Contents
What is hyperammonemia
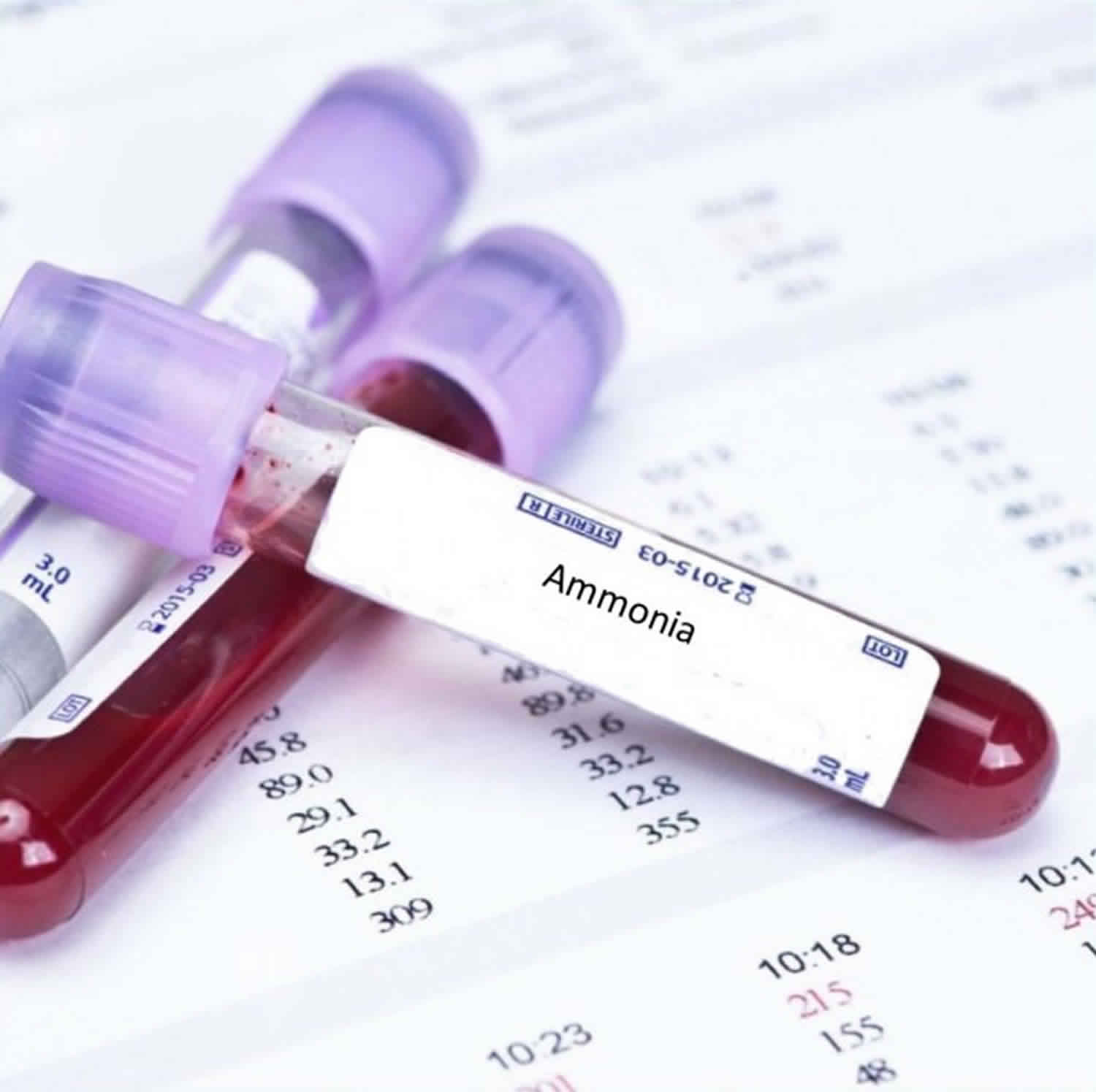
Hyperammonemia refers to a clinical condition associated with elevated ammonia levels manifested by a variety of symptoms and signs, including significant central nervous system (CNS) abnormalities. Ammonia (NH3) is an important source of nitrogen and is required for amino acid synthesis. Ammonia is also necessary for normal acid-base balance. When present in high concentrations (hyperammonemia), ammonia is toxic. Endogenous ammonia intoxication can occur when there is impaired capacity of the body to excrete nitrogenous waste, as seen with congenital enzymatic deficiencies. A variety of environmental causes and medications may also lead to ammonia toxicity.
Ammonia is also a waste product formed primarily by bacteria in the intestines during the digestion of protein. If not processed and cleared from your body appropriately, excess ammonia can accumulate in your blood. Ammonia is normally transported in the blood to the liver, where it is converted into two substances called urea and glutamine. The urea is then carried to the kidneys, where it is eliminated in the urine. If this “urea cycle” does not complete the breakdown of ammonia, ammonia builds up in your blood and can pass from your blood into your brain.
Ammonia is toxic to the brain. For example, when liver function is significantly reduced due to disorders such as cirrhosis or hepatitis, ammonia and other compounds processed by the liver can accumulate in the brain and cause a condition called hepatic encephalopathy.
Hepatic encephalopathy causes mental and neurological changes that can lead to confusion, disorientation, sleepiness, and eventually to coma and even death.
Infants and children with increased ammonia levels may vomit frequently, be irritable, and be increasingly lethargic. Left untreated, they may experience seizures, have difficulty breathing, and may lapse into a coma.
Appropriate and timely management of hyperammonemia requires a solid understanding of the fundamental pathophysiology, differential diagnosis, and treatment approaches available.
What causes hyperammonemia
Hyperammonemia can arise from conditions such as:
- Severe liver disease – damage limits the ability of the liver to process ammonia; spikes in ammonia blood levels may be seen in people with stable liver disease, especially following a triggering event such as gastrointestinal bleeding or an electrolyte imbalance.
- Decreased blood flow to the liver – ammonia is less able to get to the liver to be processed.
- Reye syndrome – a rare condition that affects the blood, brain, and liver; it is characterized by a rise in ammonia levels and a fall in glucose. It affects primarily children and young adults. In most cases, it follows and appears to be triggered by a viral infection, such as the flu or chickenpox. Children who are given aspirin are at an increased risk.
- Renal failure – the kidneys are unable to effectively rid the body of urea, leading to a build-up of ammonia in the blood.
- Urea cycle disorders – rare inherited defects in the urea cycle, a deficiency or defect in one or more of the enzymes necessary to complete the conversion of ammonia to urea.
Hyperammonemia may also be seen with:
- Gastrointestinal bleeding – blood cells are hemolyzed in the intestines, releasing protein.
- Muscular exertion – muscles produce ammonia when active and absorb it when resting.
- Tourniquet use – ammonia levels can be increased in the blood sample collected.
- Use of certain drugs, including alcohol, barbiturates, diuretics, valproic acid, and narcotics
- Cigarette smoking
Hyperammonemia indicates that the body is not effectively processing and eliminating ammonia and it may be the cause of the person’s signs and symptoms.
In infants, an extremely high ammonia level is associated with an inherited urea cycle enzyme deficiency or defect but may also be seen with hemolytic disease of the newborn. Moderate short-lived increases in ammonia are relatively common in newborns, where the level may rise and fall without causing noticeable symptoms.
Hyperammonemia and decreased glucose level may indicate the presence of Reye syndrome in symptomatic children and teens. An increased concentration may also indicate a previously undiagnosed enzymatic defect of the urea cycle.
In children and adults, hyperammonemia may indicate that severe liver or kidney damage has impacted the body’s ability to clear ammonia and that the brain may be affected. Frequently, an acute or chronic illness will act as a trigger, increasing ammonia levels to the point that an affected person has difficulty clearing the ammonia.
A normal blood ammonia level may mean that a person’s signs and symptoms are due to a cause other than excess ammonia. However, normal concentrations of ammonia do not rule out hepatic encephalopathy. Other wastes can contribute to changes in mental function and consciousness, and brain levels of ammonia may be much higher than blood levels. This can make correlation of a person’s symptoms to ammonia blood levels difficult.
Reye syndrome
Reye syndrome or Reye’s syndrome is a rare but serious illness that can affect the blood, liver, and brain of someone who has recently had a viral infection. Reye syndrome always follows another illness. Although it mostly affects children and teens (kids 4 to 14 years old), anyone can get it. Reye syndrome can develop quickly and without warning. Reye syndrome is most common in kids who are recovering from a viral infection. In rare cases, Reye syndrome can cause death within hours. Because it can be a life-threatening disorder, Reye syndrome is a medical emergency.
The cause of Reye syndrome is still not well understood, but studies have linked it to the use of aspirin (salicylates) or aspirin products during illnesses caused by viruses. The number of Reye syndrome cases has dropped greatly since doctors began advising against giving aspirin to kids and teens, especially during viral illnesses.
Most Reye’s syndrome cases happen when viral diseases are epidemic, such as during the winter months or after an outbreak of chickenpox or the flu.
Early detection and treatment are critical. The chances for a full recovery are best when Reye (pronounced: rye) syndrome is treated in its earliest stages.
Reye syndrome is most common during flu season. Symptoms include:
- Nausea and vomiting
- Listlessness
- Personality change – such as irritability, combativeness or confusion
- Delirium
- Convulsions
- Loss of consciousness
Other symptoms include changes in vision, trouble hearing, and abnormal speech. In the later stages, a child may:
- behave irrationally
- be confused
- have severe muscle weakness, seizures, and loss of consciousness (pass out)
Even though it’s rare, Reye syndrome should be considered if a child is vomiting a lot or shows a change in mental status or behavior, particularly after a recent viral illness.
If these symptoms occur soon after a viral illness, seek medical attention immediately. Reye syndrome can lead to a coma and brain death, so quick diagnosis and treatment are critical. Treatment focuses on preventing brain damage. There is no cure.
Children with Reye syndrome are usually treated in a hospital. Those who are seriously ill will be cared for in the intensive care unit (ICU).
Treatment is supportive, as there is no cure. The clinical care team will:
- Make sure the child stays hydrated and maintains electrolyte balance.
- Check nutrition intake.
- Watch the child’s heart rate and breathing.
- Watch the child’s intracranial pressure (pressure of the fluid in the brain) and blood pressure.
Tests done can include blood tests to check electrolytes and liver function and an imaging study of the brain (CAT scan or MRI).
A child also might get:
- small amounts of insulin to increase glucose metabolism
- corticosteroids to reduce brain swelling
- diuretics to get rid of excess fluid
If seizures happen, they’ll be treated with medicines. Some kids might need breathing help from a breathing machine or respirator if their breathing gets too slow or ineffective.
The outlook for children with Reye syndrome has improved thanks to earlier diagnosis and better treatment. If the late stages of the syndrome happen, they can cause brain damage, disability, or death.
Urea cycle disorders
Urea cycle disorders are genetic inborn errors of metabolism caused by a mutation that results in a deficiency of one of the six enzymes in the urea cycle or 2 transporters involved in the hepatic removal of ammonia from the bloodstream by conversion to urea which is excreted by the kidneys 1. These enzymes are responsible for removing ammonia from the blood stream. The urea cycle involves a series of biochemical steps in which nitrogen, a waste product of protein metabolism, is removed from the blood and converted to a compound called urea in the blood. Normally, the urea is transferred into the urine and removed from the body. In urea cycle disorders, the nitrogen accumulates in the form of ammonia, a highly toxic substance, resulting in hyperammonemia (elevated blood ammonia). Ammonia then reaches the brain through the blood, where it can cause irreversible brain damage, coma and/or death.
There are six enzyme disorders of the urea cycle, collectively known as inborn errors of urea synthesis, or urea cycle enzyme defects. Each is referred to by the initials of the missing enzyme.
- CPS1 – Carbamyl Phosphate Synthetase
- NAGS- N-Acetylglutamate Synthetase
- OTC Deficiency – Ornithine Transcarbamylase
- AS – Argininosuccinic Acid Synthetase (Citrullinemia)
- ASL – Argininosuccinate Lyase (Argininosuccinic Aciduria)
- AG – Arginase
Additionally, there are three transporter defects:
- Mitochondrial ornithine carrier (Hyperornithinemia-Hyperammonemia-Homocitrullinemia or HHH syndrome)
- Mitochondrial aspartate/glutamate carrier (Citrullinemia Type II, also known as Citrin Deficiency)
- Dibasic amino acid carrier (Hyperdibasic Amino Aciduria or Lysinuric Protein Intolerance)
The onset and severity of urea cycle disorders is highly variable. This depends on the specific mutation involved and correlates with the amount of urea cycle enzyme function. Severe mutations result in zero to very little enzyme function and ability to detoxify ammonia, and cause severe urea cycle disorders. Mild to moderate mutations represent a broad spectrum of enzyme function, providing some ability to detoxify ammonia, and result in mild to moderate urea cycle disorders.
Urea cycle disorders occur in both children and adults. Newborns with severe mutations become catastrophically ill within 36-48 hours of birth. Children with less severe mutations present outside the newborn period or can remain undiagnosed because symptoms are not appropriately recognized. Adults often go undiagnosed because they have mild urea cycle disorders which allow them to produce enough of the urea cycle enzymes to effectively remove ammonia until a stressor interferes with enzyme function, or causes massive amounts of ammonia to be produced. These adults may have subtle symptoms in their lifetime that go unrecognized or unheeded.
Metabolic stressors — viruses, high protein intake, excessive exercise or dieting, surgery, or a drug (valproic acid, prednisone or other corticosteroid — can create excessive ammonia in the body and overwhelm the individual’s urea cycle enzyme function, resulting in severe neurological symptoms. Seemingly normal adults with undiagnosed urea cycle disorders may present at emergency rooms with staggering, confusion, combativeness and disorientation that is mistaken for alcohol or drug intoxication. Ammonia quickly rises if untreated and causes coma and death. Some undiagnosed adults may suffer from psychiatric symptoms like schizophrenia or bipolar disorder.
Urea cycle disorders symptoms
- THE NEONATAL PERIOD: Children with severe urea cycle disorders typically show symptoms after the first 24 hours of life. The baby may be irritable at first, or refuse feedings, followed by vomiting and increasing lethargy. Soon after, seizures, hypotonia (poor muscle tone, floppiness), respiratory distress (respiratory alkalosis), and coma may occur. These symptoms are caused by rising ammonia levels in the blood. Sepsis and Reye’s syndrome are common misdiagnoses. If untreated, these severely affected infants will die. Severe neonatal symptoms are more commonly seen in both boys and girls with Ornithine Transcarbamylase and Carbamyl Phosphate Synthetase deficiency, but can also occur with citrullinemia or argininosuccinate lyase deficiency (ASA lyase).
- CHILDHOOD: Children with mild or moderate urea cycle enzyme deficiencies may not show recognizable symptoms until early childhood. Earliest symptoms may include failure to thrive, inconsolable crying, agitation or hyperactive behavior, sometimes accompanied by screaming, self-injurious behavior, and refusal to eat meat or other high-protein foods. Later symptoms may include frequent episodes of vomiting, especially following high-protein meals, lethargy and delirium, and finally, if the condition is undiagnosed and untreated, hyperammonemic coma or death may occur. Undiagnosed children may be referred to child psychologists because of their behavior, developmental delays and eating problems. Childhood episodes of hyperammonemia (high ammonia levels in the blood) may be brought on by viral illnesses including chicken pox, colds or flu, teething, growth spurts, high-protein meals, or even exhaustion. Common misdiagnoses include Reye’s syndrome. Childhood onset can be seen in both boys and girls affected by any of the urea cycle disorders. Early clinical manifestations (symptoms) of arginase deficiency (similar to those of the other disorders), may be seen as early as one year of age, but some children with AG remain asymptomatic at four years of age. Arginase symptoms are usually progressive and include growth failure, spastic tetraplegia (lower limbs more severely affected than upper limbs), seizures, psychomotor retardation and hyperactivity. Major characteristics of N-Acetylglutamate Synthetase (NAGS) deficiency, considered the rarest urea cycle disorder, include severe hyperammonemia (elevated blood ammonia), deep encephalopathy despite only mild hyperammonemia, recurrent diarrhea and acidosis, movement disorder, hypoglycemia and hyperornithinemia.1
- ADULTHOOD: Recently, the number of adults being diagnosed with urea cycle disorders has dramatically increased. These individuals have survived undiagnosed to adulthood due to mild enzyme deficiencies. Many adults are being identified due to improved diagnostics and increased awareness among medical professionals. Symptoms include episodes of disorientation, confusion, slurred speech, unusual and extreme combativeness or agitation, stroke-like symptoms, lethargy and delirium. Many may be seen by neurologists or psychiatrists because of psychiatric symptoms, including schizophrenia and bipolar disorder. Without proper diagnosis and treatment, these individuals are at risk for permanent brain damage, coma, and death. Symptoms in undiagnosed adults have been observed following viral illnesses, childbirth, excessive dieting including high-protein diets, excessive exercising, gastric bypass surgery, use of valproic acid (an anti-epileptic drug which causes excess ammonia), and chemotherapy.
- Ornithine Transcarbamylase CARRIERS: Approximately 85% of adult female carriers (heterozygotes) for Ornithine Transcarbamylase deficiency are asymptomatic (exhibit no symptoms). The remainder show symptoms including protein intolerance, headache, episodes of confusion or trouble concentrating, behavioral or neurological abnormalities, vomiting and episodes of mild hyperammonemia. Studies have shown carriers to be of normal to above-normal intelligence, but some have been shown to demonstrate subtle deficits in fine motor, visual-spatial and non-verbal functions.2Concerns are beginning to emerge about carriers with regard to common health issues (diabetes, cholesterolemia, cancer, liver disease) and potential negative effects that drugs used to treat these common conditions may have on urea cycle function.
Urea cycle disorders treatment
The treatment of urea cycle disorders consists of dietary management to limit ammonia production in conjunction with medications and/or supplements which provide alternative pathways for the removal of ammonia from the bloodstream. A careful balance of dietary protein, carbohydrates and fats is necessary to ensure that the body receives adequate calories for energy needs, as well as adequate essential amino acids (for cell growth and development). Dietary protein must be carefully monitored and some restriction is necessary; too much dietary protein causes excessive ammonia production. However, if protein intake is too restrictive or insufficient calories are provided, the body will break down lean muscle mass (called catabolism) to obtain the amino acids or energy it requires; this catabolism creates excessive ammonia. Therefore, the correct nutritional balance for each individual in each stage of growth is critical in avoiding hyperammonemic crises. Branched-chain amino acids may be reduced as a result of treatment with ammonia scavenging medication (phenylbutyrate), triggering catabolism; therefore, frequent blood tests (serum ammonia, plasma quantitative amino acids) are required to monitor the disorders and are an important tool for optimizing treatment.
- DIETARY MANAGEMENT: Treatment may include supplementation with special amino acid formulas (Cyclinex, EAA, Trio, UCD I&II), developed specifically for urea cycle disorders, which can be prescribed to provide approximately 50% of the daily dietary protein allowance. Some patients may require individual branched chain amino acid supplementation. Metabolic nutritionists routinely prescribe calorie modules such as Prophree, DuoCal, Benecalorie and ModuCal to be used in combination with the amino acid formulas. Multiple vitamins and calcium supplements are also recommended. New research also suggests that antioxidants may be helpful in minimizing free radical damage to cells in tissue and brain.
- DRUG TREATMENT: Two forms of a drug, phenylbutyrate, are FDA approved for treatment of urea cycle disorders, sodium phenylbutyrate (trade name Buphenyl, powder or pill form) and glycerol phenylbutyrate (trade name Ravicti, liquid form). The Foundation played key roles in initiating and supporting the development of these medications. Sodium benzoate is also used in some patients, solely or in combination with phenylbutyrate. All three medications serve as “ammonia scavengers” — providing alternative pathways for removal of ammonia from the bloodstream and helping to prevent hyperammonemia (elevated blood ammonia). Carglumic acid (trade name Carbaglu) is approved for treatment of NAGS deficiency. The medications are administered 3-4 times per day as prescribed in order to optimize continual removal of toxic ammonia from the bloodstream. Pharmaceutical grade (not over-the-counter) L-citrulline (for OTC and CPS deficiency) or L-arginine free base (ASA and citrullinemia) is also required. These are not to be used in Arginase Deficiency. These supplements help catalyze the urea cycle enzymes and promote optimal removal of ammonia. H-blockers (antacids) are sometimes prescribed to minimize the gastrointestinal side effects (stomachache, reflux, etc.) of drug treatment.
- FEEDING ISSUES: Children with urea cycle disorders often lack appetite (due to excess serotonin in the brain suppressing appetite) and some may benefit from receiving medications and some feedings either via gastrostomy tube (a tube surgically implanted in the stomach) or nasogastric tube (manually inserted through the nose into the stomach). The access these tubes provide often makes a critical difference in metabolic stability and in averting hyperammonemic crises; medications and formulas can still be administered when children have flu or colds, etc. Some treatment centers have reported as much as 70% reduction in hospital admissions after placement of G-tubes or parents were trained to use NG-tubes.
- MEDICAL TEAMS: Optimal treatment of urea cycle disorders requires a medical team consisting minimally of a geneticist/metabolic specialist, metabolic dietician, nurse practitioner, and developmental specialist or neuropsychologist specifically experienced in successful management of the disorders. These teams are usually found at university hospitals. Specialty consultation and second opinions from experts in the field of UCDs can be obtained by families who live in areas where optimal medical care is not available.
- LIVER TRANSPLANT: When optimal treatment fails, or for neonatal onset CPS and OTC deficiency, liver transplant becomes an option. Liver transplants have been done successfully as a cure for the disorder (although L-arginine supplementation is still necessary in ASA lyase deficiency posttransplant). The transplant alternative must be carefully considered and evaluated with medical professionals to determine the potential of success and benefits compared to the serious risks and potential for new medical concerns, including the possibility of fatal viruses (Epstein-Barr, CMV), risk of developmental delay or lymphoproliferative disease as a side effect of immunosuppression/immunosuppressants.
Hyperammonemia symptoms
Signs and symptoms of early-onset hyperammonemia (neonates) may include the following:
- Lethargy
- Irritability
- Poor feeding
- Vomiting
- Hyperventilation, grunting respiration
- Seizures
Signs and symptoms of late-onset hyperammonemia (later in life) may include the following:
- Intermittent ataxia
- Intellectual impairment
- Failure to thrive
- Gait abnormality
- Behavior disturbances
- Epilepsy
- Recurrent Reye syndrome
- Protein avoidance
- Rarely, episodic headaches and cyclic vomiting
Hyperammonemia symptoms in a newborn that arise in the first few days after birth, may include:
- Irritability
- Vomiting
- Lethargy
- Seizures
Hyperammonemia symptoms in a child develops about a week following a viral illness, such as influenza or chickenpox, or when a child have Reye syndrome.
When adults experience mental changes, disorientation, sleepiness, or lapse into a coma and may have liver disease or kidney failure, an ammonia level may be ordered to help evaluate the cause of the change in consciousness. In people with stable liver disease, an ammonia level may be ordered, along with other liver function tests, when a person suddenly becomes more acutely ill.
Hyperammonemia diagnosis
No specific physical findings are associated with hyperammonemia. Affected infants usually present with the following:
- Dehydration
- Lethargy
- Tachypnea
- Hypotonia
- Bulging fontanelle
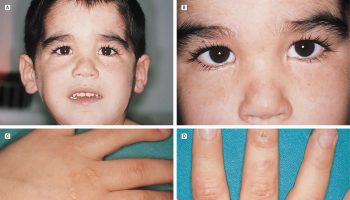
Examination occasionally reveals a peculiar finding, such as odor of “sweaty feet” in isovaleric acidemia or abnormally fragile hair in argininosuccinic aciduria. Infants with argininosuccinic lyase deficiency may present with hepatomegaly.
Lab tests
Perform the following tests in patients with suspected hyperammonemia:
- Arterial blood gas analysis
- Serum amino acid levels
- Urinary orotic acid levels
- Urinary ketone tests
- Plasma and urinary organic acid levels
- Enzyme assays
- DNA mutation analysis: Method of choice to confirm the diagnosis of urea cycle disorders 2
- Heterozygote identification in ornithine transcarbamoylase deficient pedigrees
Imaging studies
The following imaging studies may be used in evaluating patients with hyperammonemia:
- Neuroimaging: CT or MRI of the brain
- MR spectroscopy
Hyperammonemia treatment
Hyperammonemia treatment depends on treating the underlying cause of hyperammonemia. If you had a temporary condition, then it is likely that the ammonia level will return to normal. If you have a chronic condition, then it is possible that the ammonia will increase again and your health status will likely need to be monitored. Talk to your healthcare provider about what is best for you.
The therapeutic aims in patients with hyperammonemia are to correct the biochemical abnormalities and ensure adequate nutritional intake. Treatment involves compounds that increase the removal of nitrogen waste.
Pharmacotherapy
Medications used in the treatment of hyperammonemia include the following:
- Urea cycle disorder treatment agents (eg, sodium phenylbutyrate, carglumic acid, sodium phenylacetate, and sodium benzoate)
- Antiemetic agents (eg, ondansetron, granisetron, palonosetron, dolasetron)
Other treatments
Other management approaches for hyperammonemia include the following:
- Cessation of protein and/or nitrogen intake
- Hemodialysis
- Supportive care with parenteral intake of calories
Surgery
Surgical intervention for patients with hyperammonemia include liver transplantation for correction of the metabolic error and/or liver cell transplantation as an alternative or bridge to liver transplantation 3.
Medical Care
The aims are to correct biochemical abnormalities and ensure adequate nutritional intake. Treatment involves compounds that increase the removal of nitrogen waste. These compounds convert nitrogen into products other than urea, which are then excreted; hence, the load on the urea cycle is reduced. The first compounds to be used were sodium benzoate and arginine. Later, phenylacetate was used, which has now been replaced by phenylbutyrate.
Treatment of neonatal hyperammonemic coma
- Protein intake should be stopped.
- Calories should be supplied by giving hypertonic 10% glucose.
- Hemodialysis should be started promptly in all comatose neonates with plasma ammonium levels greater than 10 times reference range. Plasma ammonium levels are reduced quickly and the total dialysis time is shorter with hemodialysis than with peritoneal dialysis. Continuous arteriovenous or venovenous hemofiltration may be used as an alternative method 4.
- Intravenous sodium benzoate and phenylacetate should be started once the plasma ammonium level falls to 3-4 times the upper limit of the reference range.
- Intravenous arginine should be provided.
- Corticosteroids are not indicated for the management of increased intracranial pressure in hyperammonemia because they induce negative nitrogen balance. Mannitol is not effective in treating cerebral edema induced by hyperammonemia.
- Valproic acid should not be used to treat seizures as it decreases urea cycle function and increases serum ammonia levels.
Treatment of intercurrent hyperammonemia
- Patients with urea cycle defects may present with episodes of hyperammonemia secondary to increased protein intake, increased catabolism, or noncompliance with therapy. This should be recognized early and treated as an emergency.
- Treatment should be started if the plasma ammonium level is 3 times the reference level.
- All nitrogen intake should be stopped.
- High parenteral intake of calories from 10-15% glucose and intralipids should be provided.
- Intravenous infusion of sodium benzoate and phenylacetate should be started.
- Plasma ammonium levels should be checked at the end of the infusion and every 8 hours.
- Once the ammonia level is near normal, oral medication should be started.
- If the level does not decrease in 8 hours, hemodialysis should be started.
- Osmotic demyelination syndrome has been reported as a potential serious complication of standard therapy for hyperammonemia in patients with ornithine transcarbamylase deficiency 5.
- Stone WL, Basit H, Jaishankar GB. Urea Cycle Disorders. [Updated 2019 Apr 25]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2019 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK482363[↩]
- Haeberle J, Boddaert N, Burlina A, Chakrapani A, Dixon M, Huemer M, et al. Suggested Guidelines for the Diagnosis and Management of Urea Cycle Disorders. Orphanet J Rare Dis. 2012 May 29. 7(1):32.[↩]
- Meyburg J, Das AM, Hoerster F, Lindner M, Kriegbaum H, Engelmann G, et al. One liver for four children: first clinical series of liver cell transplantation for severe neonatal urea cycle defects. Transplantation. 2009 Mar 15. 87(5):636-41.[↩]
- Arbeiter AK, Kranz B, Wingen AM, Bonzel KE, Dohna-Schwake C, Hanssler L, et al. Continuous venovenous haemodialysis (CVVHD) and continuous peritoneal dialysis (CPD) in the acute management of 21 children with inborn errors of metabolism. Nephrol Dial Transplant. 2009 Nov 23.[↩]
- Cardenas JF, Bodensteiner JB. Osmotic Demyelination Syndrome as a Consequence of Treating Hyperammonemia in a Patient With Ornithine Transcarbamylase Deficiency. J Child Neurol. 2009 Feb 18.[↩]
{kind=link}