Contents
What is myostatin
Myostatin also known as “growth and differentiation factor 8” or GDF8, is a protein that is part of the transforming growth factor beta (TGFβ) superfamily, which is a group of proteins that help control the growth and development of tissues throughout the body 1. Myostatin is found almost exclusively in skeletal muscles that is used for movement, where it is active both before and after birth. Myostatin normally restrains muscle growth, ensuring that muscles do not grow too large. Myostatin has been studied extensively in mice, cows, and other animals, and it appears to have a similar function in humans. Researchers are studying myostatin as a potential treatment for various muscular dystrophies that cause muscle weakness and wasting (atrophy) 2.
Myostatin’s expression is mainly restricted to skeletal muscle, with low levels of mRNA reported in adipose (fat) 3 and cardiac 4 tissues. Naturally occurring and engineered mutations that reduce myostatin signaling lead to a hypermuscular phenotype 5. Conversely, myostatin overexpression in rodents induces a cachexia-like syndrome with muscle wasting and fat loss9. Because of the well-established impact of myostatin signaling on muscle mass, a number of agents that target the myostatin signaling pathway have entered the clinic for indications involving muscle atrophy.
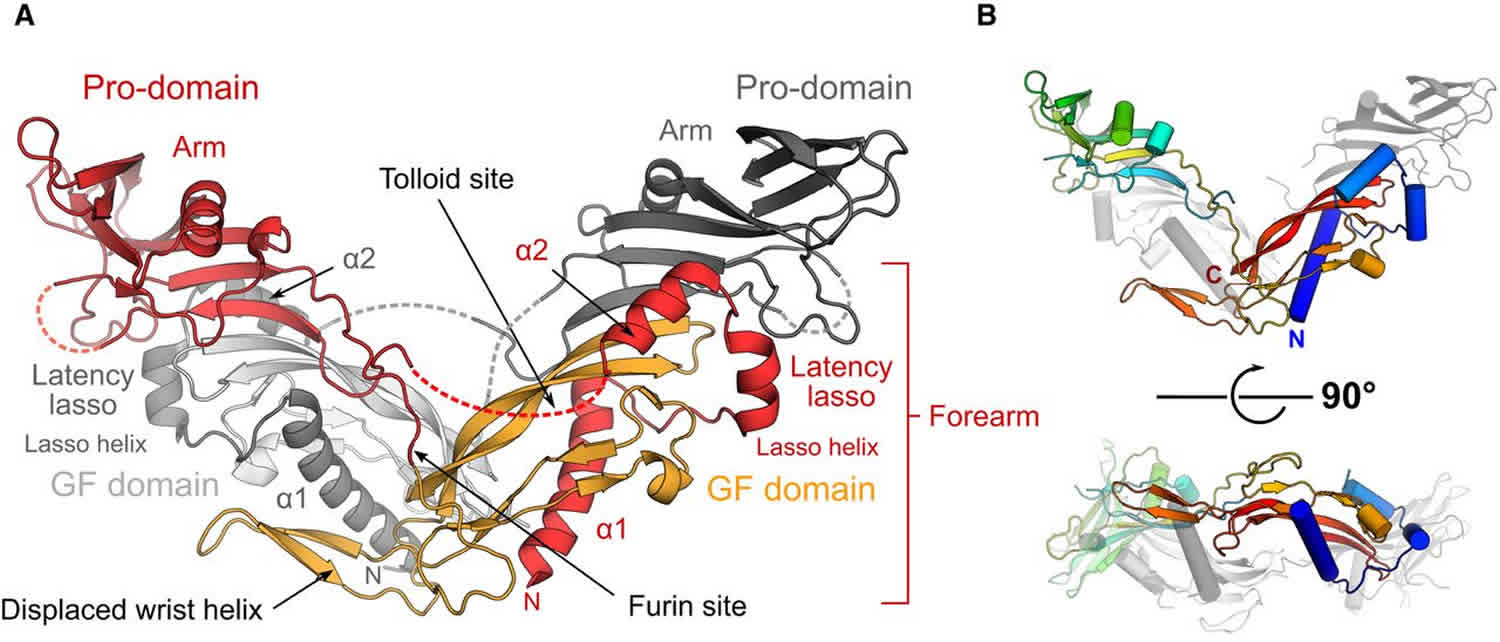
Figure 1. Human promyostatin
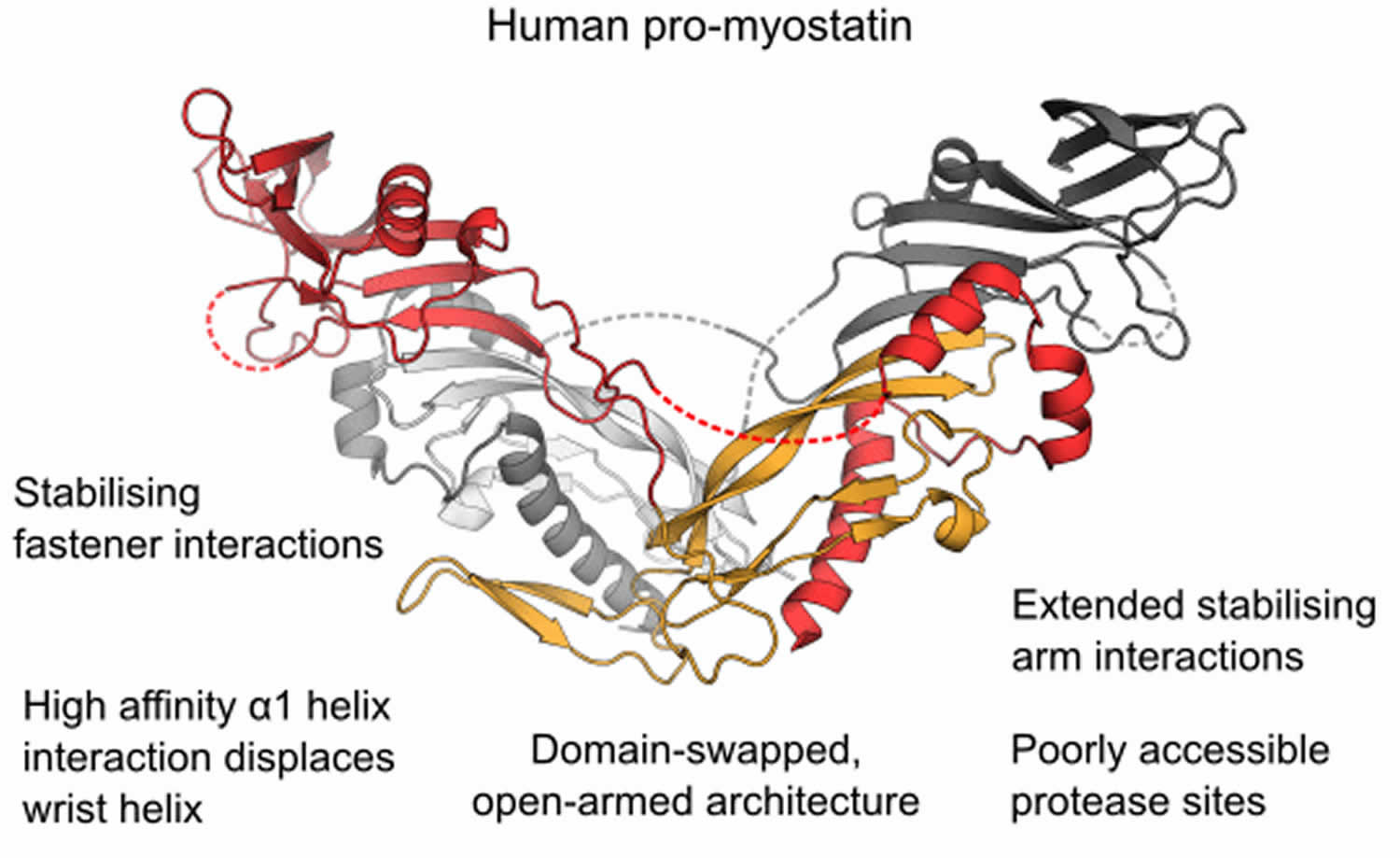
Footnote: The structure of the precursor of the muscle mass regulator myostatin/GDF8 (growth and differentiation factor 8) shows an open‐armed conformation, similar to pro‐activin A and distinct from pro‐TGF‐β1. An enhanced interface between prodomains and the mature growth factor allows pro‐myostatin to persist as a latent, antagonist resistant complex even after processing of the precursor by furin‐like proteases.
- The crystal structure of unprocessed human pro‐myostatin was determined, and is shown to have an open‐armed, domain swapped architecture.
- Following pro‐domain cleavage, pro‐myostatin persists as a stable non‐covalent complex and has significantly lower bioactivity than the mature growth factor.
- Latency of the pro‐myostatin complex is achieved by an enhanced interface between pro‐ and mature domains.
- The natural antagonist follistatin is unable to displace the prodomains of the latent myostatin complex.
Figure 2. Myostatin
Myostatin is stored extracellularly in its pro- and latent forms in vivo. Researchers have shown that pro- and latent myostatin are the predominant myostatin forms in muscle and serum, respectively 1. While Anderson et al. 7 also demonstrated that some fraction of the proMyostatin pool resides extracellularly, where it can be regulated by extracellular furin cleavage, the relative abundance of intracellular vs. extracellular proMyostatin in muscle tissue remained unclear. By immunofluorescence with a pro/latent-specific antibody, we show here that the bulk of proMyostatin is stored in the sarcolemma-associated extracellular matrix. ProMyostatin distribution overlapped with laminin, but was more focal, often detected near junctions between three sarcolemma, suggesting that additional factors may influence proMyostatin extracellular distribution. Indeed the extracellular matrix proteins latent TGFβ binding protein 3 (LTBP3), latent TGFβ binding protein 4 (LTBP4), and perlecan, have been reported to interact with myostatin and/or myostatin precursors 7, though the role of these anchoring proteins in modulating signaling is, as yet, unknown.
Finding that most proMyostatin resides extracellularly in its primary target tissue, muscle, suggests that extracellular activation is a critical step in myostatin signaling. While the follistatins and GASPs (growth and differentiation factor-associated serum proteins 1 and 2) are naturally occurring inhibitors of myostatin and GDF11, these appear to bind a small fraction of mature myostatin in circulation 7. Consistent with this hypothesis, some scientists observed changes in the balance of myostatin precursors in a model of muscle atrophy. They found that proMyostatin protein levels were upregulated in muscle by day 4 of glucocorticoid-induced atrophy, while decreased circulating latent myostatin was evident in the same timeframe. Given that changes in proMyostatin levels occurred prior to significant loss of muscle mass, some scientists believe that reduced circulating latent myostatin was not due to decreased muscle mass. Some scientists hypothesize instead that changes in serum latent myostatin reflect enhanced myostatin processing in atrophying muscle, underscoring the importance of proteolytic processing in controlling the myostatin signaling pathway.
While circulating myostatin has been examined as a biomarker of muscle atrophy and dysfunction 8, recent data highlight the importance of understanding myostatin processing and signaling in the muscle as well as in serum, as circulating myostatin levels may not adequately reflect the extent of myostatin signaling in the muscle itself. Quantitation of myostatin by ELISA or immunoaffinity mass spectrometry has been reported 8, but these measurements do not distinguish between free mature myostatin and mature myostatin in latent complexes (bound to the propeptide or other inhibitory proteins). Reagents with specificity for the various forms of myostatin, such as the antibodies could be useful in developing truly specific quantitative assays. This, coupled with additional studies to understand myostatin forms in patient samples and preclinical models, will help elucidate the broader role of extracellular processing in myostatin regulation and help to identify patient populations responsive to anti-myostatin therapy.
Myostatin signals by binding a cell surface receptor complex consisting of a type I receptor (ALK4/5) and a type II receptor (ACTRIIa/b) 9. To date, all of the anti-myostatin drug candidates in early-stage clinical studies block the interaction between mature myostatin and its receptors through antibodies, ligand traps, or overexpression of natural inhibitors such as Follistatin 10. However, because the receptor recognition surfaces of mature myostatin and other Transforming Growth Factor β (TGFβ) family members share a high degree of similarity 11, many of these myostatin-targeted biologics cross-react with other members of the TGFβ family (most commonly GDF11 or Activin A). In all of these cases, while promising results have been reported, it is too early to fully appreciate both their efficacy in patients and potential clinical toxicities associated with their lack of target specificity.
Myostatin disorder
At least one mutation in the MSTN (growth differentiation factor 8) gene has been found to cause myostatin-related muscle hypertrophy, a rare condition characterized by increased muscle mass, strength and reduced body fat 12. The mutation, which is written as IVS1+5G>A, disrupts the way the gene’s instructions are used to make myostatin. As a result, cells produce little or no functional myostatin. A loss of this protein in muscle cells leads to an overgrowth of muscle tissue. It is not known how many people are diagnosed with myostatin-related muscle hypertrophy. Unfortunately for the rare diseases, there’s often not a calculated incidence or prevalence. There is no official method for tracking these conditions.
Myostatin-related muscle hypertrophy affected individuals have up to twice the usual amount of muscle mass in their bodies. They also tend to have normal or increased muscle strength. Myostatin-related muscle hypertrophy affected individuals is not to have any other medical problems and affected individuals are intellectually normal.
Myostatin-related muscle hypertrophy has a pattern of inheritance known as incomplete autosomal dominance. People with a mutation in both copies of the MSTN gene in each cell (homozygotes) have significantly increased muscle mass and strength. People with a mutation in one copy of the MSTN gene in each cell (heterozygotes) also have increased muscle bulk, but to a lesser degree.
Clinical manifestations depend on the amount of myostatin protein present. An infant homozygous for an MSTN causative variant had muscle mass twice that of sex- and age-matched controls; intellect and cardiac function were normal. He displayed stimulus-induced myoclonus that subsided after two months 13. Heterozygotes may have increased muscle bulk and strength, but to a lesser degree 13.
Clinical manifestations of myostatin-related muscle hypertrophy appear to be dependent on the amount of myostatin protein present. Therefore both heterozygotes and homozygotes can exhibit muscle hypertrophy.
In a multigenerational family segregating a 3.4-Mb deletion of chromosome 2q32.1q32.3, which is the long (q) arm of chromosome 2 at position 32.2, including MSTN, four of seven individuals with the deletion available for examination were reported to have increased muscle strength and increased size of the gastrocnemius and soleus muscles, whereas the other three individuals with the deletion did not have increased muscle strength or size 14.
Homozygotes
A homozygous loss-of-function myostatin variant was identified in a hypermuscular infant with muscle mass approximately twice that of sex- and age-matched controls 15. At age 4.5 years, he continued to have increased muscle bulk and strength with normal intellect and normal cardiac function by echocardiography and electrocardiography.
He initially displayed stimulus-induced myoclonus that subsided after two months. The relationship between myoclonus and the MSTN causative variant is not clear.
Ultrasonography revealed normal muscle echogenicity and cross-sectional diameter of quadriceps muscle 7.2 SD (standard deviation) above the mean.
Heterozygotes
Heterozygotes may have increased muscle bulk and strength. The mother of the child identified to be homozygous for the c.506+5G>A variant was a former professional athlete with large calf muscles 15.
Myostatin-related muscle hypertrophy is diagnosed based upon the clinical signs and symptoms in the patient (i.e, reduced body fat and increased muscle size) and genetic testing. Body fat can be measured by ultrasound or with a caliper. Skeletal muscle size can be measured by ultrasound, dual-energy x-ray absorptiometry (DEXA), or MRI. Myostatin-related muscle hypertrophy is a very rare condition that is caused by mutations in the MSTN gene. Clinical genetic testing for this condition appears to be available on a limited basis.
A genetic professional would be able to evaluate a person for myostatin-related muscle hypertrophy. Genetics professionals are a source of information for individuals and families regarding genetic diagnosis, natural history, treatment, mode of inheritance, and genetic risks to other family members.
The following online resources can also help you find a genetics professional in your community:
- The National Society of Genetic Counselors (https://www.nsgc.org/) provides a searchable directory of US and international genetic counseling services.
- The American College of Medical Genetics (https://www.acmg.net/) has a searchable database of US genetics clinics.
- The University of Kansas Medical Center (http://www.kumc.edu/gec/prof/genecntr.html) provides a list of US and international genetic centers, clinics, and departments.
- The American Society of Human Genetics (http://www.ashg.org/membership/member_search.shtml) maintains a database of its members, which includes individuals who live outside the United States. Visit the link to obtain a list of the geneticists in your country, some of whom may be researchers that do not provide medical care.
Clinical diagnosis
The diagnosis of myostatin-related muscle hypertrophy is established by clinical findings of reduced subcutaneous fat pad thickness and increased muscle size in individuals with normal or increased muscle strength and an MSTN causative variant identified on molecular genetic testing.
Testing
Skeletal muscle size can be measured by ultrasound, DEXA, or MRI. It is expected to be several deviations above normal for age- and sex-matched controls.
Subcutaneous fat pad thickness can be measured by ultrasound or with a caliper at various standard locations for which normal values exist.
Creatine kinase (CK) serum concentration is expected to be normal.
Molecular genetic testing
Gene. MSTN, which encodes the protein growth differentiation factor 8 (also known as myostatin) is the only gene in which variants are known to cause myostatin-related muscle hypertrophy.
Targeted analysis of the causative variant c.506+5G>A should be performed first. If the variant is not detected and clinical suspicion is high, sequence analysis of the entire gene should be performed.
Treatment of symptoms
- Myostatin-related muscle hypertrophy is not currently known to cause any medical complications 13.
Evaluation of Relatives at Risk
Risk to Family Members
Proband is the affected individual through whom a family with a genetic disorder is ascertained; may or may not be the individual presenting for genetic counseling.
Parents of a proband who is homozygous for myostatin-related muscle hypertrophy
- The parents of a child with homozygous myostatin-related muscle hypertrophy are obligate heterozygotes and therefore have one MSTN variant.
- Heterozygotes may have increased muscle mass.
Siblings of a proband
- At conception, each sibling of a child with homozygous myostatin-related muscle hypertrophy has a 25% chance of having homozygous myostatin-related muscle hypertrophy, a 50% chance of having one MSTN causative variant with or without increased muscle mass, and a 25% chance of having normal muscle mass and no MSTN causative variants.
- Heterozygotes may have increased muscle mass.
Offspring of a proband
- The offspring of an individual with homozygous myostatin-related muscle hypertrophy are obligate heterozygotes for a causative variant in MSTN and may have increased muscle mass.
- Each child of an individual with heterozygous myostatin-related muscle hypertrophy has a 50% chance of inheriting the MSTN causative variant.
Other family members of a proband
- The chance that other family members will be affected depends on the status of the proband’s parents: if a parent has increased muscle mass, his or her family members may be affected.
- Each sibling of the proband’s parents has a 50% chance of having one MSTN causative variant and may have increased muscle mass.
Parents of a proband who is heterozygous for myostatin-related muscle hypertrophy
- Individuals diagnosed with heterozygous myostatin-related muscle hypertrophy may have a parent with an MSTN causative variant who may have increased muscle mass or may have the condition as the result of a de novo variant. The proportion of cases caused by a de novo variant is unknown.
- Recommendations for the evaluation of parents of a proband with an apparent de novo variant include clinical evaluation for evidence of muscle hypertrophy.
Note: Although individuals diagnosed with heterozygous myostatin-related muscle hypertrophy may have a parent with increased muscle mass, the family history may appear to be negative because of incomplete penetrance or failure to recognize the condition in family members.
Siblings of a proband
- The chance that the siblings of the proband will inherit the MSTN causative variant depends on the genetic status of the proband’s parents.
- If a parent of the proband has increased muscle mass, the chance that the siblings will inherit the MSTN causative variant is 50%.
Therapies Under Investigation
Search ClinicalTrials.gov (https://clinicaltrials.gov/) in the US and www.ClinicalTrialsRegister.eu (https://www.clinicaltrialsregister.eu/ctr-search/search) in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Myostatin inhibitor
Muscular dystrophies are characterized by weakness and wasting of skeletal muscle tissues. Several drugs targeting the myostatin pathway have been used in clinical trials to increase muscle mass and function but most showed limited efficacy.
Several myostatin inhibitory drugs have been designed targeting different stages of the myostatin biosynthesis or pathway among them (i) monoclonal antibodies targeting myostatin, (ii) monoclonal antibodies targeting myostatin’s receptor AcvRII, (iii) AcvRII decoys, (iv) follistatin overexpression which functions as a myostatin antagonist by preventing receptor binding 16. During the last 15 years, at least six molecules (MYO-029, BMS-986089, PF-06252616, ACE-083/-031, BYM338, FS-344) have been developed by pharmaceutical companies to block myostatin pathways (https://clinicaltrials.gov/). These molecules are/were evaluated in several neuromuscular diseases that show muscular wasting or atrophy but so far the published results were largely disappointing. Significant improvements in muscle strength or physical function have not been reached 17, 18, with the exception of two small open-label studies using an AAV vector encoding the follistatin isoform FS344 intramuscularly injected in Becker Muscular Dystrophy (n = 6) patients and in Inclusion Body Myositis (n = 6) 19, 20 and 1 small randomized controlled trial using a monoclonal antibody against the AcvRII receptor in Inclusion Body Myositis patients (n = 11 active, 3 placebo) 21. Several explanations have been proposed, among them the specificity of the drugs themselves and the possibility that they do not target the correct form of myostatin or target other growth factors besides myostatin implicated in muscle mass regulation. However, in animals, several laboratories including ours have demonstrated that myostatin pathway inhibition leads to muscle hypertrophy and enhances tetanic force in controls or in several murine models of muscle diseases such as the mdx mouse, a murine model for Duchenne Muscular Dystrophy 22.
Side effects from blocking these growth factors could limit the utility of candidate therapeutics. For example, an early clinical trial of the ActRIIb ligand trap, ACE-031, in Duchenne muscular dystrophy was terminated prematurely due to epistaxis and telangiectasis 23 attributed to BMP9 or BMP10 reactivity 24. TGFβ family prodomains universally share much less sequence conservation than the mature ligands 25 and researchers are currently applying this approach to other members of the family.
- Pirruccello-Straub M, Jackson J, Wawersik S, et al. Blocking extracellular activation of myostatin as a strategy for treating muscle wasting. Sci Rep. 2018;8(1):2292. Published 2018 Feb 2. doi:10.1038/s41598-018-20524-9 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5797207/[↩][↩]
- MSTN gene. https://ghr.nlm.nih.gov/gene/MSTN[↩]
- Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. McPherron AC, Lawler AM, Lee SJ. Nature. 1997 May 1; 387(6628):83-90.[↩]
- Myostatin represses physiological hypertrophy of the heart and excitation-contraction coupling. Rodgers BD, Interlichia JP, Garikipati DK, Mamidi R, Chandra M, Nelson OL, Murry CE, Santana LF. J Physiol. 2009 Oct 15; 587(Pt 20):4873-86.[↩]
- Mosher DS, et al. A mutation in the myostatin gene increases muscle mass and enhances racing performance in heterozygote dogs. PLoS Genet. 2007;3:e79. doi: 10.1371/journal.pgen.0030079[↩]
- Structure of the human myostatin precursor and determinants of growth factor latency. The EMBO Journal (2018) e97883. DOI 10.15252/embj.201797883 http://emboj.embopress.org/content/early/2018/01/12/embj.201797883.full[↩]
- Anderson SB, Goldberg AL, Whitman M. Identification of a novel pool of extracellular pro-myostatin in skeletal muscle. J Biol Chem. 2008;283:7027–7035. doi: 10.1074/jbc.M706678200[↩][↩][↩]
- Bergen HR, III, et al. Myostatin as a mediator of sarcopenia versus homeostatic regulator of muscle mass: insights using a new mass spectrometry-based assay. Skelet Muscle. 2015;5:21. doi: 10.1186/s13395-015-0047-5[↩][↩]
- McPherron AC, Lawler AM, Lee SJ. Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature. 1997;387:83–90. doi: 10.1038/387083a0[↩]
- Becker C, et al. Myostatin antibody (LY2495655) in older weak fallers: a proof-of-concept, randomised, phase 2 trial. The Lancet Diabetes & Endocrinology. 2015;3:948–957. doi: 10.1016/S2213-8587(15)00298-3[↩]
- Khalil AM, et al. Differential Binding Activity of TGF-beta Family Proteins to Select TGF-beta Receptors. J Pharmacol Exp Ther. 2016;358:423–430. doi: 10.1124/jpet.116.232322[↩]
- Myostatin-related muscle hypertrophy. https://ghr.nlm.nih.gov/condition/myostatin-related-muscle-hypertrophy[↩]
- Wagner KR, Cohen JS. Myostatin-Related Muscle Hypertrophy. 2005 Oct 5 [Updated 2013 Jul 3]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2018. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1498[↩][↩][↩]
- Meienberg J, Rohrbach M, Neuenschwander S, Spanaus K, Giunta C, Alonso S, Arnold E, Henggeler C, Regenass S, Patrignani A, Azzarello-Burri S, Steiner B, Nygren AO, Carrel T, Steinmann B, Mátyás G. Hemizygous deletion of COL3A1, COL5A2, and MSTN causes a complex phenotype with aortic dissection: a lesson for and from true haploinsufficiency. Eur J Hum Genet. 2010;18:1315–21[↩]
- Schuelke M, Wagner KR, Stolz LE, Hübner C, Riebel T, Kömen W, Braun T, Tobin JF, Lee SJ. Myostatin mutation associated with gross muscle hypertrophy in a child. N Engl J Med. 2004;350:2682–8.[↩][↩]
- Cohen S, Nathan JA, Goldberg AL. Muscle wasting in disease: molecular mechanisms and promising therapies. Nat. Rev. Drug. Discov. 2015;14:58–74. doi: 10.1038/nrd4467[↩]
- Garber K. No longer going to waste. Nat. Biotechnol. 2016;34:458–461. doi: 10.1038/nbt.3557[↩]
- Campbell C, et al. Myostatin inhibitor ACE-031 treatment of ambulatory boys with Duchenne muscular dystrophy: results of a randomized, placebo-controlled clinical trial. Muscle Nerve. 2016;55:458–464. doi: 10.1002/mus.25268[↩]
- Al-Zaidy SA, et al. Follistatin gene therapy improves ambulation in becker muscular dystrophy. J. Neuromuscul. Dis. 2015;2:185–192. doi: 10.3233/JND-150083[↩]
- Mendell JR, et al. Follistatin gene therapy for sporadic inclusion body myositis improves functional outcomes. Mol. Ther. 2017;25:870–879. doi: 10.1016/j.ymthe.2017.02.015[↩]
- Amato AA, et al. Treatment of sporadic inclusion body myositis with bimagrumab. Neurology. 2014;83:2239–2246. doi: 10.1212/WNL.0000000000001070[↩]
- Bechir N, et al. ActRIIB blockade increases force-generating capacity and preserves energy supply in exercising mdx mouse muscle in vivo. FASEB J. 2016;30:3551–3562. doi: 10.1096/fj.201600271RR[↩]
- Campbell, C. et al. Myostatin inhibitor ACE-031 treatment of ambulatory boys with Duchenne muscular dystrophy: Results of a randomized, placebo-controlled clinical trial. Muscle Nerve 2016, 10.1002/mus.25268[↩]
- David L, Mallet C, Mazerbourg S, Feige JJ, Bailly S. Identification of BMP9 and BMP10 as functional activators of the orphan activin receptor-like kinase 1 (ALK1) in endothelial cells. Blood. 2007;109:1953–1961. doi: 10.1182/blood-2006-07-034124[↩]
- Structural Biology and Evolution of the TGF-β Family. Hinck AP, Mueller TD, Springer TA. Cold Spring Harb Perspect Biol. 2016 Dec 1; 8(12):.[↩]


{kind=link}