Contents
- What is hematocrit
- What are red blood cells
- Red blood cell production
- What does the test result mean?
- What happens when you have low red blood cells (Anemia)
- What Causes Anemia ?
- Who Is at Risk for Anemia ?
- What Are the Signs and Symptoms of Anemia ?
- Complications of Anemia
- How Is Anemia Diagnosed ?
- How Is Anemia Treated ?
- What Is Iron-Deficiency Anemia ?
- What Is Pernicious Anemia ?
- Sickle Cell Disease
- Some Forms of Sickle Cell Disease
- What Causes Sickle Cell Disease ?
- How Is Sickle Cell Disease Inherited ?
- What Are the Signs and Symptoms of Sickle Cell Disease ?
- Major Complications of Sickle Cell Disease
- Acute Chest Syndrome
- Brain Complications
- How Is Sickle Cell Disease Diagnosed ?
- How Is Sickle Cell Disease Treated ?
- Preventing Infection
- Screening Tests and Evaluations
- Hematopoietic Stem Cell Transplantation
- What happens when you have too many red blood cells
What is hematocrit
Hematocrit is a blood test that measures the proportion of a person’s blood that is made up of red blood cells (RBCs) or the percentage of total blood volume occupied by red blood cells (RBCs). The hematocrit measures the volume of red blood cells (RBCs) compared to the total blood volume (red blood cells, white blood cells, platelets and plasma). Blood consists of red blood cells (RBCs), white blood cells (WBCs), and platelets (thrombocytes) suspended in a fluid portion called plasma (see Figure 1). The hematocrit is a ratio of the volume of red blood cells to the volume of all these components together, called whole blood. The value is expressed as a percentage or fraction. For example, a hematocrit value of 40% means that there are 40 milliliters of red blood cells in 100 milliliters of blood.
Red blood cells carry oxygen to all parts of your body. They also remove carbon dioxide (a waste product) from your body’s cells and carry it to the lungs to be exhaled.
Red blood cells are made in your bone marrow—a sponge-like tissue inside the bones. White blood cells (WBCs) and platelets (thrombocytes) also are made in your bone marrow. White blood cells help fight infection. Platelets stick together to seal small cuts or breaks on blood vessel walls and stop bleeding.
The normal hematocrit range for adult males is 40 to 54% (average = 47%); for adult females it is 38 to 46% (average = 42%). A hematocrit of 40 indicates that 40% of the volume of blood is composed of red blood cells (RBCs). The hormone testosterone, present in much higher concentration in males than in females, stimulates synthesis of erythropoietin (EPO), the hormone that in turn stimulates production of red blood cells (RBCs). Thus, testosterone contributes to higher hematocrits in males. Lower hematocrit values in women during their reproductive years also may be due to excessive loss of blood during menstruation.
The hematocrit is a fairly quick and simple way of evaluating a person’s red blood cells and checking for conditions such as anemia. It is often performed in conjunction with a hemoglobin (Hb) level and is also one component of the complete blood count (CBC), a test that is often used in the general evaluation of a person’s health. A sample is obtained by drawing blood through a needle placed in a vein in the arm or by a fingerstick (for children and adults) or a heelstick (for newborns).
The hematocrit reflects both the number of red blood cells and their volume (mean corpuscular volume or MCV). If the size of the red blood cells decreases (MCV decreases), so will the hematocrit and vice versa. In general, the hematocrit will rise when the number of red blood cells increases and the hematocrit will fall to less than normal when there is a drop in production of red blood cells by the bone marrow, an increase in the destruction of red blood cells, or if blood is lost due to bleeding. If the bone marrow is not able to produce new red blood cells fast enough, then the overall number of red blood cells and hematocrit will drop, resulting in anemia. A significant drop in hematocrit indicates anemia, a lower-than-normal number of red blood cells (RBCs). In anemia, the body does not have the capacity to deliver enough oxygen to tissues and organs, causing fatigue and weakness. In polycythemia (too many red blood cells) the percentage of red blood cells (RBCs) is abnormally high, and the hematocrit may be 65% or higher. This raises the viscosity of blood, the blood can becomes thickened which increases the resistance to flow and makes the blood more difficult for the heart to pump. Increased viscosity also contributes to high blood pressure, sluggish blood flow and increased risk of stroke. Causes of polycythemia include abnormal increases in red blood cell production (e.g., polycythemia vera or polycythemia rubra vera), tissue hypoxia, dehydration, blood doping, or the use of erythropoietin (EPO) by athletes.
The hematocrit value can be determined directly by microhematocrit centrifugation or calculated indirectly. Automated cell counters calculate the hematocrit by multiplying the red cell number (in millions/mm3) by the mean corpuscular volume (MCV, in femtoliters). When so assayed, it is subject to the vagaries inherent in obtaining an accurate measurement of the mean corpuscular volume (MCV).
Red Blood Cell Counts
The number of red blood cells in a microliter (μL or mcL or 1 mm3) of blood is called the red blood cell count (RBCC or RCC). This number varies from time to time even in healthy individuals. However, the typical range for adult males is 4,700,000 to 6,100,000 cells per microliter, and that for adult females is 4,200,000 to 5,400,000 cells per microliter.
The absolute numbers for red blood cell, white blood cell, and platelet counts can vary depending on how they are measured and the instruments used to measure them. For this reason, different sources may present different, but very similar, ranges of normal values.
An increase in the number of circulating red blood cells increases the blood’s oxygen-carrying capacity, much as a decrease in the number of circulating red blood cells decreases the blood’s oxygen-carrying capacity. Changes in this number may affect health. For this reason, red blood cell counts are routinely consulted to help diagnose and evaluate the courses of certain diseases.
Can my hematocrit be tested at home?
No. This test requires instrumentation and trained laboratory personnel. A hematocrit is typically indirectly measured [i.e., calculated from red blood cell and mean corpuscular volume (MCV)] by automated hematology analyzers. It can also be directly measured by spinning a blood-filled capillary tube in a centrifuge (so-called spun hematocrit), but this manual method is less commonly used.
How to measure hematocrit level?
For hematocrits obtained by fingerstick, wipe the fingertip pad of the fourth finger of the nondominant hand with the alcohol prep pad. Make certain the area is allowed to dry. Prick the fingertip with the lancet. Place the hematocrit tube near the incision site and allow the blood to flow via capillary action into the hematocrit tube until it is two-thirds to three-fourths full or to a predesignated mark on the tube. Avoid “milking” the finger if possible; this causes the expression of tissue fluids and may result in a falsely low hematocrit. Always fill at least three tubes. For hematocrits obtained by venipuncture, draw a sample of blood into the tube containing anticoagulant and mix well. Dip the hematocrit tube into the blood and allow the blood to rise to the desired two-thirds to three-quarters level. Because blood cells naturally sediment, a prior thorough mixing of the blood in the tube is necessary to ensure accurate reading.
After cleaning the outside of the hematocrit tubes of excess blood, invert the tube slowly so that the blood migrates just short of the bottom end of the tube. Seal the bottom of the tube with sealant. Make certain that little or no air is interspersed in the column of blood. If the seal is incomplete, leakage will occur during centrifugation and false readings will be obtained.
Place the tubes in a microhematocrit centrifuge and spin for 3 to 5 minutes at high speed. A shorter spin will not allow for complete sedimentation.
Using either a hematocrit reader or any ruled apparatus, measure the length of the column of the packed red cells and divide it by the length of the whole column of blood (cells and plasma). To obtain the hematocrit, multiply this number by 100%. Average all readings obtained from the different microhematocrit tubes.
Figure 1. Hematocrit level measurement – Note: Microhematocrit tube after sedimentation. The hematocrit is a ratio of the packed cells to total volume.

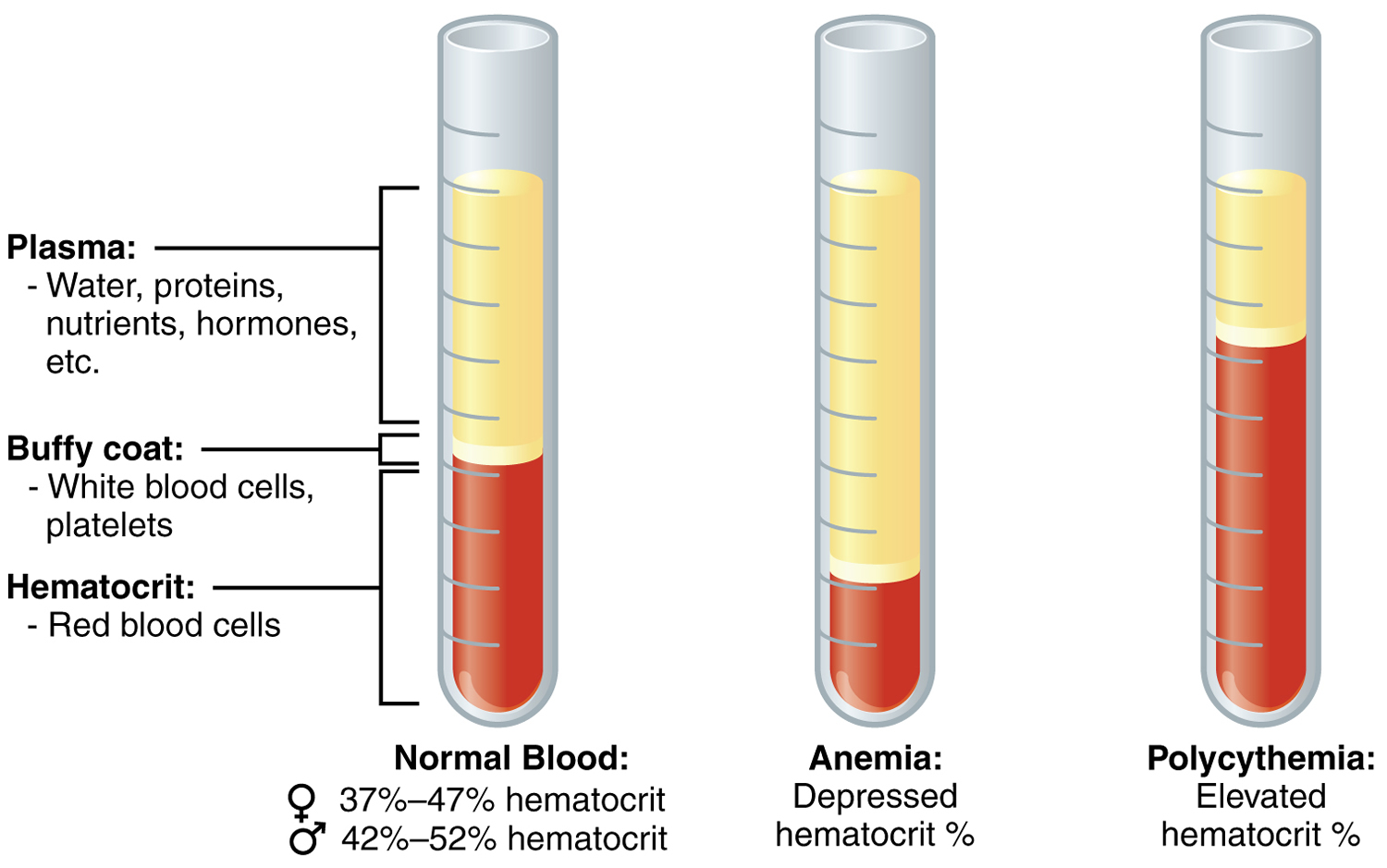
Example: If the column of packed red cells measures 20 mm and the whole blood column measures 50 mm, the hematocrit is 20/50 = 0.4 or (0.4 × 100%) = 40%.
Normal hematocrit levels
Red blood cells (RBCs) typically make up roughly 37% to 49% of the volume of blood.
Since a hematocrit is often performed as part of a complete blood count (CBC), results from other components, such as red blood cell count (RBCC or RCC), hemoglobin (Hb), reticulocyte count, and/or red blood cell indices, are taken into consideration. Age, sex, and race are other factors to be considered. In general, the hematocrit mirrors the results of the red blood cell count (RBCC or RCC) and hemoglobin (Hb).
Hemoglobin and Hematocrit
Hemoglobin (Hb) is the protein contained in red blood cells that is responsible for delivery of oxygen to the tissues. To ensure adequate tissue oxygenation, a sufficient hemoglobin level must be maintained. The amount of hemoglobin in whole blood is expressed in grams per deciliter (g/dl). The normal Hb level for males is 14 to 18 g/dl; that for females is 12 to 16 g/dl. When the hemoglobin level is low, the patient has anemia. An erythrocytosis (polycythemia) is the consequence of too many red cells; this results in hemoglobin levels above normal.
Hemoglobin determinations will usually be performed by an automated cell counter from a tube of well-mixed EDTA-anticoagulated blood filled to a predetermined level. In this assay, all forms of hemoglobins are converted to the colored protein cyanomethemoglobin and measured by a colorimeter. An inadequate sample, whether due to insufficient volume or inadequate anticoagulation, may give false readings. If it is necessary to determine the level of anemia quickly, the hematocrit is an easier, more convenient test.
Hemoglobin (Hb) is composed of two pairs of dissimilar chains, α and β, each defined by a specific amino acid sequence and incorporating an iron-containing heme group. Two α–β dimers combine to form a hemoglobin tetramer. This allows for the “heme–heme” interaction necessary for effective oxygen uptake (deoxyhemoglobin → oxyhemoglobin) and delivery (oxyhemoglobin → deoxyhemoglobin). The oxygen affinity of hemoglobin is a function of this heme–heme interaction and of pH (Bohr effect), and is a measure of how many hemoglobin molecules have oxygen bound to them for a given level of oxygen tension. In a normal individual the major hemoglobin is Hb A, constituting approximately 97% of the total hemoglobin. Variations and/or amino acid substitutions in these chains exist. Some are deleterious to the normal function of hemoglobin, whereas others may have relatively normal oxygen affinity and stability. Hemoglobins containing different types of chains make up the remainder of the hemoglobin content in red cells (α2δ2 = Hb A2 approximately 2%; α2γ2 = Hb F approximately 1%).
Figure 2. Red blood cell hemoglobin
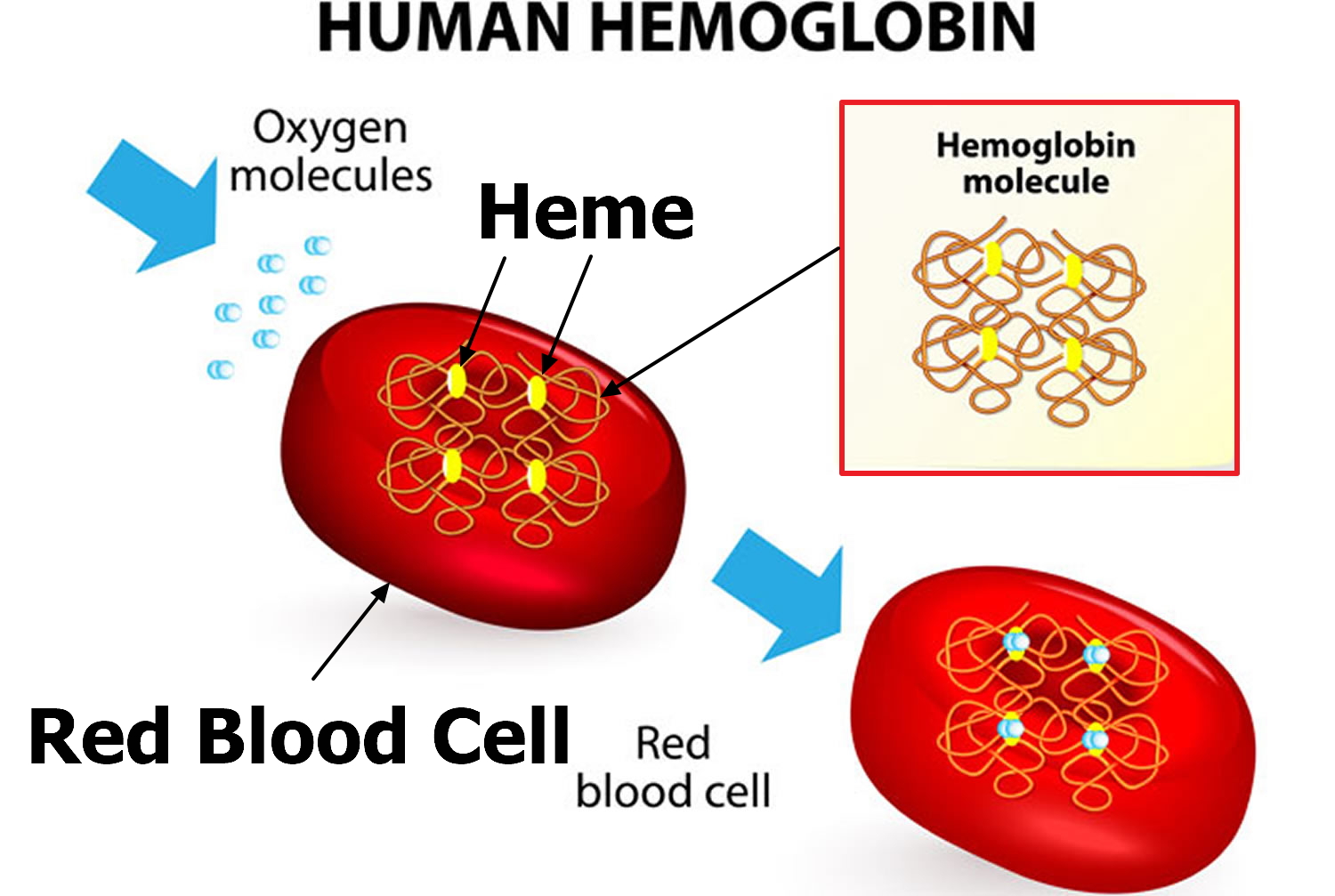
Substitutions in the normal hemoglobin amino acid sequence may result in hemoglobins that have different sub-unit interactions and varying affinities for oxygen. For example, a substitution of the sixth amino acid on the beta chain causes Hb S, or sickle hemoglobin. Hb S has a lower oxygen affinity and surrenders its oxygen more readily. Hb F, a normal minor hemoglobin constituent, has a higher oxygen affinity.
If the oxygen dissociation curve is abnormal, the body will adjust the hemoglobin level to ensure adequate oxygen distribution to the tissues. Thus in a rare disease like hemoglobin Hotel Dieu, the difficulty in extracting oxygen from a variant hemoglobin with increased oxygen affinity could result in a lack of oxygen for the tissues (tissue hypoxia) and a compensatory erythrocytosis. The smaller fraction of oxygen released from the hemoglobin is thereby offset by the increased number of hemoglobin molecules. Similarly, in sickle cell anemia, the decreased oxygen affinity allows these patients more tissue oxygen at any given hemoglobin level.
What are red blood cells
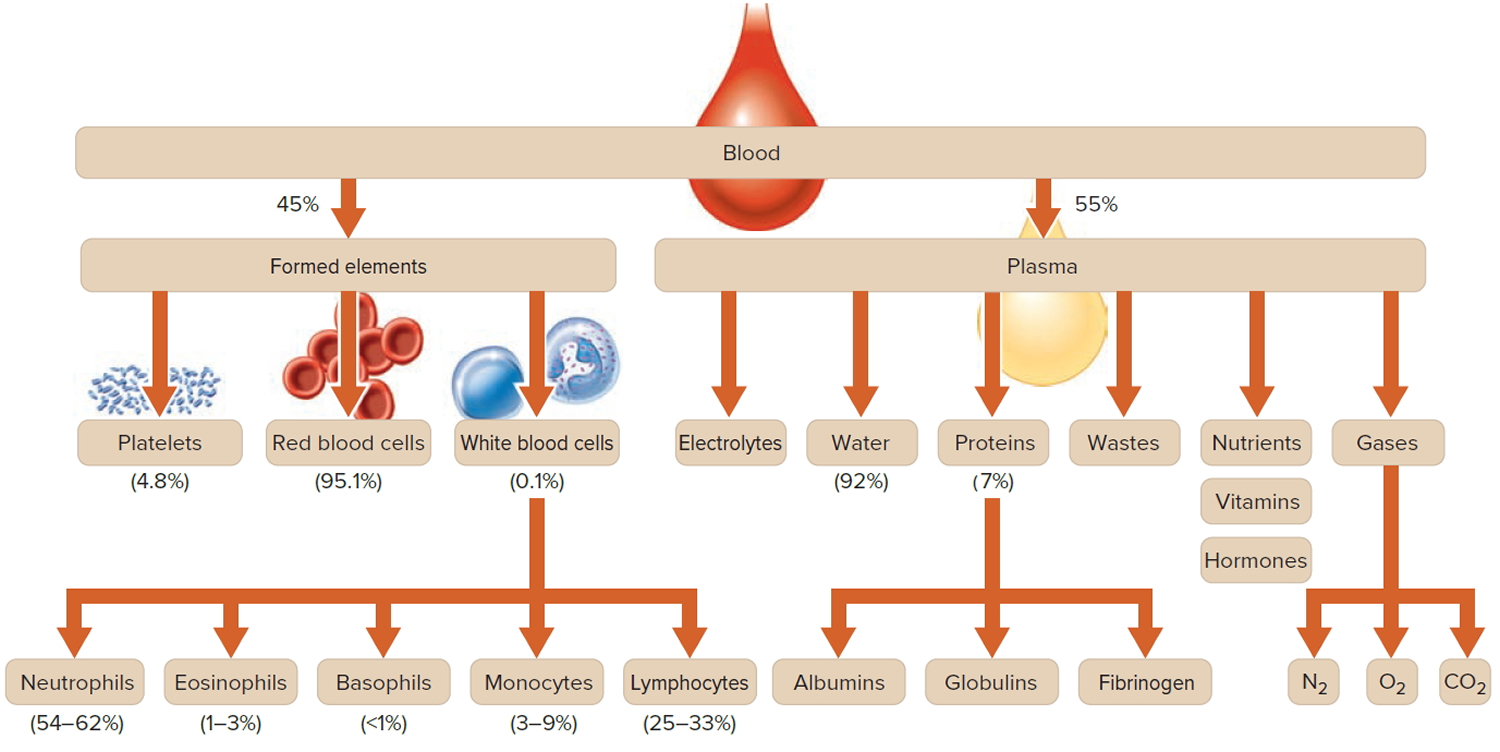
Blood transports a variety of materials between interior body cells and those that exchange substances with the external environment. In this way, blood helps maintain stable internal environmental conditions. Blood is composed of formed elements suspended in a fluid extracellular matrix called blood plasma. The “formed elements” include red blood cells, white blood cells, and cell fragments called platelets (Figure 4). Most blood cells form in red marrow within the hollow parts of certain long bones.
Most blood samples are roughly 37% to 49% red blood cells by volume – adult females is 38–46% (average = 42%) and for adult males, it is 40–54% (average = 47). This percentage is called the hematocrit. The white blood cells and platelets account for less than 1% of blood volume. The remaining blood sample, about 55%, is the plasma, a clear, straw-colored liquid. Blood plasma is a complex mixture of water, gases, amino acids, proteins, carbohydrates, lipids, vitamins, hormones, electrolytes, and cellular wastes (see Figure 3).
Blood volume varies with body size, percent adipose tissue, and changes in fluid and electrolyte concentrations. An average-size adult has a blood volume of about 5 liters (5.3 quarts), 4–5 liters in a female and 5–6 liters in a male.
Red blood cell (also called erythrocyte) is biconcave disc without a nucleus. This biconcave shape is an adaptation for transporting the gases oxygen and carbon dioxide. It increases the surface area through which oxygen and carbon dioxide can diffuse into and out of the cell. The characteristic shape of a red blood cell also places the cell membrane closer to oxygen-carrying hemoglobin molecules in the cell reducing the distance for diffusion.
Each red blood cell is about one-third hemoglobin by volume. This protein imparts the color of blood. When hemoglobin binds oxygen, the resulting oxyhemoglobin is bright red, and when oxygen is released, the resulting deoxyhemoglobin is darker.
Prolonged oxygen deficiency (hypoxia) causes cyanosis, in which the skin and mucous membranes appear bluish due to an abnormally high blood concentration of deoxyhemoglobin in the superficial blood vessels. Exposure to low temperature may also result in cyanosis by constricting superficial blood vessels. This response to environmental change slows skin blood flow. As a result, more oxygen than usual is removed from the blood flowing through the vessels, increasing the concentration of deoxyhemoglobin.
Note: Blood is a complex mixture of formed elements in a liquid extracellular matrix, called blood plasma. Note that water and proteins account for 99% of the blood plasma.
Figure 3. Blood composition
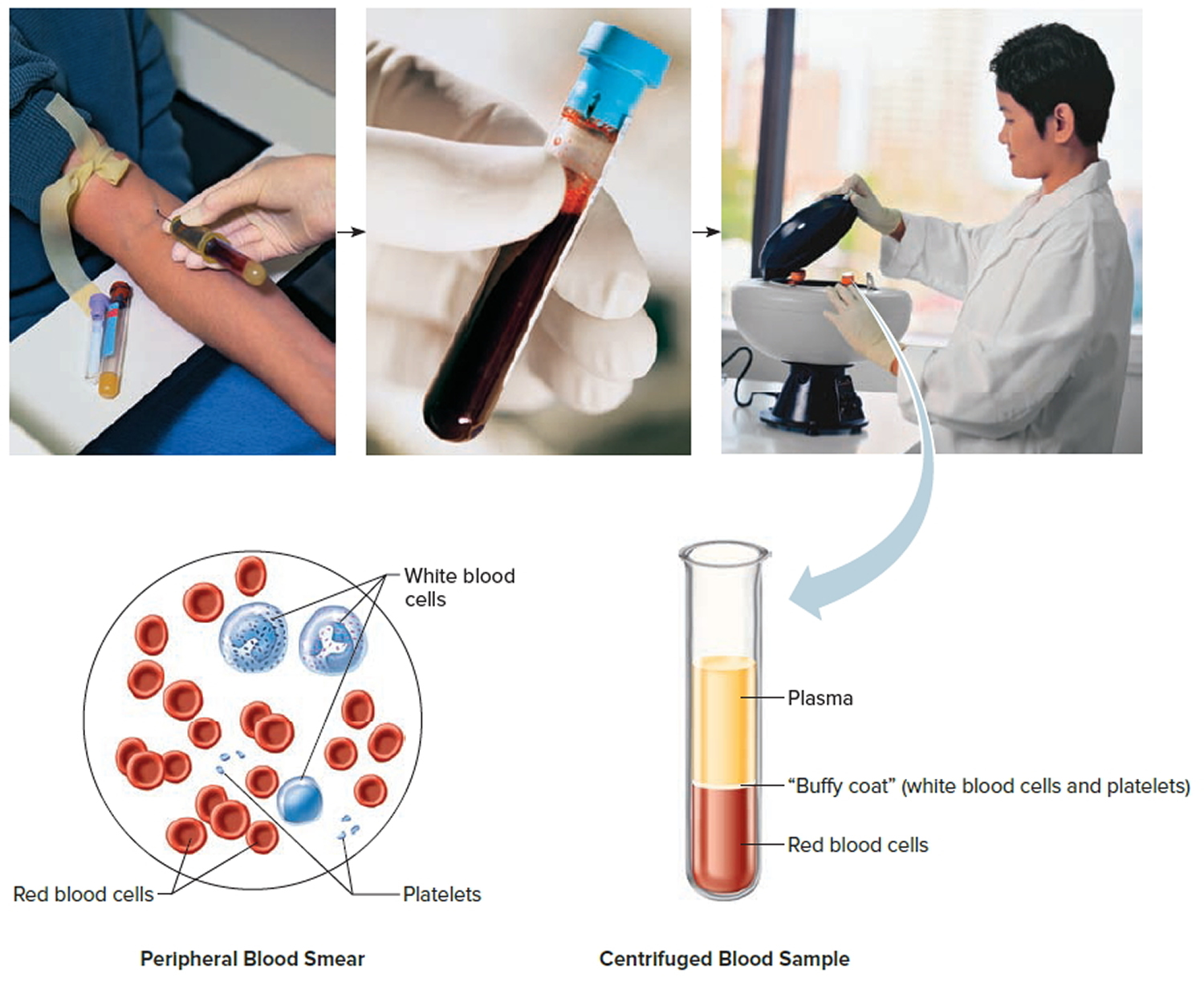
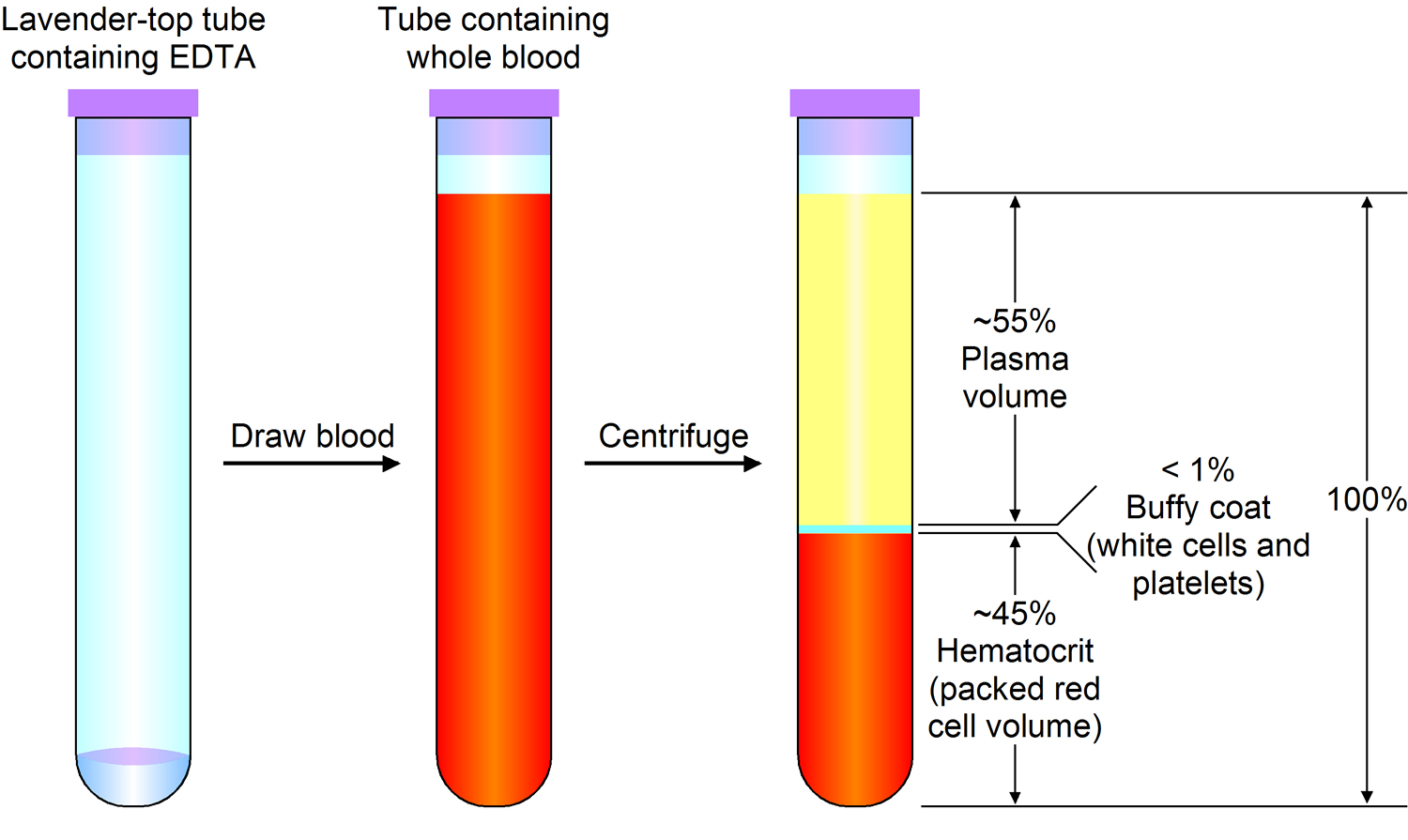
Note: Blood consists of a liquid portion called plasma and a solid portion (the formed elements) that includes red blood cells, white blood cells, and platelets. When blood components are separated by centrifugation, the white blood cells and platelets form a thin layer, called the “buffy coat,” between the plasma and the red blood cells, which accounts for about 1% of the total blood volume. Blood cells and platelets can be seen under a light microscope when a blood sample is smeared onto a glass slide.
Figure 4. Blood cells
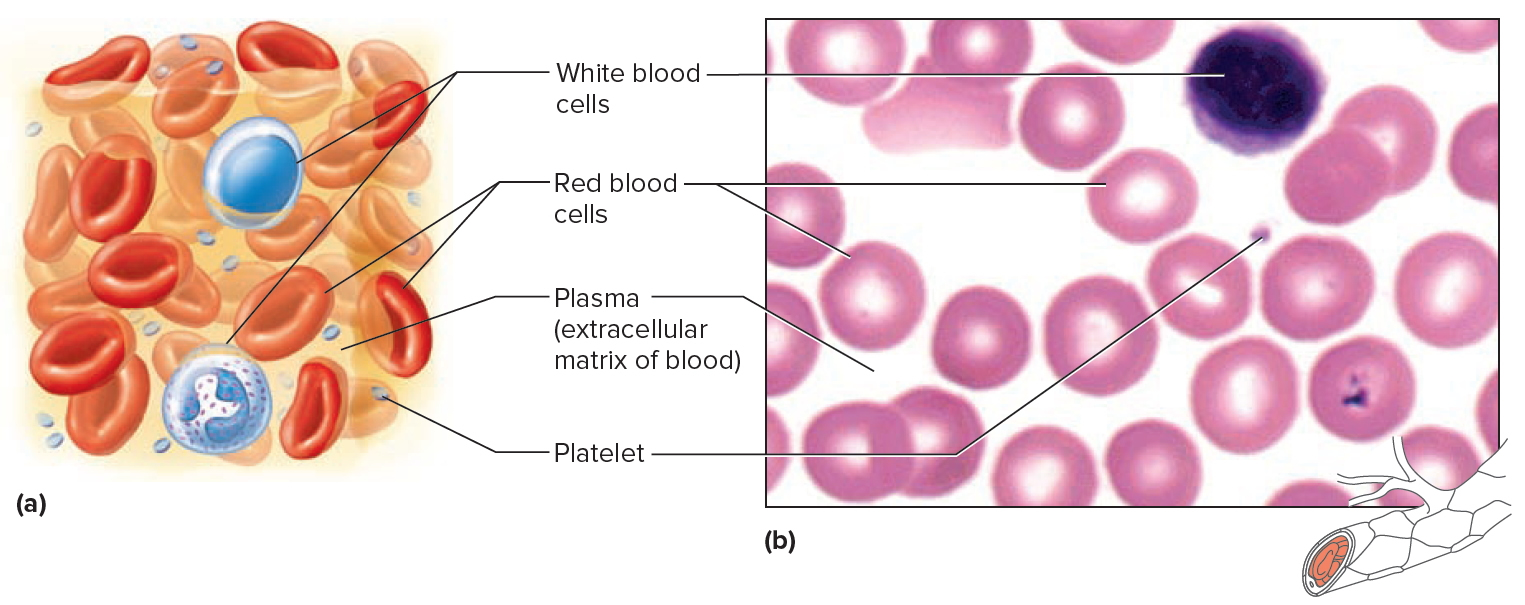
Note: Blood tissue consists of red blood cells, white blood cells, and platelets suspended in plasma. (a) Idealized representation of a sample of blood. (b) Micrograph of a sample of blood (1,000x).
Red blood cell production
Red blood cells are produced in the bone marrow and are released into the bloodstream when they are, or nearly are, mature. They typically make up roughly 37% to 49% of the volume of blood. Red blood cells contain hemoglobin, a protein that binds to oxygen. The primary function of red blood cells is to carry oxygen from the lungs to the tissues and organs of the body. They also transport a small portion of carbon dioxide, a byproduct of cell metabolism, from tissues and organs back to the lungs, where it is expelled.
The typical lifespan of an red blood cell (RBC) is 120 days and the bone marrow must continually produce new red blood cells to replace those that age and degrade or are lost through bleeding. A number of conditions can affect either the production of new red blood cells by the bone marrow or the lifespan of those in circulation or that result in significant bleeding.
Anatomy of the bone
The bone is made up of compact bone, spongy bone, and bone marrow. Compact bone makes up the outer layer of the bone. Spongy bone is found mostly at the ends of bones and contains red marrow. Bone marrow is found in the center of most bones and has many blood vessels. There are two types of bone marrow: red and yellow. Red marrow contains blood stem cells that can become red blood cells, white blood cells, or platelets. Yellow marrow is made mostly of fat.
Bone marrow is a soft, netlike mass of connective tissue within the medullary cavities of long bones, in the irregular spaces of spongy bone, and in the larger central canals of compact bone tissue. It is of two kinds: red and yellow. Red bone marrow functions in the formation of red blood cells (erythrocytes), white blood cells (leukocytes), and blood platelets. The color comes from the oxygen-carrying pigment hemoglobin in the red blood cells.
In an infant, red marrow occupies the cavities of most bones. As a person ages, yellow bone marrow, which stores fat, replaces much of the red marrow. Yellow marrow is not active in blood cell production. In an adult, red marrow is primarily found in the spongy bone of the skull, ribs, breastbone (sternum), collarbones (clavicles), backbones (vertebrae), and hip bones. If the supply of blood cells is deficient, some yellow marrow may become red marrow, which then reverts to yellow marrow when the deficiency is corrected.
Figure 5. Bone marrow anatomy
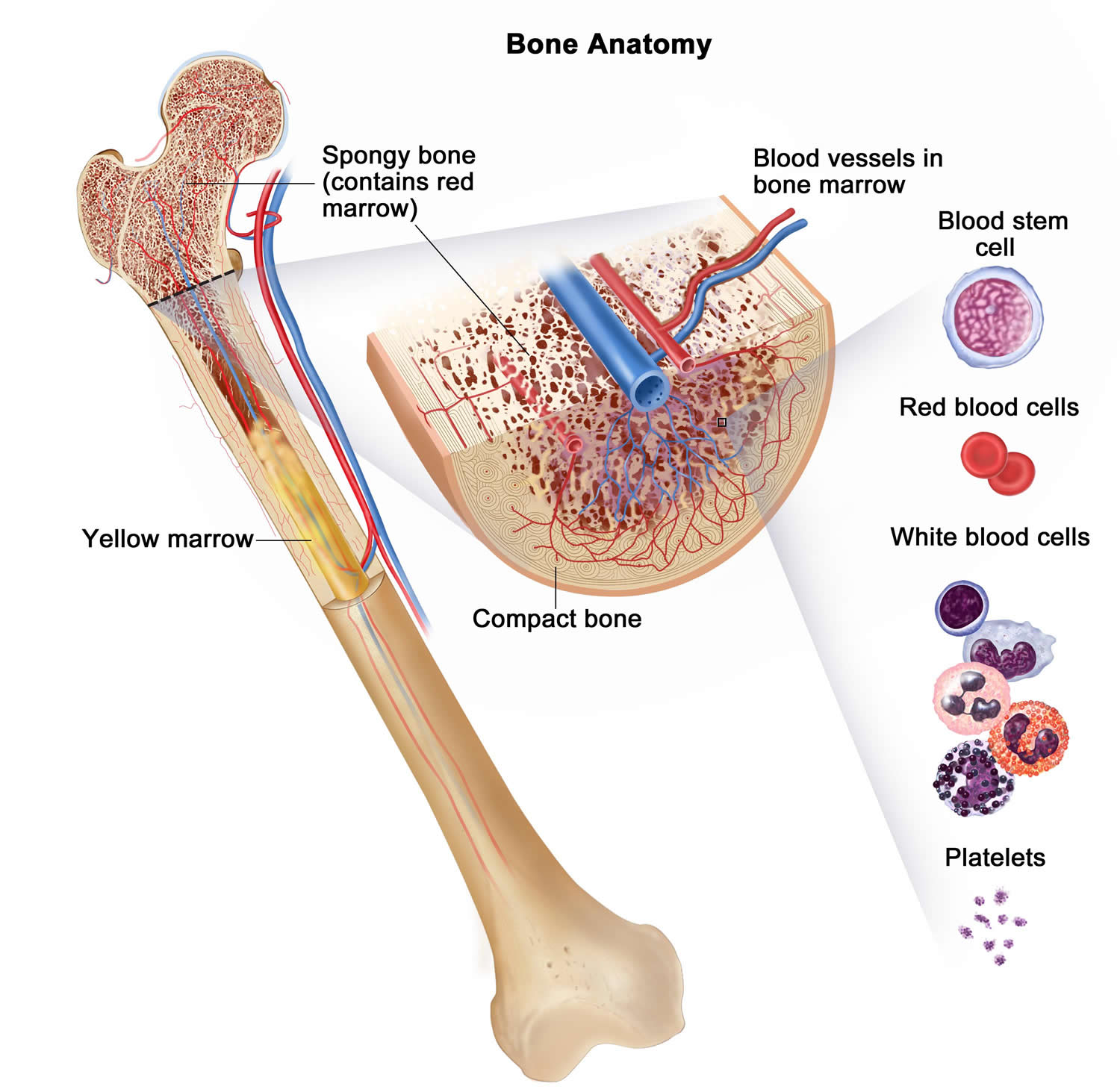
Figure 6 illustrates the stages in the formation of red blood cells from hematopoietic stem cells (blood-forming cells), which are also called hemocytoblasts.
Red blood cells have nuclei during their early stages of development but lose their nuclei as the cells mature. Losing the nuclei provides more space for hemoglobin. Because mature red blood cells do not have nuclei, they cannot divide. They use none of the oxygen they carry because they do not have mitochondria. Mature red blood cells produce ATP through glycolysis only.
The average life span of a red blood cell is 120 days. Many of these cells are removed from the circulation each day, and yet the number of cells in the circulating blood remains relatively stable. This observation suggests a homeostatic control of the rate of red blood cell production.
The hormone erythropoietin controls the rate of red blood cell formation through negative feedback. The kidneys, and to a lesser extent the liver, release erythropoietin in response to prolonged oxygen deficiency (Figure 7). At high altitudes, for example, where the amount of oxygen in the air is reduced, the blood oxygen level initially decreases. This drop in the blood oxygen level triggers the release of erythropoietin, which travels via the blood to the red bone marrow and stimulates red blood cell production.
After a few days of exposure to high altitudes, many newly formed red blood cells appear in the circulating blood. The increased rate of production continues until the number of erythrocytes in the circulation is sufficient to supply tissues with oxygen. When the availability of oxygen returns to normal, erythropoietin release decreases, and the rate of red blood cell production returns to normal as well. An excessive increase in red blood cells is called polycythemia. This condition increases blood viscosity, slowing blood flow and impairing circulation.
Figure 6. Blood cell development. A blood stem cell goes through several steps to become a red blood cell, platelet, or white blood cell
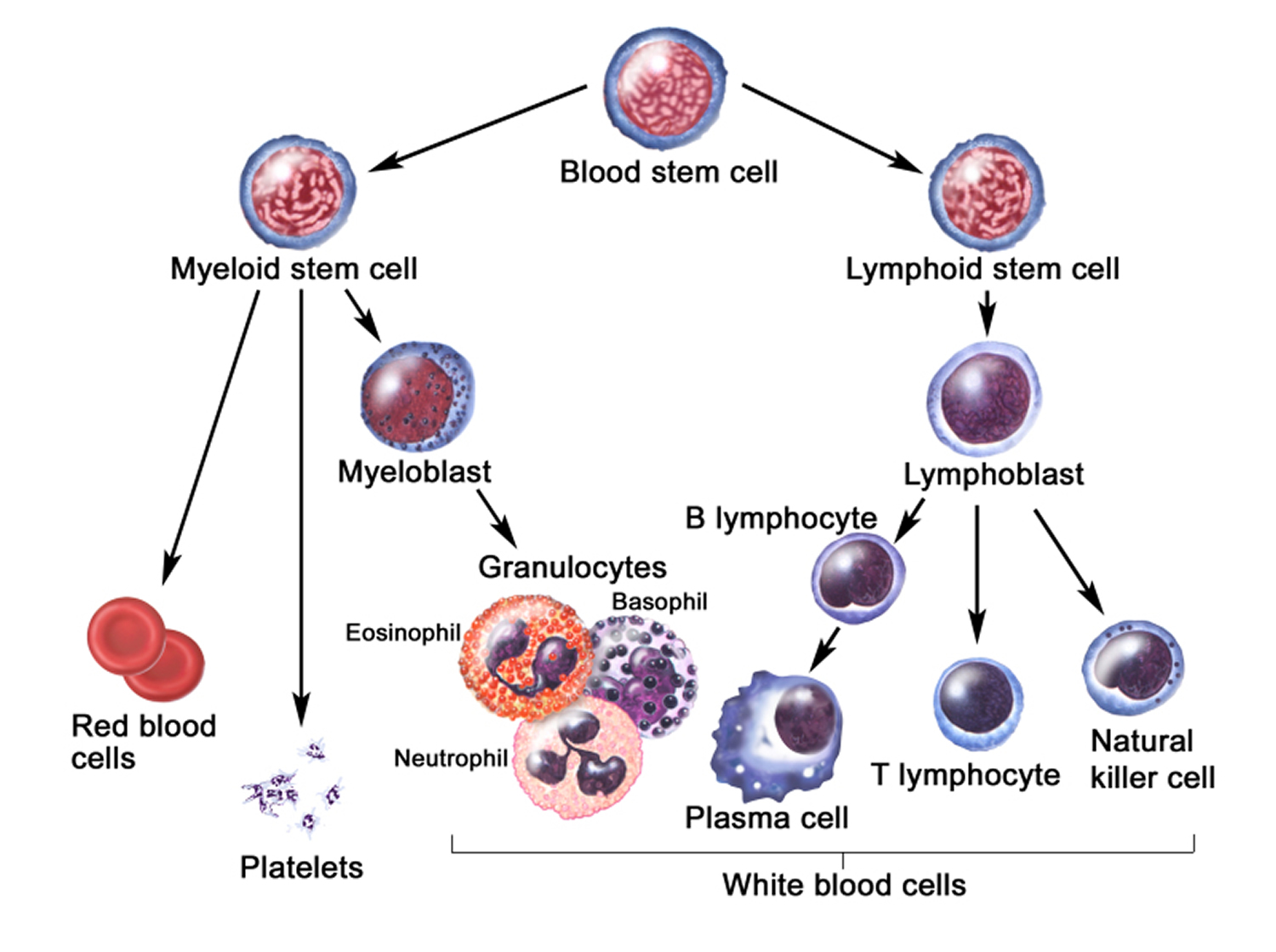
Figure 7. Red blood cell formation
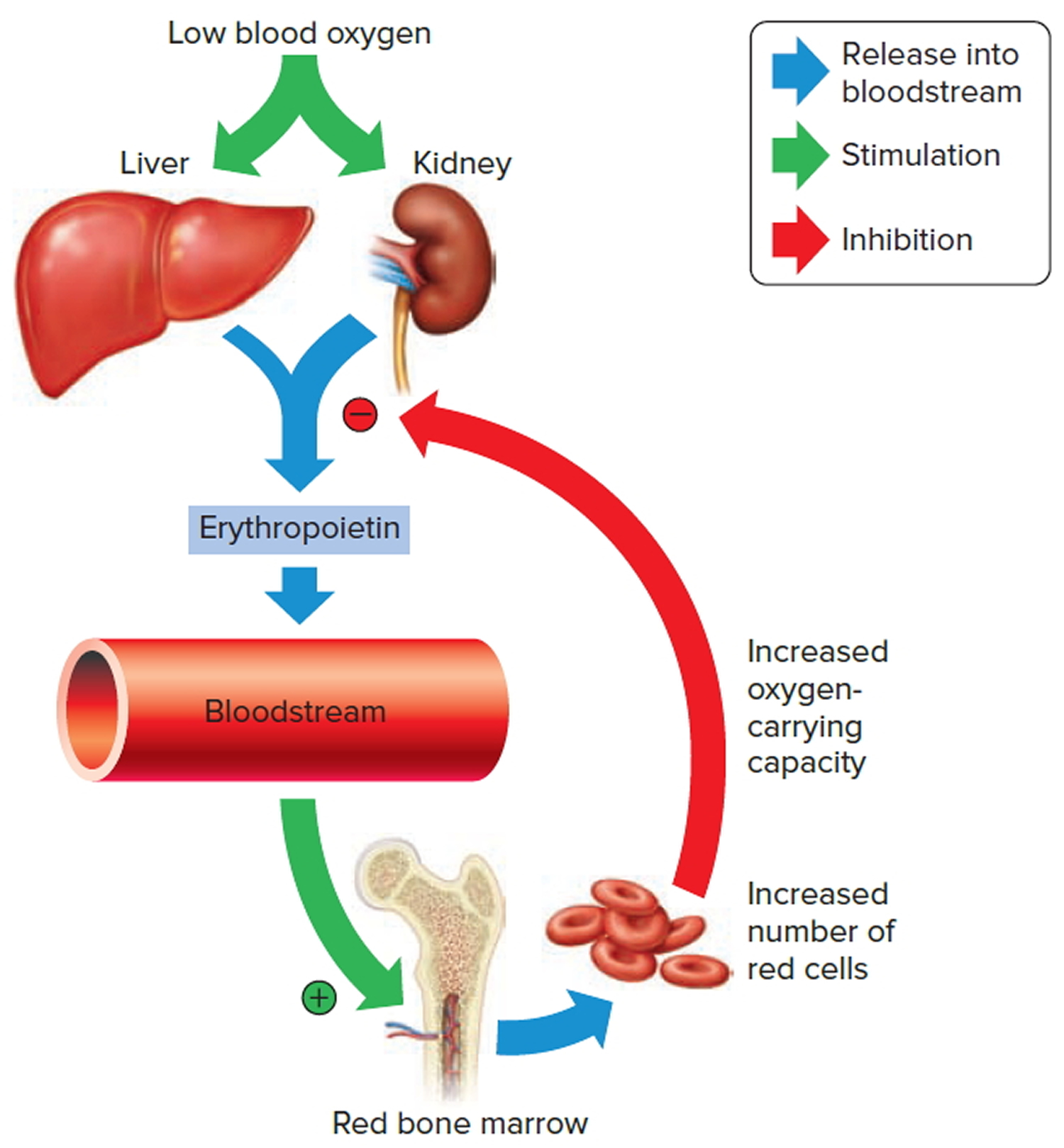
Note: Low blood oxygen causes the kidneys and to a lesser degree, the liver to release erythropoietin. Erythropoietin stimulates target cells in the red bone marrow to increase the production of red blood cells, which carry oxygen to tissues.
Dietary Factors Affecting Red Blood Cell Production
Availability of B-complex vitamins—vitamin B12 and folic acid—significantly influences red blood cell production. Because these vitamins are required for DNA synthesis, they are necessary for the growth and division of cells. Cell division is frequent in blood-forming (hematopoietic) tissue, so this tissue is especially vulnerable to a deficiency of either of these vitamins.
Hemoglobin synthesis and normal red blood cell production also require iron. The small intestine absorbs iron slowly from food. The body reuses much of the iron released by the decomposition of hemoglobin from damaged red blood cells. Nonetheless, insufficient dietary iron can reduce hemoglobin synthesis.
A deficiency of red blood cells or a reduction in the amount of hemoglobin they contain results in a condition called anemia. This reduces the oxygen-carrying capacity of the blood, and the affected person may appear pale and lack energy. A pregnant woman may have a normal number of red blood cells, but she develops a relative anemia because her plasma volume increases due to fluid retention. This shows up as a decreased hematocrit.
In contrast to anemia, the inherited disorder called hemochromatosis results in the absorption of iron in the small intestine at ten times the normal rate. Iron builds up in organs, to toxic levels. Treatment is periodic blood removal, as often as every week.
What does the test result mean?
Red blood cells (RBCs) typically make up roughly 37% to 49% of the volume of blood.
A recent blood transfusion will affect hematocrit results.
Pregnancy usually causes slightly decreased hematocrit values due to extra fluid in the blood.
Since a hematocrit is often performed as part of a complete blood count (CBC), results from other components, such as red blood cell count (RBCC or RCC), hemoglobin, reticulocyte count, and/or red blood cell indices, are taken into consideration. Age, sex, and race are other factors to be considered. In general, the hematocrit mirrors the results of the red blood cell count (RBCC or RCC) and hemoglobin.
Low hematocrit causes
A low hematocrit with low red blood cell count (RBCC or RCC) and low hemoglobin indicates anemia. Some causes include:
- Excessive loss of blood from, for example, severe trauma, or chronic bleeding from sites such as the digestive tract (e.g., ulcers, polyps, colon cancer), the bladder or uterus (in women, heavy menstrual bleeding, for example)
- Nutritional deficiencies such as iron, folate or B12 deficiency
- Damage to the bone marrow from, for example, a toxin, radiation or chemotherapy, infection or drugs
- Bone marrow disorders such as aplastic anemia, myelodysplastic syndrome, or cancers such as leukemia, lymphoma, multiple myeloma, or other cancers that spread to the marrow
- Kidney failure—severe and chronic kidney diseases lead to decreased production of erythropoietin, a hormone produced by the kidneys that stimulates RBC production by the bone marrow.
- Chronic inflammatory diseases or conditions
- Decreased production of hemoglobin (e.g., thalassemia)
- Excessive destruction of red blood cells, for example, hemolytic anemia caused by autoimmunity or defects in the red blood cell itself; the defects could be hemoglobinopathy (e.g., sickle cell anemia), abnormalities in the RBC membrane (e.g., hereditary spherocytosis) or RBC enzyme (e.g., G6PD deficiency)
High hematocrit causes
A high hematocrit with a high red blood cell count (RBCC or RCC) and high hemoglobin indicates polycythemia. Some causes of a high hematocrit include:
- Dehydration—this is the most common cause of a high hematocrit. As the volume of fluid in the blood drops, the RBCs per volume of fluid artificially rises; with adequate fluid intake, the hematocrit returns to normal.
- Lung (pulmonary) disease—if someone is unable to breathe in and absorb sufficient oxygen, the body tries to compensate by producing more red blood cells.
- Congenital heart disease—in some forms, there is an abnormal connection between the two sides of the heart, leading to reduced oxygen levels in the blood. The body tries to compensate by producing more red blood cells.
- Kidney tumor that produces excess erythropoietin
- Smoking
- Living at high altitudes (a compensation for decreased oxygen in the air)
- Genetic causes (altered oxygen sensing, abnormality in hemoglobin oxygen release)
- Polycythemia vera (polycythemia rubra vera or Vaquez disease)—a rare disease in which the body produces excess RBCs inappropriately
What happens when you have low red blood cells (Anemia)
Anemia is a condition in which your blood has a lower than normal number of red blood cells 1.
Anemia also can occur if your red blood cells don’t contain enough hemoglobin. Hemoglobin is an iron-rich protein that gives blood its red color. This protein helps red blood cells carry oxygen from the lungs to the rest of the body.
Anemia has three main causes 1:
- Blood loss,
- Lack of red blood cell production, or
- High rates of red blood cell destruction.
These causes might be the result of diseases, conditions, or other factors.
If you have anemia, your body doesn’t get enough oxygen-rich blood. As a result, you may appear pale, feel tired or weak. You also may have other symptoms, such as shortness of breath, dizziness, or headaches.
Severe or long-lasting anemia can damage your heart, brain, and other organs in your body. Very severe anemia may even cause death 1.
Many types of anemia can be mild, short term, and easily treated. You can even prevent some types with a healthy diet. Other types can be treated with dietary supplements.
However, certain types of anemia can be severe, long lasting, and even life threatening if not diagnosed and treated.
If you have signs or symptoms of anemia, see your doctor to find out whether you have the condition. Treatment will depend on the cause of the anemia and how severe it is.
There are many types of anemia with specific causes and traits.
Some of these include:
- Aplastic anemia
- Blood loss anemia
- Cooley’s anemia
- Diamond-Blackfan anemia
- Fanconi anemia
- Folate- or folic acid-deficiency anemia
- Hemolytic anemia
- Iron-deficiency anemia
- Pernicious anemia
- Sickle cell anemia
- Thalassemias; Cooley’s anemia is another name for beta thalassemia major
What Causes Anemia ?
The three main causes of anemia are:
- Blood loss.
- Lack of red blood cell production.
- High rates of red blood cell destruction.
For some people, the condition is caused by more than one of these factors.
Blood Loss
Blood loss is the most common cause of anemia, especially iron-deficiency anemia. Blood loss can be short term or persist over time.
Heavy menstrual periods or bleeding in the digestive or urinary tract can cause blood loss. Surgery, trauma, or cancer also can cause blood loss.
If a lot of blood is lost, the body may lose enough red blood cells to cause anemia.
Lack of Red Blood Cell Production
Both acquired and inherited conditions and factors can prevent your body from making enough red blood cells. “Acquired” means you aren’t born with the condition, but you develop it. “Inherited” means your parents passed the gene for the condition on to you.
Acquired conditions and factors that can lead to anemia include poor diet, abnormal hormone levels, some chronic (ongoing) diseases, and pregnancy.
Aplastic anemia also can prevent your body from making enough red blood cells. This condition can be acquired or inherited.
Diet
A diet that lacks iron, folic acid (folate), or vitamin B12 can prevent your body from making enough red blood cells. Your body also needs small amounts of vitamin C, riboflavin, and copper to make red blood cells.
Conditions that make it hard for your body to absorb nutrients also can prevent your body from making enough red blood cells.
Hormones
Your body needs the hormone erythropoietin to make red blood cells. This hormone stimulates the bone marrow to make these cells. A low level of this hormone can lead to anemia.
Diseases and Disease Treatments
Chronic diseases, like kidney disease and cancer, can make it hard for your body to make enough red blood cells.
Some cancer treatments may damage the bone marrow or damage the red blood cells’ ability to carry oxygen. If the bone marrow is damaged, it can’t make red blood cells fast enough to replace the ones that die or are destroyed.
People who have HIV/AIDS may develop anemia due to infections or medicines used to treat their diseases.
Pregnancy
Anemia can occur during pregnancy due to low levels of iron and folic acid and changes in the blood.
During the first 6 months of pregnancy, the fluid portion of a woman’s blood (the plasma) increases faster than the number of red blood cells. This dilutes the blood and can lead to anemia.
Aplastic Anemia
Some infants are born without the ability to make enough red blood cells. This condition is called aplastic anemia. Infants and children who have aplastic anemia often need blood transfusions to increase the number of red blood cells in their blood.
Acquired conditions or factors, such as certain medicines, toxins, and infectious diseases, also can cause aplastic anemia.
High Rates of Red Blood Cell Destruction
Both acquired and inherited conditions and factors can cause your body to destroy too many red blood cells. One example of an acquired condition is an enlarged or diseased spleen.
The spleen is an organ that removes wornout red blood cells from the body. If the spleen is enlarged or diseased, it may remove more red blood cells than normal, causing anemia.
Examples of inherited conditions that can cause your body to destroy too many red blood cells include sickle cell anemia, thalassemias, and lack of certain enzymes. These conditions create defects in the red blood cells that cause them to die faster than healthy red blood cells.
Hemolytic anemia is another example of a condition in which your body destroys too many red blood cells. Inherited or acquired conditions or factors can cause hemolytic anemia. Examples include immune disorders, infections, certain medicines, or reactions to blood transfusions.
Who Is at Risk for Anemia ?
Anemia is a common condition. It occurs in all age, racial, and ethnic groups. Both men and women can have anemia. However, women of childbearing age are at higher risk for the condition because of blood loss from menstruation.
Anemia can develop during pregnancy due to low levels of iron and folic acid (folate) and changes in the blood. During the first 6 months of pregnancy, the fluid portion of a woman’s blood (the plasma) increases faster than the number of red blood cells. This dilutes the blood and can lead to anemia.
During the first year of life, some babies are at risk for anemia because of iron deficiency. At-risk infants include those who are born too early and infants who are fed breast milk only or formula that isn’t fortified with iron. These infants can develop iron deficiency by 6 months of age.
Infants between 1 and 2 years of age also are at risk for anemia. They may not get enough iron in their diets, especially if they drink a lot of cow’s milk. Cow’s milk is low in the iron needed for growth.
Drinking too much cow’s milk may keep an infant or toddler from eating enough iron-rich foods or absorbing enough iron from foods.
Older adults also are at increased risk for anemia. Researchers continue to study how the condition affects older adults. Many of these people have other medical conditions as well.
Major Risk Factors
Factors that raise your risk for anemia include:
- A diet that is low in iron, vitamins, or minerals
- Blood loss from surgery or an injury
- Long-term or serious illnesses, such as kidney disease, cancer, diabetes, rheumatoid arthritis, HIV/AIDS, inflammatory bowel disease (including Crohn’s disease), liver disease, heart failure, and thyroid disease
- Long-term infections
- A family history of inherited anemia, such as sickle cell anemia or thalassemia
What Are the Signs and Symptoms of Anemia ?
The most common symptom of anemia is fatigue (feeling tired or weak). If you have anemia, you may find it hard to find the energy to do normal activities.
Other signs and symptoms of anemia include:
- Shortness of breath
- Dizziness
- Headache
- Coldness in the hands and feet
- Pale skin
- Chest pain
These signs and symptoms can occur because your heart has to work harder to pump oxygen-rich blood through your body.
Mild to moderate anemia may cause very mild symptoms or none at all.
Complications of Anemia
Some people who have anemia may have arrhythmias. Arrhythmias are problems with the rate or rhythm of the heartbeat. Over time, arrhythmias can damage your heart and possibly lead to heart failure.
Anemia also can damage other organs in your body because your blood can’t get enough oxygen to them.
Anemia can weaken people who have cancer or HIV/AIDS. This can make their treatments not work as well.
Anemia also can cause many other health problems. People who have kidney disease and anemia are more likely to have heart problems. With some types of anemia, too little fluid intake or too much loss of fluid in the blood and body can occur. Severe loss of fluid can be life threatening.
How Is Anemia Diagnosed ?
Your doctor will diagnose anemia based on your medical and family histories, a physical exam, and results from tests and procedures.
Because anemia doesn’t always cause symptoms, your doctor may find out you have it while checking for another condition.
Medical and Family Histories
Your doctor may ask whether you have any of the common signs or symptoms of anemia. He or she also may ask whether you’ve had an illness or condition that could cause anemia.
Let your doctor know about any medicines you take, what you typically eat (your diet), and whether you have family members who have anemia or a history of it.
Physical Exam
Your doctor will do a physical exam to find out how severe your anemia is and to check for possible causes. He or she may:
- Listen to your heart for a rapid or irregular heartbeat
- Listen to your lungs for rapid or uneven breathing
- Feel your abdomen to check the size of your liver and spleen
Your doctor also may do a pelvic or rectal exam to check for common sources of blood loss.
Diagnostic Tests and Procedures
You may have various blood tests and other tests or procedures to find out what type of anemia you have and how severe it is.
Complete Blood Count
Often, the first test used to diagnose anemia is a complete blood count (CBC). The CBC measures many parts of your blood.
The test checks your hemoglobin and hematocrit levels. Hemoglobin is the iron-rich protein in red blood cells that carries oxygen to the body. Hematocrit is a measure of how much space red blood cells take up in your blood. A low level of hemoglobin or hematocrit is a sign of anemia.
The normal range of these levels might be lower in certain racial and ethnic populations. Your doctor can explain your test results to you.
The CBC also checks the number of red blood cells, white blood cells, and platelets in your blood. Abnormal results might be a sign of anemia, another blood disorder, an infection, or another condition.
Finally, the CBC looks at mean corpuscular volume (MCV). MCV is a measure of the average size of your red blood cells and a clue as to the cause of your anemia. In iron-deficiency anemia, for example, red blood cells usually are smaller than normal.
Other Tests and Procedures
If the CBC results show that you have anemia, you may need other tests, such as:
- Hemoglobin electrophoresis. This test looks at the different types of hemoglobin in your blood. The test can help diagnose the type of anemia you have.
- A reticulocyte count. This test measures the number of young red blood cells in your blood. The test shows whether your bone marrow is making red blood cells at the correct rate.
- Tests for the level of iron in your blood and body. These tests include serum iron and serum ferritin tests. Transferrin level and total iron-binding capacity tests also measure iron levels.
Because anemia has many causes, you also might be tested for conditions such as kidney failure, lead poisoning (in children), and vitamin deficiencies (lack of vitamins, such as B12 and folic acid).
If your doctor thinks that you have anemia due to internal bleeding, he or she may suggest several tests to look for the source of the bleeding. A test to check the stool for blood might be done in your doctor’s office or at home. Your doctor can give you a kit to help you get a sample at home. He or she will tell you to bring the sample back to the office or send it to a laboratory.
If blood is found in the stool, you may have other tests to find the source of the bleeding. One such test is endoscopy. For this test, a tube with a tiny camera is used to view the lining of the digestive tract.
Your doctor also may want to do bone marrow tests. These tests show whether your bone marrow is healthy and making enough blood cells.
How Is Anemia Treated ?
Treatment for anemia depends on the type, cause, and severity of the condition. Treatments may include dietary changes or supplements, medicines, procedures, or surgery to treat blood loss.
Goals of Treatment
The goal of treatment is to increase the amount of oxygen that your blood can carry. This is done by raising the red blood cell count and/or hemoglobin level. (Hemoglobin is the iron-rich protein in red blood cells that carries oxygen to the body.)
Another goal is to treat the underlying cause of the anemia.
Dietary Changes and Supplements
Low levels of vitamins or iron in the body can cause some types of anemia. These low levels might be the result of a poor diet or certain diseases or conditions.
To raise your vitamin or iron level, your doctor may ask you to change your diet or take vitamin or iron supplements. Common vitamin supplements are vitamin B12 and folic acid (folate). Vitamin C sometimes is given to help the body absorb iron.
Iron
Your body needs iron to make hemoglobin. Your body can more easily absorb iron from meats than from vegetables or other foods. To treat your anemia, your doctor may suggest eating more meat—especially red meat (such as beef or liver), as well as chicken, turkey, pork, fish, and shellfish.
Nonmeat foods that are good sources of iron include:
- Spinach and other dark green leafy vegetables
- Tofu
- Peas; lentils; white, red, and baked beans; soybeans; and chickpeas
- Dried fruits, such as prunes, raisins, and apricots
- Prune juice
- Iron-fortified cereals and breads
You can look at the Nutrition Facts label on packaged foods to find out how much iron the items contain. The amount is given as a percentage of the total amount of iron you need every day.
Iron also is available as a supplement. It’s usually combined with multivitamins and other minerals that help your body absorb iron.
Doctors may recommend iron supplements for premature infants, infants and young children who drink a lot of cow’s milk, and infants who are fed breast milk only or formula that isn’t fortified with iron.
Large amounts of iron can be harmful, so take iron supplements only as your doctor prescribes.
Vitamin B12
Low levels of vitamin B12 can lead to pernicious anemia. This type of anemia often is treated with vitamin B12 supplements.
Good food sources of vitamin B12 include:
- Breakfast cereals with added vitamin B12
- Meats such as beef, liver, poultry, and fish
- Eggs and dairy products (such as milk, yogurt, and cheese)
- Foods fortified with vitamin B12, such as soy-based beverages and vegetarian burgers
Folic Acid
Folic acid (folate) is a form of vitamin B that’s found in foods. Your body needs folic acid to make and maintain new cells. Folic acid also is very important for pregnant women. It helps them avoid anemia and promotes healthy growth of the fetus.
Good sources of folic acid include:
- Bread, pasta, and rice with added folic acid
- Spinach and other dark green leafy vegetables
- Black-eyed peas and dried beans
- Beef liver
- Eggs
- Bananas, oranges, orange juice, and some other fruits and juices
Vitamin C
Vitamin C helps the body absorb iron. Good sources of vitamin C are vegetables and fruits, especially citrus fruits. Citrus fruits include oranges, grapefruits, tangerines, and similar fruits. Fresh and frozen fruits, vegetables, and juices usually have more vitamin C than canned ones.
If you’re taking medicines, ask your doctor or pharmacist whether you can eat grapefruit or drink grapefruit juice. This fruit can affect the strength of a few medicines and how well they work.
Other fruits rich in vitamin C include kiwi fruit, strawberries, and cantaloupes.
Vegetables rich in vitamin C include broccoli, peppers, Brussels sprouts, tomatoes, cabbage, potatoes, and leafy green vegetables like turnip greens and spinach.
Medicines
Your doctor may prescribe medicines to help your body make more red blood cells or to treat an underlying cause of anemia. Some of these medicines include:
- Antibiotics to treat infections.
- Hormones to treat heavy menstrual bleeding in teenaged and adult women.
- A man-made version of erythropoietin to stimulate your body to make more red blood cells. This hormone has some risks. You and your doctor will decide whether the benefits of this treatment outweigh the risks.
- Medicines to prevent the body’s immune system from destroying its own red blood cells.
- Chelation therapy for lead poisoning. Chelation therapy is used mainly in children. This is because children who have iron-deficiency anemia are at increased risk of lead poisoning.
Procedures
If your anemia is severe, your doctor may recommend a medical procedure. Procedures include blood transfusions and blood and marrow stem cell transplants.
Blood Transfusion
A blood transfusion is a safe, common procedure in which blood is given to you through an intravenous (IV) line in one of your blood vessels. Transfusions require careful matching of donated blood with the recipient’s blood.
For more information, see Blood Transfusion topic below.
Blood and Marrow Stem Cell Transplant
A blood and marrow stem cell transplant replaces your faulty stem cells with healthy ones from another person (a donor). Stem cells are made in the bone marrow. They develop into red and white blood cells and platelets.
During the transplant, which is like a blood transfusion, you get donated stem cells through a tube placed in a vein in your chest. Once the stem cells are in your body, they travel to your bone marrow and begin making new blood cells.
Surgery
If you have serious or life-threatening bleeding that’s causing anemia, you may need surgery. For example, you may need surgery to control ongoing bleeding due to a stomach ulcer or colon cancer.
If your body is destroying red blood cells at a high rate, you may need to have your spleen removed. The spleen is an organ that removes wornout red blood cells from the body. An enlarged or diseased spleen may remove more red blood cells than normal, causing anemia.
What Is Iron-Deficiency Anemia ?
Iron-deficiency anemia is a common, easily treated condition that occurs if you don’t have enough iron in your body. Low iron levels usually are due to blood loss, poor diet, or an inability to absorb enough iron from food 2.
Iron-deficiency anemia usually develops over time if your body doesn’t have enough iron to build healthy red blood cells. Without enough iron, your body starts using the iron it has stored. Soon, the stored iron gets used up.
After the stored iron is gone, your body makes fewer red blood cells. The red blood cells it does make have less hemoglobin than normal.
Iron-deficiency anemia can cause fatigue (tiredness), shortness of breath, chest pain, and other symptoms. Severe iron-deficiency anemia can lead to heart problems, infections, problems with growth and development in children, and other complications.
Infants and young children and women are the two groups at highest risk for iron-deficiency anemia.
Doctors usually can successfully treat iron-deficiency anemia. Treatment will depend on the cause and severity of the condition. Treatments may include dietary changes, medicines, and surgery.
Severe iron-deficiency anemia may require treatment in a hospital, blood transfusions, iron injections, or intravenous iron therapy.
What Causes Iron-Deficiency Anemia ?
Not having enough iron in your body causes iron-deficiency anemia. Lack of iron usually is due to blood loss, poor diet, or an inability to absorb enough iron from food.
Blood Loss
When you lose blood, you lose iron. If you don’t have enough iron stored in your body to make up for the lost iron, you’ll develop iron-deficiency anemia.
In women, long or heavy menstrual periods or bleeding fibroids in the uterus may cause low iron levels. Blood loss that occurs during childbirth is another cause of low iron levels in women.
Internal bleeding (bleeding inside the body) also may lead to iron-deficiency anemia. This type of blood loss isn’t always obvious, and it may occur slowly. Some causes of internal bleeding are:
- A bleeding ulcer, colon polyp, or colon cancer
- Regular use of aspirin or other pain medicines, such as nonsteroidal anti-inflammatory drugs (for example, ibuprofen and naproxen)
- Urinary tract bleeding
Blood loss from severe injuries, surgery, or frequent blood drawings also can cause iron-deficiency anemia.
Poor Diet
The best sources of iron are meat, poultry, fish, and iron-fortified foods (foods that have iron added). If you don’t eat these foods regularly, or if you don’t take an iron supplement, you’re more likely to develop iron-deficiency anemia.
Vegetarian diets can provide enough iron if you eat the right foods. For example, good nonmeat sources of iron include iron-fortified breads and cereals, beans, tofu, dried fruits, and spinach and other dark green leafy vegetables.
During some stages of life, such as pregnancy and childhood, it may be hard to get enough iron in your diet. This is because your need for iron increases during these times of growth and development.
Inability To Absorb Enough Iron
Even if you have enough iron in your diet, your body may not be able to absorb it. This can happen if you have intestinal surgery (such as gastric bypass) or a disease of the intestine (such as Crohn’s disease or celiac disease).
Prescription medicines that reduce acid in the stomach also can interfere with iron absorption.
Who Is at Risk for Iron-Deficiency Anemia ?
- Infants and Young Children
Infants and young children need a lot of iron to grow and develop. The iron that full-term infants have stored in their bodies is used up in the first 4 to 6 months of life.
Premature and low-birth-weight babies (weighing less than 5.5 pounds) are at even greater risk for iron-deficiency anemia. These babies don’t have as much iron stored in their bodies as larger, full-term infants.
Iron-fortified baby food or iron supplements, when used properly, can help prevent iron-deficiency anemia in infants and young children. Talk with your child’s doctor about your child’s diet.
Young children who drink a lot of cow’s milk may be at risk for iron-deficiency anemia. Milk is low in iron, and too much milk may take the place of iron-rich foods in the diet. Too much milk also may prevent children’s bodies from absorbing iron from other foods.
Children who have lead in their blood also may be at risk for iron-deficiency anemia. Lead can interfere with the body’s ability to make hemoglobin. Lead may get into the body from breathing in lead dust, eating lead in paint or soil, or drinking water that contains lead.
- Teens
Teens are at risk for iron-deficiency anemia if they’re underweight or have chronic (ongoing) illnesses. Teenage girls who have heavy periods also are at increased risk for the condition.
- Women
Women of childbearing age are at higher risk for iron-deficiency anemia because of blood loss during their monthly periods. About 1 in 5 women of childbearing age has iron-deficiency anemia.
Pregnant women also are at higher risk for the condition because they need twice as much iron as usual. The extra iron is needed for increased blood volume and for the fetus’ growth.
About half of all pregnant women develop iron-deficiency anemia. The condition can increase a pregnant woman’s risk for a premature or low-birth-weight baby.
- Adults Who Have Internal Bleeding
Adults who have internal bleeding, such as intestinal bleeding, can develop iron-deficiency anemia due to blood loss. Certain conditions, such as colon cancer and bleeding ulcers, can cause blood loss. Some medicines, such as aspirin, also can cause internal bleeding.
- Other At-Risk Groups
People who get kidney dialysis treatment may develop iron-deficiency anemia. This is because blood is lost during dialysis. Also, the kidneys are no longer able to make enough of a hormone that the body needs to produce red blood cells.
People who have gastric bypass surgery also may develop iron-deficiency anemia. This type of surgery can prevent the body from absorbing enough iron.
Certain eating patterns or habits may put you at higher risk for iron-deficiency anemia. This can happen if you:
- Follow a diet that excludes meat and fish, which are the best sources of iron. However, vegetarian diets can provide enough iron if you eat the right foods. For example, good nonmeat sources of iron include iron-fortified breads and cereals, beans, tofu, dried fruits, and spinach and other dark green leafy vegetables.
- Eat poorly because of money, social, health, or other problems.
- Follow a very low-fat diet over a long time. Some higher fat foods, like meat, are the best sources of iron.
- Follow a high-fiber diet. Large amounts of fiber can slow the absorption of iron.
What Are the Signs and Symptoms of Iron-Deficiency Anemia ?
The signs and symptoms of iron-deficiency anemia depend on its severity. Mild to moderate iron-deficiency anemia may have no signs or symptoms.
When signs and symptoms do occur, they can range from mild to severe. Many of the signs and symptoms of iron-deficiency anemia apply to all types of anemia.
- Signs and Symptoms of Anemia
The most common symptom of all types of anemia is fatigue (tiredness). Fatigue occurs because your body doesn’t have enough red blood cells to carry oxygen to its many parts.
Also, the red blood cells your body makes have less hemoglobin than normal. Hemoglobin is an iron-rich protein in red blood cells. It helps red blood cells carry oxygen from the lungs to the rest of the body.
Anemia also can cause shortness of breath, dizziness, headache, coldness in your hands and feet, pale skin, chest pain, weakness, and fatigue (tiredness).
If you don’t have enough hemoglobin-carrying red blood cells, your heart has to work harder to move oxygen-rich blood through your body. This can lead to irregular heartbeats called arrhythmias, a heart murmur, an enlarged heart, or even heart failure.
In infants and young children, signs of anemia include poor appetite, slowed growth and development, and behavioral problems.
- Signs and Symptoms of Iron Deficiency
Signs and symptoms of iron deficiency may include brittle nails, swelling or soreness of the tongue, cracks in the sides of the mouth, an enlarged spleen, and frequent infections.
People who have iron-deficiency anemia may have an unusual craving for nonfood items, such as ice, dirt, paint, or starch. This craving is called pica.
Some people who have iron-deficiency anemia develop restless legs syndrome. Restless legs syndrome is a disorder that causes a strong urge to move the legs. This urge to move often occurs with strange and unpleasant feelings in the legs. People who have restless legs syndrome often have a hard time sleeping.
Iron-deficiency anemia can put children at greater risk for lead poisoning and infections.
Some signs and symptoms of iron-deficiency anemia are related to the condition’s causes. For example, a sign of intestinal bleeding is bright red blood in the stools or black, tarry-looking stools.
Very heavy menstrual bleeding, long periods, or other vaginal bleeding may suggest that a woman is at risk for iron-deficiency anemia.
How Is Iron-Deficiency Anemia Diagnosed ?
Your doctor will diagnose iron-deficiency anemia based on your medical history, a physical exam, and the results from tests and procedures.
Once your doctor knows the cause and severity of the condition, he or she can create a treatment plan for you.
Mild to moderate iron-deficiency anemia may have no signs or symptoms. Thus, you may not know you have it unless your doctor discovers it from a screening test or while checking for other problems.
- Specialists Involved
Primary care doctors often diagnose and treat iron-deficiency anemia. These doctors include pediatricians, family doctors, gynecologists/obstetricians, and internal medicine specialists.
A hematologist (a blood disease specialist), a gastroenterologist (a digestive system specialist), and other specialists also may help treat iron-deficiency anemia.
- Medical History
Your doctor will ask about your signs and symptoms and any past problems you’ve had with anemia or low iron. He or she also may ask about your diet and whether you’re taking any medicines.
If you’re a woman, your doctor may ask whether you might be pregnant.
Physical Exam
Your doctor will do a physical exam to look for signs of iron-deficiency anemia. He or she may:
- Look at your skin, gums, and nail beds to see whether they’re pale
- Listen to your heart for rapid or irregular heartbeats
- Listen to your lungs for rapid or uneven breathing
- Feel your abdomen to check the size of your liver and spleen
- Do a pelvic and rectal exam to check for internal bleeding
Diagnostic Tests and Procedures
Many tests and procedures are used to diagnose iron-deficiency anemia. They can help confirm a diagnosis, look for a cause, and find out how severe the condition is.
Complete Blood Count
Often, the first test used to diagnose anemia is a complete blood count (CBC). The CBC measures many parts of your blood.
This test checks your hemoglobin and hematocrit levels. Hemoglobin is an iron-rich protein in red blood cells that carries oxygen to the body. Hematocrit is a measure of how much space red blood cells take up in your blood. A low level of hemoglobin or hematocrit is a sign of anemia.
The normal range of these levels varies in certain racial and ethnic populations. Your doctor can explain your test results to you.
The CBC also checks the number of red blood cells, white blood cells, and platelets in your blood. Abnormal results may be a sign of infection, a blood disorder, or another condition.
Finally, the CBC looks at mean corpuscular volume (MCV). MCV is a measure of the average size of your red blood cells. The results may be a clue as to the cause of your anemia. In iron-deficiency anemia, for example, red blood cells usually are smaller than normal.
Other Blood Tests
If the CBC results confirm you have anemia, you may need other blood tests to find out what’s causing the condition, how severe it is, and the best way to treat it.
Reticulocyte count. This test measures the number of reticulocytes in your blood. Reticulocytes are young, immature red blood cells. Over time, reticulocytes become mature red blood cells that carry oxygen throughout your body.
A reticulocyte count shows whether your bone marrow is making red blood cells at the correct rate.
Peripheral smear. For this test, a sample of your blood is examined under a microscope. If you have iron-deficiency anemia, your red blood cells will look smaller and paler than normal.
Tests to measure iron levels. These tests can show how much iron has been used from your body’s stored iron. Tests to measure iron levels include:
- Serum iron. This test measures the amount of iron in your blood. The level of iron in your blood may be normal even if the total amount of iron in your body is low. For this reason, other iron tests also are done.
- Serum ferritin. Ferritin is a protein that helps store iron in your body. A measure of this protein helps your doctor find out how much of your body’s stored iron has been used.
- Transferrin level, or total iron-binding capacity. Transferrin is a protein that carries iron in your blood. Total iron-binding capacity measures how much of the transferrin in your blood isn’t carrying iron. If you have iron-deficiency anemia, you’ll have a high level of transferrin that has no iron.
Other tests. Your doctor also may recommend tests to check your hormone levels, especially your thyroid hormone. You also may have a blood test for a chemical called erythrocyte protoporphyrin. This chemical is a building block for hemoglobin.
Children also may be tested for the level of lead in their blood. Lead can make it hard for the body to produce hemoglobin.
Tests and Procedures for Gastrointestinal Blood Loss
To check whether internal bleeding is causing your iron-deficiency anemia, your doctor may suggest a fecal occult blood test. This test looks for blood in the stools and can detect bleeding in the intestines.
If the test finds blood, you may have other tests and procedures to find the exact spot of the bleeding. These tests and procedures may look for bleeding in the stomach, upper intestines, colon, or pelvic organs.
How Is Iron-Deficiency Anemia Treated ?
Treatment for iron-deficiency anemia will depend on its cause and severity. Treatments may include dietary changes and supplements, medicines, and surgery.
Severe iron-deficiency anemia may require a blood transfusion, iron injections, or intravenous (IV) iron therapy. Treatment may need to be done in a hospital.
The goals of treating iron-deficiency anemia are to treat its underlying cause and restore normal levels of red blood cells, hemoglobin, and iron.
Dietary Changes and Supplements
- Iron
You may need iron supplements to build up your iron levels as quickly as possible. Iron supplements can correct low iron levels within months. Supplements come in pill form or in drops for children.
Large amounts of iron can be harmful, so take iron supplements only as your doctor prescribes. Keep iron supplements out of reach from children. This will prevent them from taking an overdose of iron.
Iron supplements can cause side effects, such as dark stools, stomach irritation, and heartburn. Iron also can cause constipation, so your doctor may suggest that you use a stool softener.
Your doctor may advise you to eat more foods that are rich in iron. The best source of iron is red meat, especially beef and liver. Chicken, turkey, pork, fish, and shellfish also are good sources of iron.
The body tends to absorb iron from meat better than iron from nonmeat foods. However, some nonmeat foods also can help you raise your iron levels. Examples of nonmeat foods that are good sources of iron include:
- Iron-fortified breads and cereals
- Peas; lentils; white, red, and baked beans; soybeans; and chickpeas
- Tofu
- Dried fruits, such as prunes, raisins, and apricots
- Spinach and other dark green leafy vegetables
- Prune juice
The Nutrition Facts labels on packaged foods will show how much iron the items contain. The amount is given as a percentage of the total amount of iron you need every day.
- Vitamin C
Vitamin C helps the body absorb iron. Good sources of vitamin C are vegetables and fruits, especially citrus fruits. Citrus fruits include oranges, grapefruits, tangerines, and similar fruits. Fresh and frozen fruits, vegetables, and juices usually have more vitamin C than canned ones.
If you’re taking medicines, ask your doctor or pharmacist whether you can eat grapefruit or drink grapefruit juice. Grapefruit can affect the strength of a few medicines and how well they work.
Other fruits rich in vitamin C include kiwi fruit, strawberries, and cantaloupes.
Vegetables rich in vitamin C include broccoli, peppers, Brussels sprouts, tomatoes, cabbage, potatoes, and leafy green vegetables like turnip greens and spinach.
Treatment To Stop Bleeding
If blood loss is causing iron-deficiency anemia, treatment will depend on the cause of the bleeding. For example, if you have a bleeding ulcer, your doctor may prescribe antibiotics and other medicines to treat the ulcer.
If a polyp or cancerous tumor in your intestine is causing bleeding, you may need surgery to remove the growth.
If you have heavy menstrual flow, your doctor may prescribe birth control pills to help reduce your monthly blood flow. In some cases, surgery may be advised.
Treatments for Severe Iron-Deficiency Anemia
- Blood Transfusion
If your iron-deficiency anemia is severe, you may get a transfusion of red blood cells. A blood transfusion is a safe, common procedure in which blood is given to you through an IV line in one of your blood vessels. A transfusion requires careful matching of donated blood with the recipient’s blood.
A transfusion of red blood cells will treat your anemia right away. The red blood cells also give a source of iron that your body can reuse. However, a blood transfusion is only a short-term treatment. Your doctor will need to find and treat the cause of your anemia.
Blood transfusions are usually reserved for people whose anemia puts them at a higher risk for heart problems or other severe health issues.
- Iron Therapy
If you have severe anemia, your doctor may recommend iron therapy. For this treatment, iron is injected into a muscle or an IV line in one of your blood vessels.
IV iron therapy presents some safety concerns. It must be done in a hospital or clinic by experienced staff. Iron therapy usually is given to people who need iron long-term but can’t take iron supplements by mouth. This therapy also is given to people who need immediate treatment for iron-deficiency anemia.
What Is Pernicious Anemia ?
Pernicious anemia is a condition in which the body can’t make enough healthy red blood cells because it doesn’t have enough vitamin B12 3. Without enough vitamin B12, your red blood cells don’t divide normally and are too large. They may have trouble getting out of the bone marrow—a sponge-like tissue inside the bones where blood cells are made.
Pernicious anemia is one of two major types of “macrocystic” or “megaloblastic” (large red blood cell) anemia. These terms refer to anemia in which the red blood cells are larger than normal. The other major type of macrocystic anemia is caused by folic acid deficiency.
Rarely, children are born with an inherited disorder that prevents their bodies from making intrinsic factor. This disorder is called congenital pernicious anemia.
Vitamin B12 deficiency also is called cobalamin deficiency and combined systems disease.
The term “pernicious” means “deadly.” The condition is called pernicious anemia because it often was fatal in the past, before vitamin B12 treatments were available. Now, pernicious anemia usually is easy to treat with vitamin B12 pills or shots.
Vitamin B12 is a nutrient found in some foods. The body needs this nutrient to make healthy red blood cells and to keep its nervous system working properly.
People who have pernicious anemia can’t absorb enough vitamin B12 from food. This is because they lack intrinsic factor, a protein made in the stomach 3. A lack of this protein leads to vitamin B12 deficiency.
Other conditions and factors also can cause vitamin B12 deficiency. Examples include infections, surgery, medicines, and diet. Technically, the term “pernicious anemia” refers to vitamin B12 deficiency due to a lack of intrinsic factor 3. Often though, vitamin B12 deficiency due to other causes also is called pernicious anemia.
Without enough red blood cells to carry oxygen to your body, you may feel tired and weak. Severe or long-lasting pernicious anemia can damage the heart, brain, and other organs in the body 3.
Pernicious anemia also can cause other problems, such as nerve damage, neurological problems (such as memory loss), and digestive tract problems. People who have pernicious anemia also may be at higher risk for weakened bone strength and stomach cancer 3.
With ongoing care and proper treatment, most people who have pernicious anemia can recover, feel well, and live normal lives.
Without treatment, pernicious anemia can lead to serious problems with the heart, nerves, and other parts of the body. Some of these problems may be permanent.
What Causes Pernicious Anemia ?
Pernicious anemia is caused by a lack of intrinsic factor or other causes, such as infections, surgery, medicines, or diet.
Lack of Intrinsic Factor
Intrinsic factor is a protein made in the stomach. It helps your body absorb vitamin B12. In some people, an autoimmune response causes a lack of intrinsic factor.
An autoimmune response occurs if the body’s immune system makes antibodies (proteins) that mistakenly attack and damage the body’s tissues or cells.
In pernicious anemia, the body makes antibodies that attack and destroy the parietal cells. These cells line the stomach and make intrinsic factor. Why this autoimmune response occurs isn’t known.
As a result of this attack, the stomach stops making intrinsic factor. Without intrinsic factor, your body can’t move vitamin B12 through the small intestine, where it’s absorbed. This leads to vitamin B12 deficiency.
A lack of intrinsic factor also can occur if you’ve had part or all of your stomach surgically removed. This type of surgery reduces the number of parietal cells available to make intrinsic factor.
Rarely, children are born with an inherited disorder that prevents their bodies from making intrinsic factor. This disorder is called congenital pernicious anemia.
Other Causes
Pernicious anemia also has other causes, besides a lack of intrinsic factor. Malabsorption in the small intestine and a diet lacking vitamin B12 both can lead to pernicious anemia.
Malabsorption in the Small Intestine
Sometimes pernicious anemia occurs because the body’s small intestine can’t properly absorb vitamin B12. This may be the result of:
- Too many of the wrong kind of bacteria in the small intestine. This is a common cause of pernicious anemia in older adults. The bacteria use up the available vitamin B12 before the small intestine can absorb it.
- Diseases that interfere with vitamin B12 absorption. One example is celiac disease. This is a genetic disorder in which your body can’t tolerate a protein called gluten. Another example is Crohn’s disease, an inflammatory bowel disease. HIV also may interfere with vitamin B12 absorption.
- Certain medicines that alter bacterial growth or prevent the small intestine from properly absorbing vitamin B12. Examples include antibiotics and certain diabetes and seizure medicines.
- Surgical removal of part or all of the small intestine.
- A tapeworm infection. The tapeworm feeds off of the vitamin B12. Eating undercooked, infected fish may cause this type of infection.
Diet Lacking Vitamin B12
Some people get pernicious anemia because they don’t have enough vitamin B12 in their diets. This cause of pernicious anemia is less common than other causes.
Good food sources of vitamin B12 include:
- Breakfast cereals with added vitamin B12
- Meats such as beef, liver, poultry, and fish
- Eggs and dairy products (such as milk, yogurt, and cheese)
- Foods fortified with vitamin B12, such as soy-based beverages and vegetarian burgers
Strict vegetarians who don’t eat any animal or dairy products and don’t take a vitamin B12 supplement are at risk for pernicious anemia.
Breastfed infants of strict vegetarian mothers also are at risk for pernicious anemia. These infants can develop anemia within months of being born. This is because they haven’t had enough time to store vitamin B12 in their bodies. Doctors treat these infants with vitamin B12 supplements.
Other groups, such as the elderly and people who suffer from alcoholism, also may be at risk for pernicious anemia. These people may not get the proper nutrients in their diets.
Who Is at Risk for Pernicious Anemia ?
Pernicious anemia is more common in people of Northern European and African descent than in other ethnic groups.
Older people also are at higher risk for the condition. This is mainly due to a lack of stomach acid and intrinsic factor, which prevents the small intestine from absorbing vitamin B12. As people grow older, they tend to make less stomach acid.
Pernicious anemia also can occur in younger people and other populations. You’re at higher risk for pernicious anemia if you:
- Have a family history of the condition.
- Have had part or all of your stomach surgically removed. The stomach makes intrinsic factor. This protein helps your body absorb vitamin B12.
- Have an autoimmune disorder that involves the endocrine glands, such as Addison’s disease, type 1 diabetes, Graves’ disease, or vitiligo. Research suggests a link may exist between these autoimmune disorders and pernicious anemia that’s caused by an autoimmune response.
- Have had part or all of your small intestine surgically removed. The small intestine is where vitamin B12 is absorbed.
- Have certain intestinal diseases or other disorders that may prevent your body from properly absorbing vitamin B12. Examples include Crohn’s disease, intestinal infections, and HIV.
- Take medicines that prevent your body from properly absorbing vitamin B12. Examples of such medicines include antibiotics and certain seizure medicines.
- Are a strict vegetarian who doesn’t eat any animal or dairy products and doesn’t take a vitamin B12 supplement, or if you eat poorly overall.
What Are the Signs and Symptoms of Pernicious Anemia ?
A lack of vitamin B12 (vitamin B12 deficiency) causes the signs and symptoms of pernicious anemia. Without enough vitamin B12, your body can’t make enough healthy red blood cells, which causes anemia.
Some of the signs and symptoms of pernicious anemia apply to all types of anemia. Other signs and symptoms are specific to a lack of vitamin B12.
- Signs and Symptoms of Anemia
The most common symptom of all types of anemia is fatigue (tiredness). Fatigue occurs because your body doesn’t have enough red blood cells to carry oxygen to its various parts.
A low red blood cell count also can cause shortness of breath, dizziness, headache, coldness in your hands and feet, pale or yellowish skin, and chest pain.
A lack of red blood cells also means that your heart has to work harder to move oxygen-rich blood through your body. This can lead to irregular heartbeats called arrhythmias, heart murmur, an enlarged heart, or even heart failure.
- Signs and Symptoms of Vitamin B12 Deficiency
Vitamin B12 deficiency may lead to nerve damage. This can cause tingling and numbness in your hands and feet, muscle weakness, and loss of reflexes. You also may feel unsteady, lose your balance, and have trouble walking. Vitamin B12 deficiency can cause weakened bones and may lead to hip fractures.
Severe vitamin B12 deficiency can cause neurological problems, such as confusion, dementia, depression, and memory loss.
Other symptoms of vitamin B12 deficiency involve the digestive tract. These symptoms include nausea (feeling sick to your stomach) and vomiting, heartburn, abdominal bloating and gas, constipation or diarrhea, loss of appetite, and weight loss. An enlarged liver is another symptom.
A smooth, thick, red tongue also is a sign of vitamin B12 deficiency and pernicious anemia.
Infants who have vitamin B12 deficiency may have poor reflexes or unusual movements, such as face tremors. They may have trouble feeding due to tongue and throat problems. They also may be irritable. If vitamin B12 deficiency isn’t treated, these infants may have permanent growth problems.
How Is Pernicious Anemia Diagnosed ?
Your doctor will diagnose pernicious anemia based on your medical and family histories, a physical exam, and test results.
Your doctor will want to find out whether the condition is due to a lack of intrinsic factor or another cause. He or she also will want to find out the severity of the condition, so it can be properly treated.
Specialists Involved
Primary care doctors—such as family doctors, internists, and pediatricians (doctors who treat children)—often diagnose and treat pernicious anemia. Other kinds of doctors also may be involved, including:
- A neurologist (nervous system specialist)
- A cardiologist (heart specialist)
- A hematologist (blood disease specialist)
- A gastroenterologist (digestive tract specialist)
Medical and Family Histories
Your doctor may ask about your signs and symptoms. He or she also may ask:
- Whether you’ve had any stomach or intestinal surgeries
- Whether you have any digestive disorders, such as celiac disease or Crohn’s disease
- About your diet and any medicines you take
- Whether you have a family history of anemia or pernicious anemia
- Whether you have a family history of autoimmune disorders (such as Addison’s disease, type 1 diabetes, Graves’ disease, or vitiligo). Research suggests a link may exist between these autoimmune disorders and pernicious anemia that’s caused by an autoimmune response.
Physical Exam
During the physical exam, your doctor may check for pale or yellowish skin and an enlarged liver. He or she may listen to your heart for rapid or irregular heartbeats or a heart murmur.
Your doctor also may check for signs of nerve damage. He or she may want to see how well your muscles, eyes, senses, and reflexes work. Your doctor may ask questions or do tests to check your mental status, coordination, and ability to walk.
Diagnostic Tests and Procedures
Blood tests and procedures can help diagnose pernicious anemia and find out what’s causing it.
- Complete Blood Count
Often, the first test used to diagnose many types of anemia is a complete blood count (CBC). This test measures many parts of your blood. For this test, a small amount of blood is drawn from a vein (usually in your arm) using a needle.
A CBC checks your hemoglobin and hematocrit levels. Hemoglobin is an iron-rich protein that helps red blood cells carry oxygen from the lungs to the rest of the body. Hematocrit is a measure of how much space red blood cells take up in your blood. A low level of hemoglobin or hematocrit is a sign of anemia.
The normal range of these levels may be lower in certain racial and ethnic populations. Your doctor can explain your test results to you.
The CBC also checks the number of red blood cells, white blood cells, and platelets in your blood. Abnormal results may be a sign of anemia, another blood disorder, an infection, or another condition.
Finally, the CBC looks at mean corpuscular volume (MCV). MCV is a measure of the average size of your red blood cells. MCV can be a clue as to what’s causing your anemia. In pernicious anemia, the red blood cells tend to be larger than normal.
- Other Blood Tests
If the CBC results confirm that you have anemia, you may need other blood tests to find out what type of anemia you have.
A reticulocyte count measures the number of young red blood cells in your blood. The test shows whether your bone marrow is making red blood cells at the correct rate. People who have pernicious anemia have low reticulocyte counts.
Serum folate, iron, and iron-binding capacity tests also can help show whether you have pernicious anemia or another type of anemia.
Another common test, called the Combined Binding Luminescence Test, sometimes gives false results. Scientists are working to develop a more reliable test.
Your doctor may recommend other blood tests to check:
- Your vitamin B12 level. A low level of vitamin B12 in the blood indicates pernicious anemia. However, a falsely normal or high value of vitamin B12 in the blood may occur if antibodies interfere with the test.
- Your homocysteine and methylmalonic acid levels. High levels of these substances in your body are a sign of pernicious anemia.
- For intrinsic factor antibodies and parietal cell antibodies. These antibodies also are a sign of pernicious anemia.
Bone Marrow Tests
Bone marrow tests can show whether your bone marrow is healthy and making enough red blood cells. The two bone marrow tests are aspiration and biopsy.
For aspiration, your doctor removes a small amount of fluid bone marrow through a needle. For a biopsy, your doctor removes a small amount of bone marrow tissue through a larger needle. The samples are then examined under a microscope.
In pernicious anemia, the bone marrow cells that turn into blood cells are larger than normal.
How Is Pernicious Anemia Treated ?
Doctors treat pernicious anemia by replacing the missing vitamin B12 in the body. People who have pernicious anemia may need lifelong treatment.
The goals of treating pernicious anemia include:
- Preventing or treating the anemia and its signs and symptoms
- Preventing or managing complications, such as heart and nerve damage
- Treating the cause of the pernicious anemia (if a cause can be found)
Specific Types of Treatment
Pernicious anemia usually is easy to treat with vitamin B12 shots or pills.
If you have severe pernicious anemia, your doctor may recommend shots first. Shots usually are given in a muscle every day or every week until the level of vitamin B12 in your blood increases. After your vitamin B12 blood level returns to normal, you may get a shot only once a month.
For less severe pernicious anemia, your doctor may recommend large doses of vitamin B12 pills. A vitamin B12 nose gel and spray also are available. These products may be useful for people who have trouble swallowing pills, such as older people who have had strokes.
Your signs and symptoms may begin to improve within a few days after you start treatment. Your doctor may advise you to limit your physical activity until your condition improves.
If your pernicious anemia is caused by something other than a lack of intrinsic factor, you may get treatment for the cause (if a cause can be found). For example, your doctor may prescribe medicines to treat a condition that prevents your body from absorbing vitamin B12.
If medicines are the cause of your pernicious anemia, your doctor may change the type or dose of medicine you take. Infants of strict vegetarian mothers may be given vitamin B12 supplements from birth.
Sickle Cell Disease
The term sickle cell disease describes a group of inherited red blood cell disorders. People with sickle cell disease have abnormal hemoglobin, called hemoglobin S or sickle hemoglobin, in their red blood cells 4.
Hemoglobin is a protein in red blood cells that carries oxygen throughout the body.
“Inherited” means that the disease is passed by genes from parents to their children. Sickle Cell Disease is not contagious. A person cannot catch it, like a cold or infection, from someone else.
People who have sickle cell disease inherit two abnormal hemoglobin genes, one from each parent. In all forms of sickle cell disease, at least one of the two abnormal genes causes a person’s body to make hemoglobin S. When a person has two hemoglobin S genes, Hemoglobin SS, the disease is called sickle cell anemia. This is the most common and often most severe kind of sickle cell disease.
In the United States, most people with sickle cell disease are of African ancestry or identify themselves as black 5.
- About 1 in 13 African American babies is born with sickle cell trait.
- About 1 in every 365 black children is born with sickle cell disease.
There are also many people with this disease who come from Hispanic, southern European, Middle Eastern, or Asian Indian backgrounds.
Approximately 100,000 Americans have sickle cell disease.
Hemoglobin SC disease and hemoglobin Sβ thalassemia are two other common forms of sickle cell disease.
Some Forms of Sickle Cell Disease
- Hemoglobin SS
- Hemoglobin SC
- Hemoglobin Sβ0 thalassemia
- Hemoglobin Sβ+ thalassemia
- Hemoglobin SD
- Hemoglobin SE
Cells in tissues need a steady supply of oxygen to work well. Normally, hemoglobin in red blood cells takes up oxygen in the lungs and carries it to all the tissues of the body.
Red blood cells that contain normal hemoglobin are disc shaped (like a doughnut without a hole). This shape allows the cells to be flexible so that they can move through large and small blood vessels to deliver oxygen.
Sickle hemoglobin is not like normal hemoglobin. It can form stiff rods within the red cell, changing it into a crescent, or sickle shape.
Sickle-shaped cells are not flexible and can stick to vessel walls, causing a blockage that slows or stops the flow of blood. When this happens, oxygen can’t reach nearby tissues.
Figure 8. Sickle cell anemia
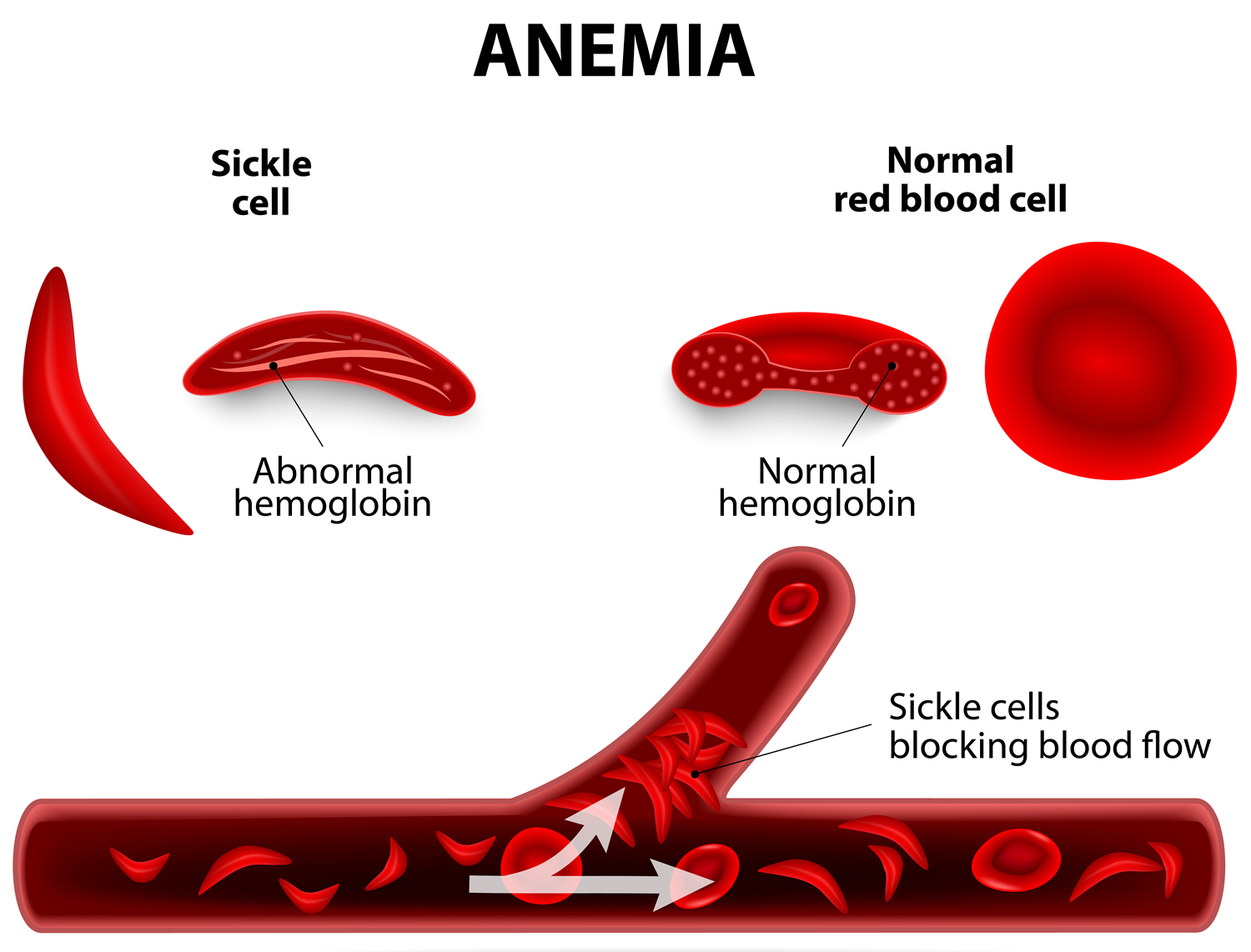
The lack of tissue oxygen can cause attacks of sudden, severe pain, called pain crises. These pain attacks can occur without warning, and a person often needs to go to the hospital for effective treatment.
Most children with sickle cell disease are pain free between painful crises, but adolescents and adults may also suffer with chronic ongoing pain.
The red cell sickling and poor oxygen delivery can also cause organ damage. Over a lifetime, sickle cell disease can harm a person’s spleen, brain, eyes, lungs, liver, heart, kidneys, penis, joints, bones, or skin.
Sickle cells can’t change shape easily, so they tend to burst apart or hemolyze. Normal red blood cells live about 90 to 120 days, but sickle cells last only 10 to 20 days.
The body is always making new red blood cells to replace the old cells; however, in sickle cell disease the body may have trouble keeping up with how fast the cells are being destroyed. Because of this, the number of red blood cells is usually lower than normal. This condition, called anemia, can make a person have less energy.
Sickle cell disease is a life-long illness. The severity of the disease varies widely from person to person.
In high-income countries like the United States, the life expectancy of a person with sickle cell disease is now about 40–60 years. In 1973, the average lifespan of a person with sickle cell disease in the United States was only 14 years. Advances in the diagnosis and care of sickle cell disease have made this improvement possible.
At the present time, hematopoietic stem cell transplantation is the only cure for sickle cell disease 4. Unfortunately, most people with sickle cell disease are either too old for a transplant or don’t have a relative who is a good enough genetic match for them to act as a donor. A well-matched donor is needed to have the best chance for a successful transplant.
There are effective treatments that can reduce symptoms and prolong life. Early diagnosis and regular medical care to prevent complications also contribute to improved well-being.
What Causes Sickle Cell Disease ?
Abnormal hemoglobin, called hemoglobin S, causes sickle cell disease.
The problem in hemoglobin S is caused by a small defect in the gene that directs the production of the beta globin part of hemoglobin. This small defect in the beta globin gene causes a problem in the beta globin part of hemoglobin, changing the way that hemoglobin works.
How Is Sickle Cell Disease Inherited ?
When the hemoglobin S gene is inherited from only one parent and a normal hemoglobin gene is inherited from the other, a person will have sickle cell trait. People with sickle cell trait are generally healthy.
Only rarely do people with sickle cell trait have complications similar to those seen in people with sickle cell disease. But people with sickle cell trait are carriers of a defective hemoglobin S gene. So, they can pass it on when they have a child.
If the child’s other parent also has sickle cell trait or another abnormal hemoglobin gene (like thalassemia, hemoglobin C, hemoglobin D, hemoglobin E), that child has a chance of having sickle cell disease.
Figure 9. Sickle cell disease inheritance
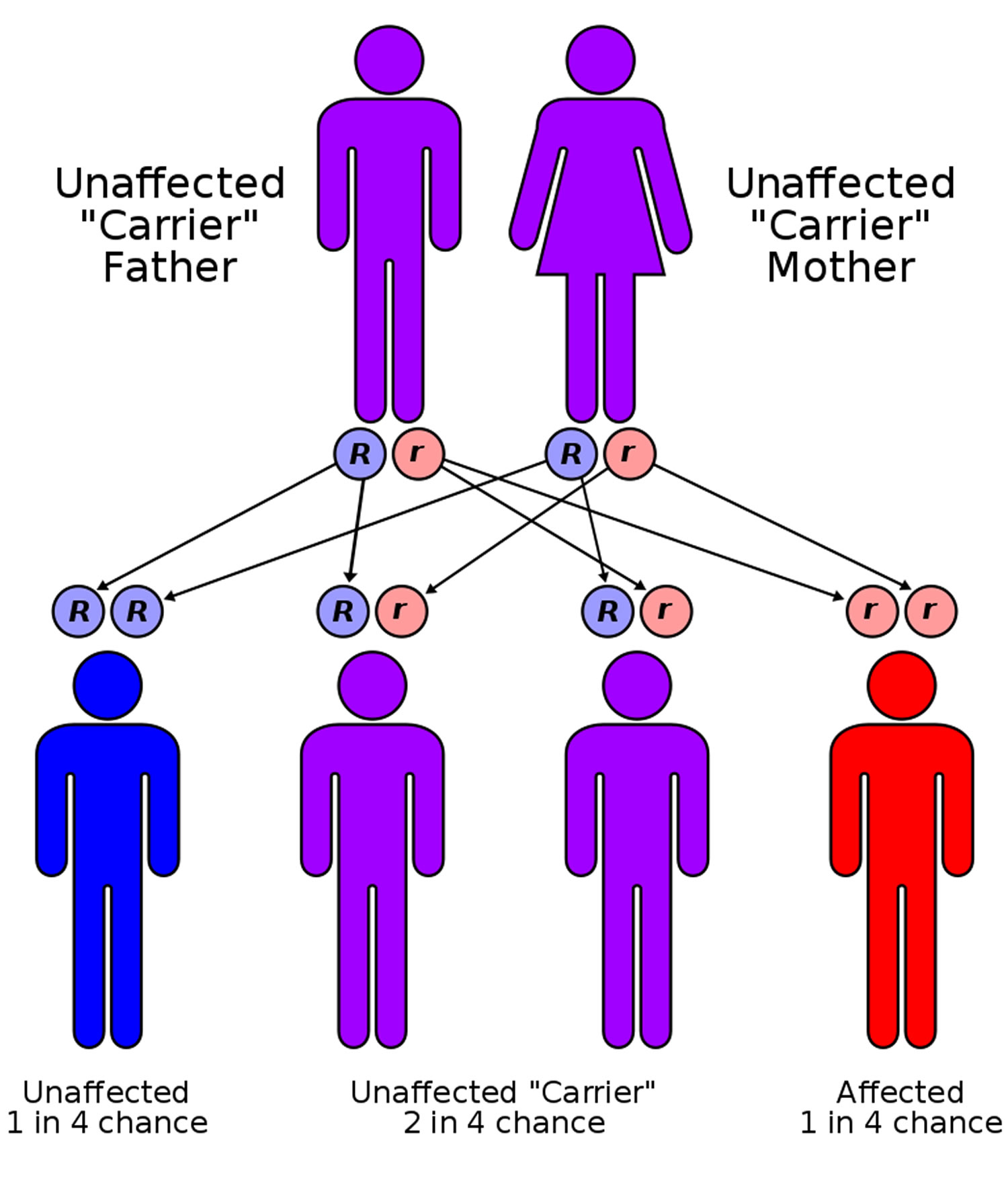
In the image above, each parent has one hemoglobin A gene and one hemoglobin S gene, and each of their children has:
- A 25 percent chance of inheriting two normal genes: In this case the child does not have sickle cell trait or disease. (Blue)
- A 50 percent chance of inheriting one hemoglobin A gene and one hemoglobin S gene: This child has sickle cell trait. (Purple)
- A 25 percent chance of inheriting two hemoglobin S genes: This child has sickle cell disease. (Red)
It is important to keep in mind that each time this couple has a child, the chances of that child having sickle cell disease remain the same. In other words, if the first-born child has sickle cell disease, there is still a 25 percent chance that the second child will also have the disease. Both boys and girls can inherit sickle cell trait, sickle cell disease, or normal hemoglobin.
If a person wants to know if he or she carries a sickle hemoglobin gene, a doctor can order a blood test to find out.
What Are the Signs and Symptoms of Sickle Cell Disease ?
Early Signs and Symptoms
If a person has sickle cell disease, it is present at birth. But most infants do not have any problems from the disease until they are about 5 or 6 months of age. Every state in the United States, the District of Columbia, and the U.S. territories requires that all newborn babies receive screening for sickle cell disease. When a child has sickle cell disease, parents are notified before the child has symptoms.
Some children with sickle cell disease will start to have problems early on, and some later. Early symptoms of sickle cell disease may include:
- Painful swelling of the hands and feet, known as dactylitis
- Fatigue or fussiness from anemia
- A yellowish color of the skin, known as jaundice, or whites of the eyes, known as icteris, that occurs when a large number of red cells hemolyze
The signs and symptoms of sickle cell disease will vary from person to person and can change over time. Most of the signs and symptoms of sickle cell disease are related to complications of the disease.
Major Complications of Sickle Cell Disease
Acute Pain (Sickle Cell or Vaso-occlusive) Crisis
Pain episodes (crises) can occur without warning when sickle cells block blood flow and decrease oxygen delivery. People describe this pain as sharp, intense, stabbing, or throbbing. Severe crises can be even more uncomfortable than post-surgical pain or childbirth.
Pain can strike almost anywhere in the body and in more than one spot at a time. But the pain often occurs in the
- Lower back
- Legs
- Arms
- Abdomen
- Chest
A crisis can be brought on by
- Illness
- Temperature changes
- Stress
- Dehydration (not drinking enough)
- Being at high altitudes
But often a person does not know what triggers, or causes, the crisis.
Chronic Pain
Many adolescents and adults with sickle cell disease suffer from chronic pain. This kind of pain has been hard for people to describe, but it is usually different from crisis pain or the pain that results from organ damage.
Chronic pain can be severe and can make life difficult. Its cause is not well understood.
Severe Anemia
People with sickle cell disease usually have mild to moderate anemia. At times, however, they can have severe anemia. Severe anemia can be life threatening. Severe anemia in an infant or child with sickle cell disease may be caused by:
- Splenic sequestration crisis
The spleen is an organ that is located in the upper left side of the belly. The spleen filters germs in the blood, breaks up blood cells, and makes a kind of white blood cell. A splenic sequestration crisis occurs when red blood cells get stuck in the spleen, making it enlarge quickly. Since the red blood cells are trapped in the spleen, there are fewer cells to circulate in the blood. This causes severe anemia.
A big spleen may also cause pain in the left side of the belly. A parent can usually palpate or feel the enlarged spleen in the belly of his or her child.
- Aplastic crisis
Aplastic crisis is usually caused by a parvovirus B19 infection, also called fifth disease or slapped cheek syndrome. Parvovirus B19 is a very common infection, but in sickle cell disease it can cause the bone marrow to stop producing new red cells for a while, leading to severe anemia.
Splenic sequestration crisis and aplastic crisis most commonly occur in infants and children with sickle cell disease. Adults with sickle cell disease may also experience episodes of severe anemia, but these usually have other causes.
No matter the cause, severe anemia may lead to symptoms that include:
- Shortness of breath
- Being very tired
- Feeling dizzy
- Having pale skin
Babies and infants with severe anemia may feed poorly and seem very sluggish.
- Infections
The spleen is important for protection against certain kinds of germs. Sickle cells can damage the spleen and weaken or destroy its function early in life.
People with sickle cell disease who have damaged spleens are at risk for serious bacterial infections that can be life-threatening. Some of these bacteria include:
- Pneumococcus
- Hemophilus influenza type B
- Meningococcus
- Salmonella
- Staphylococcus
- Chlamydia
- Mycoplasma pneumoniae
Bacteria can cause:
- Blood infection (septicemia)
- Lung infection (pneumonia)
- Infection of the covering of the brain and spinal cord (meningitis)
- Bone infection (osteomyelitis)
Acute Chest Syndrome
Sickling in blood vessels of the lungs can deprive a person’s lungs of oxygen. When this happens, areas of lung tissue are damaged and cannot exchange oxygen properly. This condition is known as acute chest syndrome. In acute chest syndrome, at least one segment of the lung is damaged.
This condition is very serious and should be treated right away at a hospital.
Acute chest syndrome often starts a few days after a painful crisis begins. A lung infection may accompany acute chest syndrome.
Symptoms may include:
- Chest pain
- Fever
- Shortness of breath
- Rapid breathing
- Cough
Brain Complications
Clinical Stroke
A stroke occurs when blood flow is blocked to a part of the brain. When this happens, brain cells can be damaged or can die. In sickle cell disease, a clinical stroke means that a person shows outward signs that something is wrong. The symptoms depend upon what part of the brain is affected. Symptoms of stroke may include:
- Weakness of an arm or leg on one side of the body
- Trouble speaking, walking, or understanding
- Loss of balance
- Severe headache
As many as 24 percent of people with hemoglobin SS and 10 percent of people with hemoglobin SC may suffer a clinical stroke by age 45.
In children, clinical stroke occurs most commonly between the ages of 2 and 9, but recent prevention strategies have lowered the risk.
When people with sickle cell disease show symptoms of stroke, their families or friends should call your local emergency number right away.
Silent Stroke and Thinking Problems
Brain imaging and tests of thinking (cognitive studies) have shown that children and adults with hemoglobin SS and hemoglobin Sβ0 thalassemia often have signs of silent brain injury, also called silent stroke. Silent brain injury is damage to the brain without showing outward signs of stroke.
This injury is common. Silent brain injury can lead to learning problems or trouble making decisions or holding down a job.
Eye Problems
Sickle cell disease can injure blood vessels in the eye.
The most common site of damage is the retina, where blood vessels can overgrow, get blocked, or bleed. The retina is the light-sensitive layer of tissue that lines the inside of the eye and sends visual messages through the optic nerve to the brain.
Detachment of the retina can occur. When the retina detaches, it is lifted or pulled from its normal position. These problems can cause visual impairment or loss.
Heart Disease
People with sickle cell disease can have problems with blood vessels in the heart and with heart function. The heart can become enlarged. People can also develop pulmonary hypertension.
People with sickle cell disease who have received frequent blood transfusions may also have heart damage from iron overload.
Pulmonary Hypertension
In adolescents and adults, injury to blood vessels in the lungs can make it hard for the heart to pump blood through them. This causes the pressure in lung blood vessels to rise. High pressure in these blood vessels is called pulmonary hypertension. Symptoms may include shortness of breath and fatigue.
When this condition is severe, it has been associated with a higher risk of death.
Kidney Problems
The kidneys are sensitive to the effects of red blood cell sickling.
Sickle cell disease causes the kidneys to have trouble making the urine as concentrated as it should be. This may lead to a need to urinate often and to have bedwetting or uncontrolled urination during the night (nocturnal enuresis). This often starts in childhood. Other problems may include:
- Blood in the urine
- Decreased kidney function
- Kidney disease
- Protein loss in the urine
Priapism
Males with sickle cell disease can have unwanted, sometimes prolonged, painful erections. This condition is called priapism.
Priapism happens when blood flow out of the erect penis is blocked by sickled cells. If it goes on for a long period of time, priapism can cause permanent damage to the penis and lead to impotence.
If priapism lasts for more than 4 hours, emergency medical care should be sought to avoid complications.
Gallstones
When red cells hemolyze, they release hemoglobin. Hemoglobin gets broken down into a substance called bilirubin. Bilirubin can form stones that get stuck in the gallbladder. The gallbladder is a small, sac-shaped organ beneath the liver that helps with digestion. Gallstones are a common problem in sickle cell disease.
Gallstones may be formed early on but may not produce symptoms for years. When symptoms develop, they may include:
- Right-sided upper belly pain
- Nausea
- Vomiting
If problems continue or recur, a person may need surgery to remove the gallbladder.
Liver Complications
There are a number of ways in which the liver may be injured in sickle cell disease.
Sickle cell intrahepatic cholestasis is an uncommon, but severe, form of liver damage that occurs when sickled red cells block blood vessels in the liver. This blockage prevents enough oxygen from reaching liver tissue.
These episodes are usually sudden and may recur. Children often recover, but some adults may have chronic problems that lead to liver failure.
People with sickle cell disease who have received frequent blood transfusions may develop liver damage from iron overload.
Leg Ulcers
Sickle cell ulcers are sores that usually start small and then get larger and larger.
The number of ulcers can vary from one to many. Some ulcers will heal quickly, but others may not heal and may last for long periods of time. Some ulcers come back after healing.
People with sickle cell disease usually don’t get ulcers until after the age of 10.
Joint Complications
Sickling in the bones of the hip and, less commonly, the shoulder joints, knees, and ankles, can decrease oxygen flow and result in severe damage. This damage is a condition called avascular or aseptic necrosis. This disease is usually found in adolescents and adults.
Symptoms include pain and problems with walking and joint movement. A person may need pain medicines, surgery, or joint replacement if symptoms persist.
Delayed Growth and Puberty
Children with sickle cell disease may grow and develop more slowly than their peers because of anemia. They will reach full sexual maturity, but this may be delayed.
Pregnancy
Pregnancies in women with sickle cell disease can be risky for both the mother and the baby.
Mothers may have medical complications including:
- Infections
- Blood clots
- High blood pressure
- Increased pain episodes
They are also at higher risk for:
- Miscarriages
- Premature births
- “Small-for-dates babies” or underweight babies
Mental Health
As in other chronic diseases, people with sickle cell disease may feel sad and frustrated at times. The limitations that sickle cell disease can impose on a person’s daily activities may cause them to feel isolated from others. Sometimes they become depressed.
People with sickle cell disease may also have trouble coping with pain and fatigue, as well as with frequent medical visits and hospitalizations.
How Is Sickle Cell Disease Diagnosed ?
Screening Tests
People who do not know whether they make sickle hemoglobin (hemoglobin S) or another abnormal hemoglobin (such as C, β thalassemia, E) can find out by having their blood tested. This way, they can learn whether they carry a gene (i.e., have the trait) for an abnormal hemoglobin that they could pass on to a child.
When each parent has this information, he or she can be better informed about the chances of having a child with some type of sickle cell disease, such as hemoglobin SS, SC, Sβ thalassemia, or others.
Newborn Screening
When a child has sickle cell disease, it is very important to diagnose it early to better prevent complications.
Every state in the United States, the District of Columbia, and the U.S. territories require that every baby is tested for sickle cell disease as part of a newborn screening program.
In newborn screening programs, blood from a heel prick is collected in “spots” on a special paper. The hemoglobin from this blood is then analyzed in special labs.
Newborn screening results are sent to the doctor who ordered the test and to the child’s primary doctor.
If a baby is found to have sickle cell disease, health providers from a special follow-up newborn screening group contact the family directly to make sure that the parents know the results. The child is always retested to be sure that the diagnosis is correct.
Newborn screening programs also find out whether the baby has an abnormal hemoglobin trait. If so, parents are informed, and counseling is offered.
Remember that when a child has sickle cell trait or sickle cell disease, a future sibling, or the child’s own future child, may be at risk. These possibilities should be discussed with the primary care doctor, a blood specialist called a hematologist, and/or a genetics counselor.
Prenatal Screening
Doctors can also diagnose sickle cell disease before a baby is born. This is done using a sample of amniotic fluid, the liquid in the sac surrounding a growing embryo, or tissue taken from the placenta, the organ that attaches the umbilical cord to the mother’s womb.
Testing before birth can be done as early as 8–10 weeks into the pregnancy. This testing looks for the sickle hemoglobin gene rather than the abnormal hemoglobin.
How Is Sickle Cell Disease Treated ?
Health Maintenance To Prevent Complications
Babies with sickle cell disease should be referred to a doctor or provider group that has experience taking care of people with this disease. The doctor might be a hematologist (a doctor with special training in blood diseases) or an experienced general pediatrician, internist, or family practitioner.
For infants, the first sickle cell disease visit should take place before 8 weeks of age.
If someone was born in a country that doesn’t perform newborn sickle cell disease screening, he or she might be diagnosed with sickle cell disease later in childhood. These people should also be referred as soon as possible for special sickle cell disease care.
All people who have sickle cell disease should see their sickle cell disease care providers regularly. Regularly means every 3 to 12 months, depending on the person’s age. The sickle cell disease doctor or team can help to prevent problems by:
- Examining the person
- Giving medicines and immunizations
- Performing tests
- Educating families about the disease and what to watch out for
Preventing Infection
In sickle cell disease, the spleen doesn’t work properly or doesn’t work at all. This problem makes people with sickle cell disease more likely to get severe infections.
Penicillin
In children with sickle cell disease, taking penicillin two times a day has been shown to reduce the chance of having a severe infection caused by the pneumococcus bacteria. Infants need to take liquid penicillin. Older children can take tablets.
Many doctors will stop prescribing penicillin after a child has reached the age of 5. Some prefer to continue this antibiotic throughout life, particularly if a person has hemoglobin SS or hemoglobin Sβ0 thalassemia, since people with sickle cell disease are still at risk. All people who have had surgical removal of the spleen, called a splenectomy, or a past infection with pneumococcus should keep taking penicillin throughout life.
Immunizations
People with sickle cell diseaseshould receive all recommended childhood vaccines. They should also receive additional vaccines to prevent other infections.
Pneumococcus. Even though all children routinely receive the vaccine against pneumococcus (PCV13), children with sickle cell disease should also receive a second kind of vaccine against pneumococcus (PPSV23). This second vaccine is given after 24 months of age and again 5 years later. Adults with sickle cell disease who have not received any pneumococcal vaccine should get a dose of the PCV13 vaccine. They should later receive the PPSV23 if they have not already received it or it has been more than 5 years since they did. A person should follow these guidelines even if he or she is still taking penicillin.
Influenza. All people with sickle cell disease should receive an influenza shot every year at the start of flu season. This should begin at 6 months of age. Only the inactivated vaccine, which comes as a shot, should be used in people with sickle cell disease.
Meningococcus. A child with sickle cell disease should receive this vaccine (Menactra or Menveo) at 2, 4, 6, and 12–15 months of age. The child should receive a booster vaccine 3 years after this series of shots, then every 5 years after that.
Screening Tests and Evaluations
Height, Weight, Blood Pressure, and Oxygen Saturation
Doctors will monitor height and weight to be sure that a child is growing properly and that a person with sickle cell disease is maintaining a healthy weight.
Doctors will also track a person’s blood pressure. When a person with sickle cell disease has high blood pressure, it needs to be treated promptly because it can increase the risk of stroke.
Oxygen saturation testing provides information about how much oxygen the blood is carrying.
Blood and Urine Testing
People with sickle cell disease need to have frequent lab tests.
Blood tests help to establish a person’s “baseline” for problems like anemia. Blood testing also helps to show whether a person has organ damage, so that it can be treated early.
Urine testing can help to detect early kidney problems or infections.
Transcranial Doppler Ultrasound Screening
Children who have hemoglobin SS or hemoglobin Sβ0 thalassemia and are between the ages of 2 and 16 should have Transcranial Doppler Ultrasound testing once a year.
This study can find out whether a child is at higher risk for stroke. When the test is abnormal, regular blood transfusions can decrease the chances of having a stroke.
The child is awake during the Transcranial Doppler Ultrasound exam. The test does not hurt at all. The Transcranial Doppler Ultrasound machine uses sound waves to measure blood flow like the ultrasound machine used to examine pregnant women.
Eye Examinations
An eye doctor, or ophthalmologist, should examine a person’s eyes every 1—2 years from the age of 10 onwards.
These exams can detect if there are sickle cell disease-related problems of the eye. Regular exams can help doctors find and treat problems early to prevent loss of vision. A person should see his or her doctor right away for any sudden change in vision.
Pulmonary Hypertension
Doctors have different approaches to screening for pulmonary hypertension. This is because studies have not given clear information as to when and how a person should receive the screening. People with sickle cell disease and their caretakers should discuss with their doctor whether screening makes sense for them.
Cognitive Screening
People with sickle cell disease can develop cognitive (thinking) problems that may be hard to notice early in life.
Sometimes these problems are caused by “silent” strokes that can only be seen with magnetic resonance imaging (MRI) of the brain.
People with sickle cell disease should tell their doctors or nurses if they have thinking problems, such as difficulties learning in school, slowed decision making, or trouble organizing their thoughts.
People can be referred for cognitive testing. This testing can identify areas in which a person could use extra help.
Children with sickle cell disease who have thinking problems may qualify for an Individualized Education Program. An Individualized Education Program is a plan that helps students to reach their educational goals. Adults may be able to enroll in vocational rehabilitation programs that can help them with job training.
Education and Guidance
Doctors and other providers will talk with people who have sickle cell disease and their caretakers about complications and also review information at every visit.
Because there are a lot of things to discuss, new topics are often introduced as a child or adult reaches an age when that subject is important to know about.
Doctors and nurses know that there is a lot of information to learn, and they don’t expect people to know everything after one discussion. People with sickle cell disease and their families should not be afraid to ask questions.
Topics that are usually covered include:
- Hours that medical staff are available and contact information to use when people with sickle cell disease or caretakers have questions
- A plan for what to do and where to get care if a person has a fever, pain, or other signs of sickle cell disease complications that need immediate attention
- How sickle cell disease is inherited and the risk of having a child with sickle cell disease
- The importance of regular medical visits, screening tests, and evaluations
- How to recognize and manage pain
- How to palpate (feel) a child’s spleen. Because of the risk of splenic sequestration crisis, caretakers should learn how to palpate a child’s spleen. They should try to feel for the spleen daily and more frequently when the child is ill. If they feel that the spleen is bigger than usual, they should call the care provider.
Transitioning Care
When children with sickle cell disease become adolescents or young adults, they often need to transition from a pediatric care team to an adult care team. This period has been shown to be associated with increased hospital admissions and medical problems. There seem to be many reasons for this.
Some of the increased risk is directly related to the disease. As people with sickle cell disease get older, they often develop more organ damage and more disabilities.
The shift in care usually occurs at the same time that adolescents are undergoing many changes in their emotional, social, and academic lives. The transition to more independent self-management may be difficult, and following treatment plans may become less likely.
When compared with pediatrics, there are often fewer adult sickle cell disease programs available in a given region. This makes it more difficult for a person with sickle cell disease to find appropriate doctors, particularly those with whom they feel comfortable.
To improve use of regular medical care by people with sickle cell disease and to reduce age-related complications, many sickle cell disease teams have developed special programs that the make transition easier. Such programs should involve the pediatric and the adult care teams. They should also start early and continue over several years.
Managing Some Complications of sickle cell disease
Acute Pain
Each person with sickle cell disease should have a home treatment regimen that is best suited to their needs. The providers on the sickle cell disease team usually help a person develop a written, tailored care plan. If possible, the person with sickle cell disease should carry this plan with them when they go to the emergency room.
When an acute crisis is just starting, most doctors will advise the person to drink lots of fluids and to take a non-steroidal anti-inflammatory (NSAID) pain medication, such as ibuprofen. When a person has kidney problems, acetaminophen is often preferred.
If pain persists, many people will find that they need a stronger medicine.
Combining additional interventions, such as massage, relaxation methods, or a heating pad, may also help.
If a person with sickle cell disease cannot control the pain at home, he or she should go to an sickle cell disease day hospital/outpatient unit or an emergency room to receive additional, stronger medicines and intravenous (IV) fluids.
Some people may be able to return home once their pain is under better control. In this case, the doctor may prescribe additional pain medicines for a short course of therapy.
People often need to be admitted to the hospital to fully control an acute pain crisis.
When taken daily, hydroxyurea has been found to decrease the number and severity of pain episodes.
Some patients may have fewer visits to the hospital or hospitalizations due to severe pain, and may have shorter hospital stays for pain crises if they are taking L-glutamine oral powder (Endari) compared to patients who are not taking this medicine. More research is needed to understand how effective L-glutamine oral powder is as a treatment and which patients may benefit from using it.
Chronic Pain
Sometimes chronic pain results from a complication, such as a leg ulcer or aseptic necrosis of the hip. In this case, doctors try to treat the complication causing the pain.
While chronic pain is common in adults with sickle cell disease, the cause is often poorly understood. Taking pain medicines daily may help to decrease the pain. Some examples of these medicines include:
- NSAID drugs, such as ibuprofen
- Duloxetine
- Gabapentin
- Amitriptyline
- Strong pain medicines, such as opiates
Other approaches, such as massage, heat, or acupuncture may be helpful in some cases. Chronic pain often comes with feelings of depression and anxiety. Supportive counseling and, sometimes, antidepressant medicines may help.
Severe Anemia
People should see their doctors or go to a hospital right away if they develop anemia symptoms from a splenic sequestration crisis or an aplastic crisis. These conditions can be life-threatening, and the person will need careful monitoring and treatment in the hospital. A person also usually needs a blood transfusion.
People with sickle cell disease and symptoms of severe anemia from other causes should also see a doctor right away.
Some patients may have fewer hospital visits due to sickle cell crises, including splenic sequestration, if they are taking L-glutamine oral powder compared to patients who are not taking this medicine. More research is needed to understand how effective L-glutamine oral powder is as a treatment and which patients may benefit from using it.
Infections
Fever is a medical emergency in sickle cell disease. All caretakers of infants and children with sickle cell disease should take their child to their doctor or go to a hospital right away when their child has a fever. Adults with sickle cell disease should also seek care for fever or other signs of infection.
All children and adults who have sickle cell disease and a fever (over 38.50 C or 101.30 F) must be seen by a doctor and treated with antibiotics right away.
Some people will need to be hospitalized, while others may receive care and follow-up as an outpatient.
Acute Chest Syndrome
People with sickle cell disease and symptoms of acute chest syndrome should see their doctor or go to a hospital right away.
They will need to be admitted to the hospital where they should receive antibiotics and close monitoring. They may need oxygen therapy and a blood transfusion.
When taken daily, the medicine hydroxyurea has been found to decrease the number and severity of acute chest events.
Some patients may have fewer hospital visits due to sickle cell crises, including acute chest syndrome, if they are taking L-glutamine oral powder compared to patients who are not taking this medicine. More research is needed to understand how effective L-glutamine oral powder is as a treatment and which patients may benefit from using it.
Clinical Stroke
People with sickle cell disease who have symptoms of stroke should be brought to the hospital right away by an ambulance. If a person is having symptoms of stroke, someone should call your local emergency number.
Symptoms of stroke may include:
- Weakness of an arm or leg on one side of the body
- Trouble speaking, walking, or understanding
- Loss of balance
- Severe headache
If imaging studies reveal that the person has had an acute stroke, he or she may need an exchange transfusion. This procedure involves slowly removing an amount of the person’s blood and replacing it with blood from a donor who does not have sickle cell disease or sickle cell trait. Afterward, the person may need to receive monthly transfusions or other treatments to help to prevent another stroke.
Silent Stroke and Cognitive Problems
Children and adults with sickle cell disease and cognitive problems may be able to get useful help based upon the results of their testing. For instance, children may qualify for an Individualized Education Program. Adults may be able to enroll in vocational, or job, training programs.
Priapism
Sometimes, a person may be able to relieve priapism by:
- Emptying the bladder by urinating
- Taking medicine
- Increasing fluid intake
- Doing light exercise
If a person has an episode that lasts for 4 hours or more, he should go to the hospital to see a hematologist and urologist.
Some patients may have fewer hospital visits due to sickle cell crises, including priapism, if they are taking L-glutamine oral powder compared to patients who are not taking this medicine. More research is needed to understand how effective L-glutamine oral powder is as a treatment and which patients may benefit from using it.
Pregnancy
Pregnant women with sickle cell disease are at greater risk for problems. They should always see an obstetrician who has experience with sickle cell disease and high-risk pregnancies and deliveries.
The obstetrician should work with a hematologist or primary medical doctor who is well informed about sickle cell disease and its complications.
Pregnant women with sickle cell disease need more frequent medical visits so that their doctors can follow them closely. The doctor may prescribe certain vitamins and will be careful to prescribe pain medicines that are safe for the baby.
A pregnant woman with sickle cell disease may need to have one or more blood transfusions during her pregnancy to treat complications, such as worsening anemia or an increased number of pain or acute chest syndrome events.
Hydroxyurea
Hydroxyurea is an oral medicine that has been shown to reduce or prevent several sickle cell disease complications.
This medicine was studied in patients with sickle cell disease because it was known to increase the amount of fetal hemoglobin (hemoglobin F) in the blood. Increased hemoglobin F provides some protection against the effects of hemoglobin S.
Hydroxyurea was later found to have several other benefits for a person with sickle cell disease, such as decreasing inflammation.
Use in adults. Many studies of adults with hemoglobin SS or hemoglobin Sβ thalassemia showed that hydroxyurea reduced the number of episodes of pain crises and acute chest syndrome. It also improved anemia and decreased the need for transfusions and hospital admissions.
Use in children. Studies in children with severe hemoglobin SS or Sβ thalassemia showed that hydroxyurea reduced the number of vaso-occlusive crises and hospitalizations. A study of very young children (between the ages of 9 and 18 months) with hemoglobin SS or hemoglobin Sβ thalassemia also showed that hydroxyurea decreased the number of episodes of pain and dactylitis.
Who Should Use Hydroxyurea ?
Since hydroxyurea can decrease several complications of sickle cell disease, most experts recommend that children and adults with hemoglobin SS or Sβ0 thalassemia who have frequent painful episodes, recurrent chest crises, or severe anemia take hydroxyurea daily.
Some experts offer hydroxyurea to all infants over 9 months of age and young children with hemoglobin SS or Sβ0 thalassemia, even if they do not have severe clinical problems, to prevent or reduce the chance of complications. There is no information about how safe or effective hydroxyurea is in children under 9 months of age.
Some experts will prescribe hydroxyurea to people with other types of sickle cell disease who have severe, recurrent pain. There is little information available about how effective hydroxyurea is for these types of sickle cell disease.
In all situations, people with sickle cell disease should discuss with their doctors whether or not hydroxyurea is an appropriate medication for them.
Pregnant women should not use hydroxyurea.
How Is Hydroxyurea Taken ?
To work properly, hydroxyurea should be taken by mouth daily at the prescribed dose. When a person does not take it regularly, it will not work as well, or it won’t work at all.
A person with sickle cell disease who is taking hydroxyurea needs careful monitoring. This is particularly true in the early weeks of taking the medicine. Monitoring includes regular blood testing and dose adjustments.
What Are the Risks of Hydroxyurea ?
Hydroxyurea can cause the blood’s white cell count or platelet count to drop. In rare cases, it can worsen anemia. These side effects usually go away quickly if a person stops taking the medication. When a person restarts it, a doctor usually prescribes a lower dose.
Other short-term side effects are less common.
It is still unclear whether hydroxyurea can cause problems later in life in people with sickle cell disease who take it for many years. Studies so far suggest that it does not put people at a higher risk of cancer and does not affect growth in children. But further studies are needed.
Red Blood Cell Transfusions
Doctors may use acute and chronic red blood cell transfusions to treat and prevent certain sickle cell disease complications. The red blood cells in a transfusion have normal hemoglobin in them.
A transfusion helps to raise the number of red blood cells and provides normal red blood cells that are more flexible than red blood cells with sickle hemoglobin. These cells live longer in the circulation. Red blood cell transfusions decrease vaso-occlusion (blockage in the blood vessel) and improve oxygen delivery to the tissues and organs.
Acute Transfusion in sickle cell disease
Doctors use blood transfusions in sickle cell disease for complications that cause severe anemia. They may also use them when a person has an acute stroke, in many cases of acute chest crises, and in multi-organ failure.
A person with sickle cell disease usually receives blood transfusions before surgery to prevent sickle cell disease-related complications afterwards.
Chronic Transfusion
Doctors recommend regular or ongoing blood transfusions for people who have had an acute stroke, since transfusions decrease the chances of having another stroke.
Doctors also recommend chronic blood transfusions for children who have abnormal Transcranial Doppler Ultrasound results because transfusions can reduce the chance of having a first stroke.
Some doctors use this approach to treat complications that do not improve with hydroxyurea. They may also use transfusions in people who have too many side effects from hydroxyurea.
What Are the Risks of Transfusion Therapy ?
Possible complications include:
- Hemolysis
- Iron overload, particularly in people receiving chronic transfusions (can severely impair heart and lung function)
- Infection
- Alloimmunization (can make it hard to find a matching unit of blood for a future transfusion)
All blood banks and hospital personnel have adopted practices to reduce the risk of transfusion problems.
People with sickle cell disease who receive transfusions should be monitored for and immunized against hepatitis. They should also receive regular screenings for iron overload. If a person has iron overload, the doctor will give chelation therapy, a medicine to reduce the amount of iron in the body and the problems that iron overload causes.
Hematopoietic Stem Cell Transplantation
At the present time, hematopoietic stem cell transplantation (HSCT) is the only cure for sickle cell disease. People with sickle cell disease and their families should ask their doctor about this procedure.
What Are Stem Cells ?
Stem cells are special cells that can divide over and over again. After they divide, these cells can go on to become blood red cells, white cells, or platelets.
A person with sickle cell disease has stem cells that make red blood cells that can sickle. People without sickle cell disease have stem cells that make red cells that usually won’t sickle.
What Stem Cells Are Used in hematopoietic stem cell transplantation ?
In hematopoietic stem cell transplantation, stem cells are taken from the bone marrow or blood of a person who does not have sickle cell disease (the donor). The donor, however, may have sickle cell trait.
The donor is often the person’s sister or brother. This is because the safest and most successful transplants use stem cells that are matched for special proteins called HLA antigens. Since these antigens are inherited from parents, a sister or brother is the most likely person to have the same antigens as the person with sickle cell disease.
What Happens During hematopoietic stem cell transplantation ?
First, stem cells are taken from the donor. After this, the person with sickle cell disease (the recipient) is treated with drugs that destroy or reduce his or her own bone marrow stem cells.
The donor stem cells are then injected into the person’s vein. The injected cells will make a home in the recipient’s bone marrow, gradually replacing the recipient’s cells. The new stem cells will make red cells that do not sickle.
Which People Receive hematopoietic stem cell transplantation ?
At the present time, most sickle cell disease transplants are performed in children who have had complications such as strokes, acute chest crises, and recurring pain crises. These transplants usually use a matched donor.
Because only about 1 in 10 children with sickle cell disease has a matched donor without sickle cell disease in their families, the number of people with sickle cell disease who get transplants is low.
Hematopoietic stem cell transplantation is more risky in adults, and that is why most transplants are done in children.
There are several medical centers that are researching new sickle cell disease hematopoietic stem cell transplantation techniques in children and adults who don’t have a matched donor in the family or are older than most recipients. Hopefully, more people with sickle cell disease will be able to receive a transplant in the future, using these new methods.
What Are the Risks ?
Hematopoietic stem cell transplantation is successful in about 85 percent of children when the donor is related and HLA matched. Even with this high success rate, hematopoietic stem cell transplantation still has risks.
Complications can include severe infections, seizures, and other clinical problems. About 5 percent of people have died. Sometimes transplanted cells attack the recipient’s organs (graft versus host disease).
Medicines are given to prevent many of the complications, but they still can happen.
What happens when you have too many red blood cells
Polycythemia vera is a rare blood disease in which your your bone marrow makes too many red blood cells and it also can make too many white blood cells and platelets 7. The disease affects people of all ages, but it’s most common in adults who are older than 60. Polycythemia vera is rare in children and young adults. Men are at slightly higher risk for polycythemia vera than women.
The extra red blood cells make your blood thicker than normal. As a result, blood clots can form more easily. These clots can block blood flow through your arteries and veins, which can cause a heart attack or stroke.
Thicker blood also doesn’t flow as quickly to your body as normal blood. Slowed blood flow prevents your organs from getting enough oxygen, which can cause serious problems, such as angina (heart pain) and heart failure. Angina is chest pain or discomfort.
A mutation, or change, in the body’s JAK2 gene is the major cause of polycythemia vera. This gene makes a protein that helps the body produce blood cells. What causes the change in the JAK2 gene isn’t known. Polycythemia vera generally isn’t inherited—that is, passed from parents to children through genes.
Polycythemia vera develops slowly and may not cause symptoms for years. The disease often is found during routine blood tests done for other reasons.
When signs and symptoms are present, they’re the result of the thick blood that occurs with polycythemia vera. This thickness slows the flow of oxygen-rich blood to all parts of your body. Without enough oxygen, many parts of your body won’t work normally.
For example, slower blood flow deprives your arms, legs, lungs, and eyes of the oxygen they need. This can cause headaches, dizziness, itching, and vision problems, such as blurred or double vision.
Polycythemia vera is a serious, chronic (ongoing) disease that can be fatal if not diagnosed and treated. Polycythemia vera has no cure, but treatments can help control the disease and its complications.
Polycythemia vera is treated with procedures, medicines, and other methods. You may need one or more treatments to manage the disease.
What Causes Polycythemia Vera ?
Primary Polycythemia
Polycythemia vera also is known as primary polycythemia. A mutation, or change, in the body’s JAK2 gene is the main cause of polycythemia vera. The JAK2 gene makes a protein that helps the body produce blood cells.
What causes the change in the JAK2 gene isn’t known. Polycythemia vera generally isn’t inherited—that is, passed from parents to children through genes. However, in some families, the JAK2 gene may have a tendency to mutate. Other, unknown genetic factors also may play a role in causing polycythemia vera.
Secondary Polycythemia
Another type of polycythemia, called secondary polycythemia, isn’t related to the JAK2 gene. Long-term exposure to low oxygen levels causes secondary polycythemia.
A lack of oxygen over a long period can cause your body to make more of the hormone erythropoietin (EPO). High levels of EPO can prompt your body to make more red blood cells than normal. This leads to thicker blood, as seen in polycythemia vera.
People who have severe heart or lung disease may develop secondary polycythemia. People who smoke, spend long hours at high altitudes, or are exposed to high levels of carbon monoxide where they work or live also are at risk.
For example, working in an underground parking garage or living in a home with a poorly vented fireplace or furnace can raise your risk for secondary polycythemia.
Rarely, tumors can make and release EPO, or certain blood problems can cause the body to make more EPO.
Sometimes doctors can cure secondary polycythemia—it depends on whether the underlying cause can be stopped, controlled, or cured.
What Are the Signs and Symptoms of Polycythemia Vera ?
Polycythemia vera develops slowly. The disease may not cause signs or symptoms for years.
When signs and symptoms are present, they’re the result of the thick blood that occurs with polycythemia vera. This thickness slows the flow of oxygen-rich blood to all parts of your body. Without enough oxygen, many parts of your body won’t work normally.
The signs and symptoms of polycythemia vera include:
- Headaches, dizziness, and weakness
- Shortness of breath and problems breathing while lying down
- Feelings of pressure or fullness on the left side of the abdomen due to an enlarged spleen (an organ in the abdomen)
- Double or blurred vision and blind spots
- Itching all over (especially after a warm bath), reddened face, and a burning feeling on your skin (especially your hands and feet)
- Bleeding from your gums and heavy bleeding from small cuts
- Unexplained weight loss
- Fatigue (tiredness)
- Excessive sweating
- Very painful swelling in a single joint, usually the big toe (called gouty arthritis)
In rare cases, people who have polycythemia vera may have pain in their bones.
Polycythemia Vera Complications
If you have polycythemia vera, the thickness of your blood and the slowed blood flow can cause serious health problems.
Blood clots are the most serious complication of polycythemia vera. Blood clots can cause a heart attack or stroke. They also can cause your liver and spleen to enlarge. Blood clots in the liver and spleen can cause sudden, intense pain.
Slowed blood flow also prevents enough oxygen-rich blood from reaching your organs. This can lead to angina (chest pain or discomfort) and heart failure. The high levels of red blood cells that polycythemia vera causes can lead to stomach ulcers, gout, or kidney stones.
Some people who have polycythemia vera may develop myelofibrosis. This is a condition in which your bone marrow is replaced with scar tissue. Abnormal bone marrow cells may begin to grow out of control.
This abnormal growth can lead to acute myelogenous leukemia (AML), a cancer of the blood and bone marrow. This disease can worsen very quickly.
How Is Polycythemia Vera Diagnosed ?
Polycythemia vera may not cause signs or symptoms for years. The disease often is found during routine blood tests done for other reasons. If the results of your blood tests aren’t normal, your doctor may want to do more tests.
Your doctor will diagnose polycythemia vera based on your signs and symptoms, your age and overall health, your medical history, a physical exam, and test results.
During the physical exam, your doctor will look for signs of polycythemia vera. He or she will check for an enlarged spleen, red skin on your face, and bleeding from your gums.
If your doctor confirms that you have polycythemia, the next step is to find out whether you have primary polycythemia or secondary polycythemia.
Your medical history and physical exam may confirm which type of polycythemia you have. If not, you may have tests that check the level of the hormone erythropoietin (EPO) in your blood.
People who have polycythemia vera have very low levels of EPO. People who have secondary polycythemia usually have normal or high levels of EPO.
Specialists Involved
If your primary care doctor thinks you have polycythemia vera, he or she may refer you to a hematologist. A hematologist is a doctor who specializes in diagnosing and treating blood diseases and conditions.
Diagnostic Tests
You may have blood tests to diagnose polycythemia vera. These tests include a complete blood count (CBC) and other tests, if necessary.
Complete Blood Count
Often, the first test used to diagnose polycythemia vera is a CBC. The CBC measures many parts of your blood.
This test checks your hemoglobin and hematocrit levels. Hemoglobin is an iron-rich protein that helps red blood cells carry oxygen from the lungs to the rest of the body. Hematocrit is a measure of how much space red blood cells take up in your blood. A high level of hemoglobin or hematocrit may be a sign of polycythemia vera.
The CBC also checks the number of red blood cells, white blood cells, and platelets in your blood. Abnormal results may be a sign of polycythemia vera, a blood disorder, an infection, or another condition.
In addition to high red blood cell counts, people who have polycythemia vera also may have high white blood cell and/or platelet counts.
Other Blood Tests
Blood smear. For this test, a small sample of blood is drawn from a vein, usually in your arm. The blood sample is examined under a microscope.
A blood smear can show whether you have a higher than normal number of red blood cells. The test also can show abnormal blood cells that are linked to myelofibrosis and other conditions related to polycythemia vera.
Erythropoietin level. This blood test measures the level of EPO in your blood. EPO is a hormone that prompts your bone marrow to make new blood cells. People who have polycythemia vera have very low levels of EPO. People who have secondary polycythemia usually have normal or high levels of EPO.
Bone Marrow Tests
Bone marrow tests can show whether your bone marrow is healthy. These tests also show whether your bone marrow is making normal amounts of blood cells.
The two bone marrow tests are aspiration and biopsy. For aspiration, your doctor removes a small amount of fluid bone marrow through a needle. For a biopsy, your doctor removes a small amount of bone marrow tissue through a larger needle. The samples are then examined under a microscope.
If the tests show that your bone marrow is making too many blood cells, it may be a sign that you have polycythemia vera.
How Is Polycythemia Vera Treated ?
Polycythemia vera doesn’t have a cure. However, treatments can help control the disease and its complications. polycythemia vera is treated with procedures, medicines, and other methods. You may need one or more treatments to manage the disease.
Goals of Treatment
The goals of treating polycythemia vera are to control symptoms and reduce the risk of complications, especially heart attack and stroke. To do this, polycythemia vera treatments reduce the number of red blood cells and the level of hemoglobin (an iron-rich protein) in the blood. This brings the thickness of your blood closer to normal.
Blood with normal thickness flows better through the blood vessels. This reduces the chance that blood clots will form and cause a heart attack or stroke.
Blood with normal thickness also ensures that your body gets enough oxygen. This can help reduce some of the signs and symptoms of polycythemia vera, such as headaches, vision problems, and itching.
Studies show that treating polycythemia vera greatly improves your chances of living longer.
The goal of treating secondary polycythemia is to control its underlying cause, if possible. For example, if the cause is carbon monoxide exposure, the goal is to find the source of the carbon monoxide and fix or remove it.
Treatments To Lower Red Blood Cell Levels
Phlebotomy
Phlebotomy is a procedure that removes some blood from your body. For this procedure, a needle is inserted into one of your veins. Blood from the vein flows through an airtight tube into a sterile container or bag. The process is similar to the process of donating blood.
Phlebotomy reduces your red blood cell count and starts to bring your blood thickness closer to normal.
Typically, a pint (1 unit) of blood is removed each week until your hematocrit level approaches normal. Hematocrit is the measure of how much space red blood cells take up in your blood.
You may need to have phlebotomy done every few months.
Medicines
Your doctor may prescribe medicines to keep your bone marrow from making too many red blood cells. Examples of these medicines include hydroxyurea and interferon-alpha.
Hydroxyurea is a medicine generally used to treat cancer. This medicine can reduce the number of red blood cells and platelets in your blood. As a result, this medicine helps improve your blood flow and bring the thickness of your blood closer to normal.
Interferon-alpha is a substance that your body normally makes. It also can be used to treat polycythemia vera. Interferon-alpha can prompt your immune system to fight overactive bone marrow cells. This helps lower your red blood cell count and keep your blood flow and blood thickness closer to normal.
Radiation Treatment
Radiation treatment can help suppress overactive bone marrow cells. This helps lower your red blood cell count and keep your blood flow and blood thickness closer to normal.
However, radiation treatment can raise your risk of leukemia (blood cancer) and other blood diseases.
Treatments for Symptoms
Aspirin can relieve bone pain and burning feelings in your hands or feet that you may have as a result of polycythemia vera. Aspirin also thins your blood, so it reduces the risk of blood clots.
Aspirin can have side effects, including bleeding in the stomach and intestines. For this reason, take aspirin only as your doctor recommends.
If your polycythemia vera causes itching, your doctor may prescribe medicines to ease the discomfort. Your doctor also may prescribe ultraviolet light treatment to help relieve your itching.
Other ways to reduce itching include:
- Avoiding hot baths. Cooler water can limit irritation to your skin.
- Gently patting yourself dry after bathing. Vigorous rubbing with a towel can irritate your skin.
- Taking starch baths. Add half a box of starch to a tub of lukewarm water. This can help soothe your skin.
Experimental Treatments
Researchers are studying other treatments for polycythemia vera. An experimental treatment for itching involves taking low doses of selective serotonin reuptake inhibitors (SSRIs). This type of medicine is used to treat depression. In clinical trials, SSRIs reduced itching in people who had polycythemia vera.
Imatinib mesylate is a medicine that’s approved for treating leukemia. In clinical trials, this medicine helped reduce the need for phlebotomy in people who had polycythemia vera. This medicine also helped reduce the size of enlarged spleens.
Researchers also are trying to find a treatment that can block or limit the effects of an abnormal JAK2 gene. A mutation, or change, in the JAK2 gene is the major cause of polycythemia vera.
Living With Polycythemia Vera
Polycythemia vera develops very slowly. It may not cause signs or symptoms for years. If you have polycythemia vera, the sooner it’s diagnosed, the sooner your doctor can begin treating you. With proper treatment, you can prevent or delay complications.
Preventing Complications
Moderate physical activities, such as walking, can safely increase your heart rate and improve blood flow to your body. Improving blood flow lowers your risk of blood clots. Leg and ankle stretching exercises also can help improve your blood flow.
Polycythemia vera may cause itching all over your body. It’s important not to scratch and damage your skin. If bathing or showering causes you to have severe itching, try using cooler water and gentler soap. Carefully and gently dry your skin after baths, and use moisturizing lotion on your skin. Starch baths also may help ease itchy skin.
Polycythemia vera causes poor blood flow in your hands and feet. As a result, you may be more prone to injuries from cold, heat, and pressure. If you have Polycythemia vera, avoid long-term exposure to extremes in temperature or pressure. For example:
- Take extra care of your hands and feet in cold weather. Wear warm gloves, socks, and shoes.
- Avoid extreme heat, and protect yourself from the sun. Drink plenty of liquids. Avoid hot tubs, heated whirlpools, or hot baths of any type. Also, tanning beds, sun lamps, and heat lamps can damage your skin if you have Polycythemia vera.
- Guard against trauma or situations where you may be at high risk of injury, such as during sports or strenuous activities. If you’re injured, seek treatment right away. Tell the person treating you that you have Polycythemia vera.
- Check your feet regularly and report any sores to your doctor.
Getting Ongoing Care
If you have polycythemia vera, you’ll need lifelong medical care for the disease. Ask your doctor how often you should schedule followup visits.
Routine care will allow your doctor to detect any changes with your Polycythemia vera and treat them early, if needed. You may need periodic blood tests to show whether the disease is getting worse.
Follow your treatment plan and take all of your medicines exactly as your doctor prescribes.
- Anemia. National Heart, Lung and Blood Institute. https://www.nhlbi.nih.gov/health/health-topics/topics/anemia[↩][↩][↩]
- Iron-Deficiency Anemia. National Heart, Lung and Blood Institute. https://www.nhlbi.nih.gov/health-topics/topics/ida[↩]
- Pernicious Anemia. National Heart, Lung and Blood Institute. https://www.nhlbi.nih.gov/health/health-topics/topics/prnanmia[↩][↩][↩][↩][↩]
- Sickle Cell Disease. National Heart, Lung and Blood Institute. https://www.nhlbi.nih.gov/health/health-topics/topics/sca[↩][↩]
- Who Is at Risk for Sickle Cell Disease ? National Heart, Lung and Blood Institute. https://www.nhlbi.nih.gov/health/health-topics/topics/sca/atrisk[↩]
- By en:User:Cburnett – Own work in Inkscape, CC BY-SA 3.0, https://commons.wikimedia.org/w/index.php?curid=1840082[↩]
- Polycythemia Vera. National Heart, Lung and Blood Institute. https://www.nhlbi.nih.gov/health/health-topics/topics/poly[↩]
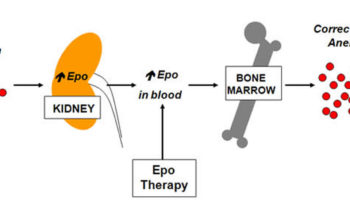
{kind=link}