Hemophilia C
Hemophilia C also called factor XI deficiency, plasma thromboplastin antecedent deficiency or Rosenthal syndrome is a very rare autosomal recessive bleeding disorder. Hemophilia C (factor XI deficiency) is the most common of the rare bleeding disorders and the second most common bleeding disorder affecting women after von Willebrand disease 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11. Hemophilia C (factor XI deficiency) was first recognized in 1953 in patients who experienced severe bleeding after dental extractions and its incidence is estimated at 1 in 100,000 in the general population. Hemophilia C (factor XI deficiency) is estimated to affect approximately 1 in 1 million people worldwide 11. The severe hemophilia C (factor XI deficiency) is much more common in people with central and eastern European Jews (Ashkenazi Jews), occurring in about 1 in 450 individuals in that population (occurs in up to 8% of Ashkenazi Jews), because of intermarriage. Researchers suggest that the actual prevalence of factor XI deficiency may be higher than reported, because mild cases of the disorder often do not come to medical attention.
Hemophilia C (factor XI deficiency) is caused by mutations in the F11 gene also called plasma thromboplastin antecedent (PTA) gene. The F11 gene provides instructions for making a protein called factor XI (factor 11). The factor XI (factor 11) protein plays a role in the coagulation cascade, which is a series of chemical reactions that forms blood clots in response to injury. After an injury, clots seal off blood vessels to stop bleeding and trigger blood vessel repair. Factor XI (factor 11) is made primarily by cells in your liver. The factor XI (factor 11) protein circulates in your bloodstream and is normally turned off (inactive) until the coagulation cascade is turned on (activated) by an injury that damages your blood vessels. When factor XI (factor 11) is activated, it interacts with other coagulation factors, resulting in conversion of an important coagulation protein called prothrombin to its active form, thrombin. Thrombin then converts a protein called fibrinogen into fibrin, which is the material that forms blood clots by trapping platelets and helps hold a clot in place.
Mutations of the F11 gene result in deficient levels of functional factor XI (factor 11). The symptoms of hemophilia C (factor XI deficiency) occur, in part, due to factor XI (factor 11) deficiency. Individuals with hemophilia C (factor XI deficiency) often have varying levels of residual factor XI (factor 11). In many disorders, the amount of residual protein activity correlates with the severity of the disease (e.g. little to no residual protein activity results in severe disease). However, in hemophilia C (factor XI deficiency) the severity of the disorder does not always correlate with the residual activity of factor XI (factor 11). For example, individuals with a severe deficiency of factor XI (factor 11) may have mild or no symptoms of the disorder, whilst individuals with a partial deficiency of factor XI (factor 11) may have more significant symptoms. This suggests that additional genetic and environmental factors play a role in the severity of hemophilia C (factor XI deficiency). This variability even exists among members of the same family.
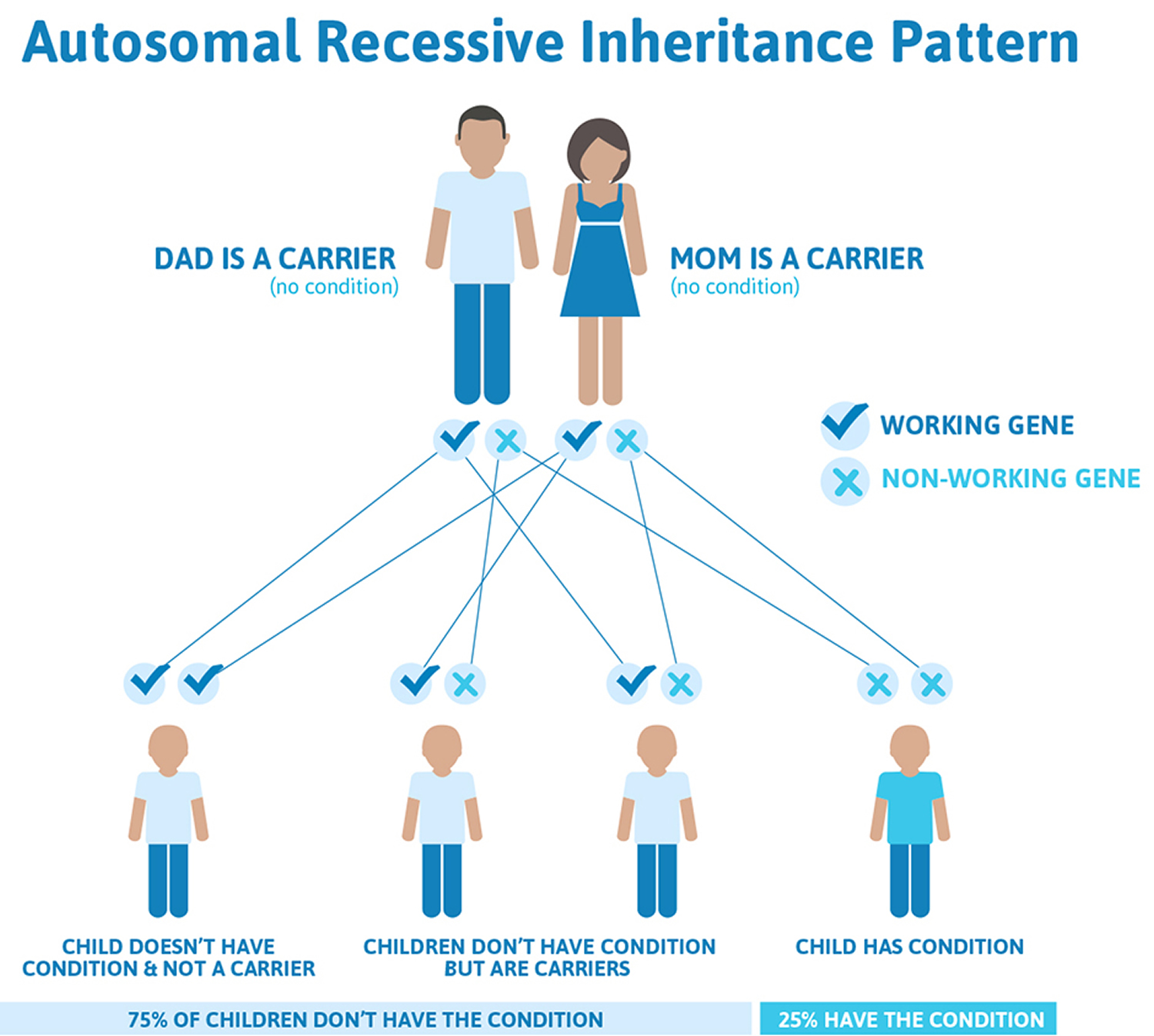
Hemophilia C (factor XI deficiency) is usually inherited in an autosomal recessive pattern. Recessive genetic disorders occur when an individual inherits a non-working gene from each parent. If an individual receives one working gene and one non-working gene for the disease, the person will be a carrier for the disease, but usually will not show symptoms. The risk for two carrier parents to both pass the non-working gene and, therefore, have an affected child is 25% with each pregnancy. The risk to have a child who is a carrier, like the parents, is 50% with each pregnancy. The chance for a child to receive working genes from both parents is 25%. Males and females are affected by hemophilia C (factor XI deficiency) equally.
Sometimes, hemophilia C (factor XI deficiency) is also inherited in an autosomal dominant pattern. Dominant genetic disorders occur when only a single copy of a non-working gene is necessary to cause a particular disease. The non-working gene can be inherited from either parent or can be the result of a mutated (changed) gene in the affected individual. The risk of passing the non-working gene from an affected parent to an offspring is 50% for each pregnancy. The risk is the same for males and females.
Some cases of hemophilia C (factor XI deficiency) are not caused by F11 gene mutations. In these cases, the condition is called acquired factor XI deficiency. It can be caused by other disorders such as conditions in which the immune system malfunctions and attacks the factor XI protein. Because factor XI is made primarily by cells in your liver, acquired factor XI deficiency can also occur as the result of severe liver disease or receiving a transplanted liver from an affected individual. In addition, approximately 25 percent of people with another disorder called Noonan syndrome have hemophilia C (factor XI deficiency). Noonan syndrome is a rare genetically inherited disorder that present from birth characterized by a wide spectrum of symptoms and physical features that vary greatly in range and severity. In many affected individuals, associated abnormalities include a distinctive facial appearance; heart problems; a broad or webbed neck; a low posterior hairline; coagulation defects; unusual chest shape with superior pectus carinatum and inferior pectus excavatum; vision and eye problems, lymphatic malformations, feeding difficulties, undescended testicles, short stature and developmental delays 12, 13. Noonan syndrome is inherited as an autosomal dominant trait. Noonan syndrome is typically inherited in an autosomal dominant pattern, with at least 8 identified gene mutations linked to the condition. The mutation in the RAS-MAPK pathway classifies Noonan syndrome as a RASopathy, with at least 8 distinct causative genetic mutations identified 14, 15. Noonan syndrome can also occur from a de novo or sporadic mutation.
Individuals with hemophilia C (factor XI deficiency) do not typically show any spontaneous bleeding or specific symptoms. Sometimes those who have hemophilia C (factor XI deficiency) are identified during special situations such as trauma or surgery. Jaw surgery is particularly associated with high risk of bleeding. Therefore, great care must be taken when treating individuals with bleeding disorders such as hemophilia C (factor XI deficiency). There are a few reports that address the management of patients with bleeding disorders during jaw surgery.
Laboratory studies for suspected hemophilia C should include the following:
- Complete blood count (CBC)
- Measurement of factor XI levels
- Measurement of factor VIII and von Willebrand factor (vWF)
- Prothrombin time (PT), activated partial thromboplastin time (aPTT), and thrombin time (TT) (usually performed before the measurement of factors).
Traditionally, the diagnosis of factor XI deficiency is based on the activated partial thromboplastin time test (aPTT), which can be prolonged. However, activated partial thromboplastin time test (aPTT) may not always correlate with the risk of bleeding, which requires the use of additional diagnostic tools 16. Major factor XI deficiency is present when the activity of factor XI (factor 11) in plasma is less than 15 IU/dL, but in contrast to hemophilia A and hemophilia B, the factor XI (factor 11) level does not reflect the severity of the bleeding risk. However, a pediatric study by Barg et al 17 suggested that in children, the severity of factor XI deficiency may be associated with such risk. Recent advances in diagnostic methodologies, particularly the use of viscoelastic testing, such as thromboelastography (TEG), rotational thromboelastometry (ROTEM), and sonorheometry (Quantra), have provided new insights into the real-time coagulation status of patients 18, 19. These tests are instrumental in evaluating the dynamic properties of clot formation and degradation, offering a more nuanced understanding of the hemostatic challenges specific to factor XI deficiency 20, 21. Viscoelastic testing has also been pivotal in guiding the administration of targeted therapies during the perioperative period, potentially reducing the risk of bleeding and thrombotic complications 22, 18.
There are several treatments available to help control bleeding in people with factor XI deficiency (factor 11 deficiency).
- Factor XI concentrate. Factor concentrates are the ideal and safest treatment for rare bleeding disorders.Factor concentrates for rare bleeding disorders are usually made from human plasma and are treated to eliminate viruses like HIV and hepatitis B and C. They are made in the laboratory and not from human plasma, so they carry no risk of infectious disease. Factor concentrates are administered intravenously.
- Antifibrinolytic drugs: The antifibrinolytic drugs tranexamic acid and aminocaproic acid are used to hold a clot in place in certain parts of the body, such as the mouth, bladder, and uterus. They are also very useful in many situations, such as during dental work, but are not effective for major internal bleeding or surgery. Antifibrinolytic drugs are particularly useful for patients with factor XI deficiency. They are also used to help control excessive menstrual bleeding. Antifibrinolytic drugs can be administered orally or by injection.
- Fibrin glue: Fibrin glue can be used to treat external wounds and during dental work, such as a tooth extraction. It is not used for major bleeding or surgery. It is applied to the bleeding site.
- Fresh frozen plasma (FFP): Plasma is the portion of blood that contains all the clotting factors, as well as other blood proteins. FFP is used to treat rare bleeding disorders when concentrates of the specific factor that is missing are not available. FFP is the usual treatment for factor V deficiency. However, it usually does not undergo viral inactivation, so the risk of transmission of infectious diseases is higher. Viral-inactivated FFP is available in some countries and is preferable. Circulatory overload is a potential problem with this treatment: since the concentration of each coagulation factor in FFP is low, a large volume of it must be given over several hours in order to achieve an adequate rise in factor level. This large amount of FFP needed can overload the circulatory system and stress the heart. Other complications of treatment with FFP can occur, particularly allergic reactions or lung problems (transfusion-related lung injury [TRALI]). These problems are much less common if viral-inactivated pooled FFP is used. FFP is administered intravenously.
Most people with hemophilia C (factor XI deficiency) never need treatment or prophylaxis for routine functions or activities 23. You are only likely to need treatment if you have an accident or to prevent bleeding before planned surgery or if you are having a baby 5, 6. For minor operations, you may have tranexamic acid tablets to take beforehand and for a few days afterwards. These help to stop the breakdown of blood clots. If you have very heavy periods, you may have tranexamic acid tablets during your periods. Or your doctor may suggest that you take the contraceptive pill to make your periods lighter.
For more serious operations you may have treatment with fresh frozen plasma (FFP), which is often used to treat rare bleeding disorders. It contains all the clotting factors, including factor XI. This is made from human blood, where the blood plasma is the straw-colored fluid that the blood cells are carried in. You have it through a drip into a vein (intravenously).
Factor XI concentrate is also available and is another possible treatment, but many doctors prefer to use fresh frozen plasma (FFP) as factor XI concentrate can cause problems with blood clots.
All blood products are now treated during manufacture to kill off any known viral infections such as hepatitis and HIV.
If you are having a baby, whether you have treatment to prevent bleeding or not will depend on the usual factor XI level in your blood. Most women will not need any treatment. If you have a low factor XI level, you may have tranexamic acid tablets to take when you go into labor. Or you may have factor XI concentrate.
You should have immunizations or other injections under your skin (subcutaneously) rather than into a muscle (intramuscularly) to reduce the risk of a painful bruise or hematoma (a localized collection of blood) developing. You should not use non-steroidal anti-inflammatory drugs (NSAIDs such as ibuprofen or aspirin) as this increases the risk of bleeding. Other methods of pain relief should be used instead. Speak to your doctor if you are unsure.
Table 1. Coagulation factors
| Coagulation factor | Other common name |
|---|---|
| I | Fibrinogen |
| II | Prothrombin |
| V | Proaccelerin or labile factor |
| VII | Proconvertin |
| VIII | Antihemophilic factor A |
| IX | Antihemophilic factor B or Christmas factor |
| X | Stuart-Prower factor |
| XI | Plasma thromboplastin antecendent |
| XIII | Fibrin stabilizing factor |
Figure 1. Overview of blood coagulation
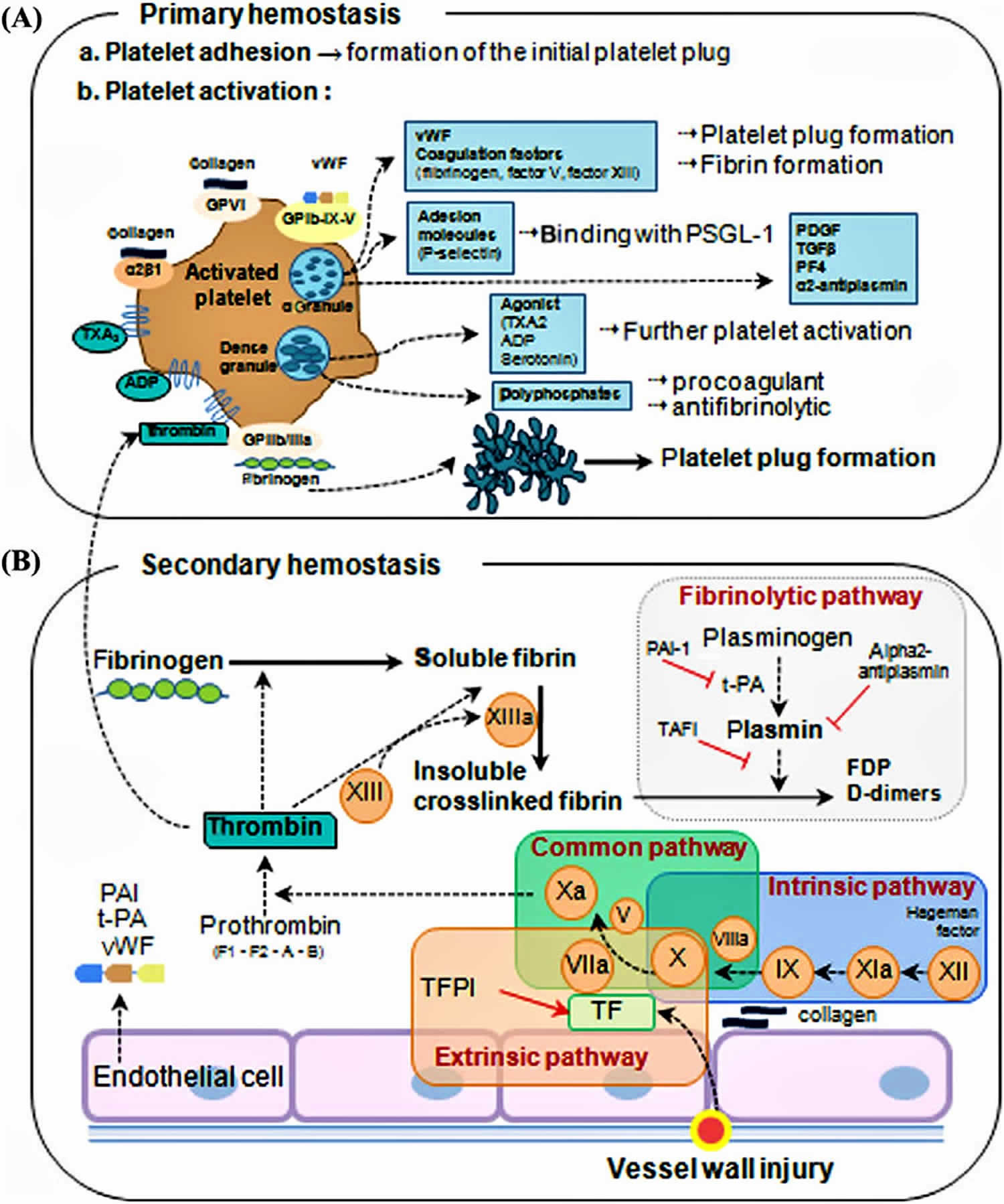
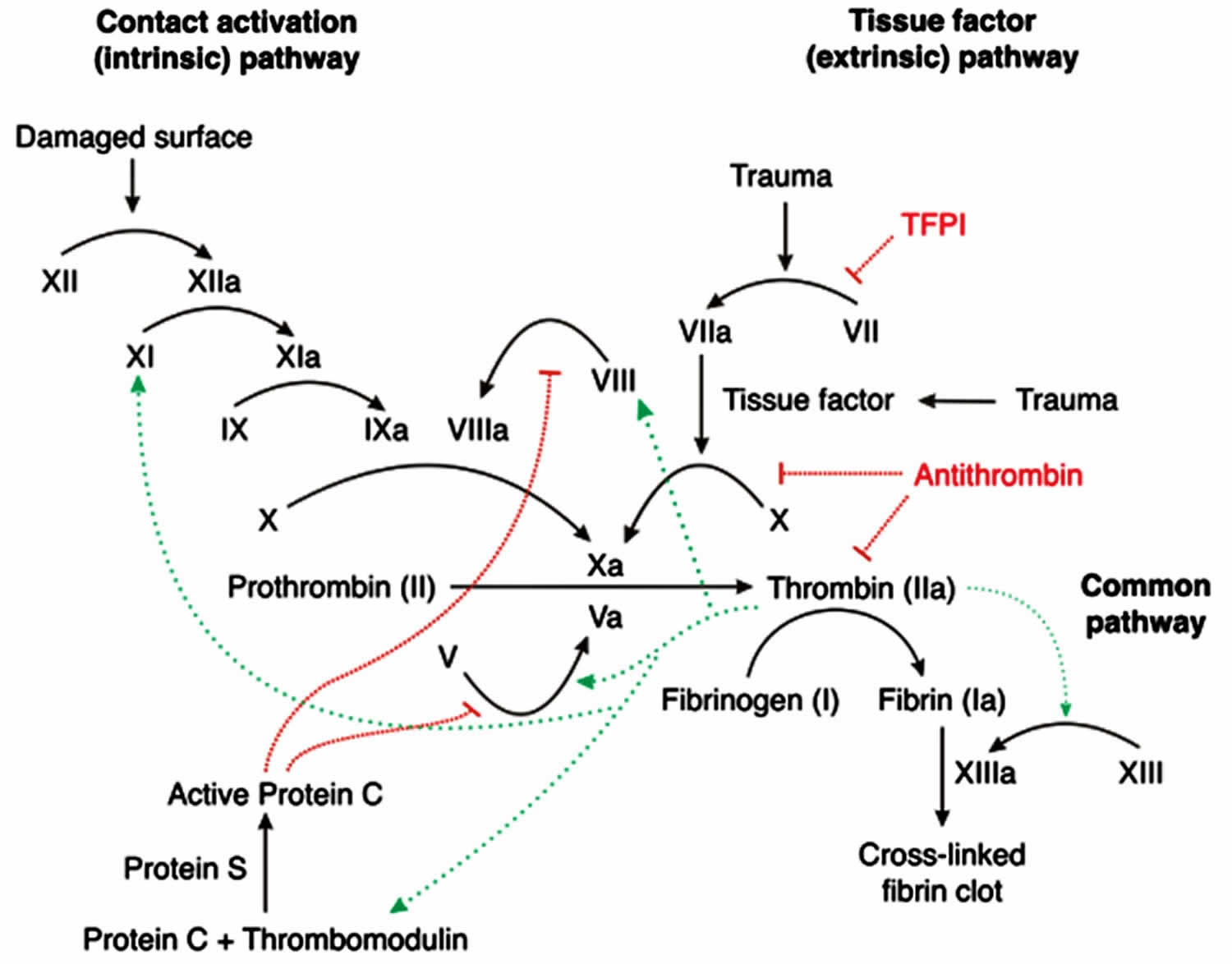
Figure 2. Coagulation cascade
Hemophilia C cause
Hemophilia C (factor XI deficiency) is caused by mutations in the F11 gene also called plasma thromboplastin antecedent (PTA) gene. The F11 gene provides instructions for making a protein called factor XI (factor 11). The factor XI (factor 11) protein plays a role in the coagulation cascade, which is a series of chemical reactions that forms blood clots in response to injury. After an injury, clots seal off blood vessels to stop bleeding and trigger blood vessel repair. Factor XI (factor 11) is made primarily by cells in your liver. The factor XI (factor 11) protein circulates in your bloodstream and is normally turned off (inactive) until the coagulation cascade is turned on (activated) by an injury that damages your blood vessels. When factor XI (factor 11) is activated, it interacts with other coagulation factors, resulting in conversion of an important coagulation protein called prothrombin to its active form, thrombin. Thrombin then converts a protein called fibrinogen into fibrin, which is the material that forms blood clots by trapping platelets and helps hold a clot in place.
Mutations of the F11 gene result in deficient levels of functional factor XI (factor 11). The symptoms of hemophilia C (factor XI deficiency) occur, in part, due to factor XI (factor 11) deficiency. Individuals with hemophilia C (factor XI deficiency) often have varying levels of residual factor XI (factor 11). In many disorders, the amount of residual protein activity correlates with the severity of the disease (e.g. little to no residual protein activity results in severe disease). However, in hemophilia C (factor XI deficiency) the severity of the disorder does not always correlate with the residual activity of factor XI (factor 11). For example, individuals with a severe deficiency of factor XI (factor 11) may have mild or no symptoms of the disorder, whilst individuals with a partial deficiency of factor XI (factor 11) may have more significant symptoms. This suggests that additional genetic and environmental factors play a role in the severity of hemophilia C (factor XI deficiency). This variability even exists among members of the same family.
Hemophilia C (factor XI deficiency) is usually inherited in an autosomal recessive pattern. Recessive genetic disorders occur when an individual inherits a non-working gene from each parent. If an individual receives one working gene and one non-working gene for the disease, the person will be a carrier for the disease, but usually will not show symptoms. The risk for two carrier parents to both pass the non-working gene and, therefore, have an affected child is 25% with each pregnancy. The risk to have a child who is a carrier, like the parents, is 50% with each pregnancy. The chance for a child to receive working genes from both parents is 25%. The risk is the same for males and females.
Sometimes, hemophilia C (factor XI deficiency) is inherited in an autosomal dominant pattern. Dominant genetic disorders occur when only a single copy of a non-working gene is necessary to cause a particular disease. The non-working gene can be inherited from either parent or can be the result of a mutated (changed) gene in the affected individual. The risk of passing the non-working gene from an affected parent to an offspring is 50% for each pregnancy. The risk is the same for males and females.
Some cases of hemophilia C (factor XI deficiency) are not caused by F11 gene mutations. In these cases, the condition is called acquired factor XI deficiency. It can be caused by other disorders such as conditions in which the immune system malfunctions and attacks the factor XI protein. Because factor XI is made primarily by cells in your liver, acquired factor XI deficiency can also occur as the result of severe liver disease or receiving a transplanted liver from an affected individual. In addition, approximately 25 percent of people with another disorder called Noonan syndrome have hemophilia C (factor XI deficiency). Noonan syndrome is a rare genetically inherited disorder that present from birth characterized by a wide spectrum of symptoms and physical features that vary greatly in range and severity. In many affected individuals, associated abnormalities include a distinctive facial appearance; heart problems; a broad or webbed neck; a low posterior hairline; coagulation defects; unusual chest shape with superior pectus carinatum and inferior pectus excavatum; vision and eye problems, lymphatic malformations, feeding difficulties, undescended testicles, short stature and developmental delays 12, 13. Noonan syndrome is inherited as an autosomal dominant trait. Noonan syndrome is typically inherited in an autosomal dominant pattern, with at least 8 identified gene mutations linked to the condition. The mutation in the RAS-MAPK pathway classifies Noonan syndrome as a RASopathy, with at least 8 distinct causative genetic mutations identified 14, 15. Noonan syndrome can also occur from a de novo or sporadic mutation.
Acquired factor XI deficiency occurs in patients who develop inhibitors to factor XI (factor 11) protein, as is sometimes observed in persons with systemic lupus erythematosus (SLE) or other immunologic diseases. However, some reagents used for factor XI (factor 11) laboratory testing are particularly sensitive to the lupus anticoagulant, and results may be falsely interpreted as an factor XI deficiency 24. Therefore, a diagnosis of factor XI deficiency must be made with caution in a patient without a family history and who is not of Jewish heritage.
Hemophilia C genetics
Hemophilia C (factor XI deficiency) is caused by mutations in the F11 gene also called plasma thromboplastin antecedent (PTA) gene. The F11 gene provides instructions for making a protein called factor XI (factor 11). The factor XI (factor 11) protein plays a role in the coagulation cascade, which is a series of chemical reactions that forms blood clots in response to injury. After an injury, clots seal off blood vessels to stop bleeding and trigger blood vessel repair. Factor XI (factor 11) is made primarily by cells in your liver. The factor XI (factor 11) protein circulates in your bloodstream and is normally turned off (inactive) until the coagulation cascade is turned on (activated) by an injury that damages your blood vessels. When factor XI (factor 11) is activated, it interacts with other coagulation factors, resulting in conversion of an important coagulation protein called prothrombin to its active form, thrombin. Thrombin then converts a protein called fibrinogen into fibrin, which is the material that forms blood clots by trapping platelets and helps hold a clot in place.
Among the Ashkenazi Jewish population, the so-called type II p.Glu117X and type III p.Phe283Leu mutations prevail 1. 1 in 450 individuals of the Ashkenazi Jewish population is expected to inherit type II homozygous, type III homozygous, or type II-III compound heterozygous factor XI deficiency 7, 25, 26.
In Israel, the mutated F11 gene frequency for Ashkenazi Jewish people is reported to be from 8% to 13.4%. One report describes 1 of 190 (0.5%) Ashkenazi Jews in Israel as being affected by severe, homozygous factor XI deficiency 27. Another estimate is that severe factor XI deficiency occurs in 1 of 450 (0.2%) Israeli Ashkenazi Jews 28. In addition, Iraqi Jewish people carry the type II (Glu117Stop) mutation at a reported frequency of 3.7%. People of Arabic background living in Israel and Jewish people of Sephardic (Spanish) background also carry the type II mutation, but at a much lower frequency. According to a report by the World Federation of Hemophilia (WFH), 17 countries have an factor XI deficiency prevalence of 0 per 1,000,000 individuals, while in the United Kingdom, the report found the prevalence to be 55.85 per 1,000,000 persons. In the United States, according to the World Federation of Hemophilia report, the prevalence is 1.44 per 1,000,000. However, many of the countries with zero prevalence in the World Federation of Hemophilia report were developing nations that probably lacked the testing and reporting resources to keep track of factor XI deficiency 29. A study estimated the prevalence of factor XI deficiency in Finnish Europeans at 0.1 per 1,000,000 individuals, while another report estimated the prevalence in French Basque Country to be 246.2 per 1,000,000 people 29. In a prospective cohort study of 112 women in the Netherlands with heavy menstrual bleeding and 28 healthy controls, Knol and colleagues found that four patients had factor XI deficiency 30.
Mutations of the F11 gene result in deficient levels of functional factor XI (factor 11). The symptoms of hemophilia C (factor XI deficiency) occur, in part, due to factor XI (factor 11) deficiency. Individuals with hemophilia C (factor XI deficiency) often have varying levels of residual factor XI (factor 11). In many disorders, the amount of residual protein activity correlates with the severity of the disease (e.g. little to no residual protein activity results in severe disease). However, in hemophilia C (factor XI deficiency) the severity of the disorder does not always correlate with the residual activity of factor XI (factor 11). For example, individuals with a severe deficiency of factor XI (factor 11) may have mild or no symptoms of the disorder, whilst individuals with a partial deficiency of factor XI (factor 11) may have more significant symptoms. This suggests that additional genetic and environmental factors play a role in the severity of hemophilia C (factor XI deficiency). This variability even exists among members of the same family.
Hemophilia C (factor XI deficiency) is usually inherited in an autosomal recessive pattern. Recessive genetic disorders occur when an individual inherits a non-working gene from each parent. If an individual receives one working gene and one non-working gene for the disease, the person will be a carrier for the disease, but usually will not show symptoms. The risk for two carrier parents to both pass the non-working gene and, therefore, have an affected child is 25% with each pregnancy. The risk to have a child who is a carrier, like the parents, is 50% with each pregnancy. The chance for a child to receive working genes from both parents is 25%. The risk is the same for males and females.
Sometimes, hemophilia C (factor XI deficiency) is inherited in an autosomal dominant pattern. Dominant genetic disorders occur when only a single copy of a non-working gene is necessary to cause a particular disease. The non-working gene can be inherited from either parent or can be the result of a mutated (changed) gene in the affected individual. The risk of passing the non-working gene from an affected parent to an offspring is 50% for each pregnancy. The risk is the same for males and females.
Some cases of hemophilia C (factor XI deficiency) are not caused by F11 gene mutations. In these cases, the condition is called acquired factor XI deficiency. It can be caused by other disorders such as conditions in which the immune system malfunctions and attacks the factor XI protein. Because factor XI is made primarily by cells in your liver, acquired factor XI deficiency can also occur as the result of severe liver disease or receiving a transplanted liver from an affected individual. In addition, approximately 25 percent of people with another disorder called Noonan syndrome have hemophilia C (factor XI deficiency). Noonan syndrome is a rare genetically inherited disorder that present from birth characterized by a wide spectrum of symptoms and physical features that vary greatly in range and severity. In many affected individuals, associated abnormalities include a distinctive facial appearance; heart problems; a broad or webbed neck; a low posterior hairline; coagulation defects; unusual chest shape with superior pectus carinatum and inferior pectus excavatum; vision and eye problems, lymphatic malformations, feeding difficulties, undescended testicles, short stature and developmental delays 12, 13. Noonan syndrome is inherited as an autosomal dominant trait. Noonan syndrome is typically inherited in an autosomal dominant pattern, with at least 8 identified gene mutations linked to the condition. The mutation in the RAS-MAPK pathway classifies Noonan syndrome as a RASopathy, with at least 8 distinct causative genetic mutations identified 14, 15. Noonan syndrome can also occur from a de novo or sporadic mutation.
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://abgc.learningbuilder.com/Search/Public/MemberRole/Verification) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (https://clinics.acmg.net/) has a searchable database of medical genetics clinic services in the United States.
Figure 3. Hemophilia C autosomal recessive inheritance pattern
Hemophilia C signs and symptoms
Most people with factor XI deficiency (factor 11 deficiency) will have little or no symptoms at all 9, 31. The relationship between the amount of factor XI deficiency (factor 11 deficiency) levels in the blood do not necessarily correlate with bleeding symptoms 9, 31. The relationship between the amount of factor XI deficiency (factor 11 deficiency) levels in a person’s blood and the severity of his/her symptoms is unclear; people with only a mild deficiency in factor XI can have serious bleeding episodes than those with higher factor XI deficiency. Symptoms of factor XI deficiency vary widely, even among family members, which can make it difficult to diagnose.
Some hemophilia C (factor XI deficiency) patients experience frequent nosebleeds or soft tissue bleeds, others first experience severe bleeding only after tooth extraction, such as for wisdom teeth, surgery or trauma. Furthermore, bleeding may be delayed after these procedures. Women may not know they have hemophilia C (factor XI deficiency) until they experience heavy menstrual periods or menorrhagia, and bleeding after childbirth (postpartum bleeding). Unlike other common hemophilia A or B, Joint and muscle bleeds are uncommon.
Hemophilia C common symptoms:
- nosebleeds (epistaxis)
- easy bruising
- heavy or prolonged menstrual bleeding (menorrhagia)
- abnormal bleeding during or after surgery, injury, or childbirth
Other reported symptoms in people with hemophilia C:
- bleeding in the gut (gastrointestinal hemorrhage)
- bleeding in the mouth, particularly after dental surgery or tooth extraction
- blood in the urine (hematuria).
Figure 4. Factor XI deficiency variation in signs and symptoms accoring to age and sex
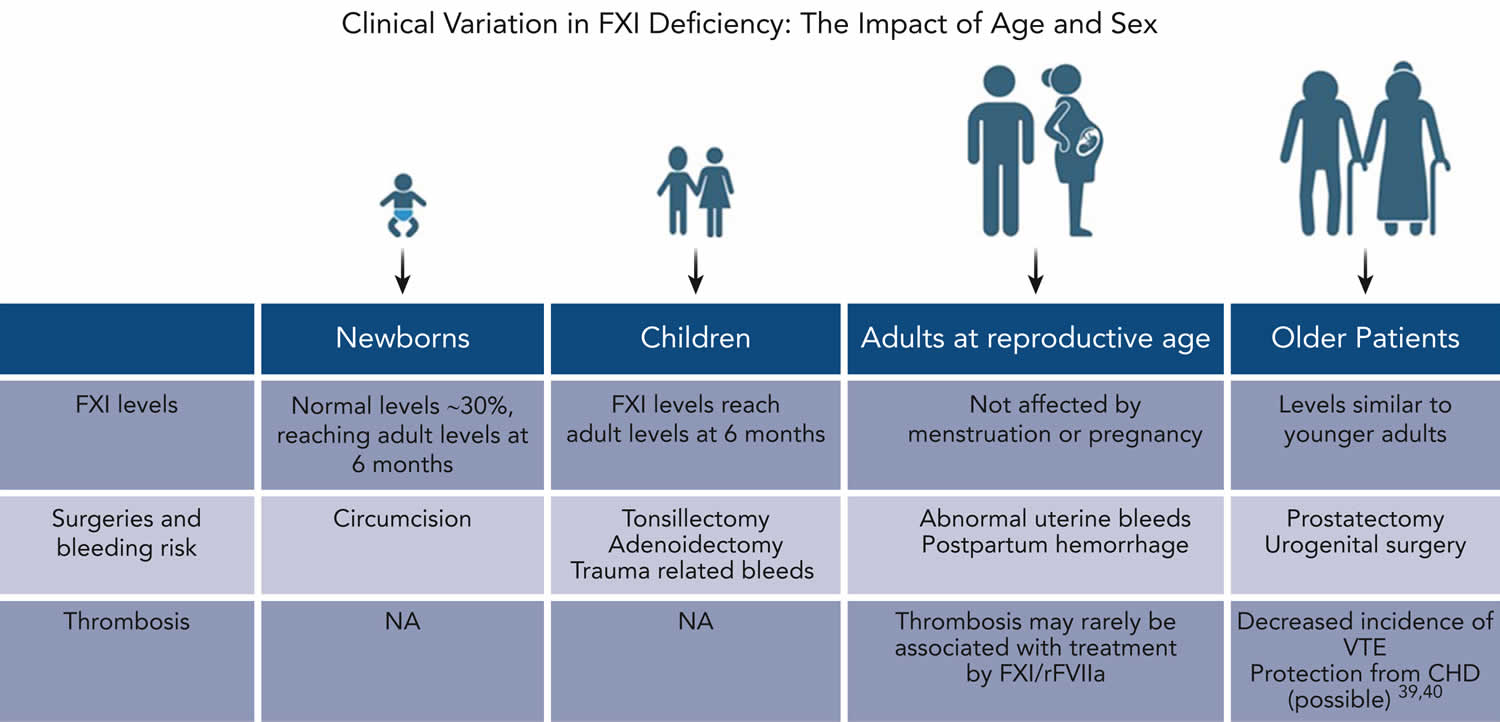
Footnotes: Clinical variation in factor XI deficiency: the impact of age and sex. This figure presents important age-related considerations that are relevant to the clinical assessment of patients with factor XI deficiency throughout the lifespan.
[Source 1 ]Hemophilia C in pregnancy
Abnormal uterine bleeding is common in women with factor XI deficiency 32. In a systematic review of 10 studies regarding gynecological bleeding in factor XI deficiency, a total of 268 women were included. Among these, in 56 (21%) women with factor XI deficiency, abnormal uterine bleeding prevailed 33. These studies included female patients with severe as well as partial factor XI deficiency.
Extensive hemorrhage may be detected after spontaneous abortions 34, 35. In general, antepartum hemorrhage is scarce among women with factor XI deficiency. However, factor XI levels do not significantly increase during pregnancy, therefore, pregnant women with factor XI deficiency may be prone to postpartum hemorrhage 36. The Rare Bleeding Disorders in the Netherlands concluded that postpartum hemorrhage was especially common in patients with factor XI deficiency (70%) 31. However, the study included only 10 women with factor XI deficiency who gave birth. Notably, this report did not delineate the mode of delivery and the severity of factor XI deficiency 31.
Several large cohort studies addressed the variability in the tendency of pregnant women who are factor XI deficient to bleed and their responses to prophylactic and therapeutic treatment. Chi et al 37 examined 61 pregnancies in 30 women (2 of 30 had severe factor XI deficiency, and 28 of 30 with partial deficiency) and observed 7 episodes of postpartum hemorrhage; notably, 2 of 7 postpartum hemorrhage cases occurred in pregnancies of women with severe factor XI deficiency who did not receive intrapartum hemostatic prophylaxis. Salomon et al 38 studied 164 pregnancies in 62 women with severe factor XI deficiency and observed no significant bleeding in women without a bleeding history. Myers et al 35 studied 105 pregnancies in 33 women (3 of 33 with severe deficiency, all of whom did not experience any postpartum hemorrhage) and observed 9 episodes of postpartum hemorrhage, mostly in females with a previously positive bleeding history. In an Italian retrospective study, the authors mention 10.5% of postpartum hemorrhage (in 4 women, of whom 2 had severe factor XI deficiency) among 31 deliveries and 8 cesarean sections performed without prophylaxis in women with factor XI deficiency 39. A US study 6 that reviewed data from 28 women with factor XI deficiency (8 of 28 with severe factor XI deficiency) disclosed 17% of postpartum hemorrhage (5 cases in women with partial factor XI deficiency, and 4 in women with severe deficiency). In another recent study there was no postpartum hemorrhage among 45 vaginal deliveries in women with partial factor XI deficiency 40. Recently, Handa et al 41 reported 198 patients with factor XI deficiency who underwent 252 procedures in the United States, including 143 vaginal deliveries and 63 cesarean section procedures. Personal history of bleeding was the strongest predictor of perioperative or obstetric bleeding, whereas FXI level of >40 U/dL predicted a lower bleeding risk. Overall, data regarding postpartum hemorrhage in women with factor XI deficiency are conflicting, and bleeding tendency seems to be associated with a positive bleeding history rather than with factor XI levels 1.
Newborns and infants with factor XI deficiency
Although factor XI levels in newborns are significantly lower than in older infants and adults, major bleeding complications are exceedingly rare, even among the youngest pediatric population with factor XI deficiency 42. One of the most feared complications in bleeding disorder, especially during the neonatal period and infancy, is intracranial hemorrhage 43. A comprehensive retrospective study conducted in the United States examined the incidence and characteristics of intracranial hemorrhage in 3717 children aged <4 years with various inherited bleeding disorders and its correlation with head trauma 44. Sixteen children with factor XI deficiency were included in this study. Although 255 (6.9%) of the study participants experienced any form of intracranial hemorrhage, 206 (5.5%) had nontraumatic intracranial hemorrhage, and none of the patients with factor XI deficiency experienced any intracranial hemorrhage. A recent pediatric cohort involving 60 children with severe factor XI deficiency reported no spontaneous bleeds or perinatal intracranial hemorrhage. However, 3 children (5%) did experience triggered intracranial hemorrhage after trauma (n = 2) or bleeding from arteriovenous malformation 45.
Among infants, especially of Jewish and Muslim origin, the first and most common surgery is circumcision 46. Barg et al 47 reported the presentation of severe factor XI deficiency as postcircumcision bleeding in 4 of 21 babies (whose factor XI level was <1%-15%) that were not previously diagnosed, whereas in 17 cases that were already identified and pretreated with tranexamic acid, no bleeding occurred. Tonsillectomies and adenoidectomies are commonly performed surgeries in children. Bleeding complications among children with bleeding disorders undergoing these operations have been reported ∼50% of procedures 48. To date, no publications have specifically addressed tonsillectomies in children with factor XI deficiency. Among 28 surgeries performed in children who were severely factor XI-deficient children, 2 were complicated by major bleeds (postcircumcision bleeding and teeth extraction). In both cases, no perioperative hemostatic treatment was applied because they were not diagnosed before the operation; in contrast, no bleeding occurred when 45 procedures were performed in previously diagnosed (and pretreated) children with factor XI deficiency. Unlike previous studies in adults, this latter pediatric study suggested an association between the severity of factor XI deficiency and the bleeding tendency among children 47.
Older patients with factor XI deficiency
Older males form a unique group that presents various challenges related to bleeding and to thrombosis as well. One of the commonest surgeries among older men is prostatectomy. There have been reports of excessive bleeding, including major bleeding events requiring prolonged hospitalization, among patients with factor XI deficiency undergoing open prostatectomy 49. However, a study that reported patients factor XI deficiency treated with replacement therapy before planned prostatectomy revealed that excessive bleeding may be prevented once a minimum level of 30% factor XI activity was maintained 50. Tranexamic acid is a viable therapeutic option in prostatic surgery as increased fibrinolytic activity is involved at this site. However, caution is warranted in case of gross hematuria.
Among the general older population, the risk of thrombosis increases 51. Consequently, antithrombotic therapy is commonly used. A recent single-center retrospective study of 269 patients with factor XI deficiency identified 15 individuals, mostly with partial factor XI deficiency, whose median age was 70 years, requiring anticoagulation, because of atrial fibrillation 52. Over >1000 months of anticoagulation, only 2 mild bleeding episodes occurred in 2 patients. The authors concluded that moderate factor XI deficiency does not interfere with anticoagulant management or bleeding risk 52.
Thrombotic events have been reported in patients after the use of factor XI concentrates, as well as after high doses of recombinant activated FVII (rFVIIa) therapy 53, 54, 55. High levels of factor XI are a risk factor for deep venous thrombosis (DVT) as well as for venous thrombotic recurrence 56, 57.
Several publications examined the potential protective role of factor XI deficiency in late adulthood by mitigating the heightened thrombotic risk associated with age. A large population study demonstrated that deep venous thrombosis is exceedingly rare among individuals with severe factor XI deficiency 58. Nonetheless, rates of myocardial infarction (heart attack) among patients with severe factor XI deficiency were similar to those observed in the general population 59. Another large retrospective population study suggested that mild and moderate factor XI deficiency confer some protection against cardiovascular events, including heart attack, stroke, and transient ischemic attacks 28. Trials on factor XI-directed inhibitors aimed at prevention of venous thromboembolism or stroke are based on such observations and show promising results 60.
Hemophilia C diagnosis
Hemophilia C or factor XI deficiency (factor 11 deficiency) is diagnosed by a variety of blood tests that should be performed by a specialist at a hemophilia/bleeding disorders treatment center. Hemophilia C diagnosis is made through a bleeding time test, platelet function tests, and prothrombin time (PT) and activated partial thromboplastin time (aPTT) tests. A factor XI (factor 11) assay helps confirm the diagnosis.
Laboratory studies for suspected hemophilia C should include the following:
- Complete blood count (CBC)
- Measurement of factor XI levels
- Measurement of factor VIII and von Willebrand factor (vWF)
- Prothrombin time (PT), activated partial thromboplastin time (aPTT), and thrombin time (TT) (usually performed before the measurement of factors).
In patients with severe factor XI deficiency, the activated partial thromboplastin time (aPTT) value will be more than two standard deviations above the normal mean; in heterozygotes, the activated partial thromboplastin time (aPTT) may be slightly prolonged or within the normal range 5.
An factor XI assay may help to confirm the diagnosis, although levels can be in the normal range 5. Homozygotes and compound heterozygotes will have an factor XI level of less than 15%. The expected factor XI level in heterozygotes is 25-70%.
A mixing study using normal pooled plasma may help to identify a factor deficiency. If the sample is incubated and, subsequently, the activated partial thromboplastin time (aPTT) is prolonged, then the presence of an inhibitor needs to be considered. Based on the data regarding high risk of inhibitor development in patients who have severe (< 1%) factor XI deficiency, checking an inhibitor titer before proceeding with surgery is recommended.
Factor assays for the intrinsic coagulation system should be performed with at least three dilutions.
In a patient who is newly diagnosed and without a previous bleeding history or family history (neither is uncommon in a patient with factor XI deficiency), care must be taken by the coagulation laboratory to separate out a nonspecific inhibitor or lupus anticoagulant versus a true factor XI deficiency.
Researchers are exploring the use of novel laboratory tests, such as thrombin generation assays and clot stability assays, to predict bleeding risk in patients with factor XI deficiency 4. For example, Gidley et al 61 reported that combining the aPTT with the rate of clot formation and area under the curve in fibrinolysis assays identifies most factor XI-deficient patients with a bleeding tendency.
Recent advances in diagnostic methodologies, particularly the use of viscoelastic testing, such as thromboelastography (TEG), rotational thromboelastometry (ROTEM), and sonorheometry (Quantra), have provided new insights into the real-time coagulation status of patients 18. These tests are instrumental in evaluating the dynamic properties of clot formation and degradation, offering a more nuanced understanding of the hemostatic challenges specific to factor XI deficiency 20, 21.. Viscoelastic testing has also been pivotal in guiding the administration of targeted therapies during the perioperative period, potentially reducing the risk of bleeding and thrombotic complications 22, 18.
Genetic testing is not required for the diagnosis of factor XI deficiency. In addition, given that genetic identity is not consistently related to phenotypic disease, genetic screening for factor XI is not routinely performed in the general population because this would be an unnecessary expense. Therefore, it may be only beneficial in selected cases, such as individuals of Ashkenazi Jewish descent, because if the patient is injured or undergoes surgery, it would allow the patient to consider prophylactic therapies as necessary. However, screening all Ashkenazi Jewish women with genetic testing might prove impossible with little yield and false confidence in the results, so we also should review the patient’s medical history focus on any previous bleeding episodes, and perform it in each case after consulting a geneticist.
Figure 5. Hemophilia C diagnostic algorithm
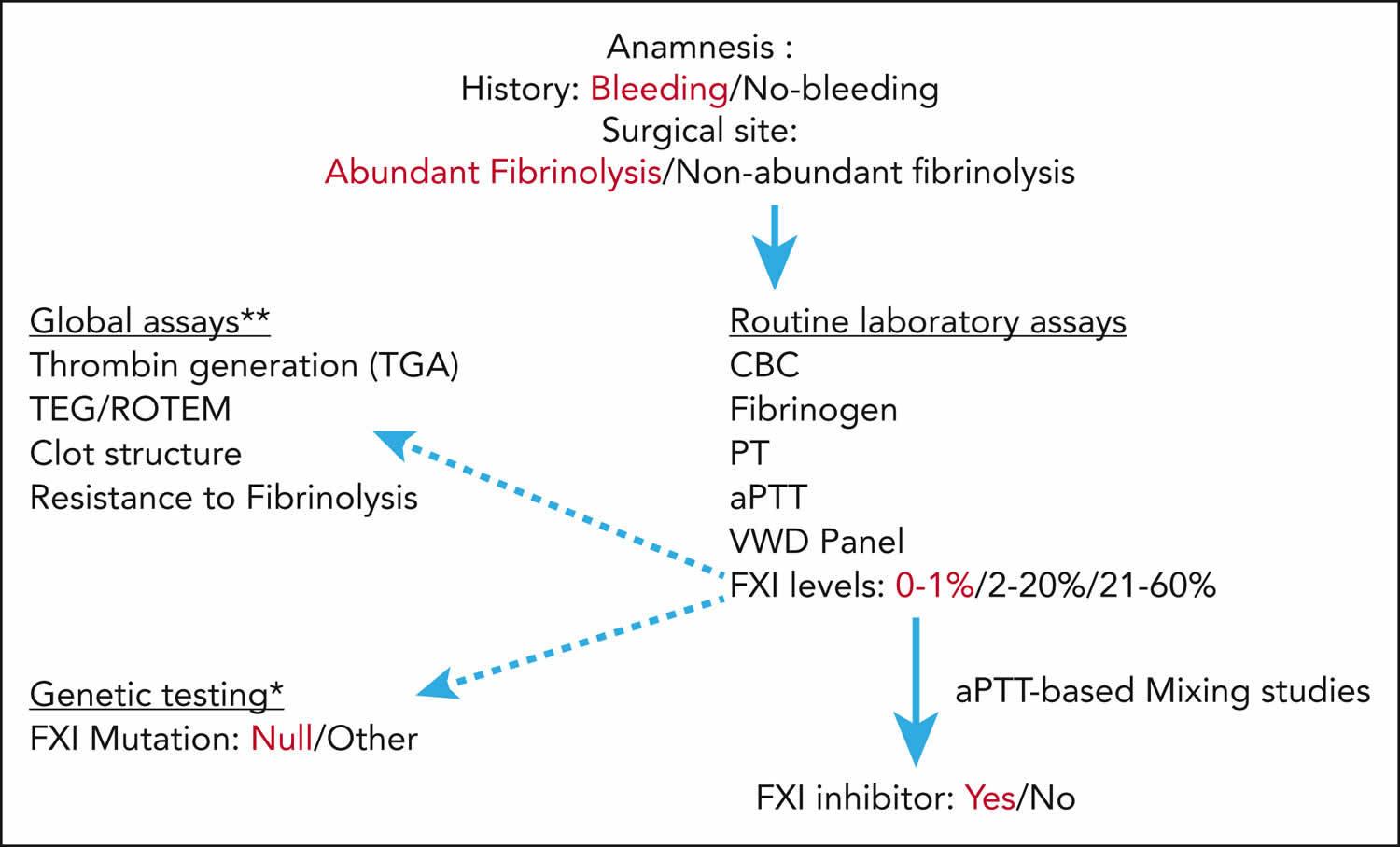
Footnotes: Assessment of bleeding risk in patient with factor XI deficiency. This figure represents our suggested stepwise approach for assessing bleeding risk for patients with factor XI deficiency before planned surgical interventions. Emphasized words demonstrate the “danger signals” increasing the risk of bleeding. Patient history and surgical site should always be considered. Sites with increased fibrinolytic activity include the genitourinary tract, and the nasal and oral cavities. The dark arrows represent the mandatory laboratory tests that should include bleeding evaluation (complete blood count (CBC), fibrinogen, prothrombin time (PT), activated partial thromboplastin time (aPTT), von Willebrand disease panel, factor XI levels, and presence of factor XI inhibitor, if relevant), whereas the dotted arrows provide additional information that may be helpful (eg, factor XI mutations∗, global assays∗∗ including assessment of clot formation/fibrinolysis).
∗ In general, because factor XI genotype and levels do not necessarily predict bleeding risk, it is not cost-effective to routinely perform factor XI molecular genetic studies. However, among patients with the severest factor XI deficiency (factor XI < 1%), we suggest confirming the presence of null mutations, because these patients may be at risk of inhibitor development.
∗∗ Global assays may be considered for additional hemostatic assessment only in patients with severe factor XI deficiency (factor XI 0%-20%).
Abbreviations: CBC = complete blood count; PT = prothrombin time; ROTEM = rotational thromboelastometry; TEG = thromboelastography; VWD panel = von Willebrand antigen and activity (cofactor ristocetin) as well as platelet aggregation.
[Source 1 ]Factor XI Inhibitors
Development of factor XI inhibitors occurs at a rate of up to 33% in patients with severe (<1%) factor XI deficiency after exposure to exogenous factor XI (factor 11), usually via plasma products 62. This complication needs to be evaluated before a planned invasive procedure.
Alpha-1 antitrypsin is a prominent inhibitor of factor XI. C1 esterase inhibitor, antithrombin III, and alpha-2 antiplasmin also cause inhibition 62.
An important complication of severe factor XI deficiency, whose frequency may increase with age and with exposure to fresh frozen plasma (FFP) or factor XI therapy, is the occurrence of factor XI inhibitors. Non-correction of a prolonged activated partial thromboplastin time (aPTT) by normal plasma should arouse suspicion of the presence of an inhibitor. Patients with severe factor XI deficiency are at a considerable risk of developing inhibitors to factor XI at any age. Inhibitors against factor XI have been described in patients with autoimmune and cancers 63, 64, 65 or as a rare complication of replacement therapy with fresh frozen plasma (FFP) in patients with severe factor XI deficiency 66, 67, 68. It was suggested that patients with factor XI deficiency with null mutations are the most susceptible to inhibitor formation after exposure to fresh frozen plasma (FFP), or to plasma-derived components 66, 67, 68. In an Israeli cohort study, 7 of 21 patients (33%) homozygous for type II mutation developed inhibitors after exposure to plasma 68. The appearance of inhibitors was not associated with any specific subtype of HLA class II. Characterization of the inhibitors revealed that they were all immunoglobulin G and inhibited activation of factor XI by thrombin or factor XIIa, and inhibited activation of FIX by factor XIa. The presence of an inhibitor during pregnancy may pose a potential hemorrhagic risk to the fetus, because anti-factor XI immunoglobulin G may cross the placenta 69.
Patients with severe factor XI deficiency who develop inhibitors directed against factor XI do not experience any spontaneous bleeds. This observation is in line with the promising results of early trials on factor XI-directed inhibitors aimed at prevention of venous thromboembolism or stroke 70, 71. Bleeding complications in patients treated by these novel anticoagulants were scarce.
Hemophilia C Differential Diagnoses
Hemophilia C differential diagnosis include:
- Hemophilia A (Factor VIII Deficiency)
- Hemophilia B (Factor IX Deficiency)
- von Willebrand disease (von Willebrand Factor Deficiency)
The differential diagnosis in patients with isolated prolongation of the activated partial thromboplastin time (aPTT) includes the following:
- Factor VIII deficiency
- Factor IX deficiency
- Factor XI deficiency
- Factor XII deficiency
- Lupus anticoagulant
- Heparin contamination
The differential diagnosis in patients with a mild or intermittent bleeding disorder includes the following:
- Von Willebrand disease
- Factor VIII deficiency/carrier state
- Factor IX deficiency/carrier state
- Factor VII deficiency (mild)
- Platelet function disorder
- Early liver dysfunction
Hemophilia C treatment
Most people with hemophilia C (factor XI deficiency) never need treatment or prophylaxis for routine functions or activities 23. You are only likely to need treatment if you have an accident or to prevent bleeding before planned surgery, dental extractions or if you are having a baby 5, 6. For minor operations, you may have tranexamic acid tablets to take beforehand and for a few days afterwards. These help to stop the breakdown of blood clots. If you have very heavy periods, you may have tranexamic acid tablets during your periods. Or your doctor may suggest that you take the contraceptive pill to make your periods lighter.
For more serious operations you may have treatment with fresh frozen plasma (FFP), which is often used to treat rare bleeding disorders. It contains all the clotting factors, including factor XI. This is made from human blood, where the blood plasma is the straw-colored fluid that the blood cells are carried in. You have it through a drip into a vein (intravenously).
Factor XI concentrate is also available and is another possible treatment, but many doctors prefer to use fresh frozen plasma (FFP) as factor XI concentrate can cause problems with blood clots.
All blood products are now treated during manufacture to kill off any known viral infections such as hepatitis and HIV.
If you are having a baby, whether you have treatment to prevent bleeding or not will depend on the usual factor XI level in your blood. Most women will not need any treatment. If you have a low factor XI level, you may have tranexamic acid tablets to take when you go into labor. Or you may have factor XI concentrate.
There are several treatments available to help control bleeding in people with factor XI deficiency (factor 11 deficiency).
- Factor XI concentrate. Factor concentrates are the ideal and safest treatment for rare bleeding disorders.Factor concentrates for rare bleeding disorders are usually made from human plasma and are treated to eliminate viruses like HIV and hepatitis B and C. They are made in the laboratory and not from human plasma, so they carry no risk of infectious disease. Factor concentrates are administered intravenously.
- Antifibrinolytic drugs: The antifibrinolytic drugs tranexamic acid and aminocaproic acid are used to hold a clot in place in certain parts of the body, such as the mouth, bladder, and uterus. They are also very useful in many situations, such as during dental work, but are not effective for major internal bleeding or surgery. Antifibrinolytic drugs are particularly useful for patients with factor XI deficiency. They are also used to help control excessive menstrual bleeding. Antifibrinolytic drugs can be administered orally or by injection.
- Fibrin glue: Fibrin glue can be used to treat external wounds and during dental work, such as a tooth extraction. It is not used for major bleeding or surgery. It is applied to the bleeding site.
- Fresh frozen plasma (FFP): Plasma is the portion of blood that contains all the clotting factors, as well as other blood proteins. FFP is used to treat rare bleeding disorders when concentrates of the specific factor that is missing are not available. FFP is the usual treatment for factor V deficiency. However, it usually does not undergo viral inactivation, so the risk of transmission of infectious diseases is higher. Viral-inactivated FFP is available in some countries and is preferable. Circulatory overload is a potential problem with this treatment: since the concentration of each coagulation factor in FFP is low, a large volume of it must be given over several hours in order to achieve an adequate rise in factor level. This large amount of FFP needed can overload the circulatory system and stress the heart. Other complications of treatment with FFP can occur, particularly allergic reactions or lung problems (transfusion-related lung injury [TRALI]). These problems are much less common if viral-inactivated pooled FFP is used. FFP is administered intravenously.
In the US there are no factor XI concentrate available 72. However, there are two factor XI concentrates manufactured in Europe, one by Bioproducts Laboratories (BPL) in the UK, the other by LFB in France, but only for limited patient use.
Fresh frozen plasma (FFP) is normally used for treatment. However, because factor XI (factor 11) is not concentrated in fresh frozen plasma (FFP), large amounts of it may be needed. This can lead to blood clots, so fresh frozen plasma (FFP) must be administered carefully.
Fibrin glue works well to maintain clots after mouth bleeds. When combined with fresh frozen plasma (FFP), it arrests bleeding after circumcision and hernia repair. Antifibrinolytics, such as aminocaproic acid, help control nosebleeds and bleeding after tooth extraction.
Excessive menstrual bleeding in women with factor XI deficiency may be controlled with hormonal contraceptives (birth control pills), intra-uterine device (IUDs), or antifibrinolytic drugs.
Table 2. Periprocedural prophylactic therapeutic options in minor and major surgeries of patients with factor XI deficiency
| Therapy | Type of intervention | |
|---|---|---|
| Minor surgery | Major surgery | |
| Antifibrinolytic agents | 3-5 days: | 7-10 days: |
| Tranexamic acid | 10-20 mg/kg per dose, 3 times daily (up to maximal dose of 1300 mg, 3 times a day) | 10-20 mg/kg per dose, 3 times daily |
| Aminocaproic acid | 50-100 mg/kg, 4 times a day (up to maximal dose of 5 g) | 50-100 mg/kg, 4 times a day (up to 5 g) |
| Fresh frozen plasma (FFP) | No | 15-20 mL/kg, repeat every 24-48 hours, as required |
| Plasma derived-factor XI concentrate (Hemoleven, Laboratoire français du Fractionnement et des Biotechnologies) (British Physical Laboratories factor XI) | No | 10-20 U/kg per dose, repeat every 48-72 hours, as needed |
| Recombinant activated factor VII (rFVIIa) NovoSeven (off-label use) | No | 10-15 μg/kg per dose, a single dose is usually sufficient |
Footnotes: In sites of increased fibrinolysis, a longer duration of antifibrinolytic treatment is suggested, even in cases of minor surgery. Caution is advised in coadministration of antifibrinolytic agents with factor XI concentrates because of potential thrombotic complications.
[Source 1 ]Fresh frozen plasma
Fresh frozen plasma (FFP) has been the most available source of factor XI. The recovery of factor XI function from plasma is excellent, and the half-life is 40-80 hours. Factor XI concentrates are produced in the United Kingdom and France but are not available in the United States 5.
Invasive surgical procedures often require replacement therapy with fresh frozen plasma (FFP). This should be continued for 7 to 14 days after surgery. Bear in mind that the half-life of factor XI is approximately 52 hours (2 days).
However, before starting fresh frozen plasma (FFP) the surgeon needs to consider the risks, which include volume overload, transmission of infectious agents, thrombosis, allergic reactions, and development of inhibitors to factor XI. In addition, even patients with severe factor XI deficiency do not inevitably bleed with surgery, and bleeding most often occurs with procedures involving tissues that exhibit fibrinolytic activity. Consequently, Salomon et al 73 suggested that surgeons may consider forgoing replacement therapy in procedures involving sites that do not have local fibrinolytic activity.
Immunization with hepatitis A virus and hepatitis B virus vaccines is recommended prior to planned surgery and plasma product replacement. Consultation with a hematologist is recommended.
Antifibrinolytic drugs
Tranexamic acid, an antifibrinolytic drug, is a prophylactic surgical option for most minor surgeries in patients with factor XI deficiency, with no need for replacement therapy. In addition, some major surgeries, such as appendectomy, can be performed using tranexamic acid, without upfront replacement therapy, although such therapy should be readily available if needed 74.
The use of desmopressin (DDAVP), a vasopressin analogue used for patients with factor VIII deficiency, von Willebrand disease, and platelet function abnormalities, has been studied in a limited number of patients with factor XI deficiency. A systematic review found an apparently statistically significant rise in factor XI levels 1 hour after desmopressin was infused in 16 patients. Another review found that in three patients with both factor XI deficiency and von Willebrand disease, factor XI levels did not improve with desmopressin therapy. Still another study reported that in two patients with heterozygous factor XI deficiency and a prior history of bleeding, a mild increase in factor XI levels was achieved with desmopressin administered prior to carpal tunnel surgery and dental extraction 75.
Salomon et al 76 reported success with a single, very low dose of recombinant factor VIIa (rFVIIa) along with tranexamic acid as prophylactic treatment for factor XI-deficient patients undergoing surgery. In their study of 12 procedures in 10 patients, recombinant factor VIIa (rFVIIa) was given in a single dose of 10-15 μg/kg at the end of surgery; tranexamic acid, 4 g/day, was started 2 hours before surgery and continued for 3 to 5 days 76.
Minami et al 77 reported that emicizumab potentiates coagulation function in factor XI-deficient plasma. These authors suggest that this agent may provide possibilities for clinical application in patients with factor XI deficiency. Emicizumab, the bispecific antibody to factors IX/IXa and X/Xa, is currently approved for routine prophylaxis in patients with hemophilia A.
Treatment of patients with acquired antibodies to factor XI has not been standardized because of the rarity of this occurrence.
Unless needed for another medical indication, aspirin products should be avoided by patients with factor XI deficiency.
Treatment in pregnancy
Pregnant women will need fresh frozen plasma (FFP) if cesarean delivery is planned. In a case-control study of 40 women with mild factor XI deficiency, the rate of postpartum hemorrhage was significantly higher among the 26 cesarean deliveries compared with 75 control cesarean deliveries in women with no known bleeding disorder 40. There were no episodes of postpartum hemorrhage among the vaginal deliveries in women with factor XI deficiency 40.
Without treatment, however, excessive bleeding will occur at the time of delivery in about 20% of pregnant persons with severe factor XI deficiency 32.
Peripartum treatment of women with factor XI deficiency is controversial. One group treats patients to maintain factor XI levels above 50% during labor and then continues treatment for 3-4 days after vaginal delivery and 7 days after cesarean delivery. This is recommended because of the high incidence of postpartum hemorrhage. The recommendation to treat expectantly must be understood in the context of the known variability of bleeding manifestations based on patient history and factor XI level, as well as the unpredictable risk of exposure to blood-borne pathogens with the use of FFP.
Thrombosis
Although factor XI deficiency offers some antithrombotic protection, patients may still develop indications for anticoagulation therapy, such as atrial fibrillation. A retrospective review by Bravo-Perez et al 52 of 15 patients with mild-to-moderate factor XI deficiency who required anticoagulation therapy principally for atrial fibrillation recorded two mild bleeding episodes in two patients, and no major or fatal events. Although four of the patients were on direct oral anticoagulants by the end of the follow-up period, 14 started therapy with vitamin K antagonists (VKA), and vitamin K antagonists dosing and management were standard in those cases, unaltered by factor XI deficiency 52.
Thrombotic events are a risk in some patients who receive factor XI concentrates, particularly those with preexisting risk factors such as older age, known peripheral or central vascular disease, or morbid obesity. A study by Batty et al 78 reported thrombotic events such as transient ischemic attack (TIA) and pulmonary emboli (PE) in two of 29 patients with factor XI deficiency who were treated with plasma-derived factor XI concentrate. However, the investigators concluded that the concentrate was safe and effective in most cases of the disorder.
Long-Term Monitoring
Annual visits to a hemophilia treatment center are recommended to provide the following care:
- Monitoring of bleeding episodes
- Planning for any elective surgical procedures
- Monitoring for the development of hepatitis
- Administering preventive immunizations as needed.
Worldwide hemophilia treatment centers, see World Federation of Haemophilia 79 at https://wfh.org/find-local-support/#HTCs
US only, see National Bleeding Disorders Foundation 80 at https://www.bleeding.org/
Hemophilia C prognosis
The prognosis for hemophilia C (factor XI deficiency) is generally good, with most people living a normal life span and often requiring no treatment unless they need to undergo surgery or have an injury 23. Bleeding is typically less severe than in hemophilia A or hemophilia B and usually involves mucosal tissues, such as the nose and mouth, rather than joint or muscle bleeding. However, bleeding severity can be unpredictable, and individuals may experience heavy menstrual periods or postpartum bleeding. You should have immunizations or other injections under your skin (subcutaneously) rather than into a muscle (intramuscularly) to reduce the risk of a painful bruise or hematoma (a localized collection of blood) developing. You should not use non-steroidal anti-inflammatory drugs (NSAIDs such as ibuprofen or aspirin) as this increases the risk of bleeding. Other methods of pain relief should be used instead. Speak to your doctor if you are unsure.
- Barg AA, Livnat T, Kenet G. Factor XI deficiency: phenotypic age-related considerations and clinical approach towards bleeding risk assessment. Blood. 2024 Apr 11;143(15):1455-1464. https://doi.org/10.1182/blood.2023020721[↩][↩][↩][↩][↩][↩]
- Sucker C, Geisen C, Litmathe J, Zawislak B. Two cases of Factor XI deficiency: Use of Thrombin Generation Assays (TGA) to detect a non-bleeding phenotype. Arch Clin Cases. 2024 Apr 17;11(1):1-4. doi: 10.22551/2024.42.1101.10277[↩]
- Lewandowska M.D., Connors J.M. Factor XI Deficiency. Hematol. Oncol. Clin. North Am. déc 2021;35(6):1157–1169. doi: 10.1016/j.hoc.2021.07.012[↩]
- Wheeler AP, Gailani D. Why factor XI deficiency is a clinical concern. Expert Rev Hematol. 2016 Jul;9(7):629-37. doi: 10.1080/17474086.2016.1191944[↩][↩]
- Seligsohn U. Factor XI deficiency in humans. J Thromb Haemost. 2009 Jul;7 Suppl 1:84-7. doi: 10.1111/j.1538-7836.2009.03395.x[↩][↩][↩][↩][↩][↩]
- Gerber GF, Klute KA, Chapin J, Bussel J, DeSancho MT. Peri- and Postpartum Management of Patients With Factor XI Deficiency. Clin Appl Thromb Hemost. 2019 Jan-Dec;25:1076029619880262. doi: 10.1177/1076029619880262[↩][↩][↩][↩]
- Asselta R, Paraboschi EM, Rimoldi V, Menegatti M, Peyvandi F, Salomon O, Duga S. Exploring the global landscape of genetic variation in coagulation factor XI deficiency. Blood. 2017 Jul 27;130(4):e1-e6. doi: 10.1182/blood-2017-04-780148[↩][↩]
- Duga S, Salomon O. Congenital factor XI deficiency: an update. Semin Thromb Hemost. 2013 Sep;39(6):621-31. doi: 10.1055/s-0033-1353420[↩]
- Peyvandi F, Palla R, Menegatti M, et al. European Network of Rare Bleeding Disorders Group. Coagulation factor activity and clinical bleeding severity in rare bleeding disorders: results from the European Network of Rare Bleeding Disorders. J Thromb Haemost. 2012 Apr;10(4):615-21. doi: 10.1111/j.1538-7836.2012.04653.x[↩][↩][↩]
- Martinez-Lopez PR, Barroso-Gonzalez A. Anesthetic Implications of Factor XI Deficiency: A Clinical Case Study and Review of Literature. Cureus. 2024 Oct 28;16(10):e72594. doi: 10.7759/cureus.72594[↩]
- Factor XI deficiency. https://medlineplus.gov/genetics/condition/factor-xi-deficiency[↩][↩]
- Roberts AE. Noonan Syndrome. 2001 Nov 15 [Updated 2025 Jun 5]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1124[↩][↩][↩]
- Sharma L, Winters R, Morales A. Noonan Syndrome. [Updated 2025 Aug 2]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK532269[↩][↩][↩]
- Rauen KA. The RASopathies. Annu Rev Genomics Hum Genet. 2013;14:355-69. doi: 10.1146/annurev-genom-091212-153523[↩][↩][↩]
- Myers A, Bernstein JA, Brennan ML, Curry C, Esplin ED, Fisher J, Homeyer M, Manning MA, Muller EA, Niemi AK, Seaver LH, Hintz SR, Hudgins L. Perinatal features of the RASopathies: Noonan syndrome, cardiofaciocutaneous syndrome and Costello syndrome. Am J Med Genet A. 2014 Nov;164A(11):2814-21. doi: 10.1002/ajmg.a.36737[↩][↩][↩]
- Anaesthesia management of caesarean section in a patient with severe factor XI deficiency. Bhoi D, Sreekumar E, Anand R, Baidya D, Chhabra A. J Obstet Anaesth Crit Care. 2013;3:37–39.[↩]
- Barg AA, Levy-Mendelovich S, Budnik I, et al. Pediatric severe factor XI deficiency: A multicenter study. Pediatr Blood Cancer. 2022 Mar;69(3):e29545. doi: 10.1002/pbc.29545[↩]
- Favaloro EJ, Amgalan A, Allen T, Othman M, Ahmadzia HK. “Systematic review of viscoelastic testing (TEG/ROTEM) in obstetrics and recommendation from the women’s SSC of the ISTH”: Response to comment from Kitchen et al. J Thromb Haemost. 2020 Sep;18(9):2420-2422. doi: 10.1111/jth.15007[↩][↩][↩][↩]
- Ladib A, Ladib J, Jebali F, Korbi A, Ghaddab I, Sassi M. Optimizing the anesthetic management of factor XI deficiency in obstetrics: A case report using ROTEM. Int J Surg Case Rep. 2025 Aug;133:111566. doi: 10.1016/j.ijscr.2025.111566[↩]
- Management of rare coagulation disorders in 2018. Jain S, Acharya SS. Transfus Apher Sci. 2018;57:705–712. doi: 10.1016/j.transci.2018.10.009[↩][↩]
- Perioperative use of modified thrombelastography in factor XI deficiency: a helpful method to assess drug effects. Dirkmann D, Hanke AA, Görlinger K, Peters J. Acta Anaesthesiol Scand. 2007;51:640–643. doi: 10.1111/j.1399-6576.2007.01284.x[↩][↩]
- Viscoelastic testing: Critical appraisal of new methodologies and current literature. Wool GD, Carll T. Int J Lab Hematol. 2023;45:643–658. doi: 10.1111/ijlh.14144[↩][↩]
- Factor XI Deficiency. https://haemophilia.org.uk/wp-content/uploads/pdf/factsheet_factorXI.pdf[↩][↩][↩]
- Hemophilia C (Factor XI Deficiency). https://emedicine.medscape.com/article/209984-overview#a8[↩]
- Seligsohn U. High gene frequency of factor XI (PTA) deficiency in Ashkenazi Jews. Blood. 1978 Jun;51(6):1223-8.[↩]
- Asakai R, Chung DW, Davie EW, Seligsohn U. Factor XI deficiency in Ashkenazi Jews in Israel. N Engl J Med. 1991 Jul 18;325(3):153-8. doi: 10.1056/NEJM199107183250303[↩]
- Asakai R, Chung DW, Ratnoff OD, Davie EW. Factor XI (plasma thromboplastin antecedent) deficiency in Ashkenazi Jews is a bleeding disorder that can result from three types of point mutations. Proc Natl Acad Sci U S A. 1989 Oct;86(20):7667-71. doi: 10.1073/pnas.86.20.7667[↩]
- Preis M, Hirsch J, Kotler A, Zoabi A, Stein N, Rennert G, Saliba W. Factor XI deficiency is associated with lower risk for cardiovascular and venous thromboembolism events. Blood. 2017 Mar 2;129(9):1210-1215. doi: 10.1182/blood-2016-09-742262[↩][↩]
- Zhang X, Lewandowska M, Aldridge M, Iglay K, Wolford E, Shapiro A. Global epidemiology of factor XI deficiency: A targeted review of the literature and foundation reports. Haemophilia. 2023 Mar;29(2):423-434. doi: 10.1111/hae.14687[↩][↩]
- Knol HM, Mulder AB, Bogchelman DH, Kluin-Nelemans HC, van der Zee AG, Meijer K. The prevalence of underlying bleeding disorders in patients with heavy menstrual bleeding with and without gynecologic abnormalities. Am J Obstet Gynecol. 2013 Sep;209(3):202.e1-7. doi: 10.1016/j.ajog.2013.05.059[↩]
- Saes JL, Verhagen MJA, Meijer K, Cnossen MH, Schutgens REG, Peters M, Nieuwenhuizen L, van der Meer FJM, Kruis IC, van Heerde WL, Schols SEM. Bleeding severity in patients with rare bleeding disorders: real-life data from the RBiN study. Blood Adv. 2020 Oct 27;4(20):5025-5034. doi: 10.1182/bloodadvances.2020002740[↩][↩][↩][↩]
- Wheeler AP, Hemingway C, Gailani D. The clinical management of factor XI deficiency in pregnant women. Expert Rev Hematol. 2020 Jul;13(7):719-729. doi: 10.1080/17474086.2020.1772745[↩][↩]
- Wiewel-Verschueren S, Arendz IJ, M Knol H, Meijer K. Gynaecological and obstetrical bleeding in women with factor XI deficiency – a systematic review. Haemophilia. 2016 Mar;22(2):188-195. doi: 10.1111/hae.12856[↩]
- Kadir RA, Lee CA, Sabin CA, Pollard D, Economides DL. Pregnancy in women with von Willebrand’s disease or factor XI deficiency. Br J Obstet Gynaecol. 1998 Mar;105(3):314-21. doi: 10.1111/j.1471-0528.1998.tb10093.x[↩]
- Myers B, Pavord S, Kean L, Hill M, Dolan G. Pregnancy outcome in Factor XI deficiency: incidence of miscarriage, antenatal and postnatal haemorrhage in 33 women with Factor XI deficiency. BJOG. 2007 May;114(5):643-6. doi: 10.1111/j.1471-0528.2007.01296.x[↩][↩]
- Hammerova L, Chabada J, Drobny J, Batorova A. Longitudinal evaluation of markers of hemostasis in pregnancy. Bratisl Lek Listy. 2014;115(3):140-4. doi: 10.4149/bll_2014_030[↩]
- Chi C, Kulkarni A, Lee CA, Kadir RA. The obstetric experience of women with factor XI deficiency. Acta Obstet Gynecol Scand. 2009;88(10):1095-100. doi: 10.1080/00016340903144238[↩]
- Salomon O, Steinberg DM, Tamarin I, Zivelin A, Seligsohn U. Plasma replacement therapy during labor is not mandatory for women with severe factor XI deficiency. Blood Coagul Fibrinolysis. 2005 Jan;16(1):37-41. doi: 10.1097/00001721-200501000-00006[↩]
- Santoro C, Di Mauro R, Baldacci E, De Angelis F, Abbruzzese R, Barone F, Bochicchio RA, Ferrara G, Guarini A, Foà R, Mazzucconi MG. Bleeding phenotype and correlation with factor XI (FXI) activity in congenital FXI deficiency: results of a retrospective study from a single centre. Haemophilia. 2015 Jul;21(4):496-501. doi: 10.1111/hae.12628[↩]
- Stoeckle JH, Bogue T, Zwicker JI. Postpartum haemorrhage in women with mild factor XI deficiency. Haemophilia. 2020 Jul;26(4):663-666. doi: 10.1111/hae.14081[↩][↩][↩]
- Handa S, Sterpi M, Sacchi De Camargo Correia G, Frankel DS, Beilin Y, Cytryn L, Hawkins K, Frankel E. Obstetric and perioperative management of patients with factor XI deficiency: a retrospective observational study. Blood Adv. 2023 May 23;7(10):1967-1975. doi: 10.1182/bloodadvances.2022008648[↩]
- Toulon P, Berruyer M, Brionne-François M, Grand F, Lasne D, Telion C, Arcizet J, Giacomello R, De Pooter N. Age dependency for coagulation parameters in paediatric populations. Results of a multicentre study aimed at defining the age-specific reference ranges. Thromb Haemost. 2016 Jul 4;116(1):9-16. doi: 10.1160/TH15-12-0964[↩]
- Tabibian S, Motlagh H, Naderi M, Dorgalaleh A. Intracranial hemorrhage in congenital bleeding disorders. Blood Coagul Fibrinolysis. 2018 Jan;29(1):1-11. doi: 10.1097/MBC.0000000000000660[↩]
- Anderst JD, Carpenter SL, Presley R, Berkoff MC, Wheeler AP, Sidonio RF Jr, Soucie JM. Relevance of Abusive Head Trauma to Intracranial Hemorrhages and Bleeding Disorders. Pediatrics. 2018 May;141(5):e20173485. doi: 10.1542/peds.2017-3485[↩]
- Barg AA, Brutman-Barazani T, Avishai E, Budnik I, Cohen O, Dardik R, Levy-Mendelovich S, Livnat T, Kenet G. Anti-TFPI for hemostasis induction in patients with rare bleeding disorders, an ex vivo thrombin generation (TG) guided pilot study. Blood Cells Mol Dis. 2022 Jul;95:102663. doi: 10.1016/j.bcmd.2022.102663[↩]
- American Academy of Pediatrics Task Force on Circumcision. Male circumcision. Pediatrics. 2012 Sep;130(3):e756-85. doi: 10.1542/peds.2012-1990[↩]
- Barg AA, Levy-Mendelovich S, Budnik I, Mandel-Shorer N, Dardik R, Avishai E, Brutman-Barazani T, Ifrah AD, Oren-Malek L, Yacobovich J, Gilad O, Nakav S, Fruchtman Y, Revel-Vilk S, Miskin H, Kenet G. Pediatric severe factor XI deficiency: A multicenter study. Pediatr Blood Cancer. 2022 Mar;69(3):e29545. doi: 10.1002/pbc.29545[↩][↩]
- Warad D, Hussain FTN, Rao AN, Cofer SA, Rodriguez V. Haemorrhagic complications with adenotonsillectomy in children and young adults with bleeding disorders. Haemophilia. 2015 May;21(3):e151-e155. doi: 10.1111/hae.12577[↩]
- Kaufman JM. Prostatectomy in factor XI deficiency. J Urol. 1977 Jan;117(1):75-8. doi: 10.1016/s0022-5347(17)58343-3[↩]
- Sidi A, Seligsohn U, Jonas P, Many M. Factor XI deficiency: detection and management during urological surgery. J Urol. 1978 Apr;119(4):528-30. doi: 10.1016/s0022-5347(17)57537-0[↩]
- Silverstein RL, Bauer KA, Cushman M, Esmon CT, Ershler WB, Tracy RP. Venous thrombosis in the elderly: more questions than answers. Blood. 2007 Nov 1;110(9):3097-101. doi: 10.1182/blood-2007-06-096545[↩]
- Bravo-Pérez C, Serna MJ, Esteban J, Fernandez-Mellid E, Fontanes-Trabazo E, Lorenzo A, Calviño-Suárez M, Miñano A, Padilla J, Roldán V, Vicente V, Corral J, de la Morena-Barrio ME. Anticoagulant therapy in patients with congenital FXI deficiency. Blood Adv. 2021 Oct 26;5(20):4083-4086. doi: 10.1182/bloodadvances.2021005695[↩][↩][↩][↩]
- Pike GN, Cumming AM, Hay CR, Bolton-Maggs PH, Burthem J. Sample conditions determine the ability of thrombin generation parameters to identify bleeding phenotype in FXI deficiency. Blood. 2015 Jul 16;126(3):397-405. doi: 10.1182/blood-2014-12-616565[↩]
- O’Connell NM, Riddell AF, Pascoe G, Perry DJ, Lee CA. Recombinant factor VIIa to prevent surgical bleeding in factor XI deficiency. Haemophilia. 2008 Jul;14(4):775-81. doi: 10.1111/j.1365-2516.2008.01663.x[↩]
- Kenet G, Lubetsky A, Luboshitz J, Ravid B, Tamarin I, Varon D, Martinowitz U. Lower doses of rFVIIa therapy are safe and effective for surgical interventions in patients with severe FXI deficiency and inhibitors. Haemophilia. 2009 Sep;15(5):1065-73. doi: 10.1111/j.1365-2516.2009.02043.x[↩]
- Meijers JC, Tekelenburg WL, Bouma BN, Bertina RM, Rosendaal FR. High levels of coagulation factor XI as a risk factor for venous thrombosis. N Engl J Med. 2000 Mar 9;342(10):696-701. doi: 10.1056/NEJM200003093421004[↩]
- Kyrle PA, Eischer L, Šinkovec H, Eichinger S. Factor XI and recurrent venous thrombosis: an observational cohort study. J Thromb Haemost. 2019 May;17(5):782-786. doi: 10.1111/jth.14415[↩]
- Salomon O, Steinberg DM, Zucker M, Varon D, Zivelin A, Seligsohn U. Patients with severe factor XI deficiency have a reduced incidence of deep-vein thrombosis. Thromb Haemost. 2011 Feb;105(2):269-73. doi: 10.1160/TH10-05-0307[↩]
- Salomon O, Steinberg DM, Dardik R, Rosenberg N, Zivelin A, Tamarin I, Ravid B, Berliner S, Seligsohn U. Inherited factor XI deficiency confers no protection against acute myocardial infarction. J Thromb Haemost. 2003 Apr;1(4):658-61. doi: 10.1046/j.1538-7836.2003.00195.x[↩]
- Gailani D, Gruber A. Targeting factor XI and factor XIa to prevent thrombosis. Blood. 2024 Apr 11;143(15):1465-1475. doi: 10.1182/blood.2023020722[↩]
- Gidley GN, Holle LA, Burthem J, Bolton-Maggs PHB, Lin FC, Wolberg AS. Abnormal plasma clot formation and fibrinolysis reveal bleeding tendency in patients with partial factor XI deficiency. Blood Adv. 2018 May 22;2(10):1076-1088. doi: 10.1182/bloodadvances.2017015123[↩]
- Hemophilia C (Factor XI Deficiency). https://emedicine.medscape.com/article/209984-overview#a5[↩][↩]
- Vercellotti GM, Mosher DF. Acquired factor XI deficiency in systemic lupus erythematosus. Thromb Haemost. 1982 Dec 27;48(3):250-2.[↩]
- Goodrick MJ, Prentice AG, Copplestone JA, Pamphilon DH, Boon RJ. Acquired factor XI inhibitor in chronic lymphocytic leukaemia. J Clin Pathol. 1992 Apr;45(4):352-3. doi: 10.1136/jcp.45.4.352[↩]
- Reece EA, Clyne LP, Romero R, Hobbins JC. Spontaneous factor XI inhibitors. Seven additional cases and a review of the literature. Arch Intern Med. 1984 Mar;144(3):525-9. doi: 10.1001/archinte.144.3.525[↩]
- Schnall SF, Duffy TP, Clyne LP. Acquired factor XI inhibitors in congenitally deficient patients. Am J Hematol. 1987 Dec;26(4):323-8. doi: 10.1002/ajh.2830260405[↩][↩]
- Zucker M, Zivelin A, Teitel J, Seligsohn U. Induction of an inhibitor antibody to factor XI in a patient with severe inherited factor XI deficiency by Rh immune globulin. Blood. 2008 Feb 1;111(3):1306-8. doi: 10.1182/blood-2007-08-108449[↩][↩]
- Salomon O, Zivelin A, Livnat T, Seligsohn U. Inhibitors to Factor XI in patients with severe Factor XI deficiency. Semin Hematol. 2006 Jan;43(1 Suppl 1):S10-2. doi: 10.1053/j.seminhematol.2005.11.018[↩][↩][↩]
- Ginsberg SS, Clyne LP, McPhedran P, Duffy TP, Hanson T. Successful childbirth by a patient with congenital factor XI deficiency and an acquired inhibitor. Br J Haematol. 1993 May;84(1):172-4. doi: 10.1111/j.1365-2141.1993.tb03044.x[↩]
- Weitz JI, Strony J, Ageno W, Gailani D, Hylek EM, Lassen MR, Mahaffey KW, Notani RS, Roberts R, Segers A, Raskob GE; AXIOMATIC-TKR Investigators. Milvexian for the Prevention of Venous Thromboembolism. N Engl J Med. 2021 Dec 2;385(23):2161-2172. doi: 10.1056/NEJMoa2113194[↩]
- Shoamanesh A, Mundl H, Smith EE, et al. Factor XIa inhibition with asundexian after acute non-cardioembolic ischaemic stroke (PACIFIC-Stroke): an international, randomised, double-blind, placebo-controlled, phase 2b trial. Lancet. 2022 Sep 24;400(10357):997-1007. doi: 10.1016/S0140-6736(22)01588-4[↩]
- Factor XI. https://www.bleeding.org/bleeding-disorders-a-z/types/other-factor-deficiencies/factor-xi[↩]
- Salomon O, Steinberg DM, Seligshon U. Variable bleeding manifestations characterize different types of surgery in patients with severe factor XI deficiency enabling parsimonious use of replacement therapy. Haemophilia. 2006 Sep;12(5):490-3. doi: 10.1111/j.1365-2516.2006.01304.x[↩]
- Barg AA, Livnat T, Kenet G. Factor XI deficiency: phenotypic age-related considerations and clinical approach towards bleeding risk assessment. Blood. 2024 Apr 11;143(15):1455-1464. doi: 10.1182/blood.2023020721[↩]
- Mohinani A, Patel S, Tan V, Kartika T, Olson S, DeLoughery TG, Shatzel J. Desmopressin as a hemostatic and blood sparing agent in bleeding disorders. Eur J Haematol. 2023 May;110(5):470-479. doi: 10.1111/ejh.13930[↩]
- Salomon O, Budnik I, Avishai E, Tamarin I, Bashari D, Dardik R, Livnat T. Single Low Dose of rFVIIa Combined with Antifibrinolytic Agent is a Simple and Safe Treatment for Factor XI-Deficient Patients undergoing Surgery. Thromb Haemost. 2019 Dec;119(12):1927-1932. doi: 10.1055/s-0039-1696685[↩][↩]
- Minami H, Nogami K, Yada K, Ogiwara K, Furukawa S, Soeda T, Kitazawa T, Shima M. Emicizumab, the bispecific antibody to factors IX/IXa and X/Xa, potentiates coagulation function in factor XI-deficient plasma in vitro. J Thromb Haemost. 2019 Jan;17(1):126-137. doi: 10.1111/jth.14334[↩]
- Batty P, Honke A, Bowles L, Hart DP, Pasi KJ, Uprichard J, Austin SK. Ongoing risk of thrombosis with factor XI concentrate: 5 years experience in two centres. Haemophilia. 2015 Jul;21(4):490-5. doi: 10.1111/hae.12682[↩]
- Search the Global Treatment Centre Directory. World Federation of Hemophilia https://wfh.org/find-local-support/#HTCs[↩]
- National Bleeding Disorders Foundation https://www.bleeding.org/[↩]


{kind=link}