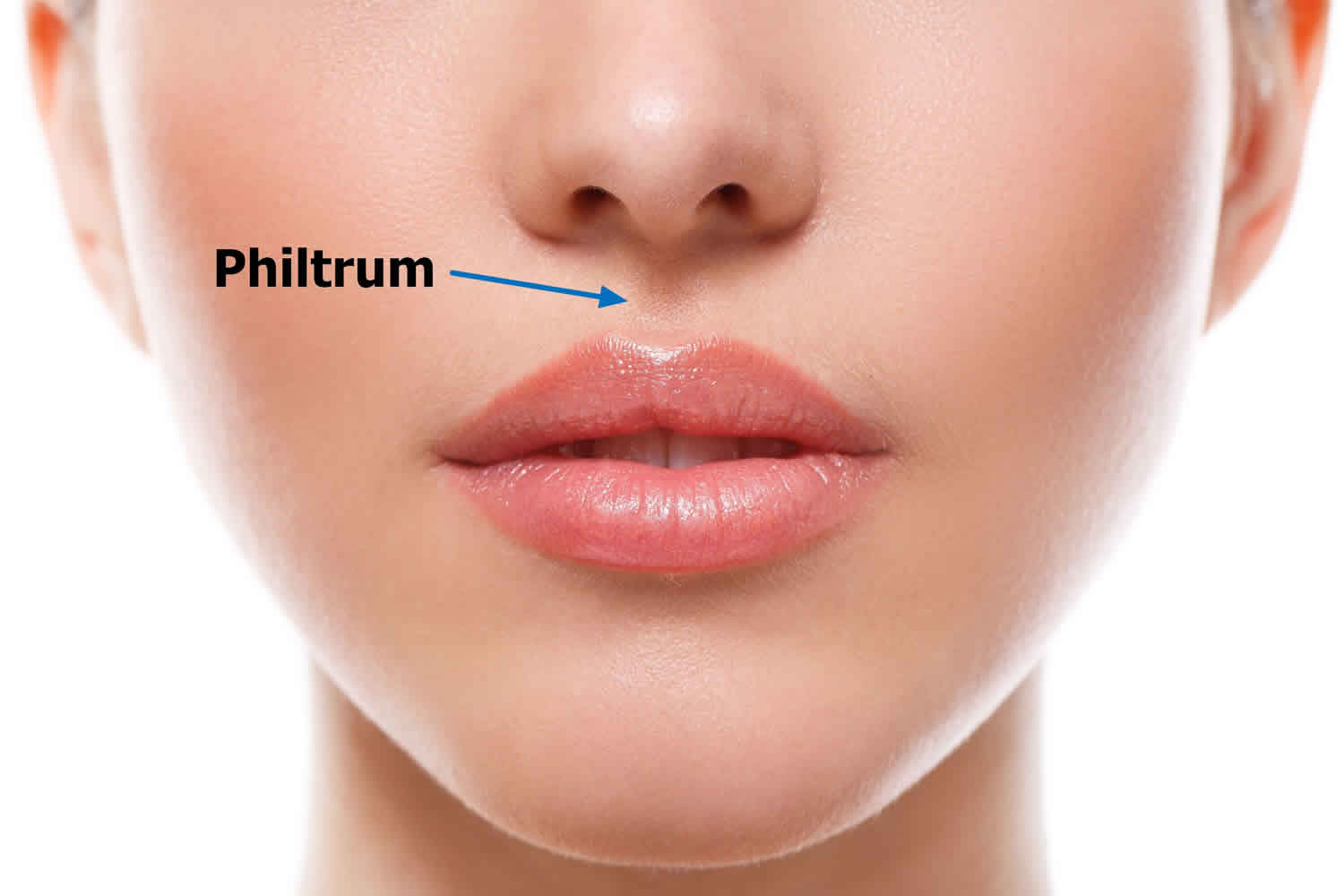
What is a philtrum
Philtrum is the central depression or the vertical groove between the nose and the upper lip. Variations in the anatomy of the lips and philtrum can be indicative of developmental abnormalities. The philtral ridges and the philtrum are formed by a unique collection of dermal collagen and dense elastic tissue. As elasticity is diminished with age, the philtrum takes on a less prominent appearance. The inferior margin of the philtrum forms the downward arch of the cupid’s bow, while the underlying fleshy fullness is known as the tubercle or procheilon. Outlining the vermilion borders of the upper and lower lips is a 2 to 3 mm pale convexity known as the white roll, formed by the bulging of the orbicularis oris muscle laying beneath. The upper and lower lips connect to the gums by the frenulum labii superioris and frenulum labii inferioris, respectively.
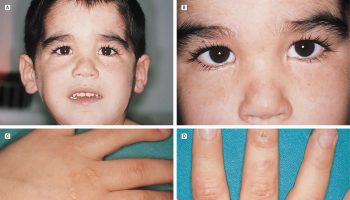
Astley and colleagues 1, 2 introduced a 5-point Likert-scaled philtrum guide based on Caucasian North American subjects as an objective tool for the evaluation of the facial dysmorphology in fetal alcohol syndrome and fetal alcohol spectrum disorders. This Caucasian guide has been incorporated into all current diagnostic schemes for fetal alcohol spectrum disorders. However, broad international clinical experience with fetal alcohol syndrome indicates racial and ethnic differences with respect to the facial morphology 3.
Figure 1. Normal philtrum
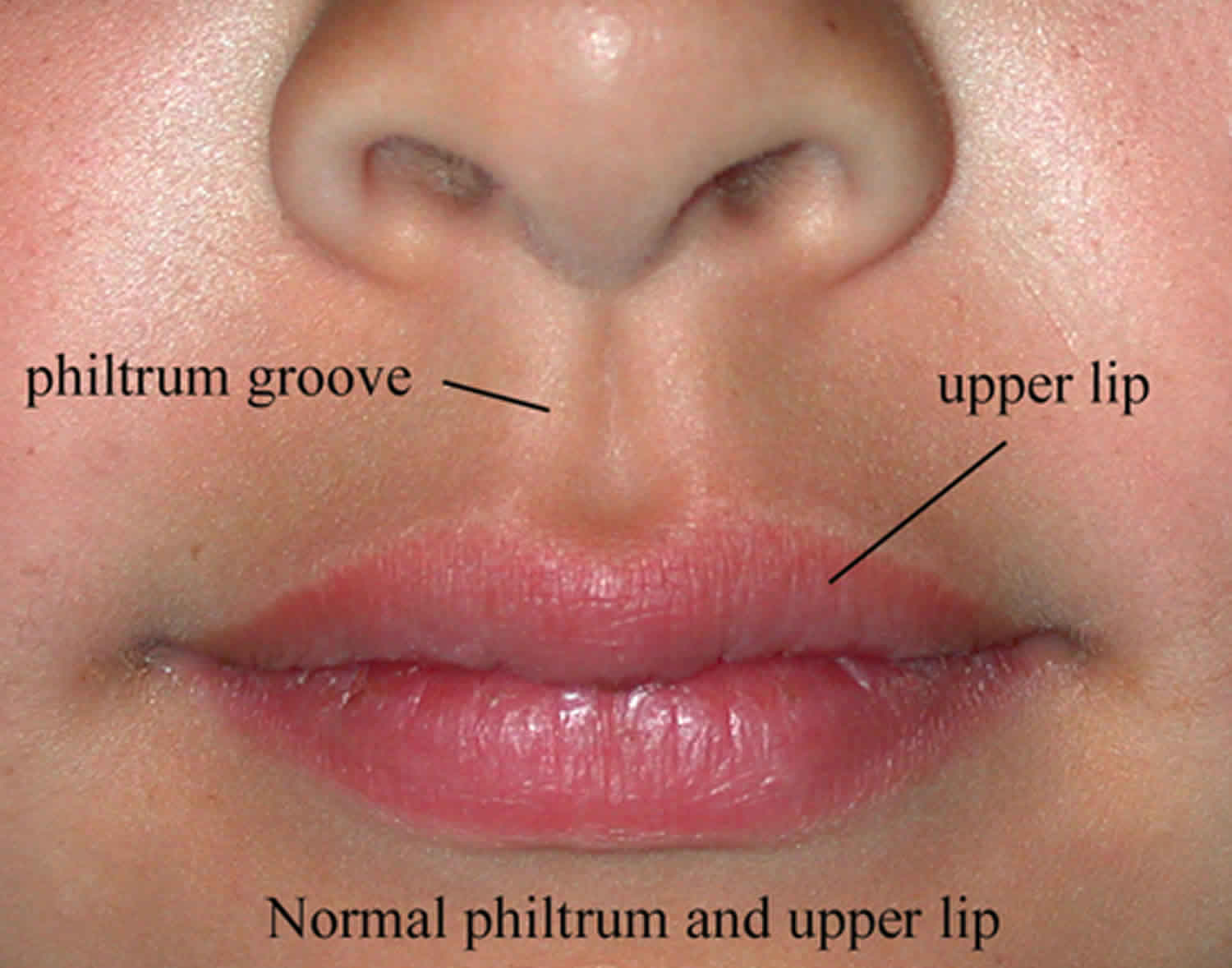
Figure 2. Philtrum guide (Likert-scaled philtrum guide)
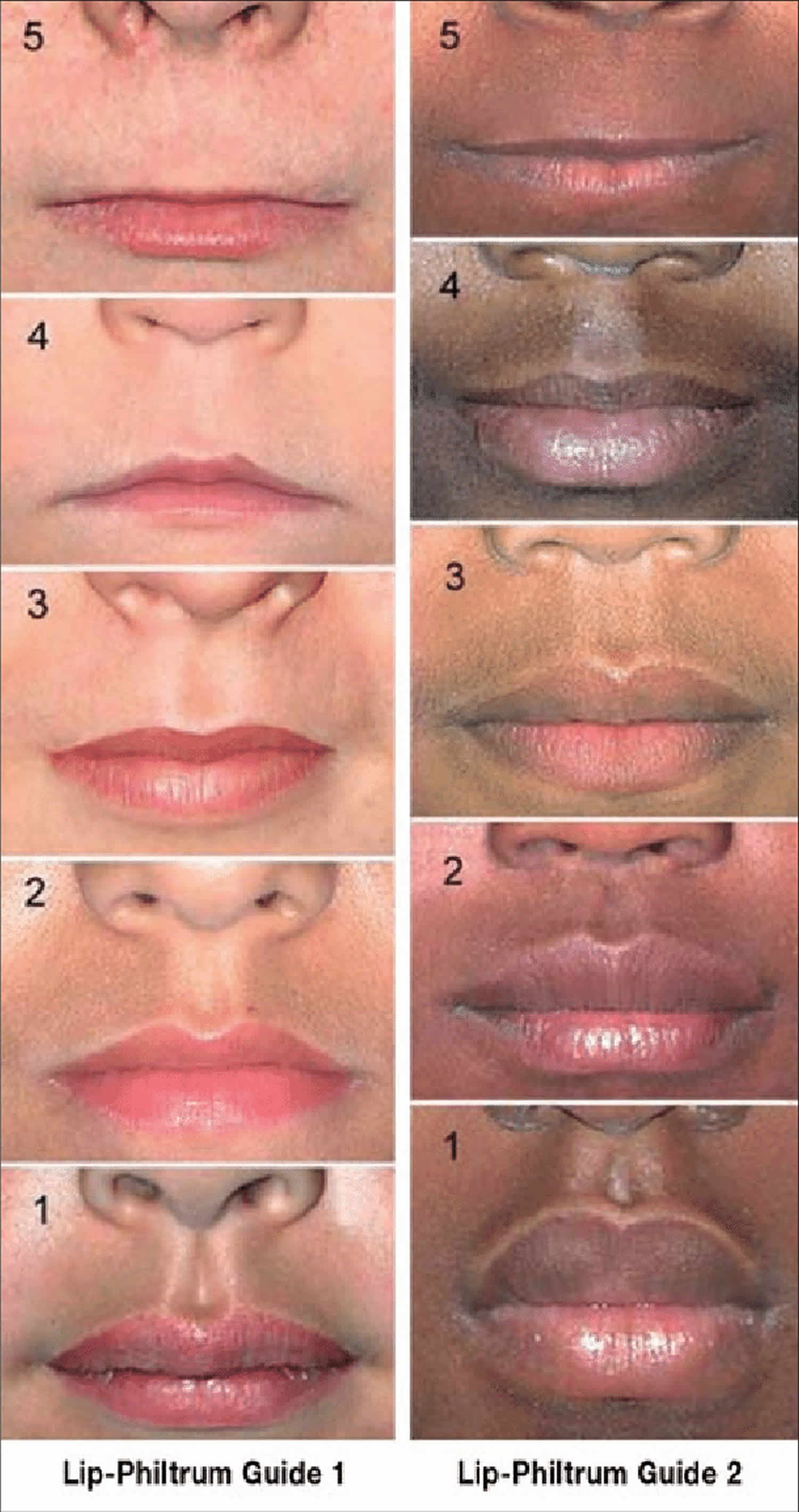
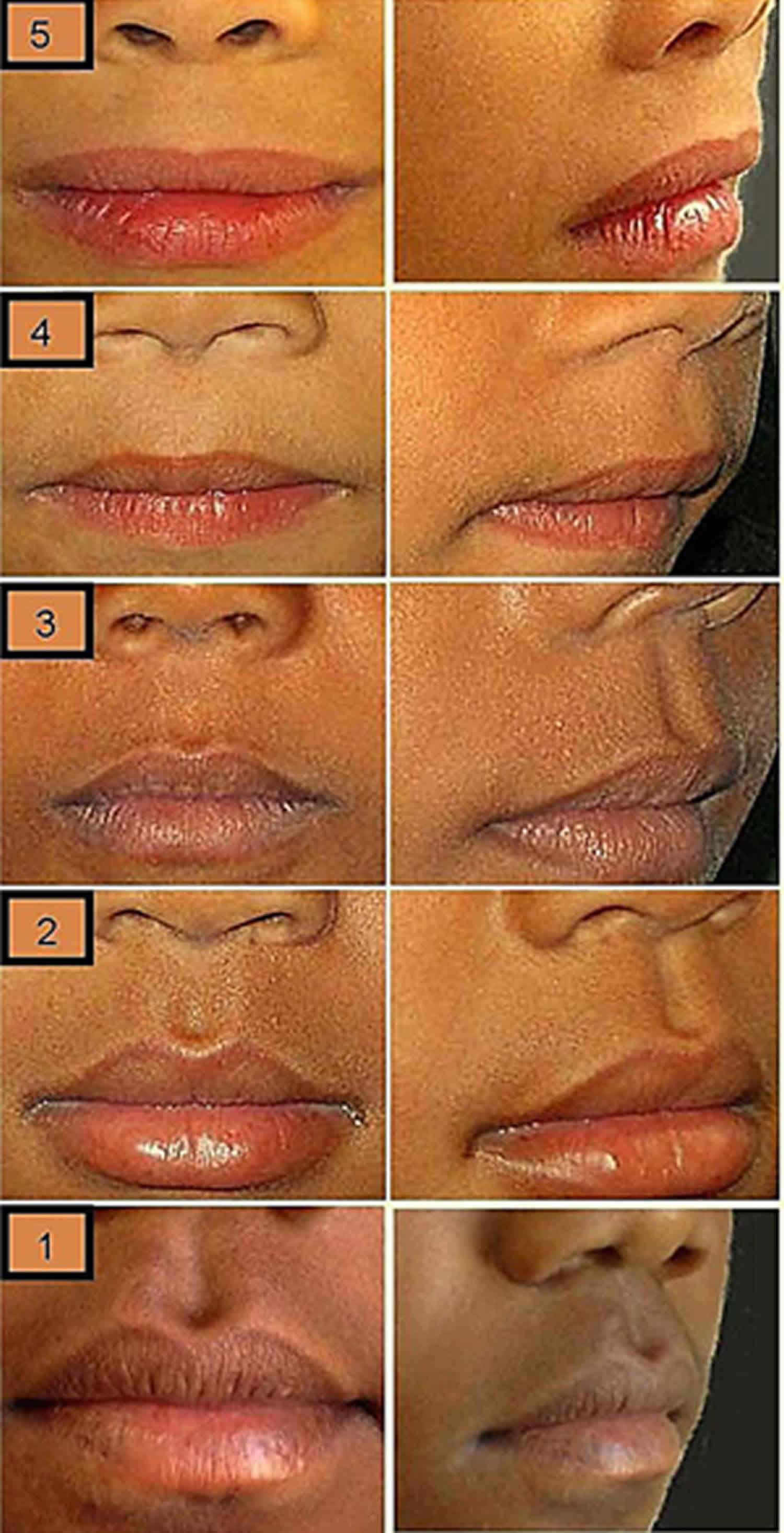
Footnote: Lip–philtrum guide (left Caucasian, right African ethnicity) for assessment of thickness of the upper lip and smoothness of the philtrum (the vertical groove between nose and upper lip). Grade 3 = average appearance in the normal population. Grade 4 and 5 = thin upper lip and smooth philtrum characteristic of fetal alcohol syndrome.
[Source 2 ]Figure 3. Philtrum guide (mixed-race philtrum guide)
Philtrum purpose
The philtrum is believed to serve as a supply of additional skin to be recruited for oral movements requiring stretching of the upper lip 4. The principal muscle of the lips is the circumferential orbicularis oris, functioning primarily as a sphincter for the oral aperture. Rather uniquely, the orbicularis oris bears no direct bony attachments but is appended by the other oral muscles that attach to it. At the philtrum, the fibers of the orbicularis oris decussate to insert into the opposite philtral ridge. Interdigitation of the muscle’s fibers at the commissures allows for a scissor-like closure. The perpendicular force vectors formed by the contraction of the orbicular oris results in the generation of relaxed skin tension lines angled radially outward from the oral orifice. Adequate function of the orbicularis oris is requisite for the closure of the mouth, chewing and creating an oral seal.
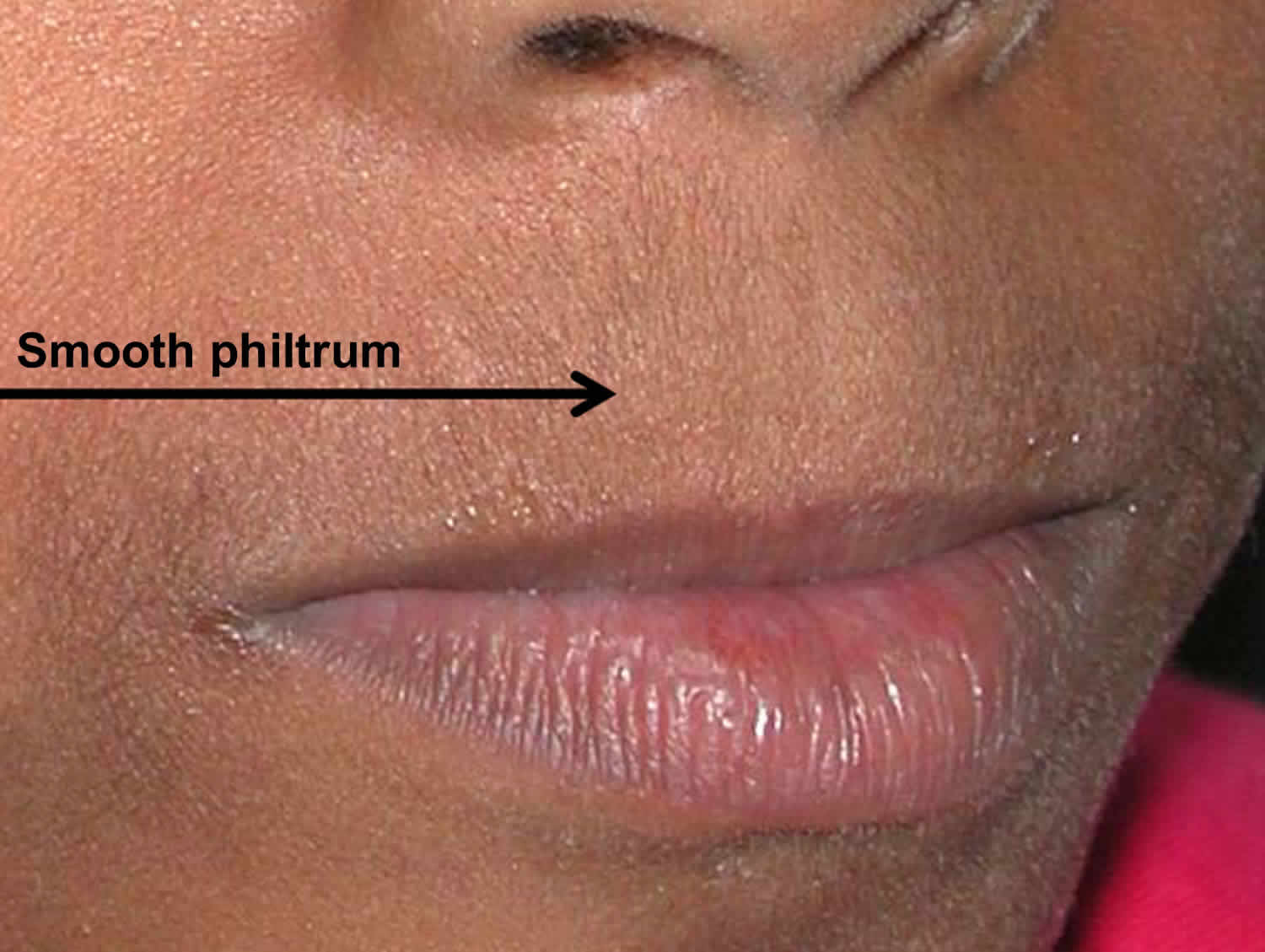
Smooth philtrum
Smooth philtrum is flat skin surface, with no ridge formation in the central region of the upper lip between the nasal base and upper vermilion border. Short palpebral fissures, a smooth philtrum, and a thin vermilion border of the upper lip comprise the three cardinal facial features of fetal alcohol syndrome 3.
The fetal alcohol spectrum disorder includes:
- Fetal alcohol syndrome: Fetal alcohol syndrome (FAS) is diagnosed by the presence of all of the following criteria: two of the three characteristic facial features (short palpebral fissures, thin vermillion border, and a smooth philtrum), growth retardation (prenatally and/or postnatally), and central nervous system defects. Because all of these criteria are met for diagnosis, fetal alcohol syndrome does not require documentation of prenatal alcohol exposure.
- Partial fetal alcohol syndrome: Partial fetal alcohol syndrome (pFAS) has two of the characteristic facial features plus, depending on where alcohol exposure was documented, varies in its other criteria.
- Alcohol-related neurodevelopmental disorder: Alcohol-related neurodevelopmental disorder describes those with neurobehavioral impairment in the setting of documented prenatal alcohol exposure but have minimal to no physical findings and cannot be diagnosed before three years of age.
- Alcohol-related birth defects: Alcohol-related birth defects is the term used to describe those with the physical defects secondary to known fetal alcohol exposure, but who do not have neurobehavioral deficits. On the opposite end of the spectrum.
- Neurobehavioral disorder associated with prenatal alcohol exposure is very similar to alcohol-related congenital disabilities but may involve some physical features.
All of the conditions that comprise fetal alcohol spectrum disorders stem from one common cause which is prenatal exposure to alcohol 5. Alcohol is extremely teratogenic to a fetus. Its effects are wide-ranging and irreversible. Although higher amounts of prenatal alcohol exposure have been linked to increased incidence and severity of fetal alcohol spectrum disorders, there are no studies that demonstrate a safe amount of alcohol that can be consumed during pregnancy. There is also no safe time during pregnancy in which alcohol can be consumed without risk to the fetus. Alcohol is teratogenic during all three trimesters. In summary, any amount of alcohol consumed at any point during pregnancy has the potential cause irreversible damage that can lead to a fetal alcohol spectrum disorder.
Figure 4. Smooth philtrum (Absolutely smooth philtrum. No hint of a philtrum depression, no matter what angle you view it at)
Long philtrum
Long philtrum or elongated philtrum, which is the distance between nasal base and midline upper lip vermilion border more than 2 SD (standard deviation) above the mean. Alternatively, an apparently increased distance between nasal base and midline upper lip vermilion border.
Diseases associated with long philtrum
- 14q11.2 Microdeletion Syndrome
- 15q14 Microdeletion Syndrome
- 15q24 Microdeletion Syndrome
- 15q26 Overgrowth Syndrome
- 16q24.3 Microdeletion Syndrome
- 19p13.12 Microdeletion Syndrome
- 1p36 Deletion Syndrome
- 1q21.1 Microdeletion Syndrome
- 20p12.3 Microdeletion Syndrome
- 22q11.2 Deletion Syndrome
- 2p15p16.1 Microdeletion Syndrome
- 2q31.1 Microdeletion Syndrome
- 2q32q33 Microdeletion Syndrome
- 3-hydroxyisobutyric Aciduria
- 3m Syndrome
- 3q13 Microdeletion Syndrome
- 48,xyyy Syndrome
- 6q25 Microdeletion Syndrome
- 8p Inverted Duplication/deletion Syndrome
- 8p23.1 Duplication Syndrome
- 8q12 Microduplication Syndrome
- 8q22.1 Microdeletion Syndrome
- Aarskog Syndrome, Autosomal Dominant
- Aarskog-scott Syndrome
- Achondrogenesis
- Achondrogenesis Type 1a
- Achondrogenesis Type 1b
- Achondrogenesis Type 2
- Acrocallosal Syndrome
- Acromicric Dysplasia
- Acromicric Dysplasia
- Acroosteolysis Dominant Type
- Adenylosuccinase Deficiency
- Adenylosuccinate Lyase Deficiency
- Agenesis Of The Corpus Callosum And Congenital Lymphedema
- Al Kaissi Syndrome
- Alazami-yuan Syndrome
- Alg13-cdg
- Alpha-thalassemia/mental Retardation Syndrome, Chromosome 16-related
- Anophthalmia-megalocornea-cardiopathy-skeletal Anomalies Syndrome
- Antley-bixler Syndrome
- Antley-bixler Syndrome Without Genital Anomalies Or Disordered Steroidogenesis
- Aplasia Cutis Congenita, Reticulolinear, With Microcephaly, Facialdysmorphism, And Other Congenital Anomalies
- Arterial Tortuosity Syndrome
- Arthrogryposis, Distal, Type 2a
- Arthrogryposis, Distal, Type 2b
- Atypical Rett Syndrome
- Autosomal Dominant Omodysplasia
- Autosomal Dominant Robinow Syndrome
- Autosomal Recessive Cutis Laxa Type 2
- Autosomal Recessive Cutis Laxa Type 2, Classic Type
- Autosomal Recessive Faciodigitogenital Syndrome
- Autosomal Recessive Multiple Pterygium Syndrome
- Autosomal Recessive Omodysplasia
- Autosomal Recessive Robinow Syndrome
- Autosomal Recessive Spondylocostal Dysostosis
- Ayme-gripp Syndrome
- Band-like Calcification With Simplified Gyration And Polymicrogyria
- Bannayan-riley-ruvalcaba Syndrome
- Baraitser-winter Cerebrofrontofacial Syndrome
- Baraitser-winter Syndrome 1
- Baraitser-winter Syndrome 2
- Blepharonasofacial Malformation Syndrome
- Blepharophimosis With Facial And Genital Anomalies And Mental Retardation
- Blomstrand Lethal Chondrodysplasia
- Bone Fragility With Contractures, Arterial Rupture, And Deafness
- Brachycephaly, Deafness, Cataract, Microstomia, And Mental Retardation
- Brachymorphism-onychodysplasia-dysphalangism Syndrome
- Brain Malformation-congenital Heart Disease-postaxial Polydactyly Syndrome
- C Syndrome
- Camptodactyly Syndrome, Guadalajara, Type Ii
- Camptodactyly With Muscular Hypoplasia, Skeletal Dysplasia, And Abnormalpalmar Creases
- Cardiofaciocutaneous Syndrome
- Cardiospondylocarpofacial Syndrome
- Carey-fineman-ziter Syndrome
- Cataracts, Growth Hormone Deficiency, Sensory Neuropathy, Sensorineural Hearing Loss, And Skeletal Dysplasia
- Cerebellar Ataxia, Nonprogressive, With Mental Retardation
- Cerebrocostomandibular Syndrome
- Cerebrooculofacioskeletal Syndrome 1
- Cerebrooculonasal Syndrome
- Chromosome 10q26 Deletion Syndrome
- Chromosome 14q11-q22 Deletion Syndrome
- Chromosome 1p36 Deletion Syndrome
- Chromosome 2p16.1-p15 Deletion Syndrome
- Chromosome 3pter-p25 Deletion Syndrome
- Chromosome 5q12 Deletion Syndrome
- Chromosome 6q11-q14 Deletion Syndrome
- Chromosome 9p Deletion Syndrome
- Chst3-related Skeletal Dysplasia
- Combined Oxidative Phosphorylation Deficiency 25
- Congenital Contractures Of The Limbs And Face, Hypotonia, And Developmental Delay
- Congenital Disorder Of Glycosylation, Type Ih
- Congenital Disorder Of Glycosylation, Type Il
- Cornelia De Lange Syndrome
- Cornelia De Lange Syndrome 1
- Cornelia De Lange Syndrome 4
- Cornelia De Lange Syndrome 5
- Cortical Blindness, Retardation, And Postaxial Polydactyly
- Cortical Blindness-intellectual Disability-polydactyly Syndrome
- Craniofaciofrontodigital Syndrome
- Craniofaciofrontodigital Syndrome
- Craniolenticulosutural Dysplasia
- Craniolenticulosutural Dysplasia
- Craniosynostosis-hydrocephalus-arnold-chiari Malformation Type I-radioulnar Synostosis Syndrome
- Craniosynostosis-mental Retardation Syndrome Of Lin And Gettig
- Crisponi Syndrome
- Crisponi/cold-induced Sweating Syndrome 1
- Cutis Laxa
- Cutis Laxa, Autosomal Recessive, Type Ic
- Cutis Laxa, Autosomal Recessive, Type Iia
- Cutis Laxa, Autosomal Recessive, Type Iic
- D-bifunctional Protein Deficiency
- Deafness, Onychodystrophy, Osteodystrophy, Mental Retardation, And Seizures Syndrome
- Delayed Speech-facial Asymmetry-strabismus-ear Lobe Creases Syndrome
- Dend Syndrome
- Desbuquois Dysplasia 2
- Developmental Delay, Intellectual Disability, Obesity, And Dysmorphic Features
- Diabetes Insipidus, Neurohypophyseal Type
- Diabetes Mellitus, Neonatal, With Congenital Hypothyroidism
- Diabetes Mellitus, Permanent Neonatal
- Distal 22q11.2 Microduplication Syndrome
- Distal Monosomy 3p
- Distal Trisomy 15q
- Distal Trisomy 5q
- Ehlers-danlos Syndrome, Musculocontractural Type, 1
- Ehlers-danlos Syndrome, Musculocontractural Type, 2
- Emanuel Syndrome
- Emanuel Syndrome
- Emery-nelson Syndrome
- Encephalopathy Due To Sulfite Oxidase Deficiency
- Epilepsy-telangiectasia Syndrome
- Epileptic Encephalopathy, Early Infantile, 63
- Epiphyseal Dysplasia-hearing Loss-dysmorphism Syndrome
- Faciocardiomelic Syndrome
- Faciothoracogenital Syndrome
- Femoral-facial Syndrome
- Femoral-facial Syndrome
- Fetal Akinesia Deformation Sequence
- Fetal Valproate Syndrome
- Fibrochondrogenesis 1
- Flat Face-microstomia-ear Anomaly Syndrome
- Fontaine Progeroid Syndrome
- Freeman-sheldon Syndrome
- Fryns Syndrome
- Fryns Syndrome
- Gapo Syndrome
- Geleophysic Dysplasia 1
- Geleophysic Dysplasia 2
- Geleophysic Dysplasia 3
- Genitopatellar Syndrome
- Gm1 Gangliosidosis
- Hajdu-cheney Syndrome
- Hamamy Syndrome
- Hermansky-pudlak Syndrome 2
- Histidinuria Due To A Renal Tubular Defect
- Histidinuria-renal Tubular Defect Syndrome
- Hypertelorism And Other Facial Dysmorphism, Brachydactyly, Genital Abnormalities, Mental Retardation, And Recurrent Inflammatory Episodes
- Hypertelorism And Tetralogy Of Fallot
- Hypertelorism, Teebi Type
- Hypertelorism-hypospadias-polysyndactyly Syndrome
- Hypertrichotic Osteochondrodysplasia
- Hypertrichotic Osteochondrodysplasia, Cantu Type
- Hypoparathyroidism-retardation-dysmorphism Syndrome
- Immunodeficiency 26 With Or Without Neurologic Abnormalities
- Infantile-onset X-linked Spinal Muscular Atrophy
- Insulin-like Growth Factor I, Resistance To
- Intellectual Developmental Disorder With Dysmorphic Facies And Behavioral Abnormalities
- Intellectual Developmental Disorder With Dysmorphic Facies And Ptosis
- Intellectual Developmental Disorder With Dysmorphic Facies, Seizures, And Distal Limb Anomalies
- Intellectual Developmental Disorder With Speech Delay, Dysmorphic Facies, And T-cell Abnormalities
- Intellectual Disability-short Stature-hypertelorism Syndrome
- Intellectual Disability-sparse Hair-brachydactyly Syndrome
- Intermediate Nemaline Myopathy
- Jacobsen Syndrome
- Kagami-ogata Syndrome
- Kbg Syndrome
- Klippel-feil Syndrome 4, Autosomal Recessive, With Nemaline Myopathy And Facial Dysmorphism
- Lateral Meningocele Syndrome
- Lathosterolosis
- Lathosterolosis
- Lethal Congenital Contracture Syndrome 10
- Leukodystrophy, Hypomyelinating, 10
- Lissencephaly, X-linked, 2
- Macrocephaly, Benign Familial
- Macrocephaly, Macrosomia, Facial Dysmorphism Syndrome
- Macrocephaly/autism Syndrome
- Mandibulofacial Dysostosis With Macroblepharon And Macrostomia
- Marden-walker Syndrome
- Marshall Syndrome
- Marshall Syndrome
- Megalocornea-mental Retardation Syndrome
- Mehmo Syndrome
- Meier-gorlin Syndrome 5
- Melanocytic Nevus Syndrome, Congenital
- Mental Retardation And Microcephaly With Pontine And Cerebellar Hypoplasia
- Mental Retardation, Autosomal Dominant 19
- Mental Retardation, Autosomal Dominant 49
- Mental Retardation, Autosomal Recessive 35
- Mental Retardation, Autosomal Recessive 44
- Mental Retardation, Autosomal Recessive 48
- Mental Retardation, Microcephaly, Growth Retardation, Joint Contractures,and Facial Dysmorphism
- Mental Retardation, X-linked 99, Syndromic, Female-restricted
- Mental Retardation, X-linked, Syndromic 33
- Mental Retardation, X-linked, Syndromic, 35
- Mesomelia-synostoses Syndrome
- Methylmalonate Semialdehyde Dehydrogenase Deficiency
- Microcephaly With Or Without Chorioretinopathy, Lymphedema, Or Mental Retardation
- Microcephaly, Cerebellar Hypoplasia, And Cardiac Conduction Defect Syndrome
- Microcephaly-lymphedema-chorioretinopathy Syndrome
- Microlissencephaly-micromelia Syndrome
- Microphthalmia With Limb Anomalies
- Microphthalmia, Syndromic 2
- Mitochondrial Complex V (atp Synthase) Deficiency, Nuclear Type 2
- Mitochondrial Myopathy And Sideroblastic Anemia
- Mitochondrial Pyruvate Carrier Deficiency
- Molybdenum Cofactor Deficiency, Complementation Group A
- Molybdenum Cofactor Deficiency, Complementation Group B
- Momo Syndrome
- Momo Syndrome
- Monosomy 22
- Monosomy 9p
- Monosomy 9q22.3
- Mosaic Variegated Aneuploidy Syndrome 1
- Moyamoya Disease 4 With Short Stature, Hypergonadotropic Hypogonadism,and Facial Dysmorphism
- Mucolipidosis Ii Alpha/beta
- Mucolipidosis Type Ii
- Multiple Benign Circumferential Skin Creases On Limbs
- Multiple Congenital Anomalies-hypotonia-seizures Syndrome 1
- Multiple Congenital Anomalies-hypotonia-seizures Syndrome 3
- Multiple Pterygium Syndrome, Escobar Variant
- Multiple Pterygium-malignant Hyperthermia Syndrome
- Nablus Mask-like Facial Syndrome
- Nemaline Myopathy 2
- Neurodevelopmental Disorder With Cerebellar Atrophy And With Or Without Seizures
- Neurodevelopmental Disorder With Progressive Microcephaly, Spasticity, And Brain Anomalies
- Nicolaides-baraitser Syndrome
- Non-distal Trisomy 13q
- Non-progressive Cerebellar Ataxia With Intellectual Disability
- Non-rhizomelic Chondrodysplasia Punctata
- Noonan Syndrome-like Disorder With Or Without Juvenile Myelomonocytic Leukemia
- Occipital Horn Syndrome
- Occipital Horn Syndrome
- Oculocerebrorenal Syndrome Of Lowe
- Oculodentodigital Dysplasia, Autosomal Recessive
- Oculofaciocardiodental Syndrome
- Ohdo Syndrome
- Ohdo Syndrome, X-linked
- Omodysplasia
- Omodysplasia 1
- Opitz-kaveggia Syndrome
- Opsismodysplasia
- Osteogenesis Imperfecta, Type Vii
- Osteogenesis Imperfecta, Type Xiii
- Osteoglophonic Dysplasia
- Otodental Dysplasia
- Otodental Syndrome
- Overgrowth-macrocephaly-facial Dysmorphism Syndrome
- Pallister-killian Syndrome
- Palmoplantar Carcinoma, Multiple Self-healing
- Peroxisomal Fatty Acyl-coa Reductase 1 Disorder
- Peroxisome Biogenesis Disorder 7a (zellweger)
- Peters Plus Syndrome
- Peters-plus Syndrome
- Phelan-mcdermid Syndrome
- Phocomelia-ectrodactyly, Ear Malformation, Deafness, And Sinus Arrhythmia
- Phocomelia-ectrodactyly-deafness-sinus Arrhythmia Syndrome
- Piebaldism
- Pontocerebellar Hypoplasia, Type 3
- Ptosis-upper Ocular Movement Limitation-absence Of Lacrimal Punctum Syndrome
- Puerto Rican Infant Hypotonia Syndrome
- Pyruvate Dehydrogenase Deficiency
- Pyruvate Dehydrogenase E1-alpha Deficiency
- Rin2 Syndrome
- Ring Chromosome 1 Syndrome
- Ring Chromosome 10 Syndrome
- Robinow Syndrome, Autosomal Dominant
- Robinow Syndrome, Autosomal Dominant 2
- Robinow Syndrome, Autosomal Dominant 3
- Robinow Syndrome, Autosomal Recessive
- Roifman Syndrome
- Sanjad-sakati Syndrome
- Scarf Syndrome
- Scarf Syndrome
- Schwartz-jampel Syndrome
- Short Rib-polydactyly Syndrome, Verma-naumoff Type
- Short Stature, Brachydactyly, Intellectual Developmental Disability, And Seizures
- Short Stature, Facial Dysmorphism, And Skeletal Anomalies With Or Without Cardiac Anomalies
- Short Stature, Hearing Loss, Retinitis Pigmentosa, And Distinctive Facies
- Short Stature, Optic Nerve Atrophy, And Pelger-huet Anomaly
- Short-rib Thoracic Dysplasia 19 With Or Without Polydactyly
- Sialuria
- Simosa Craniofacial Syndrome
- Skeletal Dysplasia And Progressive Central Nervous System Degeneration, Lethal
- Smith-kingsmore Syndrome
- Smith-lemli-opitz Syndrome
- Smith-lemli-opitz Syndrome
- Speech Development, Delayed, With Facial Asymmetry, Strabismus, Andtransverse Earlobe Crease
- Spinocerebellar Ataxia, Autosomal Recessive 20
- Spondylo-ocular Syndrome
- Spondyloepimetaphyseal Dysplasia With Joint Laxity
- Spondyloepimetaphyseal Dysplasia With Joint Laxity, Type 1, With Or Without Fractures
- Spondyloepiphyseal Dysplasia With Congenital Joint Dislocations
- Spondyloepiphyseal Dysplasia, Cantu Type
- Spondyloepiphyseal Dysplasia, Nishimura Type
- Spondyloepiphyseal Dysplasia-brachydactyly And Distinctive Speech
- Spondylometaphyseal Dysplasia With Dentinogenesis Imperfecta
- Stickler Syndrome
- Stickler Syndrome Type 1
- Stickler Syndrome Type 3
- Takenouchi-kosaki Syndrome
- Temple-baraitser Syndrome
- Temtamy Syndrome
- Tetrasomy 12p
- Tetrasomy 18p
- Tetrasomy 5p
- Three M Syndrome 1
- Three M Syndrome 2
- Trichohepatoenteric Syndrome 1
- Trichorhinophalangeal Syndrome Type 1 And 3
- Trichorhinophalangeal Syndrome Type 2
- Trichorhinophalangeal Syndrome, Type I
- Trichorhinophalangeal Syndrome, Type Iii
- Trisomy 13
- Verheij Syndrome
- Visceral Neuropathy-brain Anomalies-facial Dysmorphism-developmental Delay Syndrome
- Weaver Syndrome
- Weaver Syndrome
- Whistling Face Syndrome, Recessive Form
- Wieacker-wolff Syndrome
- Wiedemann-steiner Syndrome
- Wiedemann-steiner Syndrome
- Williams Syndrome
- Williams-beuren Syndrome
- Witteveen-kolk Syndrome
- Wrinkly Skin Syndrome
- Wrinkly Skin Syndrome
- X Small Rings
- X-linked Intellectual Disability, Najm Type
Pallister Killian syndrome is a rare genetic disorder with a distinct phenotype caused by tissue- limited mosaicism tetrasomy of the short arm of chromosome 12 (chromosome 12p), which usually cytogenetically presents as an extra isochromosome 12p 6. Wide phenotypic variability in Pallister Killian syndrome has been reported, ranging from pre-to perinatal death due to multiple congenital anomalies, especially diaphragmatic hernia, and classic phenotypes including seizures, severe developmental delay, macrosomia at birth, deafness, and distinct dysmorphic features, such as coarse face, temporal alopecia, a small nose with anteverted nostrils, long philtrum, and hypo−/hyper- pigmented streaks on the skin.
Short philtrum
Short philtrum is associated with cleft lip 7 or Cri-du-chat syndrome 8 a rare genetic disorder caused by either a partial or complete deletion of the short arm of chromosome 5 (chromosome 5p). The name of the Cri-du-chat syndrome, meaning cat cry was coined after the main clinical finding of a high-pitched, monochromatic cat-like cry. The clinical picture, severity, and progression of the disease vary depending on the region of the chromosome that is deleted and whether it is terminal or interstitial. In other words, differences in phenotype can be attributed to the differences in genotype. Cri-du-chat syndrome is often characterized by distinctive facial features, delayed development, and intellectual disability.
Cri-du-chat syndrome morbidity and mortality rates decrease after the first few years of life. It is reported that 75% of deaths occur during the first month of life, and about 90% of deaths occur during the first year. It is important to note that the type, size, and location of the deletion(s) greatly influence the prognosis.
One of the most important factors in the prognosis of Cri-du-chat syndrome is an early diagnosis. Early diagnosis allows for the implementation of therapeutic measures early on to improve the outcome of physical as well as psychomotor development and helps with social adaptation.
The most characteristic feature of Cri-du-chat syndrome is high-pitched crying, which normally disappears within the first few months of life. The cry is not limited to this syndrome alone and has been reported in a few other neurological disorders. Newborns also exhibit low birthweight and microcephaly as well as asphyxia, muscle hypotonia, and impaired suction. These lead to impaired growth and development during the first few years of life. Recurrent respiratory, as well as intestinal infections, have been reported.
Cri-du-chat syndrome general characteristics:
(a) Craniofacial malformations:
- Microcephaly
- Moon face
- Hypertelorism
- Prominent epicanthal folds
- Large nasal bridge
- Downturned corners of the mouth
- Short philtrum
- Premature gray hair
- Abnormal transverse flexion creases
Uncommonly:
- Downward slanting palpebral fissures
- Low-set ears
- Narrow auditory ducts
- Preauricular tags
- Deafness
- Myopia and cataracts
- Hypersensitivity of pupils to methacholine
- Hypospadias and cryptorchidism
With increasing age, the following features change:
- Hypotonia in neonatal period is replaced with hypertonia
- Prominent microcephaly
- Prominent supraorbital arch
- Dental malocclusions
- Moon face changes into a more narrow vertical face in adulthood
(b) Other anomalies that might be present:
- Hypersensitivity to sound
- Cardiac disorders including congenital heart defects
- Cutaneous hemangioma
- Renal pathology
(c) Orofacial abnormalities:
- High palate
- Mandibular microretrognathia
- Hypoplasia of the enamel
- Chronic periodontitis
(d) Developmental and behavioral manifestations:
- Hyperactivity
- Self-injurious behavior
- Repetitive movements
- Gentle personality
- Obsessive attachment to objects
- Comprehension of speech is better than their ability to express or communicate.
Other causes of short philtrum:
- 15q14 Microdeletion Syndrome
- 17q21.31 Microduplication Syndrome
- 20q11.2 Microdeletion Syndrome
- 22q11.2 Deletion Syndrome
- 2q24 Microdeletion Syndrome
- 3p25.3 Microdeletion Syndrome
- 3q29 Microdeletion Syndrome
- 3q29 Microdeletion Syndrome
- 4q21 Microdeletion Syndrome
- 5q14.3 Microdeletion Syndrome
- 7q11.23 Microduplication Syndrome
- 8q21.11 Microdeletion Syndrome
- Acro-renal-mandibular Syndrome
- Acrocallosal Syndrome
- Acrocraniofacial Dysostosis
- Acrocraniofacial Dysostosis
- Acrofacial Dysostosis Syndrome Of Rodriguez
- Aicardi Syndrome
- Alagille Syndrome
- Alazami Syndrome
- Anonychia-onychodystrophy With Hypoplasia Or Absence Of Distal Phalanges
- Autosomal Dominant Robinow Syndrome
- Autosomal Recessive Robinow Syndrome
- Axenfeld-rieger Syndrome, Type 1
- Axenfeld-rieger Syndrome, Type 2
- Basel-vanagaite-smirin-yosef Syndrome
- Bifid Nose With Or Without Anorectal And Renal Anomalies
- Birk-barel Mental Retardation Dysmorphism Syndrome
- Branchioskeletogenital Syndrome
- Burn-mckeown Syndrome
- Cataract-intellectual Disability-hypogonadism Syndrome
- Cenani-lenz Syndrome
- Cerebellar Atrophy, Visual Impairment, And Psychomotor Retardation
- Cerebrooculofacioskeletal Syndrome 4
- Char Syndrome
- Char Syndrome
- Charlie M Syndrome
- Chime Syndrome
- Chromosome 15q14 Deletion Syndrome
- Chromosome 18q Deletion Syndrome
- Chromosome 19q13.11 Deletion Syndrome, Distal
- Chromosome 3q13.31 Deletion Syndrome
- Chromosome 3q29 Deletion Syndrome
- Chromosome 4q21 Deletion Syndrome
- Chromosome 4q32.1-q32.2 Triplication Syndrome
- Chromosome 5p13 Duplication Syndrome
- Chromosome 8q21.11 Deletion Syndrome
- Cloverleaf Skull-multiple Congenital Anomalies Syndrome
- Coffin-siris Syndrome
- Coffin-siris Syndrome 5
- Coffin-siris Syndrome 6
- Cohen Syndrome
- Cohen Syndrome
- Coloboma, Congenital Heart Disease, Ichthyosiform Dermatosis, Mental Retardation, And Ear Anomalies Syndrome
- Congenital Disorder Of Glycosylation, Type Ig
- Congenital Heart Defects, Dysmorphic Facial Features, And Intellectual Developmental Disorder
- Craniofacial Abnormalities, Cataracts, Congenital Heart Disease, Sacralneural Tube Defects, And Growth And Developmental Retardation
- Craniofacial Dyssynostosis
- Craniofacioskeletal Syndrome
- Cri-du-chat Syndrome
- Cubitus Valgus With Mental Retardation And Unusual Facies
- Deaf Blind Hypopigmentation Syndrome, Yemenite Type
- Deafness-craniofacial Syndrome
- Dentinogenesis Imperfecta-short Stature-hearing Loss-intellectual Disability Syndrome
- Developmental Delay, Intellectual Disability, Obesity, And Dysmorphic Features
- Digeorge Syndrome
- Disorder Of Sex Development-intellectual Disability Syndrome
- Distal 22q11.2 Microduplication Syndrome
- Distal Monosomy 15q
- Distal Monosomy 19p13.3
- Distal Monosomy 6p
- Distal Trisomy 17q
- Duplication/inversion 15q11
- Ectodermal Dysplasia 12, Hypohidrotic/hair/tooth/nail Type
- Ectodermal Dysplasia, Sensorineural Hearing Loss, And Distinctive Facial Features
- Epidermolysis Bullosa Simplex With Anodontia/hypodontia
- Epidermolysis Bullosa, Late-onset Localized Junctional, With Mentalretardation
- Epileptic Encephalopathy, Early Infantile, 23
- Epileptic Encephalopathy, Early Infantile, 49
- Filippi Syndrome
- Filippi Syndrome
- Floating-harbor Syndrome
- Focal Facial Dermal Dysplasia Type Iii
- Frank-ter Haar Syndrome
- Fried Syndrome
- Frontometaphyseal Dysplasia 2
- Fryns Macrocephaly
- Fryns-smeets-thiry Syndrome
- Gms Syndrome
- Goldberg-shprintzen Syndrome
- Hennekam Syndrome
- Holoprosencephaly 9
- Hunter-macdonald Syndrome
- Hyperphosphatasia With Mental Retardation Syndrome 1
- Hypertrichosis, Hyperkeratosis, Mental Retardation, And Distinctive Facial Features
- Hypotonia, Infantile, With Psychomotor Retardation And Characteristic Facies 2
- Intellectual Developmental Disorder With Dysmorphic Facies And Ptosis
- Joubert Syndrome 14
- Keppen-lubinsky Syndrome
- Keppen-lubinsky Syndrome
- Lujan-fryns Syndrome
- Macrocephaly-spastic Paraplegia-dysmorphism Syndrome
- Marshall-smith Syndrome
- Martsolf Syndrome
- Mcdonough Syndrome
- Mcdonough Syndrome
- Megalocornea-intellectual Disability Syndrome
- Mental Retardation, Autosomal Dominant 18
- Mental Retardation, Autosomal Dominant 20
- Mental Retardation, Autosomal Dominant 26
- Mental Retardation, Autosomal Dominant 27
- Mental Retardation, Autosomal Dominant 40
- Mental Retardation, Autosomal Recessive 13
- Mental Retardation, Autosomal Recessive 15
- Mental Retardation, Autosomal Recessive 5
- Mental Retardation, X-linked 98
- Mental Retardation, X-linked, Syndromic, Bain Type
- Mental Retardation, X-linked, Syndromic, Snyder-robinson Type
- Mental Retardation, X-linked, With Cerebellar Hypoplasia And Distinctive Facial Appearance
- Metaphyseal Dysplasia With Maxillary Hypoplasia And Brachydactyly
- Micro Syndrome
- Microform Holoprosencephaly
- Microtriplication 11q24.1
- Monosomy 18p
- Monosomy 18q
- Müllerian Aplasia And Hyperandrogenism
- Multiple Synostoses Syndrome 1
- Myelodysplasia, Immunodeficiency, Facial Dysmorphism, Short Stature,
- Myhre Syndrome
- Oculocerebrofacial Syndrome, Kaufman Type
- Orofaciodigital Syndrome Type 4
- Orofaciodigital Syndrome Xviii
- Paternal 20q13.2q13.3 Microdeletion Syndrome
- Pfeiffer Syndrome
- Pitt-hopkins Syndrome
- Pitt-hopkins Syndrome
- Polyvalvular Heart Disease Syndrome
- Potocki-shaffer Syndrome
- Potocki-shaffer Syndrome
- Progeroid Facial Appearance With Hand Anomalies
- Progressive Non-infectious Anterior Vertebral Fusion
- Renpenning Syndrome
- Renpenning Syndrome 1
- Ring Chromosome 13 Syndrome
- Ring Chromosome 7 Syndrome
- Ritscher-schinzel Syndrome 2
- Rothmund-thomson Syndrome
- Severe Intellectual Disability And Progressive Spastic Paraplegia
- Severe Intellectual Disability-epilepsy-anal Anomalies-distal Phalangeal Hypoplasia
- Shoulder And Girdle Defects-familial Intellectual Disability Syndrome
- Smith-magenis Syndrome
- Spastic Paraplegia 47, Autosomal Recessive
- Spastic Paraplegia 50, Autosomal Recessive
- Spastic Paraplegia 51, Autosomal Recessive
- Spastic Paraplegia 52, Autosomal Recessive
- Spondyloepiphyseal Dysplasia Tarda With Characteristic Facies
- Sweeney-cox Syndrome
- Takenouchi-kosaki Syndrome
- Temple Syndrome
- Tetraploidy
- Tetrasomy 9p
- Tmem70-related Mitochondrial Encephalo-cardio-myopathy
- Transaldolase Deficiency
- Trisomy 20p
- Van Regemorter-pierquin-vamos Syndrome
- White-sutton Syndrome
- Wiedemann-rautenstrauch Syndrome
- Williams-beuren Region Duplication Syndrome
- Wolf-hirschhorn Syndrome
- Wolf-hirschhorn Syndrome
- X-linked Dominant Chondrodysplasia, Chassaing-lacombe Type
- X-linked Intellectual Disability With Marfanoid Habitus
- X-linked Intellectual Disability, Armfield Type
- X-linked Intellectual Disability, Cabezas Type
- X-linked Intellectual Disability, Cantagrel Type
- X-linked Intellectual Disability-cubitus Valgus-dysmorphism Syndrome
- X-linked Intellectual Disability-hypogammaglobulinemia-progressive Neurological Deterioration Syndrome
- Yunis-varon Syndrome
- Yunis-varon Syndrome
- Zttk Syndrome
- A fetal alcohol syndrome screening tool. Astley SJ, Clarren SK. Alcohol Clin Exp Res. 1995 Dec; 19(6):1565-71. https://www.ncbi.nlm.nih.gov/pubmed/8749828/[↩]
- Lip-Philtrum Guides. https://depts.washington.edu/fasdpn/htmls/lip-philtrum-guides.htm[↩][↩]
- Hoyme HE, Hoyme DB, Elliott AJ, et al. A South African mixed race lip/philtrum guide for diagnosis of fetal alcohol spectrum disorders. Am J Med Genet A. 2015;167A(4):752-5. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4507499/[↩][↩][↩]
- Piccinin MA, Zito PM. Anatomy, Head and Neck, Lips. [Updated 2018 Oct 27]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2018 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK507900[↩]
- Vorgias D, Bernstein B. Fetal Alcohol Syndrome. [Updated 2018 Oct 27]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2018 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK448178[↩]
- Karaman B, Kayserili H, Ghanbari A, et al. Pallister-Killian syndrome: clinical, cytogenetic and molecular findings in 15 cases. Mol Cytogenet. 2018;11:45. Published 2018 Aug 17. doi:10.1186/s13039-018-0395-z https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6098576/[↩]
- Walker NJ, Podda S. Cleft Lip. [Updated 2018 Nov 18]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2018 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK482262[↩]
- Ajitkumar A, Mathai JK. Cri Du Chat Syndrome. [Updated 2018 Oct 27]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2018 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK482460[↩]
{kind=link}