Contents
What is polyarteritis nodosa
Polyarteritis nodosa is a serious but rare form of inflammation of blood vessels (vasculitis) that involves small and medium-sized arteries where they become swollen and damaged, preventing them from bringing oxygen and food to organs 1. Arteries are the blood vessels that carry oxygen-rich blood to organs and tissues. Polyarteritis nodosa occurs when certain immune cells attack the affected arteries. The tissues that are fed by the affected arteries DO NOT get the oxygen and nourishment they need. Damage occurs as a result. Blood vessels involvement occurs preferentially at blood vessel bifurcations, resulting in microaneurysm formation, aneurysmal rupture with hemorrhage (bleeding), thrombosis (blood clots), and consequently organ ischemia or infarction (tissue death). Polyarteritis nodosa is a systemic necrotizing vasculitis first described in 1866 by Adolph Kussmaul and Rudolph Maier 2. Unlike other small-sized arterial vessel vasculitides, polyarteritis nodosa (PAN) is not typically associated with anti-neutrophil cytoplasmic antibodies (ANCA). Polyarteritis nodosa is a systemic disease process though there is a limited form of the disease called cutaneous polyarteritis nodosa (CPAN). Even with the limited form of the disease, there is a significant morbidity secondary to digital ulcerations, ischemia, and painful skin nodules.
Polyarteritis nodosa is a rare disease, with an incidence of about 3 to 4.5 cases per 100,000 people annually in the United States (about 1 in 22,000 to 1 in 33,000 people). Internationally, the annual estimated incidence of polyarteritis nodosa ranges from 1.6 cases per million in south Sweden, for example, to 30.7 cases per million adults in Paris 3. Polyarteritis nodosa affects men more frequently than women. While it has been diagnosed in people of every age, it predominantly occurs in people between the ages of 45-65 years 3.
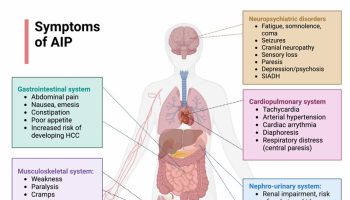
Polyarteritis nodosa symptoms are caused by damage to affected organs. Systemic polyarteritis nodosa is a vasculitis that causes destructive inflammation of medium-sized muscular arteries of multiple systems including the liver, kidney, heart, lung, gastrointestinal tract, musculoskeletal and nervous systems. Systemic polyarteritis nodosa is a potentially life-threatening form of vasculitis, whereas cutaneous polyarteritis nodosa (CPAN) usually runs a chronic but relatively benign course.
Polyarteritis nodosa symptoms include fever, fatigue, weakness, loss of appetite, and weight loss. Muscle and joint aches are common. The skin may show rashes, swelling, ulcers, and lumps. Other symptoms include abdominal pain and gastrointestinal bleeding (occasionally is mistaken for inflammatory bowel disease). Nerve involvement may cause sensory changes with numbness, pain, burning, and weakness. Central nervous system involvement may cause strokes or seizures. Kidney involvement can produce varying degrees of renal failure. Involvement of the arteries of the heart may cause a heart attack, heart failure, and inflammation of the sack around the heart (pericarditis).
There is no specific test to diagnose polyarteritis nodosa. Diagnosis is based upon physical examination, lab tests and biopsy of affected area. Most patients with polyarteritis nodosa have elevated erythrocyte sedimentation rate (ESR). Proteinuria (protein in the urine) is common among patients with kidney involvement.
Treatment involves medicines to suppress inflammation and the immune system. These may include steroids, such as prednisone. Similar medicines, such as azathioprine, methotrexate or mycophenolate that allow for reducing the dose of steroids are often used as well. Cyclophosphamide is used in severe cases.
For polyarteritis nodosa related to hepatitis, treatment may involve plasmapheresis and antiviral medicines.
Current treatments with steroids and other drugs that suppress the immune system (such as azathioprine or cyclophosphamide) can improve symptoms and the chance of long-term survival.
The most serious complications most often involve the kidneys and gastrointestinal tract.
Without treatment, the outlook is poor.
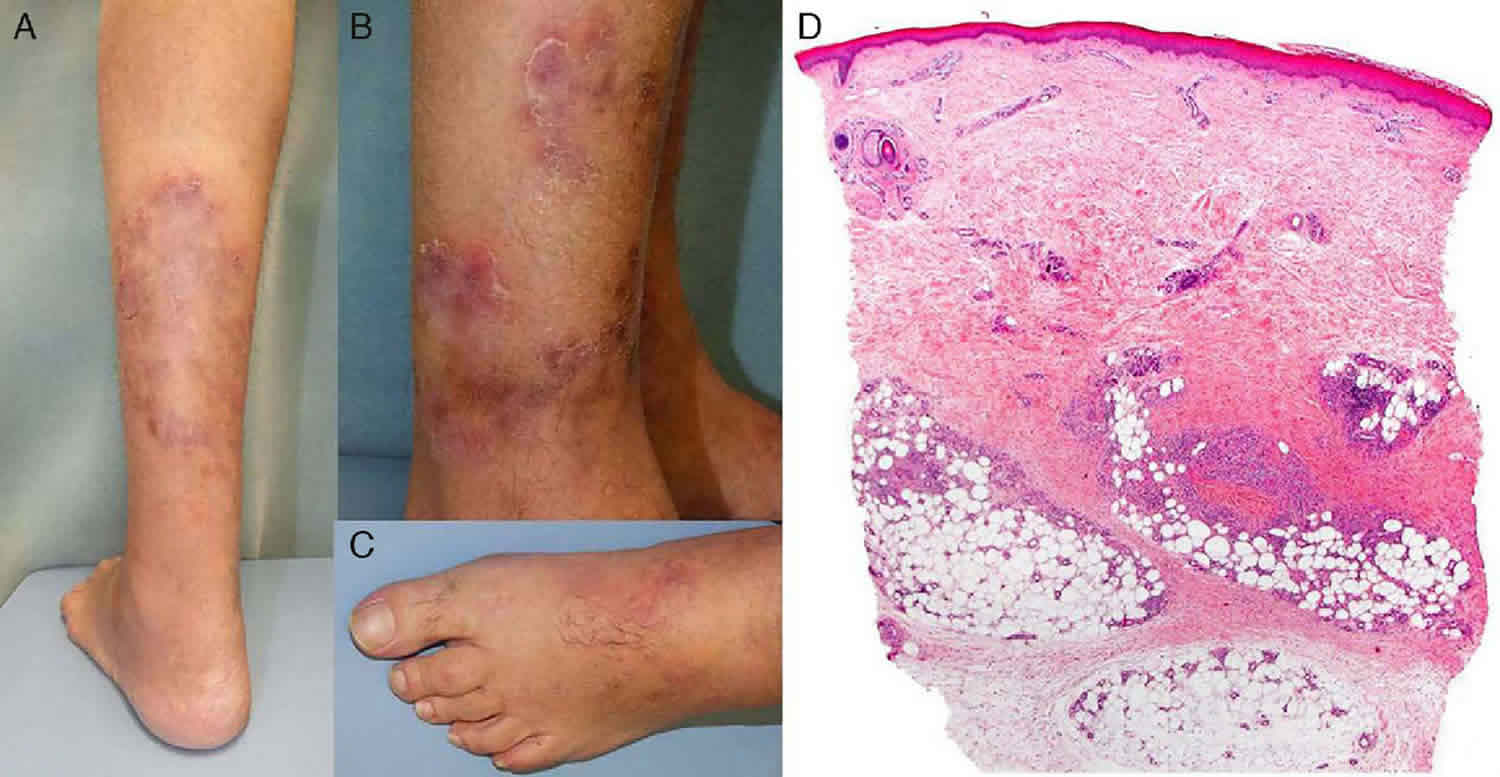
Figure 1. Polyarteritis nodosa rash
How often does polyarteritis nodosa return (relapse) after initial treatment?
One study 4 of 348 individuals with polyarteritis nodosa found that approximately 22% of people experienced a return of symptoms (a relapse) within 5.5 years of their initial diagnosis and treatment. Individuals in whom the polyarteritis nodosa was associated with a hepatitis B virus (HBV) infection were less likely to have a relapse than those in whom the polyarteritis nodosa was not associated with HBV. Another factor that seemed to increase the chance of a relapse included having skin (cutaneous) findings at diagnosis, such as nodules, or purple or bluish patches in the skin (purpura or livedo, respectively).
How might polyarteritis nodosa affect the gastrointestinal tract?
Gastrointestinal (GI) involvement in polyarteritis nodosa usually manifests as nonspecific symptoms such as abdominal pain, nausea, and/or vomiting, with or without obvious GI bleeding (blood in the stools) 3. Abdominal discomfort may be intermittent or continuous and may be most prominent after meals 5. Weight loss may follow due to decreased food intake and/or malabsorption 5.
In some cases, symptoms may depend on the exact location of the affected part of the GI tract. For example, vasculitis affecting the liver or gallbladder may cause right upper quadrant pain 6.
More serious complications of polyarteritis nodosa involving the GI tract are rare but include bowel infarction (obstruction of blood) and perforation, cholecystitis, hepatic infarction, or pancreatic infarction 3.
Does polyarteritis nodosa have anything to do with collagen?
Polyarteritis nodosa has sometimes been referred to as a “collagen disease” or a “collagen vascular disease”. Historically, the term “collagen vascular disease” has been used to describe similar autoimmune disorders associated with inflammation of the connective tissues (including the arteries). Connective tissues are made up of collagen fibers, in addition to other materials. However, now that we have names for these individual disorders, the term is not used as frequently for these disorders. Additionally, the term “collagen disease” has more recently been used to describe diseases caused by defects in collagen (eg, Ehlers-Danlos syndrome), rather than diseases affecting structures made of collagen. To our knowledge, there is not currently evidence that polyarteritis nodosa is due to a defect in collagen. Besides polyarteritis nodosa, other disorders that have been considered collagen vascular diseases include ankylosing spondylitis, dermatomyositis, psoriatic arthritis, rheumatoid arthritis, scleroderma, and systemic lupus erythematosus.
Cutaneous polyarteritis nodosa
Cutaneous polyarteritis nodosa (CPAN) is a rare form of vasculitis (inflammation of blood vessels) that involves small and medium-sized arteries of the dermis and subcutaneous tissue. Although identical skin lesions are common in systemic polyarteritis nodosa (PAN), cutaneous polyarteritis nodosa (CPAN) should be considered a separate disease and distinguished from systemic polyarteritis nodosa, as the clinical course and management of these conditions differ from each other. Though rare, patients with cutaneous polyarteritis nodosa (CPAN) can progress to systemic polyarteritis nodosa (PAN).
Cutaneous polyarteritis nodosa often starts in childhood or adolescence. It is rare.
In most cases of cutaneous polyarteritis nodosa, the disease is triggered by certain infections, particularly Group A streptococcus, hepatitis B, hepatitis C, human immunodeficiency virus, parvovirus B19 (the cause of fifth disease). Genetic defects lead to over-reaction to the infection.
Cutaneous polyarteritis nodosa causes
Cutaneous polyarteritis nodosa results from a complex interaction of autoinflammatory and autoimmune factors, and immunodeficiency.
Autosomal recessive mutations in the CERC1 gene have been implicated in some patients with cutaneous polyarteritis nodosa. This genetic abnormality results in deficiency in the ADA2 protein (DADA2). DADA2 can also cause immunodeficiency. ADA2 is a plasma protein essential for development of endothelial cells (these line the blood vessel wall), and leucocytes (white blood cells). DADA2 leads to uncontrolled, chronic activity of neutrophils and damages endothelial cells.
What are the clinical features of cutaneous polyarteritis nodosa?
Clinical features of cutaneous polyarteritis nodosa relate to inflammation or occlusion of small and medium-sized blood vessels in the skin and sometimes in other organs. It tends to have periods of activity and remission.
Vasculitic lesions are most often found on the legs and feet. Other areas that may be affected include the arms, trunk, buttocks, and head and neck. They are most likely on pressure points such as the knees, back of the foot and lower leg.
- Tender lumps appear under the skin, especially on the thighs and lower legs. These usually measure between 4–15 mm in diameter and follow along the course of medium-sized arteries.
- Larger inflammatory plaques may be seen. These tend to have nodules along the edges. As the plaques heal, they leave patches of postinflammatory pigmentation.
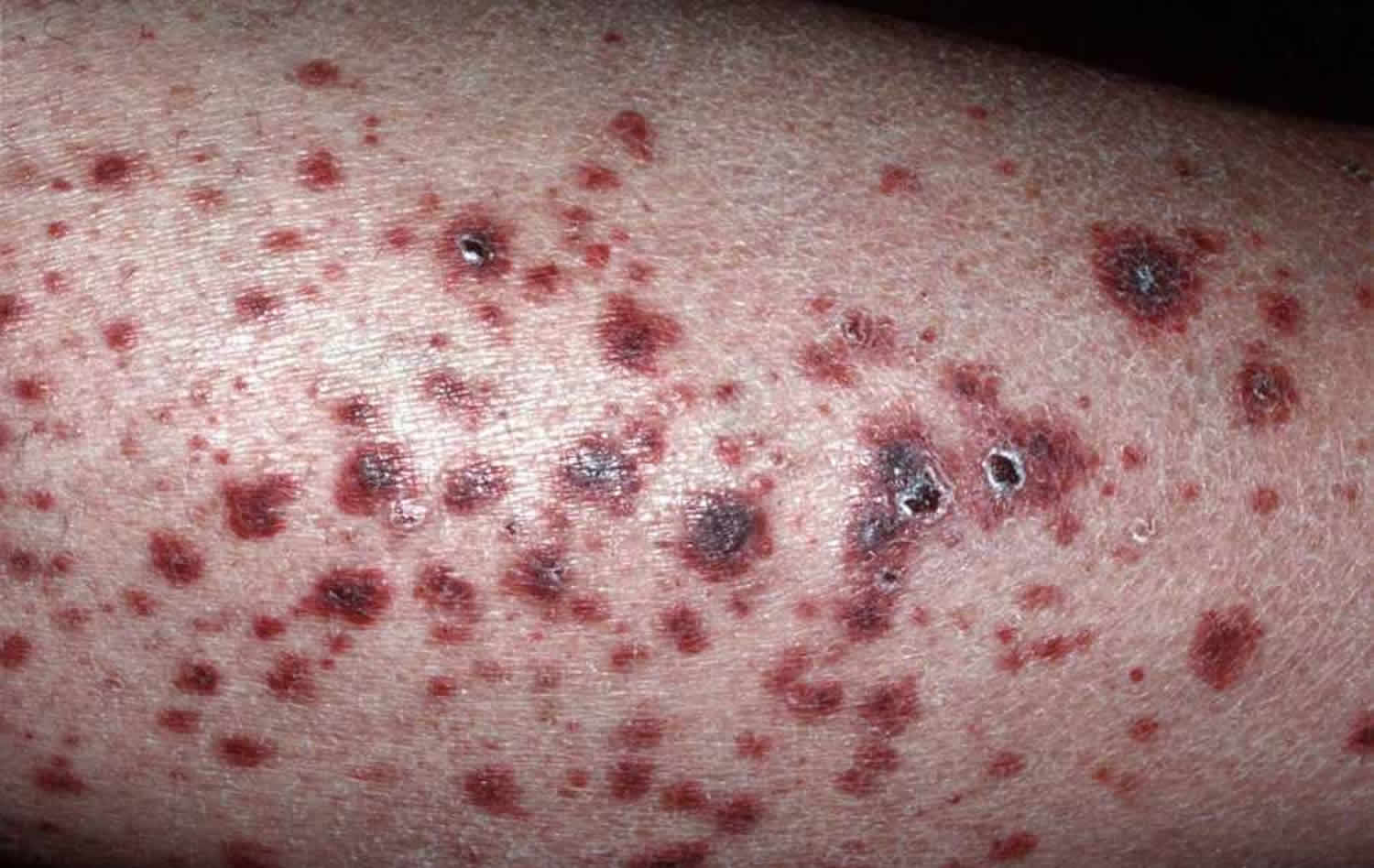
- Infarcts in the skin present as purple or black patches or blood-filled blisters. They are dead areas of skin due to blocked blood vessels.
- Small vessel vasculitis may present as palpable purpura
- Blistering and ulceration may occur.
- Livedo reticularis may appear (a starburst dusky discoloration).
Systemic symptoms in cutaneous polyarteritis nodosa
In addition to the skin problems, patients with cutaneous polyarteritis nodosa may also have generalized symptoms such as malaise, fever, sore throat, and joint and muscle aches and pains. Neurological symptoms may also be present and include numbness, tingling, sensory disturbances, weakness, and absent reflexes.
Cutaneous polyarteritis nodosa diagnosis
Skin biopsy of a typical lesion is often performed to make an accurate diagnosis of cutaneous polyarteritis nodosa. A specimen showing panarteritis (inflammation of all blood vessels in the skin sample) is the only definitive proof of polyarteritis nodosa.
Laboratory tests of blood samples are generally unhelpful in diagnosing or monitoring cutaneous polyarteritis nodosa, as blood counts and chemistry are often normal. They are initially required to determine the cause of vasculitis or to exclude other organ involvement as occurs in systemic polyarteritis nodosa.
Levels of ADA2 protein and/or CERC1 gene can be measured in some centers.
Cutaneous polyarteritis nodosa treatment
As cutaneous polyarteritis nodosa is rare, there are no randomized trials to guide treatment. Standard treatment follows guidelines for other forms of vasculitis. The mainstay of treatment is often with oral corticosteroids (prednisone).
Other active treatments for cutaneous polyarteritis nodosa may include:
- Colchicine
- Cyclophosphamide
- Intravenous immunoglobulin
- Penicillin to prevent flares after streptococcal tonsillitis or cellulitis
Warfarin (with an international normalized ratio [INR] of 3) has been reported to be effective in 3 cases of cutaneous polyarteritis nodosa with improvement in livedo reticularis and healing of ulcers.
New therapies are being developed for patients with cutaneous polyarteritis nodosa that have DADA2. These unproven treatments include:
- Recombinant ADA2 protein
- Fresh frozen plasma
- Haematopoietic cell transplantation
- Anti-TNFα biologics (if TNFα is elevated)
- Anti-interleukin 6 monoclonal antibodies (eg, tocilizumab)
Symptomatic treatment
- Non-steroidal anti-inflammatory drugs are used for pain relief and anti-inflammatory action.
- Pain can be severe, necessitating regular paracetamol, narcotics and anticonvulsant medications.
- Ulcerating skin lesions may be treated with bland topical preparations such as active manuka honey and covered with special dressings to improve healing.
- Occasionally skin grafts may be advised for slow-healing ulcers, but they may fail because of the damage to the blood vessels supplying nutrition to the skin.
Cutaneous polyarteritis nodosa prognosis
Cutaneous polyarteritis nodosa usually runs a chronic course lasting from months to years with exacerbations and remissions. Neurological symptoms and muscular aches and pains usually resolve over a matter of months, whilst skin lesions take longer to heal.
Remissions may occur spontaneously or as a result of treatment.
Polyarteritis nodosa causes
The exact cause of polyarteritis nodosa is not known, and in most cases, no predisposing cause has been found (it is idiopathic). More adults than children get polyarteritis nodosa. Many scientists believe that it is an autoimmune disease. Research has suggested that an abnormal immune response to an initial infection may trigger the development of polyarteritis nodosa. However, the reasons that many smaller arteries and capillaries are spared is not understood 5.
Hepatitis B virus (HBV), hepatitis C, and hairy cell leukemia have been associated with some cases of polyarteritis nodosa. In one report from France, hepatitis B virus accounted for a third of the cases of polyarteritis nodosa. Hepatitis B virus-related polyarteritis nodosa typically occurs within four months after the onset of hepatitis B virus infection 5. Polyarteritis nodosa has also been seen in drug abusers (particularly those using amphetamines). It has also appeared to occur as an allergic reaction to some drugs and vaccines.
Polyarteritis nodosa symptoms
The specific symptoms of polyarteritis nodosa are due to ischemia or infarction of tissues and organs. Thickening of the walls of affected vessels causes narrowing of the inside of the vessels, reducing blood flow and predisposing to blood clots in affected vessels. The skin, joints, muscle, gastrointestinal tract, heart, kidneys, and nervous system are often affected.
Polyarteritis nodosa symptoms include:
- Abdominal pain
- Decreased appetite
- Fatigue
- Fever
- Joint aches
- Muscle aches
- Unintentional weight loss
- Weakness
- High blood pressure
- Blood in the stool
- Testicular pain in men
- Chest pain
- Difficulty breathing
- Numbness or tingling of the hands or feet
- Sudden loss of strength in the hands or feet
If nerves are affected, you may have numbness, pain, burning, and weakness. Damage to the nervous system may cause strokes or seizures.
Polyarteritis nodosa possible complications
Polyarteritis nodosa complications may include:
- Heart attack
- Intestinal necrosis and perforation
- Kidney failure
- Stroke
Polyarteritis nodosa diagnosis
No specific lab tests are available to diagnose polyarteritis nodosa. There are a number of disorders that have features similar to polyarthritis nodosa. These are known as “mimics.”
You will have a complete physical exam.
Lab tests that can help make the diagnosis and rule out mimics include:
- Complete blood count (CBC) with differential, creatinine, tests for hepatitis B and C, and urinalysis
- Erythrocyte sedimentation rate (ESR) or C-reactive protein (CRP)
- Serum protein electrophoresis, cryoglobulins
- Serum complement levels
- Arteriogram
- Tissue biopsy
- Other blood tests will be done to rule out similar conditions, such as systemic lupus erythematosus (ANA) or granulomatosis with polyangiitis (ANCA)
- Test for HIV
- Cryoglobulins
- Anti-phospholipid antibodies
- Blood cultures
The American College of Rheumatology 1990 criteria for the classification of polyarteritis nodosa:
- Weight loss of > 4 kg since beginning of illness
- Livedo reticularis
- Testicular pain or tenderness
- Myalgias, weakness, or leg tenderness
- Mononeuropathy or polyneuropathy
- Development of hypertension
- Elevated BUN or creatinine unrelated to dehydration or obstruction
- Presence of hepatitis B surface antigen or antibody in serum
- Arteriogram demonstrating aneurysms or occlusions of the visceral arteries
- Biopsy of small or medium-sized artery containing granulocytes
Polyarteritis nodosa treatment
There is no cure for polyarteritis nodosa, but the disease and its symptoms can be managed. Treatment will vary based on patient symptoms, disease activity, organ involvement and lab test results. The goal of treatment is to prevent disease progression and further organ damage 7. The exact treatment depends on the severity in each person 6. While many people do well with treatment, relapses can occur 7.
When the cause of polyarteritis nodosa is unknown (idiopathic), treatment involves corticosteroids and immunosuppressive medications7. If there are no serious neurologic, renal, gastrointestinal, or heart symptoms, corticosteroids may initially be sufficient. For severe disease with these symptoms, cyclophosphamide may also be used. Hypertension should be treated aggressively 6.
When polyarteritis nodosa is related to hepatitis B, treatment often involves steroids, anti-viral medications and sometimes plasma exchange (also called plasmapheresis) 7.
Treatment of polyarteritis nodosa has improved dramatically in the past couple of decades. Before the availability of effective therapy, untreated polyarteritis nodosa was usually fatal within weeks to months. Most deaths occurred as a result of kidney failure, heart or gastrointestinal complications. However, effective treatment is now available for polyarteritis nodosa. After diagnosis, patients are treated with high doses of corticosteroids. Other immunosuppressive drugs are also added for patients who are especially ill. In most cases of polyarteritis nodosa now, if diagnosed early enough the disease can be controlled, and often cured.
The newly proposed regimen for patients with polyarteritis nodosa associated with hepatitis B, consists of 2 weeks of prednisone to control the vasculitis, followed by plasmapheresis to remove immune complexes, and accompanied by antiviral therapy with lamivudine to rid the patient of the hepatitis B infection. The long–term value of anti–viral therapy for polyarteritis nodosa associated with hepatitis C is not established.
Polyarteritis nodosa life expectancy
Without treatment, 5-year survival is < 15%. With treatment, 5-year survival is > 80% but may be lower for patients with hepatitis B. Prognosis is better if disease remission is achieved within 18 months after diagnosis. Relapses are less common than in other vasculitic disorders.
The following findings are associated with a poor prognosis:
- Renal insufficiency
- Gastrointestinal involvement
- Neurologic involvement
In one study 8 examined the overall mortality of a group of individuals with polyarteritis nodosa. Mortality is a measure of the proportion of individuals in a group who die in a given time period. Of 348 individual with polyarteritis nodosa, approximately 20% had died within 5 years of initial diagnosis and treatment; approximately 32% had died within 10 years 8. Only a third of these deaths was directly caused by severe symptoms of polyarteritis nodosa. Factors which increased the risk of death included being older than 65 years, being recently diagnosed with high blood pressure (hypertension), or having gastrointestinal symptoms that required surgery at the time of diagnosis (for example, abdominal pain, internal bleeding, pancreatitis, cholecystitis, appendicitis).
- Polyarteritis nodosa. https://medlineplus.gov/ency/article/001438.htm[↩]
- De Virgilio A, Greco A, Magliulo G, Gallo A, Ruoppolo G, Conte M, Martellucci S, de Vincentiis M. Polyarteritis nodosa: A contemporary overview. Autoimmun Rev. 2016 Jun;15(6):564-70.[↩]
- Polyarteritis nodosa. https://emedicine.medscape.com/article/330717-overview[↩][↩][↩][↩]
- Pagnoux C, Seror R, Henegar C, Mahr A, Cohen P, Le Guern V, Bienvenu B, Mouthon L, Guillevin L; French Vasculitis Study Group. Clinical features and outcomes in 348 patients with polyarteritis nodosa: a systematic retrospective study of patients diagnosed between 1963 and 2005 and entered into the French Vasculitis Study Group Database. Arthritis and Rheumatism. 2010; 62:616-626. http://www.ncbi.nlm.nih.gov/pubmed/20112401[↩]
- Peter A Merkel. Clinical manifestations and diagnosis of polyarteritis nodosa in adults. UpToDate. Waltham, MA: UpToDate; October, 2016[↩][↩][↩][↩]
- Polyarteritis Nodosa (PAN). https://www.merckmanuals.com/professional/musculoskeletal-and-connective-tissue-disorders/vasculitis/polyarteritis-nodosa-pan?qt=Polyarteritis%20nodosa&alt=sh[↩][↩][↩]
- Polyarteritis nodosa. https://www.vasculitisfoundation.org/education/forms/polyarteritis-nodosa/[↩][↩][↩][↩]
- Pagnoux C, Seror R, Henegar C, Mahr A, Cohen P, Le Guern V, Bienvenu B, Mouthon L, Guillevin L; French Vasculitis Study Group. Clinical features and outcomes in 348 patients with polyarteritis nodosa: a systematic retrospective study of patients diagnosed between 1963 and 2005 and entered into the French Vasculitis Study Group Database. Arthritis and Rheumatism. 2010; 62:616-626.[↩][↩]


{kind=link}