Contents
Rotor syndrome
Rotor syndrome also called Rotor type hyperbilirubinemia, is a rare, benign, autosomal recessive inherited genetic disorder characterized by hyperbilirubinemia due to elevated levels of both unconjugated bilirubin (also known as indirect bilirubin or toxic bilirubin) and conjugated bilirubin (also known as direct bilirubin or nontoxic bilirubin) with the majority of the bilirubin being the conjugated bilirubin (nontoxic bilirubin) in the blood 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12. Rotor syndrome is caused by mutation in the SLCO1B1 and SLCO1B3 genes, which are located near each other on chromosome 12p 13. Mutations in both SLCO1B1 and SLCO1B3 genes are required for Rotor syndrome to occur. The SLCO1B1 and SLCO1B3 genes provide instructions for making similar proteins, called organic anion transporting polypeptide 1B1 (OATP1B1) and organic anion transporting polypeptide 1B3 (OATP1B3), respectively 14, 15. Both OATP1B1 and OATP1B3 proteins are found in your liver cells where they transport bilirubin and other compounds from your blood into your liver so that they can be cleared from your body. In your liver, bilirubin is dissolved in a digestive fluid called bile and then excreted from your body. Recently, a study looked at the insertion of long interspersed nuclear element-1 (LINE-1) in genes as potential of causing Rotor syndrome 16. Rotor syndrome is inherited in an autosomal recessive pattern. In one study, Rotor syndrome was found to be coexistent with other inherited disorders, such as glucose-6-phosphate dehydrogenase deficiency (G6PD deficiency) and heterozygous beta-thalassemia, which suggests a potential interaction between co-inherited genes 17.
In people with Rotor syndrome, jaundice (a yellow discoloration of the body tissue resulting from the accumulation of excess bilirubin) is usually evident shortly after birth or in childhood and may come and go; yellowing of the whites of your eyes also called conjunctival icterus is often the only symptom. Rotor syndrome is a benign and harmless condition that typically does not require treatment and does not affect life expectancy. Jaundice may be noticeable shortly after birth or in childhood, but often it’s an incidental finding.
Rotor syndrome was first described in 1948 by Rotor et al. in the Philippines 18. Since then, cases have been reported worldwide, including in the US, Japan, France, Mexico, Papua New Guinea, and Italy. The exact prevalence of Rotor syndrome is not known but is very low (<1:1,000,000) 3. As most patients are asymptomatic, Rotor syndrome may be picked up based on incidental findings on investigations done for other reasons. Rotor syndrome is the second rare hereditary cause of hyperbilirubinemia, the first being Crigler-Najjar syndrome 19. Furthermore, there is no gender predisposition seen with Rotor syndrome. While Rotor syndrome often presents in adolescence or early adulthood, it has been reported to present shortly after birth or during childhood 20, 10.
Rotor syndrome should be suspected in individuals with the following signs and symptoms, family history, laboratory, and hepatobiliary iminodiacetic acid (HIDA) scan findings. Rotor syndrome diagnosis can be established by genetic testing identifying the SLCO1B1 and SLCO1B3 genes mutations. A liver biopsy is not required to diagnose Rotor syndrome, but if done, a liver biopsy in patients with Rotor syndrome reveals normal histology. Liver biopsy may help distinguish Rotor syndrome from other, more serious liver diseases. Since Rotor syndrome is clinically similar to Dubin-Johnson syndrome, it is imperative to distinguish between these two conditions; the absence of dark melanin-like pigments on liver biopsy distinguishes Rotor Syndrome from Dubin-Johnson syndrome 3, 10, 21. Immunohistologic staining does not detect hepatic proteins SLCO1B1 and SLCO1B3 at the sinusoidal membrane of hepatocytes 3. Expression of MRP2, frequently absent in Dubin-Johnson syndrome, is normal 22 and dark melanin-like pigment in liver cells typical of Dubin-Johnson syndrome is not present 3.
Rotor syndrome clinical features:
- Mild jaundice (may be intermittent)
- Conjunctival icterus (in some affected individuals)
- Otherwise normal physical examination
Laboratory findings
- Conjugated hyperbilirubinemia with serum total bilirubin concentration usually between 2 and 5 mg/dL but possibly higher. Conjugated bilirubin usually exceeds 50% of total bilirubin.
- Presence of bilirubin in the urine
- Absence of hemolysis*
- Normal serum alanine transaminase (ALT), aspartate transaminase (AST), alkaline phosphatase (ALP), and gamma-glutamyl transferase (GGT) activity*
- Note: * Tests for hemolysis and measurements of alanine transaminase (ALT), aspartate transaminase (AST), alkaline phosphatase (ALP), and gamma-glutamyl transferase (GGT) activity are needed to evaluate for hemolytic anemia and hepatobiliary diseases that are considered in the differential diagnosis of Rotor syndrome.
- Total urinary porphyrins: elevated coproporphyrin. Coproporphyrin is a metabolite synthesized in the heme biosynthetic pathway. Coproporphyrin is produced from uroporphyrinogen by uroporphyrinogen decarboxylase activity 23. Increased levels of coproporphyrins can indicate various conditions, most commonly liver disease, alcohol abuse, certain medications, sideroblastic anemia and exposure to heavy metals like lead. Increased levels of coproporphyrins can also be a sign of rare inherited genetic disorders called porphyrias:
- The best method of diagnosing Rotor syndrome is the analysis of urine coproporphyrin excretion. Total coproporphyrin excretion in the urine is markedly increased in Rotor syndrome. Dubin-Johnson syndrome patients excreted 88.9% as coproporphyrin 1, whereas this value was 64.8% in Rotor syndrome homozygotes and 42.9% in Rotor syndrome heterozygotes. The standard errors of these values were such that the differences were highly significant 24.
Serological abnormalities in Rotor syndrome only include elevated total serum bilirubin (typically elevated between 2 to 5 mg/dL but may be as high as 20 mg/dL). Most of the time, alanine aminotransferase (ALT), aspartate aminotransferase (AST), gamma-glutamyl transferase (GGT), and alkaline phosphatase (ALP) levels are normal, but mild elevations can be seen. If any of these lab values are markedly elevated, investigation for other, more serious conditions is warranted.
Imaging test findings
A hepatobiliary iminodiacetic acid (HIDA) scan also known as cholescintigraphy or hepatobiliary scintigraphy, is an imaging procedure to assess the anatomy and function of the biliary system and the liver indirectly. A hepatobiliary iminodiacetic acid (HIDA) scan uses a radioactive tracer (Tc-99m-IDA) that is injected into a vein in your arm 25, 26. The radioactive tracer (Tc-99m-IDA) travels through your bloodstream to your liver, where the bile-producing cells take it up. The tracer then travels with the bile into your gallbladder and through the bile ducts to your small intestine. A nuclear medicine scanner, called a gamma camera, tracks the flow of the tracer from the liver into the gallbladder and small intestine and creates computer images.
Hepatobiliary iminodiacetic acid (HIDA) scan or hepatobiliary scintigraphy is an inexpensive and straightforward method and represents the imaging test of choice for Rotor syndrome, allowing accurate differential diagnosis 27. Even without considering clinic–laboratory findings, this too-faint hepatic visualization, although suggestive of hepatocellular disease, is unlikely to be consistent with cirrhosis or hepatitis 9. The abnormal, instead of normal, hepatic perfusion phase (increased or anticipated) due to liver arterialization is more likely in cirrhosis or hepatitis 9. Dubin-Johnson syndrome, a similar syndrome of conjugated hyperbilirubinemia, presents, in contrast, hepatobiliary scintigraphy findings of intrahepatic cholestasis (no biliary phase, associated with prolonged, rapid, and intensive, instead of slow and negligible, liver radiotracer uptake) 27, 28.
It is important to differentiate Rotor syndrome from other diseases causing hyperbilirubinemia. Normal alkaline phosphatase (ALP) levels and gamma-glutamyl transpeptidase (GGT) help distinguish Rotor syndrome from disorders associated with biliary obstruction. Abnormal urinary coproporphyrin excretion and normal liver histology help distinguish this entity from Dubin-Johnson syndrome 21, 3.
Rotor syndrome can be distinguished from Dubin-Johnson syndrome by a lack of pigment deposits in liver cells biopsy, delayed plasma clearance of the unconjugated anionic dye bromosulphthalein (BSP), poor liver visualization on certain radiographic imaging studies, and prominent urinary excretion of coproporphyrin 1 27.
No adverse drug effects have been documented in people with Rotor syndrome; however, the absence of the liver proteins OATP1B1 and OATP1B3 may have serious consequences for liver uptake and toxicity of numerous commonly used drugs and/or their metabolites, which enter the liver via either of the two OATP1B transporters. OATP1B1 plays a role in drug detoxification, and with reduced activity of this protein, certain drugs such as anticancer agents, methotrexate, and statins can accumulate and result in drug toxicity 29. Caution should be taken before administering these drugs 20, 30, 31, 32:
- Statins – simvastatin, atorvastatin, pravastatin, pitavastatin, rosuvastatin
- Ezetimibe
- Anticancer drugs – methotrexate and irinotecan, cabazitaxel, some tyrosine kinase inhibitors (e.g., sunitinib)
- Sartans – olmesartan and valsartan
- Rifampicin
- Mycophenolic acid
- Torsemide
- Thiazolidine diones – pioglitazone and rosiglitazone
- Glinides – nateglinide and repaglinide
- Lopinavir
- Fexofenadine
- Cyclosporin A.
Figure 1. Rotor syndrome hepatobiliary scintigraphy
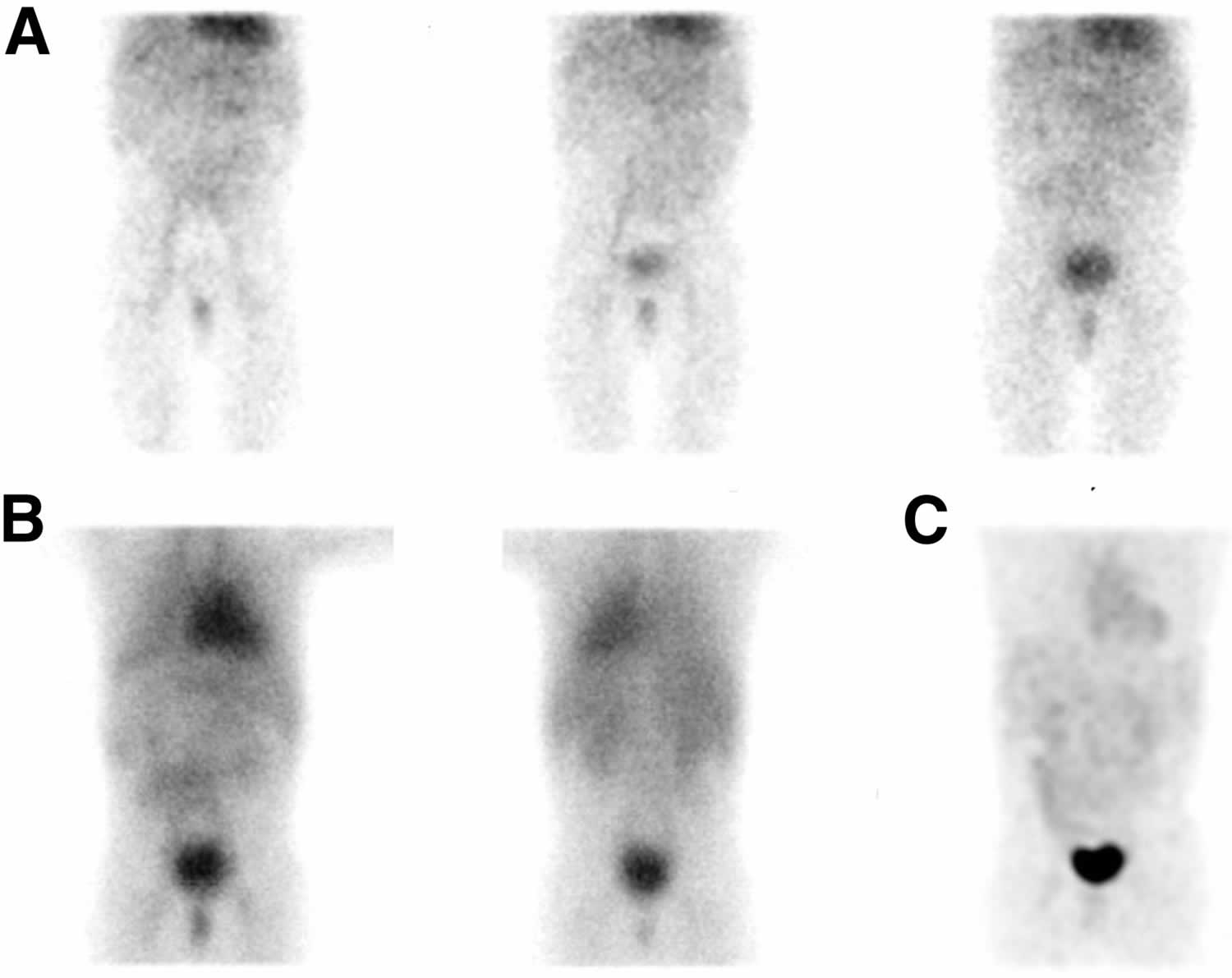
Footnotes: A 3 year old boy presented with persistent jaundice since birth. The physical examination was unremarkable, except for mild icterus of the sclerae. Serum total biliburin was elevated (2.59 mg/dL), with a conjugated biliburin of 2.41 mg/dL. The results of liver function tests, abdominal ultrasound, serology for viral and autoimmune hepatitis, and tests for genetic disorders, such as cystic fibrosis and Wilson disease, were normal. Additional investigation revealed elevated coproporphyrin I (97 nmol/L; reference range, <40 nmol/L) and III (109 nmol/L; reference range, <75 nmol/L). Hepatobiliary iminodiacetic acid (HIDA) scan or hepatobiliary scintigraphy showing abnormal 99mTc-mebrofenin findings consistent with Rotor syndrome. (A) No succession of the normal scintigraphic phases (hepatic, biliary, and intestinal) was noticed, as 99mTc-mebrofenin liver uptake was too slow. Left to right: 10th-, 30th-, and 50th-minute frames of dynamic acquisition. (B) Delayed static images at 1.5 hour (anterior [left]/posterior [right]). (C) Delayed maximum-intensity-projection 3-dimensional anterior reconstructed image at 3 hour. (B and C) The biliary duct system and gallbladder were not seen, whereas a faint visualization of the liver and small bowel was more evident on the delayed static images. In contrast, an extremely prolonged blood-pool phase (visualization of the heart and spleen) and prominent renal excretion, mimicking liver failure, were noticed. A molecular genetic analysis with next-generation sequencing was performed, but no sequence of the SLCO1B1 and SLCO1B3 genes involved in Rotor syndrome was detected. During a follow-up of 2 years, the boy had a favorable clinical course without any complications.
[Source 9 ]What is bilirubin?
Bilirubin is an orange-yellow pigment, a waste product primarily produced by the normal breakdown of heme (ferroprotoporphyrin IX) 33, 34, 35, 36, 37. Approximately 85 percent of the heme is from the hemoglobin (Hb; an iron-containing protein in red blood cells that transports oxygen from the lungs to the body’s tissues) following the degradation of red blood cells (RBCs), while the remaining heme derives from the ineffective erythropoiesis and the breakdown of other hemoproteins such as cytochromes, myoglobin, and catalase 38. Bilirubin is ultimately processed by the liver to allow its elimination from the body.
There are 2 forms of bilirubin in the body: a toxic form called unconjugated bilirubin and a nontoxic form called conjugated bilirubin. Both forms can be measured or estimated by laboratory tests, and a total bilirubin result (a sum of these) may also be reported. Bilirubin is ultimately processed by the liver to allow its elimination from the body.
Red blood cells normally degrade after about 120 days in the blood circulation. As heme is released from hemoglobin, it is converted to bilirubin. The conversion of heme to bilirubin is a 2-step reaction, in the first step the microsomal heme oxygenase enzyme of the reticuloendothelial system (a network of phagocytic cells, including monocytes and macrophages, that is part of the immune system whose function is to engulf and destroy foreign particles, such as bacteria and toxins, and cellular debris, a process known as phagocytosis), converts heme to biliverdin, which in turn is reduced to unconjugated bilirubin (toxic form of bilirubin) by a second enzyme biliverdin reductase 39. The unconjugated bilirubin (toxic form of bilirubin) is lipophilic (dissolves readily in fats). Unconjugated bilirubin (toxic form of bilirubin) tightly bound to albumin is transported to the liver. The entry of unconjugated bilirubin into the liver has not been elucidated, and the best candidate appears to be a bilirubin transporter 34. In liver cells, unconjugated bilirubin dissociates from albumin and binds to proteins of the glutathione-S-transferases family that present it for conjugation and prevent it from effluxing from the liver 34. Next, unconjugated bilirubin gets conjugated (attached) with one or two molecules of glucuronic acid by the enzyme uridine diphospho-glucuronate glucuronosyltransferase (UGT1A1), which forms bilirubin monoglucuronide and bilirubin diglucuronide respectively 39, 40. Conjugation increases the solubility of bilirubin in plasma and thereby enhances its elimination from the body. The conjugated bilirubin (nontoxic bilirubin) is then pumped into bile via an energy-requiring process requiring the multidrug resistance-associated protein 2 (MRP2) 41. This process also reduces the ability of bilirubin to diffuse across the blood-brain barrier. Therefore, unconjugated hyperbilirubinemia can result from dysfunction of any of these conjugation steps. Conjugated bilirubin enters the bile and passes from the liver to the small intestines; there, it is further broken down by bacteria and eventually eliminated in the stool. Therefore, the breakdown products of bilirubin give stool its characteristic brown color. In newborns, inefficient conjugation of bilirubin leads to unconjugated hyperbilirubinemia (physiologic neonatal jaundice).
A small amount (approximately 250 to 350 milligrams) of bilirubin is produced daily in a normal, healthy adult. Most (85%) of bilirubin is derived from damaged or degraded red blood cells, with the remaining amount derived from the bone marrow or liver. Normally, small amounts of unconjugated bilirubin are released into the blood, but virtually no conjugated bilirubin is present.
Bilirubin concentrations tend to be slightly higher in males than females. African Americans routinely show lower bilirubin concentrations than non-African Americans. Strenuous exercise may increase bilirubin levels.
Drugs that can decrease total bilirubin include barbiturates, caffeine, penicillin, and high doses of salicylates. The drug atazanavir increases unconjugated (indirect) bilirubin.
If the bilirubin level increases in the blood, a person may appear jaundiced, with a yellowing of the skin and/or whites of the eyes. The pattern of bilirubin test results can give your doctor information regarding the condition that may be present. For example, unconjugated bilirubin may be increased when there is an unusual amount of red blood cell destruction (hemolysis) or when the liver is unable to process bilirubin (i.e., with liver diseases such as cirrhosis or inherited problems). Conversely, conjugated bilirubin can increase when the liver is able to process bilirubin but is not able to pass the conjugated bilirubin to the bile for removal; when this happens, the cause is often acute hepatitis or blockage of the bile ducts.
Rotor syndrome cause
Rotor syndrome is a rare, autosomal recessive inherited genetic disorder caused by both the SLCO1B1 (solute carrier organic anion transporter family member 1B1) and SLCO1B3 (solute carrier organic anion transporter family member 1B3) genes, which are located near each other on chromosome 12p 13. In Rotor syndrome, the homozygous inactivation of both genes results in nonfunctional or absent proteins and increased bilirubinemia and porphyrinuria 42, 27. Recently, a study looked at the insertion of long interspersed nuclear element-1 (LINE-1) in genes as potential of causing Rotor syndrome 16. Researchers in Korea reported cases with an intronic LINE-1 insertion in SLCO1B3 gene, supporting it was a prevalent pathogenic haplotype in the East Asian population 7.
Rotor syndrome is a special form of autosomal recessive genetic disorder involving two linked SLCO1B1 and SLCO1B3 genes on chromosome 12. Therefore, 4 sites of genetic mutations (bi-allelic pathological variants in each gene) are required for a confirmative genetic diagnosis. Up to now, four haplotypes have been reported: R1 haplotype with SLCO1B1:c.1738C>T linked with a 7.2 kb deletion removes exon 13 of SLCO1B3, R2 haplotype with 405 kb deletion encompasses exons 4–16 of SLCO1B3 and the whole of SLCO1B1, R3 haplotype with SLCO1B1:c.757C>T linked with SLCO1B3:c.1747+1G>A, and JP haplotype with SLCO1B1:c.1738C>T linked with a LINE-1 insertion in intron 6 of SLCO1B3 27, 42.
SLCO1B1 gene provides instructions for making a protein called organic anion transporting polypeptide 1B1 or OATP1B1 14. The OATP1B1 (organic anion transporting polypeptide 1B1) protein is found in your liver cells where it transports compounds from your blood into your liver so that they can be cleared from your body. For example, the OATP1B1 protein transports bilirubin, which is a yellowish substance that is produced when red blood cells are broken down. In your liver, bilirubin is dissolved in a digestive fluid called bile and then excreted from the body. The OATP1B1 protein also transports certain hormones, toxins, and drugs into the liver for removal from your body 14. Drugs transported by the OATP1B1 protein include statins, which are used to treat high cholesterol; heart disease medications; certain antibiotics; and some drugs used for the treatment of cancer.
SLCO1B3 gene provides instructions for making a protein called organic anion transporting polypeptide 1B3 or OATP1B3 15. The OATP1B3 (organic anion transporting polypeptide 1B3) protein is found in your liver cells where it transports compounds from your blood into your liver so that they can be cleared from your body. For example, the OATP1B3 protein transports bilirubin, which is a yellowish substance that is produced when red blood cells are broken down. In your liver, bilirubin is dissolved in a digestive fluid called bile and then excreted from the body. The OATP1B3 protein also transports certain hormones, toxins, and drugs into the liver for removal from your body 15. Some of the drugs transported by the OATP1B3 protein include statins, which are used to treat high cholesterol; heart disease medications; certain antibiotics; and some drugs used for the treatment of cancer.
In a normal liver, a majority of bilirubin is conjugated by liver cells and secreted back into the blood. It is then reabsorbed in downstream liver cells by the OATP1B1 and OATP1B3 proteins 43. In Rotor syndrome, the OATP1B1 and OATP1B3 proteins are abnormally short; therefore, the bilirubin is less efficiently taken up by the liver and removed from the body, causing a buildup of bilirubin in the blood and urine, which results in jaundice and dark urine 44, 45, 46, 5. The jaundice (icterus) is caused by conjugated hyperbilirubinemia, excess coproporphyrin in urine, and a deficiency in the intra-cellular capacity of the liver to store organic anions, such as bilirubin diglucuronide 47, 48.
A study of eight families of Rotor syndrome found that it was associated with mutations that are predicted to cause deficiencies of the organic anion transporters OATP1B1 and OATP1B3. These crucial detoxification-limiting proteins carry out the uptake and clearance of a large number of drugs and drug conjugates through the sinusoidal hepatocyte membrane. The observation suggested the individuals with Rotor syndrome maybe a risk of life-threatening toxicities with the use of certain medications 27.
Rotor syndrome inheritance pattern
Rotor syndrome is inherited in an autosomal recessive pattern. In autosomal recessive inheritance, both copies of a gene in each cell have mutations. In Rotor syndrome, an affected individual must have mutations in both the SLCO1B1 and the SLCO1B3 gene, so both copies of the two genes are altered. The parents of an individual with Rotor syndrome each carry one altered copy of both genes, but they do not show signs and symptoms of the condition.
It is rare to see any history of autosomal recessive conditions within a family because if someone is a carrier for one of these conditions, they would have to have a child with someone who is also a carrier for the same condition. Autosomal recessive conditions are individually pretty rare, so the chance that you and your partner are carriers for the same recessive genetic condition are likely low. Even if both partners are a carrier for the same condition, there is only a 25% chance that they will both pass down the non-working copy of the gene to the baby, thus causing a genetic condition. This chance is the same with each pregnancy, no matter how many children they have with or without the condition.
- If both partners are carriers of the same abnormal gene, they may pass on either their normal gene or their abnormal gene to their child. This occurs randomly.
- Each child of parents who both carry the same abnormal gene therefore has a 25% (1 in 4) chance of inheriting a abnormal gene from both parents and being affected by the condition.
- This also means that there is a 75% ( 3 in 4) chance that a child will not be affected by the condition. This chance remains the same in every pregnancy and is the same for boys or girls.
- There is also a 50% (2 in 4) chance that the child will inherit just one copy of the abnormal gene from a parent. If this happens, then they will be healthy carriers like their parents.
- Lastly, there is a 25% (1 in 4) chance that the child will inherit both normal copies of the gene. In this case the child will not have the condition, and will not be a carrier.
These possible outcomes occur randomly. The chance remains the same in every pregnancy and is the same for boys and girls.
A high carrier frequency of an insertion of a ~6.1-kb L1 retrotransposon in intron 5 of SLCO1B3 resulting in aberrant splicing was discovered in East Asian populations (10.1%), especially in Southern Han Chinese (18.5%), but this pathogenic variant was almost absent in other studied populations 7, 42.
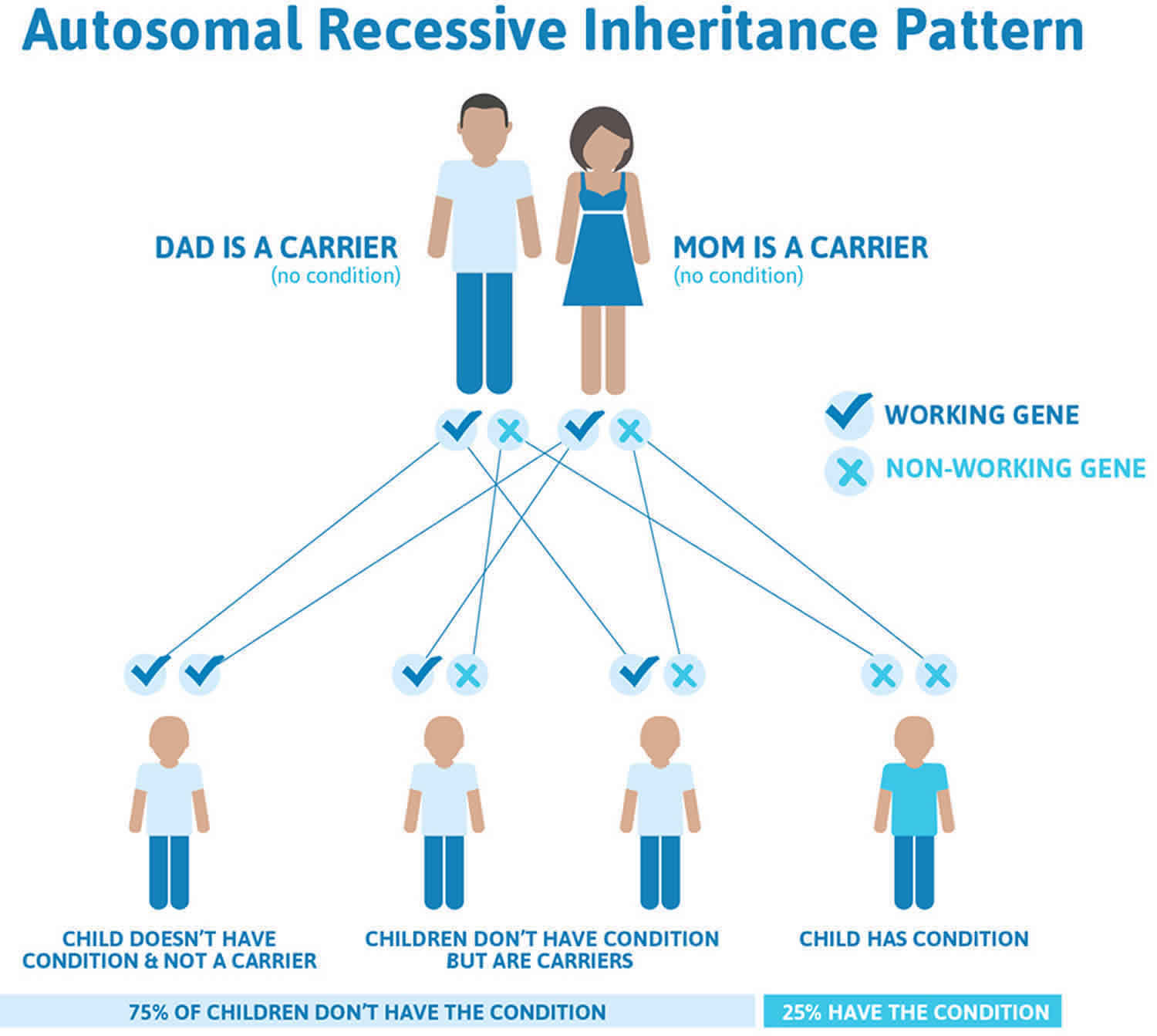
Figure 2 illustrates autosomal recessive inheritance. The example below shows what happens when both dad and mum is a carrier of the abnormal gene, there is only a 25% chance that they will both pass down the abnormal gene to the baby, thus causing a genetic condition.
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://abgc.learningbuilder.com/Search/Public/MemberRole/Verification) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (https://clinics.acmg.net/) has a searchable database of medical genetics clinic services in the United States.
Figure 2. Rotor syndrome autosomal recessive inheritance pattern
Rotor syndrome signs and symptoms
Individuals with Rotor syndrome are typically asymptomatic; however, generalized non-pruritic mild jaundice due to conjugated and unconjugated hyperbilirubinemia can present at birth or early childhood. Jaundice may be intermittent. Conjunctival icterus may be the only clinical feature.
Individuals with Rotor syndrome may also complain about passing dark-colored urine, long-term jaundice, and fatigue. Additionally, around 5% to 30% of individuals with Rotor syndrome may also experience abdominal pain, gastric mucosal abnormalities, and fever 3, 20.
Rotor syndrome diagnosis
Individuals with Rotor syndrome are typically asymptomatic. Jaundice may be noticeable shortly after birth or in childhood, but often it’s an incidental finding.
Rotor syndrome is largely a diagnosis of exclusion. Physical examination is typically normal, except for mild jaundice. Excess bilirubin results in a yellow-orange tinge of the tissues that most easily appears as icteric (yellowish) discoloration of the sclera. Unlike other disorders with a severely reduced ability to produce and release a digestive fluid called bile (cholestasis), itch (pruritus) is absent. Another feature to remember in Rotor syndrome is that there will be no hepatosplenomegaly in the presence of jaundice which can help narrow down the list of differentials.
Rotor syndrome clinical features:
- Mild jaundice (may be intermittent)
- Conjunctival icterus (in some affected individuals)
- Otherwise normal physical examination.
Laboratory findings
- Conjugated hyperbilirubinemia with serum total bilirubin concentration usually between 2 and 5 mg/dL but possibly higher. Conjugated bilirubin usually exceeds 50% of total bilirubin.
- Presence of bilirubin in the urine
- Absence of hemolysis*
- Normal serum alanine transaminase (ALT), aspartate transaminase (AST), alkaline phosphatase (ALP), and gamma-glutamyl transferase (GGT) activity*
- Note: * Tests for hemolysis and measurements of alanine transaminase (ALT), aspartate transaminase (AST), alkaline phosphatase (ALP), and gamma-glutamyl transferase (GGT) activity are needed to evaluate for hemolytic anemia and hepatobiliary diseases that are considered in the differential diagnosis of Rotor syndrome.
- Total urinary porphyrins: elevated coproporphyrin. Coproporphyrin is a metabolite synthesized in the heme biosynthetic pathway. Coproporphyrin is produced from uroporphyrinogen by uroporphyrinogen decarboxylase activity 23. Increased levels of coproporphyrins can indicate various conditions, most commonly liver disease, alcohol abuse, certain medications, sideroblastic anemia and exposure to heavy metals like lead. Increased levels of coproporphyrins can also be a sign of rare inherited genetic disorders called porphyrias:
- The best method of diagnosing Rotor syndrome is the analysis of urine coproporphyrin excretion. Total coproporphyrin excretion in the urine is markedly increased in Rotor syndrome. Dubin-Johnson syndrome patients excreted 88.9% as coproporphyrin 1, whereas this value was 64.8% in Rotor syndrome homozygotes and 42.9% in Rotor syndrome heterozygotes. The standard errors of these values were such that the differences were highly significant 24.
Serological abnormalities in Rotor syndrome only include elevated total serum bilirubin (typically elevated between 2 to 5 mg/dL but may be as high as 20 mg/dL). Most of the time, alanine aminotransferase (ALT), aspartate aminotransferase (AST), gamma-glutamyl transferase (GGT), and alkaline phosphatase (ALP) levels are normal, but mild elevations can be seen. If any of these lab values are markedly elevated, investigation for other, more serious conditions is warranted.
The total urine coproporphyrin excretion in Rotor syndrome has a two to five-fold elevation, with 65% constituting coproporphyrin 1 20, 3.
- Total coproporphyrin excretion is greatly elevated in both Rotor syndrome and Dubin-Johnson syndrome
- The ratio of coproporphyrin 1 to 3 in urine allows the differentiation of these two conditions.
- Around 90% is coproporphyrin 1 in Dubin-Johnson syndrome but a much lower proportion in Rotor syndrome.
- The plasma disappearance of injected bromosulphthalein (BSP) is delayed, with no secondary rise (which is seen in Dubin-Johnson syndrome).
- The hepatic biopsy will show pigment deposition in Dubin-Johnson syndrome but not Rotor syndrome.
Imaging test findings
Imaging studies cannot diagnose Rotor syndrome but can help rule out other diseases that cause hyperbilirubinemia. For example, an ultrasound of the liver and biliary tree can help investigate the causes of extra-hepatic biliary obstruction. The gallbladder is visualized on oral cholecystography or hepatobiliary iminodiacetic acid (HIDA) scan in Rotor syndrome, while it is not visualized in Dubin-Johnson syndrome 28.
A hepatobiliary iminodiacetic acid (HIDA) scan also known as cholescintigraphy or hepatobiliary scintigraphy, is an imaging procedure to assess the anatomy and function of the biliary system and the liver indirectly. A hepatobiliary iminodiacetic acid (HIDA) scan uses a radioactive tracer (Tc-99m-IDA) that is injected into a vein in your arm 25, 26. The radioactive tracer (Tc-99m-IDA) travels through your bloodstream to your liver, where the bile-producing cells take it up. The tracer then travels with the bile into your gallbladder and through the bile ducts to your small intestine. A nuclear medicine scanner, called a gamma camera, tracks the flow of the tracer from the liver into the gallbladder and small intestine and creates computer images.
Hepatobiliary iminodiacetic acid (HIDA) scan or hepatobiliary scintigraphy is an inexpensive and straightforward method and represents the imaging test of choice for Rotor syndrome, allowing accurate differential diagnosis 27. Even without considering clinic–laboratory findings, this too-faint hepatic visualization, although suggestive of hepatocellular disease, is unlikely to be consistent with cirrhosis or hepatitis 9. The abnormal, instead of normal, hepatic perfusion phase (increased or anticipated) due to liver arterialization is more likely in cirrhosis or hepatitis 9. Dubin-Johnson syndrome, a similar syndrome of conjugated hyperbilirubinemia, presents, in contrast, hepatobiliary scintigraphy findings of intrahepatic cholestasis (no biliary phase, associated with prolonged, rapid, and intensive, instead of slow and negligible, liver radiotracer uptake) 27, 28.
Rotor syndrome can be distinguished from Dubin-Johnson syndrome by a lack of pigment deposits in liver cells biopsy, delayed plasma clearance of the unconjugated anionic dye bromosulphthalein (BSP), poor liver visualization on certain radiographic imaging studies, and prominent urinary excretion of coproporphyrin 1 27.
Table 1. Laboratory Findings in Rotor Syndrome
| Laboratory Finding | Rotor Syndrome | Normal | |
|---|---|---|---|
| Blood | Total bilirubin | 2-5 mg/dL 1 | 0.3-1.0 mg/dL2 |
| Conjugated:total bilirubin ratio | >50% | <20% | |
| Liver enzymes | Normal | Normal | |
| Hemolysis | None | None | |
| Urine | Bilirubin | Present | Not detected |
| Coproporphyrins | ↑ 2.5 to 5x normal 3 | ||
Footnotes:
1 Rarely, levels exceeding 20 mg/dL are possible 49.
2 For total and direct bilirubin in persons older than age one year. Note: Although normal levels of total and direct bilirubin may be higher in the neonatal period and infancy, Rotor syndrome is not usually diagnosed in this age group.
3 Coproporphyrinuria is frequently observed in those with parenchymal liver diseases. It is not specific to Rotor syndrome.
[Source 3 ]Rotor syndrome differential diagnosis
Inherited disorders of bilirubin clearance can present with either conjugated or unconjugated hyperbilirubinemia. Distinguishing Rotor syndrome from other more serious disorders is important to avoid unnecessary workup and interventions.
Some conditions that can cause hyperbilirubinemia to include 1:
- Dubin-Johnson syndrome
- Gilbert syndrome
- Crigler-Najjar syndrome (type 1 and type 2)
- Extra-hepatic biliary obstruction
- Familial intra-hepatic cholestasis
- Benign recurrent intrahepatic cholestasis (BRIC)
- Drug-induced hepatotoxicity
- Hemolysis
- Cholestasis of pregnancy
- Viral hepatitis
- Autoimmune hepatitis
- Wilson disease
- Hemochromatosis
- Alpha-1-antitrypsin deficiency
- Cirrhosis
Dubin-Johnson syndrome, a benign conjugated hyperbilirubinemia similar to Rotor syndrome, is caused by decreased secretion of conjugated bilirubin into bile. Defects in bilirubin conjugation resulting in increased levels of unconjugated bilirubin are represented by Gilbert syndrome, Crigler-Najjar syndrome type 2, and Crigler-Najjar syndrome type 1 (a rare, severe, life-threatening disease associated with kernicterus typically manifesting within the first days after birth). Since Rotor syndrome is usually diagnosed after the neonatal period, only benign forms of genetic jaundice are included in the differential diagnosis.
Cholestatic liver diseases and/or bile duct obstruction should be suspected whenever hyperbilirubinemia is accompanied by clinical signs other than jaundice and by elevation of serum alanine transaminase (ALT), aspartate transaminase (AST), alkaline phosphatase (ALP), and gamma-glutamyl transferase (GGT) activity. The same holds true for any abnormal findings in the gallbladder and the biliary tree obtained by imaging and/or endoscopy techniques.
Hemolytic jaundice is characterized by predominantly unconjugated hyperbilirubinemia and signs of increased hemolysis.
Table 2. Rotor Syndrome differential diagnosis
| Gene(s) | Disorder | Clinical & Laboratory Findings | Comment |
|---|---|---|---|
| UGT1A1 | Gilbert syndrome 1 (OMIM 143500) |
| Gilbert syndrome and Crigler-Najjar syndrome type 2 have quantitatively different consequences on UGT1A1 enzyme activity. Since plasma bilirubin level is not stable, the phenotypes may overlap in the same person. However, unconjugated hyperbilirubinemia associated with homozygous A(TA)7TAA genotype and no pathogenic variants in the UGT1A1 coding region should always be diagnosed as Gilbert syndrome. |
| Crigler-Najjar syndrome type 2 (Arias syndrome) 2 (OMIM 606785) |
| ||
| ABCC2 | Dubin-Johnson syndrome (DJS) 3 (OMIM 237500) |
| See Table 3 for comparison of laboratory findings in Dubin-Johnson syndrome & Rotor syndrome. |
Footnotes:
1 Caused by pathogenic variants in UGT1A1 that decrease the rate of bilirubin conjugation catalyzed by UDP-glucuronosyltransferase 1A1 (UGT1A1). Associated pathogenic variants include the promoter TATA repeat variation A(TA)7TAA (normal: A(TA)6TAA), which is often combined with the promoter SNP c.[-3279T>G], or missense variants in the coding region of UGT1A1, which are frequent in the Japanese population but rare in Europeans.
2 Caused by pathogenic variants in the coding region of UGT1A1.
3 Disorder of secretion of conjugated bilirubin into bile.
Table 3. Comparison between Rotor Syndrome and Dubin‐Johnson Syndrome
| Finding | Rotor Syndrome | Dubin-Johnson Syndrome | Normal | |
|---|---|---|---|---|
| Blood | Total bilirubin | 2-5 mg/dL 1 | 2-5 mg/dL 1 | 0.3-1.0 mg/dL2 |
| Conjugated:total bilirubin ratio | >50% | >50% | <20% | |
| Liver enzymes 3 | Normal | Normal | Normal | |
| Hemolysis 4 | None | None | None | |
| Urine | Bilirubin | Present; urine may be dark. | Present; urine may be dark. | Not detected |
| Porphyrins | Total porphyrin output ↑; coproporphyrin ↑ 2.5-5x normal | Total porphyrin output normal 5 | <200 μg in 24 hrs 6 | |
| Disappearance of plasma anionic compounds 7 | Severely delayed | Delayed | Rapid | |
| Cholescintigraphy | Scarcely visualized on cholescintigraphy, with slow liver uptake, persistent visualization of cardiac blood pool, & prominent kidney excretion | Visualization of liver is normal or somewhat delayed but filling of gallbladder is absent or delayed. | Normal | |
| Pruritus | Absent | Absent | Absent | |
| Precipitating factors | Intercurrent illness, drugs | Pregnancy, intercurrent illness, drugs | Normal | |
| Histology | Normal | Gross pathology: Black liver; histology: Lysosomal pigment | Normal | |
| Molecular mechanism | OATP1B1 and OATP1B3 transporters affected; hepatic storage deficiency of conjugated bilirubin | ABCC2 transporter affected; biliary transport deficiency of nonbile acid organic anions | Normal | |
| Prognosis | Benign | Benign | Normal | |
Footnotes:
1 Rarely, levels exceeding 20 mg/dL are possible 49
2 For total and direct bilirubin in persons older than age one year. Note: Although normal levels of total and direct bilirubin may be higher in the neonatal period and infancy, Rotor syndrome is not usually diagnosed in this age group.
3 Serum alanine transaminase (ALT), aspartate transaminase (AST), alkaline phosphatase (ALP), and gamma-glutamyl transferase (GGT) activity.
4 Red blood count and reticulocyte count.
5 Total urinary porphyrin output is normal; however, predominance of coproporphyrin isomer I among urinary porphyrin species is observed on chromatography.
6 Total urinary porphyrin output.
7 Includes bromosulfophthalein (BSP), indocyanine green, and cholescintigraphy radiotracers (99mTc-HIDA/99mTc-N [2,6-dimethylphenyl-carbamoylmethyl] iminodiacetic acid, 99mTc-DISIDA/disofenin, 99mTc-BrIDA/mebrofenin). Note: In Dubin-Johnson syndrome, bromosulfophthalein (BSP) conjugates reappear in the blood after administration of unconjugated bromosulfophthalein (BSP); this is not the case in Rotor syndrome.
Rotor syndrome treatment
Rotor syndrome is a benign lifelong condition that does not require treatment 27, 20. No clinical practice guidelines for Rotor syndrome have been published as no treatment or surveillance is recommended.
Agents and circumstances to avoid
No adverse drug effects have been documented in people with Rotor syndrome; however, the absence of the liver proteins OATP1B1 and OATP1B3 may have serious consequences for liver uptake and toxicity of numerous commonly used drugs and/or their metabolites, which enter the liver via either of the two OATP1B transporters. OATP1B1 plays a role in drug detoxification, and with reduced activity of this protein, certain drugs such as anticancer agents, methotrexate, and statins can accumulate and result in drug toxicity 29.
No adverse drug effects have been documented in people with Rotor syndrome; however, the absence of the liver proteins OATP1B1 and OATP1B3 may have serious consequences for liver uptake and toxicity of numerous commonly used drugs and/or their metabolites, which enter the liver via either of the two OATP1B transporters. OATP1B1 plays a role in drug detoxification, and with reduced activity of this protein, certain drugs such as anticancer agents, methotrexate, and statins can accumulate and result in drug toxicity 29. Caution should be taken before administering these drugs 20, 30, 31, 32:
- Statins – simvastatin, atorvastatin, pravastatin, pitavastatin, rosuvastatin
- Ezetimibe
- Anticancer drugs – methotrexate and irinotecan, cabazitaxel, some tyrosine kinase inhibitors (e.g., sunitinib)
- Sartans – olmesartan and valsartan
- Rifampicin
- Mycophenolic acid
- Torsemide
- Thiazolidine diones – pioglitazone and rosiglitazone
- Glinides – nateglinide and repaglinide
- Lopinavir
- Fexofenadine
- Cyclosporin A.
Pregnancy management
No special pregnancy management issues from the perspective of either an affected mother or an affected fetus are known.
Of note, during pregnancy the hyperbilirubinemia of Rotor syndrome may complicate the diagnosis and management of liver disease related to pregnancy (e.g., intrahepatic cholestasis of pregnancy) and liver disease not related to pregnancy.
Genetic Counseling
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://abgc.learningbuilder.com/Search/Public/MemberRole/Verification) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (https://clinics.acmg.net/) has a searchable database of medical genetics clinic services in the United States.
Rotor syndrome prognosis
Rotor syndrome is a benign lifelong condition that does not require treatment 27, 20. Rotor syndrome generally begins shortly after birth or during childhood. Generally, individuals with Rotor syndrome are asymptomatic, and jaundice is usually an incidental finding and conjunctival icterus could be the only clinical feature. There are no recommended management options for patients suffering from Rotor syndrome. There is no sickness or death associated with Rotor syndrome; hence its prognosis is good 1. Mortality and morbidity occur if there is another coexisting liver disease.
- Hashmi MF, Mehta D. Rotor Syndrome. [Updated 2023 Feb 19]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK532306[↩][↩][↩]
- Rotor syndrome. https://medlineplus.gov/genetics/condition/rotor-syndrome[↩]
- Jirsa M, Knisely AS, Schinkel A, et al. Rotor Syndrome. 2012 Dec 13 [Updated 2025 Feb 27]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK114805[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Morais M, Couvert P, Jéru I, Machado MV. Rotor Syndrome Presenting as Dubin-Johnson Syndrome. Case Rep Gastroenterol. 2022 Aug 16;16(2):452-455. doi: 10.1159/000525517[↩]
- Kimura A, Kagawa T, Takei H, Maruo Y, Sakugawa H, Sasaki T, Murai T, Naritaka N, Takikawa H, Nittono H. Rotor Syndrome: Glucuronidated Bile Acidemia From Defective Reuptake by Hepatocytes. Hepatol Commun. 2020 Dec 31;5(4):629-633. doi: 10.1002/hep4.1660[↩][↩]
- Cheng YY, Chang KC, Chen PL, Yeung CY, Liou BY, Chen HL. SLCO1B1 and SLCO1B3 genetic mutations in Taiwanese patients with Rotor syndrome. J Formos Med Assoc. 2023 Jul;122(7):648-652. https://doi.org/10.1016/j.jfma.2023.03.003[↩]
- Kim YG, Sung H, Shin HS, Kim MJ, Lee JS, Park SS, Seong MW. Intronic LINE-1 insertion in SLCO1B3 as a highly prevalent cause of rotor syndrome in East Asian population. J Hum Genet. 2022 Feb;67(2):71-77. doi: 10.1038/s10038-021-00967-1[↩][↩][↩]
- Zhao C, Huang H. Recurrent SLCO1B1 and SLCO1B3 mutations identified in three patients with Rotor syndrome. Front Med (Lausanne). 2025 Aug 18;12:1630360. doi: 10.3389/fmed.2025.1630360[↩]
- Dimopoulou D, Lyra V, Dimopoulou A, Papaevangelou V, Fessatou S. Is Hepatobiliary Scintigraphy Sufficient to Diagnose Rotor Syndrome in a 3-Year-Old Boy? J Nucl Med Technol. 2021 Jun;49(2):193-194. https://tech.snmjournals.org/content/49/2/193.long[↩][↩][↩][↩][↩][↩]
- Memon N, Weinberger BI, Hegyi T, Aleksunes LM. Inherited disorders of bilirubin clearance. Pediatr Res. 2016 Mar;79(3):378-86. doi: 10.1038/pr.2015.247[↩][↩][↩]
- Zhang W, He YJ, Gan Z, et al. OATP1B1 polymorphism is a major determinant of serum bilirubin level but not associated with rifampicin-mediated bilirubin elevation. Clin Exp Pharmacol Physiol. 2007;34:1240–4. doi: 10.1111/j.1440-1681.2007.04798.x[↩]
- Sanna S, Busonero F, Maschio A, et al. Common variants in the SLCO1B3 locus are associated with bilirubin levels and unconjugated hyperbilirubinemia. Hum Mol Genet. 2009;18:2711–8. doi: 10.1093/hmg/ddp203[↩]
- HYPERBILIRUBINEMIA, ROTOR TYPE; HBLRR. https://omim.org/entry/237450[↩][↩]
- SLCO1B1 gene. https://medlineplus.gov/genetics/gene/slco1b1[↩][↩][↩]
- SLCO1B3 gene. https://medlineplus.gov/genetics/gene/slco1b3[↩][↩][↩]
- Zhou D, Qi S, Zhang W, Wu L, Xu A, Li X, Zhang B, Li Y, Jia S, Wang H, Jia J, Ou X, Huang J, You H. Insertion of LINE-1 Retrotransposon Inducing Exon Inversion Causes a Rotor Syndrome Phenotype. Front Genet. 2020 Jan 31;10:1399. doi: 10.3389/fgene.2019.01399[↩][↩]
- Fretzayas A, Koukoutsakis P, Moustaki M, Stavrinadis C, Karpathios T. Coinheritance of Rotor syndrome, G-6-PD deficiency, and heterozygous beta thalassemia: a possible genetic interaction. J Pediatr Gastroenterol Nutr. 2001 Aug;33(2):211-3. doi: 10.1097/00005176-200108000-00023[↩]
- A.B. Rotor, L. Manahan, A. Florentin. Familial nonhemolytic jaundice with direct van den Bergh reaction. Acta Med Philipp, 5 (1948), pp. 37-49.[↩]
- The familial conjugated hyperbilirubinemias. Semin Liver Dis. 1994 Nov;14(4):386-94. doi: 10.1055/s-2007-1007329[↩]
- Erlinger S, Arias IM, Dhumeaux D. Inherited disorders of bilirubin transport and conjugation: new insights into molecular mechanisms and consequences. Gastroenterology. 2014 Jun;146(7):1625-38. doi: 10.1053/j.gastro.2014.03.047[↩][↩][↩][↩][↩][↩][↩]
- Nisa AU, Ahmad Z. Dubin-Johnson syndrome. J Coll Physicians Surg Pak. 2008 Mar;18(3):188-9.[↩][↩]
- Hrebícek M, Jirásek T, Hartmannová H, Nosková L, Stránecký V, Ivánek R, Kmoch S, Cebecauerová D, Vítek L, Mikulecký M, Subhanová I, Hozák P, Jirsa M. Rotor-type hyperbilirubinaemia has no defect in the canalicular bilirubin export pump. Liver Int. 2007 May;27(4):485-91. doi: 10.1111/j.1478-3231.2007.01446.x[↩]
- Bonkovsky HL, Ma CD, Araque M, Tiley JB, Brouwer KLR, Stölzel U. Understanding Coproporphyrins and Their Disposition: Coproporphyrinuria is Common, of Diverse Cause, and Rarely Indicates Porphyria. Am J Med. 2025 Sep;138(9):1214-1226.e3. doi: 10.1016/j.amjmed.2025.04.004[↩][↩]
- Wolkoff AW, Wolpert E, Pascasio FN, Arias IM. Rotor’s syndrome. A distinct inheritable pathophysiologic entity. Am J Med. 1976 Feb;60(2):173-9. doi: 10.1016/0002-9343(76)90426-5[↩][↩]
- HIDA scan. https://www.mayoclinic.org/tests-procedures/hida-scan/about/pac-20384701[↩][↩]
- Cholescintigraphy. https://radiopaedia.org/articles/cholescintigraphy[↩][↩]
- van de Steeg E, Stránecký V, Hartmannová H, Nosková L, Hřebíček M, Wagenaar E, van Esch A, de Waart DR, Oude Elferink RP, Kenworthy KE, Sticová E, al-Edreesi M, Knisely AS, Kmoch S, Jirsa M, Schinkel AH. Complete OATP1B1 and OATP1B3 deficiency causes human Rotor syndrome by interrupting conjugated bilirubin reuptake into the liver. J Clin Invest. 2012 Feb;122(2):519-28. doi: 10.1172/JCI59526[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Sirucek P, Sulakova A, Jirsa M, Mrhac L, Havel M, Kraft O. Radionuclide cholescintigraphy in genetically confirmed Rotor syndrome. Pediatr Int. 2015 Oct;57(5):981-5. doi: 10.1111/ped.12676[↩][↩][↩]
- Sticova E, Jirsa M. New insights in bilirubin metabolism and their clinical implications. World J Gastroenterol. 2013 Oct 14;19(38):6398-407. doi: 10.3748/wjg.v19.i38.6398[↩][↩][↩]
- Niemi M, Pasanen MK, Neuvonen PJ. Organic anion transporting polypeptide 1B1: a genetically polymorphic transporter of major importance for hepatic drug uptake. Pharmacol Rev. 2011 Mar;63(1):157-81. doi: 10.1124/pr.110.002857[↩][↩]
- Garrison DA, Talebi Z, Eisenmann ED, Sparreboom A, Baker SD. Role of OATP1B1 and OATP1B3 in Drug-Drug Interactions Mediated by Tyrosine Kinase Inhibitors. Pharmaceutics. 2020 Sep 9;12(9):856. doi: 10.3390/pharmaceutics12090856[↩][↩]
- Anabtawi N, Drabison T, Hu S, Sparreboom A, Talebi Z. The role of OATP1B1 and OATP1B3 transporter polymorphisms in drug disposition and response to anticancer drugs: a review of the recent literature. Expert Opin Drug Metab Toxicol. 2022 Jul-Aug;18(7-8):459-468. doi: 10.1080/17425255.2022.2113380[↩][↩]
- Kalakonda A, Jenkins BA, John S. Physiology, Bilirubin. [Updated 2022 Sep 12]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK470290[↩]
- Singh A, Koritala T, Jialal I. Unconjugated Hyperbilirubinemia. [Updated 2023 Feb 20]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK549796[↩][↩][↩]
- Kosmachevskaya OV, Topunov AF. Alternate and Additional Functions of Erythrocyte Hemoglobin. Biochemistry (Mosc). 2018 Dec;83(12):1575-1593. doi: 10.1134/S0006297918120155[↩]
- Dosch AR, Imagawa DK, Jutric Z. Bile Metabolism and Lithogenesis: An Update. Surg Clin North Am. 2019 Apr;99(2):215-229. doi: 10.1016/j.suc.2018.12.003[↩]
- Shen H, Zeng C, Wu X, Liu S, Chen X. Prognostic value of total bilirubin in patients with acute myocardial infarction: A meta-analysis. Medicine (Baltimore). 2019 Jan;98(3):e13920. doi: 10.1097/MD.0000000000013920[↩]
- Franchini M, Targher G, Lippi G. Serum bilirubin levels and cardiovascular disease risk: a Janus Bifrons? Adv Clin Chem. 2010;50:47-63. doi: 10.1016/s0065-2423(10)50003-9[↩]
- Vítek L, Schwertner HA. The heme catabolic pathway and its protective effects on oxidative stress-mediated diseases. Adv Clin Chem. 2007;43:1-57. doi: 10.1016/s0065-2423(06)43001-8[↩][↩]
- Vítek L, Ostrow JD. Bilirubin chemistry and metabolism; harmful and protective aspects. Curr Pharm Des. 2009;15(25):2869-83. doi: 10.2174/138161209789058237[↩]
- Jedlitschky G, Hoffmann U, Kroemer HK. Structure and function of the MRP2 (ABCC2) protein and its role in drug disposition. Expert Opin Drug Metab Toxicol. 2006 Jun;2(3):351-66. doi: 10.1517/17425255.2.3.351[↩]
- Kagawa T, Oka A, Kobayashi Y, Hiasa Y, Kitamura T, Sakugawa H, Adachi Y, Anzai K, Tsuruya K, Arase Y, Hirose S, Shiraishi K, Shiina T, Sato T, Wang T, Tanaka M, Hayashi H, Kawabe N, Robinson PN, Zemojtel T, Mine T. Recessive inheritance of population-specific intronic LINE-1 insertion causes a rotor syndrome phenotype. Hum Mutat. 2015 Mar;36(3):327-32. doi: 10.1002/humu.22745[↩][↩][↩]
- Keppler D. The roles of MRP2, MRP3, OATP1B1, and OATP1B3 in conjugated hyperbilirubinemia. Drug Metab Dispos. 2014 Apr;42(4):561-5. doi: 10.1124/dmd.113.055772[↩]
- Pratt E, Sissung TM, Figg WD. Loss of OATP1B3 function causes Rotor syndrome: implications for potential use of inhibitors in cancer. Cancer Biol Ther. 2012 Dec;13(14):1374-5. doi: 10.4161/cbt.22010[↩]
- Kagawa T, Adachi Y, Hashimoto N, Mitsui H, Ohashi T, Yoneda M, Hasegawa I, Hirose S, Tsuruya K, Anzai K, Mine T. Loss of organic anion transporting polypeptide 1B3 function causes marked delay in indocyanine green clearance without any clinical symptoms. Hepatology. 2017 Mar;65(3):1065-1068. doi: 10.1002/hep.28950[↩]
- Dhumeaux D, Erlinger S. Hereditary conjugated hyperbilirubinaemia: 37 years later. J Hepatol. 2013 Feb;58(2):388-90. doi: 10.1016/j.jhep.2012.08.025[↩]
- Bar-Meir S, Baron J, Seligson U, Gottesfeld F, Levy R, Gilat T. 99mTc-HIDA cholescintigraphy in Dubin-Johnson and Rotor syndromes. Radiology. 1982 Mar;142(3):743-6. doi: 10.1148/radiology.142.3.7063695[↩]
- Zimniak P. Dubin-Johnson and Rotor syndromes: molecular basis and pathogenesis. Semin Liver Dis. 1993 Aug;13(3):248-60. doi: 10.1055/s-2007-1007353[↩]
- Strassburg CP. Hyperbilirubinemia syndromes (Gilbert-Meulengracht, Crigler-Najjar, Dubin-Johnson, and Rotor syndrome). Best Pract Res Clin Gastroenterol. 2010 Oct;24(5):555-71. doi: 10.1016/j.bpg.2010.07.007[↩][↩]





{kind=link}