Contents
- What is malignant hyperthermia
- Disorders associated with malignant hyperthermia
- Can known malignant hyperthermia susceptible have surgery?
- Malignant hyperthermia medication
- Malignant hyperthermia causes
- Malignant hyperthermia prevention
- Malignant hyperthermia signs and symptoms
- Malignant hyperthermia complications
- Malignant hyperthermia diagnosis
- Malignant hyperthermia treatment
What is malignant hyperthermia
Malignant hyperthermia is a pharmacogenetic disorder of skeletal muscle that presents as a hypermetabolic response to potent volatile anesthetic gases such as halothane, sevoflurane, desflurane, isoflurane and the depolarizing muscle relaxant succinylcholine, and rarely, to stressors such as vigorous exercise and heat 1. The risk of an exercise-induced event is remote and patients should be advised to continue with a normal lifestyle although patients should be cautioned regarding the remote, but conceivable possibility of heat stroke in environments in which exposure to high heat and humidity is possible 1. Malignant hyperthermia is called a pharmacogenetic disorder because the reaction is caused by specific drugs. Specifically, malignant hyperthermia occurs in response to some anesthetic gases, which are used to block the sensation of pain, and with a muscle relaxant that is used to temporarily paralyze a person during a surgical procedure. If given these drugs, people at risk for malignant hyperthermia may experience muscle rigidity, breakdown of muscle fibers (rhabdomyolysis), a high fever that may exceed 110 degrees F (43.3 °C), increased acid levels in the blood and other tissues (acidosis), and a rapid heart rate. Severe complications include: cardiac arrest, brain damage, internal bleeding or failure of other body systems. Thus, death, primarily due to a secondary cardiovascular collapse, can result. An increase in end-tidal carbon dioxide despite increased minute ventilation provides an early diagnostic clue. Without prompt treatment, the complications of malignant hyperthermia can be life-threatening. Treatments for malignant hyperthermia include medication and ice packs to cool the body temperature. However, even when treated properly, malignant hyperthermia crisis can cause death 2. Survivors might be left with brain damage, failed kidneys, muscle damage or impaired function of other major organs 2.
In most cases, the genetic defect that causes malignant hyperthermia is inherited in an autosomal dominant pattern. If you have a parent, sibling or child with malignant hyperthermia, there is a 50 percent chance that you have the condition as well. Other close relatives, such as aunts, uncles and grandchildren, have a 25 percent chance. Men are more likely to have an episode of malignant hyperthermia than are women. Children with malignant hyperthermia also are susceptible to reactions during surgery. Genetic testing can reveal whether you have these mutations.
The incidence of malignant hyperthermia reactions ranges from 1:5,000 to 1: 250,000 anesthetics. However, the prevalence of the genetic abnormalities may be as great as one in 400 individuals. Susceptibility to malignant hyperthermia is probably more frequent, because many people with an increased risk of malignant hyperthermia are never exposed to anesthetic drugs that trigger a reaction.
People at increased risk for malignant hyperthermia are said to have malignant hyperthermia susceptibility. Affected individuals may never know they have the condition unless they undergo testing or have a severe reaction to anesthesia during a surgical procedure. While malignant hyperthermia often occurs in people without other serious medical problems, certain inherited muscle diseases (including central core disease and multiminicore disease) are associated with malignant hyperthermia susceptibility.
Uncontrolled rise of myoplasmic calcium, which activates biochemical processes related to muscle activation leads to the pathophysiologic changes. In most cases, the malignant hyperthermia syndrome is caused by a defect in the ryanodine receptor. Over 400 variants have been identified in the RYR1 gene located on chromosome 19q13.1, and at least 34 are causal for malignant hyperthermia. Less than 1 % of variants have been found in CACNA1S but not all of these are causal. Diagnostic testing involves the in vitro contracture response of biopsied muscle to halothane, caffeine, and in some centers ryanodine and 4-chloro-m-cresol. Elucidation of the genetic changes has led to the introduction of DNA testing for susceptibility to malignant hyperthermia. Dantrolene sodium is a specific antagonist and should be available wherever general anesthesia is administered. Increased understanding of the clinical manifestation and pathophysiology of the syndrome, has lead to the mortality decreasing from 80 % thirty years ago to <5 % in 2006.
Disorders associated with malignant hyperthermia
Succinylcholine induced masseter muscle rigidity occurs in 1 in 100 children with anesthesia induced by halothane and given succinylcholine 3. The incidence is probably the same following induction with sevoflurane, but much less following induction with thiopental 4. The clinical incidence of malignant hyperthermia as defined by arterial blood gas changes is about 15 % after masseter muscle rigidity. However, muscle biopsy reveals that 50 % of patients experiencing MMR are malignant hyperthermia susceptible 5. Patients with generalized rigidity along with masseter muscle rigidity are at much greater risk for malignant hyperthermia. Kaplan (personal communication,) has hypothesized that children with “jaws of steel” as opposed to mild rigidity after administration are at greater risk for malignant hyperthermia. He has hypothesized that the children with the more dramatic masseter rigidity are more often referred for biopsy and hence the high incidence of positive biopsies.
Central Core Disease is a rare non-progressive myopathy with mainly autosomal dominant inheritance, presenting in infancy and characterized by hypotonia and proximal muscle weakness. A few families demonstrate autosomal recessive inheritance. Histological examination of affected muscles shows a predominance of type I fibres containing clearly defined areas (cores) lacking oxidative enzyme activity 6.
Central Core Disease patients are often susceptible to malignant hyperthermia as confirmed by accepted muscle biopsy caffeine-halothane contracture testing (either in vitro contracture test or the caffeine halothane contracture test), but malignant hyperthermia and Central Core Disease phenotypes do not always co-segregate within families. Patients with malignant hyperthermia may present with cores despite being clinically asymptomatic and with some RYR1 variants (specifically some of those in the C-terminal transmembrane domain of the protein) specific to Central Core Disease. DNA Sequencing showed that RYR1 variants occurred in over 93 % (25 out of 27) of Japanese patients with Central Core Disease 7. While this is of importance, it may not reflect the incidence of RYR1 mutations in other populations. Another study indicated that the distribution and frequency of RYR1 variants differed markedly in the Japanese malignant hyperthermia susceptible population as compared to the North American and European malignant hyperthermia susceptible population 8. Although RYR1 variants are the most common identified cause of Central Core Disease, it does show genetic heterogeneity, with several rare susceptibility loci known (the ACTA1 gene, in association with nemaline myopathy, and the MYH7 gene, in association with hypertrophic cardiomyopathy), with further loci yet to be identified 9.
Other myopathies that have been suggested to be associated with malignant hyperthermia susceptibility include multiminicore myopathy and centronuclear myopathy. Multiminicore myopathy is an early onset congenital myopathy that may affect bulbar, respiratory and extraocular muscles and has autosomal recessive inheritance 10. Recessive variants in RYR1 have been associated with MmD, some of which result in altered Ca2+ release from intracellular stores and others that do not 11. Taken together, these observations suggest that there may be a subset of RYR1 variants that result in both malignant hyperthermia and multiminicore myopathy and a subset that are associated only with multiminicore myopathy, similar to the situation with malignant hyperthermia and Central Core Disease. Consequently, it will be important to distinguish between RYR1 variants that result in multiminicore myopathy, and those that do not.
King (or King-Denborough) syndrome 12 is a rare myopathy characterized by dysmorphic facies, ptosis, down-slanting palpebral fissures, hypertelorism, epicanthic folds, low-set ears, malar hypoplasia, micrognathia, high-arched palate, clinodactyly, palmar simian line, pectus excavatum, winging of the scapulae, lumbar lordosis and mild thoracic scoliosis. The patients with King-Denborough syndrome also present congenital hypotonia, slightly delayed motor development, diffuse joint hyperextensibility and mild proximal weakness. Such patients are malignant hyperthermia susceptible. Gillies et al. 13 identified a causative mutation in one family affected with King-Denborough syndrome. Dowling however, did not find a causative mutation to be a consistent feature in this syndrome 14.
Can known malignant hyperthermia susceptible have surgery?
Yes. Surgery can be safely performed in the known malignant hyperthermia-susceptible patients. However, only those anesthetics that do not trigger the malignant hyperthermia reaction must be used. In addition, close monitoring of appropriate vital functions is necessary. When dealing with an malignant hyperthermia susceptible, the anesthesiologist should:
- Avoid the use of malignant hyperthermia-triggering anesthetics.
- Be familiar with the signs and treatment of malignant hyperthermia, e.g., re-review the routine information.
- Continuously monitor the patient’s exhaled carbon dioxide concentration and minute ventilation.
- Continuously monitor the patient’s temperature (also during recovery). Skin temperature is not optimal in this situation.
- Have an malignant hyperthermia kit or cart within the operating room suite stocked with an adequate supply of dantrolene.
Can malignant hyperthermia occur outside of the operating room?
Yes. While most cases of malignant hyperthermia occur during general anesthesia, the one-hour period immediately following surgery (including the recovery room) is also a critical time. In addition, malignant hyperthermia can occur if trigger anesthetics and/or succinylcholine are used in any location, such as emergency rooms, dental surgeries, surgeon’s offices or intensive care units.
Malignant hyperthermia medication
Unsafe anesthetics 15:
- Inhaled General Anesthetics
- Desflurane
- Enflurane
- Ether
- Halothane
- Isoflurane
- Methoxyflurane
- Sevoflurane
- Succinylcholine
All other anesthetic agents not in the above categories of volatile anesthetic agents and depolarizing muscle relaxants are considered safe 15.
Malignant hyperthermia triggers
Numerous factors could be involved in triggering malignant hyperthermia – age, type of anesthetic, environmental temperature, mitigating drugs administered simultaneously, genetic makeup and degree and type of stress 16.
All inhalation anesthetics except nitrous oxide are triggers for malignant hyperthermia. The muscle relaxant succinylcholine is also a trigger for malignant hyperthermia. No other anesthetic drugs appear to be triggers, including propofol and ketamine. Neither are catecholamines, nondepolarizing muscle relaxants, catechol congeners, digitalis or similar agents 17.
Another potential risk factor is the use of inhalational sedation devices postoperatively in the intensive care unit (ICU) for a range of different conditions 18. Patients susceptible to malignant hyperthermia also resident in the ICU may be at risk from such exposure, although administration of sevoflurane via the AnaConDa® device was found to be safe for healthcare workers with the caveat that a gas extraction system should be used in conjunction with such devices to reduce occupational exposure 19. A case of malignant hyperthermia triggered by sevoflurane administration via an AnaConDa® was reported in a patient admitted to ICU for lumbalgia. malignant hyperthermia susceptibility was confirmed at a later date, highlighting the significance of malignant hyperthermia differential diagnosis in intensive care patients admitted for other conditions, if these types of sedation devices are used 20.
Can anything other than anesthetic drugs trigger malignant hyperthermia?
Studies have shown that a small percent of people who develop muscle breakdown following exercise only, or after heat stroke, harbor the genetic changes associated with malignant hyperthermia susceptibility. It is still unclear if the muscle breakdown and other changes result from these non-anesthetic incidences. In the absence of a personal or family history of heat stroke or exercise-induced muscle breakdown or evidence of muscle disorders, ask your personal physician to consult with an malignant hyperthermia expert.
Safe Medications
All Local Anesthetics are Safe 15:
- Barbiturates / Intravenous Anesthetics
- Diazepam
- Etomidate (Amidate)
- Hexobarbital
- Ketamine (Ketalar)
- Methohexital (Brevital)
- Midazolam
- Pentobarbital
- Propofol (Diprivan)
- Thiopental (Pentothal)
- Inhaled Non-Volatile General Anesthetic
- Nitrous Oxide
- Narcotics (Opioids)
- Alfentanil (Alfenta)
- Anileridine
- Codeine (Methyl Morphine)
- Diamorphine
- Fentanyl (Sublimaze)
- Hydromorphone (Dilaudid)
- Meperidine (Demerol)
- Methadone
- Morphine
- Naloxone
- Oxycodone
- Phenoperidine
- Remifentanil
- Sufentanil (Sufenta)
- Safe Muscle Relaxants
- Arduan (Pipecuronium)
- Curare (The active ingredient is Tubocurraine)
- Gallamine
- Methocarbamol (Robaxin, Robaxin-750, Carbacot, Skelex)
- Metocurine
- Mivacron (Mivacurium)
- Neuromax (Doxacurium)
- Nimbex (Cisatracurium)
- Norcuron (Vecuronium)
- Pavulon (Pancuronium)
- Tracrium (Atracurium)
- Zemuron (Rocuronium)
- Anxiety Relieving Medications
- Ativan (Lorazepam)
- Centrax
- Dalmane (Flurazepam)
- Halcion (Triazolam)
- Klonopin
- Librax
- Librium (Chlordiazepoxide)
- Midazolam (Versed)
- Paxil (paroxetine)
- Paxipam (Halazepam)
- Restoril (Temazepam)
- Serax (Oxazepam)
- Tranxene (Clorazepate)
- Valium (Diazepam)
Malignant hyperthermia causes
Malignant hyperthermia is caused by genes that you have at birth. If someone in your family has malignant hyperthermia and you need to have surgery, it’s important to tell your doctor. Other drugs may be used instead. Variations of the CACNA1S and RYR1 genes increase the risk of developing malignant hyperthermia. It is important to know that not everyone who has a gene defect linked to malignant hyperthermia develops the malignant hyperthermia crisis upon each exposure to the triggering anesthetics 2.
Malignant hyperthermia may not trigger a reaction during a person’s first surgery. However, the risk of a crisis remains for future surgeries. For those at risk of having a reaction, other safe medications are available.
In rare cases, people with malignant hyperthermia have shown signs of a reaction after intense physical activity.
Researchers have described at least six forms of malignant hyperthermia susceptibility, which are caused by mutations in different genes. Mutations in the RYR1 gene are responsible for a form of the condition known as MHS1. These mutations account for most cases of malignant hyperthermia susceptibility. Another form of the condition, MHS5, results from mutations in the CACNA1S gene. These mutations are less common, causing less than 1 percent of all cases of malignant hyperthermia susceptibility.
The RYR1 and CACNA1S genes provide instructions for making proteins that play essential roles in muscles used for movement (skeletal muscles). For the body to move normally, these muscles must tense (contract) and relax in a coordinated way. Muscle contractions are triggered by the flow of certain charged atoms (ions) into muscle cells. The proteins produced from the RYR1 and CACNA1S genes are involved in the movement of calcium ions within muscle cells. In response to certain signals, the CACNA1S protein helps activate the RYR1 channel, which releases stored calcium ions within muscle cells. The resulting increase in calcium ion concentration inside muscle cells stimulates muscle fibers to contract.
Mutations in the RYR1 or CACNA1S gene cause the RYR1 channel to open more easily and close more slowly in response to certain drugs. As a result, large amounts of calcium ions are released from storage within muscle cells. An overabundance of available calcium ions causes skeletal muscles to contract abnormally, which leads to muscle rigidity in people with malignant hyperthermia. An increase in calcium ion concentration within muscle cells also activates processes that generate heat (leading to increased body temperature) and produce excess acid (leading to acidosis).
The genetic causes of several other types of malignant hyperthermia (MHS2, MHS4, and MHS6) are still under study. A form of the condition known as MHS3 has been linked to the CACNA2D1 gene. This gene provides instructions for making a protein that plays an essential role in activating the RYR1 channel to release calcium ions into muscle cells. Although this gene is thought to be related to malignant hyperthermia in a few families, no causative mutations have been identified.
Malignant hyperthermia inheritance pattern
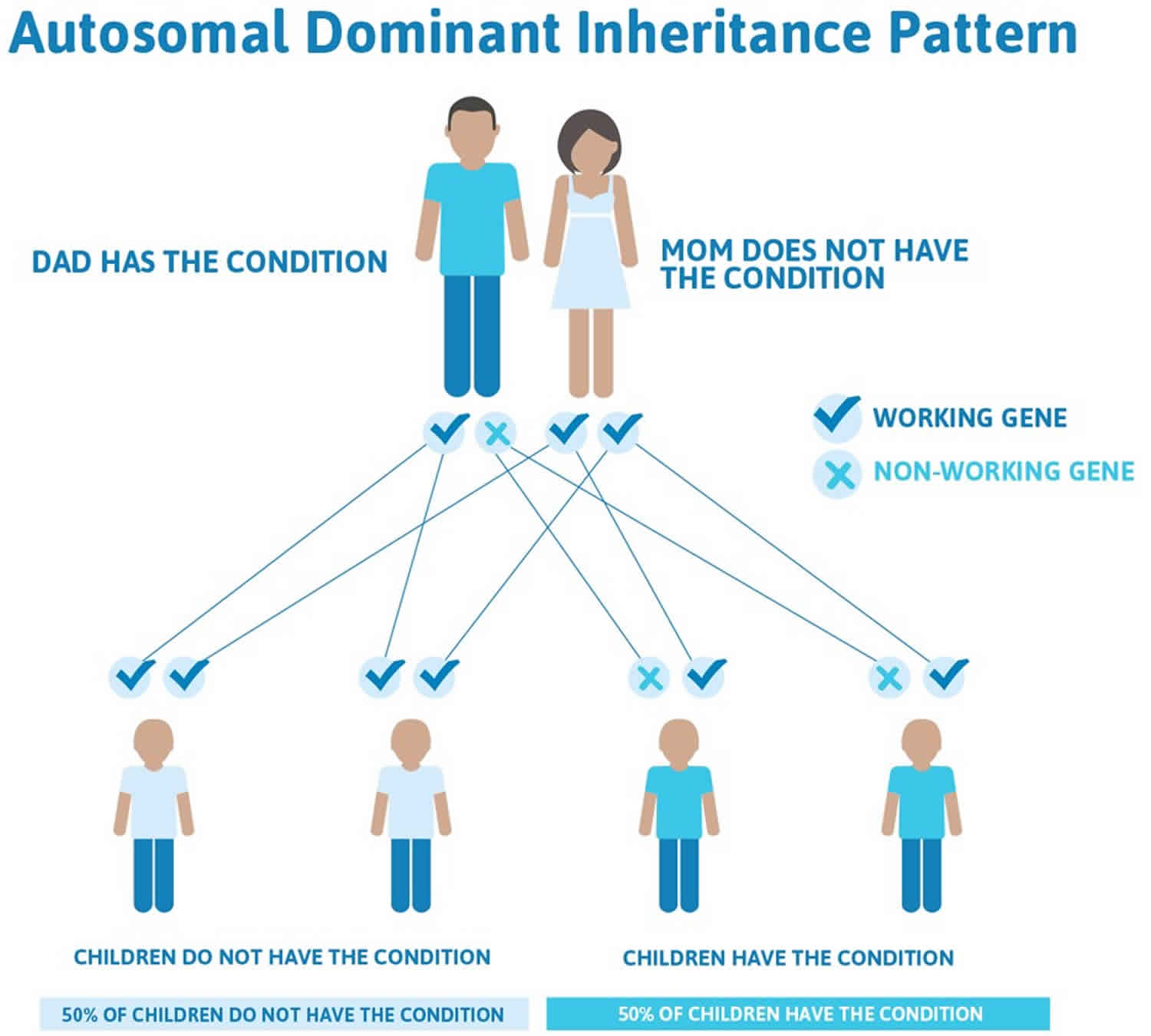
Malignant hyperthermia susceptibility is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to increase the risk of a severe reaction to certain drugs used during surgery. In most cases, an affected person inherits the altered gene from a parent who is also at risk for the condition.
- A person only needs to inherit one copy of the abnormal gene in order to be affected by malignant hyperthermia (50% chance). These outcomes occur randomly. They remain the same in every pregnancy and are the same for boys and girls.
In cases where the autosomal dominant condition does run in the family, the chance for an affected person to have a child with the same condition is 50% regardless of whether it is a boy or a girl. These possible outcomes occur randomly. The chance remains the same in every pregnancy and is the same for boys and girls.
- When one parent has the abnormal gene, they will pass on either their normal gene or their abnormal gene to their child. Each of their children therefore has a 50% (1 in 2) chance of inheriting the changed gene and being affected by the condition.
- There is also a 50% (1 in 2) chance that a child will inherit the normal copy of the gene. If this happens the child will not be affected by the disorder and cannot pass it on to any of his or her children.
Figure 1. Malignant hyperthermia autosomal dominant inheritance pattern
Risk factors for malignant hyperthermia
Your risk of having malignant hyperthermia is higher if someone in your family has the condition. If a parent, sibling or child has the condition, you have a 50 percent chance of having it too. You have a 25 percent chance of having the condition if a close relative like an aunt, uncle, or grandchild has it. Men are more likely than women to have malignant hyperthermia. If someone in your family has malignant hyperthermia and you need to have surgery, it’s important to tell your doctor. Other drugs may be used instead.
Malignant hyperthermia prevention
Tell your health care provider if you or anyone in your family has malignant hyperthermia, especially before having surgery with general anesthesia.
Avoid stimulant drugs such as cocaine, amphetamine (speed), and ecstasy. These drugs may cause problems similar to malignant hyperthermia in people who are prone to this condition.
Genetic counseling is recommended for anyone with a family history of myopathy, muscular dystrophy, or malignant hyperthermia.
Genetic counseling
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Malignant hyperthermia signs and symptoms
Signs and symptoms of malignant hyperthermia reaction include a dangerously high body temperature, severe muscle spasms and a fast heart rate.
Symptoms of malignant hyperthermia include:
- Bleeding
- Dark brown urine
- Muscle ache without an obvious cause, such as exercise or injury
- Muscle rigidity and stiffness
- Rise in body temperature to 105°F (40.6°C) or higher (hyperthermia)
Signs of malignant hyperthermia
The classic signs of malignant hyperthermia include hyperthermia, tachycardia (fast heart rate), tachypnea (rapid breathing), increased carbon dioxide production, increased oxygen consumption, acidosis, hyperkalemia (high blood potassium), muscle rigidity, and rhabdomyolysis (breakdown of skeletal muscle, which is associated with excretion of myoglobin in the urine), all related to a hypermetabolic response 1.
Several reports of isolated rhabdomyolysis apparent immediately following anesthesia or developing up to 24 h post anesthesia have been reported 21. Increased creatine kinase (CK) measurement and a positive in vitro contracture test have been obtained in these patients, indicating malignant hyperthermia susceptibility. Malignant hyperthermia-like muscle responses however, can represent false positive diagnoses and an underlying myopathic process may produce a positive vitro contracture test 21 so there must remain some doubt on the validity of this feature i.e., rhabdomyolysis as an expression of malignant hyperthermia. Burns et al. 22 stated however that malignant hyperthermia should be considered in all patients presenting with rhabdomyolysis where the degree of muscle necrosis exceeds that expected for the severity of the accompanying disorder. The most prudent diagnostic course, therefore, is contracture testing for malignant hyperthermia susceptibility.
Malignant hyperthermia complications
Malignant hyperthermia possible complications:
- Amputation
- Breakdown of muscle tissue
- Swelling of the hands and feet and problems with blood flow and nerve function (compartment syndrome)
- Death
- Abnormal blood clotting and bleeding
- Heart rhythm problems
- Kidney failure
- Buildup of acid in the body fluids (metabolic acidosis)
- Fluid buildup in the lungs
- Weak or deformed muscles (myopathy or muscular dystrophy)
A recent report from the North American Malignant Hyperthermia Registry of the Malignant Hyperthermia Association of the United States (MHAUS) demonstrated that early recognition of the signs of malignant hyperthermia and routine use of core temperature monitoring are essential in minimizing morbidity and mortality from malignant hyperthermia 1. Larach and colleagues 23 showed that in analyzing deaths from malignant hyperthermia, in 8 of 84 patients the risk of dying from malignant hyperthermia was about 14 times greater in those patients where core temperature monitoring was not used and 9.7 times greater where only skin temperature monitoring was used. The data also showed that the likelihood of any complication increased 2.9 times per 2o °C in maximum temperature and 1.6 times per 30 min delay in dantrolene use. Furthermore, the time interval between anesthetic induction to maximum end-tidal carbon dioxide (ETCO2) was longer in cases with cardiac arrest/death compared with the others (216 versus 87 min). Other signs include acidosis, tachypnea and hyperkalemia. The progression of the syndrome may be rapid and dramatic, particularly if precipitated by succinylcholine, or more slowly and not become manifest until after several hours after induction of anesthesia.
Malignant hyperthermia diagnosis
Malignant hyperthermia is often discovered after a person is given anesthesia during surgery.
There may be a family history of malignant hyperthermia or unexplained death during anesthesia.
The earliest signs are tachycardia, rise in end-expired carbon dioxide concentration despite increased minute ventilation, accompanied by muscle rigidity, especially following succinylcholine administration 1. Body temperature elevation can be a dramatic sign of malignant hyperthermia. Larach et al. 24 found that increased temperature was the first to third earliest sign in 63.5 % of malignant hyperthermia reactions. This confirms Sessler’s comment 25 that core temperature should be monitored in most patients undergoing general anesthesia for periods lasting more than 30 min and in all patients with anesthesia lasting 60 mins.
Although end-tidal carbon dioxide (ETCO2) is a sensitive early sign of malignant hyperthermia 26, in recent years, with a decline in the use of succinylcholine, rather than an abrupt rise in CO2, a more gradual rise is often noted. Indeed, by increasing minute ventilation it is possible to mask this rise 27.
Hyperthermia can be marked, with an increase in core temperature at a rate of 1–2 °C every five minutes. Severe hyperthermia (core temperature greater than 44 °C) may occur, and lead to a marked increase in oxygen consumption, CO2 production, widespread vital organ dysfunction, and disseminated intravascular coagulation (DIC) 28.
Uncontrolled hypermetabolism leads to respiratory and in most cases metabolic acidosis due to rapid consumption of energy stores and ATP. If untreated, continuing myocyte death and rhabdomyolysis result in life-threatening hyperkalemia; myoglobinuria may lead to acute renal failure. Additional life-threatening complications include DIC, congestive heart failure, bowel ischemia, and compartment syndrome of the limbs secondary to profound muscle swelling. Indeed, when body temperature exceeds approximately 41 °C, DIC is the usual cause of death.
A clinical grading scale was developed by Larach and colleagues 29 in order to assist in clinical diagnosis. The elements of the scale are given in Table 1. Differential weighting is given to each of the manifestations of the syndrome. The scale lacks sensitivity however, since not all tests may be performed in an individual episode.
Each process is weighted and scored according to its significance in differentiating malignant hyperthermia from other causes of change in the physiologic process. Only one element in each process need be present to qualify for scoring. A score is then generated assessing the likelihood of the episode being an malignant hyperthermia episode on a scale from almost never to almost certain. Being a clinical scale and depending on the presence of laboratory tests, its value resides mainly in identifying those subjects with the most convincing episodes of malignant hyperthermia for subsequent evaluation of the sensitivity and specificity of the diagnostic tests.
Table 1. Criteria used in the Clinical Grading Scale for Malignant Hyperthermia
| Process | Indicator |
|---|---|
| 1: Rigidity | a. Generalized muscular rigidity (in absence of shivering due to hypothermia, or during or immediately following emergence from inhalational anesthesia) |
| b. Masseter spasm shortly following succinylcholine administration | |
| 2: Muscle Breakdown | a. Elevated creatine kinase >20,000 IU after anesthetic that included succinylcholine |
| b. Elevated creatine kinase >10,000 IU after anesthetic without succinylcholine | |
| c. Cola colored urine in perioperative period | |
| d. Myoglobin in urine >60 μg/L | |
| e. Myoglobin in serum >170 μg/L | |
| f. Blood/plasma/serum K+ > 6 mEq/L (in absence of renal failure) | |
| 3: Respiratory Acidosis | a. PETCO2 > 55 mmHg with appropriately controlled ventilation |
| b. Arterial PaCO2 > 60 mmHg with appropriately controlled ventilation | |
| c. PETCO2 > 60 mmHg with spontaneous ventilation | |
| d. Arterial PaCO2 > 65 mmHg with spontaneous ventilation | |
| e. Inappropriate hypercarbia (in anesthesiologist’s judgment) | |
| f. Inappropriate tachypnea | |
| 4: Temperature Increase | a. Inappropriately rapid increase in temperature (in anesthesiologist’s judgement) |
| b. Inappropriately increased temperature > 38.8 °C (101.8 °F) in the perioperative period (in anesthesiologist’s judgment) | |
| 5: Cardiac Involvement | a. Inappropriate sinus tachycardia |
| b. Ventricular tachycardia or ventricular fibrillation |
Tests for malignant hyperthermia may include:
- Blood clotting studies (PT, or prothombin time; PTT, or partial thrombloplastin time)
- Blood chemistry panel, including CPK (creatinine phosphokinase, which is higher in the blood when muscle is destroyed during a bout of the illness)
- Genetic testing to look for defects in the genes that are linked with the disease
- Muscle biopsy
- Urine myoglobin (muscle protein)
Malignant hyperthermia testing
There are two types of testing for malignant hyperthermia: genetic testing and muscle biopsy. Consult with a genetic counselor to determine the type of test that is best for your situation.
Laboratory diagnostic methods
The “gold standard” for diagnosis of malignant hyperthermia is currently an in vitro contracture test, which is based on contracture of muscle fibers in the presence of halothane or caffeine. Two widely used forms of this test have been developed; one (in vitro contracture test) by the European Malignant Hyperthermia group 30 and the other (Caffeine Halothane Contracture Test) by the North American Malignant Hyperthermia Group 31. Using the European Malignant Hyperthermia group protocol, an individual is considered susceptible to malignant hyperthermia (malignant hyperthermia susceptible) when both caffeine and halothane test results are positive. An individual is considered not susceptible to malignant hyperthermia (not susceptible to malignant hyperthermia) when both tests are negative. An individual is also diagnosed as malignant hyperthermia susceptible when either a positive halothane or caffeine test alone is obtained and these individuals are designated malignant hyperthermia susceptible(h) or malignant hyperthermia susceptible(c). This nomenclature was determined at the 32nd European Malignant Hyperthermia group meeting in Basel, Switzerland, 2013. This test is similar to the North American Malignant Hyperthermia Group protocol but there are differences in the concentrations used and mode of testing agents. Sensitivity of 99 % and a specificity of 94 % are obtained with the European Malignant Hyperthermia group protocol 32 while figures of 97 % sensitivity and 78 % specificity are reported for the North American Malignant Hyperthermia Group protocol 33, which provide some confidence to the results obtained. The specificity of either protocol may be affected by neuromuscular disorders unrelated to malignant hyperthermia, which have an associated increase in myoplasmic Ca2+ concentration 31. Studies based on results from monozygotic twins however, indicate that the In Vitro Contracture Test has acceptable reproducibility 34. A third variation of the In Vitro Contracture Test, the caffeine skinned fiber test, does not appear to be used diagnostically outside of Japan, and has lower specificity and sensitivity than either the European Malignant Hyperpyrexia Group or North American Malignant Hyperthermia Group protocols 35.
In Vitro Contracture Test is expensive, confined to specialized testing centers, requires a surgical procedure and can yield false positive or negative results. Modifications of the European Malignant Hyperpyrexia Group protocol include the use of ryanodine 36 or 4-chloro-m-cresol 37 (but to date these agents have not been included in the standard protocol). A possible alternative testing agent is the fluorinated ether sevoflurane, however trials with this agent have not found responses consistent with halothane 38.
Other biochemical, hematological and physical tests lack significant sensitivity and specificity to be used diagnostically. A further caveat with these tests is that the results may be difficult to interpret in a patient suffering from a myopathy other than malignant hyperthermia such as Duchenne Muscular Dystrophy where intracellular Ca2+ is elevated at baseline.
A variety of minimally invasive diagnostic tests have been investigated. These include nuclear magnetic resonance spectroscopy to evaluate ATP depletion 39, metabolite assays and microdialysis of caffeine to elicit an enhanced release of carbon dioxide from the muscle tissue 40. The ethics of injecting a triggering agent, even a small volume into a potentially susceptible individual have to be questioned and determination of cutoff points would be difficult.
DNA analysis, however, offers an alternative to the In Vitro Contracture Test, requiring only a blood specimen, which can be sent to an accredited diagnostic laboratory. To date 50 to 70 % of malignant hyperthermia susceptibility has been linked to RYR1 with over 400 variants associated with malignant hyperthermia being identified within this gene 41. While the majority of variants lead to a single amino acid change in the receptor, deletions or truncations have also been reported. A number of recessive variants result in malignant hyperthermia, Central Core Disease or related disorders 42.
At least 44 variants have been reported in the RYR1 gene in association with Central Core Disease. In general terms, a single point RYR1 variant can cause (a) Central Core Disease only, (b) malignant hyperthermia only, (c) malignant hyperthermia with variable Central Core Disease penetrance. In this latter case, the likelihood of an RYR1 mutation resulting in both malignant hyperthermia and Central Core Disease depends on a number of factors including sensitivity of mutant protein to agonists, size of the intracellular Ca2+ pool and the level of abnormality in channel-gating 43. All individuals with the variant should be considered as malignant hyperthermia susceptible, while they may or may not have Central Core Disease. If a variant specific to Central Core Disease is identified in a family, malignant hyperthermia is not automatically excluded as a second variant may be present and malignant hyperthermia susceptibility needs to be assessed by IVCT or CHCT or family members treated as if they are malignant hyperthermia susceptible 44. An malignant hyperthermia negative parent eliminates susceptibility in the children although Central Core Disease may still be present.
While traditional DNA sequencing from either genomic DNA or complementary DNA prepared from muscle biopsy tissue are time consuming and laborious, the advent of massively parallel sequencing (or next generation sequencing, NGS) provides potentially cost effective, rapid and high throughput platforms for both variant discovery and diagnosis at the whole genome level 45. A number of RYR1 or CACNA1S variants have been identified using next generation sequencing (NGS) 46. Some caution in this approach should however, be exercised as none of the currently available platforms for sequencing, or chemistry for sample preparation, or analysis software are able to yield 100 % coverage of all exons in the human genome 47. Pathogenicity prediction is problematic (see below) and an additional consideration is the ethical dilemma associated with the reporting of incidental findings 48.
The European Malignant Hyperpyrexia Group has established criteria including functional studies of DNA variants to establish that the variant is clinically significant 49. Thirty-four mutations within RYR1 have been shown to cause an alteration in Ca2+ release from intracellular stores. A number of functional tests have been used successfully to assess the role of RYR1 variants in Ca2+ release. These include the use of lymphoblastoid cell lines generated from malignant hyperthermiaS individuals 50, COS-7 or HEK293 cells transfected with the cDNA for rabbit or human 51 RYR1 carrying point mutations introduced by site-directed mutagenesis, myotubes generated from muscle biopsy tissue and 1B5 dyspedic myotubes transduced with wild type or mutated RYR1 cDNA 52. Ca2+ release can be monitored and quantified directly using Ca2+-specific indicators or indirectly using [3H] ryanodine binding assays 53 or by proton release 54. Systems using 1B5 dyspedic myotubes are more physiological as they constitutively express all the components of the skeletal muscle with the exception of RYR1 55. To date, all mutations functionally characterized have been shown to cause alterations in Ca2+ flux through the ryanodine receptor Ca2+ release channel.
Malignant hyperthermia treatment
If you have a family history of malignant hyperthermia or have a family member who has problems with anesthesia, tell your surgeon and anesthesiologist prior to surgery. This step allows your doctors to prepare for and respond quickly to any reactions.
A drug called dantrolene (Dantrium) is used to treat the reaction. Ice packs, cooling blankets and fans may also be used to help reduce body temperature.
Dantrolene is the only drug known to specifically treat malignant hyperthermia. Dantrolene inhibits the dihydropyridine receptor in an RyR1-dependent manner [156], has been found to bind to a specific site on the RyR1 protein 56 and reduces RyR1 channel activity in intact muscle cells. The drug, introduced in 1979, has been responsible for lowering the mortality from malignant hyperthermia to 1.4 % in North America. The original preparation called Dantrium contains 20 mg of a lyophilized form of the drug per vial, which must be reconstituted before injection.
During an episode of malignant hyperthermia, dantrolene is often given. Wrapping the person in a cooling blanket can help reduce fever and the risk of serious complications.
To preserve kidney function during an episode, the person may receive fluids through a vein.
Acute malignant hyperthermia crisis treatment
The essential points in the treatment of an acute malignant hyperthermia crisis are the immediate discontinuation of trigger agents, hyperventilation, administration of dantrolene in doses of 2.5 mg/kg repeated pro re nata to limit malignant hyperthermia, cooling by all routes available (intravenous saline at 4o C, topical ice to all exposed areas, peritoneal exchange). Nasogastric lavage and bladder irrigation are contraindicated as complications such as gastric rupture can occur. Hyperkalaemia should be managed in a standard fashion. Ca2+ blockers viz verapamil should not be used along with dantrolene, since hyperkalemia and profound hypotension may occur with such a drug combination [16, 158]. The steps in the treatment of acute malignant hyperthermia are shown in Table 2.
Table 2. Managing an malignant hyperthermia crisis
| Action | Notes |
|---|---|
| Stop potent inhalation agents | Turn vaporisers “OFF” and /or activated charcoal filters inserted into the circuit |
| Do not repeat succinylcholine if it has been previously administered | |
| Increase minute ventilation to lower ETCO2 | Eliminate the inhalational agent |
| Get help | • Duty anesthestist |
| • Consultant anesthetist | |
| Prepare and administer dantrolene | • 2.5 mg/kg initial dose |
| • Every 10–15 min until acidosis, pyrexia, muscle rigidity are resolving | |
| Begin cooling measures if hyperthermic | • Tissue destruction will occur at 41.5 °C |
| • Use intravenous normal saline at 4 °C. | |
| • Ice Packs to all exposed areas | |
| • More aggressive measures as needed | |
| Stop cooling measures at 38.5 °C | |
| Treat arrhythmias as needed | • Amiodarone is the first choice |
| • Lignocaine | |
| • Do not use calcium channel blockers | |
| Secure blood gases, electrolytes, creatine kinase, blood and urine for myoglobin | • Coagulation profile check values regularly |
| • Treat hyperkalemia with hyperventilation, glucose and insulin as needed | |
| • Once crisis is under control, an MH hotline should be contacted for further guidance | |
| Continue dantrolene | • 1 mg/kg every 4–8 h for 24–48 h |
| • Alternatively and only if recrudescence occurs, dantrolene at 2.5 mg/kg bolus | |
| Ensure urine output of 2 mL/kg/h with | • Mannitol |
| • Furosemide | |
| • Fluids as needed | |
| Evaluate need for invasive monitoring and continued mechanical ventilation. | |
| Observe patient in Intensive Care Unit | At least 24 h |
| Refer patient and family for MH Testing | Contracture or DNA testing |
Emergency Treatment for An Acute malignant hyperthermia Event 57
The following four things should be done as soon as possible:
- Notify surgeon to halt the procedure ASAP: Discontinue volatile agents and succinylcholine.
- If surgery must be continued, maintain general anesthesia with IV non-triggering anesthetics (e.g., IV sedatives, narcotics, amnestics and non-depolarizing neuromuscular blockers as needed)
- Get dantrolene/malignant hyperthermia cart.
- Call for help within your institution; also, call the MHAUS (Malignant Hyperthermia Association of the United States) Hotline (1-800-644-9737) for additional advice. (Outside the US, please call: 001-209-417-3722)
- Hyperventilate with 100% oxygen at flows of 10L/min to flush volatile anesthetics and lower ETCO2. If available, insert activated charcoal filters (Vapor-Clean™, Dynasthetics, Salt Lake City, UT) into the inspiratory and expiratory limbs of the breathing circuit. The Vapor-Clean™ filter may become saturated after one hour; therefore, a replacement set of filters should be substituted after each hour of use.
- Give IV dantrolene 2.5 mg/kg rapidly through large-bore IV, if possible. Repeat as frequently as needed until the patient responds with a decrease in ETCO2, decreased muscle rigidity, and/or lowered heart rate. Large doses (>10mg/kg) may be required for patients with persistent contractures or rigidity.
- DANTRIUM®/REVONTO® – Each 20 mg vial should be reconstituted by adding 60 ml of sterile water for injection, USP (without a bacteriostatic agent) and the vial shaken until the solution is clear.
- RYANODEX®– Each 250 mg vial should be reconstituted with 5 ml of sterile water for injection, USP (without a bacteriostatic agent) and shaken to ensure an orange-colored uniform, opaque suspension.
If giving large doses (> 10 mg/kg) without symptom resolution, consider alternative diagnoses.
Obtain blood gas (venous or arterial) to determine degree of metabolic acidosis. Consider administration of sodium bicarbonate, 1-2 mEq/kg dose, for base excess greater than -8 (maximum dose 50 mEq).
Cool the patient if core temperature is >39°C or less if rapidly rising. Stop cooling when the temperature has decreased to <38°C.
If hyperkalemia (K > 5.9 or less with ECG changes) is present, treat with:
- Calcium chloride 10 mg/kg (maximum dose 2,000 mg) or calcium gluconate 30 mg/kg (maximum dose 3,000 mg) for life-threatening hyperkalemia
- Sodium bicarbonate
- 1-2 mEq/kg IV (maximum dose 50 mEq)
- Glucose/insulin
- For pediatric patients: 0.1 units regular insulin/kg IV and 0.5 grams/kg dextrose (% in formulation not important)
- For adult patients: 10 units regular insulin IV and 50 ml 50% dextrose
- Check glucose levels hourly
For refractory hyperkalemia, consider albuterol (or other beta-agonist), kayexelate, dialysis, or ECMO if patient is in cardiac arrest.
Treat dysrhythmias with standard medication but avoid calcium channel blockers. Treat acidosis and hyperkalemia if present. (See above)
- Diurese to >1ml/kg/hr urine output. If CK or K+ rise, assume myoglobinuria and give bicarbonate infusion of 1 mEq/kg/hr, to alkalinize urine
Institute appropriate monitoring including: core temperature, urine output with bladder catheter, and consider arterial and/or central venous monitoring if warranted by the clinical severity of the patient.
Follow: HR, core temperature, ETCO2, minute ventilation, blood gases, K+, CK, urine myoglobin and coagulation studies as warranted by the clinical severity of the patient.
When stable, transfer to post anesthesia care unit or intensive care unit for at least 24 hours. Key indicators of stability include:
- ETCO2 is declining or normal
- Heart rate is stable or decreasing with no signs of ominous dysrhythmias
- Hyperthermia is resolving
- If present, generalized muscular rigidity has resolved.
How does the antidote dantrolene work?
Dantrolene is the only currently accepted specific treatment for malignant hyperthermia. In an episode of malignant hyperthermia, muscle metabolism is dramatically increased secondary to an increase in calcium within the muscle. This causes muscles to contract, ATP hydrolysis, and heat production. Dantrolene directly interferes with muscle contraction; decreasing calcium in muscle cells.
Dantrolene does not block neuromuscular transmission nor interfere with reversal of muscle relaxants. Although it does not block neuromuscular transmission, the mechanical response to nerve stimulation will be depressed, with subsequent potentiation of the non-depolarizing neuromuscular blockade. When dantrolene is used with non-depolarizing muscle relaxants, care should be taken to ensure muscle strength has returned prior to extubation.
Dantrolene may cause significant muscle weakness in patients with pre-existing muscle disease and should be used with extreme caution in those patients. Sterile phlebitis may follow administration of dantrolene, and should be infused through the largest possible vein. The sterile phlebitis can be later treated with warm soaks and elevation. When used with calcium channel blockers (verapamil or diltiazem), dantrolene may produce life-threatening hyperkalemia and myocardial depression. Otherwise there does not appear to be significant negative interaction with other drugs.
Once a patient has been successfully treated for 48 hours with intravenous dantrolene may be stopped and the blood tested daily until the CK level is trending down.
NOTE: Dantrolene should not be mixed with any other diluent other than sterile water. The drug will not completely dissolve in crystalloid-containing solutions.
How quickly must dantrolene be accessible?
Dantrolene must be available for all anesthetizing locations within 10 minutes of the decision to treat for malignant hyperthermia. Dantrolene must be available for all anesthetizing locations where malignant hyperthermia trigger agents are used.” This is a slight modification of the current recommendation that the drug be available within five minutes because the five minute recommendation was not made based on consensus discussion and it is often not practical to have a large supply of dantrolene in every area where anesthesia is administered. For example anesthesia administration is now common in locations far from the operating rooms such as interventional radiology suites. This comment and others were made at the malignant hyperthermia Hotline – Professional Advisory Council meeting held on May 14, 2011.
The Professional Advisory Council of MHAUS strongly recommends that an adequate supply of dantrolene be available wherever general anesthesia is administered. Responsibility for treatment rests with the facility where the surgery is performed.
- Rosenberg H, Pollock N, Schiemann A, Bulger T, Stowell K. Malignant hyperthermia: a review. Orphanet J Rare Dis. 2015;10:93. Published 2015 Aug 4. doi:10.1186/s13023-015-0310-1 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4524368/[↩][↩][↩][↩][↩][↩][↩]
- Malignant Hyperthermia Frequently Asked Questions. https://www.mhaus.org/faqs/[↩][↩][↩]
- Schwartz L, Rockoff MA, Koka BV. Masseter spasm with anesthesia: incidence and implications. Anesthesiology. 1984;61(6):772–5.[↩]
- Lazzell VA, Carr AS, Lerman J, Burrows FA, Creighton RE. The incidence of masseter muscle rigidity after succinylcholine in infants and children. Can J Anaesth. 1994;41(6):475–9.[↩]
- O’Flynn RP, Shutack JG, Rosenberg H, Fletcher JE. Masseter muscle rigidity and malignant hyperthermia susceptibility in pediatric patients. An update on management and diagnosis. Anesthesiology. 1994;80(6):1228–33.[↩]
- Jungbluth H, Sewry CA, Muntoni F. Core myopathies. Semin Pediatr Neurol. 2011;18(4):239–49.[↩]
- Wu S, Ibarra MC, Malicdan MC, Murayama K, Ichihara Y, Kikuchi H, et al. Central core disease is due to RYR1 mutations in more than 90 % of patients. Brain. 2006;129(Pt 6):1470–80.[↩]
- Ibarra MC, Wu S, Murayama K, Minami N, Ichihara Y, Kikuchi H, et al. Malignant hyperthermia in Japan: mutation screening of the entire ryanodine receptor type 1 gene coding region by direct sequencing. Anesthesiology. 2006;104(6):1146–54.[↩]
- Jungbluth H. Central core disease. Orphanet J Rare Dis. 2007;2:25[↩]
- Wei L, Dirksen RT. Ryanodinopathies: RyR-Linked Muscle Diseases. Curr Top Membr. 2010;66:139–67.[↩]
- Ducreux S, Zorzato F, Ferreiro A, Jungbluth H, Muntoni F, Monnier N, et al. Functional properties of ryanodine receptors carrying three amino acid substitutions identified in patients affected by multi-minicore disease and central core disease, expressed in immortalized lymphocytes. Biochem J. 2006;395(2):259–66.[↩]
- King JO, Denborough MA. Anesthetic-induced malignant hyperpyrexia in children. J Pediatr. 1973;83(1):37–40.[↩]
- D’Arcy CE, Bjorksten A, Yiu EM, Bankier A, Gillies R, McLean CA, et al. King-denborough syndrome caused by a novel mutation in the ryanodine receptor gene. Neurology. 2008;71(10):776–7.[↩]
- Dowling JJ, Lillis S, Amburgey K, Zhou H, Al-Sarraj S, Buk SJ, et al. King-Denborough syndrome with and without mutations in the skeletal muscle ryanodine receptor (RYR1) gene. Neuromuscul Disord. 2011;21(6):420–7.[↩]
- Safe and Unsafe Anesthetics. https://www.mhaus.org/healthcare-professionals/be-prepared/safe-and-unsafe-anesthetics[↩][↩][↩]
- Ording H. Incidence of malignant hyperthermia in Denmark. Anesth Analg. 1985;64(7):700–4.[↩]
- Hopkins PM. Malignant hyperthermia: advances in clinical management and diagnosis. Br J Anaesth. 2000;85(1):118–28.[↩]
- Bomberg H, Glas M, Groesdonk VH, Bellgardt M, Schwarz J, Volk T, et al. A novel device for target controlled administration and reflection of desflurane–the Mirus. Anaesthesia. 2014;69(11):1241–50.[↩]
- Gonzalez-Rodriguez R, Munoz Martinez A, Galan Serrano J, Moral Garcia MV. Health worker exposure risk during inhalation sedation with sevoflurane using the (AnaConDa®) anaesthetic conserving device. Rev Esp Anestesiol Reanim. 2014;61(3):133–9.[↩]
- Johannsen S, Mögele S, Roewer N, Schuster F. Malignant hyperthermia on ICU – sudden attack of the “snake” BMC Anesthesiol. 2014;14:A11.[↩]
- McKenney KA, Holman SJ. Delayed postoperative rhabdomyolysis in a patient subsequently diagnosed as malignant hyperthermia susceptible. Anesthesiology. 2002;96(3):764–5.[↩][↩]
- Burns AP, Hopkins PM, Hall G, Pusey CD. Rhabdomyolysis and acute renal failure in unsuspected malignant hyperpyrexia. Q J Med. 1993;86(7):431–4.[↩]
- Larach MG, Brandom BW, Allen GC, Gronert GA, Lehman EB. Malignant hyperthermia deaths related to inadequate temperature monitoring, 2007–2012: a report from the North American malignant hyperthermia registry of the malignant hyperthermia association of the United States. Anesth Analg. 2014;119(6):1359–66.[↩]
- Larach MG, Allen GC, Brandom BW, Lehman EB. Temperature changes are not late signs of malignant hyperthermia: A NAMH Registry of MHAUS study. Anesthesiology. 2008;109:A374.[↩]
- Sessler DI. Temperature monitoring and perioperative thermoregulation. Anesthesiology. 2008;109(2):318–38.[↩]
- Pollock AN, Langton EE, Couchman K, Stowell KM, Waddington M. Suspected malignant hyperthermia reactions in New Zealand. Anaesth Intensive Care. 2002;30(4):453–61.[↩]
- Karan SM, Crowl F, Muldoon SM. Malignant hyperthermia masked by capnographic monitoring. Anesth Analg. 1994;78(3):590–2.[↩]
- Nelson TE. Porcine malignant hyperthermia: critical temperatures for in vivo and in vitro responses. Anesthesiology. 1990;73(3):449–54.[↩]
- Larach MG, Localio AR, Allen GC, Denborough MA, Ellis FR, Gronert GA, et al. A clinical grading scale to predict malignant hyperthermia susceptibility. Anesthesiology. 1994;80(4):771–9.[↩]
- A protocol for the investigation of malignant hyperpyrexia (MH) susceptibility. The European Malignant Hyperpyrexia Group. British Journal of Anaesthesia. 1984; 56(11):1267–9.[↩]
- Larach MG, Landis JR, Bunn JS, Diaz M. Prediction of malignant hyperthermia susceptibility in low-risk subjects. An epidemiologic investigation of caffeine halothane contracture responses. The North American Malignant Hyperthermia Registry. Anesthesiology. 1992;76(1):16–27.[↩][↩]
- Ording H, Brancadoro V, Cozzolino S, Ellis FR, Glauber V, Gonano EF, et al. In vitro contracture test for diagnosis of malignant hyperthermia following the protocol of the European MH Group: results of testing patients surviving fulminant MH and unrelated low-risk subjects. The European Malignant Hyperthermia Group. Acta Anaesthesiol Scand. 1997;41(8):955–66.[↩]
- Allen GC, Larach MG, Kunselman AR. The sensitivity and specificity of the caffeine-halothane contracture test: a report from the North American Malignant Hyperthermia Registry. The North American Malignant Hyperthermia Registry of MHAUS. Anesthesiology. 1998;88(3):579–88.[↩]
- Islander G, Ranklev TE. Results of in vitro contracture tests for the diagnosis of malignant hyperthermia susceptibility in monozygote twins. Acta Anaesthesiol Scand. 1997;41(6):731–5.[↩]
- Endo M, Yagi S, Ishizuka T, Horiuti K, Koga Y, Amaha K. Changes in the Ca-induced Ca release mechanism in the sarcoplasmic reticulum of the muscle from a patient with malignant hyperthermia. Biomed Res. 1983;4:83–92.[↩]
- Bendahan D, Guis S, Monnier N, Kozak-Ribbens G, Lunardi J, Ghattas B, et al. Comparative analysis of in vitro contracture tests with ryanodine and a combination of ryanodine with either halothane or caffeine: a comparative investigation in malignant hyperthermia. Acta Anaesthesiol Scand. 2004;48(8):1019–27.[↩]
- Baur CP, Bellon L, Felleiter P, Fiege M, Fricker R, Glahn K, et al. A multicenter study of 4-chloro-m-cresol for diagnosing malignant hyperthermia susceptibility. Anesth Analg. 2000;90(1):200–5.[↩]
- Johannsen S, Klingler W, Schneiderbanger D, Heiderich S, Roewer N, Schuster F. Sevoflurane is less sensitive than halothane for in vitro detection of malignant hyperthermia susceptibility. Acta Anaesthesiol Scand. 2013;57(9):1161–6.[↩]
- Payen JF, Bosson JL, Bourdon L, Jacquot C, Le Bas JF, Stieglitz P, et al. Improved noninvasive diagnostic testing for malignant hyperthermia susceptibility from a combination of metabolites determined in vivo with 31P-magnetic resonance spectroscopy. Anesthesiology. 1993;78(5):848–55.[↩]
- Metterlein T, Schuster F, Kranke P, Roewer N, Anetseder M. Minimally invasive metabolic testing for malignant hyperthermia susceptibility: a systematic review of the methodology and results. Expert Opinion Medical Diagn. 2010;4(2):149–58.[↩]
- ClinVar-National Center for Biotechnology Information http://www.ncbi.nlm.nih.gov/clinvar[↩]
- Bevilacqua JA, Monnier N, Bitoun M, Eymard B, Ferreiro A, Monges S, et al. Recessive RYR1 mutations cause unusual congenital myopathy with prominent nuclear internalization and large areas of myofibrillar disorganization. Neuropathol Appl Neurobiol. 2011;37(3):271–84.[↩]
- McCarthy TV, Quane KA, Lynch PJ. Ryanodine receptor mutations in malignant hyperthermia and central core disease. Hum Mutat. 2000;15(5):410–7.[↩]
- Robinson RL, Brooks C, Brown SL, Ellis FR, Halsall PJ, Quinnell RJ, et al. RYR1 mutations causing central core disease are associated with more severe malignant hyperthermia in vitro contracture test phenotypes. Hum Mutat. 2002;20(2):88–97.[↩]
- Stowell KM. DNA testing for malignant hyperthermia: the reality and the dream. Anesth Analg. 2014;118(2):397–406.[↩]
- Broman M, Kleinschnitz I, Bach JE, Rost S, Islander G, Muller CR. Next-generation DNA sequencing of a Swedish malignant hyperthermia cohort. Clinical Genetics. 2014. Sep 25. 10.1111/cge.12508.[↩]
- Rieber N, Zapatka M, Lasitschka B, Jones D, Northcott P, Hutter B, et al. Coverage bias and sensitivity of variant calling for four whole-genome sequencing technologies. PLoS One. 2013;8(6):e66621[↩]
- Burke W, Antommaria AH, Bennett R, Botkin J, Clayton EW, Henderson GE, et al. Recommendations for returning genomic incidental findings? We need to talk! Genet Med. 2013;15(11):854–9[↩]
- European Malignant Hyperthermia Group. https://www.emhg.org/[↩]
- Schiemann AH, Paul N, Parker R, Pollock N, Bulger TF, Stowell KM. Functional characterization of 2 known ryanodine receptor mutations causing malignant hyperthermia. Anesth Analg. 2014;118(2):375–80.[↩]
- Roesl C, Sato K, Schiemann A, Pollock N, Stowell KM. Functional characterisation of the R2452W ryanodine receptor variant associated with malignant hyperthermia susceptibility. Cell Calcium. 2014;56(3):195–201[↩]
- Lefebvre R, Legrand C, Groom L, Dirksen RT, Jacquemond V. Ca2+ release in muscle fibers expressing R4892W and G4896V type 1 ryanodine receptor disease mutants. PLoS One. 2013;8(1):e54042[↩]
- Sato K, Pollock N, Stowell KM. Functional studies of RYR1 mutations in the skeletal muscle ryanodine receptor using human RYR1 complementary DNA. Anesthesiology. 2010;112(6):1350–4.[↩]
- Zullo A, Klingler W, De Sarno C, Ferrara M, Fortunato G, Perrotta G, et al. Functional characterization of ryanodine receptor (RYR1) sequence variants using a metabolic assay in immortalized B-lymphocytes. Hum Mutat. 2009;30(4):E575–90.[↩]
- Buck ED, Nguyen HT, Pessah IN, Allen PD. Dyspedic mouse skeletal muscle expresses major elements of the triadic junction but lacks detectable ryanodine receptor protein and function. J Biol Chem. 1997;272(11):7360–7.[↩]
- Bannister RA. Dantrolene-induced inhibition of skeletal L-type Ca2+ current requires RyR1 expression. BioMed Res Int. 2013;2013:390493.[↩]
- Managing A Crisis. https://www.mhaus.org/healthcare-professionals/managing-a-crisis/[↩]
{kind=link}