Contents
What is Russell-Silver syndrome
Russell-Silver syndrome or Silver-Russell syndrome, is a growth disorder characterized by slow growth before and after birth. Babies with Russell-Silver syndrome have a low birth weight and often fail to grow and gain weight at the expected rate (failure to thrive). Head growth is normal, however, so the head may appear unusually large compared to the rest of the body. Affected children are thin and have poor appetites, and some develop recurrent episodes of low blood sugar (hypoglycemia) as a result of feeding difficulties. Adults with Russell-Silver syndrome are short; the average height for affected men is about 151 centimeters (4 feet, 11 inches) and the average height for affected women is about 140 centimeters (4 feet, 7 inches).
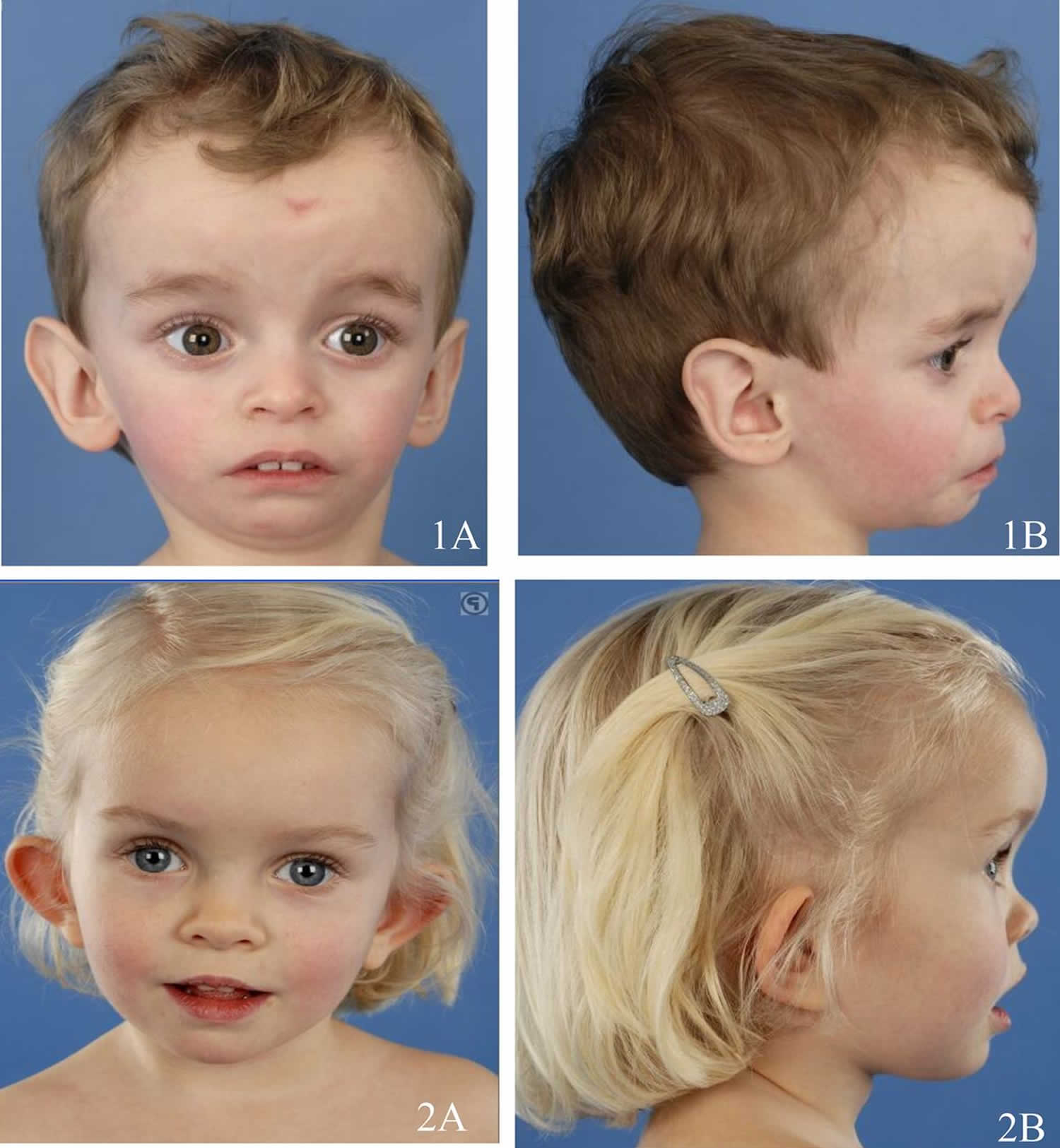
Many children with Russell-Silver syndrome have a small, triangular face with distinctive facial features including a prominent forehead, a narrow chin, a small jaw, and downturned corners of the mouth. Other features of this disorder can include an unusual curving of the fifth finger (clinodactyly), asymmetric or uneven growth of some parts of the body, and digestive system abnormalities. Russell-Silver syndrome is also associated with an increased risk of delayed development, speech and language problems, and learning disabilities.
Russell-Silver syndrome is a genetic disorder that usually results from the abnormal regulation of certain genes that control growth 1. Two genetic causes have been found to result in about 60% of the cases 2:
- Abnormalities at an imprinted region on chromosome 11p15 – for some genes, only the copy inherited from a person’s father (paternal copy) or mother (maternal copy) is “turned on,” or expressed. These parent-specific differences in gene expression are caused by a phenomenon called genomic imprinting.
- Abnormalities involving genes that undergo imprinting are responsible for many cases of Russell-Silver syndrome.
- Maternal disomy of chromosome 7 (written as matUPD7) – this occurs when a child inherits both copies of chromosome 7 from the mother, instead of one copy from the mother and one copy from the father.
Other chromosome abnormalities have also been described as causing Russell-Silver syndrome, or Russell-Silver syndrome-like syndromes 3.
In many people with Russell-Silver syndrome, the cause of the condition is unknown. Researchers are working to identify additional genetic changes that cause Russell-Silver syndrome 1.
The exact incidence of Russell-Silver syndrome is unknown. Worldwide estimates range from 1 in 30,000 to 1 in 100,000 people 1.
It was way back in 1953 and 1954 that Dr. Silver and Dr. Russell independently described groups of small-for-gestational-age children whose pregnancies had been complicated by intrauterine growth restriction 4. Their common findings were short stature without catch-up growth, normal head size for age, a distinctive triangular face, low-set ears and incurving fifth fingers. These two groups of patients are now considered to have had variations of the same disorder that experts now call Russell-Silver syndrome in North America, and Silver-Russell syndrome in Europe.
Russell-Silver syndrome support groups:
There are a number of groups and advocacy organizations that help people with Russell-Silver syndrome and their families connect with others who have been impacted by this syndrome.
The two largest groups for English-speaking families are:
- MAGIC Foundation (https://www.magicfoundation.org/)
- Russell-Silver Syndrome Support (https://silverrussellsyndrome.org/)
Read the Description sections on the above pages for information about how to join each group.
Do children with Russell-Silver syndrome have some kind of speech disorders?
Detailed information about the speech and language problems in children with Russell-Silver syndrome is limited. Specific problems with speech delay have been reported, particularly in people with a certain genetic cause for Russell-Silver syndrome called maternal uniparental disomy of chromosome 7 (matUPD7) 5. When only maternal copies of chromosome 7 are inherited, there are no chromosome 7 genes inherited from the father. Developmental verbal dyspraxia has been reported in people without a paternal copy (or working copy) of a specific gene on chromosome 7 called FOXP2 6. People with Russell-Silver syndrome due to matUPD7 also do not have a paternal copy of the FOXP2 gene; this may be the cause of developmental verbal dyspraxia in these cases.
A few people with Russell-Silver syndrome and speech delay have been described in the medical literature. One study noted poor perceptual motor skills, and expressive and receptive language problems 7. Another study described 4 people with uniparental disomy for chromosome 7 that all had speech delay, particularly with problems in articulation. The first person described had dysphasia-like problems caused by difficulties in articulation, mostly because of dyspraxia in the vocal muscles, and significant problems in finding the appropriate words. The second person had normal psychomotor development, and speech delay with multiple articulation problems. A third person had marked speech delay, mainly in producing speech. She understood speech well, but produced only rare one- or two-word sentences, and communicated mostly by gestures and sign language. The fourth person described had normal oral motor functions, somewhat delayed psychomotor development, and speech delay with articulation problems 8.
Is someone with Russell-Silver syndrome at an increased risk for being a carrier of cystic fibrosis (CF)?
Experts were not able to find any reports that a person with Russell-Silver syndrome (Russell-Silver syndrome) had a higher or lower risk of being a carrier of cystic fibrosis (CF).
There are several case reports of children with Russell-Silver syndrome also having cystic fibrosis 9, 10. In these cases, the genetic cause was even a bit more complicated than stated above. The cause of the Russell-Silver syndrome was maternal monosomy of chromosome 7, however in these cases, the child did not inherit two different chromosome 7s from their mother (called heterodisomy), but instead has two copies of the same chromosome 7 (called isodisomy). Furthermore, the mother happened to be a carrier of cystic fibrosis and the inherited chromosome 7 (which the child now has two copies) of had this mutation. Therefore the child ended up with two copies of the mutation causing cystic fibrosis 9, 10.
Russell-Silver syndrome causes
The genetic causes of Russell-Silver syndrome are complex 1. Russell-Silver syndrome often results from the abnormal regulation of certain genes that control growth. Research has focused on genes located in particular regions of chromosome 7 and chromosome 11.
People normally inherit one copy of each chromosome from their mother and one copy from their father. For most genes, both copies are expressed, or “turned on,” in cells. For some genes, however, only the copy inherited from a person’s father (the paternal copy) is expressed. For other genes, only the copy inherited from a person’s mother (the maternal copy) is expressed. These parent-specific differences in gene expression are caused by a phenomenon called genomic imprinting. Both chromosome 7 and chromosome 11 contain groups of genes that normally undergo genomic imprinting; some of these genes are active only on the maternal copy of the chromosome, while others are active only on the paternal copy. Abnormalities involving these genes appear to be responsible for many cases of Russell-Silver syndrome.
Researchers suspect that 30 to 50 percent of all cases of Russell-Silver syndrome result from changes in a process called methylation on the short (p) arm of chromosome 11 at position 15 (11p15 LOM) 11. Methylation is a chemical reaction that attaches small molecules called methyl groups to certain segments of DNA. In genes that undergo genomic imprinting, methylation is one way that a gene’s parent of origin is marked during the formation of egg and sperm cells. Russell-Silver syndrome has been associated with changes in methylation involving the H19 and IGF2 genes, which are located near one another at 11p15. These genes are thought to be involved in directing normal growth. A loss of methylation disrupts the regulation of these genes, which leads to slow growth and the other characteristic features of this disorder.
Abnormalities involving genes on chromosome 7 can also cause Russell-Silver syndrome. In 7 percent to 10 percent of cases, people inherit both copies of chromosome 7 from their mother instead of one copy from each parent. This phenomenon is called maternal uniparental disomy (UPD). Maternal uniparental disomy causes people to have two active copies of some imprinted genes and no active copies of others. An imbalance in certain active paternal and maternal genes on chromosome 7 underlies the signs and symptoms of the disorder.
In about 40 percent of people with Russell-Silver syndrome, the cause of the condition is unknown. It is likely that changes involving imprinted genes on chromosomes other than 7 and 11 play a role. Researchers are working to identify additional genetic changes that underlie this disorder.
Russell-Silver syndrome inheritance pattern
Most cases of Russell-Silver syndrome are sporadic (not inherited), which means they occur in people with no history of the disorder in their family.
Rarely, Russell-Silver syndrome can run in families. In some affected families, the condition appears to have an autosomal dominant pattern of inheritance. Autosomal dominant inheritance means one copy of a genetic change in each cell is sufficient to cause the disorder. In other families, the condition appears to have an autosomal recessive pattern of inheritance. Autosomal recessive inheritance means both copies of a gene are altered in each cell. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
When 2 carriers of an autosomal recessive condition have children, each child has a:
- 25% (1 in 4) chance to be affected
- 50% (1 in 2) chance to be an unaffected carrier like each parent
- 25% chance to be unaffected and not be a carrier
Genetic counseling
Accurate genetic counseling depends on the underlying molecular cause. Methylation on the short (p) arm of chromosome 11 at position 15 (11p15 LOM) is associated with a low recurrence risk (with parents of a child with Russell-Silver syndrome being unlikely to have another affected child) 12. The offspring risk is also low (meaning that individuals with Russell-Silver syndrome are unlikely to pass the condition on to their children). However, empirical figures are not available. Only three sibships with 11p15 LOM are reported in the literature 13, and the underlying mechanism is unknown in all three.
The potential for a familial trans-acting gene mutation suggests that the recurrence risk in patients with Russell-Silver syndrome and MLID could be higher than in other patients with Russell-Silver syndrome; however, evidence to support this supposition does not yet exist.
Rare familial cases of Russell-Silver syndrome have been reported with underlying mechanisms including: maternally inherited 11p15 duplication 14; maternally inherited CDKN1C gain-of-function mutations 15; and paternally inherited IGF2 loss-of-function mutations 16. In these families, the risk of recurrence might be as high as 50%24,26,60,61. Investigation for underlying copy number variants in patients with 11p15 LOM is, therefore, important. upd(7)mat is associated with a low recurrence and offspring risk (if the karyotype of the patient is normal)50. Data are limited regarding the risk of parents of children with clinically diagnosed Russell-Silver syndrome having another child with Russell-Silver syndrome; however, the overall risk is probably low. Similarly, the offspring risk for individuals with clinically diagnosed Russell-Silver syndrome is likely to be low.
Genetic counseling should be performed by a health professional experienced in the field of imprinting disorders. As the recurrence risk associated with copy number variants is dependent on their size, location and parental origin, these should be taken into consideration during counseling for the family.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
What are the chances for a person with Russell Silver syndrome to have an affected child?
The chance for a person with Russell-Silver syndrome to have an affected child depends on the genetic cause of Russell-Silver syndrome in the parent and the family history. In some cases, the genetic cause is not identified.
If a parent has Russell-Silver syndrome due to an imprinting defect on chromosome 11p15, the risk to his/her children is thought to be low. However to our knowledge, with the exception of one report of father-to-daughter transmission of the imprinting defect, no data to determine this exact risk are available 2.
If a parent has Russell-Silver syndrome due to maternal disomy of chromosome 7, the risk to his/her children is thought to be low. However in this case as well, there is no data we are aware of regarding exact recurrence risks 2.
View the information in our Inheritance Section for information on the recurrence risk when Russell-Silver syndrome is known to be an autosomal dominant or autosomal recessive disorder (which rarely occur).
Because most cases of Russell-Silver syndrome occur only in a single family member, most pregnancies are not considered to be at increased risk for the disorder 2. However, people with personal questions about the genetic cause of Russell-Silver syndrome and risks to family members are strongly encouraged to speak with a genetic counselor or other genetics professional. A genetics professional can help by:
- thoroughly evaluating the family history
- addressing questions and concerns
- assessing recurrence risks
- facilitating genetic testing if desired
- discussing reproductive options
Russell-Silver syndrome symptoms
Features of Russell-Silver syndrome can vary. Some people with Russell-Silver syndrome have many features, while others have very few features 2. Signs and symptoms may include 12:
- Intrauterine growth restriction (poor growth before birth)
- Low birth weight
- Head that appears large in relation to body size (relative macrocephaly)
- Poor appetite and feeding difficulties
- Hypoglycemia
- Poor growth after birth, leading to short stature
- Scoliosis
- Curving of the pinky finger (clinodactyly)
- Characteristic facial features (prominent forehead; small, triangular-shaped face; a small jaw; a narrow chin; and down-turned corners of the mouth)
- Arms and legs of different lengths (body asymmetry)
- Delayed bone age
- Gastroesophageal reflux disease or other digestive problems
- Kidney problems
- Developmental delay and/or learning disabilities
- Psychosocial challenges
Typical Russell-Silver syndrome physical characteristics
Characteristics That Occur More Frequently in Russell-Silver Syndrome Children than Other Small-for-Gestational-Age Children 17:
The characteristics that Azzi et al 17 found to be statistically different between specific molecular Russell-Silver syndrome groups and Small-for-Gestational-Age-non Russell-Silver syndrome subjects are marked below with an asterisk *.
- body asymmetry – LARGE side is the “normal” side*
- large head size for body size*
- broad protruding forehead*
- low-set, posteriorly rotated &/or prominent ears
- clinodactyly (inward curving) of the 5th finger (pinky)*
- syndactyly (webbing) of the 2nd and 3rd toes*
- inadequate catch-up growth in first 2 years
- persistently low weight-for-height*
- lack of muscle mass and/or poor muscle tone*
- hypoplastic (underdeveloped) chin & midface
- downturned corners of mouth*
- thin upper lip
- high-arched palate
- small, crowded teeth
- unusually high-pitched voice in early years
- café-au-lait (coffee-with-milk) birth marks
- dimples in the posterior shoulders and hips*
- narrow, flat feet
- scoliosis (curved spine, associated with spinal asymmetry & accentuated by a short leg)
- prominent heels [mUPD7 primarily]*
- autism [mUPD7 children primarily]*
- myoclonus dystonia (movement disorder) [mUPD7 primarily]
Characteristics of both Small-for-Gestational-Age Children and Russell-Silver Syndrome Patients:
- lack of interest in eating
- fasting hypoglycemia & mild metabolic acidosis
- generalized intestinal movement abnormalities:
- esophageal reflux resulting in movement of food up from stomach into food tube
- delayed stomach emptying resulting in vomiting or frequent spitting up
- slow movement of the small intestine &/or large intestine (constipation)
- late closure of the anterior fontanel (soft spot)
- frequent ear infections or chronic fluid in ears
- congenital absence of the second premolars
- delay of gross and fine motor development
- delay of speech and oral motor development
- kidney abnormalities
- delayed bone age early, later fast advancement
- early pubic hair and underarm odor (adrenarche)
- early puberty or rarely true precocious puberty
- classical or neurosecretory growth hormone deficiency
- Attention Deficit Disorder (ADD) and specific learning disabilities
- blue sclera (bluish tinge in white of eye)
- hypospadias (abnormal opening of the penis)
- cryptorchidism (undescended testicles)
Are there other rare syndromes with symptoms similar to those of Russell-Silver syndrome?
Symptoms of several disorders can be similar to those of Russell-Silver syndrome 3. For this reason, when Russell-Silver syndrome is suspected, it may be useful compare the symptoms of these disorders with a person’s symptoms to narrow the differential diagnosis. The differential diagnosis of Russell-Silver syndrome generally includes any condition that can cause intrauterine growth restriction (IUGR) and short stature 2.
- Intrauterine growth restriction (IUGR) may occur in a number of congenital disorders, including Mulibrey nanism and many chromosome disorders 2. Chromosome abnormalities to consider in the differential diagnosis of Russell-Silver syndrome include 3, 2:
- mosaic Turner syndrome
- diploid/triploid mixoploidy (because of limb asymmetry)
- Yq deletions
- other chromosome deletions (involving 12p14 , 15q26.3, and a distal deletion of 22q11.2)
- rearrangements of chromosome 17q25
- Three M syndrome is an extremely rare genetic disorder with features that include low birth weight, short stature, characteristic head and facial features, and distinctive bone abnormalities 3.
- Disorders of DNA repair (chromosome breakage disorders), including Fanconi anemia, Bloom syndrome, and Nijmegen breakage syndrome, are often associated with IUGR and short stature. In these conditions, additional features such as small head size (microcephaly), limb abnormalities, and abnormal sensitivity to sunlight (photosensitivity), are often evident 3.
- Fetal alcohol spectrum disorders (FASDs) may be characterized by mental and physical birth defects from maternal use of alcohol during pregnancy. The range and severity of symptoms vary greatly. In some cases, learning delays or intellectual disability occurs without any obvious physical abnormalities 3.
- IMAGe syndrome is characterized by IUGR, metaphyseal dysplasia, adrenal hypoplasia congenita, and genital abnormalities. A skeletal survey should be performed to exclude a skeletal dysplasia that may mimic Russell-Silver syndrome 2.
- One condition that has been confused with Russell-Silver syndrome is an X-linked disorder of short stature with skin hyperpigmentation. It has sometimes been referred to as X-linked Russell-Silver syndrome. This condition may be difficult to distinguish from classic Russell-Silver syndrome in the absence of a positive family history 2.
The presence of other particular features (especially those atypical for Russell-Silver syndrome) should prompt consideration of a diagnosis other than Russell-Silver syndrome. Atypical features may include microcephaly; notable global developmental delay or intellectual disability (without a related explanation); absence of severe feeding difficulties; the presence of additional congenital anomalies; and facial dysmorphism. Disproportionate short stature is suggestive of skeletal dysplasia, and photosensitivity or recurrent respiratory infections may be suggestive of chromosome breakage disorders. Because Russell-Silver syndrome is generally sporadic (not inherited), a family history of growth failure and/or consanguinity might suggest a different diagnosis.[3]
People with a questions about a possible genetic diagnosis are encouraged to meet with a genetics professional who can provide detailed information and support.
Russell-Silver syndrome diagnosis
Russell-Silver syndrome is currently a clinical diagnosis, based on a combination of characteristic features 12. Because the condition varies widely in severity and many of its features are nonspecific, making a diagnosis can be difficult 3. Consensus guidelines including a summary of recommendations for clinical diagnosis have recently been published and can be viewed here.
Molecular genetic testing can confirm the diagnosis in around 60% of patients, and may be useful in guiding management. However, genetic testing results are negative (“normal”) in a notable proportion of patients with the characteristic features of Russell-Silver syndrome. Therefore, a negative genetic test result does not exclude the diagnosis of Russell-Silver syndrome 12.
Molecular testing
Russell-Silver syndrome can be diagnosed with genetic testing; but negative genetic testing does not rule out a clinical diagnosis. Currently, genetic testing can be run for known causes of Russell-Silver Syndrome involving chromosomes 7 and 11. Chromosome 7 is involved in the cause of the maternal uniparental disomy of chromosome 7 [matUPD7] type of Russell-Silver syndrome, which can be either heterodisomy (rare) or isodisomy (most common).
Chromosome 11 at band p15 is involved in four different Russell-Silver syndrome mechanisms representing approximately 44-60% of Russell-Silver syndrome cases:
- A) hypomethylation of the paternal imprinting control region 1 (ICR1) due to an imprinting error in up to 40-55% of cases;
- B) duplication of ICR1 and ICR2 in about 2-3% of cases;
- C) duplication of ICR2 in about 1% of cases;
- D) mutation of CDKN1C gene in about 1% of cases.
In the rare mechanisms B through D above, which are chromosomal copy number variations, there can be a risk of transmission either to the patient’s own children or with the patient’s siblings. Genetic testing of the child’s parents will need to be completed and the results discussed with the geneticist.
There are other chromosomal abnormalities that have overlapping physical characteristics with Russell-Silver syndrome such as matUPD16, matUPD20 and matUPD14 (Temple Syndrome). As additional research is published in the future, we hope to know if these molecular abnormalities represent additional causes of Russell-Silver syndrome.
Clinical diagnosis
These above molecular causes account for only 60-70% of Russell-Silver syndrome cases; the other 30-40% are “idiopathic” at this time, meaning the cause is unknown. For these children, doctors will generally base their diagnosis on characteristic, clinical findings that make up the Russell-Silver syndrome phenotype. It is easy to diagnose the “textbook” Russell-Silver syndrome phenotype; other cases are more difficult to classify and may require a specialist highly experienced in Russell-Silver syndrome in order to rule out another syndrome or another cause of short stature. The Netchine-Harbison Russell-Silver syndrome clinical scoring system found that the following six clinical characteristics are statistically correlated with either a positive molecular result or a clinical Russell-Silver syndrome diagnosis by an experienced specialist:
- Small for gestational age (SGA) in birth for weight and/or length
- Large head size for body size at BIRTH (1.5 standard deviations score (SDS) difference between birth head circumference and birth weight or length)
- Postnatal growth failure (length <-2 standard deviations score (SDS) by 24 months)
- Feeding difficulties and/or low BMI (<-2 standard deviations score (SDS) at 24 months)
- Prominent protruding forehead (age 1-3 years)
- Body asymmetry
Until 2005, the only known molecular cause of Russell-Silver syndrome was maternal uniparental disomy of chromosome 7, which is present in less than 10% of cases. As such, many older teens and young adults may have a clinical Russell-Silver syndrome diagnosis with no confirmed molecular testing.
What types of tests may provide additional information for an undiagnosed person with features of Russell-Silver syndrome?
The following types of evaluations may be recommended for a person suspected of having Russell-Silver syndrome (Russell-Silver syndrome):
- karyotype and chromosomal microarrays to exclude other disorders
- evaluation of patient and parental blood for uniparental disomy of chromosome 7 and hypomethylation (a form of DNA modification) at chromosome 11p15 by polymerase chain reaction (PCR)
- hand x-ray to determine bone age – findings may include:
- delayed bone age
- white or ivory appearance on parts of the fingers (ivory epiphyses of distal phalanges)
- short middle bone of the pinky (small middle phalanx of the fifth finger), which occurs in 80% of people with Russell-Silver syndrome
- extra bone formation at the base of the pointer finger (pseudoepiphyses at base of second metacarpal)[4]
Russell-Silver syndrome life expectancy
The long-term outlook associated with Russell-Silver syndrome is generally good 18, but may depend on how severely affected a person is and whether complications arise. People with Russell-Silver syndrome may face challenges from birth to adulthood. While some people with Russell-Silver syndrome believe that they are not at risk for associated health issues once they reach their adult height, recent research has shown there may be increased risks for certain health issues in adulthood. These possible risks include 19:
- Metabolic syndrome
- Uterine and vaginal dysgenesis (in females with 11p15-related Russell-Silver syndrome)
- Gonadal hypofunction or testicular cancer in males
- Low muscle mass or low bone mineral density
- Myoclonus dystonia (in people with chromosome 7-related Russell-Silver syndrome)
Russell-Silver syndrome treatment
Russell-Silver syndrome leads to a wide spectrum of abnormal physical characteristics and functional abnormalities. Multidisciplinary follow up and early, specific, intervention are necessary for optimum management of this group of patients.
Patients with Russell-Silver syndrome should receive multidisciplinary care in a center of expertise in Russell-Silver syndrome in coordination with their local centre. The multidisciplinary team should be composed of paediatric subspecialists such as an endocrinologist (coordinator), gastroenterologist, dietician, clinical geneticist, craniofacial team, orthopaedic surgeon, neurologist, speech and language therapist and psychologist.
Early feeding and nutritional support
The typical neonate with Russell-Silver syndrome has length standard deviations score (SDS) below weight standard deviations score (SDS); but after birth, due to poor appetite, feeding difficulties and gastrointestinal problems, weight standard deviations score (SDS) drops below the length standard deviations score (SDS) 11. Over time, progressive failure to thrive can result in a calorie-related length deficit 11.
Feeding difficulties and failure to thrive are considerably more frequent in patients with Russell-Silver syndrome than in children with small for gestational age (SGA) but not Russell-Silver syndrome 11. Failure to thrive in children with Russell-Silver syndrome is probably due to a combination of factors, including feeding difficulties (poor appetite, oromotor issues and the resulting low caloric intake) as well as functional and structural gastrointestinal problems. Digestive problems or malnutrition occur in over 70% of patients with Russell-Silver syndrome 20, including severe gastrooesophageal reflux in 55%, which often results in persistent vomiting after the age of 1 year. Constipation is also common, particularly after age 2 years 20. Cyproheptadine used as an appetite stimulant improves weight gain in other paediatric conditions 21; however, specific studies of its use in Russell-Silver syndrome are needed before it can be recommended in these patients.
The main therapeutic goals for the first 2 years of life in patients with Russell-Silver syndrome are nutritional support, prevention of hypoglycaemia and recovery of any calorie-related length or height deficit, which should be addressed before initiation of growth hormone (GH) therapy. However, careful monitoring is needed, especially during nonvolitional feeding, because rapid catch-up weight gain in children born small for gestational age has been associated with an increased risk of metabolic and cardiovascular disease in later life 22.
Children with Russell-Silver syndrome have an abnormal body composition with low muscle mass, and are typically light for their length or height 23. The target for healthy nutritional status is narrow, and is dependent on individual innate muscle mass and even slight overnourishment (for example, weight >90% of ideal weight for length or height) can rapidly increase relative fat mass. Suggested targets for children aged 2–4 years preparing for growth hormone (GH) therapy are: weight 75–85% of the 50th centile weight for length or height and/or BMI 12–14 kg/m2, using height measurements on the longer side if notable leg length discrepancy is found. A weight below 70% of the ideal weight for length or height compromises growth velocity, despite growth hormone (GH) treatment. For children >4 years old, the optimal target BMI will depend on their muscle mass. Two groups of patients are exceptions to this observation. Firstly, in patients with 11p15 LOM who have a very low muscle mass and considerable body asymmetry, a lower BMI might be adequate (11–12 kg/m²). Secondly, for patients with upd(7)mat with near normal muscle mass, a higher BMI might be acceptable (14–15 kg/m²).
Recommendations
- For nutritional goals in the first years of life, we recommend nutritional repletion* with awareness of possible hazards of rapid postnatal catch-up leading to subsequent increased metabolic risk.
- Ask for and/or screen early for gut dysmotility (gastrooesophageal reflux, delayed gastric emptying and constipation) in all children.
- Diagnose and treat any oromotor and/or sensory issues that affect oral intake of food.
- In patients with severe feeding failure who are unresponsive to standard care, anatomical or functional disorders of the gastrointestinal tract, such as malrotation, should be excluded.
- Avoid enteral feeding by nasogastric or gastrostomy tube in a child capable of eating where there is adequate nutritional repletion.
- In cases of extreme feeding difficulties or gastrooesophageal reflux, consider enteral feeding by gastrostomy tube (with or without fundoplication) or low-profile transgastric jejunostomy as a last resort to protect against hypoglycaemia and/or malnutrition.
- In the case of enteral feeding, prevent excessive weight gain in both volitionally and nonvolitionally fed children.
*Low muscle mass makes typical BMI targets excessive in this population. Targets currently used in some centres include: Waterlow score 75–85% 24; weight-for-length standard deviations score −2 to −1 in first year of life; BMI target standard deviations score between −2 to −1 after first year of life.
Prevention of hypoglycaemia
Young children with Russell-Silver syndrome, particularly under age 5 years, have low muscle and liver mass, a disproportionately large brain-for-body size and feeding difficulties, all of which increase their risk of fasting hypoglycaemia and its potential neurocognitive consequences. The incidence of hypoglycaemia in these children is approximately 27% 13, with a high frequency of spontaneous, asymptomatic nocturnal hypoglycaemia 25.
Monitoring of levels of urinary ketones is usually effective in pre-empting hypoglycaemia related to fasting, activity or illness. This measurement can be used to determine the ‘safe fasting time’ for a child, which will change with age. Night time hypoglycaemia can be prevented by adding either high molecular weight glucose polymer (for infants under 10 months) or uncooked corn starch (for older infants and children particularly at risk) to the last evening feed. Dental hygiene is important as complex carbohydrates can promote cavities 26. Severe, non-fasting and non-ketotic hypoglycaemia should always be identified and investigated further.
For episodes of preoperative fasting or febrile illness, intravenous glucose (10% dextrose) might be required. Children with Russell-Silver syndrome might need longer periods of gut rest than children with small for gestational age but not Russell-Silver syndrome before oral or enteral feeding because of their gut dysmotility and intrinsic feeding defects. Before discharge, it is advisable to achieve an absence of ketonuria following at least 12 hours of feeding, without intravenous support. When hypoglycaemia remains a problem, early growth hormone (GH) therapy should be considered 27.
Recommendations
- Monitoring for ketonuria at home is useful to determine which children need intervention for impending hypoglycaemia.*
- Develop a plan with the child’s local paediatrician and emergency room for rapid admission and intravenous dextrose treatment when the child is ill.
- Admit children with Russell-Silver syndrome to hospital early in the course of an illness associated with ketonuria or hypoglycaemia and do not discharge them until they are metabolically stable and can be adequately fed.
- Glucagon is not recommended to correct hypoglycaemia, because of poor glycogen stores and limited ability for gluconeogenesis.
- Provide parents with an emergency guidance plan for illnesses.
- Teach parents how to recognize signs of hypoglycaemia, measure ketones, determine the ‘safe fasting time’ for their child, prevent hypoglycaemia using complex carbohydrates and avoid fasting outside a controlled environment.
- In severe cases of fasting hypoglycaemia, where other causes have been excluded and if other alternatives are ineffective, consider:
- Early start of growth hormone (GH) therapy to support glucose sources (increase in muscle mass and gluconeogenesis)
- Placement of a gastrostomy tube or jejunostomy tube.
*Children with a history of hypoglycaemia who do not have an appropriate ketone response will require formal fasting studies.
Surgery and anesthesia
Any surgery should be carefully planned due to the increased risk of fasting hypoglycemia in patients with Russell-Silver syndrome 28. As a result of their diminished weight-for-height ratio, low BMI and large head, young patients with Russell-Silver syndrome are at risk of hypothermia in a cool operating room 29. Many children with Russell-Silver syndrome also have abnormal tooth distribution and a small mandible, which affects airway visualization and intubation 30. Finally, young children with Russell-Silver syndrome who are malnourished might not heal well following surgery 31.
Recommendations
- Review issues related to Russell-Silver syndrome with the anaesthetist and surgeon in advance.
- Consider admission the night before surgery for early administration of intravenous dextrose before surgery to avoid ketonuria and hypoglycemia.
- Schedule first on the surgical list where possible.
- Monitor blood glucose and administer intravenous dextrose during and after surgery. Do not discharge until ketonuria is absent and the child can sustain themselves on oral or enteral feeding.
- Follow the intraoperative temperature maintenance protocol appropriate for the patient’s size, not age.
- Delay elective surgery until the child is adequately nourished.
- Be aware of the high risk of malnutrition after surgery and follow appropriate guidelines.
Growth hormone treatment
Data on adult height in untreated patients with Russell-Silver syndrome are limited; however, Russell-Silver syndrome is associated with a significant reduction in adult height (around −3 standard deviations score) 32. Russell-Silver syndrome is an indication for growth-promoting growth hormone treatment under the small for gestational age registered licence. It is worth noting that Russell-Silver syndrome was the only syndrome to be included in the clinical trials of growth hormone in short children born small for gestational age that led to the US FDA and the European Medicines Agency (EMA) small for gestational age indications for growth hormone therapy in 2001 and 2003, respectively 33. The results of these clinical trials, therefore, validate the use of growth hormone for patients with Russell-Silver syndrome.
Overall, clinical trials of growth hormone treatment in patients with small for gestational age (in which patients with Russell-Silver syndrome were included) demonstrated a satisfactory growth response and an increase in predicted adult height of 7–11 cm at pharmacological doses of growth hormone 33. However, the response in patients with Russell-Silver syndrome was not investigated until a Dutch longitudinal study analysed the response to growth hormone in 62 children with a clinical diagnosis of Russell-Silver syndrome using the NH-CSS compared with 227 short, non-syndromic children born small for gestational age. Overall, the study showed a similar response to growth hormone in patients with Russell-Silver syndrome compared with non-Russell-Silver syndrome children born small for gestational age (mean total height gains of 1.30 standard deviations score and 1.26 standard deviations score, respectively); however, the final adult height attained in patients with Russell-Silver syndrome was lower (mean adult height −2.17 standard deviations score versus −1.65 standard deviations score for non-Russell-Silver syndrome children born small for gestational age) 23. Although the mean height at the start of growth hormone treatment in patients with Russell-Silver syndrome was statistically significantly lower than in those without Russell-Silver syndrome, it was shown that patients with all Russell-Silver syndrome subtypes benefited from growth hormone treatment, with a trend towards increased height gain in patients with upd(7)mat or clinical Russell-Silver syndrome. In addition, some interim 34 and long-term 35 studies have focused on the response to growth hormone specifically in patients with Russell-Silver syndrome, albeit without a control group of non-Russell-Silver syndrome short children born small for gestational age. Strong predictors of the short-term and long-term responses to growth hormone were age and height standard deviations score at the start of growth hormone treatment (both inversely related) 35. However, the study by Rakover et al. 36 of 33 patients with Russell-Silver syndrome lacked data on adult height. Mean total height gain ranged from +1.2 to +1.4 standard deviations score for growth hormone doses of 35–70 µg/kg per day, which is similar to that achieved in patients with non-syndromic small for gestational age 33. In 2007, an small for gestational age consensus statement advocated early treatment with growth hormone for children born small for gestational age, including those with Russell-Silver syndrome, who had severe growth retardation (height standard deviations score ≤2.5; age 2–4 years; dose 35–70 µg/kg per day) 37.
Additional potential benefits of growth hormone treatment are increases in appetite, lean body mass and muscle power, which can result in improved mobility 38. In patients with Prader–Willi syndrome, another imprinting disorder, growth hormone treatment started in infancy results in increased lean body mass and motor development, as well as decreased fat mass 39; consequently, growth hormone treatment is now recommended from infancy in this condition. Children with Russell-Silver syndrome who are <2 years old typically present with low muscle mass and hypotonia, similarly to patients with Prader–Willi syndrome 40, and could also benefit from early growth hormone treatment. Further studies are necessary to investigate this option in patients with Russell-Silver syndrome.
Classic growth hormone deficiency is neither a common nor a relevant cause of short stature in Russell-Silver syndrome, nor is it predictive of the response to growth hormone treatment in children born small for gestational age 41. Furthermore, given the risk of hypoglycaemia associated with fasting required for growth hormone testing, testing children with Russell-Silver syndrome might carry added risks.
For most children with Russell-Silver syndrome, an increase in height velocity of ≥3 cm per year is the lower limit of an effective response range 37. The growth response depends on the patient’s age, growth hormone dose, height deficit, rate of weight gain and confounding problems such as intercurrent illness and scoliosis.
Levels of insulin-like growth factor 1 (IGF1) in response to growth hormone treatment in patients with Russell-Silver syndrome are difficult to interpret. Children with 11p15 LOM have significantly higher IGF1 levels than children with upd(7)mat and other children born small for gestational age, which suggests an element of IGF1 resistance in patients with 11p15 LOM 42. Basal serum levels of IGF1 in the upper quartile of the normal age-related range or higher can be expected in children with Russell-Silver syndrome, especially those with 11p15 LOM 42. In children with 11p15 LOM, serum levels of insulin-like growth factor-binding protein 3 (IGFBP3) are also elevated 43. IGF1 levels might rise significantly above the reference range in children with Russell-Silver syndrome on standard doses of growth hormone 43. Further studies are needed to understand how best to use IGF1 and IGFBP3 serum levels to monitor growth hormone doses in children with Russell-Silver syndrome and IGF1 resistance.
Comprehensive reviews on the use of growth hormone in children born small for gestational age have concluded that growth hormone treatment seems to be safe and effective 44. Adverse effects due to growth hormone treatment are no more frequent in children with Russell-Silver syndrome than in those with non-syndromic small for gestational age 23and no specific precautions are advised.
Recommendations
- Defer growth hormone treatment until caloric deficits are addressed.
- Avoid growth hormone stimulation testing.
- Goals of growth hormone treatment are to improve body composition (especially lean body mass), psychomotor development and appetite, to reduce the risk of hypoglycemia, and to optimize linear growth.
- Treat with growth hormone as soon as possible; starting at age 2–4 years is adequate for the majority of patients; however, due consideration should be given to the exceptions listed below*.
- Start growth hormone at a dose of approximately 35 µg/kg per day. Use the lowest dose that results in catch-up growth.
- Terminate growth hormone therapy when height velocity is <2 cm per year over a 6-month period and bone age is >14 years (female patients) or >17 years (male patients).
- If response to growth hormone is poor, re-evaluate the underlying diagnosis, growth hormone dose, IGF1 response, adherence to therapy and other confounding systemic problems.
- Monitor circulating levels of IGF1 and IGFBP3 at least yearly during growth hormone treatment.
*Growth hormone treatment does not have a specific indication for Russell-Silver syndrome and is prescribed under the small for gestational age indication (height standard deviations score −2.5; age >2–4 years; dose 35–70 µg/kg per day) 37. Exemptions from the current small for gestational age licensed indication used in some centers include starting growth hormone therapy below the age of 2 years in case of: severe fasting hypoglycemia; severe malnutrition, despite nutritional support, which will lead to gastrostomy if no improvement is seen; and severe muscular hypotonia.
Bone age advancement and puberty
The published literature on the natural history of bone age progression in patients with Russell-Silver syndrome is limited. Early bone age delay is followed by rapid advancement typically at around 8–9 years of age 45, but sometimes much younger, especially in nonvolitionally overfed children. Onset of puberty is usually within the normal range (8–13 years in girls and 9–14 years in boys) 46 but at the younger end of the spectrum 32. Adrenarche can be early and aggressive in comparison with children born with non-Russell-Silver syndrome SGA, particularly in those with 11p15 LOM 47.
In patients with Russell-Silver syndrome and early adrenarche, the onset of central puberty might be earlier and the tempo faster than expected. In the past few decades, population studies analysing the timing of normal puberty observed a mean age of puberty onset of 9.7–10.0 years in girls 46. As a group, girls with Russell-Silver syndrome seem to start central puberty at a mean age of 9.1 years. This early puberty further accelerates bone age maturation, which leads to an attenuated pubertal growth spurt and compromised adult height. Children with upd(7)mat are likely to progress to central puberty at an even younger age than patients with Russell-Silver syndrome and 11p15 LOM (mean starting age 8.5 years in girls and 9.5 years in boys) (I. Netchine, unpublished work). A rapid increase in BMI might also exacerbate the tendency to early adrenarche and central puberty 48.
The window for effective growth hormone treatment seems to be shorter in patients with Russell-Silver syndrome than in non-Russell-Silver syndrome patients with small for gestational age. In a study comparing a cohort of patients with Russell-Silver syndrome and a cohort of patients born small for gestational age but without Russell-Silver syndrome, puberty started significantly earlier in the former (at 10.2 years versus 11.2 years in girls with Russell-Silver syndrome and non-Russell-Silver syndrome small for gestational age, respectively, and at 11.4 years versus 12.0 years in boys with Russell-Silver syndrome and non-Russell-Silver syndrome small for gestational age, respectively) 49. Furthermore, a steeper decline in height standard deviations score from the onset of puberty until adult height was seen in patients with Russell-Silver syndrome, which contributed to a lower adult height and a larger distance to target height than in non-Russell-Silver syndrome patients with small for gestational age. However, in 17 patients with Russell-Silver syndrome in this study, puberty was postponed for 2 years with gonadotropin-releasing hormone analogue (GnRHa) due to a low predicted adult height. The effect of GnRHa on final height has been analysed in a cohort of patients with small for gestational age, including patients with Russell-Silver syndrome 50. This analysis suggested that the combination of GnRHa, started at the initiation of puberty and continued for at least 2 years, along with growth hormone treatment, improves adult height in patients born small for gestational age with a poor adult height prognosis. A retrospective study of GnRHa treatment specifically in patients with Russell-Silver syndrome did not detect an effect of GnRHa on adult height, but this therapy was used in only 16 of 37 patients and was not standardized 35. Further studies are required to specifically look at its effects in patients with Russell-Silver syndrome.
Aromatase catalyses the rate-limiting step in the conversion of androstenedione to estrone and testosterone to estradiol. In patients with adrenarche with advancing bone age, but without central puberty, third-generation aromatase inhibitors (such as anastrozole) might be helpful in preventing rapid bone maturation, but are currently not licensed for growth disorders 51. An 18-month double-blind clinical trial is currently underway to study the efficacy and tolerance of treatment with anastrozole to slow bone maturation related to pathological adrenarche in patients with Russell-Silver syndrome and Prader–Willi syndrome 52.
Recommendations
- Monitor for signs of premature adrenarche, fairly early and accelerated central puberty, and insulin resistance.
- Monitor and anticipate acceleration of bone age especially from mid childhood.
- Consider personalized treatment with GnRHa for at least 2 years in children with evidence of central puberty (starting no later than age 12 years in girls and age 13 years in boys) to preserve adult height potential.
Long-term metabolic complications
Individuals born with a low birth weight are at increased risk of adult health problems including coronary heart disease 53, hypertension, dyslipidaemia, insulin resistance and obesity (the metabolic syndrome) 54. Studies of children born small for gestational age indicate that those who have rapid or disproportionate catch-up in weight are at particularly high risk 55.
Insulin resistance in young, pre-pubertal, children with Russell-Silver syndrome can be atypical and difficult to detect in the fasting state; however, impaired glucose tolerance can be confirmed on formal oral glucose tolerance testing 56. Insulin resistance becomes more classic in the pubertal or post-pubertal age groups with elevation in fasting levels of glucose and insulin, and possibly the development of type 2 diabetes mellitus 57.
Overall, growth hormone therapy seems to have positive metabolic effects in children born small for gestational age 58, but specific data on such effects in Russell-Silver syndrome are lacking. Many studies of long-term growth hormone treatment in children born small for gestational age have shown positive outcomes, including increased lean body mass, reduced fat mass, decreased blood pressure and an improved lipid profile 59, which might last after discontinuation of therapy 60.
In a study of 110 children born small for gestational age treated with growth hormone, those with the highest baseline levels of IGF1 were the least insulin sensitive. Gains in height and IGF1 response were positively associated with insulin secretion 61. In Russell-Silver syndrome, children with 11p15 LOM seem to be at a higher metabolic risk than children who have upd(7)mat and other children born small for gestational age due to poor muscle mass and raised levels of IGF1 40. Further research is, therefore, required on the long-term effects of growth hormone therapy on body composition and metabolic parameters in Russell-Silver syndrome and its various genotypes.
Recommendations
- Avoid excessive or rapid weight gain to prevent increased insulin resistance, which is associated with early and rapidly advancing adrenarche, early central puberty, and, in girls, a future risk of developing polycystic ovary syndrome.
- Raise awareness among gastroenterologists, dieticians, neonatologists, paediatricians and primary health-care providers of the importance of not overfeeding this group of children.
- Advise parents, grandparents and care-givers about the risk of insulin resistance associated with intrauterine growth retardation and overfeeding.
- Screen for physical and biochemical indicators of insulin resistance during growth hormone treatment, especially in children with low muscle mass and high baseline levels of IGF1.
- In patients with clinical signs of insulin resistance, consider formal assessment of insulin sensitivity with a 2-h oral glucose tolerance test including measurement of insulin and C-peptide levels.
- Advocate a healthy diet and lifestyle in older children and young adults with particular emphasis on protein calorie balance and regular exercise to avoid disproportionate weight gain, particularly after discontinuation of growth hormone treatment.
Neurocognitive problems
Motor and speech delay are common in children with Russell-Silver syndrome 11. Motor delay might be related to reduced muscle bulk and fairly large head size. Verbal dyspraxia and more global developmental delay or learning difficulties, usually mild, have been described in some children with Russell-Silver syndrome, particularly those with upd(7)mat 62. Autistic spectrum disorder has also been reported more frequently in this subgroup than in the other subgroups of Russell-Silver syndrome 40. Myoclonus dystonia in patients with upd(7)mat is probably associated with altered expression of the paternally expressed SGCE on chromosome 7q21 13.
Recommendations
- Refer infants and children with Russell-Silver syndrome for a developmental assessment when necessary to ensure appropriate intervention as early as possible.
- In patients with upd(7)mat, check for symptoms of myoclonus dystonia at each clinical appointment and refer early to a pediatric neurologist if required.
- Monitor children with upd(7)mat for signs of verbal or oromotor dyspraxia and/or signs of autistic spectrum disorders.
- Inform parents about increased risk of speech, oromotor and learning disabilities (especially in those with upd(7)mat).
- Follow up school-age children for any learning difficulties, psychosocial challenges and/or cognitive delay, to enable appropriate intervention.
Orthopedic problems
Orthopedic problems seen in association with Russell-Silver syndrome include limb or body asymmetry, scoliosis, hip dysplasia and hand and/or foot anomalies.
Limb asymmetry can affect the arms, legs or both. In seven patients with clinically diagnosed Russell-Silver syndrome, limb length discrepancy was not significantly affected by growth hormone treatment 63. Limb lengthening surgery performed to equalize limb lengths in patients with Russell-Silver syndrome has shown positive results 64.
Scoliosis has been reported in 9–36% of individuals with Russell-Silver syndrome 13. The causal relationship to leg length asymmetry is not clear146,147. Associated back pain has been reported inconsistently 65. growth hormone therapy might be associated with worsening of existing scoliosis; however, causality has not been established (Farber, R. S. & Kerrigan, J. R. The multiple indications for growth hormone treatment of pediatric patients. Pediatr. Ann. 35, 926–932; 2006.()). A study in a large group of children with Prader–Willi syndrome (an imprinting disorder with clinical features that overlap with those of Russell-Silver syndrome: growth failure; infant hypotonia; early feeding difficulties; and an increased risk of scoliosis) has clearly shown that growth hormone therapy does not influence onset and progression of scoliosis 66; however, specific studies are required to determine whether growth hormone therapy modifies the risk of scoliosis in patients with Russell-Silver syndrome.
Recommendations
- Where necessary, refer to a paediatric orthopaedic surgeon for collaborative management of body asymmetry, limb length discrepancy and scoliosis.
- Routinely examine all patients with Russell-Silver syndrome for scoliosis.
- Before initiation of growth hormone therapy, refer patients with scoliosis to the orthopaedic team and monitor while receiving growth hormone.
- Evaluate leg length asymmetry regularly and consider orthopedic management if necessary.
Maxillofacial abnormalities
Russell-Silver syndrome is characterized by craniofacial disproportion, which results in a triangular-shaped face 30. Delayed dental eruption, microdontia, absence of secondary teeth and blunted condyles have all been reported in patients with Russell-Silver syndrome 67.
In our experience, the upper jaw arch is frequently narrow and crowded, but crowding might be severe in the lower arch, with displacement of lower incisors into a lingual position. Micrognathia is frequent, with lack of mandibular growth, which results in a small, pointed chin and an overbite. Children with notable facial asymmetry might have a crossbite that impairs normal chewing. Velopharyngeal insufficiency with or without a submucous cleft is quite common in patients with 11p15 LOM Russell-Silver syndrome 13. Otitis media is frequent in young children with Russell-Silver syndrome7 and seems to be improved by orthodontic treatment 13.
Orthodontic intervention in children with Russell-Silver syndrome can help normalize oropharangeal function and facial appearance. An experienced craniofacial team, including orthodontists, plastic surgeons and ear, nose and throat surgeons is ideal. Multiple orthodontic techniques have been used successfully 68. Currently, rapid palatal expansion is the most effective technique to change the shape of the face 69.
Many patients with Russell-Silver syndrome report excessive daytime fatigue, snoring and/or disrupted sleep. However, data are very limited regarding sleep problems, including sleep disordered breathing, in association with Russell-Silver syndrome. A retrospective study identified mild sleep disordered breathing in 74% of patients with Russell-Silver syndrome (not exacerbated with growth hormone therapy) 70. Further studies are necessary.
Recommendations
- Develop a referral relationship with a maxillofacial team or orthodontist who has experience caring for patients with Russell-Silver syndrome.
- Refer patients to the maxillofacial team for assessment after eruption of primary dentition when necessary.
- Encourage early orthodontic intervention and compliance with follow-up.
- Screen for symptoms of sleep disordered breathing (such as snoring, apneas, excessive daytime fatigue, disrupted sleep and agitation).
- Refer patients with suspected sleep disordered breathing to the appropriate specialist for evaluation of obstructive sleep apnea.
Other congenital anomalies
Congenital anomalies have been described in a minority of patients with Russell-Silver syndrome, particularly those with 11p15 LOM. Genital abnormalities, including cryptorchidism and hypospadias, occur frequently in boys 71. Mayer–Rokitansky–Kuster–Hauser syndrome in female patients is characterized by congenital hypoplasia or aplasia of the uterus and upper part of the vagina 71. Structural renal anomalies 72 and congenital heart defects 72 have also been reported.
Recommendations
- Investigate genital abnormalities in boys.
- Investigate girls with primary amenorrhea for Mayer–Rokitansky–Kuster–Hauser syndrome.
Adulthood
Very little information exists in the literature regarding the long-term natural history of Russell-Silver syndrome. The majority of individuals with Russell-Silver syndrome are not routinely followed up, and the small numbers of adults reported have few medical problems. However, it is well recognized that being small for gestational age at birth with accelerated gain in weight for length, particularly during early life, increases the risk of metabolic problems in adulthood 55. Medical problems reported in adult patients with 11p15 LOM include hypertension, dilated cardiomyopathy, type 2 diabetes mellitus, hypercholesterolemia, fatty liver infiltration, elevated glucose levels and raised HbA1c levels 57; however, these reports might not be representative of the population as a whole.
Recommendations
- Consider medical follow-up of adolescents and young adult patients with Russell-Silver syndrome or develop collaboration with a general or internal medicine team for follow-up.
- Avoid losing contact with adult patients with Russell-Silver syndrome, to facilitate their participation in, and potential benefit from, future clinical research.
- Russell-Silver syndrome. https://ghr.nlm.nih.gov/condition/russell-silver-syndrome[↩][↩][↩][↩]
- Saal HM. Russell-Silver Syndrome. 2002 Nov 2 [Updated 2011 Jun 2]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2019. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1324[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Netchine I and Salem J. Russell Silver syndrome. National Organization for Rare Disorders. 2017; http://rarediseases.org/rare-diseases/russell-silver-syndrome[↩][↩][↩][↩][↩][↩][↩]
- Russell-Silver syndrome. https://www.magicfoundation.org/Growth-Disorders/Russell-Silver-Syndrome/[↩]
- Wakeling EL. Silver-Russell syndrome. Arch Dis Child. December, 2011; 96(12):1156-1161.[↩]
- Feuk L et. al. Absence of a Paternally Inherited FOXP2 Gene in Developmental Verbal Dyspraxia. Am J Hum Genet. November, 2006; 79(5):965-972.[↩]
- Kelly Y C Lai, David Skuse, Richard Stanhope, Peter Hindmarsh. Cognitive abilities associated with the Silver-Russell syndrome. Archives of Disease in Childhood. 1994; 71:490-496. [↩]
- Katarina Hannula, Juha Kere, Sinikka Pirinen, Christer Holmberg, Marita Lipsanen-Nyman. Do patients with maternal uniparental disomy for chromosome 7 have a distinct mild Silver-Russell phenotype?. J Med Genet. 2001; 38:273-278[↩]
- Sonnappa S, Prescott K, Adler B, Dinwiddie R, and Wallis C. Cystic fibrosis and Russell-Silver syndrome in a child with maternal isodisomy of chromosome 7. Pediatr Pulmonol. August 2005; 40(2):166-168. https://www.ncbi.nlm.nih.gov/pubmed/15965898[↩][↩]
- Hehr U, Dörr S, Hagemann M, Hansmann I, Preiss U, and Brömme S. Silver-Russell syndrome and cystic fibrosis associated with maternal uniparental disomy 7. Am J Med Genet. March 20 2000; 91(3):237-239. https://www.ncbi.nlm.nih.gov/pubmed/10756351[↩][↩]
- Netchine, I. et al. 11p15 imprinting center region 1 loss of methylation is a common and specific cause of typical Russell–Silver syndrome: clinical scoring system and epigenetic-phenotypic correlations. J. Clin. Endocrinol. Metab. 92, 3148–3154; 2007.[↩][↩][↩][↩][↩]
- Wakeling EL, Brioude F, Lokulo-Sodipe O, et al. Diagnosis and management of Silver-Russell syndrome: first international consensus statement. Nat Rev Endocrinol. February, 2017; 13(2):105-124. https://www.nature.com/articles/nrendo.2016.138[↩][↩][↩][↩]
- Wakeling, E. L. et al. Epigenotype–phenotype correlations in Silver–Russell syndrome. J. Med. Genet. 47, 760–768; 2010.[↩][↩][↩][↩][↩][↩]
- Demars, J. & Gicquel, C. Epigenetic and genetic disturbance of the imprinted 11p15 region in Beckwith–Wiedemann and Silver–Russell syndromes. Clin. Genet. 81, 350–361; 2012.[↩]
- Brioude, F. et al. CDKN1C mutation affecting the PCNA-binding domain as a cause of familial Russell Silver syndrome. J. Med. Genet. 50, 823–830; 2013.[↩]
- Begemann, M. et al. Paternally inherited IGF2 mutation and growth restriction. N. Engl. J. Med. 373, 349–356; 2015.[↩]
- Azzi S, Salem J, Thibaud N, et al. A prospective study validating a clinical scoring system and demonstrating phenotypical-genotypical correlations in Silver-Russell syndrome. J Med Genet. 2015;52(7):446-53. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4501172/[↩][↩]
- Silver-Russell syndrome. https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=en&Expert=813[↩]
- Russell Silver Syndrome. https://www.magicfoundation.org/Growth-Disorders/Russell-Silver-Syndrome/[↩]
- Marsaud, C., Rossignol, S., Tounian, P., Netchine, I. & Dubern, B. Prevalence and management of gastrointestinal manifestations in Silver–Russell syndrome. Arch. Dis. Child 100, 353–358; 2015.[↩][↩]
- Chinuck, R., Dewar, J., Baldwin, D. R. & Hendron, E. Appetite stimulants for people with cystic fibrosis. Cochrane Database Syst. Rev. 7, CD008190; 2014.[↩]
- Ezzahir, N. et al. Time course of catch-up in adiposity influences adult anthropometry in individuals who were born small for gestational age. Pediatr. Res. 58, 243–247; 2005.[↩]
- Smeets, C. C., Zandwijken, G. R., Renes, J. S. & Hokken-Koelega, A. C. Long-term results of GH treatment in Silver–Russell syndrome (SRS): do they benefit the same as non-SRS short-SGA? J. Clin. Endocrinol. Metab. 101, 2105–2112; 2016.[↩][↩][↩]
- Waterlow, J. C. Classification and definition of protein-calorie malnutrition. BMJ 3, 566–569; 1972.[↩]
- Azcona, C. & Stanhope, R. Hypoglycaemia and Russell–Silver syndrome. J. Pediatr. Endocrinol. Metab. 18, 663–670; 2005.[↩]
- O’Connell, A. C., O’Connell, S. M., O’Mullane, E. & Hoey, H. M. Oral health of children born small for gestational age. Ir. Med. J. 103, 275–278; 2010.[↩]
- Boonstra, V. H. et al. Food intake of children with short stature born small for gestational age before and during a randomized GH trial. Horm. Res. 65, 23–30; 2006.[↩]
- Tomiyama, H., Ibuki, T., Nakajima, Y. & Tanaka, Y. Late intraoperative hypoglycemia in a patient with Russell–Silver syndrome. J. Clin. Anesth. 11, 80–82; 1999.[↩]
- Scarlett, M. D. & Tha, M. W. Russell–Silver syndrome: anaesthetic implications and management. West Indian Med. J. 55, 127–129; 2006.[↩]
- Kotilainen, J. et al. Craniofacial and dental characteristics of Silver–Russell syndrome. Am. J. Med. Genet. 56, 229–236; 1995.[↩][↩]
- Canada, N. L., Mullins, L., Pearo, B. & Spoede, E. Optimizing perioperative nutrition in pediatric populations. Nutr. Clin. Pract. 31, 49–58; 2016.[↩]
- Wollmann, H. A., Kirchner, T., Enders, H., Preece, M. A. & Ranke, M. B. Growth and symptoms in Silver–Russell syndrome: review on the basis of 386 patients. Eur. J. Pediatr. 154, 958–968; 1995.[↩][↩]
- Ranke, M. B. & Lindberg, A. Height at start, first-year growth response and cause of shortness at birth are major determinants of adult height outcomes of short children born small for gestational age and Silver–Russell syndrome treated with growth hormone: analysis of data from KIGS. Horm. Res. Paediatr. 74, 259–266; 2010.[↩][↩][↩]
- Jensen, R. B. et al. A randomised controlled trial evaluating IGF1 titration in contrast to current GH dosing strategies in children born small for gestational age: the North European Small-for-Gestational-Age Study. Eur. J. Endocrinol. 171, 509–518; 2014.[↩]
- Binder, G. et al. Adult height and epigenotype in children with Silver–Russell syndrome treated with GH. Horm. Res. Paediatr. 80, 193–200; 2013.[↩][↩][↩]
- Rakover, Y. et al. Growth hormone therapy in Silver Russell syndrome: 5 years experience of the Australian and New Zealand Growth database (OZGROW). Eur. J. Pediatr. 155, 851–857; 1996.[↩]
- Clayton, P. E. et al. Management of the child born small for gestational age through to adulthood: a consensus statement of the International Societies of Pediatric Endocrinology and the Growth Hormone Research Society. J. Clin. Endocrinol. Metab. 92, 804–810; 2007.[↩][↩][↩]
- Schweizer, R., Martin, D. D., Schonau, E. & Ranke, M. B. Muscle function improves during growth hormone therapy in short children born small for gestational age: results of a peripheral quantitative computed tomography study on body composition. J. Clin. Endocrinol. Metab. 93, 2978–2983; 2008.[↩]
- Myers, S. E. et al. Two years of growth hormone therapy in young children with Prader–Willi syndrome: physical and neurodevelopmental benefits. Am. J. Med. Genet. A 143A, 443–448; 2007.[↩]
- Azzi, S. et al. A prospective study validating a clinical scoring system and demonstrating phenotypical-genotypical correlations in Silver–Russell syndrome. J. Med. Genet. 52, 446–453; 2015.[↩][↩][↩]
- Toumba, M., Albanese, A., Azcona, C. & Stanhope, R. Effect of long-term growth hormone treatment on final height of children with Russell–Silver syndrome. Horm. Res. Paediatr. 74, 212–217; 2010.[↩]
- Binder, G. et al. The endocrine phenotype in Silver–Russell syndrome is defined by the underlying epigenetic alteration. J. Clin. Endocrinol. Metab. 93, 1402–1407; 2008.[↩][↩]
- Dufourg, M. N. et al. Silver Russell syndrome: a cause of partial IGF1 resistance? Horm. Res. Paediatr. 84, 235; 2015.[↩][↩]
- Saenger, P., Czernichow, P., Hughes, I. & Reiter, E. O. Small for gestational age: short stature and beyond. Endocr. Rev. 28, 219–251; 2007.[↩]
- Vu-Hong, T. A. et al. Russell–Silver syndrome with 11p15 epimutation: analysis of growth, bone maturation, puberty and response to GH treatment on a large series of 101 patients. Horm. Res. 72, 447–448; 2009.[↩]
- Latronico, A. C., Brito, V. N. & Carel, J. C. Causes, diagnosis, and treatment of central precocious puberty. Lancet Diabetes Endocrinol. 4, 265–274; 2016.[↩][↩]
- Vu-Hong, T. A., Rossignol, S., Chivu, O., Cabrol, S. & Netchine, I. Aggressive adrenarche in Silver–Russell Syndrome compromises final height despite GH treatment. Horm. Res. Paediatr. 76 (Suppl. 2), 234; 2011.[↩]
- Verkauskiene, R., Petraitiene, I. & Albertsson Wikland, K. Puberty in children born small for gestational age. Horm. Res. Paediatr. 80, 69–77; 2013.[↩]
- Smeets, C. C., Zandwijken, G. R., Renes, J. S. & Hokken-Koelega, A. C. Long-term results of growth hormone treatment in Silver–Russell syndrome (SRS): do they benefit the same as non-SRS short-small for gestational age? J. Clin. Endocrinol. Metab. 101, 2105–2112; 2016.[↩]
- van der Steen, M. et al. Metabolic health in short children born small for gestational age treated with growth hormone and gonadotropin-releasing hormone analog: results of a randomized, dose-response trial. J. Clin. Endocrinol. Metab. 100, 3725–3734; 2015.[↩]
- Wit, J. M., Hero, M. & Nunez, S. B. Aromatase inhibitors in pediatrics. Nat. Rev. Endocrinol. 8, 135–147; 2012.[↩]
- US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT01520467 (2016).[↩]
- Barker, D. J., Winter, P. D., Osmond, C., Margetts, B. & Simmonds, S. J. Weight in infancy and death from ischaemic heart disease. Lancet 2, 577–580; 1989.[↩]
- Newsome, C. A. et al. Is birth weight related to later glucose and insulin metabolism? — A systematic review. Diabet Med. 20, 339–348; 2003.[↩]
- Leunissen, R. W., Kerkhof, G. F., Stijnen, T. & Hokken-Koelega, A. Timing and tempo of first-year rapid growth in relation to cardiovascular and metabolic risk profile in early adulthood. JAMA 301, 2234–2242; 2009.[↩][↩]
- Milovanovic, I. et al. SGA children with moderate catch-up growth are showing the impaired insulin secretion at the age of 4. PLoS ONE 9, e100337; 2014.[↩]
- Takenouchi, T., Awazu, M., Eggermann, T. & Kosaki, K. Adult phenotype of Russell–Silver syndrome: a molecular support for Barker–Brenner’s theory. Congenit. Anom. (Kyoto) 55, 167–169; 2015.[↩][↩]
- Sas, T., Mulder, P. & Hokken-Koelega, A. Body composition, blood pressure, and lipid metabolism before and during long-term growth hormone (GH) treatment in children with short stature born small for gestational age either with or without GH deficiency. J. Clin. Endocrinol. Metab. 85, 3786–3792; 2000.[↩]
- Willemsen, R. H. et al. Long-term effects of growth hormone (GH) treatment on body composition and bone mineral density in short children born small-for-gestational-age: six-year follow-up of a randomized controlled GH trial. Clin. Endocrinol. (Oxf.) 67, 485–492; 2007.[↩]
- Van Pareren, Y. et al. Effect of discontinuation of growth hormone treatment on risk factors for cardiovascular disease in adolescents born small for gestational age. J. Clin. Endocrinol. Metab. 88, 347–353; 2003.[↩]
- Jensen, R. B. et al. Baseline IGF-I levels determine insulin secretion and insulin sensitivity during the first year on growth hormone therapy in children born small for gestational age. Results from a North European Multicentre Study (NESGAS). Horm. Res. Paediatr. 80, 38–46; 2013.[↩]
- Lai, K. Y., Skuse, D., Stanhope, R. & Hindmarsh, P. Cognitive abilities associated with the Silver–Russell syndrome. Arch. Dis. Child 71, 490–496; 1994.[↩]
- Rizzo, V., Traggiai, C. & Stanhope, R. Growth hormone treatment does not alter lower limb asymmetry in children with Russell–Silver syndrome. Horm. Res. 56, 114–116; 2001.[↩]
- Goldman, V., McCoy, T. H., Harbison, M. D., Fragomen, A. T. & Rozbruch, S. R. Limb lengthening in children with Russell–Silver syndrome: a comparison to other etiologies. J. Child. Orthop. 7, 151–156; 2013.[↩]
- Abraham, E., Altiok, H. & Lubicky, J. P. Musculoskeletal manifestations of Russell–Silver syndrome. J. Pediatr. Orthop. 24, 552–564; 2004.[↩]
- de Lind van Wijngaarden, R. F. et al. Randomized controlled trial to investigate the effects of growth hormone treatment on scoliosis in children with Prader–Willi syndrome. J. Clin. Endocrinol. Metab. 94, 1274–1280; 2009.[↩]
- Bergman, A., Kjellberg, H. & Dahlgren, J. Craniofacial morphology and dental age in children with Silver–Russell syndrome. Orthod. Craniofac. Res. 6, 54–62; 2003.[↩]
- Kisnisci, R. S., Fowel, S. D. & Epker, B. N. Distraction osteogenesis in Silver Russell syndrome to expand the mandible. Am. J. Orthod. Dentofacial Orthop. 116, 25–30; 1999.[↩]
- Metzler, P., Obwegeser, J. A., Jacobsen, C. & Zemann, W. Anterior alveolar segmental osteodistraction with a bone-borne device: clinical and radiographic evaluation. J. Oral Maxillofac. Surg. 70, 2549–2558; 2012.[↩]
- Giabicani, E., Boule, M., Galliani, E. & Netchine, I. Sleep apneas in Silver Russell syndrome: a constant finding. Horm. Res. Paediatr. 84 (Suppl. 1), 262; 2015.[↩]
- Bruce, S., Hannula-Jouppi, K., Peltonen, J., Kere, J. & Lipsanen-Nyman, M. Clinically distinct epigenetic subgroups in Silver–Russell syndrome: the degree of H19 hypomethylation associates with phenotype severity and genital and skeletal anomalies. J. Clin. Endocrinol. Metab. 94, 579–587; 2009.[↩][↩]
- Bliek, J. et al. Hypomethylation of the H19 gene causes not only Silver–Russell syndrome (SRS) but also isolated asymmetry or an SRS-like phenotype. Am. J. Hum. Genet. 78, 604–614; 2006.[↩][↩]

{kind=link}