Contents
Tangier disease
Tangier disease is a rare inherited disorder characterized by significantly reduced levels of high-density lipoproteins (HDL) in the blood. HDL transports cholesterol and certain fats called phospholipids from the body’s tissues to the liver, where they are removed from the blood. HDL-cholesterol (HDL-C) is often referred to as the “good cholesterol” as it can facilitate the removal of cholesterol out of the walls of arteries, particularly the coronary (heart) arteries, reducing the chances of developing heart and blood vessel (cardiovascular) disease. Because people with Tangier disease have very low levels of HDL, they have a moderately increased risk of cardiovascular disease.
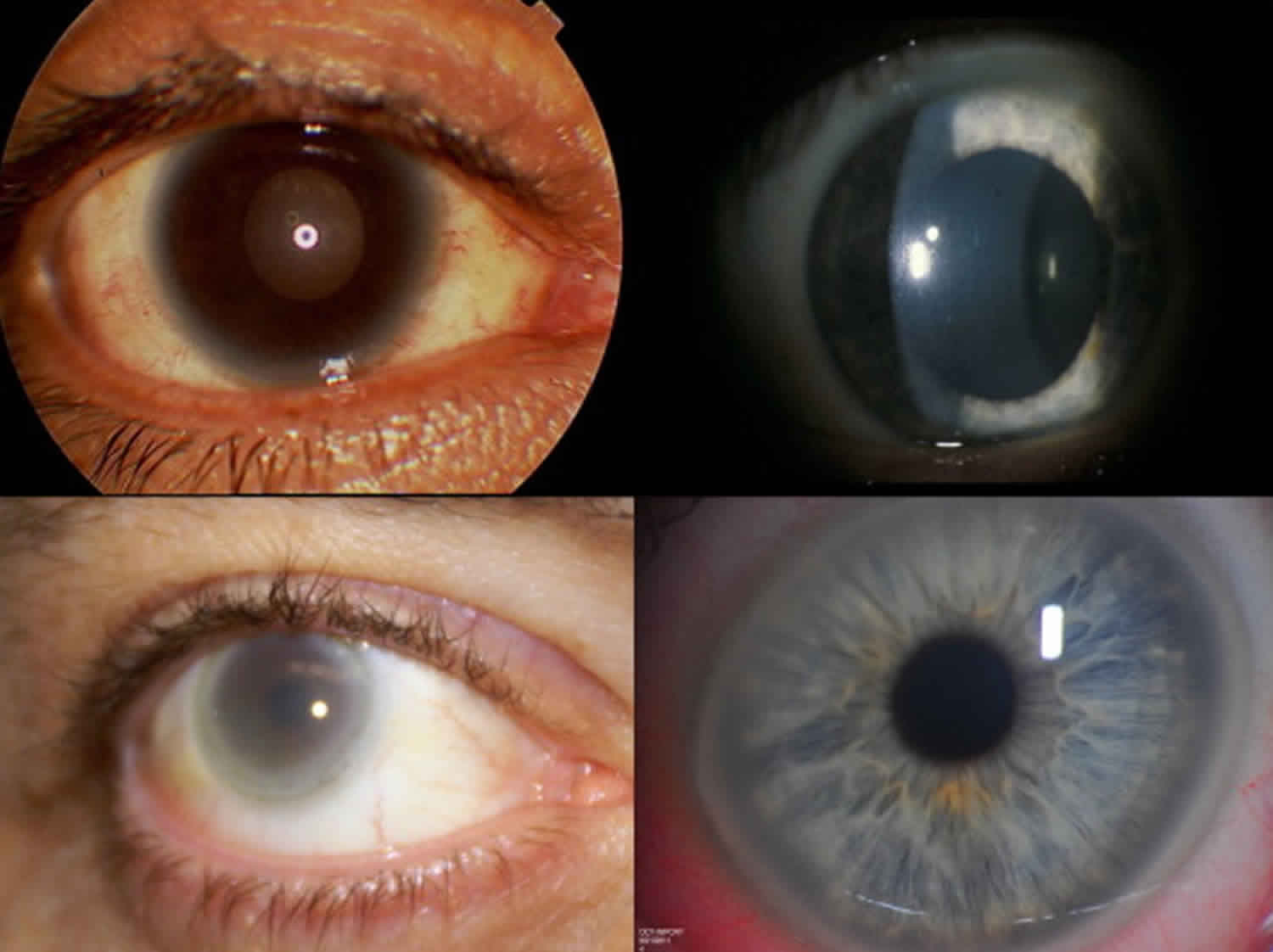
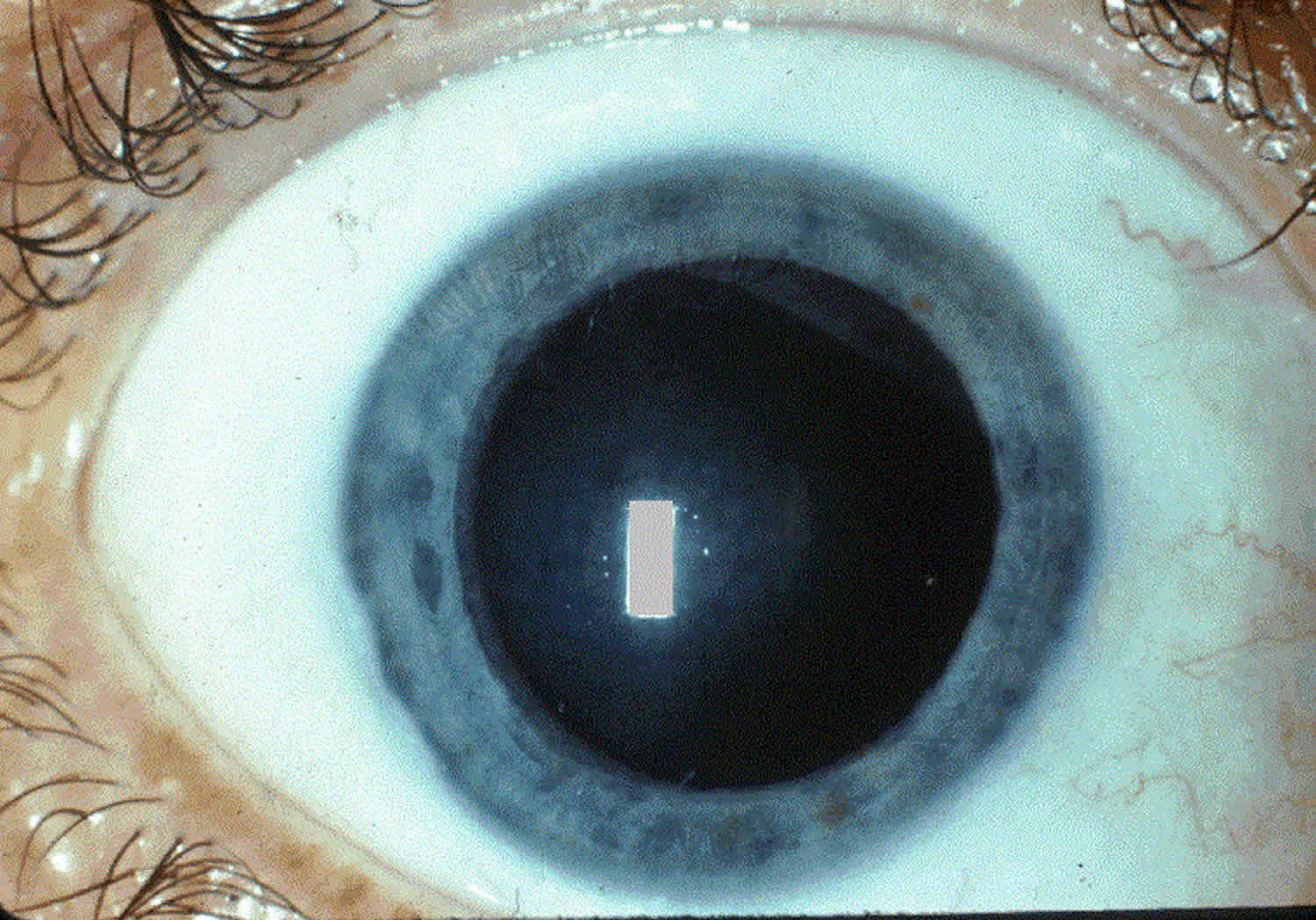
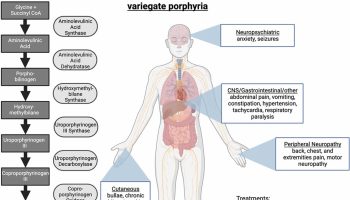
Classic features of Tangier disease include fatty accumulations that present as enlarged and yellow- or orange-colored tonsils, or enlarged liver (hepatomegaly), enlarged spleen (splenomegaly), or lymph nodes 1. Tangier disease may also be associated with an increased risk of cardiovascular disease, moderate elevation in triglycerides (hypertriglyceridemia), disturbances in nerve function (neuropathy), and rarely an opaqueness in the covering of the eye (corneal clouding) and type 2 diabetes. Affected individuals often develop atherosclerosis, which is an accumulation of fatty deposits and scar-like tissue in the lining of the arteries.
Tangier disease is an extremely rare disorder with approximately 100 cases identified worldwide. More cases are likely undiagnosed. This condition is named after an island off the coast of Virginia where the first affected individuals were identified.
Tangier disease was originally named after the location in which it was first discovered – Tangier Island in the Chesapeake Bay. Later, the disease was further characterized as more individuals were found to have the disease in other areas of the United States and around the globe.
Tangier disease is caused by mutations in the ABCA1 gene. It is inherited in an autosomal recessive pattern 2.
Figure 1. Tangier disease tonsils
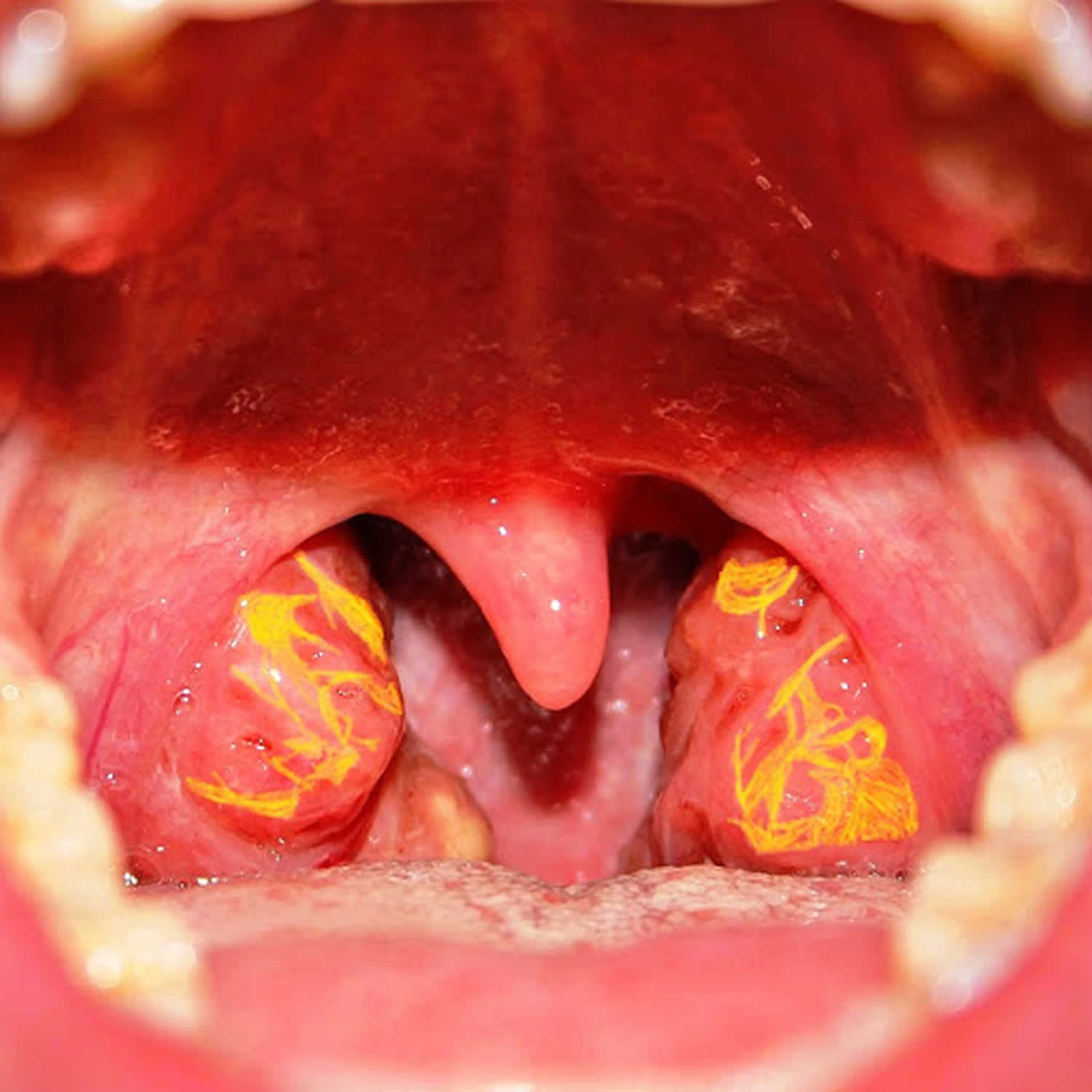
Figure 2. Tangier disease eye (diffuse hazy opacity of the right cornea in the patient with Tangier disease)
Tangier disease causes
Mutations in the ABCA1 (ATP-binding cassette transporter A1) gene cause Tangier disease. ABCA1 (ATP-binding cassette transporter A1) gene provides instructions for making a protein that releases cholesterol and phospholipids from cells. These substances are used to make HDL, which transports them to the liver. ABCA1 codes for a cell surface protein that is important in the process of reverse cholesterol transport, which allows the movement of cholesterol from inside the cell to apolipoprotein AI (apoA-I), the major protein constituent of HDL. When two ABCA1 gene mutations are present, the cell is no longer able to efflux cholesterol out of the cell to ApoA-I.
Mutations in the ABCA1 gene prevent the release of cholesterol and phospholipids from cells. As a result, these substances accumulate within cells, causing certain body tissues to enlarge and the tonsils to acquire a yellowish-orange color. A buildup of cholesterol can be toxic to cells, leading to impaired cell function or cell death. In addition, the inability to transport cholesterol and phospholipids out of cells results in very low HDL levels, which increases the risk of cardiovascular disease. These combined factors cause the signs and symptoms of Tangier disease.
Cholesterol is a soft, waxy substance found among the lipids (fats) in the bloodstream and in all the cells in our body. Cholesterol is essential to the formation of cell membranes, hormones, and other cellular functions. Cholesterol and other fats cannot dissolve in the blood and have to be transported to and from the cells by special carriers called lipoproteins. There are several types of lipoproteins that vary in density, but the most clinically important types are low-density lipoprotein (LDL) and HDL. About one-third to one-fourth of blood cholesterol is carried by HDL. HDL is commonly called the “good” cholesterol because it may be involved with the removal of cholesterol from the artery walls and ultimate disposal to the liver. On the other hand, LDL deposits cholesterol in the artery walls, causing the formation of cholesterol plaque.
Patients with Tangier disease have been found to have a severe reduction in HDL levels. Without sufficient HDL to help clear arteries of plaques, individuals are more susceptible to having excess lipid deposits on organs of the body such as liver, heart, spleen, lymph nodes, and brain.
Tangier disease inheritance pattern
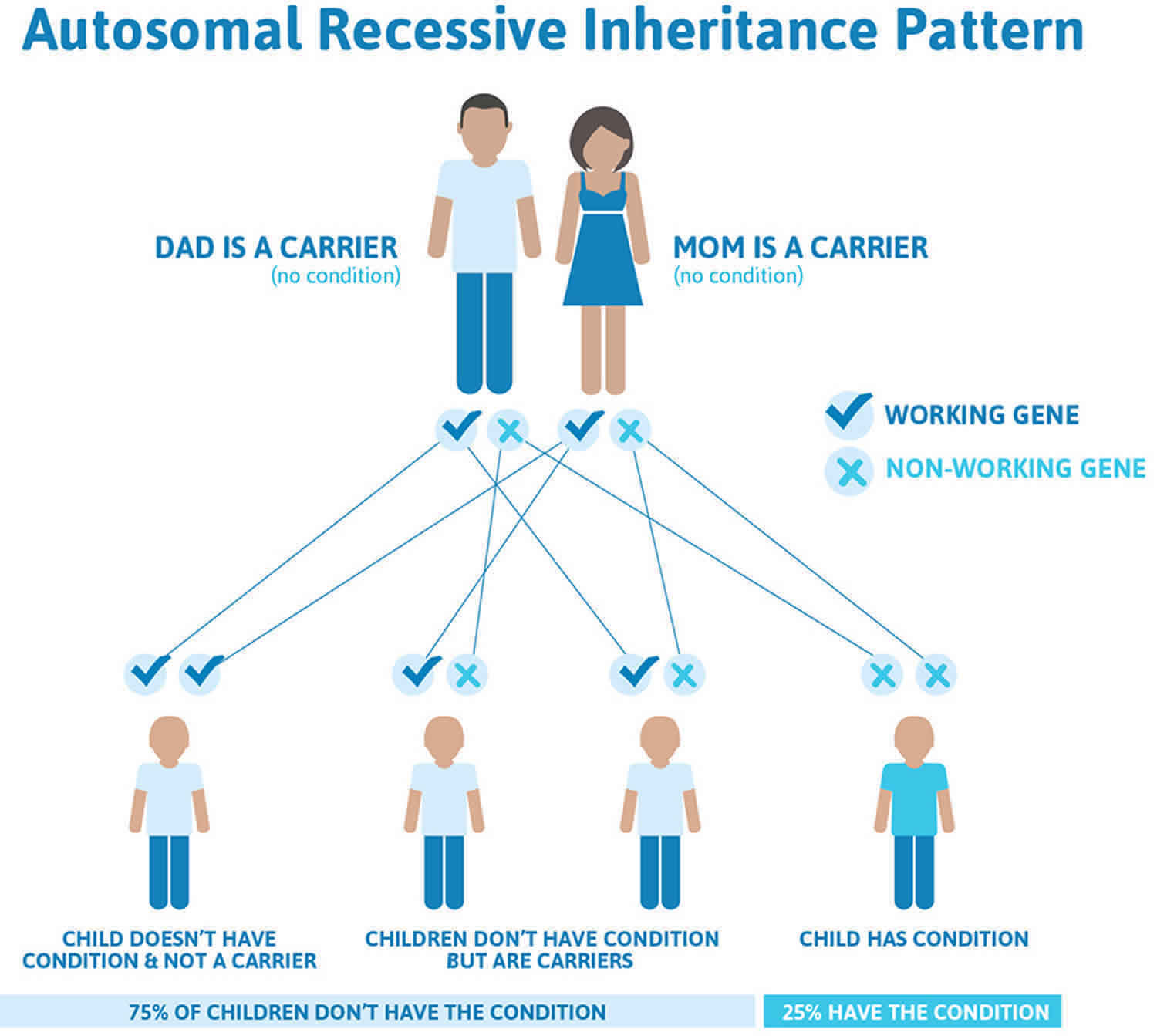
Tangier disease is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
It is rare to see any history of autosomal recessive conditions within a family because if someone is a carrier for one of these conditions, they would have to have a child with someone who is also a carrier for the same condition. Autosomal recessive conditions are individually pretty rare, so the chance that you and your partner are carriers for the same recessive genetic condition are likely low. Even if both partners are a carrier for the same condition, there is only a 25% chance that they will both pass down the non-working copy of the gene to the baby, thus causing a genetic condition. This chance is the same with each pregnancy, no matter how many children they have with or without the condition.
- If both partners are carriers of the same abnormal gene, they may pass on either their normal gene or their abnormal gene to their child. This occurs randomly.
- Each child of parents who both carry the same abnormal gene therefore has a 25% (1 in 4) chance of inheriting a abnormal gene from both parents and being affected by the condition.
- This also means that there is a 75% ( 3 in 4) chance that a child will not be affected by the condition. This chance remains the same in every pregnancy and is the same for boys or girls.
- There is also a 50% (2 in 4) chance that the child will inherit just one copy of the abnormal gene from a parent. If this happens, then they will be healthy carriers like their parents.
- Lastly, there is a 25% (1 in 4) chance that the child will inherit both normal copies of the gene. In this case the child will not have the condition, and will not be a carrier.
These possible outcomes occur randomly. The chance remains the same in every pregnancy and is the same for boys and girls.
Figure 3 illustrates autosomal recessive inheritance. The example below shows what happens when both dad and mum is a carrier of the abnormal gene, there is only a 25% chance that they will both pass down the abnormal gene to the baby, thus causing a genetic condition.
Figure 3. Tangier disease autosomal recessive inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Tangier disease symptoms
Tangier disease affects many different body systems. Symptoms of Tangier disease are variable and depend on which organs are involved and the severity of those manifestations. Tangier disease is most often characterized by enlarged orange- or yellow-colored tonsils. This discoloration is due to fatty deposits accumulating in the tonsils. People with Tangier disease often suffer from tonsillitis, which may interfere with swallowing food. Many patients undergo tonsillectomy. Even so, yellow or orange material is often visible in the backs of the throats of patients who have had tonsillectomies. Young children may also have yellow/orange patches on the adenoids, which are small glands that sit at the back of the nasal cavity. These glands are part of the immune system and they disappear by the teenage years.
Tangier disease commonly affects the cardiovascular system and certain internal organs. In particular, fatty deposits can also form in other organs causing enlargement of the throat, liver, spleen, or lymph nodes. Fat accumulations in nerves can cause disturbances and loss-of-sensation called peripheral neuropathy. Discoloration may also occur in the digestive system, particularly the rectum and large intestine. Cardiovascular disease has been reported in adults with Tangier disease. Roughly half of patients have cardiac abnormalities. In rare cases, a clouding of the cornea of the eye can occur, but is generally mild and does not cause vision impairment.
Common features of Tangier disease are listed below.
Common clinical features of Tangier disease:
- Yellowish/orange tonsils that may frequently become enlarged
- Very low serum levels of HDL cholesterol
- Very low levels of apolipoprotein A1
- Elevated levels of triglycerides
- Cloudiness of the cornea
- Low platelet count
- Enlarged spleen
- Enlarged liver
- Chest pains
- Atherosclerosis
Note that while many patients with Tangier disease have elevated levels of triglycerides in their blood, levels may be normal in some.
Signs of Tangier disease may not be obvious at birth or in childhood. However, any child with yellow/orange tonsils should be checked for Tangier disease and monitored carefully for abnormalities in the blood.
Tangier disease neuropathy
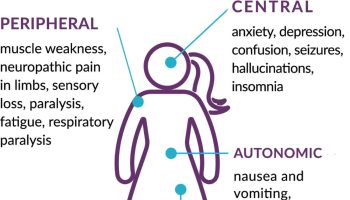
In addition to the problems noted above, roughly half of people with Tangier disease also develop neurological problems. Their specific problems tend to be among the clinical features in the list below, and tend to fit the definition of a condition called polyneuropathy. This term refers to disease that affects the peripheral nerves. Common problems include weakness, numbness, and burning pain. However, the way that these problems occur varies, especially in Tangier disease.
A 1985 study proposed that there are 3 subtypes of polyneuropathy in Tangier disease 3:
- Type I Tangier disease polyneuropathy is transient or relapsing. These term means that the symptoms like weakness may come and go, and may not last for long periods when they occur. The symptoms are asymmetrical, meaning that they may only occur on one side of the body. In addition, a process called nerve conduction velocity (NCV) is usually, but not always, normal in Type I Tangier disease neuropathy. NCV is a measure of how quickly nerve transmit impulses. Patients with this form of Tangier disease polyneuropathy are described in references 3 and 4.
- Type II Tangier disease polyneuropathy is a slowly progressing form. Thus, once problems begin, they do not tend to go away for a while, and they continue to get worse as time passes. Unlike type I, in type II, problems occur on both sides of the body. These problems are most marked in the lower extremities, and like type I, nerve conduction velocity is normal. A patient with this form of Tangier disease is described in reference 3.
- Type III Tangier disease polyneuropathy is also a slowly progressing symmetrical condition. Unlike type II, atrophy and weakeness tend to occur in the upper extremities (especially in the distal parts, or hands and other parts of the arms that are furthest from the shoulders). Nerve conduction velocity is not normal in type III Tangier disease polyneuropathy and is slower or much slower than normal. A patient with this condition is described in reference 2.
Patients with all three neurological forms of Tangier disease may lose the sensations of pain and temperature. This problem makes them susceptible to burns, which may occur without their knoweldge until damage has been done.
Neurological features of Tangier disease:
- Peripheral neuropathy (weakness, numbness, pain, usually in hands and feet)
- Abnormal results of test of motor nerve conduction velocity
- Muscle weakness (may get worse or relapse and remit)
- Lost/reduced deep tendon reflexes (e.g. knee jerks)
- Myelin abnormalities in peripheral nerves
- Reduced temperature sensitivity
- Reduced pain sensitivity
- Muscle wasting
Tangier disease diagnosis
Diagnosis of Tangier disease is achieved through clinical evaluation and can be confirmed through genetic testing involving the sequencing of the ABCA1 gene. HDL-C deficiency and an extremely low apolipoprotein A1 (ApoA1) level are typical diagnostic criteria.
Talk to your health care provider or a genetic professional to learn more about your testing options.
GeneTests lists laboratories (https://www.ncbi.nlm.nih.gov/gtr) offering clinical genetic testing for this condition. Clinical genetic tests are ordered to help diagnose a person or family and to aid in decisions regarding medical care or reproductive issues.
Orphanet lists international laboratories (https://www.orpha.net/consor/cgi-bin/ClinicalLabs.php?lng=EN) offering diagnostic testing for this condition.
Tangier disease treatment
There are no known specific treatments for Tangier disease. Treatment of Tangier disease is supportive and based on specific disease manifestations in a given individual. Drugs known to increase high density lipoprotein levels in unaffected people, such as estrogens, nicotinic acid, statins, or phenytoin, do not work in people with Tangier disease 4.
To reduce the risk for heart and blood vessel disease, people with this condition should maintain a low fat (especially saturated fat) diet and overall healthy lifestyle. Heart disease risk factors such as smoking, high blood pressure, diabetes, obesity, high level of triglycerides and homocysteine in the blood should receive prompt treatment. Fibrates can be used to help lower triglycerides 4.
To date, no treatment has been found to prevent the progression of this disease, including trials of omega-3-fatty acids, antioxidants, and vitamin E 4.
Individuals with Tangier disease may benefit from referral to specialized lipid centers for advanced management. Consultation with the following specialists may be required 5:
- Lipidologist
- Endocrinologist
- Cardiologist
- Vascular specialist
- Cardiovascular surgeon
- Dietitian
Surgical removal of the spleen, tonsils, or other enlarged tissues may become necessary in some patients. It is suggested that management of Tangier disease should include regular assessment of cardiovascular risk and neurological and ophthalmological examination.
Genetic counseling is recommended for families of patients with Tangier disease.
Tangier disease life expectancy
Tangier disease is an extremely rare condition and, as such, the published literature around its range of clinical manifestations, including life expectancy is limited.
- Tangier disease. https://rarediseases.org/rare-diseases/tangier-disease[↩]
- Tangier disease. https://ghr.nlm.nih.gov/condition/tangier-disease[↩]
- Gibbels E et al. (1985) Severe polyneuropathy in Tangier disease mimicking syringomyelia or leprosy. Clinical, biochemical, electrophysiological, and morphological evaluation, including electron microscopy of nerve, muscle, and skin biopsies. J Neurol 232(5):283-294.[↩]
- Assmann G, von Eckardstein A, Brewer HB. Familial analphalipoproteinemia: Tangier disease. In: Scriver et al., eds.. The Metabolic & Molecular Basis of Inherited Disease. 8th Ed. 2001[↩][↩][↩]
- Low HDL Cholesterol (Hypoalphalipoproteinemia) Treatment & Management. https://emedicine.medscape.com/article/127943-treatment#showall[↩]

{kind=link}