertilized egg implants, the corpus luteum secretes progesterone in early pregnancy until the placenta develops and takes over progesterone production for the remainder of the pregnancy 63.
Luteinizing hormone (LH) and Testicular Dysfunction in Chronic Kidney Disease
Low libido, erectile dysfunction, and smaller testicular size are all signs of testicular dysfunction. All these signs can be present in end-stage kidney disease. Testosterone concentration in the plasma and how quickly testosterone production takes place are usually low in patients with chronic kidney disease. Spermatogenesis (sperm production) has been noted to be either lowered or completely absent as well 11. After kidney transplantation, the low libido, erectile dysfunction, and smaller testicular size changes can be reversed and return to normal 11. Studies have shown that this testicular dysfunction and altered testosterone concentration results from higher levels of luteinizing hormone (LH) in the plasma and lower amounts of secretory luteinizing hormone (LH) pulses seen in men with end-stage kidney disease when compared to healthy subjects or men who underwent a successful renal transplant. This is significant because LH’s pulsatile secretion is necessary for the testes’ gonadotropin receptors to function properly. Furthermore, sustained high levels of LH in the blood and testes can cause a loss of gonadotropin receptors in the testes 64.
Luteinizing hormone (LH) diagnostic use
Women use ovulation predictor kits to determine the exact time of ovulation while trying to get pregnant. These ovulation predictor kits quantify luteinizing hormone (LH) levels in the urine 63.
Human chorionic gonadotropin (hCG)
Human chorionic gonadotropin (hCG) is a pregnancy hormone produced primarily by syncytiotrophoblastic cells of the placenta of a pregnant woman 65, 66, 67. Smaller amounts of human chorionic gonadotropin (hCG) are also produced in the pituitary gland, the liver, and the colon 68. Human chorionic gonadotropin (hCG) plays an important role in synchronizing fetal and endometrial developments. Early in pregnancy, the level of human chorionic gonadotropin (hCG) increases in the blood and is eliminated in the urine. A pregnancy test detects human chorionic gonadotropin (hCG) in the blood or urine and confirms or rules out pregnancy 67. Throughout pregnancy, human chorionic gonadotropin (hCG) is also a marker of placental function.
In everyday clinical practice, human chorionic gonadotropin (hCG) is mainly used to diagnose pregnancy and to supervise first trimester adverse pregnancy outcomes. Abnormalities in the production and the circulating levels of hCG during specific periods of gestation have been associated with a large array of pregnancy complications, such as miscarriages 69, fetal chromosomal anomalies 70, pre-eclampsia 71, 72, disturbances in fetal growth and development 73 and gestational trophoblastic diseases 74. Trophoblastic cancers (hydatidiform mole, choriocarcinoma, and germ cell tumors) are associated with high serum levels of hCG-related molecules 65, 75.
Nevertheless, the persistence of low hCG concentrations in a non-pregnant woman is not always malignant and can even be benign 76, 77. In addition, very high concentrations of hCG have been shown to have deleterious effects on fetal tissues, notably on fetal gonadal steroidogenesis 78. To avoid this, the human fetal tissue macrophages are thought to eliminate excess hCG. Katabuchi et al. 79 have shown that hCG induces the formation of vacuoles in human monocytes and hypothesized that these vacuoles would be involved in the protection of fetal tissues.
Multiple factors influence hCG levels during pregnancy. Among them, endocrine disruptive chemicals (EDCs), particularly bisphenol A and para-nonylphenol, can modulate hCG production and cause fetal damage as well as long-lasting consequences in adult life 80.
Human chorionic gonadotropin (hCG) stimulates the corpus luteum to produce progesterone to maintain the pregnancy. Human chorionic gonadotropin (hCG), is crucially involved in processes such as implantation and placentation, two milestones of pregnancy whose successful progress is a prerequisite for adequate fetal growth. Moreover, hCG determines fetal fate by regulating maternal innate and adaptive immune responses allowing the acceptance of the foreign fetal antigens 81. As one of the first signals provided by the embryo to its mother, human chorionic gonadotropin (hCG) has the potential to regulate very early pregnancy-driven immune responses, allowing the establishment and preservation of fetal tolerance.
During the early weeks of pregnancy, human chorionic gonadotropin (hCG) is important in maintaining function of the corpus luteum. Circulating human chorionic gonadotropin (hCG) interacts with the luteinizing hormone receptors (LHRs) of the ovary, promoting the corpus luteum and its production of progesterone which is necessary to maintain pregnancy and support the growth of the fetus. Production of human chorionic gonadotropin (hCG) increases steadily during the first trimester (8-10 weeks) of a normal pregnancy, peaking around the 10th week after the last menstrual cycle. Levels then fall slowly during the remainder of the pregnancy. Human chorionic gonadotropin is no longer detectable within a few weeks after delivery.
When a pregnancy occurs outside of the uterus (ectopic pregnancy), the level of human chorionic gonadotropin (hCG) in the blood increases at a slower rate. When an ectopic pregnancy is suspected, measuring the level of human chorionic gonadotropin (hCG) in the blood (quantitative test) over time may be useful in helping to make a diagnosis of ectopic pregnancy.
Similarly, the human chorionic gonadotropin (hCG) blood level may be abnormal when the developing baby (fetus) has a chromosome defect such as Down syndrome. An human chorionic gonadotropin (hCG) test is used routinely in conjunction with a few other tests as part of screening for fetal abnormalities.
Injections of human chorionic gonadotropin (hCG) mimic the surge in luteinizing hormone (LH) that is necessary for ovulation and are used in the therapy of female infertility, in assisted reproduction techniques (ART). In clinical trials, hCG resulted in pregnancies in approximately 30% of women. Human chorionic gonadotropin (hCG) prepared from urine of pregnant women and was approved for use in the United States in 1967 as treatment of ovulatory dysfunction in women desiring pregnancy. Subsequently, recombinant forms of hCG have been developed and licensed for use. Currently, hCG is available as a powder or in solution generically and under trade names such as Novarel and Pregnyl. Recombinant hCG is available as Overle. The dose and regimen of hCG therapy varies by indication and it should be used only by physicians with expertise in the management of infertility and hypogonadism. Common side effects include headache, nausea, anorexia, and local injection reactions. Uncommon, but potentially severe adverse events include ovarian hyperstimulation syndrome.
Figure 7. Human chorionic gonadotropin (hCG)
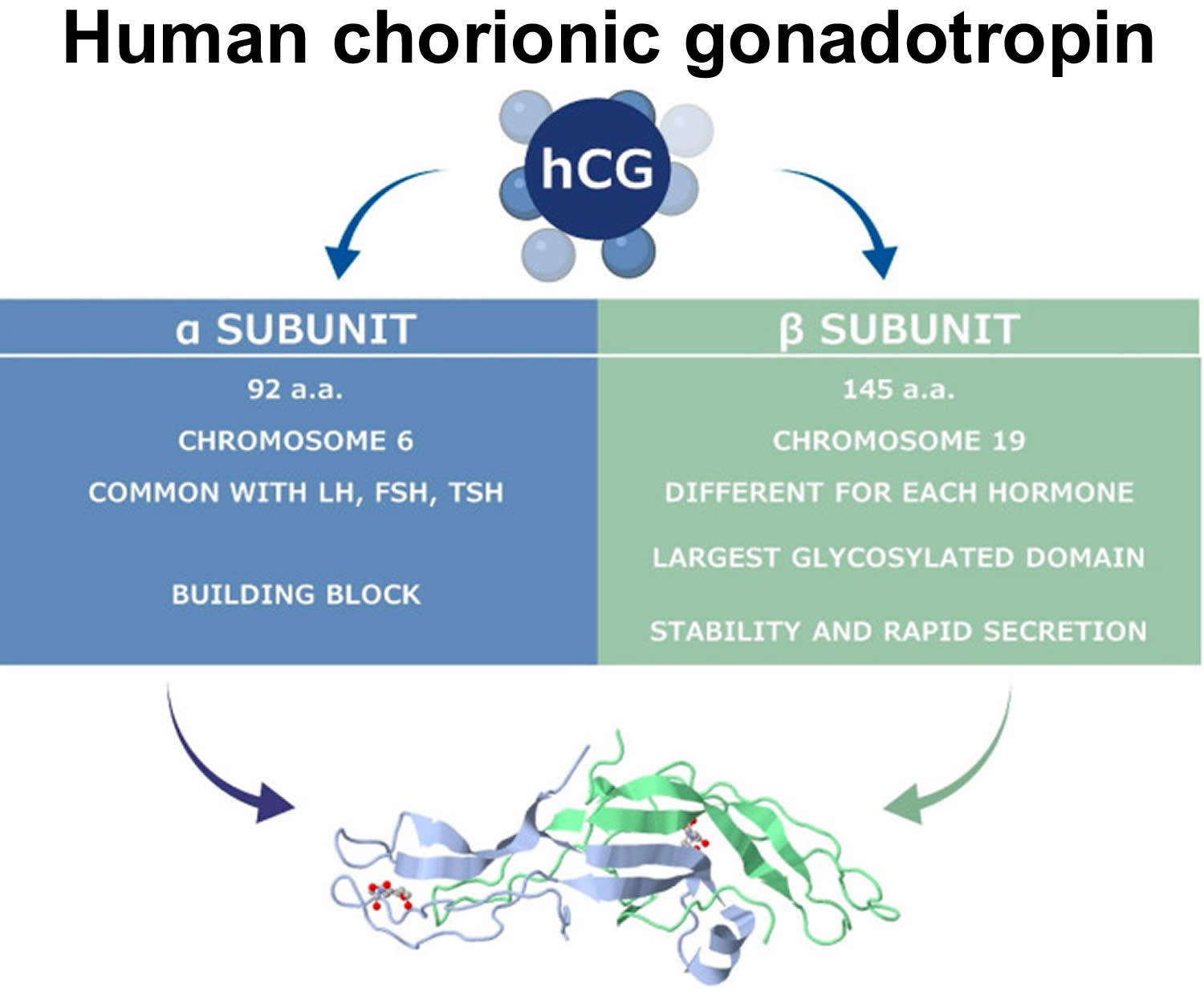
Figure 8. Human chorionic gonadotropin (hCG) actions during pregnancy and in non-pregnant woman
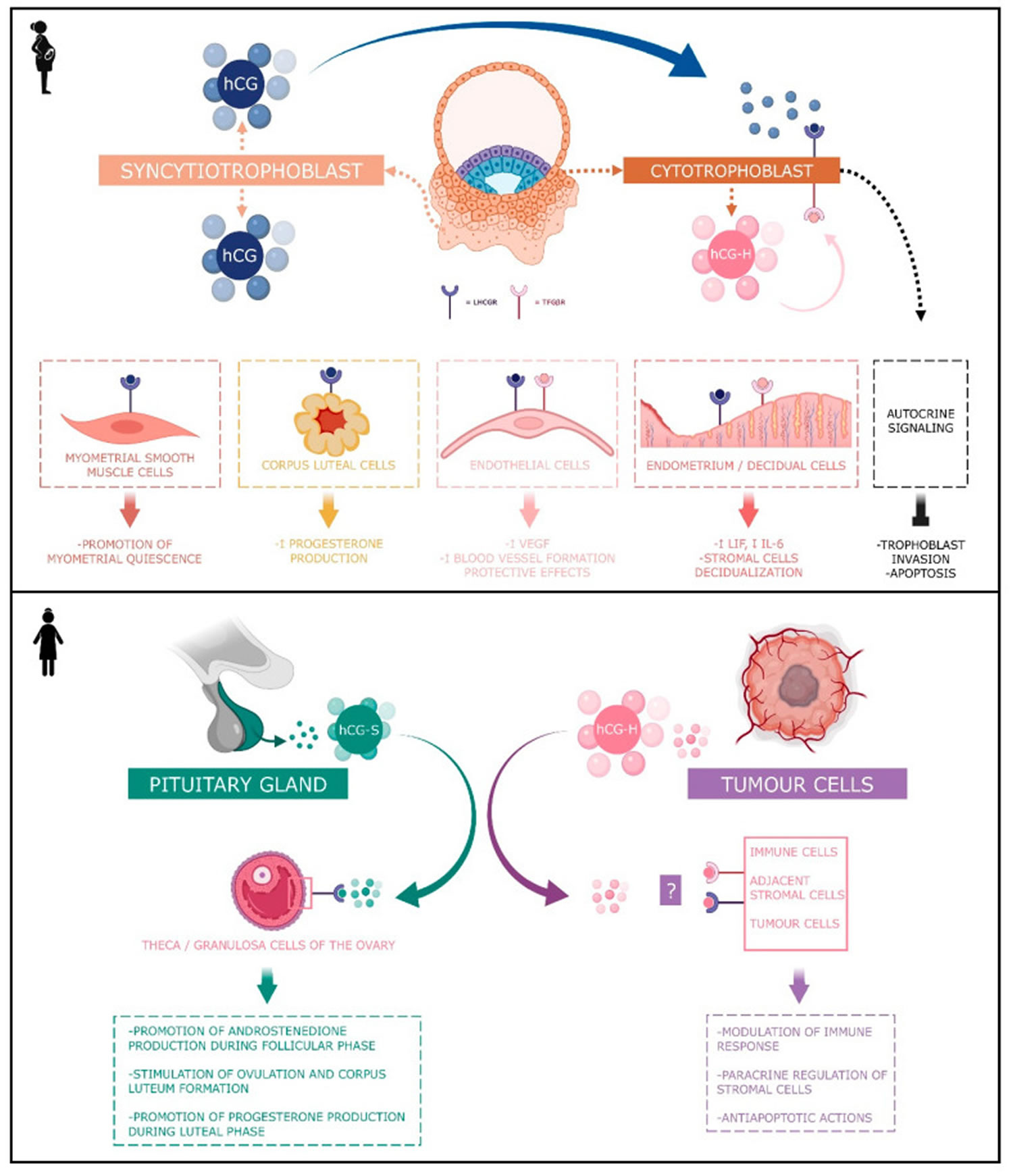
Footnotes: Human chorionic gonadotropin (hCG) has 4 major isoforms found in the serum and urine during pregnancy: classical hCG, hyperglycosylated hCG, free beta (β) subunit, and sulphated hCG 82. The classical form of hCG is schematized by a blue dot, the hyperglycosylated hCG by a pink dot and the sulphated form of hCG by a green dot. The blue receptor is the LH-hCG receptor (LHCGR) and the pink receptor is the transforming growth factor β receptor (TGFβR).
[Source 82 ]Human chorionic gonadotropin function
Human chorionic gonadotropin (hCG) has 4 major isoforms found in the serum and urine during pregnancy: classical hCG, hyperglycosylated hCG, free beta subunit, and sulphated hCG 82:
- Classical human chorionic gonadotropin (hCG) is one of the first molecules secreted by the embryo. Its RNA is transcribed as early as the eight-cells stage 83 and the blastocyst produces the protein before implantation 84, 85. During the implantation, human chorionic gonadotropin (hCG) is mainly secreted by the syncytiotrophoblast and less by the cytotrophoblast 82. Human chorionic gonadotropin (hCG) can be detected in the maternal blood 10 days after ovulation. Its concentration reaches its top level around the 10th and 11th weeks of gestation 82. Afterwards, this level decreases and remains basal from the 12th week of gestation onwards until the end of the pregnancy. However, it remains significantly higher than in non-pregnant women (Betz D, Fane K. Human Chorionic Gonadotropin. [Updated 2023 Aug 14]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK532950)), 86. By binding to its receptor called LH-hCG receptor (LHCGR), classical hCG acts on multiple types of cells: corpus luteum cells, myometrial smooth muscle cells, endothelial cells, and decidual cells. During the fifth and sixth days of embryogenesis, the blastocyst secretes hCG into the uterine cavity. This hormone binds to its LH-hCG receptor (LHCGR) on the deciduous surface. In response, the decidua prepares for implantation 87, 88, 89. Human chorionic gonadotropin (hCG) influences stromal cells by underpinning the decidualization and the prolactin secretion 90. D’Hauterive et al 88 have shown that the hCG–LHCGR complex also increases the secretion of leukemia inhibitory factor (LIF) and decreases the secretion of interleukine-6 (IL-6) by endometrial cells, factors affecting embryo implantation. This complex also promotes the differentiation of cytotrophoblasts into syncytiotrophoblasts 91. The hCG–LHCGR complex also regulates prostaglandin synthesis 92 and the formation of cAMP 93. Human chorionic gonadotropin (hCG) encourages trophoblast invasion and interstitial theca cell proliferation by over-modulating ERK and AKT signals 94, 95. Aside from this hCG-LHCGR complex, it has been shown that multiple hCG isoforms could stimulate trophoblastic invasion without regard to the LH-hCG receptor (LHCGR) 96.
- Hyperglycosylated hCG. Hyperglycosylated hCG β subunit has four oligosaccharide-linked instead of two in the classical form of the hCG β subunit 97. Hyperglycosylated hCG is massively produced during the first trimester of pregnancy, particularly by the extravillous cytotrophoblasts. Hyperglycosylated hCG represents the majority of the total hCG in the third week of gestation and the half during the fourth week 82. Then, it decreases rapidly until it completely disappears from the maternal blood circulation at the end of the first trimester 98. Hyperglycosylated hCG is useful for predicting pregnancy outcomes in women, with a first trimester suspicion of abortion. However, it is not considered as a better tool than the classical form of hCG 99. Hyperglycosylated hCG acts through autocrine instead of endocrine action. It decreases the apoptosis of trophoblast cells 100 and induces the implantation of the embryo 101 and trophoblastic invasion 102. Hyperglycosylated hCG is also massively secreted by choriocarcinomas and germ cell tumors 102, 103, 97. Its anti-apoptotic action would be achieved by its binding with the TGF-β receptor and independently of LH-hCG receptor (LHCGR). Hyperglycosylated hCG monitoring is useful in predicting Down’s syndrome 102, pre-eclampsia 104, and the therapeutic response to trophoblastic diseases, as well as in pregnancy predictions performed in in vitro fertilization (IVF) 105.
- Free beta subunit human chorionic gonadotropin (hCG). Free β subunit human chorionic gonadotropin (hCG) acts as an agonist of LH-hCG receptor (LHCGR) and an antagonist of the TGF-β receptor. Gestational hypertension could also be predicted by the abnormal rise in the circulating free β subunit of hCG. However, the association of β-hCG and inflammation, and oxidative stress in a pregnancy-caused hypertensive disorder, on the perinatal stage remains unclear. However, Wang et al. 106 demonstrated via a case–control study that the correlation of circulating free β subunit levels with inflammatory and oxidative stress markers in patients with pregnancy-induced hypertension in perinatal stage was statistically significant. Like hyperglycosylated hCG, maternal serum free β-hCG is also used as a biomarker in first trimester screening for fetal Down’s syndrome 107. The free β subunit also has a promotive action on cancer: germ cell malignancies, epithelial malignancies or carcinomas, adenocarcinomas, sarcomas, teratomas, blastomas, leukemias and lymphomas 108. For example, this action on the bladder carcinoma is exerted through the inhibition of apoptosis 109. According to Sirikunalai et al. 69, abnormally low (<0.5 MoM) or high (>2.0 MoM) free β subunit human chorionic gonadotropin (hCG) levels during gestation are generally associated with an increased risk of adverse pregnancy outcome (spontaneous abortion, preterm birth, low Apgar score, etc.).
- Sulphated human chorionic gonadotropin (hCG). Sulphated human chorionic gonadotropin (hCG) is produced by the pituitary gland in non-pregnant women and is secreted at the same time as LH during the cycle. Hence, its concentration ranges around one-fiftieth of the LH concentration 76, 110, 111, 112. While these levels are low, sulphated hCG is exactly 50 times more potent than LH 70 and acts the same way by stimulating androstenedione production during the follicular phase of the cycle as well as stimulating ovulation and corpus luteum formation. During the luteal phase, it may help stimulate progesterone production 76, 110, 111, 112.
Human chorionic gonadotropin (hCG) is a glycoprotein composed of two subunits, the alpha (α) and beta (β) subunits 68. The alpha-subunit is common to hCG, to the autocrine/paracrine hyperglycosylated hCG, to the hormone pituitary hCG, and to the hormones luteinizing hormone (LH), follicle stimulating hormone (FSH), and thyroid stimulating hormone (TSH), and to the common free alpha-subunit formed in excess 113. The beta-subunit of hCG, while structurally somewhat similar to the beta-subunit of luteinizing hormone (LH), differentiates hCG, hyperglycosylated hCG, and pituitary hCG from other molecules. Both human chorionic gonadotropin (hCG) and luteinizing hormone (LH) bind and function through a common hCG/LH receptor 113. The marked distinction between the human chorionic gonadotropin (hCG) and luteinizing hormone (LH) besides the absence of a beta subunit in LH is the difference in half-life. LH has a half-life of approximately 25 to 30 minutes 114, while human chorionic gonadotropin (hCG) has a much longer half-life at 24 to 37 hours 115, 116. This difference in half-life is critical to hCG’s function as a type of “super LH” during pregnancy to support the maintenance of an optimal intrauterine environment 117, 115, 118.
Human chorionic gonadotropin (hCG) is primarily catabolized by the liver, although about 20% is excreted in the urine 119. The beta-subunit is degraded in the kidney to make hCG β-core fragment (hCGβcf) which is measured by urine hCG tests 65.
The most well-known function of human chorionic gonadotropin (hCG) is the promotion of progesterone production during pregnancy. Human chorionic gonadotropin (hCG) stimulates ovarian corpus luteal cells to produce progesterone, therefore reinforcing the endometrial walls and preventing menstrual bleeding. This promotion of progesterone production is active in approximately 10% of the total length of the pregnancy or around 3 to 4 weeks following implantation. In a non-pregnant female, LH promotes progesterone production 113, 120, 121.
Studies done over recent years have shown hCG to be involved in a plethora of functions supporting the placenta, uterus, and fetus throughout pregnancy. These functions include the promotion of angiogenesis, immunosuppression, and blockage of phagocytosis of invading trophoblasts, promotion of growth and differentiation of fetal organs, and involvement in the adult brain and brainstem 122, 121.
Human chorionic gonadotropin (hCG) promotes the formation of new blood vessels (angiogenesis) and vasculogenesis through the upregulation of endocrine gland-derived vascular endothelial growth factor (EG-VEGF) 123. Uterine spinal arteries have hCG receptors that, when acted upon by hCG, undergo growth, and support the adequate blood supply and nutrition to the placenta. Human chorionic gonadotropin (hCG) also promotes the fusion of cytotrophoblast cells and their subsequent differentiation into syncytiotrophoblasts 122, 113.
Human chorionic gonadotropin (hCG) achieves many of its functions through the regulation of the expression of endocrine gland-derived vascular endothelial growth factor (EG-VEGF) and its receptors 123. The EG-VEGF receptors are G protein-coupled receptors (GPCRs), prokineticin 1 (PROKR1), and prokineticin 2. EG-VEGF is an angiogenic factor specific to endocrine tissues, including the placenta. EG-VEGF expression peaks around the same time as the peak of hCG concentration at approximately 8 to 11 weeks gestation 123. As an angiogenic factor, EG-VEGF expression increases in conditions of hypoxia. EG-VEGF and its receptors are regulators of both pathological and normal development of the fetus. EG-VEGF, PROKR1, and PROKR2 levels are significantly higher in fetal growth-restricted patients. Some have proposed that increases in EG-VEGF expression and its receptors brought on by increased levels of hCG are a form of compensation in fetal growth restriction 121, 124, 118, 120.
Maternal human chorionic gonadotropin (hCG) has implications in the development of fetal organs during development. There are hCG receptors in the fetal liver and kidney that are completely absent in adult organs. Human chorionic gonadotropin (hCG) has also been shown to support the growth and development of the umbilical cord 113, 118, 120.
Researchers have found hCG receptors in various areas of the adult female brain, including the hippocampus, hypothalamus, and brain stem. Speculation is that the presence of these receptors in the brain are involved in the pathophysiology of hyperemesis gravidarum. Other contributing factors may involve a combination of rising hormone levels overall, including estrogen, progesterone, and serum thyroxine, in addition to elevated hCG 113, 120, 125.
Human chorionic gonadotropin angiogenic actions
Classical human chorionic gonadotropin (hCG) has angiogenic actions through the LHCGR (LH-hCG receptor) and achieves many of its functions through the regulation of the expression of endocrine gland-vascular endothelial growth factor (EG-VEGF) and its receptors 126, 127, 127, 66.
Human chorionic gonadotropin (hCG) increases blood vessel formation (angiogenesis) and the migration and maturation of pericytes in different in vitro and in vivo models. Through this action, the trophoblast can form plugs that prevent maternal blood from bleeding into the intervillous spaces during early pregnancy 66, 128, 129, 130, 131.
Human chorionic gonadotropin (hCG) also enhances the secretion of VEGF through the activation of NF-κB on angiogenesis during the luteal phase (Berndt S., D’Hauterive S.P., Blacher S., Pequeux C., Lorquet S., Munaut C., Applanat M., Hervé M.A., Lamandé N., Corvol P., et al. Angiogenic activity of human chorionic gonadotropin through LH receptor activation on endothelial and epithelial cells of the endometrium. FASEB J. 2006;20:2630–2632. doi: 10.1096/fj.06-5885fje)), 132, 133. In addition, hCG shields vascular endothelial cells against oxidative stress through the inhibition of apoptosis, activation of cell survival signaling, and mitochondrial function retention 134. Jing et al. 135 have shown that the decreased production of the β subunit human chorionic gonadotropin in early pregnant women could act on the expression of VEGF-MEK/ERK signal pathway by down-regulating it. It reduces angiogenesis and eventually leads to the abnormal angiogenesis of the villosities, a mechanism which may be an important factor of missed abortion 135.
As hyperglycosylated hCG subunit still presents a potent angiogenic effect but is acting regardless of LHCGR signaling pathways 136, 137. Gallardo et al. 138 have suggested that the striking overlapping of hCG and Heme oxygenase-1 (HO-1) functions in pregnancy could indicate that hCG hormonal effects are mediated by HO-1 activity, which may be affected by a HMOX1polymorphism in humans.
Human chorionic gonadotropin (hCG) and its hyperglycosylated isoform are accordingly considered pro-angiogenic molecules granting adequate fetal perfusion and fetal-maternal exchanges.
Human chorionic gonadotropin immunological actions
The immunomodulatory properties of human chorionic gonadotropin (hCG) are various and important for maternal tolerance of the embryo, an essential mechanism for the embryonic implantation and development 127, 139, 140. Immune cells situated in the uterine cavity play a key role in the embryo implantation 141, 142.
Several studies have supported the function of hCG in the prevention of fetoplacental tissue rejection through inhibitory immune-mediated mechanisms 143, 144. Some groups have shown that an anti-macrophage inhibitory factor is upregulated by human chorionic gonadotropin (hCG) activity during pregnancy, thus reducing macrophage activity at the uterine-placental interface 145, 146, 147 Other studies support a more proximate mechanism of action in which hCG directly suppresses immune actions taken against the fetus 122, 121, 148.
Human chorionic gonadotropin test
Human chorionic gonadotropin (hCG) is an important hormone in pregnancy, and its clinical utility is primarily centered around its detection in early pregnancy, along with serial measurement during pregnancy and pregnancy-related complications. Early in pregnancy, the level of human chorionic gonadotropin (hCG) increases in the blood and is eliminated in the urine. A pregnancy test detects human chorionic gonadotropin (hCG) in the blood or urine and confirms or rules out pregnancy 67. Levels of hCG can vary widely between women with normal pregnancies. Typically, serum and urine concentrations of hCG rise exponentially in the first trimester of pregnancy, doubling about every 24 hours during the first 8 weeks 65. The peak is usually around 10 weeks of gestation and then levels decrease until about the 16th week of gestation where they remain fairly constant until term 149. Patients who have hCG levels that plateau prior to 8 weeks or that fail to double commonly have a nonviable pregnancy, whether intra-uterine or extra-uterine. Extra-uterine (ectopic) pregnancies usually have a rate-of-rise that is low without the typical doubling. However, given the large range of normal hCG levels and inconsistent rates-of-rise of this hormone, checking serum levels is typically paired with ultrasound evaluation to improve sensitivity and specificity 150.
Some signs and symptoms of ectopic pregnancy include:
- Abnormal vaginal bleeding—because a woman is pregnant, she may not have a regular period but then may have light bleeding or spotting with an ectopic pregnancy
- Low back pain
- Pain or cramping in the lower abdomen or on one side of the pelvis
If untreated ectopic pregnancy, signs and symptoms may get worse and may include:
- Dizziness, weakness
- Feeling faint or fainting
- Low blood pressure
- Pain in the shoulder area
- Sudden, sharp pain in the pelvic area
- Fever, flu-like symptoms
- Vomiting
The area around an ectopic pregnancy may rupture and start to bleed, and, if undiagnosed, can lead to cardiac arrest and death.
An human chorionic gonadotropin (hCG) test may be ordered prior to a medical procedure or treatment that might be harmful during pregnancy.
Certain cancers can also produce either human chorionic gonadotropin (hCG) or hCG-related hormone. Trophoblastic cancers (hydatidiform mole, choriocarcinoma, and germ cell tumors) are associated with high serum levels of hCG-related molecules 65, 75. Detection of hCG is also useful in the evaluation of trophoblastic disease, including complete and partial hydatidiform mole, postmolar tumor, gestational choriocarcinoma, testicular choriocarcinoma, and placental site trophoblastic disease. All of these entities produce hCG, varying levels of which are reported on commercial assays. A total hCG level of greater than 100,000 mIU/mL in early pregnancy, for example, is highly suggestive of a complete hydatidiform mole, although many normal pregnancies may reach this level at their peak around weeks 8 to 11 of gestation 151. Precise hCG measurements are important to assess the tumor mass, the successful treatment of trophoblastic cancer and to test for recurrence or persistence of trophoblastic disease 152.
The development of molar pregnancy correlates with fluxes in the levels of free beta-subunit of hCG. Return of hCG to zero following delivery or termination of pregnancy ranges from 7 to 60 days 153. Trending the fall of hCG levels can be important in termination of molar pregnancies and also following the termination of normal or ectopic pregnancies to be assured that the therapy has been successful. In a complete molar pregnancy, it is not uncommon to see large theca-lutein cysts as a result of increased stimulation of the ovaries by excess free beta-subunit hCG 154, 155, 156.
A molar pregnancy or hydatidiform mole, is a tumor arising from the trophoblast, which surrounds a blastocyst and subsequently develops into the chorion and amnion 155, 156. Hydatidiform mole may manifest as a complete or partial molar pregnancy. A complete hydatidiform mole is usually diploid with a 46 XX karyotype. There is trophoblastic hyperplasia producing a mass of multiple vesicles with little evidence of fetal and embryonic development. A partial hydatidiform mole is usually triploid due to dispermic fertilization or fertilization with an unreduced diploid sperm. In contrast to the complete mole, there is usually evidence of fetal development with an enlarged placenta 121, 155.
Patients with a history of prior molar pregnancy are at a 10-fold greater risk of a second hydatidiform pregnancy compared to the general population. The recommendation is that these women have their hCG levels monitored throughout pregnancy, as well as undergo evaluation by early ultrasonography 155, 156, 67.
Abnormal levels of hCG are associated with adverse pregnancy outcomes such as molar pregnancies and fetal growth restrictions. The intrauterine conditions are dependent upon placental function as the placenta is the main source of fetal nourishment. Suboptimal conditions due to an atrophic placenta may contribute to the risk of low birth weight. Several studies support the correlation between low birth weight and the risk of developing chronic conditions such as diabetes and hypertension later in life 157, 154, 155, 113.
Several clinical studies support the association of hCG concentration abnormalities with adverse fetal outcomes. This association varies with gestational age as hCG levels fluctuate throughout the pregnancy 155, 156, 154.
In the first trimester, low levels of hCG have correlated with spontaneous abortion and preeclampsia. Some studies have shown an association between low hCG concentrations (especially of the free beta-subunit of hCG) during the latter half of the first trimester and low birth weight due to attenuated fetal growth. Interestingly, some studies show that higher maternal hCG concentrations at the end of the first trimester are associated with fetal growth acceleration only in female-sex fetuses 157, 154.
In the second trimester, high levels of hCG have associations with gestational hypertension, spontaneous abortion, preeclampsia, fetal growth restriction (low birth weight), and pre-term delivery; this is in contrast to the association of low levels of hCG and low birth weight observed in the first trimester of pregnancy 154, 158.
How is human chorionic gonadotropin used?
Qualitative human chorionic gonadotropin (hCG) testing detects the presence of human chorionic gonadotropin (hCG) and is routinely used to screen for a pregnancy. This test may be performed by a laboratory, at a doctor’s office, or at home using a home pregnancy test kit. Methods will vary slightly but for most, a test strip is dipped into a collected cup of urine or exposed to a woman’s urine stream. A colored line (or other color change) appears within the time allotted per instructions, usually about 5 minutes. For accurate test results, it is important to carefully follow the test directions. (See the article on Home Testing: Avoiding Errors for more on this.) If the test is negative, it is often repeated several days later. Since human chorionic gonadotropin (hCG) rises rapidly, an initial negative test can turn positive within this time period.
Quantitative human chorionic gonadotropin (hCG) testing, often called beta human chorionic gonadotropin (β-hCG), measures the amount of human chorionic gonadotropin present in the blood. It may be used to confirm a pregnancy. It may also be used, along with a progesterone test, to help diagnose an ectopic pregnancy, to help diagnose and monitor a pregnancy that may be failing, and/or to monitor a woman after a miscarriage.
Human chorionic gonadotropin (hCG) blood measurements may also be used, along with a few other tests, as part of screening for fetal abnormalities.
Occasionally, an human chorionic gonadotropin test is used to screen for pregnancy if a woman is to undergo a medical treatment, be placed on certain drugs, or have other testing, such as x-rays, that might harm the developing baby. This is usually done to help confirm that the woman is not pregnant. It has become standard practice at most institutions to screen all female patients for pregnancy using a urine or blood human chorionic gonadotropin test before a medical intervention, such as an operation, that could potentially harm a fetus.
What does human chorionic gonadotropin test result mean?
- A negative human chorionic gonadotropin result means that it is unlikely that a woman is pregnant. However, tests performed too early in a pregnancy, before there is a significant human chorionic gonadotropin (hCG) level, may give false-negative results. The test may be repeated a few days later if there is a strong possibility of pregnancy.
- A positive human chorionic gonadotropin means that a woman is likely pregnant.
- However, blood or protein in the urine may cause false-positive pregnancy results. Urine human chorionic gonadotropin (hCG) tests may give a false-negative result if the urine is too diluted or if testing is done too soon in the pregnancy.
- Certain drugs such as diuretics and promethazine (an antihistamine) may cause false-negative urine results. Other drugs such as anti-convulsants, anti-parkinson drugs, hypnotics, and tranquilizers may cause false-positive results. The presence of protein in the urine (proteinuria), blood in the urine (hematuria), or excess pituitary gonadotropin may also cause a false positive.
- There are reports of false-positive blood human chorionic gonadotropin (hCG) results due to the presence of certain types of antibodies that some individuals produce or fragments of the human chorionic gonadotropin (hCG) molecule. Generally, if results are questionable, they may be confirmed by testing with a different method.
The blood level of human chorionic gonadotropin in a woman with an ectopic pregnancy usually rises at a slower rate than normal. Typically, human chorionic gonadotropin (hCG) levels double about every two days for the first four weeks of a normal pregnancy, then slow to every 31/2 days by six weeks. Those with failing pregnancies will also frequently have a longer doubling time early on or may even show falling human chorionic gonadotropin (hCG) concentrations during the doubling period. Human chorionic gonadotropin (hCG) concentrations will drop rapidly following a miscarriage. If human chorionic gonadotropin (hCG) does not fall to undetectable levels, it may indicate remaining human chorionic gonadotropin-producing tissue that will need to be removed (dilation and curettage – D&C).
How does the test that I do at home myself compare with the results of a test done in a lab?
Home pregnancy testing is very similar to qualitative urine human chorionic gonadotropin testing performed in the laboratory, but there are factors surrounding its use that are important to note.
- Home tests come with very specific directions that must be followed explicitly. If you are using a home test, follow the directions extremely carefully. There can be variability in sensitivity to detecting the presence of human chorionic gonadotropin with different brands of home pregnancy kits.
- Home tests are sometimes done too soon after the missed menstrual cycle to result in a positive test. It typically takes 10 days after a missed menstrual period before the presence of human chorionic gonadotropin can be detected by the urine test.
- All urine human chorionic gonadotropin tests should be done on a first morning urine sample, if possible. Urine becomes more dilute after ingestion of liquids (coffee, juice, water, etc.) and urine human chorionic gonadotropin concentrations may become too low to register as positive.
Generally, when used correctly, the home test should produce the same result as the urine human chorionic gonadotropin test done by your health practitioner. Blood testing for human chorionic gonadotropin is more sensitive than urine human chorionic gonadotropin testing, so sometimes a blood test will indicate pregnancy when the urine test is negative.
When is a blood human chorionic gonadotropin test ordered instead of a urine human chorionic gonadotropin?
Since human chorionic gonadotropin is not normally detected in the urine of a non-pregnant woman, a urine human chorionic gonadotropin is enough to confirm a pregnancy. This can also be done with a qualitative blood human chorionic gonadotropin test. Sometimes, however, it is important to know how much human chorionic gonadotropin is present to evaluate a suspected ectopic pregnancy or to monitor a woman following a miscarriage. In these circumstances, a health practitioner will order a quantitative blood human chorionic gonadotropin test.
How many days after a miscarriage would it take for a urine pregnancy test to show a negative result?
Urine human chorionic gonadotropin decreases at about the same rate as serum human chorionic gonadotropin, which can take anywhere from 9 to 35 days, with a median of 19 days. However, the timeframe for when an human chorionic gonadotropin result will be negative is dependent on what the human chorionic gonadotropin level was at the time of the miscarriage. Frequently, miscarriages are monitored with quantitative blood human chorionic gonadotropin testing. If the levels of human chorionic gonadotropin do not fall to undetectable levels, some human chorionic gonadotropin-producing tissue may remain and have to be removed.
What is an ectopic pregnancy?
An ectopic pregnancy occurs when the fertilized egg (ovum) implants somewhere other than in the uterus. This is a serious condition needing immediate treatment. Women with ectopic pregnancies often have abdominal pain and uterine bleeding. Usually, abnormally low levels of human chorionic gonadotropin are produced in ectopic pregnancies with slower-than-normal rates of increase.
Gonadotropin deficiency
Isolated gonadotropin-releasing hormone (GnRH) deficiency is characterized by inappropriately low serum concentrations of the gonadotropins – LH (luteinizing hormone) and FSH (follicle-stimulating hormone) in the presence of low circulating concentrations of sex steroids 159. Isolated gonadotropin-releasing hormone (GnRH) deficiency is associated with a normal sense of smell (normosmic isolated gonadotropin-releasing hormone deficiency) in approximately 40% of affected individuals and an impaired sense of smell (Kallmann syndrome) in approximately 60% 159. Isolated gonadotropin-releasing hormone (GnRH) deficiency can first become apparent in infancy, adolescence, or adulthood. Infant boys with congenital isolated gonadotropin-releasing hormone deficiency often have micropenis and cryptorchidism. Adolescents and adults with isolated gonadotropin-releasing hormone deficiency have clinical evidence of hypogonadism and incomplete sexual maturation on physical examination. Adult males with isolated gonadotropin-releasing hormone (GnRH) deficiency tend to have prepubertal testicular volume (i.e., <4 mL), absence of secondary sexual features (e.g., facial and axillary hair growth, deepening of the voice), decreased muscle mass, diminished libido, erectile dysfunction, and infertility. Adult females have little or no breast development and primary amenorrhea. Although skeletal maturation is delayed, the rate of linear growth is usually normal except for the absence of a distinct pubertal growth spurt.
Typically, a definitive diagnosis of isolated gonadotropin-releasing hormone (GnRH) deficiency is made around age 18 years. Occasionally, however, a high clinical suspicion of isolated gonadotropin-releasing hormone (GnRH) deficiency may be present in an adolescent presenting with anosmia and delayed puberty or in an infant with microphallus and cryptorchidism.
The term “hypogonadism” refers to impaired sexual development based on findings from the individual’s clinical history (e.g., amenorrhea, hot flashes, erectile dysfunction) as well as physical examination (e.g., small testes, vaginal pallor).
With greater understanding of the hypothalamo-pituitary-gonadal axis (see Figures 1 to 3) and the introduction of urinary gonadotropin measurements, the term “hypergonadotropic” hypogonadism was used to identify those with a primary gonadal defect, while “hypogonadotropic” hypogonadism identified those with a central (i.e., pituitary or hypothalamic) defect.
When anatomic (and later functional) causes of central hypogonadism were identified, “idiopathic” or “isolated” hypogonadotropic hypogonadism came into use to indicate those individuals in whom secondary causes of hypogonadotropic hypogonadism had been excluded.
Subsequently the ability to measure the effect of exogenous gonadotropin-releasing hormone (GnRH) administration demonstrated that the vast majority of individuals with “idiopathic” hypogonadotropic hypogonadism had a functional deficiency of gonadotropin-releasing hormone (GnRH) resulting from a defect in GnRH biosynthesis, secretion, and/or action (hence “isolated GnRH deficiency” [IGD]). Aside from hypothalamic gonadotropin-releasing hormone (GnRH) deficiency, individuals with isolated gonadotropin-releasing hormone (GnRH) deficiency typically have normal pituitary function tests and their hypogonadism typically responds to a physiologic regimen of exogenous gonadotropin-releasing hormone (GnRH) 160.
At this point, the term “isolated GnRH deficiency” (IGD) more properly reflects the current understanding of the clinical entity rather than the previous biochemical description of isolated hypogonadotropic hypogonadism and, thus, is the better term for what was previously called isolated or idiopathic hypogonadotropic hypogonadism.
Isolated gonadotropin-releasing hormone (GnRH) deficiency: Included Phenotypes
- Normosmic (with a normal sense of smell) gonadotropin-releasing hormone (GnRH) deficiency ~ 40 percent of cases
- Kallmann syndrome with impaired sense of smell ~ 60 percent of cases. The impaired olfactory function in Kallmann syndrome can be either hyposmia or complete anosmia) 161. The difference between hyposmia and anosmia is quantitative and not qualitative (i.e., odorants can be variably affected in persons with hyposmia). Most individuals with impaired smell do not have any physical or social impairment and the finding often goes unnoticed until isolated gonadotropin-releasing hormone (GnRH) deficiency is diagnosed.
A recent epidemiologic study in Finland showed a minimal incidence of Kallmann syndrome of 1:30,000 in males and 1:125,000 in females 162.
In the Seminara et al. 163 cohort of 250 individuals with IGD, males predominate with a male-to-female ratio of nearly 4:1.
Kallmann syndrome accounts for nearly two thirds of individuals with isolated GnRH deficiency (IGD).
Gonadotropin deficiency diagnosis and testing
Isolated gonadotropin-releasing hormone (GnRH) deficiency is typically diagnosed in adolescents presenting with absent or partial puberty using biochemical testing that reveals low serum testosterone or estradiol (hypogonadism) that results from complete or partial absence of gonadotropin-releasing hormone (GnRH)-mediated release of luteinizing hormone (LH) and follicle-stimulating hormone (FSH) (hypogonadotropic hypogonadism) in the setting of otherwise normal anterior pituitary anatomy and function and in the absence of secondary causes of hypogonadotropic hypogonadism. Pathogenic variants in more than 25 genes account for about half of all isolated gonadotropin-releasing hormone (GnRH) deficiency; the genetic cause for the remaining cases of isolated gonadotropin-releasing hormone (GnRH) deficiency is unknown.
In individuals with isolated gonadotropin-releasing hormone (GnRH) deficiency, analyses of the pulsatile pattern of gonadotropins have demonstrated a rather broad spectrum of abnormal developmental patterns varying from completely absent gonadotropin-releasing hormone (GnRH)-induced luteinizing hormone (LH) pulses to sleep-entrained gonadotropin-releasing hormone (GnRH) release that is indistinguishable from that of early puberty 164. This broad spectrum of neuroendocrine activity accounts for the variable reproductive phenotypes observed in persons with isolated gonadotropin-releasing hormone (GnRH) deficiency (IGD).
Suggestive findings
Isolated gonadotropin-releasing hormone (GnRH) deficiency should be suspected in individuals with the following:
- Absent or partial puberty at presentation in adolescents; low serum testosterone or estradiol on biochemical testing
- Findings of incomplete sexual maturation on physical examination as determined by Tanner staging (see Table 1):
- Men with isolated gonadotropin-releasing hormone (GnRH) deficiency typically have Tanner stage I-II genitalia (prepubertal testicular volumes; i.e., <4 mL); however, some males show evidence of partial pubertal maturation 165.
- Women with isolated gonadotropin-releasing hormone (GnRH) deficiency typically have Tanner stage I breast development and amenorrhea; however, some have spontaneous breast development and occasional menses 166.
- Both men and women with isolated gonadotropin-releasing hormone (GnRH) deficiency typically have Tanner stage II-III pubic hair, since pubic hair is controlled in part by adrenal androgens.
- In rare males, isolated gonadotropin-releasing hormone (GnRH) deficiency may present later in adulthood (i.e., adult-onset isolated gonadotropin-releasing hormone deficiency). However, in these patients, as puberty was not disrupted, sexual maturation is complete and secondary sexual characteristics may be fully developed. Diagnosis of adult-onset isolated gonadotropin-releasing hormone (GnRH) deficiency relies on documentation of hypogonadotropic hypogonadism and absence of other secondary causes of hypogonadotropic hypogonadism.
Laboratory findings of isolated gonadotropin-releasing hormone (GnRH) deficiency (see Figure 4 and Figure 5 for algorithm)
- Total testosterone (T) <100 ng/dL in males and estradiol (E2) <50 pg/mL in females
- Inappropriately low or normal serum concentration of LH (luteinizing hormone) and FSH (follicle stimulating hormone) in the presence of low circulating concentrations of sex steroids. Levels of other anterior pituitary hormones are typically normal.
Table 1. Tanner Staging
| Stage | Normal Findings | ||
|---|---|---|---|
| Pubic Hair | Male Genitalia | Female Breast Development | |
| 1 | None | Childhood appearance of testes, scrotum, and penis (testicular volume <4 mL) | No breast bud, small areola, slight elevation of papilla |
| 2 | Sparse hair that is long and slightly pigmented | Enlargement of testes; reddish discoloration of scrotum | Formation of the breast bud; areolar enlargement |
| 3 | Darker, coarser, curly hair | Continued growth of testes and elongation of penis | Continued growth of the breast bud and areola; areola confluent with breast |
| 4 | Adult hair covering pubis | Continued growth of testes, widening of the penis with growth of the glans penis; scrotal darkening | Continued growth; areola and papilla form secondary mound projecting above breast contour |
| 5 | Laterally distributed adult-type hair | Mature adult genitalia (testicular volume >15 mL) | Mature (areola again confluent with breast contour; only papilla projects) |
Figure 9. Testing algorithm to establish the diagnosis of isolated GnRH deficiency (IGD) in males
Figure 10. Testing algorithm to establish the diagnosis of isolated GnRH deficiency (IGD) in females
Imaging findings of isolated gonadotropin-releasing hormone (GnRH) deficiency
- In persons with isolated gonadotropin-releasing hormone (GnRH) deficiency: typically, normal-appearing hypothalamus and pituitary on MRI exam
- In persons with Kallmann Syndrome: typically, aplasia or hypoplasia of the olfactory bulbs/sulci/tracts.
Olfactory findings
Olfactory function is evaluated by history and by formal diagnostic smell tests, such as the University of Pennsylvania smell identification test (UPSIT), a “scratch and sniff” test that evaluates an individual’s ability to identify 40 microencapsulated odorants and can be easily performed in most clinical settings 167. Anosmia, hyposmia, or normosmia is identified using the University of Pennsylvania smell identification test (UPSIT) manual normogram, which incorporates an individual’s score, age at testing, and gender.
Individuals with isolated gonadotropin-releasing hormone (GnRH) deficiency with either self-reported complete anosmia or a score of hyposmia/anosmia on University of Pennsylvania smell identification test (UPSIT) are diagnosed with Kallmann Syndrome, while those with normal olfactory function are diagnosed with normosmic IGD (nIGD) 168.
Establishing the Diagnosis
The diagnosis of isolated gonadotropin-releasing hormone (GnRH) deficiency is established in a proband based on clinical and biochemical investigations above; a genetic diagnosis can be made with identification of pathogenic variant(s) in one of the genes listed in Table 2 and Table 3.
See Table 2 for the most common genetic causes (i.e., pathogenic variants of any one of the genes included in this table account for >2% of isolated gonadotropin-releasing hormone (GnRH) deficiency) and Table 3 for less common genetic causes (i.e., pathogenic variants of any one of the genes included in this table are reported in only a few families).
Molecular testing approaches can include serial single-gene testing, use of a multi-gene panel, and more comprehensive genomic testing.
Serial single-gene testing can be considered based on mode of inheritance and clinical findings, especially non-reproductive phenotypic features that indicate that pathogenic variation of a particular gene is most likely. Sequence analysis of the gene of interest is performed first, followed by gene-targeted deletion/duplication analysis if no pathogenic variant is found.
To help prioritize the order of serial single-gene testing, the following can be considered (see Figure 6 and Figure 7):
Sense of smell
- Pathogenic variants in CHD7, FGF8, FGF17, FGFR1, HS6ST1, NSMF (NELF), PROK2, PROKR2, and WDR11 cause both Kallmann syndrome (KS) and normosmic IGD (nIGD).
- Pathogenic variants in ANOS1 (KAL1), CCDC141, FEZF1, IL17RD, SEMA3A, SEMA3E, and SOX10 cause Kallmann syndrome (KS).
- Pathogenic variants in GNRH1, GNRHR, KISS1, KISS1R (GPR54), TAC3, and TACR3 cause nIGD.
Mode of inheritance
- X-linked. Sequence analysis of ANOS1 (KAL1) is the highest-yield molecular genetic test.
- Autosomal dominant. In families with clear autosomal dominant inheritance, testing of CHD7, FGFR1, FGF8 and SOX10 can be considered.
- Autosomal recessive. Testing of GNRH1, GNRHR, KISS1, KISS1R, TAC3, and TACR3 can be considered in families with autosomal recessive normosmic isolated gonadotropin-releasing hormone (GnRH) deficiency; testing of FEZF1, PROK2 and PROKR2 can be considered in families with autosomal recessive Kallmann syndrome (KS).
Associated phenotypic features
- The presence of some associated clinical phenotypic features may also help prioritize genetic testing in isolated gonadotropin-releasing hormone (GnRH) deficiency 169. See Figure 7.
Figure 11. Genes associated with isolated GnRH deficiency (IGD) by sense of smell and mode of inheritance
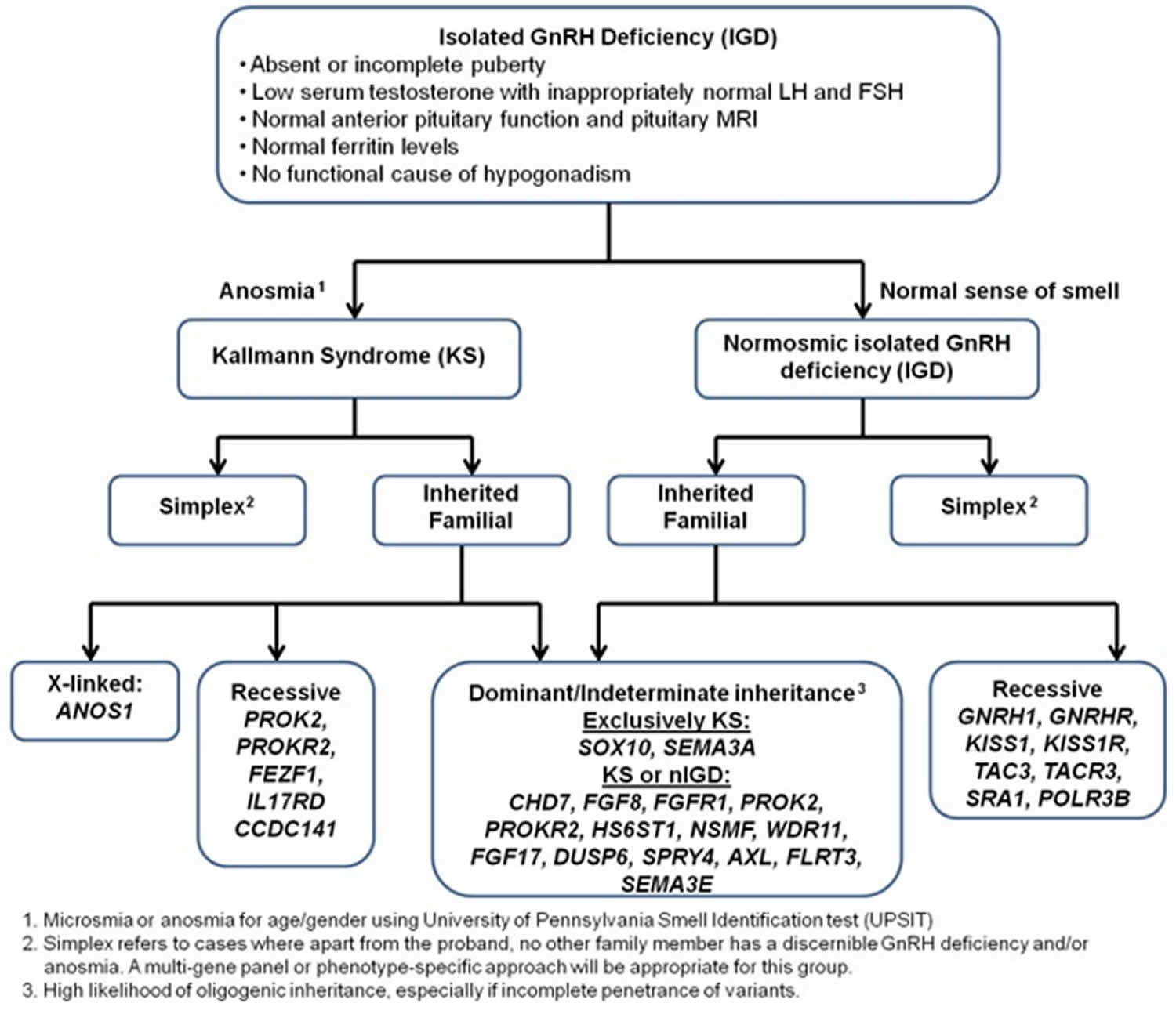
Figure 12. Suggested guidelines for prioritization of genetic testing for persons with isolated GnRH deficiency (IGD) based on phenotype
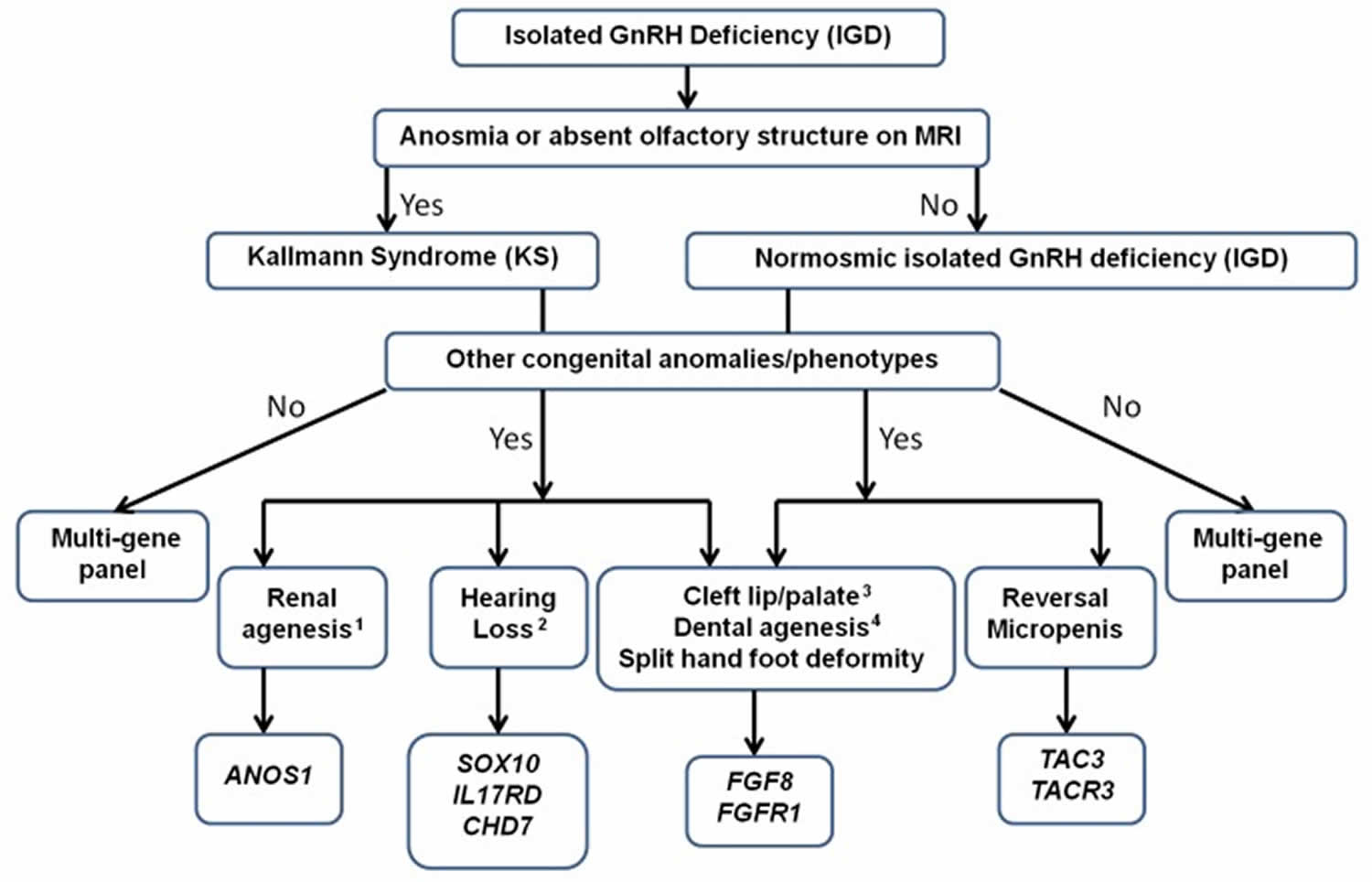
Table 2. Summary of Molecular Genetic Testing Used in Isolated Gonadotropin-Releasing Hormone (GnRH) Deficiency: Most Common Genetic Causes
| Gene 1, 2 | % of IGD Attributed to Pathogenic Variants in This Gene 3 | Proportion of Pathogenic Variants 4 Detected by Test Method | |
|---|---|---|---|
| Sequence analysis 5 | Gene-targeted deletion/duplication analysis 6 | ||
| ANOS1 (KAL1) | 5%-10% (KS) | ~88%-99% | ≤12% in one study (4/33 persons w/KS) 7 |
| CHD7 | 5%-10% (KS or nIGD) | ~100% | Unknown 8 |
| FGFR1 | ~10% (KS or nIGD) | ~99% | Rare 9 |
| GNRHR | 5%-10% (nIGD) | ~100% | Unknown 8 |
| IL17RD | 2%-5% (KS or nIGD) | ~100% | Unknown 8 |
| PROKR2 | ~5% (KS or nIGD) | ~100% | Unknown8 |
| SOX10 | 2%-5% (KS) | ~100% | Unknown 8 |
| TACR3 | ~5% (nIGD) | ~100% | Unknown 8 |
Footnotes: Pathogenic variants of any one of the genes included in this table account for >2% of isolated gonadotropin-releasing hormone (GnRH) deficiency (IGD).
KS = Kallmann syndrome
nIGD = normosmic isolated gonadotropin-releasing hormone deficiency
- Genes are listed in alphabetic order.
- See Table A. Genes and Databases for chromosome locus and protein (https://www.ncbi.nlm.nih.gov/books/NBK1334/#kms.molgen.TA).
- Proportion of IGD attributed to these genes is determined from the author’s cohort of 950 probands with IGD who were screened for rare sequence variants (<1% of control cohort).
- See Molecular Genetics for information on pathogenic allelic variants detected.
- Sequence analysis detects variants that are benign, likely benign, of uncertain significance, likely pathogenic, or pathogenic. Pathogenic variants may include small intragenic deletions/insertions and missense, nonsense, and splice site variants; typically, exon or whole-gene deletions/duplications are not detected. For issues to consider in interpretation of sequence analysis results, click here.
- Gene-targeted deletion/duplication analysis detects intragenic deletions or duplications. Methods that may be used include: quantitative PCR, long-range PCR, multiplex ligation-dependent probe amplification (MLPA), and a gene-targeted microarray designed to detect single-exon deletions or duplications.
- 12% of persons with KS harbored intragenic deletions in ANOS1 170.
- No data on detection rate of gene-targeted deletion/duplication analysis are available.
- FGFR1 deletions are rare 171.
Table 3. Molecular Genetics of Isolated Gonadotropin-Releasing Hormone (GnRH) Deficiency (IGD): Less Common Genetic Causes
| Gene 1, 2, 3 | Comments |
|---|---|
| AXL | Described in 1 report: 4/104 persons with Kallmann syndrome or normosmic isolated gonadotropin-releasing hormone deficiency |
| CCDC141 | Described in 1 report: 1/20 persons with Kallmann syndrome |
| DUSP6 | Described in 1 report: 5/386 persons with Kallmann syndrome or normosmic isolated gonadotropin-releasing hormone deficiency |
| FEZF1 | Described in 1 report: 2/30 persons with Kallmann syndrome |
| FGF8 | <2% 4 of persons with Kallmann syndrome or normosmic isolated gonadotropin-releasing hormone deficiency |
| FGF17 | Described in 1 report: 3/386 persons with Kallmann syndrome or normosmic isolated gonadotropin-releasing hormone deficiency |
| FLRT3 | Described in 1 report: 3/386 persons with Kallmann syndrome or normosmic isolated gonadotropin-releasing hormone deficiency |
| GNRH1 | Typically autosomal recessive; <2% 4 of persons with normosmic isolated gonadotropin-releasing hormone deficiency |
| HS6ST1 | <2% of persons with Kallmann syndrome or normosmic isolated gonadotropin-releasing hormone deficiency 4, 5 |
| KISS1 | Typically autosomal recessive; <2% of persons with normosmic isolated gonadotropin-releasing hormone deficiency 4 |
| KISS1R | Typically autosomal recessive; <2% of persons w/normosmic isolated gonadotropin-releasing hormone deficiency 4 |
| POLR3B | Described in 1 report: 3/565 persons with Kallmann syndrome or normosmic isolated gonadotropin-releasing hormone deficiency |
| PROK2 | Typically autosomal recessive; <2% 4 of persons with Kallmann syndrome or normosmic isolated gonadotropin-releasing hormone deficiency 4 |
| SEMA3A | <2% of persons with Kallmann syndrome or normosmic isolated gonadotropin-releasing hormone deficiency 4, 5 |
| SEMA3E | Described in 1 report: 1/121 persons with Kallmann syndrome or normosmic isolated gonadotropin-releasing hormone deficiency |
| SPRY4 | Described in 1 report: 14/386 persons with Kallmann syndrome or normosmic isolated gonadotropin-releasing hormone deficiency |
| SRA1 | Described in 1 report: 3/136 persons w/normosmic isolated gonadotropin-releasing hormone deficiency |
| TAC3 | Typically autosomal recessive; <2% of persons w/normosmic isolated gonadotropin-releasing hormone deficiency 4 |
| WDR11 | Described in 1 report: 1 person w/balanced translocation; 6/201 persons with Kallmann syndrome or normosmic isolated gonadotropin-releasing hormone deficiency |
Footnotes: Pathogenic variants of any one of the genes listed in this table are reported in only a few families [i.e., <2% of isolated gonadotropin-releasing hormone (GnRH) deficiency (IGD)]
KS = Kallmann syndrome
AR = autosomal recessive
nIGD = normosmic isolated gonadotropin-releasing hormone deficiency
- Genes are listed in alphabetic order.
- See Table A. Genes and Databases for chromosome locus and protein (https://www.ncbi.nlm.nih.gov/books/NBK1334/#kms.molgen.TA)
- Genes are not described in detail in Molecular Genetics but may be included here (https://www.ncbi.nlm.nih.gov/books/NBK1334/bin/kms-less_common_genes.pdf).
- Proportion of isolated gonadotropin-releasing hormone (GnRH) deficiency attributed to these genes is determined from the author’s cohort of 950 probands with isolated gonadotropin-releasing hormone (GnRH) deficiency who were screened for rare sequence variants (<1% of control cohort).
- Pathogenic variants in this gene are not thought to cause isolated gonadotropin-releasing hormone (GnRH) deficiency without contributions from other isolated gonadotropin-releasing hormone (GnRH) deficiency-related genes; thus, the proportion of isolated gonadotropin-releasing hormone (GnRH) deficiency caused by pathogenic variants in this gene is unknown.
Genetic counseling
Isolated gonadotropin-releasing hormone (GnRH) deficiency can be inherited in an X-linked, autosomal dominant, or autosomal recessive manner. Almost all isolated gonadotropin-releasing hormone (GnRH) deficiency-related genes have also been associated with indeterminate or oligogenic inheritance. Recurrence risk counseling is based on family history and the results of molecular genetic testing when available. Carrier testing for at-risk relatives in families with X-linked isolated gonadotropin-releasing hormone (GnRH) deficiency or autosomal recessive isolated gonadotropin-releasing hormone (GnRH) deficiency is possible if the pathogenic variant(s) in the family are known. Prenatal testing for pregnancies at increased risk is possible if the pathogenic variant(s) in the family are known.
Gonadotropin deficiency treatment
Treatment of manifestations: To induce and maintain secondary sex characteristics, gradually increasing doses of testosterone or human chorionic gonadotropin (hCG) injections in males or estrogen and progestin in females; to stimulate spermatogenesis or folliculogenesis, either combined gonadotropin therapy (hCG and human menopausal gonadotropins [hMG] or recombinant FSH) or pulsatile gonadotropin-releasing hormone (GnRH) therapy. If conception fails despite spermatogenesis in a male or ovulation induction in a female, in vitro fertilization may be an option.
Males with isolated gonadotropin-releasing hormone deficiency Age ≥18 Years
Treatment options include sex steroids, gonadotropins, and pulsatile gonadotropin-releasing hormone (GnRH) administration. Choice of therapy in adults is determined by the goal(s) of treatment (i.e., to induce and maintain secondary sex characteristics and/or to induce and maintain fertility). The selection of hormone replacement therapy is also based on the preference of the individual being treated; however, when fertility is not immediately desired, replacement with testosterone therapy is the most practical option. As the majority of individuals with isolated gonadotropin-releasing hormone (GnRH) deficiency have not progressed through puberty at the time of diagnosis, initial therapy should be started at low doses and gradually increased to adult doses once the development of secondary sexual characteristics is achieved.
Hormone replacement therapy for males not desiring fertility
Testosterone therapy
Testosterone therapy in the form of injectable and transdermal routes of testosterone administration is typically used to both induce puberty and maintain adult levels of testosterone. Recently nasal testosterone has become available but use has not been reported in patients with isolated gonadotropin-releasing hormone (GnRH) deficiency.
The injectable testosterone preparations have a “roller-coaster” pharmacokinetic effect, with peak and trough levels that can go to extraphysiologic levels; thus, the transdermal preparations have the added benefit of offering a more favorable pharmacokinetic profile. A typical adult dose of testosterone replacement is 200 mg of testosterone ester injection every two weeks or 5 g of a 1% testosterone gel every day. Doses do vary with newer testosterone preparations; manufacturer’s instructions should be followed for individual testosterone preparations.
Men using topical androgen replacement must take care to avoid exposing other individuals to treated skin. Anecdotal reports suggest that the transmission of clinically effective levels of testosterone from the patient to other family members (including women and children) is possible with undesirable side effects.
Once puberty is initiated, testosterone replacement therapy is usually required indefinitely to ensure normal sexual function and maintenance of proper muscle, bone, and red blood cell mass. However, in approximately 10% of males, reversal of isolated gonadotropin-releasing hormone (GnRH) deficiency may occur; thus, if clinical evidence shows endogenous activity of the hypothalamo-pituitary-axis (e.g., testicular growth on testosterone, maintained testosterone levels despite missing/withholding therapy), a brief washout of testosterone therapy should be done with monitoring of testosterone levels. If testosterone levels fall, therapy should be reinitiated. If levels are normal, no further testosterone therapy will be required; serial monitoring of levels should be undertaken, as some individuals may require reinitiation of therapy.
Human chorionic gonadotropin injections
Human chorionic gonadotropin injections is an alternative to testosterone therapy, human chorionic gonadotropin (hCG) injections promote testicular growth, normalize serum concentration of testosterone, and induce development of secondary sexual characteristics.
In adults, treatment with human chorionic gonadotropin (hCG) is usually initiated at 1,500 IU intramuscularly or subcutaneously every other day to normalize serum testosterone concentrations. Dose should be increased by increments of 250 IU if serum testosterone levels remain low.
Treatment with human chorionic gonadotropin (hCG) must be weighed against the increased risk of developing gynecomastia (resulting from the estrogen produced by stimulation of the testes with human chorionic gonadotropin). To some extent the risk of gynecomastia can be minimized by gradually reducing the dose of human chorionic gonadotropin (hCG) to the minimum required to sustain a serum testosterone concentration in the mid-normal range (~500 ng/dL).
Male Infants/Adolescents with Suspicion of isolated gonadotropin-releasing hormone deficiency
If isolated gonadotropin-releasing hormone (GnRH) deficiency is clinically suspected (e.g., low testosterone levels with low/normal gonadotropins) low-dose testosterone or hCG therapy can be given in early infancy to boys with microphallus to increase penile length 172.
Since a definitive diagnosis of isolated gonadotropin-releasing hormone (GnRH) deficiency may not be possible until age 18 years, after infancy these boys do not generally need to be treated until around the time of puberty. At this time, if a high suspicion of isolated gonadotropin-releasing hormone (GnRH) deficiency remains (e.g., associated anosmia and delay in onset puberty), these subjects may benefit from early initiation of hormonal replacement therapy with either testosterone or human chorionic gonadotropin (hCG) treatment early in the pubertal period. A suggestive puberty induction regimen in adolescents is to start a long-acting testosterone ester at a dose of 25-50 mg, given intramuscularly every two weeks. The doses can be gradually increased by 25-50 mg every two to three months until full virilization is achieved. Once adult doses (~200 mg/2 weeks) are reached, further adjustments are based on serum testosterone levels.
Hormone replacement therapy for males desiring fertility (fertility induction in males)
As testosterone replacement therapy suppresses spermatogenesis in the testes, gonadotropins or pulsatile gonadotropin-releasing hormone (GnRH) therapy is usually required to realize the fertility potential in males.
Gonadotropin therapy
In most males with isolated gonadotropin-releasing hormone (GnRH) deficiency, a combination of gonadotropins (hCG along with either human menopausal gonadotropins [hMG] or recombinant FSH) is used to stimulate spermatogenesis. In males with very low testicular volumes (≤~8 mL) the initiating dose of hCG is usually 1,500 IU intramuscularly or subcutaneously every other day; follicle-stimulating hormone (FSH) is added at doses ranging from 37.5 to 75 IU as either human menopausal gonadotropins [hMG] or recombinant formulation. Trough serum testosterone concentrations (target: mid-normal range [~500 ng/dL]), trough serum FSH levels (target: mid-normal reference range), and sperm count are monitored to assess response. Recent trials show that in those with lower testicular volumes, priming with FSH prior to combination therapy may improve spermatogenic outcomes 173.
In males with higher baseline testicular volumes, treatment with human chorionic gonadotropin alone may be sufficient to achieve spermatogenesis and conception 174. However, if after six to nine months, semen analysis reveals persistent azoospermia or marked oligospermia, follicle-stimulating hormone (FSH) is added to the regimen at doses ranging from 37.5 to 75 IU as either human menopausal gonadotropins [hMG] or a recombinant formulation.
In either treatment, testicular volume must be tracked, as this is one of the primary determinants of successful spermatogenesis. In fact, sperm are rarely seen in the semen analysis until testicular volume reaches 8 mL 175. In most males without a history of cryptorchidism, sperm function is usually normal and conception can occur even with relatively low sperm counts.
Note: Liu et al 176 have noted that previous treatment with gonadotropins may reduce the period of subsequent gonadotropin treatment required for initiation of spermatogenesis.
If a pituitary defect exists, gonadotropin therapy becomes the treatment of choice.
Pulsatile GnRH stimulation vs. gonadotropin therapy
While either gonadotropin therapy or pulsatile GnRH stimulation can induce spermatogenesis in approximately 90%-95% of men with isolated gonadotropin-releasing hormone (GnRH) deficiency, some men have a better response to pulsatile GnRH stimulation than to gonadotropin therapy.
Subcutaneous administration of gonadotropin-releasing hormone (GnRH) in a pulsatile manner through a portable pump that delivers a GnRH bolus every two hours is an efficient way of inducing testicular growth and spermatogenesis 177. As the primary defect of isolated gonadotropin-releasing hormone (GnRH) deficiency is typically localized to the hypothalamus, the pituitary responds appropriately to physiologic doses of GnRH.
Note: In the US, pulsatile GnRH therapy is not currently approved by the Food and Drug Administration for the treatment of infertility in men and, thus, is available for such treatment only at specialized research centers.
Females with isolated gonadotropin-releasing hormone deficiency
Hormone replacement therapy for females not desiring fertility. Although a definitive diagnosis of isolated gonadotropin-releasing hormone (GnRH) deficiency in females is usually made around age 18 years, occasionally a high clinical suspicion of isolated gonadotropin-releasing hormone (GnRH) deficiency may be present in an adolescent presenting with anosmia and delayed puberty, and therapy may need to be initiated earlier (age ~14 years)
- To allow optimal breast development, initial treatment should consist of unopposed estrogen replacement via oral or topical preparations. Many formulations of estrogens are available; a suggested oral regimen is using premarin 0.3 mg daily to be increased gradually to an adult replacement dose of 1-1.25 mg daily.
- Once breast development is optimal, a progestin should be added for endometrial protection (e.g., cyclical Prometrium® 200 mg daily for 10-12 days).
- Although preference of the individual plays an important role in choice of treatment plan, low-estrogen formulations should be considered in women with clotting abnormalities (e.g., Factor V Leiden Thrombophilia and Prothrombin Thrombophilia).
Hormone replacement therapy for females desiring fertility (fertility induction in females). Pulsatile GnRH stimulation and exogenous gonadotropins are FDA approved for folliculogenesis in women with isolated gonadotropin-releasing hormone (GnRH) deficiency. Either therapy should be administered with close supervision by physicians specializing in ovulation induction. Intravenous administration of GnRH at various frequencies throughout the menstrual cycle closely mimics the dynamics of normal menstrual cycles resulting in ovulation of a single follicle 178. This therapy offers a clear advantage over the traditional treatment with exogenous gonadotropins, which results in higher rates of both multiple gestation and ovarian hyperstimulation syndrome. For either approach, however, the rate of conception is approximately 30% per ovulatory cycle 179.
Fertility Options in Patients with isolated gonadotropin-releasing hormone (GnRH) deficiency if Fertility Induction is Unsuccessful
In vitro fertilization (IVF). Although successful spermatogenesis can be obtained in most males with isolated gonadotropin-releasing hormone (GnRH) deficiency through pulsatile GnRH therapy or combined gonadotropin therapy, some men with Kallmann syndrome caused by an ANOS1 (KAL1) pathogenic variant may have an atypical response to therapy 180. In those who respond to therapy, low sperm numbers can often result in conception; however, if infertility continues despite successful spermatogenesis or if spermatogenesis fails to occur, in vitro fertilization (IVF) is an option.
Similarly, if spontaneous conception fails to occur in women with isolated gonadotropin-releasing hormone (GnRH) deficiency who have undergone ovulation induction, IVF may be an option.
Prevention of secondary complications
Optimal calcium and vitamin D intake should be encouraged and specific treatment for decreased bone mass as needed.
Surveillance
For children of both sexes with findings suggestive of isolated gonadotropin-releasing hormone (GnRH) deficiency, monitor at regular intervals after age 11 years:
- sexual maturation (by Tanner staging on physical examination);
- gonadotropin and sex hormone levels (measurement of serum concentrations of LH, FSH, and total testosterone (T) in males and estradiol (E2) in females);
- bone age.
In individuals with confirmed isolated gonadotropin-releasing hormone (GnRH) deficiency, monitor at regular intervals: serum sex steroid levels (to guide optimal hormone replacement); bone mineral density.
Evaluation of relatives at risk
If the pathogenic variant(s) in a family are known, genetic testing of prepubertal at-risk relatives may be indicated to clarify their genetic status. Because of variable expressivity, a prepubertal child with a known pathogenic variant may progress through puberty in a normal or delayed fashion, or not at all; therefore, clinical reevaluation over time is necessary.
Gonadotropin treatment
Highly purified and man-made (synthetic) gonadotropins have been developed and used in the treatment of hypogonadism and infertility. Synthetic forms of gonadotropin-releasing hormone (GnRH) have been used with the gonadotropins in assisted reproductive techniques (ART) and in vitro fertilization (IVF).
Follicle stimulating hormone (FSH) therapeutic uses
Therapeutic preparations of follicle stimulating hormone (FSH) are widely used in the treatment of infertility (Table 4). Partially and highly purified human menopausal urine derived FSH (Menotropins or human menopausal gonadotropin [hMG] or Menopur which also has LH activity); industrial production of therapeutic grade urinary FSH (urofollitropin, Bravelle) and recombinant DNA (rDNA)-derived human FSH (follitropin alpha, Follistim, Gonal F) are available and approved for use in treatment of infertility and hypogonadism 26. They are generally given by subcutaneous injection daily or several times weekly. The dose and appropriate regimen vary by indication. These agents should be used only by doctors with expertise in management of infertility and hypogonadism.
Therapeutic preparations of follicle stimulating hormone (FSH) use in assisted reproduction technology can be divided into 3 categories 36:
- Induction of ovulation when a single healthy oocyte is required.
- Induction of multiple ovulation or superovulation to maximize efficiency when assisted reproductive technologies are used that allow replacement of a fixed number of embryos.
- Stimulation of spermatogenesis.
Treatment of female infertility is a situation in which patients are otherwise generally healthy and common disorders of reproduction such as anovulatory infertility can be treated in a safe and effective way 181. However, there is a narrow dose-range for use of FSH between a threshold level required to stimulate growth of a follicle(s) and the maximal dose (ceiling dose) above which overstimulation can occur 182. Therefore there is a significant risk to health due to the iatrogenic induction of ovarian hyperstimulation syndrome or multiple pregnancies. Different physiological and clinical states can affect the levels of the threshold and ceiling for FSH treatment. Thus careful dose adjustment and monitoring of FSH levels and ovarian responses are required, particularly for women with polycystic ovary syndrome (PCOS) 183. This cannot be achieved without accurate and reproducible calibration of therapeutic products. However, the end point used for patient response to therapeutic preparations should also be carefully considered.
Table 4. Common causes of infertility and treatments that require follicle stimulating hormone (FSH)
| Cause | Treatment |
|---|---|
| Female infertility | |
| Ovulatory failure (oligo- or amenorrhea) | Ovulation induction |
| Primary ovarian failure | Superovulation followed by In vitro fertilization (IVF) using donated oocytes |
| Tubal or pelvic damage a | Superovulation and In vitro fertilization (IVF) |
| Endometriosis a | Superovulation and In vitro fertilization (IVF), gamete intra-fallopian tube transfer (GIFT), intra-uterine insemination (IUI) |
| Cervical mucus dysfunction or defects | Superovulation and intra-uterine insemination (IUI), gamete intra-fallopian tube transfer (GIFT), In vitro fertilization (IVF), zygote intra-fallopian tube transfer (ZIFT) |
| Antisperm antibodies | Superovulation and In vitro fertilization (IVF), intra-uterine insemination (IUI) |
| Idiopathic infertility | Superovulation and In vitro fertilization (IVF), gamete intra-fallopian tube transfer (GIFT), intra-uterine insemination (IUI) |
| Male infertility | |
| Sperm dysfunction | In vitro fertilization (IVF), intra-cytoplasmic sperm injection (ICSI) |
| Azoospermia | Stimulation of spermatogenesis with FSH if due to hypogonadotrophic hypogonadism or pituitary failure |
Footnote: a May be treated surgically in appropriate cases.
[Source 36 ]Luteinizing hormone (LH) use Infertility and Assisted Reproductive Technology
Infertility is defined clinically as the inability to become clinically pregnant after at least 12 months of unprotected sexual intercourse. Female factors, male factors, or both can cause it. In women, it can be the result of ovulatory issues (ie, anovulation), obstructions of the fallopian tubes, and endometriosis. To become pregnant, many women undergo assisted reproductive technologies (ARTs), like intrauterine insemination (IUI) and in vitro fertilization (IVF) 184.
Proper development of a ovarian follicle and ovulation involves the combined effects of follicle-stimulating hormone (FSH) and luteinizing hormone (LH) and their bodily activities. This interplay between FSH and LH has also been shown to be important in assisted reproductive technology (ART). It has been found that low LH levels in the body can result in poor outcomes in Assisted Reproductive Technology (ART). Therefore, patients who have low endogenous LH, such as those with hypogonadotropic hypogonadism, can have an increase in the efficacy of ART with exogenous LH treatment 185. A study found that with supplementation of LH during the mid-follicular phase, there were better pregnancy results in women who had not responded optimally to conventional Assisted Reproductive Technology (ART). This outcome was thought to be due to the increased production of 17-beta-estradiol 60. Another study reported that fertilization, implantation, and clinical pregnancy rates were higher in patients who underwent Assisted Reproductive Technology (ART) that included recombinant human LH (rhLH) when compared to patients who underwent ART without recombinant human LH (rhLH). A lower apoptosis rate was also present in patients who underwent ART, which included recombinant human LH (rhLH) 12.
Although there are demonstrable benefits of LH supplementation during Assisted Reproductive Technology (ART), research also shows that LH levels can have unfavorable effects on ART 185. These adverse effects are thought to result from inhibition of Granulosa cell proliferation, atresia of immature ovarian follicles, and luteinization of preovulatory follicles before they would be under physiological conditions. Additionally, increased LH before ovulation has been shown to influence the conception and implantation of the embryo negatively 60.
Gonadotropins use in Assisted Reproductive Technology
To increase likelihood of pregnancy through assisted reproductive technology (ART), multiple eggs must be produced. This is accomplished through the administration of gonadotropins medications that directly stimulate the ovaries. Stimulation can be achieved with a variety of drug regimens. Gonadotropin medications come in several forms; Repronex® (menotropins for injection) and Menopur® (menotropins for injection) are a combination of luteinizing hormone (LH) and follicle-stimulating hormone (FSH). They replace a woman’s own LH and FSH which are normally produced by the anterior pituitary gland. Bravelle®, Follistim® AQ Cartridge for use with Follistim Pen®, Follistim® AQ Vial, Gonal-F®, and Gonal-F® RFF Pen are preparations that contain only FSH. Follistim® AQ Cartridge for use with Follistim Pen®, Follistim® AQ Vial, Gonal-F®, and Gonal-F® RFF Pen are recombinant products which are made by genetically engineered cells. This process ensures uniform purity and potency. Because the dose of hormones that are used in ART is greater than what the body normally produces, the ovaries typically develop more than one oocyte as occurs in a natural cycle.
Gonadotropins act directly on the ovary to stimulate the growth of follicles (the structures in the ovaries which contain eggs). Granulosa cells within the follicles grow and develop which cause the follicles to enlarge and fill with follicular fluid. These developing follicles can be counted and measured using transvaginal ultrasound. As the follicles grow, they produce increasing amounts of estrogen, which can be measured with a laboratory blood test. Some physicians prefer one formulation or another. Your doctor can discuss this with you in more detail.
Gonadotropins Dosage and Monitoring
Gonadotropins are packaged in vials containing 37.5, 75 or 150 International Units (IU). Follistim AQ Pens and Gonal-F RFF Pens are packaged in pre-mixed injectable pens. Multi-dose vials of some medications are also available. Dosage may vary depending on the patient’s history. Patients will then have regularly scheduled transvaginal ultrasound examinations and serum estradiol tests. The dose of gonadotropins is then determined by the result of the ultrasound and estradiol tests. Most women require between seven to 10 days of gonadotropin therapy.
Bravelle®, and Repronex® are administered subcutaneously or by intramuscular injection, usually into the muscles of the buttocks. Gonal-F,® Follistim,® Follistim AQ Pens, and Gonal-F RFF Pens are administered subcutaneously, like an insulin or allergy shot.
Human Chorionic Gonadotropin (hCG) in Assisted Reproductive Technology
Human chorionic gonadotropin (hCG) is an injectable medication that is administered to complete egg maturation. Human chorionic gonadotropin (hCG) is structurally similar to the LH that is produced by a woman’s pituitary gland. It acts on the ovary in a manner similar to a woman’s own LH. Human chorionic gonadotropin (hCG), like LH, stimulates the final maturation of the eggs in the follicle. It also stimulates progesterone production from the ovary after egg retrieval. This progesterone is important to prepare the uterus for implantation of the embryo.
Human chorionic gonadotropin (hCG) Dosage and Administration
Human chorionic gonadotropin (hCG) medications are Profasi®, Ovidrel®, Novarel®,and Pregnyl® can be administered several different ways. The commonly administered dose is a single injection of 10,000 units. Once hCG is administered, ovulation usually occurs in approximately 36 to 40 hours. Therefore oocyte retrieval is routinely scheduled at 34-36 hours after hCG. This helps ensure maximal egg maturity, which is important for fertilization and embryo development. Occasionally, several doses of 2,500 units (usually every three days) are administered after egg retrieval to stimulate progesterone production. If your response to stimulation is particularly exuberant, the dose of hCG can be reduced to 5,000 units in an attempt to reduce the risk of ovarian hyperstimulation syndrome.
It typically takes 8-10 days for single injection of 10,000 units of hCG to be cleared from the blood stream. As hCG is the same hormone that is produced by a developing pregnancy, patients should not have a blood or urine pregnancy test sooner than 10 days following the hCG injection. If a pregnancy test is performed earlier, it may measure the hCG that was given by injection rather than measure hCG produced by a pregnancy.
Hypogonadotropic Hypogonadism Treatment in Males
Hypogonadism is impaired testicular function; this can occur due to a problem with the testes (primary hypogonadism or hypergonadotropic hypogonadism) or due to a problem with the hypothalamic-pituitary-gonadal axis (secondary hypogonadism or hypogonadotropic hypogonadism). These two entities can be distinguished by measuring serum LH and FSH concentrations 186. Primary hypogonadism or hypergonadotropic hypogonadism is characterized by a low serum testosterone level and oligo- or azoospermia in the presence of elevated serum LH and FSH concentrations 186. In contrast, secondary hypogonadism or hypogonadotropic hypogonadism is diagnosed in the setting of a low testosterone level and sperm count in association with low or inappropriately normal serum LH and FSH concentrations 186.
Men with secondary hypogonadism or hypogonadotropic hypogonadism have low levels of androgens in the plasma as well as a lack or delay of sexual maturity, which can cause symptoms such as a lack of libido, depression, increase in adipose tissue, and diminished erectile function 187.
Patients with secondary hypogonadism or hypogonadotropic hypogonadism usually have an issue with gonadotropin-releasing hormone (GnRH) signaling, which then causes a decrease in follicle-stimulating hormone (FSH) and luteinizing hormone (LH) secretion. This decreased FSH and LH contributes to decreased androgen levels and reduced spermatogenesis. Studies have shown that giving these patients pulsatile GnRH or LH (or hCG) and FSH can help increase spermatogenesis and thus increase the sperm concentration in the ejaculate. Even then, most couples will need assisted reproductive technology (ART) to achieve pregnancy 188.
Testosterone therapy, whether by exogenous testosterone replacement or induction of endogenous testosterone production by human chorionic gonadotropin (hCG) is needed in all hypogonadotropic hypogonadism patients 186. Testosterone plays a number of important physiologic roles in the human and are required not only for virilization and normal sexual function, but also for maintenance of both muscle and bone mass, as well as normal mood and cognition.
While testosterone is the primary treatment modality used to induce and maintain secondary sexual characteristics and sexual function in men with hypogonadism, treatment with testosterone does not restore fertility. Therefore, in patients in whom fertility is the treatment goal, induction of gonadotropin secretion by pulsatile GnRH or exogenous gonadotropins is necessary. While hormone therapy is the mainstay of treatment for congenital hypogonadotropic hypogonadism; undescended testicle (cryptorchidism), if present, should be treated surgically with orchidopexy, ideally before the age of 2 years to improve fertility outcomes and reduce the risk of future testicular malignancy 189.
Exogenous Gonadotropin Therapy
The typical gonadotropin regimen for induction of spermatogenesis in men comprises human chorionic gonadotropin (hCG) in combination with follicle-stimulating hormone (FSH). Purified human chorionic gonadotropin (hCG) is an effective substitute for luteinizing hormone (LH) given the structural homology between these 2 hormones which act through the same receptor on Leydig cells 186. While a variety of FSH formulations are now available in different countries, there is little to choose between them in terms of therapeutic efficacy. Traditionally, FSH has been administered in the form of human menopausal gonadotropins (hMG) derived from the urine of postmenopausal women. Although human menopausal gonadotropins (hMG) has both FSH and LH activity, FSH activity predominates and LH activity is so low that combined administration with hCG is necessary to achieve fertility. Subsequently, highly purified urinary FSH preparations were developed, which have enhanced specific activity in comparison to hMG (10,000 IU/mg of protein vs 150 IU/mg of protein for hMG) 186. In the early 1990s, recombinant human follicle stimulating hormone (r-hFSH) formulations were developed, which have greater purity and specific activity than any of the urinary preparations and no intrinsic LH activity 190, 191. Recombinant human follicle stimulating hormone (r-hFSH) is produced in genetically engineered Chinese hamster ovary cells, in which the genes encoding the alpha and beta subunits have been introduced using recombinant DNA technology 190. Pharmacokinetic studies of r-hFSH indicate a half-life of 48 ± 5 hour and a dose-dependent increase in the serum level of FSH 190.
The subcutaneous route of administration is as effective as the intramuscular route for both gonadotropins and significantly increases patient compliance. Therapy is typically initiated with human chorionic gonadotropin (hCG) alone at a dose of 1,000 IU on alternate days and the dose titrated based on trough testosterone levels and testicular growth 192. Alternatively, recombinant human hCG can also be administered subcutaneously from a prefilled syringe. In some patients the dose of hCG can be decreased over time as testicular size increases. In the majority of patients with larger testes at baseline, spermatogenesis can be initiated with hCG alone most likely due to residual FSH secretion 193, 194. Once there is a plateau in the response to hCG which typically occurs at around 6 months, therapy with FSH (in one of the three forms described above) should be added at a dose of 75 IU on alternate days. If sperm output and testicular growth remain suboptimal the dose of FSH can be gradually increased to 150 U daily. Continuation of this combined regimen for 12-24 months induces testicular growth in almost all patients, spermatogenesis in approximately 80% and pregnancy rates in the range of 50% 195, 196, 197. In an Australian study of 75 men with hypogonadotropic hypogonadism treated with gonadotropins the median time for sperm to appear in the ejaculate was 7.1 months and for conception was just over 28 months 196. Similar data were reported in a compilation of clinical trial data from Asian, European, Australian and American patients 197. Factors predictive of better outcome include larger baseline testicular size and absence of cryptorchidism. The Australian study reported that prior androgen use is also a negative prognostic indicator of response 196. While the study investigators propose that gonadotropin therapy be considered to induce puberty based on their results, confirmation that such an approach is superior to the conventional practice of giving testosterone would require a large clinical trial the logistics of which would be challenging given the rarity of this patient population. Gynecomastia is the most common side effect of gonadotropin therapy and is due to human chorionic gonadotropin (hCG) stimulation of aromatase causing increased secretion of estradiol. This undesirable side effect can be prevented by using the lowest dose of human chorionic gonadotropin (hCG) capable of maintaining serum testosterone levels towards the lower end of the normal range.
In the majority of hypogonadotropic hypogonadism patients treated with gonadotropins sperm density remains below the normal range. However, failure to achieve a normal sperm density does not preclude fertility. Indeed, the median sperm concentrations reported at conception range from 5-8 million/mL 193, 196. While spermatogenesis can be initiated even in patients with very small testes 193, 198, a longer duration of therapy is typically required and it may take up to 24 months for spermatogenesis to be induced. Accordingly when discussing the issue of fertility with patients, experts recommend that they start treatment at least 6 to 12 months prior to the time at which fertility is desired. Once pregnancy is achieved, experts advise continuing therapy until at least the second trimester. If the couple plans to have another child in the near future, then hCG monotherapy should be continued. However, if a long interval is expected to elapse before the next pregnancy, it may be more convenient for the patient to resume testosterone therapy. Patients should also be offered the option of storing sperm for subsequent use in intrauterine insemination or intracytoplasmic sperm injection. In patients in whom the combination of hCG and FSH is required to induce spermatogenesis initially, treatment with hCG alone may be sufficient for subsequent pregnancies due to larger testicular size.
In patients with panhypopituitarism who fail to respond to gonadotropin therapy, the addition of recombinant growth hormone (rGH) therapy should be considered. It is thought that a direct effect of growth hormone on Leydig cells may play a role in the delayed puberty encountered commonly in patients with isolated growth hormone deficiency 199, 200. While small non-randomized studies suggest that recombinant growth hormone (rGH) may enhance the testosterone response to human chorionic gonadotropin (hCG) administration 199, larger, randomized studies are needed before a definitive decision about the benefit of recombinant growth hormone (rGH) in inducing spermatogenesis in men with hypopituitarism can be reached.
Pulsatile GnRH Therapy
The alternative to gonadotropin therapy is pulsatile administration of gonadotropin-releasing hormone (GnRH), which may be administered by a programmable, portable mini-infusion pump 201. While intravenous administration produces the most physiologic GnRH pulse contour and ensuing LH response 202, the subcutaneous route is clearly more practical for the long term treatment required to stimulate spermatogenesis. Based on the normative data 203, the frequency of GnRH administration that we employ is every 2 hours. The dose of gonadotropin-releasing hormone (GnRH) is titrated for each individual to ensure normalization of testosterone, LH and FSH and varies from 25 to 600 ng/kg per bolus. Patients on longterm therapy are monitored with serum testosterone and gonadotropin levels at monthly intervals. Once testicular volume reaches 6-8 mL, regular semen analyses are obtained. The majority of patients require treatment for 18-24 months to maximize testicular growth and achieve spermatogenesis, although the time taken to reach these endpoints tends to be shorter in those with a larger initial gonadal size. In an analysis of 76 men with idiopathic hypogonadotropic hypogonadism, pulsatile GnRH restored normal testosterone levels in 93% of cases and was successful in inducing spermatogenesis in 77% by 12 months and 82% by 24 months 204. As with gonadotropin therapy, testicular size is an important predictor of successful treatment while negative predictors include a history of cryptorchidism and a pre-treatment inhibin B level <60 pg/mL 204.
If pulsatile GnRH treatment fails, a mutation of GnRH receptor should be considered 205 and genetic testing arranged, if available. A second cause of failure of pulsatile GnRH treatment is the appearance of anti-GnRH antibodies, which typically occur in the setting of erratic compliance and are associated with a progressive decrease in T and gonadotropins levels.
Both exogenous gonadotropins and pulsatile GnRH are very effective in stimulating spermatogenesis. Most studies have no shown no advantage of either therapy in terms of testicular growth, onset of spermatogenesis, final sperm counts or pregnancy rates 206, 195. However, pulsatile GnRH therapy is not approved for induction of spermatogenesis by the Food and Drug Administration (FDA) in the United States, and its use is thus confined to specialist centers.
Human chorionic gonadotropin therapy
Human chorionic gonadotropin (hCG) is a hormone that supports the normal development of an egg in a woman’s ovary, and stimulates the release of the egg during ovulation. Human chorionic gonadotropin (hCG) is used to cause ovulation and to treat infertility in women, and to increase sperm count in men. Human chorionic gonadotropin (hCG) is also used in young boys when their testicles have not dropped down into the scrotum normally. This can be caused by a pituitary gland disorder. Although hCG can help you become pregnant, it should not be used during pregnancy. Tell your doctor right away if you become pregnant during treatment.
Circulating human chorionic gonadotropin interacts with the luteinizing hormone receptors (LHR) of the ovary, promoting the corpus luteum and its production of progesterone which is necessary to maintain pregnancy and support the growth of the fetus. Injections of hCG mimic the surge in LH that is necessary for ovulation and are used in the therapy of female infertility, in assisted reproduction techniques. In clinical trials, hCG resulted in pregnancies in approximately 30% of women. hCG prepared from urine of pregnant women and was approved for use in the United States in 1967 as treatment of ovulatory dysfunction in women desiring pregnancy. Subsequently, recombinant forms of hCG have been developed and licensed for use. Currently, hCG is available as a powder or in solution generically and under trade names such as Novarel and Pregnyl. Recombinant hCG is available as Overle. The dose and regimen of hCG therapy varies by indication and it should be used only by physicians with expertise in the management of infertility and hypogonadism. Common side effects include headache, nausea, anorexia, and local injection reactions. Uncommon, but potentially severe adverse events include ovarian hyperstimulation syndrome.
- Marshall JC, Kelch RP. Gonadotropin-releasing hormone: role of pulsatile secretion in the regulation of reproduction. N Engl J Med. 1986 Dec 4;315(23):1459-68. doi: 10.1056/NEJM198612043152306[↩][↩][↩]
- Sadiq NM, Tadi P. Physiology, Pituitary Hormones. [Updated 2023 May 1]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK557556[↩][↩][↩][↩]
- LiverTox: Clinical and Research Information on Drug-Induced Liver Injury [Internet]. Bethesda (MD): National Institute of Diabetes and Digestive and Kidney Diseases; 2012-. Gonadotropins. [Updated 2018 Mar 26]. Available from: https://www.ncbi.nlm.nih.gov/books/NBK548856[↩][↩]
- Plant TM, Marshall GR. The functional significance of FSH in spermatogenesis and the control of its secretion in male primates. Endocr Rev. 2001;22(6):764–786. doi: 10.1210/edrv.22.6.0446[↩]
- Griswold MD. Action of FSH on mammalial Sertoli cells. In: Russell LD, Griswold MD, editors. The Sertoli cell. Clearwater: Cache River Press; 1993. pp. 493–508.[↩]
- Zirkin BR, Awoniyi C, Griswold MD, Russell LD, Sharpe R. Is FSH required for adult spermatogenesis? J Androl. 1994 Jul-Aug;15(4):273-6.[↩]
- Simorangkir DR, Ramaswamy S, Marshall GR, Pohl CR, Plant TM. A selective monotropic elevation of FSH, but not that of LH, amplifies the proliferation and differentiation of spermatogonia in the adult rhesus monkey (Macacamulatta) Hum Reprod. 2009;24:1584–1595. doi: 10.1093/humrep/dep052[↩]
- Ciccone NA, Kaiser UB. The biology of gonadotroph regulation. Curr Opin Endocrinol Diabetes Obes. 2009 Aug;16(4):321-7. doi: 10.1097/MED.0b013e32832d88fb[↩]
- Gharib SD, Wierman ME, Shupnik MA, Chin WW. Molecular biology of the pituitary gonadotropins. Endocr Rev. 1990 Feb;11(1):177-99. doi: 10.1210/edrv-11-1-177[↩]
- Goodman HM. Discovery of the luteinizing hormone of the anterior pituitary gland. Am J Physiol Endocrinol Metab. 2004 Nov;287(5):E818-9. doi: 10.1152/classicessays.00006.2004[↩]
- Nedresky D, Singh G. Physiology, Luteinizing Hormone. [Updated 2022 Sep 26]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK539692[↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Ezcurra D, Humaidan P. A review of luteinising hormone and human chorionic gonadotropin when used in assisted reproductive technology. Reprod Biol Endocrinol. 2014 Oct 3;12:95. doi: 10.1186/1477-7827-12-95[↩][↩]
- Crowley WF Jr, Filicori M, Spratt DI, Santoro NF. The physiology of gonadotropin-releasing hormone (GnRH) secretion in men and women. Recent Prog Horm Res. 1985;41:473–531.[↩]
- Waldhauser F, Weissenbacher G, Frisch H, Pollak A. Pulsatile secretion of gonadotropins in early infancy. Eur J Pediatr. 1981;137:71–4[↩]
- Gupta, Priya & Mahapatra, Archisman & Suman, Anjali & Singh, Rahul. (2021). Effect of Endocrine Disrupting Chemicals on HPG Axis: A Reproductive Endocrine Homeostasis. https://www.researchgate.net/publication/350037153_Effect_of_Endocrine_Disrupting_Chemicals_on_HPG_Axis_A_Reproductive_Endocrine_Homeostasis[↩]
- Tsutsumi R, Webster NJ. GnRH pulsatility, the pituitary response and reproductive dysfunction. Endocr J. 2009;56(6):729-37. doi: 10.1507/endocrj.k09e-185[↩][↩]
- Orlowski M, Sarao MS. Physiology, Follicle Stimulating Hormone. [Updated 2023 May 1]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK535442[↩]
- Stamatiades GA, Kaiser UB. Gonadotropin regulation by pulsatile GnRH: Signaling and gene expression. Mol Cell Endocrinol. 2018 Mar 5;463:131-141. doi: 10.1016/j.mce.2017.10.015[↩][↩][↩][↩][↩]
- Barbieri RL. The endocrinology of the menstrual cycle. Methods Mol Biol. 2014;1154:145-69. doi: 10.1007/978-1-4939-0659-8_7[↩][↩][↩][↩][↩][↩][↩][↩]
- Clarke H, Dhillo WS, Jayasena CN. Comprehensive Review on Kisspeptin and Its Role in Reproductive Disorders. Endocrinol Metab (Seoul). 2015 Jun;30(2):124-41. doi: 10.3803/EnM.2015.30.2.124[↩][↩]
- Yonkers KA, Simoni MK. Premenstrual disorders. Am J Obstet Gynecol. 2018 Jan;218(1):68-74. doi: 10.1016/j.ajog.2017.05.045[↩]
- Shaw ND, Histed SN, Srouji SS, Yang J, Lee H, Hall JE. Estrogen negative feedback on gonadotropin secretion: evidence for a direct pituitary effect in women. J Clin Endocrinol Metab. 2010 Apr;95(4):1955-61. doi: 10.1210/jc.2009-2108[↩]
- Boepple PA, Hayes FJ, Dwyer AA, Raivio T, Lee H, Crowley WF Jr, Pitteloud N. Relative roles of inhibin B and sex steroids in the negative feedback regulation of follicle-stimulating hormone in men across the full spectrum of seminiferous epithelium function. J Clin Endocrinol Metab. 2008 May;93(5):1809-14. doi: 10.1210/jc.2007-2450[↩]
- Richards JS, Russell DL, Robker RL, Dajee M, Alliston TN. Molecular mechanisms of ovulation and luteinization. Mol Cell Endocrinol. 1998 Oct 25;145(1-2):47-54. doi: 10.1016/s0303-7207(98)00168-3[↩]
- Reed BG, Carr BR. The Normal Menstrual Cycle and the Control of Ovulation. [Updated 2018 Aug 5]. In: Feingold KR, Anawalt B, Blackman MR, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK279054[↩]
- Lunenfeld B, Lunenfeld E. Gonadotropic preparations–lessons learned. Fertil Steril. 1997 May;67(5):812-4. https://www.fertstert.org/article/S0015-0282(97)81389-1/pdf[↩][↩][↩]
- Lanciotti L, Cofini M, Leonardi A, Penta L, Esposito S. Up-To-Date Review About Minipuberty and Overview on Hypothalamic-Pituitary-Gonadal Axis Activation in Fetal and Neonatal Life. Front Endocrinol (Lausanne). 2018 Jul 23;9:410. doi: 10.3389/fendo.2018.00410[↩][↩][↩][↩][↩]
- Grinspon RP, Urrutia M, Rey RA. Male Central Hypogonadism in Paediatrics – the Relevance of Follicle-stimulating Hormone and Sertoli Cell Markers. Eur Endocrinol. 2018 Sep;14(2):67-71. doi: 10.17925/EE.2018.14.2.67[↩][↩]
- McLachlan RI, Wreford NG, O’Donnell L, de Kretser DM, Robertson DM. The endocrine regulation of spermatogenesis: independent roles for testosterone and FSH. J Endocrinol. 1996 Jan;148(1):1-9. doi: 10.1677/joe.0.1480001[↩]
- Kumar TR, Wang Y, Lu N, Matzuk MM. Follicle stimulating hormone is required for ovarian follicle maturation but not male fertility. Nat Genet. 1997 Feb;15(2):201-4. doi: 10.1038/ng0297-201[↩]
- Richards JS. Hormonal control of gene expression in the ovary. Endocr Rev. 1994 Dec;15(6):725-51. doi: 10.1210/edrv-15-6-725[↩]
- Christenson LK, Stouffer RL. Follicle-stimulating hormone and luteinizing hormone/chorionic gonadotropin stimulation of vascular endothelial growth factor production by macaque granulosa cells from pre- and periovulatory follicles. J Clin Endocrinol Metab. 1997 Jul;82(7):2135-42. doi: 10.1210/jcem.82.7.4169[↩]
- Li R, Phillips DM, Moore A, Mather JP. Follicle-stimulating hormone induces terminal differentiation in a predifferentiated rat granulosa cell line (ROG). Endocrinology. 1997 Jul;138(7):2648-57. doi: 10.1210/endo.138.7.5154[↩]
- Sharma OP, Flores JA, Leong DA, Veldhuis JD. Cellular basis for follicle-stimulating hormone-stimulated calcium signaling in single rat Sertoli cells: possible dissociation from effects of adenosine 3′,5′-monophosphate. Endocrinology. 1994 Apr;134(4):1915-23. doi: 10.1210/endo.134.4.8137759[↩]
- Nieschlag E, Simoni M, Gromoll J, Weinbauer GF. Role of FSH in the regulation of spermatogenesis: clinical aspects. Clin Endocrinol (Oxf). 1999 Aug;51(2):139-46. doi: 10.1046/j.1365-2265.1999.00846.x[↩][↩]
- Matthew P. Rose, Rose E. Gaines Das, Adam H. Balen, Definition and Measurement of Follicle Stimulating Hormone, Endocrine Reviews, Volume 21, Issue 1, 1 February 2000, Pages 5–22, https://doi.org/10.1210/edrv.21.1.0388[↩][↩][↩]
- Manasco PK, Umbach DM, Muly SM, Godwin DC, Negro-Vilar A, Culler MD, Underwood LE. Ontogeny of gonadotropin, testosterone, and inhibin secretion in normal boys through puberty based on overnight serial sampling. J Clin Endocrinol Metab. 1995 Jul;80(7):2046-52. doi: 10.1210/jcem.80.7.7608253[↩]
- Balen AH , Jacobs HS 1997 Male factor infertility. In: Balen AH, Jacobs HS (eds) Infertility in Practice. Churchill Livingstone, London, pp 213 –240[↩]
- Balen AH , Jacobs HS 1997 Assisted conception. In: Balen AH, Jacobs HS (eds) Infertility in Practice. Churchill Livingstone, London, pp 255 –286[↩]
- Muasher SJ, Oehninger S, Simonetti S, Matta J, Ellis LM, Liu HC, Jones GS, Rosenwaks Z. The value of basal and/or stimulated serum gonadotropin levels in prediction of stimulation response and in vitro fertilization outcome. Fertil Steril. 1988 Aug;50(2):298-307. doi: 10.1016/s0015-0282(16)60077-8[↩]
- Balen AH , Jacobs HS 1997 Investigating infertility. In: Balen AH, Jacobs HS (eds) Infertility in Practice. Churchill Livingstone, London, pp 39 –114[↩]
- Scott RT, Toner JP, Muasher SJ, Oehninger S, Robinson S, Rosenwaks Z. Follicle-stimulating hormone levels on cycle day 3 are predictive of in vitro fertilization outcome. Fertil Steril. 1989 Apr;51(4):651-4. doi: 10.1016/s0015-0282(16)60615-5[↩]
- Cameron IT, O’Shea FC, Rolland JM, Hughes EG, de Kretser DM, Healy DL. Occult ovarian failure: a syndrome of infertility, regular menses, and elevated follicle-stimulating hormone concentrations. J Clin Endocrinol Metab. 1988 Dec;67(6):1190-4. doi: 10.1210/jcem-67-6-1190[↩]
- Koskinen P, Penttilä TA, Anttila L, Erkkola R, Irjala K. Optimal use of hormone determinations in the biochemical diagnosis of the polycystic ovary syndrome. Fertil Steril. 1996 Mar;65(3):517-22. doi: 10.1016/s0015-0282(16)58146-1[↩]
- Balen AH, Conway GS, Kaltsas G, Techatrasak K, Manning PJ, West C, Jacobs HS. Polycystic ovary syndrome: the spectrum of the disorder in 1741 patients. Hum Reprod. 1995 Aug;10(8):2107-11. doi: 10.1093/oxfordjournals.humrep.a136243[↩]
- Santoro N, Brown JR, Adel T, Skurnick JH. Characterization of reproductive hormonal dynamics in the perimenopause. J Clin Endocrinol Metab. 1996 Apr;81(4):1495-501. doi: 10.1210/jcem.81.4.8636357[↩]
- Klein NA, Battaglia DE, Fujimoto VY, Davis GS, Bremner WJ, Soules MR. Reproductive aging: accelerated ovarian follicular development associated with a monotropic follicle-stimulating hormone rise in normal older women. J Clin Endocrinol Metab. 1996 Mar;81(3):1038-45. doi: 10.1210/jcem.81.3.8772573[↩]
- Bódis J, Török A, Tinneberg HR. LH/FSH ratio as a predictor of ovarian hyperstimulation syndrome. Hum Reprod. 1997 Apr;12(4):869-70. doi: 10.1093/humrep/12.4.869[↩]
- Krishnan A, Muthusami S. Hormonal alterations in PCOS and its influence on bone metabolism. J Endocrinol. 2017 Feb;232(2):R99-R113. doi: 10.1530/JOE-16-0405[↩]
- Hypogonadotropic hypogonadism. https://medlineplus.gov/ency/article/000390.htm[↩]
- Kallmann Syndrome. https://rarediseases.org/rare-diseases/kallmann-syndrome[↩][↩][↩]
- Kallmann syndrome. https://medlineplus.gov/genetics/condition/kallmann-syndrome[↩][↩][↩]
- Turner syndrome. https://www.mayoclinic.org/diseases-conditions/turner-syndrome/symptoms-causes/syc-20360782[↩]
- Turner Syndrome. https://medlineplus.gov/turnersyndrome.html[↩]
- Turner Syndrome. https://rarediseases.org/rare-diseases/turner-syndrome/[↩]
- Pouresmaeili F, Fazeli Z. Premature ovarian failure: a critical condition in the reproductive potential with various genetic causes. Int J Fertil Steril. 2014 Apr;8(1):1-12. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3973172[↩]
- Kihara M, Sugita T, Nagai Y, Saeki N, Tatsuno I, Seki K. Ovarian hyperstimulation caused by gonadotroph cell adenoma: a case report and review of the literature. Gynecol Endocrinol. 2006 Feb;22(2):110-3. doi: 10.1080/09513590600581665[↩]
- Pouwer AW, Farquhar C, Kremer JA. Long-acting FSH versus daily FSH for women undergoing assisted reproduction. Cochrane Database Syst Rev. 2015 Jul 14;2015(7):CD009577. doi: 10.1002/14651858.CD009577.pub3[↩]
- Ilahi S, Ilahi TB. Anatomy, Adenohypophysis (Pars Anterior, Anterior Pituitary) [Updated 2022 Oct 3]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK519039[↩]
- Kumar P, Sait SF. Luteinizing hormone and its dilemma in ovulation induction. J Hum Reprod Sci. 2011 Jan;4(1):2-7. doi: 10.4103/0974-1208.82351[↩][↩][↩][↩][↩]
- Choi J, Smitz J. Luteinizing hormone and human chorionic gonadotropin: distinguishing unique physiologic roles. Gynecol Endocrinol. 2014 Mar;30(3):174-81. doi: 10.3109/09513590.2013.859670[↩][↩][↩][↩]
- El Sayed SA, Fahmy MW, Schwartz J. Physiology, Pituitary Gland. [Updated 2023 May 1]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK459247[↩]
- Holesh JE, Bass AN, Lord M. Physiology, Ovulation. [Updated 2023 May 1]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK441996[↩][↩][↩]
- Rodger RS, Morrison L, Dewar JH, Wilkinson R, Ward MK, Kerr DN. Loss of pulsatile luteinising hormone secretion in men with chronic renal failure. Br Med J (Clin Res Ed). 1985 Dec 7;291(6509):1598-600. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1418441/pdf/bmjcred00477-0008.pdf[↩]
- Betz D, Fane K. Human Chorionic Gonadotropin. [Updated 2023 Aug 14]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK532950[↩][↩][↩][↩][↩]
- Ogino MH, Tadi P. Physiology, Chorionic Gonadotropin. [Updated 2022 Nov 7]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK556118[↩][↩][↩]
- Cole LA, Laidler LL. Inherited human chorionic gonadotropin. J Reprod Med. 2010 Mar-Apr;55(3-4):99-102.[↩][↩][↩][↩]
- Montagnana M, Trenti T, Aloe R, Cervellin G, Lippi G. Human chorionic gonadotropin in pregnancy diagnostics. Clin Chim Acta. 2011 Aug 17;412(17-18):1515-20. doi: 10.1016/j.cca.2011.05.025[↩][↩]
- Sirikunalai P., Wanapirak C., Sirichotiyakul S., Tongprasert F., Srisupundit K., Luewan S., Traisrisilp K., Tongsong T. Associations between maternal serum free beta human chorionic gonadotropin (β-hCG) levels and adverse pregnancy outcomes. J. Obstet. Gynaecol. 2016;36:178–182. doi: 10.3109/01443615.2015.1036400[↩][↩]
- Craig W.Y., Haddow J.E., Palomaki G.E., Roberson M. Major fetal abnormalities associated with positive screening tests for Smith-Lemli-Opitz syndrome (SLOS) Prenat. Diagn. 2007;27:409–414. doi: 10.1002/pd.1699[↩][↩]
- Norris W., Nevers T., Sharma S., Kalkunte S. Review: hCG, preeclampsia and regulatory T cells. Placenta. 2011;32:S182–S185. doi: 10.1016/j.placenta.2011.01.009[↩]
- Barjaktarovic M., Korevaar T.I.M., Jaddoe V.W.V., de Rijke Y.B., Peeters R.P., Steegers E.A.P. Human chorionic gonadotropin and risk of pre-eclampsia: Prospective population-based cohort study. Ultrasound Obstet. Gynecol. 2019;54:477–483. doi: 10.1002/uog.20256[↩]
- Barjaktarovic M., Korevaar T.I., Jaddoe V.W., de Rijke Y.B., Visser T.J., Peeters R.P., Steegers E.A. Human chorionic gonadotropin (hCG) concentrations during the late first trimester are associated with fetal growth in a fetal sex-specific manner. Eur. J. Epidemiol. 2017;32:135–144. doi: 10.1007/s10654-016-0201-3[↩]
- Stevens F.T., Katzorke N., Tempfer C., Kreimer U., Bizjak G.I., Fleisch M.C., Fehm T.N. Gestational Trophoblastic Disorders: An Update in 2015. Geburtshilfe Und Frauenheilkd. 2015;75:1043–1050. doi: 10.1055/s-0035-1558054[↩]
- Cole LA, Butler S. Detection of hCG in trophoblastic disease. The USA hCG reference service experience. J Reprod Med. 2002 Jun;47(6):433-44.[↩][↩]
- Cole L.A. “Background” Human Chorionic Gonadotropin in Healthy, Nonpregnant Women. Clin. Chem. 2005;51:1765–1766. doi: 10.1373/clinchem.2005.056507[↩][↩][↩]
- De Backer B., Goffin F., Nisolle M., Minon J.M. Élévation faible d’hCG en dehors d’un contexte gravidique: À propos de deux cas et revue de la littérature [Persistent low hCG levels beyond pregnancy: Report of two cases and review of the literature] Ann. Biol. Clin. 2013;71:496–502. doi: 10.1684/abc.2013.0876[↩]
- Katabuchi H., Ohba T. Human chorionic villous macrophages as a fetal biological shield from maternal chorionic gonadotropin. Dev. Growth Differ. 2008;50:299–306. doi: 10.1111/j.1440-169X.2008.01030.x[↩]
- Yamaguchi M., Ohba T., Tashiro H., Yamada G., Katabuchi H. Human Chorionic Gonadotropin Induces Human Macrophages to Form Intracytoplasmic Vacuoles Mimicking Hofbauer Cells in Human Chorionic Villi. Cells Tissues Organs. 2013;197:127–135. doi: 10.1159/000342806[↩]
- Paulesu L., Rao C., Ietta F., Pietropolli A., Ticconi C. hCG and Its Disruption by Environmental Contaminants during Human Pregnancy. Int. J. Mol. Sci. 2018;19:914. doi: 10.3390/ijms19030914[↩]
- Human Chorionic Gonadotropin as a Pivotal Endocrine Immune Regulator Initiating and Preserving Fetal Tolerance. Int. J. Mol. Sci. 2017, 18(10), 2166; doi:10.3390/ijms18102166 http://www.mdpi.com/1422-0067/18/10/2166/htm[↩]
- d’Hauterive SP, Close R, Gridelet V, Mawet M, Nisolle M, Geenen V. Human Chorionic Gonadotropin and Early Embryogenesis: Review. Int J Mol Sci. 2022 Jan 26;23(3):1380. doi: 10.3390/ijms23031380[↩][↩][↩][↩][↩][↩][↩]
- Jurisicova A., Antenos M., Kapasi K., Meriano J., Casper R.F. Variability in the expression of trophectodermal markers β-human chorionic gonadotrophin, human leukocyte antigen-G and pregnancy specific β-1 glycoprotein by the human blastocyst. Hum. Reprod. 1999;14:1852–1858. doi: 10.1093/humrep/14.7.1852[↩]
- Bonduelle M.-L., Dodd R., Liebaers I., Van Steirteghem A., Williamson R., Akhurst R. Chorionic gonadotrophin-β mRNA, a trophoblast marker, is expressed in human 8-cell embryos derived from tripronucleate zygotes. Hum. Reprod. 1988;3:909–914. doi: 10.1093/oxfordjournals.humrep.a136808[↩]
- Lopata A., Hay D.L. The potential of early human embryos to form blastocysts, hatch from their zona and secrete HCG in culture. Hum. Reprod. 1989;4:87–94. doi: 10.1093/humrep/4.suppl_1.87[↩]
- Braunstein G.D., Rasor J., Danzer H., Adler D., Wade M.E. Serum human chorionic gonadotropin levels throughout normal pregnancy. Am. J. Obstet. Gynecol. 1976;126:678–681. doi: 10.1016/0002-9378(76)90518-4[↩]
- Ohlsson R., Larsson E., Nilsson O., Wahlström T., Sundström P. Blastocyst implantation precedes induction of insulin-like growth factor II gene expression in human trophoblasts. Development. 1989;106:555–559. doi: 10.1242/dev.106.3.555[↩]
- D’Hauterive S.P., Charlet-Renard C., Berndt S., Dubois M., Munaut C., Goffin F., Hagelstein M.-T., Noel A., Hazout A., Foidart J.-M., et al. Human chorionic gonadotropin and growth factors at the embryonic–endometrial interface control leukemia inhibitory factor (LIF) and interleukin 6 (IL-6) secretion by human endometrial epithelium. Hum. Reprod. 2004;19:2633–2643. doi: 10.1093/humrep/deh450[↩][↩]
- Srisuparp S., Strakova Z., Fazleabas A.T. The Role of Chorionic Gonadotropin (CG) in Blastocyst Implantation. Arch. Med. Res. 2001;32:627–634. doi: 10.1016/S0188-4409(01)00330-7[↩]
- Lobo S.C., Srisuparp S., Peng X., Fazleabas A.T. Uterine Receptivity in the Baboon: Modulation by Chorionic Gonadotropin. Semin. Reprod. Med. 2001;19:069–074. doi: 10.1055/s-2001-13913[↩]
- Shi Q.J., Lei Z.M., Rao C.V., Lin J. Novel role of human chorionic gonadotropin in differentiation of human cytotrophoblasts. Endocrinology. 1993;132:1387–1395. doi: 10.1210/endo.132.3.7679981[↩]
- North R.A., Whitehead R., Larkins R.G. Stimulation by Human Chorionic Gonadotropin of Prostaglandin Synthesis by Early Human Placental Tissue. J. Clin. Endocrinol. Metab. 1991;73:60–70. doi: 10.1210/jcem-73-1-60[↩]
- Weedon-Fekjær M., Taskén K. Review: Spatiotemporal dynamics of hCG/cAMP signaling and regulation of placental function. Placenta. 2012;33:S87–S91. doi: 10.1016/j.placenta.2011.11.003[↩]
- Prast J., Saleh L., Husslein H., E Sonderegger S., Helmer H., Knöfler M. Human Chorionic Gonadotropin Stimulates Trophoblast Invasion through Extracellularly Regulated Kinase and AKT Signaling. Endocrinology. 2007;149:979–987. doi: 10.1210/en.2007-1282[↩]
- Palaniappan M., Menon K. Human Chorionic Gonadotropin Stimulates Theca-Interstitial Cell Proliferation and Cell Cycle Regulatory Proteins by a cAMP-Dependent Activation of AKT/mTORC1 Signaling Pathway. Mol. Endocrinol. 2010;24:1782–1793. doi: 10.1210/me.2010-0044[↩]
- Lee C.-L., Chiu C.N., Hautala L., Salo T., Yeung S.B.W., Stenman U.-H., Koistinen H. Human chorionic gonadotropin and its free β-subunit stimulate trophoblast invasion independent of LH/hCG receptor. Mol. Cell. Endocrinol. 2013;375:43–52. doi: 10.1016/j.mce.2013.05.009[↩]
- Cole LA, Butler SA. Hyperglycosylated human chorionic gonadotropin and human chorionic gonadotropin free beta-subunit: tumor markers and tumor promoters. J Reprod Med. 2008 Jul;53(7):499-512.[↩][↩]
- Guibourdenche J., Handschuh K., Tsatsaris V., Gerbaud P., Leguy M.C., Müller F., Brion D.E., Fournier T. Hyperglycosylated hCG Is a Marker of Early Human Trophoblast Invasion. J. Clin. Endocrinol. Metab. 2010;95:E240–E244. doi: 10.1210/jc.2010-0138[↩]
- Salas A., Gastón B., Barrenetxea J., Sendino T., Jurado M., Alcázar J.L. Predictive value of hyperglycosylated human chorionic gonadotropin for pregnancy outcomes in threatened abortion in first-trimester viable pregnancies. An. Sist. Sanit. Navar. 2021;44:23–31. doi: 10.23938/assn.0933[↩]
- Hamada A.L., Nakabayashi K., Sato A., Kiyoshi K., Takamatsu Y., Laoag-Fernandez J.B., Ohara N., Maruo T. Transfection of Antisense Chorionic Gonadotropin β Gene into Choriocarcinoma Cells Suppresses the Cell Proliferation and Induces Apoptosis. J. Clin. Endocrinol. Metab. 2005;90:4873–4879. doi: 10.1210/jc.2004-2458[↩]
- Sasaki Y., Ladner D.G., Cole L.A. Hyperglycosylated human chorionic gonadotropin and the source of pregnancy failures. Fertil. Steril. 2008;89:1781–1786. doi: 10.1016/j.fertnstert.2007.03.010[↩]
- Cole L. Hyperglycosylated hCG. Placenta. 2007;28:977–986. doi: 10.1016/j.placenta.2007.01.011[↩][↩][↩]
- Cole L.A., Dai D., Butler S.A., Leslie K.K., Kohorn E.I. Gestational trophoblastic diseases: 1. Pathophysiology of hyperglycosylated hCG. Gynecol. Oncol. 2006;102:145–150. doi: 10.1016/j.ygyno.2005.12.047[↩]
- Kovalevskaya G., Kakuma T., Schlatterer J., O’Connor J.F. Hyperglycosylated HCG expression in pregnancy: Cellular origin and clinical applications. Mol. Cell. Endocrinol. 2007;260–262:237–243. doi: 10.1016/j.mce.2006.02.021[↩]
- Bersinger N.A., Wunder D.M., Nicolas M., Birkhäuser M.H., Porquet D., Guibourdenche J. Serum Hyperglycosylated Human Chorionic Gonadotropin to Predict the Gestational Outcome in in vitro Fertilization/Intracytoplasmic Sperm Injection Pregnancies. Fetal Diagn. Ther. 2008;24:74–78. doi: 10.1159/000132412[↩]
- Wang R., Chen L., Wang X., Liu Y. Association between serum beta-human chorionic gonadotropin and inflammation, oxidative stress in pregnancy-induced hypertension. Microvasc. Res. 2021;135:104130. doi: 10.1016/j.mvr.2020.104130[↩]
- Ballantyne A., Rashid L., Pattenden R. Stability of maternal serum free β-hCG following whole blood sample transit: First trimester Down’s syndrome screening in Scotland. Ann. Clin. Biochem. 2022;59:87–91. doi: 10.1177/00045632211045250[↩]
- Cole L.A., Butler S. Hyperglycosylated hCG, hCGβ and Hyperglycosylated hCGβ: Interchangeable cancer promoters. Mol. Cell. Endocrinol. 2012;349:232–238. doi: 10.1016/j.mce.2011.10.029[↩]
- Butler S.A., Ikram M.S., Mathieu S., Iles R.K. The increase in bladder carcinoma cell population induced by the free beta subunit of human chorionic gonadotrophin is a result of an anti-apoptosis effect and not cell proliferation. Br. J. Cancer. 2000;82:1553–1556. doi: 10.1054/bjoc.2000.1177[↩]
- Cole L.A., Laidler L.L., Muller C.Y. USA hCG reference service, 10-year report. Clin. Biochem. 2010;43:1013–1022. doi: 10.1016/j.clinbiochem.2010.05.006[↩][↩]
- Birken S., Maydelman Y., Gawinowicz M.A., Pound A., Liu Y., Hartree A.S. Isolation and characterization of human pituitary chorionic gonadotropin. Endocrinology. 1996;137:1402–1411. doi: 10.1210/endo.137.4.8625917[↩][↩]
- Cole LA, Gutierrez JM. Production of human chorionic gonadotropin during the normal menstrual cycle. J Reprod Med. 2009 Apr;54(4):245-50.[↩][↩]
- Cole LA. Biological functions of hCG and hCG-related molecules. Reprod Biol Endocrinol. 2010 Aug 24;8:102. doi: 10.1186/1477-7827-8-102[↩][↩][↩][↩][↩][↩][↩]
- Schalch DS, Parlow AF, Boon RC, Reichlin S. Measurement of human luteinizing hormone in plasma by radioimmunoassay. J Clin Invest. 1968 Mar;47(3):665-78. doi: 10.1172/JCI105762[↩]
- Faiman C, Ryan RJ, Zwirek SJ, Rubin ME. Serum FSH and HCG during human pregnancy and puerperium. J Clin Endocrinol Metab. 1968 Sep;28(9):1323-9. doi: 10.1210/jcem-28-9-1323[↩][↩]
- Rao CV. Differential properties of human chorionic gonadotrophin and human luteinizing hormone binding to plasma membranes of bovine corpora lutea. Acta Endocrinol (Copenh). 1979 Apr;90(4):696-710. doi: 10.1530/acta.0.0900696[↩]
- Cole LA, Dai D, Butler SA, Leslie KK, Kohorn EI. Gestational trophoblastic diseases: 1. Pathophysiology of hyperglycosylated hCG. Gynecol Oncol. 2006 Aug;102(2):145-50. doi: 10.1016/j.ygyno.2005.12.047[↩]
- Strott CA, Yoshimi T, Ross GT, Lipsett MB. Ovarian physiology: relationship between plasma LH and steroidogenesis by the follicle and corpus luteum; effect of HCG. J Clin Endocrinol Metab. 1969 Sep;29(9):1157-67. doi: 10.1210/jcem-29-9-1157[↩][↩][↩]
- Montagnana M., Trenti T., Aloe R., Cervellin G., Lippi G. Human chorionic gonadotropin in pregnancy diagnostics. Clin. Chim. Acta. 2011;412:1515–1520. doi: 10.1016/j.cca.2011.05.025[↩]
- Toth P, Lukacs H, Gimes G, Sebestyen A, Pasztor N, Paulin F, Rao CV. Clinical importance of vascular LH/hCG receptors–a review. Reprod Biol. 2001 Nov;1(2):5-11.[↩][↩][↩][↩]
- Iles RK. Ectopic hCGbeta expression by epithelial cancer: malignant behaviour, metastasis and inhibition of tumor cell apoptosis. Mol Cell Endocrinol. 2007 Jan 2;260-262:264-70. doi: 10.1016/j.mce.2006.02.019[↩][↩][↩][↩][↩]
- Toth P, Li X, Rao CV, Lincoln SR, Sanfilippo JS, Spinnato JA 2nd, Yussman MA. Expression of functional human chorionic gonadotropin/human luteinizing hormone receptor gene in human uterine arteries. J Clin Endocrinol Metab. 1994 Jul;79(1):307-15. doi: 10.1210/jcem.79.1.8027246[↩][↩][↩]
- Berndt S, Blacher S, Perrier d’Hauterive S, Thiry M, Tsampalas M, Cruz A, Péqueux C, Lorquet S, Munaut C, Noël A, Foidart JM. Chorionic gonadotropin stimulation of angiogenesis and pericyte recruitment. J Clin Endocrinol Metab. 2009 Nov;94(11):4567-74. doi: 10.1210/jc.2009-0443[↩][↩][↩]
- Butler SA, Ikram MS, Mathieu S, Iles RK. The increase in bladder carcinoma cell population induced by the free beta subunit of human chorionic gonadotrophin is a result of an anti-apoptosis effect and not cell proliferation. Br J Cancer. 2000 May;82(9):1553-6. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2363404/pdf/82-6691177a.pdf[↩]
- Lei ZM, Rao CV, Kornyei JL, Licht P, Hiatt ES. Novel expression of human chorionic gonadotropin/luteinizing hormone receptor gene in brain. Endocrinology. 1993 May;132(5):2262-70. doi: 10.1210/endo.132.5.8477671[↩]
- Tsampalas M., Gridelet V., Berndt S., Foidart J.-M., Geenen V., D’Hauterive S.P. Human chorionic gonadotropin: A hormone with immunological and angiogenic properties. J. Reprod. Immunol. 2010;85:93–98. doi: 10.1016/j.jri.2009.11.008[↩]
- Polese B., Gridelet V., Araklioti E., Martens H., Perrier d’Hauterive S., Geenen V. The Endocrine Milieu and CD4 T-Lymphocyte Polarization during Pregnancy. Front. Endocrinol. 2014;5:106. doi: 10.3389/fendo.2014.00106[↩][↩][↩]
- Berndt S., D’Hauterive S.P., Blacher S., Pequeux C., Lorquet S., Munaut C., Applanat M., Hervé M.A., Lamandé N., Corvol P., et al. Angiogenic activity of human chorionic gonadotropin through LH receptor activation on endothelial and epithelial cells of the endometrium. FASEB J. 2006;20:2630–2632. doi: 10.1096/fj.06-5885fje[↩]
- Berndt S., Blacher S., D’Hauterive S.P., Thiry M., Tsampalas M., Cruz A., Péqueux C., Lorquet S., Munaut C., Noël A., et al. Chorionic Gonadotropin Stimulation of Angiogenesis and Pericyte Recruitment. J. Clin. Endocrinol. Metab. 2009;94:4567–4574. doi: 10.1210/jc.2009-0443[↩]
- Herr F., Baal N., Reisinger K., Lorenz A., McKinnon T., Preissner K., Zygmunt M. hCG in the Regulation of Placental Angiogenesis. Results of an In Vitro Study. Placenta. 2007;28:S85–S93. doi: 10.1016/j.placenta.2007.02.002[↩]
- Bourdiec A., Bédard D., Rao C.V., Akoum A. Human Chorionic Gonadotropin Regulates Endothelial Cell Responsiveness to Interleukin 1 and Amplifies the Cytokine-Mediated Effect on Cell Proliferation, Migration and the Release of Angiogenic Factors. Am. J. Reprod. Immunol. 2013;70:127–138. doi: 10.1111/aji.12080[↩]
- Reisinger K., Baal N., McKinnon T., Münstedt K., Zygmunt M. The gonadotropins: Tissue-specific angiogenic factors? Mol. Cell. Endocrinol. 2007;269:65–80. doi: 10.1016/j.mce.2006.11.015[↩]
- Zhang Z., Huang Y., Zhang J., Liu Z., Lin Q., Wang Z. Activation of NF-κB signaling pathway during HCG-induced VEGF expression in luteal cells. Cell Biol. Int. 2019;43:344–349. doi: 10.1002/cbin.11090[↩]
- Surico D., Farruggio S., Marotta P., Raina G., Mary D., Surico N., Vacca G., Grossini E. Human Chorionic Gonadotropin Protects Vascular Endothelial Cells from Oxidative Stress by Apoptosis Inhibition, Cell Survival Signalling Activation and Mitochondrial Function Protection. Cell. Physiol. Biochem. 2015;36:2108–2120. doi: 10.1159/000430178[↩]
- Jing G., Yao J., Dang Y., Liang W., Xie L., Chen J., Li Z. The role of β-HCG and VEGF-MEK/ERK signaling pathway in villi angiogenesis in patients with missed abortion. Placenta. 2021;103:16–23. doi: 10.1016/j.placenta.2020.10.005[↩][↩]
- Fournier T., Guibourdenche J., Evain-Brion D. Review: hCGs: Different sources of production, different glycoforms and functions. Placenta. 2015;36:S60–S65. doi: 10.1016/j.placenta.2015.02.002[↩]
- Berndt S., Blacher S., Munaut C., Detilleux J., D’Hauterive S.P., Huhtaniemi I., Evain-Brion D., Noël A., Fournier T., Foidart J. Hyperglycosylated human chorionic gonadotropin stimulates angiogenesis through TGF-β receptor activation. FASEB J. 2013;27:1309–1321. doi: 10.1096/fj.12-213686[↩]
- Gallardo V., González M., Toledo F., Sobrevia L. Role of heme oxygenase 1 and human chorionic gonadotropin in pregnancy associated diseases. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2020;1866:165522. doi: 10.1016/j.bbadis.2019.07.016[↩]
- Raghupathy R. Th 1-type immunity is incompatible with successful pregnancy. Immunol. Today. 1997;18:478–482. doi: 10.1016/S0167-5699(97)01127-4[↩]
- Schumacher A., Heinze K., Witte J., Poloski E., Linzke N., Woidacki K., Zenclussen A.C. Human Chorionic Gonadotropin as a Central Regulator of Pregnancy Immune Tolerance. J. Immunol. 2013;190:2650–2658. doi: 10.4049/jimmunol.1202698[↩]
- Lea R.G., Sandra O. Immunoendocrine aspects of endometrial function and implantation. Reproduction. 2007;134:389–404. doi: 10.1530/REP-07-0167[↩]
- Fujiwara H. Do circulating blood cells contribute to maternal tissue remodeling and embryo-maternal cross-talk around the implantation period? Mol. Hum. Reprod. 2009;15:335–343. doi: 10.1093/molehr/gap027[↩]
- Akoum A, Metz CN, Morin M. Marked increase in macrophage migration inhibitory factor synthesis and secretion in human endometrial cells in response to human chorionic gonadotropin hormone. J Clin Endocrinol Metab. 2005 May;90(5):2904-10. doi: 10.1210/jc.2004-1900[↩]
- Herrmann-Lavoie C, Rao CV, Akoum A. Chorionic gonadotropin down-regulates the expression of the decoy inhibitory interleukin 1 receptor type II in human endometrial epithelial cells. Endocrinology. 2007 Nov;148(11):5377-84. doi: 10.1210/en.2007-0368[↩]
- Matsuura T, Sugimura M, Iwaki T, Ohashi R, Kanayama N, Nishihira J. Anti-macrophage inhibitory factor antibody inhibits PMSG-hCG-induced follicular growth and ovulation in mice. J Assist Reprod Genet. 2002 Dec;19(12):591-5. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3455831/pdf/10815_2004_Article_450330.pdf[↩]
- Kamada M, Ino H, Naka O, Irahara M, Daitoh T, Mori K, Maeda N, Maegawa M, Hirano K, Aono T. Immunosuppressive 30-kDa protein in urine of pregnant women and patients with trophoblastic diseases. Eur J Obstet Gynecol Reprod Biol. 1993 Aug;50(3):219-25. doi: 10.1016/0028-2243(93)90204-p[↩]
- Majumdar S, Bapna BC, Mapa MK, Gupta AN, Devi PK, Subrahmanyam D. Pregnancy specific proteins: suppression of in vitro blastogenic response to mitogen by these proteins. Int J Fertil. 1982;27(2):66-9.[↩]
- CEDARD L, VARANGOT J, YANNOTTI S. [The metabolism of estrogens in human placentas artificially maintained in survival by perfusion in vitro]. C R Hebd Seances Acad Sci. 1962 Mar 5;254:1870-1. French.[↩]
- Cole LA. Immunoassay of human chorionic gonadotropin, its free subunits, and metabolites. Clin Chem. 1997 Dec;43(12):2233-43.[↩]
- Davies S, Byrn F, Cole LA. Human chorionic gonadotropin testing for early pregnancy viability and complications. Clin Lab Med. 2003 Jun;23(2):257-64, vii. doi: 10.1016/s0272-2712(03)00026-x[↩]
- Menczer J, Modan M, Serr DM. Prospective follow-up of patients with hydatidiform mole. Obstet Gynecol. 1980 Mar;55(3):346-9. doi: 10.1097/00006250-198003000-00015[↩]
- Cole LA, Shahabi S, Butler SA, Mitchell H, Newlands ES, Behrman HR, Verrill HL. Utility of commonly used commercial human chorionic gonadotropin immunoassays in the diagnosis and management of trophoblastic diseases. Clin Chem. 2001 Feb;47(2):308-15.[↩]
- Butts SF, Guo W, Cary MS, Chung K, Takacs P, Sammel MD, Barnhart KT. Predicting the decline in human chorionic gonadotropin in a resolving pregnancy of unknown location. Obstet Gynecol. 2013 Aug;122(2 Pt 1):337-343. doi: 10.1097/AOG.0b013e31829c6ed6[↩]
- Barjaktarovic M, Korevaar TI, Jaddoe VW, de Rijke YB, Visser TJ, Peeters RP, Steegers EA. Human chorionic gonadotropin (hCG) concentrations during the late first trimester are associated with fetal growth in a fetal sex-specific manner. Eur J Epidemiol. 2017 Feb;32(2):135-144. doi: 10.1007/s10654-016-0201-3[↩][↩][↩][↩][↩]
- Cavaliere A, Ermito S, Dinatale A, Pedata R. Management of molar pregnancy. J Prenat Med. 2009 Jan;3(1):15-7. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3279094[↩][↩][↩][↩][↩][↩]
- Brouillet S, Murthi P, Hoffmann P, Salomon A, Sergent F, De Mazancourt P, Dakouane-Giudicelli M, Dieudonné MN, Rozenberg P, Vaiman D, Barbaux S, Benharouga M, Feige JJ, Alfaidy N. EG-VEGF controls placental growth and survival in normal and pathological pregnancies: case of fetal growth restriction (FGR). Cell Mol Life Sci. 2013 Feb;70(3):511-25. doi: 10.1007/s00018-012-1141-z[↩][↩][↩][↩]
- Visconti F, Quaresima P, Chiefari E, Caroleo P, Arcidiacono B, Puccio L, Mirabelli M, Foti DP, Di Carlo C, Vero R, Brunetti A. First Trimester Combined Test (FTCT) as a Predictor of Gestational Diabetes Mellitus. Int J Environ Res Public Health. 2019 Sep 28;16(19):3654. doi: 10.3390/ijerph16193654[↩][↩]
- Butler SA, Ikram MS, Mathieu S, Iles RK. The increase in bladder carcinoma cell population induced by the free beta subunit of human chorionic gonadotrophin is a result of an anti-apoptosis effect and not cell proliferation. Br J Cancer. 2000 May;82(9):1553-6. doi: 10.1054/bjoc.2000.1177[↩]
- Balasubramanian R, Crowley WF Jr. Isolated Gonadotropin-Releasing Hormone (GnRH) Deficiency. 2007 May 23 [Updated 2017 Mar 2]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2018. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1334[↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Hoffman AR, Crowley WF Jr. Induction of puberty in men by long-term pulsatile administration of low-dose gonadotropin-releasing hormone. N Engl J Med. 1982;307:1237–41[↩]
- Bianco SD, Kaiser UB. The genetic and molecular basis of idiopathic hypogonadotropic hypogonadism. Nat Rev Endocrinol. 2009;5:569–76.[↩]
- Laitinen EM, Vaaralahti K, Tommiska J, Eklund E, Tervaniemi M, Valanne L, Raivio T. Incidence, phenotypic features and molecular genetics of Kallmann syndrome in Finland. Orphanet J Rare Dis. 2011;6:41[↩]
- Seminara SB, Hayes FJ, Crowley WF Jr. Gonadotropin-releasing hormone deficiency in the human (idiopathic hypogonadotropic hypogonadism and Kallmann’s syndrome): pathophysiological and genetic considerations. Endocr Rev. 1998;19:521–39[↩]
- Raivio T, Falardeau J, Dwyer A, Quinton R, Hayes FJ, Hughes VA, Cole LW, Pearce SH, Lee H, Boepple P, Crowley WF Jr, Pitteloud N. Reversal of idiopathic hypogonadotropic hypogonadism. N Engl J Med. 2007;357:863–73[↩]
- Pitteloud N, Boepple PA, DeCruz S, Valkenburgh SB, Crowley WF Jr, Hayes FJ. The fertile eunuch variant of idiopathic hypogonadotropic hypogonadism: spontaneous reversal associated with a homozygous mutation in the gonadotropin-releasing hormone receptor. J Clin Endocrinol Metab. 2001;86:2470–5[↩]
- Shaw ND, Seminara SB, Welt CK, Au MG, Plummer L, Hughes VA, Dwyer AA, Martin KA, Quinton R, Meriq V, Merino PM, Gusella JF, Crowley WF Jr, Pitteloud N, Hall JE. Expanding the phenotype and genotype of female GnRH deficiency. J. Clin. Endocrinol. Metab. 2011;96:E566–76[↩]
- Doty RL. Office procedures for quantitative assessment of olfactory function. Am J Rhinol. 2007;21:460–73[↩]
- Lewkowitz-Shpuntoff HM, Hughes VA, Plummer L, Au MG, Doty RL, Seminara SB, Chan YM, Pitteloud N, Crowley WF Jr, Balasubramanian R. Olfactory phenotypic spectrum in idiopathic hypogonadotropic hypogonadism: pathophysiological and genetic implications. J Clin Endocrinol Metab. 2012;97:E136–44[↩]
- Costa-Barbosa FA, Balasubramanian R, Keefe KW, Shaw ND, Al-Tassan N, Plummer L, Dwyer A, Buck CL, Choi J-H, Seminara SB, Quinton R, Monies D, Meyer B, Hall JE, Pitteloud N, Crowley WF. Prioritizing Genetic Testing in Patients With Kallmann Syndrome Using Clinical Phenotypes. J Clin Endocrinol Metab. 2013;98:E943–53.[↩]
- Pedersen-White JR, Chorich LP, Bick DP, Sherins RJ, Layman LC. The prevalence of intragenic deletions in patients with idiopathic hypogonadotropic hypogonadism and Kallmann syndrome. Mol Hum Reprod. 2008;14:367–70.[↩]
- Trarbach EB, Teles MG, Costa EM, Abreu AP, Garmes HM, Guerra G Jr, Baptista MT, de Castro M, Mendonca BB, Latronico AC. Screening of autosomal gene deletions in patients with hypogonadotropic hypogonadism using multiplex ligation-dependent probe amplification: detection of a hemizygosis for the fibroblast growth factor receptor 1. Clin Endocrinol (Oxf). 2010b;72:371–6.[↩]
- Young J. Approach to the male patient with congenital hypogonadotropic hypogonadism. J Clin Endocrinol Metab. 2012;97:707–18.[↩]
- Dwyer AA, Sykiotis GP, Hayes FJ, Boepple PA, Lee H, Loughlin KR, Dym M, Sluss PM, Crowley WF Jr, Pitteloud N. Trial of recombinant follicle-stimulating hormone pretreatment for GnRH-induced fertility in patients with congenital hypogonadotropic hypogonadism. J Clin Endocrinol Metab. 2013;98:E1790–5[↩]
- Burris AS, Rodbard HW, Winters SJ, Sherins RJ. Gonadotropin therapy in men with isolated hypogonadotropic hypogonadism: the response to human chorionic gonadotropin is predicted by initial testicular size. J Clin Endocrinol Metab. 1988;66:1144–51.[↩]
- Whitcomb RW, Crowley WF Jr. Clinical review 4: Diagnosis and treatment of isolated gonadotropin-releasing hormone deficiency in men. J Clin Endocrinol Metab. 1990;70:3–7.[↩]
- Liu PY, Baker HW, Jayadev V, Zacharin M, Conway AJ, Handelsman DJ. Induction of spermatogenesis and fertility during gonadotropin treatment of gonadotropin-deficient infertile men: predictors of fertility outcome. J Clin Endocrinol Metab. 2009;94:801–8.[↩]
- Pitteloud N, Hayes FJ, Dwyer A, Boepple PA, Lee H, Crowley WF Jr. Predictors of outcome of long-term GnRH therapy in men with idiopathic hypogonadotropic hypogonadism. J Clin Endocrinol Metab. 2002b;87:4128–36[↩]
- Santoro N, Filicori M, Crowley WF Jr. Hypogonadotropic disorders in men and women: diagnosis and therapy with pulsatile gonadotropin-releasing hormone. Endocr Rev. 1986;7:11–23[↩]
- Martin K, Santoro N, Hall J, Filicori M, Wierman M, Crowley WF Jr (1990) Clinical review 15: Management of ovulatory disorders with pulsatile gonadotropin-releasing hormone. J Clin Endocrinol Metab. 71:1081A-G[↩]
- Sykiotis GP, Hoang XH, Avbelj M, Hayes FJ, Thambundit A, Dwyer A, Au M, Plummer L, Crowley WF Jr, Pitteloud N. Congenital idiopathic hypogonadotropic hypogonadism: evidence of defects in the hypothalamus, pituitary, and testes. J Clin Endocrinol Metab. 2010a;95:3019–27[↩]
- Balen AH, Braat DD, West C, Patel A, Jacobs HS. Cumulative conception and live birth rates after the treatment of anovulatory infertility: safety and efficacy of ovulation induction in 200 patients. Hum Reprod. 1994 Aug;9(8):1563-70. doi: 10.1093/oxfordjournals.humrep.a138750[↩]
- Ben-Rafael Z, Levy T, Schoemaker J. Pharmacokinetics of follicle-stimulating hormone: clinical significance. Fertil Steril. 1995 Apr;63(4):689-700. doi: 10.1016/s0015-0282(16)57467-6[↩]
- White DM, Polson DW, Kiddy D, Sagle P, Watson H, Gilling-Smith C, Hamilton-Fairley D, Franks S. Induction of ovulation with low-dose gonadotropins in polycystic ovary syndrome: an analysis of 109 pregnancies in 225 women. J Clin Endocrinol Metab. 1996 Nov;81(11):3821-4. doi: 10.1210/jcem.81.11.8923819[↩]
- Nardelli AA, Stafinski T, Motan T, Klein K, Menon D. Assisted reproductive technologies (ARTs): evaluation of evidence to support public policy development. Reprod Health. 2014 Nov 7;11(1):76. doi: 10.1186/1742-4755-11-76[↩]
- Raju GA, Chavan R, Deenadayal M, Gunasheela D, Gutgutia R, Haripriya G, Govindarajan M, Patel NH, Patki AS. Luteinizing hormone and follicle stimulating hormone synergy: A review of role in controlled ovarian hyper-stimulation. J Hum Reprod Sci. 2013 Oct;6(4):227-34. doi: 10.4103/0974-1208.126285[↩][↩]
- Hayes F, Dwyer A, Pitteloud N. Hypogonadotropic Hypogonadism (HH) and Gonadotropin Therapy. [Updated 2013 Nov 25]. In: Feingold KR, Anawalt B, Blackman MR, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK279078[↩][↩][↩][↩][↩][↩]
- Fraietta R, Zylberstejn DS, Esteves SC. Hypogonadotropic hypogonadism revisited. Clinics (Sao Paulo). 2013;68 Suppl 1(Suppl 1):81-8. doi: 10.6061/clinics/2013(sup01)09[↩]
- Clavijo RI, Hsiao W. Update on male reproductive endocrinology. Transl Androl Urol. 2018 Jul;7(Suppl 3):S367-S372. doi: 10.21037/tau.2018.03.25[↩]
- Canavese F, Mussa A, Manenti M, Cortese MG, Ferrero L, Tuli G, Macchieraldo R, Lala R. Sperm count of young men surgically treated for cryptorchidism in the first and second year of life: fertility is better in children treated at a younger age. Eur J Pediatr Surg. 2009 Dec;19(6):388-91. doi: 10.1055/s-0029-1241171[↩]
- Recombinant follicle stimulating hormone: development of the first biotechnology product for the treatment of infertility. Recombinant Human FSH Product Development Group. Hum Reprod Update. 1998 Nov-Dec;4(6):862-81. doi: 10.1093/humupd/4.6.862[↩][↩][↩]
- Mannaerts B, Fauser B, Lahlou N, Harlin J, Shoham Z, Bennink HC, Bouchard P. Serum hormone concentrations during treatment with multiple rising doses of recombinant follicle stimulating hormone (Puregon) in men with hypogonadotropic hypogonadism. Fertil Steril. 1996 Feb;65(2):406-10. doi: 10.1016/s0015-0282(16)58108-4[↩]
- King TF, Hayes FJ. Long-term outcome of idiopathic hypogonadotropic hypogonadism. Curr Opin Endocrinol Diabetes Obes. 2012 Jun;19(3):204-10. doi: 10.1097/MED.0b013e328353565b[↩]
- Burris AS, Rodbard HW, Winters SJ, Sherins RJ. Gonadotropin therapy in men with isolated hypogonadotropic hypogonadism: the response to human chorionic gonadotropin is predicted by initial testicular size. J Clin Endocrinol Metab. 1988 Jun;66(6):1144-51. doi: 10.1210/jcem-66-6-1144[↩][↩][↩]
- Vicari E, Mongioì A, Calogero AE, Moncada ML, Sidoti G, Polosa P, D’Agata R. Therapy with human chorionic gonadotrophin alone induces spermatogenesis in men with isolated hypogonadotrophic hypogonadism–long-term follow-up. Int J Androl. 1992 Aug;15(4):320-9. doi: 10.1111/j.1365-2605.1992.tb01131.x[↩]
- Büchter D, Behre HM, Kliesch S, Nieschlag E. Pulsatile GnRH or human chorionic gonadotropin/human menopausal gonadotropin as effective treatment for men with hypogonadotropic hypogonadism: a review of 42 cases. Eur J Endocrinol. 1998 Sep;139(3):298-303. doi: 10.1530/eje.0.1390298[↩][↩]
- Liu PY, Baker HW, Jayadev V, Zacharin M, Conway AJ, Handelsman DJ. Induction of spermatogenesis and fertility during gonadotropin treatment of gonadotropin-deficient infertile men: predictors of fertility outcome. J Clin Endocrinol Metab. 2009 Mar;94(3):801-8. doi: 10.1210/jc.2008-1648[↩][↩][↩][↩]
- Warne DW, Decosterd G, Okada H, Yano Y, Koide N, Howles CM. A combined analysis of data to identify predictive factors for spermatogenesis in men with hypogonadotropic hypogonadism treated with recombinant human follicle-stimulating hormone and human chorionic gonadotropin. Fertil Steril. 2009 Aug;92(2):594-604. doi: 10.1016/j.fertnstert.2008.07.1720[↩][↩]
- Whitcomb RW, Crowley WF Jr. Clinical review 4: Diagnosis and treatment of isolated gonadotropin-releasing hormone deficiency in men. J Clin Endocrinol Metab. 1990 Jan;70(1):3-7. doi: 10.1210/jcem-70-1-3[↩]
- Kulin HE, Samojlik E, Santen R, Santner S. The effect of growth hormone on the Leydig cell response to chorionic gonadotrophin in boys with hypopituitarism. Clin Endocrinol (Oxf). 1981 Nov;15(5):463-72. doi: 10.1111/j.1365-2265.1981.tb00689.x[↩][↩]
- Chatelain PG, Sanchez P, Saez JM. Growth hormone and insulin-like growth factor I treatment increase testicular luteinizing hormone receptors and steroidogenic responsiveness of growth hormone deficient dwarf mice. Endocrinology. 1991 Apr;128(4):1857-62. doi: 10.1210/endo-128-4-1857[↩]
- Hoffman AR, Crowley WF Jr. Induction of puberty in men by long-term pulsatile administration of low-dose gonadotropin-releasing hormone. N Engl J Med. 1982 Nov 11;307(20):1237-41. doi: 10.1056/NEJM198211113072003[↩]
- Spratt DI, Crowley WF Jr, Butler JP, Hoffman AR, Conn PM, Badger TM. Pituitary luteinizing hormone responses to intravenous and subcutaneous administration of gonadotropin-releasing hormone in men. J Clin Endocrinol Metab. 1985 Nov;61(5):890-5. doi: 10.1210/jcem-61-5-890[↩]
- Spratt DI, O’Dea LS, Schoenfeld D, Butler J, Rao PN, Crowley WF Jr. Neuroendocrine-gonadal axis in men: frequent sampling of LH, FSH, and testosterone. Am J Physiol. 1988 May;254(5 Pt 1):E658-66. doi: 10.1152/ajpendo.1988.254.5.E658[↩]
- Pitteloud N, Hayes FJ, Dwyer A, Boepple PA, Lee H, Crowley WF Jr. Predictors of outcome of long-term GnRH therapy in men with idiopathic hypogonadotropic hypogonadism. J Clin Endocrinol Metab. 2002 Sep;87(9):4128-36. doi: 10.1210/jc.2002-020518[↩][↩]
- Caron P, Chauvin S, Christin-Maitre S, Bennet A, Lahlou N, Counis R, Bouchard P, Kottler ML. Resistance of hypogonadic patients with mutated GnRH receptor genes to pulsatile GnRH administration. J Clin Endocrinol Metab. 1999 Mar;84(3):990-6. doi: 10.1210/jcem.84.3.5518[↩]
- Liu L, Banks SM, Barnes KM, Sherins RJ. Two-year comparison of testicular responses to pulsatile gonadotropin-releasing hormone and exogenous gonadotropins from the inception of therapy in men with isolated hypogonadotropic hypogonadism. J Clin Endocrinol Metab. 1988 Dec;67(6):1140-5. doi: 10.1210/jcem-67-6-1140[↩]





{kind=link}