Contents
What is Myeloneuropathy
Myeloneuropathy also called myelopathy is any disease affecting the spinal cord and peripheral nerves in the lower limbs 1. Myeloneuropathy signs and symptoms include difficulty in walking, weakness of lower limbs, ataxic gait, and sensory manifestations in glove and stocking distribution 1. On examination, there are myelopathic signs such as hyperreflexia, spasticity, extensor plantar responses, and infrequently, bladder bowel disturbances 1. Romberg sign indicates involvement of the posterior column 1. Classical neuropathic signs include glove and stocking sensory loss, absent or diminished ankle jerk, and distal limb atrophy 1. Cognitive impairment and vision loss, because of optic nerve damage may occasionally dominate the clinical picture in a patient with myeloneuropathy 1. All of these patients have gait difficulty primarily due to severe sensory ataxia 2, 3.
A correct diagnosis of myeloneuropathy is often challenging. Vitamin B12, folate, copper, and vitamin E deficiencies can lead to myeloneuropathy. The clinical picture in all these conditions is similar to that of mimics subacute combined degeneration of the spinal cord. The pattern of neurologic involvement and results obtained from biochemical tests and imaging studies can help in establishing the correct diagnosis.
Figure 1. Myeloneuropathy causes
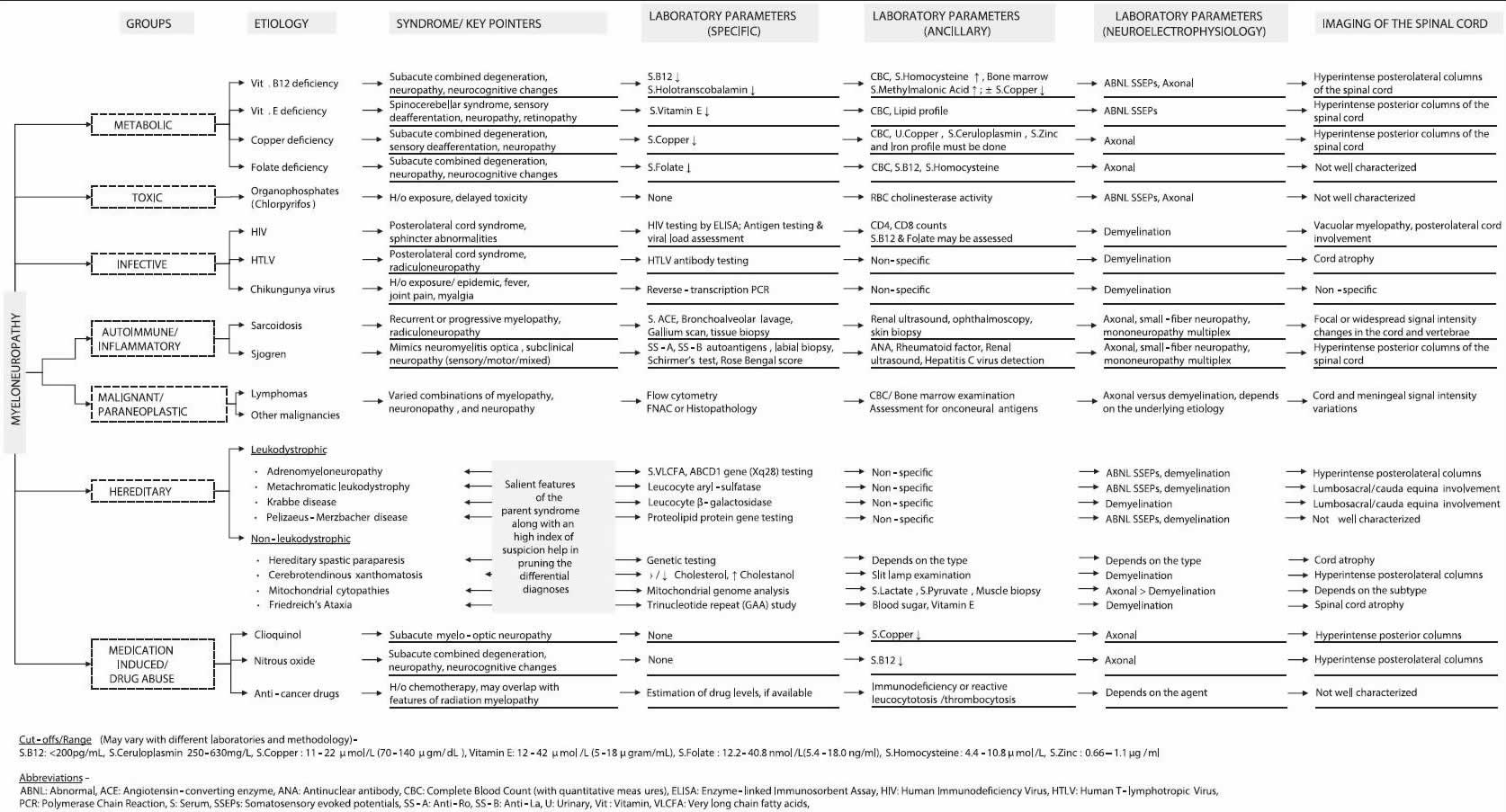
Figure 2. Common nutritional, metabolic, and toxic causes of myeloneuropathy
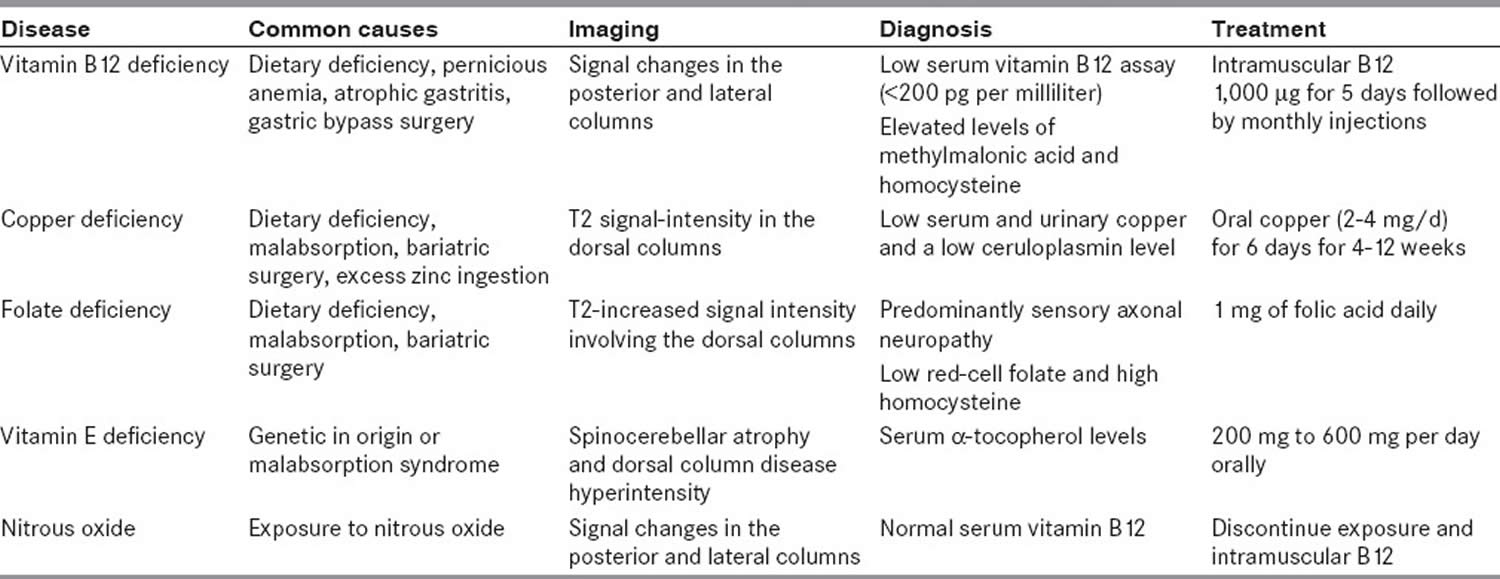
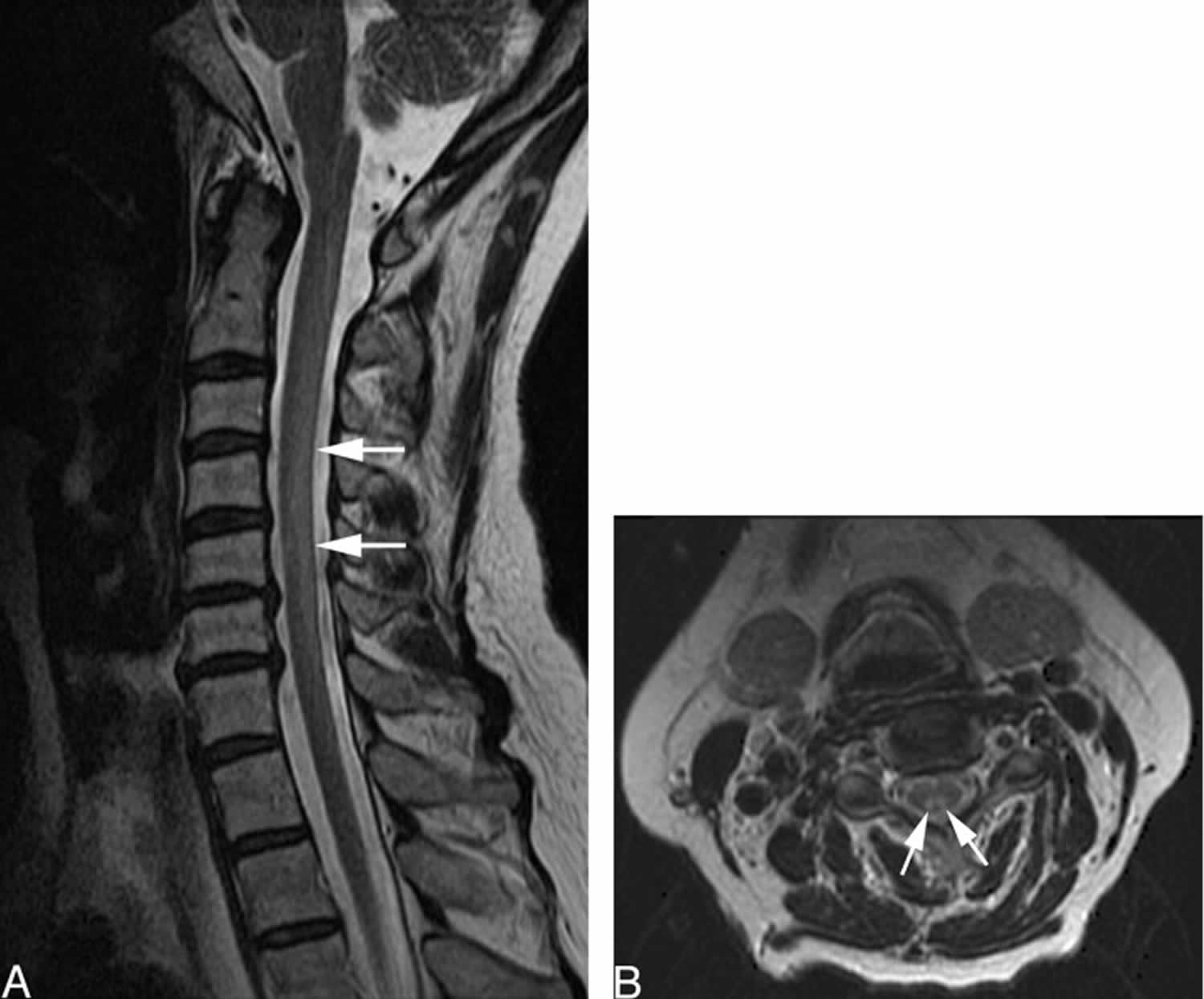
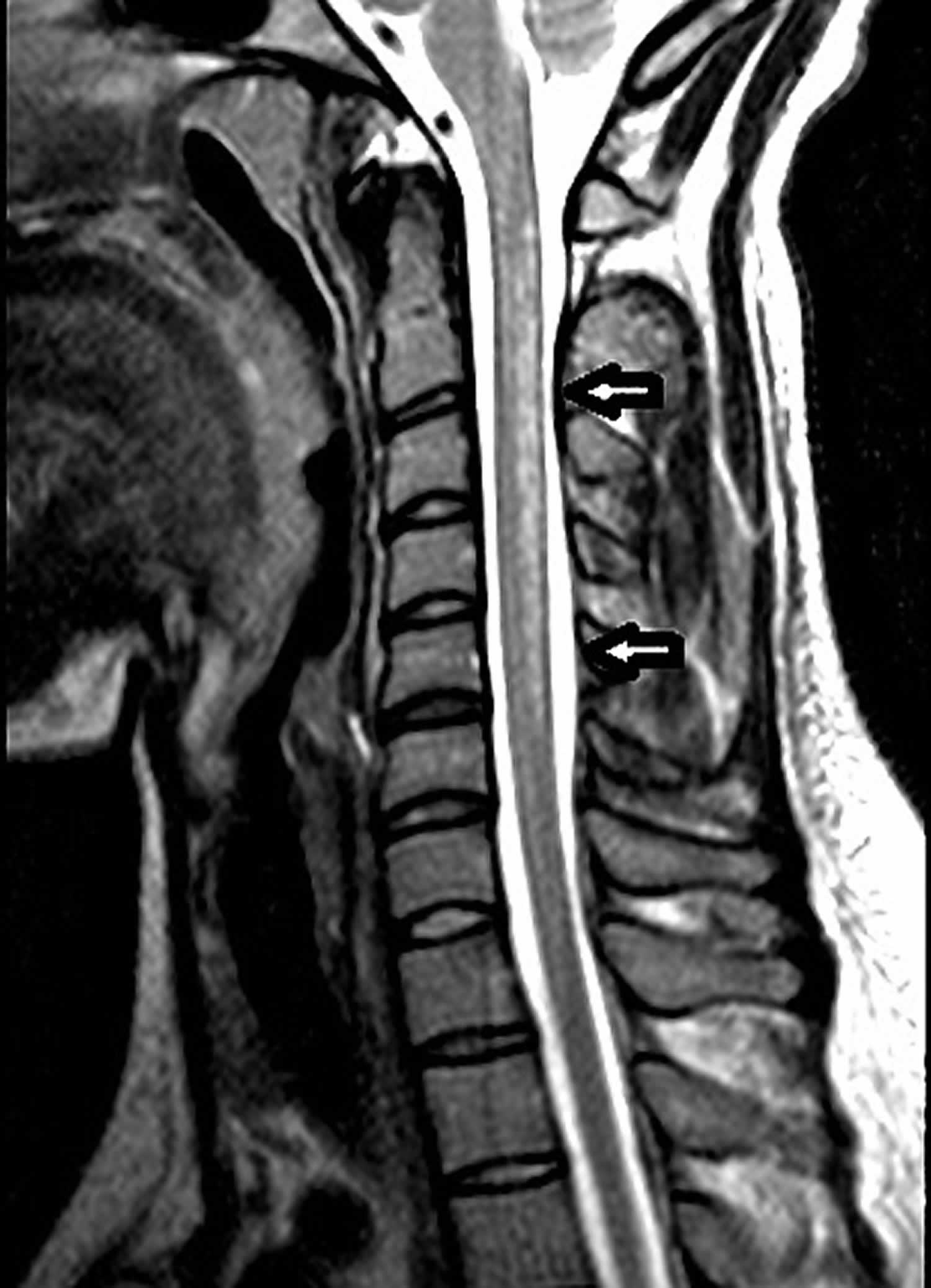
Figure 3. Copper deficiency myeloneuropathy
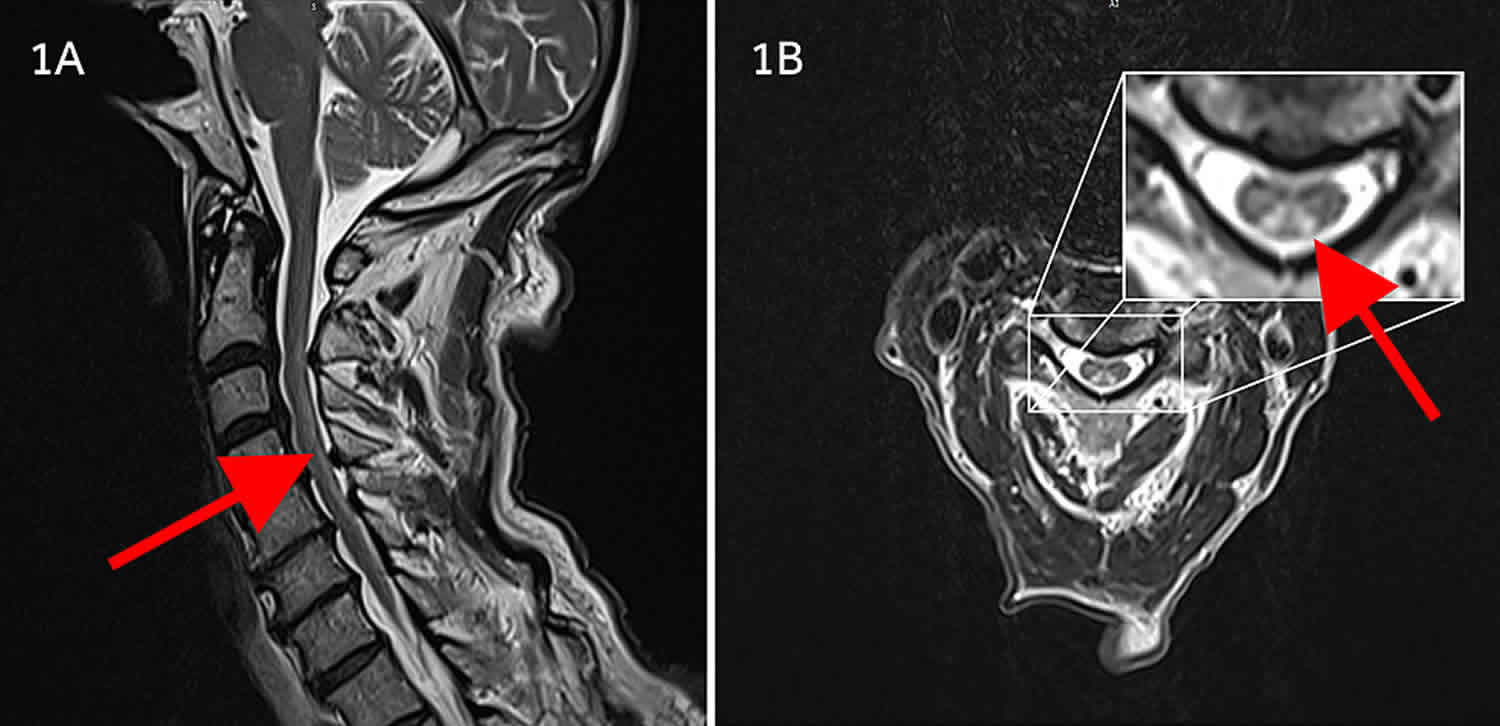
Footnotes: Sagittal and axial fast spin-echo MRI T2-weighted images of the cervical spine. Sagittal (A) and axial (B) fast spin-echo MRI of the cervical spine before treatment demonstrate diffuse increase in signal intensity (arrows) involving the dorsal columns of the cervical spinal cord extending from C1 to C7. The thoracic levels (not shown) were not involved. Findings of MR imaging of the brain were normal.
[Source 4 ]Figure 4. Nitrous oxide induced myeloneuropathy
Footnotes: A 20 year old female with a history of nitrous oxide abuse, she used about 5-10 canisters of nitrous oxide per week. She presented with tingling sensation in all extremities and burning sensation in bilateral soles; weakness and difficulty walking for 1 week. Physical exam reveals decreased light touch and pinprick sensation in all extremities with distal to proximal gradient. Decreased vibration sense in both feet. Hyporeflexia in upper limbs and absent reflexes in both lower limbs. Rhomberg sign weakly positive. No bladder or bowel incontinence. Her blood tests revealed: vitamin B12 = 279pg/mL, homocysteine = 58 nmol/mL and methylmalonic acid (MMA) = 1.59 nmol/mL. MRI of her spinal cord showed patchy increased signal intensity from C1-C5 affecting dorsal column. Axial T2 mid-cervical demonstrates bilateral hyperintensity in the dorsal column (inverted V sign). No cord compression or edema seen.
[Source 5 ]Myeloneuropathy causes
A variety of nutritional, toxic, metabolic, infective, inflammatory, and paraneoplastic disorders can present with myeloneuropathy 1. Deficiencies of vitamin B12, folic acid, copper, and vitamin E may lead to myeloneuropathy with a clinical picture of subacute combined degeneration of the spinal cord 1. Chikungunya virus has been shown to produce a syndrome similar to myeloneuropathy 1. Vacuolar myelopathy seen in human immunodeficiency virus (HIV) infection is clinically very similar to subacute combined degeneration 1. A paraneoplastic myeloneuropathy, an immune-mediated disorder associated with an underlying cancer, may rarely be seen with breast cancer 1. Tropical myeloneuropathies are classified into two overlapping clinical entities — tropical ataxic neuropathy and tropical spastic paraparesis 1.
Copper deficiency myeloneuropathy
Copper deficiency is a rare and potentially treatable cause of myeloneuropathy 6, 7. Copper is an essential mineral that function as a cofactor (a compound that is essential for the activity of an enzyme) for several enzymes known as copper-dependent enzymes (cuproenzymes) involved in energy (ATP) production, iron metabolism, neuropeptide activation, connective tissue synthesis, and neurotransmitter synthesis 8, 9, 10, 11, 12. One abundant copper-dependent enzyme is ceruloplasmin, which plays a role in iron metabolism and carries more than 95% of the total copper in healthy human plasma 13. Copper is also involved in many physiologic processes, such as angiogenesis; neurohormone homeostasis; and regulation of gene expression, brain development, brain and nervous system physiological processes including neurotransmitter synthesis and formation and maintenance of myelin, pigmentation, and immune system functioning 10, 14, 15. In addition, defense against oxidative damage depends mainly on the copper-containing superoxide dismutases 16, 17.
Clinically evident, or frank, dietary copper deficiency is relatively uncommon in humans 11. Based on studies in animals and humans, the effects of copper deficiency include anemia, hypopigmentation, hypercholesterolemia, connective tissue disorders, osteoporosis and other bone defects, abnormal lipid metabolism, central nervous system demyelination, ataxia, polyneuropathy, myelopathy, inflammation of the optic nerve, and increased risk of infection 10, 18, 19.
The harmful effects of copper deficiency 20:
- Increased cholesterol, decreased glucose tolerance and abnormal electrocardiograms (ECGs) 21.
- Increased LDL and triglycerides and decreased HDL 22
- Increased susceptibility of lipoproteins and tissues to oxidation 23.
- Increased apolipoprotein B 23.
- Increased blood pressure 24.
- Increased plasminogen activator inhibitor type 1 25
- Increased early and advanced glycation end-products 26
- Increased inflammation43 and increase in the expression of genes involved in inflammation and fibrinogenesis 27
- Ultrastructural irregularities of elastin and abnormal endothelial cells, subendothelial space, collagen fibres and smooth muscle cells 28
- Increased atherosclerosis 29
- Myelodysplastic syndrome 30
- Hepatic iron overload 31
- Fatty liver disease 32
- Cardiac hypertrophy 33, cardiomyopathy 34, 35
- Optic neuropathy, myeloneuropathy, anemia and neutropenia 36
- Atrial thrombosis, abnormal ECGs and sudden death 37
Common causes of acquired copper deficiency include 38:
- Malabsorption of copper
- Gastric surgery, including gastric bypass or gastrectomy
- Enteropathies such as inflammatory bowel disease, cystic fibrosis, and celiac disease
- Excessive use of copper chelators
- Zinc supplement overuse, parenteral overdosing, denture cream ingestion
- Chronic total parenteral nutrition, prolonged jejunal enteral feeding
- Diet low in copper
- Cause unknown
The most common causes of acquired copper deficiency include malabsorption as a consequence of prior gastric surgery or enteropathy (e.g., celiac disease, cystic fibrosis, and inflammatory bowel disease) and excessive zinc supplementation 39, 40, 38, 41. Given that most dietary copper is absorbed in the duodenum, the potential harmful effect of gastric surgery on absorption is obvious. Furthermore, zinc and copper compete for absorption across the intestinal brush border through the binding of an intracellular protein called metallothionein. Consequently, excessive zinc supplementation can impair the absorption of copper into enterohepatic circulation.
The following groups are most likely to have inadequate copper status 42, 43, 39, 44:
- People with celiac disease. In a study of 200 adults and children with celiac disease, of which 69.9% claimed to maintain a gluten-free diet, 15% had copper deficiency (less than 70 mcg/dL in serum in men and girls younger than 12 years and less than 80 mcg/dL in women older than 12 years and/or ceruloplasmin less than 170 mg/L) as a result of intestinal malabsorption resulting from the intestinal lining alterations associated with celiac disease 45. In its 2009 clinical guidelines for celiac disease, the American College of Gastroenterology notes that people with celiac disease appear to have an increased risk of copper deficiency and that copper levels normalize within a month of adequate copper supplementation while eating a gluten-free diet 46.
- People with Menkes disease. Menkes disease is a rare, X-linked, recessive disorder of copper homeostasis caused by ATP7A mutations, which encode a copper-transporting ATPase 47. In individuals with Menkes disease, intestinal absorption of dietary copper drops sharply, leading to signs of copper deficiency, including low serum copper and ceruloplasmin levels 47, 48. The typical signs and symptoms of Menkes disease include failure to thrive, impaired cognitive development, aortic aneurysms, seizures, and unusually kinky hair 47, 49. The life span of patients with Menkes disease is difficult to predict, although the majority of these children do not live past the age of three years, but subcutaneous injections of copper starting in the first few weeks after birth can reduce mortality risk and improve development 47, 50.
- People taking high doses of zinc supplements. High dietary intakes of zinc can interfere with copper absorption, and excessive use of zinc supplements can lead to copper deficiency. Reductions in erythrocyte copper-zinc superoxide dismutase, a marker of copper status, have been reported with even moderately high zinc intakes of approximately 60 mg/day for up to 10 weeks 12. People who regularly consume high doses of zinc from supplements or use excessive amounts of zinc-containing denture creams can develop copper deficiency because zinc can inhibit copper absorption. This is one reason the Food and Nutrition Board established the Tolerable Upper Intake Level (UL) for zinc at 40 mg/day for adults 12, 10.
The pathophysiology underlying the myeloneuropathy of copper deficiency remains unclear 51. It has been theorized that copper may play an integral role in methylation via methionine synthase, a necessary step in the production of purines and myelin proteins 52. Copper is also a cofactor for the enzyme superoxide dismutase, which is involved in the scavenging of free radicals. Additionally, copper is a known cofactor in cytochrome C oxidase-driven electron transport and oxidative phosphorylation, both crucial processes in the production of adenosine triphosphate (ATP). It remains unclear whether copper deficiency in a combination of the above processes are responsible for the damage that precipitates myelopathy 51.
Currently, there is not a sensitive and specific biomarker that accurately and reliably assess copper inadequacy in humans 9, 53, 54, 55. Copper status is not routinely assessed in clinical practice 11. Human studies typically measure copper and copper-dependent enzymes (cuproenzyme) activity in plasma and blood cells because individuals with known copper deficiency often have low blood levels of copper and ceruloplasmin 11. However, both the plasma ceruloplasmin and copper levels can be influenced by other factors, such as estrogen status, pregnancy, infection, inflammation, and some cancers, therefore limiting the usefulness of these assays to estimate body copper status 11, 9. Normal serum copper concentrations are 10–25 mcmol/L (63.5–158.9 mcg/dL) and 180–400 mg/L for ceruloplasmin 56. Experimental work has recently identified other copper-related biomarkers, including erythrocyte copper Cu/Zn superoxide dismutase (SOD1) and copper chaperone for superoxide dismutase 57, 58, 59, but further experimental validation is required, including clinical testing in humans.
Oral copper supplementation is efficacious, acceptable and practical mode of therapy for copper deficiency myeloneuropathy. In some cases, oral copper maintenance therapy was preceded by parenteral supplementation, presumably in order to rapidly replenish copper stores and avert any further deterioration. Although some reports refer to copper parenteral treatment only 60, 61, 62, the efficacy of oral copper supplements should make prolonged parenteral therapy unnecessary.
Typical daily copper doses were equivalent to 2–4 mg of elemental copper. In one case attributed to non-weight loss gastrointestinal surgery, administration of 2 mg of oral copper per day normalised blood parameters and improved neurological symptoms. However, a subsequent symptomatic and biochemical relapse necessitated that the dose be incremented to 4 mg and then 6 mg, which achieved sustained remission 63. Another patient with idiopathic zinc overload was poorly compliant with oral copper supplementation at 2 mg/day. His serum copper levels remained low and the neurological deficit progressed, though serum caeruloplasmin increased to a value at the low end of the normal range and the anaemia resolved. The copper dose was gradually escalated to 8 mg/day but compliance remained poor and he continued to deteriorate neurologically. Subsequently, the patient reported better compliance and his neurological status improved. However, his highest recorded serum copper level remained below the normal range, and it was unclear whether this reflected insufficient dosage, poor compliance or other factors. Copper doses of up to 9 mg per day have been reported 64.
Treatment may need to be continued indefinitely where the underlying risk factor cannot be eliminated. Regular follow-up is necessary to ensure normalisation of biochemical and haematological parameters and stabilisation or improvement of neurological features.
Therapeutic efficacy does not appear to be influenced by the type of copper compound administered. Oral copper gluconate did not influence serum copper levels in normal volunteers 65. Whilst this finding was previously interpreted as signifying poor bioavailability 66, 62, it appears to merely reflect intact copper homeostasis through biliary excretion in a copper-replete study population.
Since zinc can interfere with copper absorption, care must be taken to avoid copper preparations that also contain significant quantities of zinc, as may be the case with multivitamin tablets.
Nitrous oxide induced myeloneuropathy
Nitrous oxide (N2O) also known as laughing gas misuse through use of whipped cream chargers or ‘whippets’ bought from ‘head shops’ or online can cause a severe but potentially reversible nitrous oxide induced myeloneuropathy through interference with vitamin B12 metabolism, leading to megaloblastic anemia (characterized by large, abnormally nucleated red blood cells as well as low counts of white and red blood cells, platelets; glossitis of the tongue; fatigue; palpitations; pale skin; dementia; weight loss; and infertility) and subacute combined degeneration of the spinal cord 41, 67, 68, 69, 70, 71, 72, 73, 74, 75, 68, which itself can be irreversible 76. Progressive demyelination or degeneration of the spinal cord can cause neurological signs and symptoms such as sensory disturbance in the lower (± upper) limbs, areflexia (absence of neurologic reflexes such as the knee-jerk reaction) and the loss of proprioception (the sense that lets you perceive the location, movement, and action of parts of you body) and vibratory sense along with gait abnormalities. Areflexia can be permanent if neuronal death occurs in the posterior and lateral spinal cord tracts 77, 78, 79, 80. Dementia-like disease, including episodes of psychosis, is possible with more severe and chronic vitamin B12 deficiency 77, 80.
Vitamin B12 also known as cobalamin is required for the development, myelination, and function of the central nervous system (brain and spinal cord); healthy red blood cell formation; and DNA synthesis 81, 82, 71. Vitamin B12 functions as a cofactor (a compound that is essential for the activity of an enzyme) for two enzymes, methionine synthase and L-methylmalonyl-CoA mutase 81, 82, 71, 70, 83. Methionine synthase catalyzes the conversion of homocysteine to the essential amino acid methionine 81, 70. Methionine is required for the formation of S-adenosyl-L-methionine (SAMe), a universal methyl donor for almost 100 different substrates, including DNA, RNA, proteins, and lipids 82, 71, 71. L-methylmalonyl-CoA mutase converts L-methylmalonyl-CoA to succinyl-CoA in the metabolism of propionate, a short-chain fatty acid 70.
The mechanism involves nitrous oxide-induced inactivation of vitamin B12 and inhibition of methionine synthetase, disrupting methylation and DNA synthesis and leading to injury of the neuronal axons. Patients with a pre-existing B12 deficiency are particularly prone to developing acute life-threatening neurological symptoms on exposure to nitrous oxide, which can be fatal 84, 85.
Non-functioning vitamin B12 leads to accumulation of methylmalonic acid (MMA) and homocystine, which can be tested in the patient serum when B12 levels appear normal, suggesting a ‘functional’ B12 disorder. Serum B12 and active B12 levels may be normal because in this situation the myeloneuropathy is triggered by functional rather than absolute B12 deficiency.
A high index of suspicion is required to prompt appropriate investigation, make the diagnosis and commence treatment early.
MRI imaging of the spine showed the characteristic features of dorsal column hyperintensity on T2 weighted sequences strongly suggest the diagnosis.
The management of patients with nitrous oxide induced myeloneuropathy includes educating them about the risks of nitrous oxide and vitamin B12 replacement using high-dose intramuscular hydroxocobalamin (1 mg on alternate days until no further neurological improvement, followed by 1 mg every 2 months) 86. Guidelines from the British Society for Haematology recommend injections three times per week for two weeks in patients without neurologic deficits 87. If neurologic deficits are present, injections should be given every other day for up to three weeks or until no further improvement is noted 88. In general, patients with an irreversible cause should be treated indefinitely, whereas those with a reversible cause should be treated until the deficiency is corrected and symptoms resolve 77. If vitamin B12 deficiency coexists with folate deficiency, vitamin B12 should be replaced first to prevent subacute combined degeneration of the spinal cord 77. The British Society for Haematology does not recommend retesting vitamin B12 levels after treatment has been initiated, and no guidelines address the optimal interval for screening high-risk patients 87.
A 2018 Cochrane review involving 108 patients with vitamin B12 deficiency found that high-dose oral vitamin B12 replacement (1 mg to 2 mg per day) was as effective as high-dose intramuscular administration for correcting anemia and neurologic symptoms 89. However, oral therapy does not improve serum methylmalonic acid (MMA) levels as well as intramuscular therapy, although the clinical relevance is unclear 90. There is also a lack of data on the long-term benefit of oral therapy when patients do not take daily doses 78. There is insufficient data to recommend other formulations of vitamin B12 replacement (e.g., nasal, sublingual, subcutaneous) 78. The British Society for Haematology recommends intramuscular vitamin B12 for severe deficiency and malabsorption syndromes, whereas oral replacement may be considered for patients with asymptomatic, mild disease with no absorption or compliance concerns 87.
Neurological deficits can improve with abstinence and vitamin B12 supplementation, even in the most severely affected patients.
Folic acid deficiency
Folic acid deficiency can also produce a clinical picture similar to that of subacute combined degeneration of the spinal cord caused by vitamin B12 deficiency. Many other deficiencies such as vitamin B12 deficiency are concomitantly present. Folic acid deficiency is usually caused by dietary deficiency, malabsorption syndromes, pregnancy and lactation, usage of anticancer, antiepileptic, or oral contraceptive drugs, and long-term alcoholism. Folic acid deficiency-associated myeloneuropathy partially responds to folic acid supplementation 91, 92.
Vitamin E deficiency
Vitamin E deficiency is classically characterized with a spinocerebellar syndrome; however, it can also present with myeloneuropathy. Ataxia with vitamin E deficiency is an autosomal recessive disorder. The clinical features of vitamin E deficiency are similar to Friedreich’s ataxia. Patients present with cerebellar ataxia, loss of deep jerks, loss of vibration sense, dysarthria, and Babinski sign. Head titubation, retinopathy and dystonia are more common in these patients. Early diagnosis is crucial for successful treatment 91, 92, 93.
Chlorpyrifos poisoning
Chlorpyrifos is an organophosphate insecticide, which can cause delayed neurological toxicity following a high dose exposure 94, 95. A delayed myeloneuropathy following chlorpyrifos poisoning has been reported in an isolated case 96.
Chikungunya virus
Chikungunya virus in India has been shown to produce a syndrome similar to myeloneuropathy. A study during a chikungunya epidemic noted that among 90 laboratory-confirmed cases, 12 patients had myeloneuropathy, with or without encephalitis. The outcome of the neurological complications was good. In an another report of 300 patients with chikungunya, in 2006, 14% (7/49) patients had myeloneuropathy 97, 98.
HIV or AIDS
Vacuolar myelopathy in HIV infection is clinically very similar to subacute combined degeneration. Vacuolar myelopathy manifests with a posterolateral spinal cord syndrome with bladder and bowel disturbances. HIV myelopathy appears in the late stages of HIV infection when CD4+ cell counts are very low. Most of these patients concomitantly have other complications of HIV infection such as encephalopathy, opportunistic infections, or malignancies. Spinal cord pathology, including vacuolar myelopathy, has rarely been reported in Indian HIV-infected patients 99, 100.
Paraneoplastic myeloneuropathy
A paraneoplastic myeloneuropathy is an infrequent immune-mediated disorder that is associated with an underlying cancer. In some case reports, paraneoplastic myeloneuropathy has been reported with breast cancer. Antineuronal nuclear antibody 1 (anti-Hu) is frequently positive in these patients and they respond well to corticosteroids 101, 102. Reports on paraneoplastic myelopathies have demonstrated a characteristic magnetic resonance imaging (MRI) pattern of longitudinally extensive signal changes in the spinal cord.
Sjögren’s syndrome
Sjögren’s syndrome can manifest with myeloneuropathy 103. Sjögren’s syndrome is an autoimmune disorder characterized by decreased secretions of lacrimal and salivary glands (sicca symptoms). The central and peripheral nervous systems’ involvement in Sjögren’s syndrome is a result of vasculitis as well as direct immunological injury to neurons. Clinical spectrum of Sjögren’s syndrome-associated neuropathy includes sensory ataxic neuropathy, trigeminal neuropathy, multiple mononeuropathy, radiculoneuropathy, painful sensory neuropathy without sensory ataxia, autonomic neuropathy with anhidrosis, and multiple cranial neuropathy. Neurological symptoms occur in approximately 20% of patients with Sjögren’s syndrome, and may be the presenting manifestations of the disease 103.
Hashimoto’s disease
In Hashimoto’s disease, antithyroid antibodies can also be associated with acute myeloneuropathy or myelopathy 104, 105. The exact role of antithyroid antibodies in the pathogenesis of the myelopathy is not precisely clear; however, a vasculitic process has been suggested. There is often a good response to corticosteroids 104, 105.
Sarcoidosis
Sarcoidosis is a granulomatous disorder that can affect virtually any organ of the human body and nearly any component of the nervous system. A variety of neurological manifestations are possible including myelopathic signs and symptoms, and signs of segmental radiculopathy at the affected levels. Polyradiculopathy, including cauda equina syndrome and peripheral neuropathy, may accompany myelopathic finding. The diagnosis of sarcoidosis is quite challenging in such patients.
Tropical myeloneuropathies
Tropical myeloneuropathies were described initially in tropical countries and are classified into 2 overlapping clinical syndromes that can have overlapping features — tropical spastic paraparesis (TSP) and tropical ataxic neuropathy (TAN) 106. Tropical spastic paraparesis (TSP), a chronic cause of myeloneuropathy, has frequently been reported from South India. It is caused by an infection with human T-cell lymphotropic virus-1 (HTLV-1). Tropical ataxic neuropathy (TAN) is a slowly progressive cause of myeloneuropathy seen in cassava-eating countries. This disease characteristically occurs in an endemic form and is clinically characterized by sensory polyneuropathy, gait ataxia, optic atrophy, and nerve deafness. Tropical ataxic neuropathy often starts with dysesthesias in the lower limbs followed by unsteadiness of the gait. Romberg test is characteristically present. Reflexes in the lower limbs are either diminished or absent. Plantar response is usually flexor. Tropical ataxic neuropathy is seen in populations that use large quantities of cassava in their diets for very long periods 107, 108.
Inherited myeloneuropathies
A variety of hereditary myeloneuropathies have been described. Adrenomyeloneuropathy, a variant of adrenoleukodystrophy, is a noninflammatory involvement of the spinal cord that involves the descending corticospinal tracts, dominantly in the thoracic and lumbosacral regions, and the posterior columns, in the cervical spinal segments. The clinical features of adrenomyeloneuropathy include a slowly progressive spastic paraparesis and mild polyneuropathy in adult men, with or without sensory manifestations and sphincter disturbances 109.
Recently, Motley et al 110 from Belgium described five siblings in a family with dominantly inherited myeloneuropathy. All affected family members had a mild axonal neuropathy and three out of four family members had lower extremity hyperreflexia, suggestive of a superimposed myelopathy. A nerve biopsy showed evidence of chronic axonal loss. All affected family members had a heterozygous missense mutation in the alanyl-tRNA synthetase gene 110.
Myeloneuropathy symptoms
Myeloneuropathy symptoms vary depending on the underlying cause and the relative degree of involvement of the corticospinal tracts, spinocerebellar tracts, the dorsal columns, and peripheral nerves. Paresthesias (pins-and-needles sensation) and numbness involving the digits of the upper and/or lower limbs represent the most common complaints. In addition to spinal cord and peripheral nerve involvement, patients may also have visual deficits, and neuropsychiatric disease (depression and dementia).
Dorsal column involvement leads to impaired tactile discrimination, proprioception, and vibration sense. The earliest symptoms of dorsal column involvement are paresthesia, observed in the form of tingling, burning, and sensory loss of the distal extremities. Either the upper or lower limbs are involved first, or all four limbs are affected simultaneously. In addition, Lhermitte’s sign which is a transient sensation of an electric shock that extends down the spine and extremities upon flexion and/or movement of the neck may be present. Loss of proprioception usually presents as a difficulty in maintaining balance in the absence of visual cues (e.g., in the dark or with closed eyes).
Lateral corticospinal tract dysfunction causes muscle weakness, hyperreflexia, and spasticity. Stiffness is often the initial symptom of lateral cord involvement. Diffuse hyperreflexia can occur, although ankle reflexes are usually absent. Other signs of upper motor neuron damage such as ankle clonus and Babinski sign may be present. Spasticity can progress to paraplegia or quadriplegia if the condition remains untreated. Sphincter involvement in advanced cases can lead to bowel and bladder incontinence.
Spinocerebellar tract degeneration causes gait abnormalities in the form of sensory ataxia. Romberg’s test is a simple bedside test to determine the integrity of the dorsal column pathway of your brain and spinal cord (the neural pathways that carry proprioception sense by which sensory information from the peripheral nerves is transmitted to the cerebral cortex), which controls proprioception 111. Proprioception is the sense that lets you perceive the location, movement, and action of parts of your body 112. Proprioception encompasses a complex of sensations, including perception of joint position and movement, muscle force, and effort 112. These sensations arise from signals of sensory receptors in your muscle, skin, and joints, and from central signals related to motor output. Proprioception enables you to judge limb movements and positions, force, heaviness, stiffness, and viscosity. It combines with other senses to locate external objects relative to the body and contributes to body image. Romberg sign is positive in a patient who can stand with his feet placed together and eyes open but paradoxically sways or falls while closing his eyes, thereby eliminating his visual cues 113, 114. During a Romberg test, your doctor will ask you to close your eyes while standing with your feet together and your arms to your side, this removes the visual and vestibular components that contribute to maintaining balance. If you feel unbalanced or unsteady, positive Romberg’s sign, it could mean that you have an issue with your central nervous system (your brain or spinal cord) 111.
Physical examination may reveal decreased sensation to light touch and pain/temperature, hyperreflexia, spasticity, increased tone, disorders of gait, and the emergence of an extensor plantar response. Urinary and/or fecal incontinence or retention may also be seen 115.
Myeloneuropathy diagnosis
If you have myeloneuropathy symptoms, your doctor will look for a treatable cause. Besides conducting a physical exam and a neurological exam, including checking your vision, balance, coordination and reflexes, your doctor might request tests, including:
- Blood tests. Blood levels of vitamin B12, folic acid, methylmalonic acid (MMA), homocysteine, vitamins A, D, E, and K, iron, and calcium should be assessed to detect other nutritional deficiencies
- Imaging studies. Imaging of the brain and spinal cord, with contrast, is always needed.
- Lumbar puncture (spinal tap). In some cases of myeloneuropathy, this may be a helpful test. A needle is inserted into the lower back (lumbar region) between two lumbar bones (vertebrae) to remove a small sample of cerebrospinal fluid. The fluid, which surrounds and protects your brain and spinal cord, is sent to a laboratory for testing.
- Genetic testing. Your doctor might recommend genetic testing to determine whether a gene mutation causes one of the inherited myeloneuropathy conditions.
- Electromyography (EMG) and nerve conduction study. Electromyography and nerve conduction study results are often consistent with axonal sensorimotor polyneuropathy in the legs; somatosensory evoked potentials may provide an electrophysiological evidence of dysfunction in the central pathways.
Myeloneuropathy treatment
Myeloneuropathy treatment involves treating the underlying cause such as vitamin B12 supplements for nitrous oxide induced vitamin B12 deficiency and copper for copper deficiency.
Because there are so many causes of myeloneuropathy and each case is different, your doctor is the best person to tell you what kind of treatments are possible and likely to help you. The information they provide will be the most relevant to your particular situation.
Myeloneuropathy prognosis
Myeloneuropathy prognosis depends on the underlying cause. Some patients are left with a permanent deficit, including lifelong paraparesis despite optimal treatment.
Neurological deficits can improve with nitrous oxide abstinence and vitamin B12 supplementation, even in the most severely affected patients. Neurological recovery may be incomplete, particularly when patients continue to use nitrous oxide (N2O) 116.
In patients with copper deficiency myeloneuropathy, copper supplementation can arrest further neurological deterioration and even reverse symptoms and imaging findings 4.
The life span of patients with Menkes disease is difficult to predict, although the majority of these children do not live past the age of three years, but subcutaneous injections of copper starting in the first few weeks after birth can reduce mortality risk and improve development 47, 50.
- Garg RK, Malhotra HS, Kumar N. Approach to a case of myeloneuropathy. Ann Indian Acad Neurol. 2016 Apr-Jun;19(2):183-7. doi: 10.4103/0972-2327.182303[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Goodman BP. Diagnostic approach to myeloneuropathy. Continuum (Minneap Minn). 2011 Aug;17(4):744-60. doi: 10.1212/01.CON.0000403793.13291.f3[↩]
- Kumar N. Metabolic and toxic myelopathies. Semin Neurol. 2012 Apr;32(2):123-36. doi: 10.1055/s-0032-1322583[↩]
- Goodman BP, Chong BW, Patel AC, Fletcher GP, Smith BE. Copper deficiency myeloneuropathy resembling B12 deficiency: partial resolution of MR imaging findings with copper supplementation. AJNR Am J Neuroradiol. 2006 Nov-Dec;27(10):2112-4. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7977229[↩][↩]
- Nitrous oxide induced myeloneuropathy. https://radiopaedia.org/cases/nitrous-oxide-induced-myeloneuropathy?lang=us[↩]
- Lanska DJ, Remler B: Myelopathy among zinc-smelter workers in Upper Silesia during the late 19th century. Neurology. 2014, 82:1175-9. 10.1212/WNL.0000000000000270[↩]
- Jaiser SR, Winston GP. Copper deficiency myelopathy. J Neurol. 2010 Jun;257(6):869-81. doi: 10.1007/s00415-010-5511-x[↩]
- Prohaska JR. Impact of copper limitation on expression and function of multicopper oxidases (ferroxidases). Adv Nutr. 2011 Mar;2(2):89-95. doi: 10.3945/an.110.000208[↩]
- Collins JF. Copper nutrition and biochemistry and human (patho)physiology. Adv Food Nutr Res. 2021;96:311-364. doi: 10.1016/bs.afnr.2021.01.005[↩][↩][↩]
- Collins JF. Copper. In: Ross AC, Caballero B, Cousins RJ, Tucker KL, Ziegler TR, eds. Modern Nutrition in Health and Disease. 11th ed. Baltimore, MD: Lippincott Williams & Wilkins; 2014:206-16.[↩][↩][↩][↩]
- Prohaska JR. Copper. In: Erdman JW, Macdonald IA, Zeisel SH, eds. Present Knowledge in Nutrition. 10th ed. Washington, DC: Wiley-Blackwell; 2012:540-53.[↩][↩][↩][↩][↩]
- Institute of Medicine, Food and Nutrition Board. Dietary Reference Intakes for Vitamin A, Vitamin K, Arsenic, Boron, Chromium, Copper, Iodine, Iron, Manganese, Molybdenum, Nickel, Silicon, Vanadium, and Zinc. Washington, DC: National Academies Press; 2001.[↩][↩][↩]
- Hellman NE, Gitlin JD. Ceruloplasmin metabolism and function. Annu Rev Nutr. 2002;22:439-58. doi: 10.1146/annurev.nutr.22.012502.114457[↩]
- Turnlund JR. Copper. In: Shils ME, Shike M, Ross A, Caballero B, Cousins RA, eds. Modern Nutrition in Health and Disease. 10th ed. Baltimore: Lipincott Williams & Wilkins; 2006:289-299.[↩]
- Harris E. Copper. In: O’Dell B, Sunde R, eds. Handbook of Nutritionally Essential Minerals. New York: Marcel Dekker, Inc.; 1997:231-273.[↩]
- Owen CAJ. Biochemical Aspects of Copper: Copper Proteins, Ceruloplasmin, and Copper Protein Binding. Park Ridge, NJ: Noyes Publications; 1982.[↩]
- Allen KG, Klevay LM. Copper: an antioxidant nutrient for cardiovascular health. Curr Opin Lipidol. 1994 Feb;5(1):22-8. doi: 10.1097/00041433-199402000-00005[↩]
- Fairweather-Tait SJ, Harvey LJ, Collings R. Risk-benefit analysis of mineral intakes: case studies on copper and iron. Proc Nutr Soc. 2011 Feb;70(1):1-9. doi: 10.1017/S0029665110003873[↩]
- Prohaska JR. Impact of copper deficiency in humans. Ann N Y Acad Sci. 2014 May;1314:1-5. doi: 10.1111/nyas.12354[↩]
- DiNicolantonio JJ, Mangan D, O’Keefe JH. Copper deficiency may be a leading cause of ischaemic heart disease. Open Heart. 2018 Oct 8;5(2):e000784. doi: 10.1136/openhrt-2018-000784[↩]
- Klevay LM. Ischemic heart disease. A major obstacle to becoming old. Clin Geriatr Med. 1987 May;3(2):361-72.[↩]
- Alarcón-Corredor OM, Guerrero Y, Ramírez de Fernández M, D’Jesús I, Burguera M, Burguera JL, Di Bernardo ML, García MY, Alarcón AO. Efecto de la suplementación oral con cobre en el perfil lipídico de pacientes Venezolanos hiperlipémicos [Effect of copper supplementation on lipid profile of Venezuelan hyperlipemic patients]. Arch Latinoam Nutr. 2004 Dec;54(4):413-8. Spanish.[↩]
- Rayssiguier Y, Gueux E, Bussiere L, Mazur A. Copper deficiency increases the susceptibility of lipoproteins and tissues to peroxidation in rats. J Nutr. 1993 Aug;123(8):1343-8. doi: 10.1093/jn/123.8.1343[↩][↩]
- Alarcón OM, Guerrero Y, Ramírez de Fernández M, D’Jesús I, Burguera M, Burguera JL, Di Bernardo ML. Efecto de la suplementación con cobre sobre los valores de presión arterial en pacientes con hipertensión moderada estable [Effect of cooper supplementation on blood pressure values in patients with stable moderate hypertension]. Arch Latinoam Nutr. 2003 Sep;53(3):271-6. Spanish.[↩]
- Bügel S, Harper A, Rock E, et al.. Effect of copper supplementation on indices of copper status and certain CVD risk markers in young healthy women. Br J Nutr 2005;94:231–6. 10.1079/BJN20051470[↩]
- Saari JT, Dahlen GM. Early and advanced glycation end-products are increased in dietary copper deficiency. J Nutr Biochem 1999;10:210–4. 10.1016/S0955-2863(98)00100-4[↩]
- Tallino S, Duffy M, Ralle M, et al.. Nutrigenomics analysis reveals that copper deficiency and dietary sucrose up-regulate inflammation, fibrosis and lipogenic pathways in a mature rat model of nonalcoholic fatty liver disease. J Nutr Biochem 2015;26:996–1006. 10.1016/j.jnutbio.2015.04.009[↩]
- Hunsaker H, Morita M, Allen K. Marginal copper deficiency in rats aortal morphology of elastin and cholesterol values in first-generation adult males. Atherosclerosis 1984;51:1–19. 10.1016/0021-9150(84)90140-0[↩]
- Hamilton IM, Gilmore WS, Strain JJ. Marginal copper deficiency and atherosclerosis. Biol Trace Elem Res 2000;78:179–90. 10.1385/BTER:78:1-3:179[↩]
- Gregg XT, Reddy V, Prchal JT. Copper deficiency masquerading as myelodysplastic syndrome. Blood 2002;100:1493–5. 10.1182/blood-2002-01-0256[↩]
- Thackeray EW, Sanderson SO, Fox JC, et al.. Hepatic iron overload or cirrhosis may occur in acquired copper deficiency and is likely mediated by hypoceruloplasminemia. J Clin Gastroenterol 2011;45:153–8. 10.1097/MCG.0b013e3181dc25f7[↩]
- Mendoza M, Caltharp S, Song M, et al.. Low hepatic tissue copper in pediatric nonalcoholic fatty liver disease. J Pediatr Gastroenterol Nutr 2017;65:89–92. 10.1097/MPG.0000000000001571[↩]
- Zhou Z, Johnson WT, Kang YJ. Regression of copper-deficient heart hypertrophy: reduction in the size of hypertrophic cardiomyocytes. J Nutr Biochem 2009;20:621–8. 10.1016/j.jnutbio.2008.06.007[↩]
- Li Y, Wang L, Schuschke DA, et al.. Marginal dietary copper restriction induces cardiomyopathy in rats. J Nutr 2005;135:2130–6. 10.1093/jn/135.9.2130[↩]
- Elsherif L, Wang L, Saari JT, et al.. Regression of dietary copper restriction-induced cardiomyopathy by copper repletion in mice. J Nutr 2004;134:855–60. 10.1093/jn/134.4.855[↩]
- Yarandi SS, Griffith DP, Sharma R, Mohan A, Zhao VM, Ziegler TR. Optic neuropathy, myelopathy, anemia, and neutropenia caused by acquired copper deficiency after gastric bypass surgery. J Clin Gastroenterol. 2014 Nov-Dec;48(10):862-5. doi: 10.1097/MCG.0000000000000092[↩]
- Klevay LM. Atrial thrombosis, abnormal electrocardiograms and sudden death in mice due to copper deficiency. Atherosclerosis 1985;54:213–24. 10.1016/0021-9150(85)90180-7[↩]
- Wazir SM, Ghobrial I. Copper deficiency, a new triad: anemia, leucopenia, and myeloneuropathy. J Community Hosp Intern Med Perspect. 2017 Sep 19;7(4):265-268. doi: 10.1080/20009666.2017.1351289[↩][↩]
- Rowin J, Lewis SL. Copper deficiency myeloneuropathy and pancytopenia secondary to overuse of zinc supplementation. J Neurol Neurosurg Psychiatry. 2005 May;76(5):750-1. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1739637/pdf/v076p00750.pdf[↩][↩]
- Spinazzi M, De Lazzari F, Tavolato B, Angelini C, Manara R, Armani M. Myelo-optico-neuropathy in copper deficiency occurring after partial gastrectomy. Do small bowel bacterial overgrowth syndrome and occult zinc ingestion tip the balance? J Neurol. 2007 Aug;254(8):1012-7. doi: 10.1007/s00415-006-0479-2[↩]
- Qudsiya Z, De Jesus O. Subacute Combined Degeneration of the Spinal Cord. [Updated 2023 Feb 12]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK559316[↩][↩]
- Copper. https://ods.od.nih.gov/factsheets/Copper-HealthProfessional[↩]
- Kumar N, Ahlskog JE, Gross JB Jr. Acquired hypocupremia after gastric surgery. Clin Gastroenterol Hepatol. 2004 Dec;2(12):1074-9. doi: 10.1016/s1542-3565(04)00546-4[↩]
- Ashkenazi A, Levin S, Djaldetti M, Fishel E, Benvenisti D. The syndrome of neonatal copper deficiency. Pediatrics. 1973 Oct;52(4):525-33.[↩]
- Botero-López JE, Araya M, Parada A, Méndez MA, Pizarro F, Espinosa N, Canales P, Alarcón T. Micronutrient deficiencies in patients with typical and atypical celiac disease. J Pediatr Gastroenterol Nutr. 2011 Sep;53(3):265-70. doi: 10.1097/MPG.0b013e3181f988fc[↩]
- Rubio-Tapia A, Hill ID, Kelly CP, Calderwood AH, Murray JA; American College of Gastroenterology. ACG clinical guidelines: diagnosis and management of celiac disease. Am J Gastroenterol. 2013 May;108(5):656-76; quiz 677. doi: 10.1038/ajg.2013.79[↩]
- Ramani PK, Parayil Sankaran B. Menkes Disease. [Updated 2023 Nov 14]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK560917[↩][↩][↩][↩][↩]
- Costa LS, Pegler SP, Lellis RF, Krebs VL, Robertson S, Morgan T, Honjo RS, Bertola DR, Kim CA. Menkes disease: importance of diagnosis with molecular analysis in the neonatal period. Rev Assoc Med Bras (1992). 2015 Sep-Oct;61(5):407-10. doi: 10.1590/1806-9282.61.05.407[↩]
- Hordyjewska A, Popiołek Ł, Kocot J. The many “faces” of copper in medicine and treatment. Biometals. 2014 Aug;27(4):611-21. doi: 10.1007/s10534-014-9736-5[↩]
- Kaler SG. Neurodevelopment and brain growth in classic Menkes disease is influenced by age and symptomatology at initiation of copper treatment. J Trace Elem Med Biol. 2014 Oct;28(4):427-30. doi: 10.1016/j.jtemb.2014.08.008[↩][↩]
- Grossman JT, Ruiz S. Copper Deficiency Myeloneuropathy in Autoimmune Disease. Cureus. 2021 Jul 23;13(7):e16591. doi: 10.7759/cureus.16591[↩][↩]
- Zatta P, Frank A. Copper deficiency and neurological disorders in man and animals. Brain Res Rev. 2007 Apr;54(1):19-33. doi: 10.1016/j.brainresrev.2006.10.001[↩]
- Bost M, Houdart S, Oberli M, Kalonji E, Huneau JF, Margaritis I. Dietary copper and human health: Current evidence and unresolved issues. J Trace Elem Med Biol. 2016 May;35:107-15. doi: 10.1016/j.jtemb.2016.02.006[↩]
- Harvey LJ, McArdle HJ. Biomarkers of copper status: a brief update. Br J Nutr. 2008 Jun;99 Suppl 3:S10-3. doi: 10.1017/S0007114508006806[↩]
- Olivares M, Méndez MA, Astudillo PA, Pizarro F. Present situation of biomarkers for copper status. Am J Clin Nutr. 2008 Sep;88(3):859S-62S. doi: 10.1093/ajcn/88.3.859S[↩]
- Institute of Medicine. Food and Nutrition Board. Dietary Reference Intakes: Thiamin, Riboflavin, Niacin, Vitamin B6, Folate, Vitamin B12, Pantothenic Acid, Biotin, and Choline. Washington, DC: National Academy Press; 1998.[↩]
- Lassi KC, Prohaska JR. Rapid alteration in rat red blood cell copper chaperone for superoxide dismutase after marginal copper deficiency and repletion. Nutr Res. 2011 Sep;31(9):698-706. doi: 10.1016/j.nutres[↩]
- Lassi KC, Prohaska JR. Erythrocyte copper chaperone for superoxide dismutase is increased following marginal copper deficiency in adult and postweanling mice. J Nutr. 2012 Feb;142(2):292-7. doi: 10.3945/jn.111.150755[↩]
- Dirksen K, Roelen YS, van Wolferen ME, Kruitwagen HS, Penning LC, Burgener IA, Spee B, Fieten H. Erythrocyte copper chaperone for superoxide dismutase and superoxide dismutase as biomarkers for hepatic copper concentrations in Labrador retrievers. Vet J. 2016 Dec;218:1-6. doi: 10.1016/j.tvjl.2016.10.007[↩]
- Bertfield DL, Jumma O, Pitceathly RD, Sussman JD. Copper deficiency: an unusual case of myelopathy with neuropathy. Ann Clin Biochem. 2008;45:434–435. doi: 10.1258/acb.2008.007218[↩]
- Schleper B, Stuerenburg HJ. Copper deficiency-associated myelopathy in a 46-year-old woman. J Neurol. 2001;248:705–706. doi: 10.1007/s004150170118[↩]
- Wu J, Ricker M, Muench J. Copper deficiency as cause of unexplained hematologic and neurologic deficits in patient with prior gastrointestinal surgery. J Am Board Fam Med. 2006;19:191–194. doi: 10.3122/jabfm.19.2.191[↩][↩]
- Prodan CI, Bottomley SS, Holland NR, Lind SE. Relapsing hypocupraemic myelopathy requiring high-dose oral copper replacement. J Neurol Neurosurg Psychiatry. 2006;77:1092–1093. doi: 10.1136/jnnp.2006.096883[↩]
- Bartner R, Will M, Conrad J, Engelhardt A, Schwarz-Eywill M. Pancytopenia, arthralgia and myeloneuropathy due to copper deficiency. Med Klin (Munich) 2005;100:497–501. doi: 10.1007/s00063-005-1072-7[↩]
- Pratt WB, Omdahl JL, Sorenson JR. Lack of effects of copper gluconate supplementation. Am J Clin Nutr. 1985 Oct;42(4):681-2. doi: 10.1093/ajcn/42.4.681[↩]
- Kumar N. Copper deficiency myelopathy (human swayback) Mayo Clin Proc. 2006;81:1371–1384. doi: 10.4065/81.10.1371[↩]
- Swart G, Blair C, Lu Z, Yogendran S, Offord J, Sutherland E, Barnes S, Palavra N, Cremer P, Bolitho S, Michael Halmagyi G. Nitrous oxide-induced myeloneuropathy. Eur J Neurol. 2021 Dec;28(12):3938-3944. doi: 10.1111/ene.15077[↩]
- Layzer R. Myeloneuropathy after prolonged exposure to nitrous oxide. Lancet. 1978;312:1227–1230. doi: 10.1016/S0140-6736(78)92101-3[↩][↩]
- Keddie S, Adams A, Kelso ARC, Turner B, Schmierer K, Gnanapavan S, Malaspina A, Giovannoni G, Basnett I, Noyce AJ. No laughing matter: subacute degeneration of the spinal cord due to nitrous oxide inhalation. J Neurol. 2018 May;265(5):1089-1095. doi: 10.1007/s00415-018-8801-3[↩]
- Carmel R. Cobalamin (vitamin B12). In: Ross AC, Caballero B, Cousins RJ, Tucker KL, Ziegler TR, eds. Modern Nutrition in Health and Disease. 11th ed. Baltimore, MD: Lippincott Williams & Wilkins; 2014:369-89.[↩][↩][↩][↩]
- Stabler SP. Vitamin B12. In: Marriott BP, Birt DF, Stallings VA, Yates AA, eds. Present Knowledge in Nutrition. 11th ed. Washington, DC: Elsevier; 2020:257-71.[↩][↩][↩][↩][↩]
- Langan RC, Goodbred AJ. Vitamin B12 Deficiency: Recognition and Management. Am Fam Physician. 2017 Sep 15;96(6):384-389. https://www.aafp.org/pubs/afp/issues/2017/0915/p384.html[↩]
- Thompson AG, Leite MI, Lunn MP, Bennett DLH. Whippits, nitrous oxide and the dangers of legal highs. Pract Neurol. 2015;15:207–209. doi: 10.1136/practneurol-2014-001071[↩]
- Ng J, O’Grady G, Pettit T, Frith R. Nitrous oxide use in first-year students at Auckland University. Lancet. 2003;361:1349–1350. doi: 10.1016/S0140-6736(03)13045-0[↩]
- Morris N, Lynch K, Greenberg SA. Severe motor neuropathy or neuronopathy due to nitrous oxide toxicity after correction of vitamin B12 deficiency. Muscle Nerve. 2015;51:614–616. doi: 10.1002/mus.24482[↩]
- Vasconcelos OM, Poehm EH, McCarter RJ, Campbell WW, Quezado ZMN. Potential outcome factors in subacute combined degeneration: review of observational studies. J Gen Intern Med. 2006;21:1063–1068. doi: 10.1111/j.1525-1497.2006.00525.x[↩]
- Hunt A, Harrington D, Robinson S. Vitamin B12 deficiency. BMJ. 2014 Sep 4;349:g5226. doi: 10.1136/bmj.g5226[↩][↩][↩][↩]
- Stabler SP. Clinical practice. Vitamin B12 deficiency. N Engl J Med. 2013 Jan 10;368(2):149-60. doi: 10.1056/NEJMcp1113996[↩][↩][↩]
- Dali-Youcef N, Andrès E. An update on cobalamin deficiency in adults. QJM. 2009 Jan;102(1):17-28. doi: 10.1093/qjmed/hcn138[↩]
- Reynolds E. Vitamin B12, folic acid, and the nervous system. Lancet Neurol. 2006 Nov;5(11):949-60. doi: 10.1016/S1474-4422(06)70598-1[↩][↩]
- Institute of Medicine (US) Standing Committee on the Scientific Evaluation of Dietary Reference Intakes and its Panel on Folate, Other B Vitamins, and Choline. Dietary Reference Intakes for Thiamin, Riboflavin, Niacin, Vitamin B6, Folate, Vitamin B12, Pantothenic Acid, Biotin, and Choline. Washington (DC): National Academies Press (US); 1998. Available from: https://www.ncbi.nlm.nih.gov/books/NBK114310[↩][↩][↩]
- Allen LH. Vitamin B-12. Adv Nutr. 2012 Jan;3(1):54-5. doi: 10.3945/an.111.001370[↩][↩][↩]
- Allen LH. Vitamin B12. In: Coates PM, Betz JM, Blackman MR, et al., eds. Encyclopedia of Dietary Supplements. 2nd ed. London and New York: Informa Healthcare; 2010:812-20.[↩]
- Flippo TS, Holder WD Jr. Neurologic degeneration associated with nitrous oxide anesthesia in patients with vitamin B12 deficiency. Arch Surg. 1993 Dec;128(12):1391-5. doi: 10.1001/archsurg.1993.01420240099018[↩]
- Lin RJ, Chen HF, Chang YC, Su JJ. Subacute combined degeneration caused by nitrous oxide intoxication: case reports. Acta Neurol Taiwan. 2011 Jun;20(2):129-37.[↩]
- Joint Formulary Committee (2017) British National Formulary, 74th edn. BMJ Group and Pharmaceutical Press, London[↩]
- Devalia V, Hamilton MS, Molloy AM; British Committee for Standards in Haematology. Guidelines for the diagnosis and treatment of cobalamin and folate disorders. Br J Haematol. 2014 Aug;166(4):496-513. doi: 10.1111/bjh.12959[↩][↩][↩]
- Carmel R. How I treat cobalamin (vitamin B12) deficiency. Blood. 2008 Sep 15;112(6):2214-21. doi: 10.1182/blood-2008-03-040253[↩]
- Wang H, Li L, Qin LL, Song Y, Vidal-Alaball J, Liu TH. Oral vitamin B12 versus intramuscular vitamin B12 for vitamin B12 deficiency. Cochrane Database Syst Rev. 2018 Mar 15;3(3):CD004655. doi: 10.1002/14651858.CD004655.pub3[↩]
- Kuzminski AM, Del Giacco EJ, Allen RH, Stabler SP, Lindenbaum J. Effective treatment of cobalamin deficiency with oral cobalamin. Blood. 1998 Aug 15;92(4):1191-8. https://doi.org/10.1182/blood.V92.4.1191[↩]
- Goodman BP. Metabolic and toxic causes of myelopathy. Continuum (Minneap Minn). 2015 Feb;21(1 Spinal Cord Disorders):84-99. doi: 10.1212/01.CON.0000461086.79241.3b[↩][↩]
- Román GC. Nutritional disorders in tropical neurology. Handb Clin Neurol. 2013;114:381-404. doi: 10.1016/B978-0-444-53490-3.00030-3[↩][↩]
- Vorgerd M, Tegenthoff M, Kühne D, Malin JP. Spinal MRI in progressive myeloneuropathy associated with vitamin E deficiency. Neuroradiology. 1996 May;38 Suppl 1:S111-3. doi: 10.1007/BF02278134[↩]
- Thivakaran T, Gamage R, Gunarathne KS, Gooneratne IK. Chlorpyrifos-induced delayed myelopathy and pure motor neuropathy: a case report. Neurologist. 2012 Jul;18(4):226-8. doi: 10.1097/NRL.0b013e318261035b[↩]
- Nand N, Aggarwal HK, Bharti K, Chakrabarti D. Organophosphate induced delayed neuropathy. J Assoc Physicians India. 2007 Jan;55:72-3.[↩]
- Ostwal P, Dabadghao VS, Sharma SK, Dhakane AB. Chlorpyrifos toxicity causing delayed myeloneuropathy. Ann Indian Acad Neurol. 2013 Oct;16(4):736. doi: 10.4103/0972-2327.120443[↩]
- Tandale BV, Sathe PS, Arankalle VA, Wadia RS, Kulkarni R, Shah SV, Shah SK, Sheth JK, Sudeep AB, Tripathy AS, Mishra AC. Systemic involvements and fatalities during Chikungunya epidemic in India, 2006. J Clin Virol. 2009 Oct;46(2):145-9. doi: 10.1016/j.jcv.2009.06.027[↩]
- Chandak NH, Kashyap RS, Kabra D, Karandikar P, Saha SS, Morey SH, Purohit HJ, Taori GM, Daginawala HF. Neurological complications of Chikungunya virus infection. Neurol India. 2009 Mar-Apr;57(2):177-80. doi: 10.4103/0028-3886.51289[↩]
- Modi G, Ranchhod J, Hari K, Mochan A, Modi M. Non-traumatic myelopathy at the Chris Hani Baragwanath Hospital, South Africa–the influence of HIV. QJM. 2011 Aug;104(8):697-703. doi: 10.1093/qjmed/hcr038[↩]
- Shankar SK, Mahadevan A, Satishchandra P, Kumar RU, Yasha TC, Santosh V, Chandramuki A, Ravi V, Nath A. Neuropathology of HIV/AIDS with an overview of the Indian scene. Indian J Med Res. 2005 Apr;121(4):468-88.[↩]
- Alsharabati M, Oh SJ. Paraneoplastic myeloneuropathy in a man with breast cancer. Muscle Nerve. 2015 Oct;52(4):685-6. doi: 10.1002/mus.24706[↩]
- Rajabally YA, Qaddoura B, Abbott RJ. Steroid-responsive paraneoplastic demyelinating neuropathy and myelopathy associated with breast carcinoma. J Clin Neuromuscul Dis. 2008 Dec;10(2):65-9. doi: 10.1097/CND.0b013e31818e952b[↩]
- Verma R, Lalla R, Patil TB, Mehta V. Acute myeloneuropathy: An uncommon presentation of Sjögren’s syndrome. Ann Indian Acad Neurol. 2013 Oct;16(4):696-8. doi: 10.4103/0972-2327.120462[↩][↩]
- Kayal AK, Basumatary LJ, Dutta S, Mahanta N, Islam S, Mahanta A. Myeloneuropathy in a case of Hashimoto’s disease. Neurol India. 2013 Jul-Aug;61(4):426-8. doi: 10.4103/0028-3886.117591[↩][↩]
- Turkoglu R, Tuzun E. Steroid-responsive myeloneuropathy associated with antithyroid antibodies. J Spinal Cord Med. 2010;33(3):278-80. doi: 10.1080/10790268.2010.11689708[↩][↩]
- Tropical Myeloneuropathies. https://emedicine.medscape.com/article/1166055-overview[↩]
- Oomman A, Madhusoodanan M. Tropical spastic paraparesis in Kerala. Neurol India. 2003 Dec;51(4):493-6.[↩]
- Ajdukiewicz A, Yanagihara R, Garruto RM, Gajdusek DC, Alexander SS. HTLV-1 myeloneuropathy in the Solomon Islands. N Engl J Med. 1989 Aug 31;321(9):615-6. doi: 10.1056/NEJM198908313210913[↩]
- Chafale VA, Lahoti SA, Biswas A, Roy A, Senapati AK. Adrenomyeloneuropathy with bulbar palsy: A rare association. Ann Indian Acad Neurol. 2014 Jul;17(3):361-3. doi: 10.4103/0972-2327.138530[↩]
- Motley WW, Griffin LB, Mademan I, Baets J, De Vriendt E, De Jonghe P, Antonellis A, Jordanova A, Scherer SS. A novel AARS mutation in a family with dominant myeloneuropathy. Neurology. 2015 May 19;84(20):2040-7. doi: 10.1212/WNL.0000000000001583[↩][↩]
- Forbes J, Munakomi S, Cronovich H. Romberg Test. [Updated 2023 Aug 13]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK563187[↩][↩]
- Proprioception. Encyclopedia of Neuroscience 2009, Pages 1143-1149. https://doi.org/10.1016/B978-008045046-9.01907-0[↩][↩]
- Lanska DJ, Goetz CG. Romberg’s sign: development, adoption, and adaptation in the 19th century. Neurology. 2000 Oct 24;55(8):1201-6. https://doi.org/10.1212/WNL.55.8.1201[↩]
- Berge JE, Goplen FK, Aarstad HJ, Storhaug TA, Nordahl SHG. The Romberg sign, unilateral vestibulopathy, cerebrovascular risk factors, and long-term mortality in dizzy patients. Front Neurol. 2022 Aug 5;13:945764. doi: 10.3389/fneur.2022.945764[↩]
- Robert S. Hoffman, Lewis S. Nelson, MD, Lewis R. Goldfrank et al. Goldfrank’s Toxicologic Emergencies, Eleventh Edition. (2019) ISBN: 9781259859618[↩]
- Vasconcelos OM, Poehm EH, McCarter RJ, Campbell WW, Quezado ZMN. Potential outcome factors in subacute combined degeneration: review of observational studies. J Gen Intern Med. 2006;21:1063–1068. doi: 10.1111/j.1525-1497.2006.00525.x[↩]

{kind=link}