Contents
- What is Treacher Collins syndrome
- Treacher Collins syndrome facts
- Does this mean that Treacher Collins syndrome can happen again in my family?
- If my child, who has Treacher Collins syndrome, marries and has children, will all the children have it, too?
- What are the risks that my other children will transmit Treacher Collins syndrome to their own children?
- Is there a treatment for Treacher Collins syndrome?
- Will my child be intellectually disabled?
- Will my child be deaf?
- What kinds of problems can I expect?
- What about other areas of development: social, educational, etc.?
- Treacher Collins syndrome facts
- Treacher Collins syndrome life expectancy
- Treacher Collins syndrome symptoms
- Treacher Collins syndrome causes
- Treacher Collins syndrome diagnosis
- Treacher Collins syndrome treatment
What is Treacher Collins syndrome
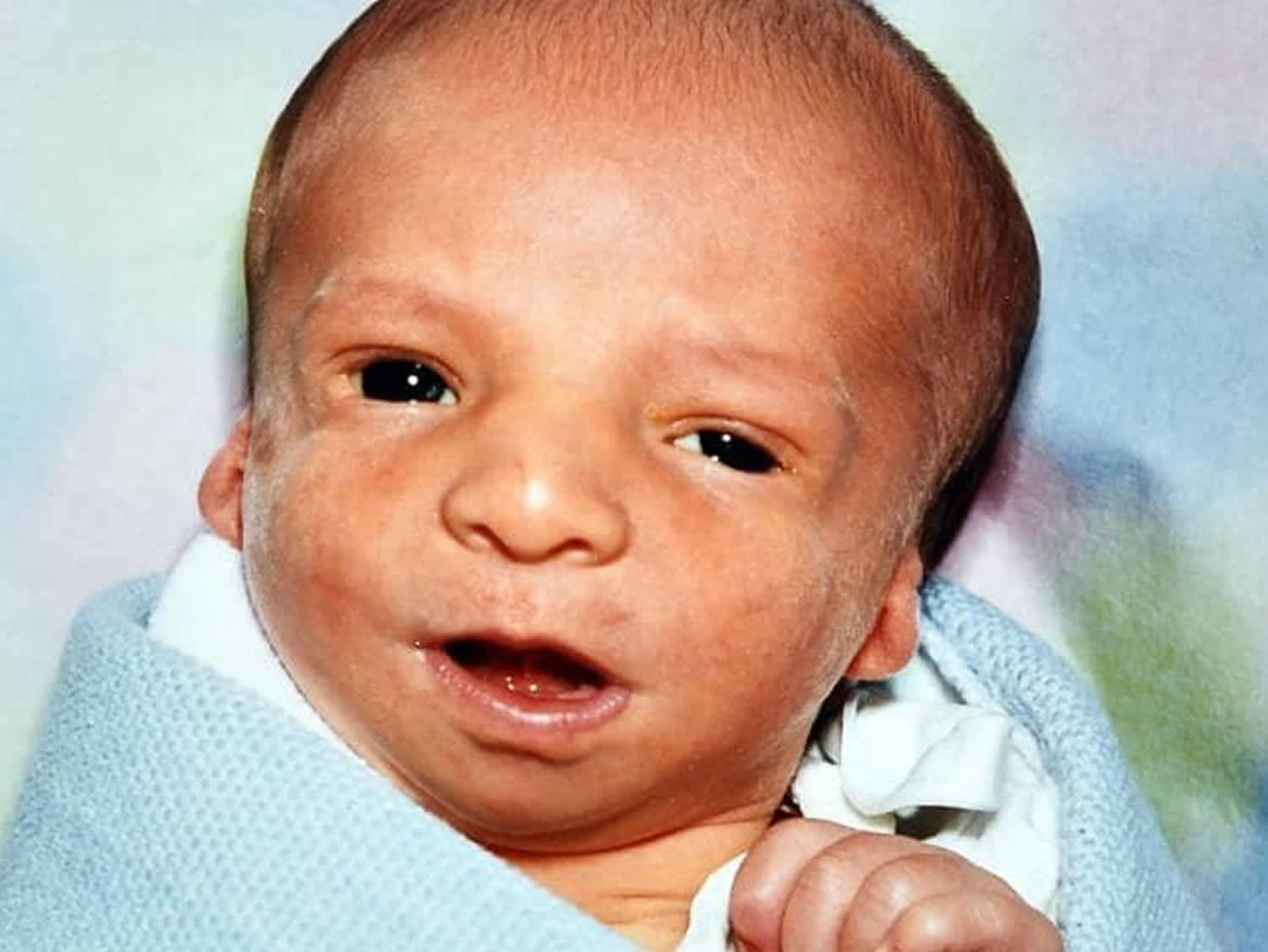
Treacher Collins syndrome is also called mandibulofacial dysostosis or Treacher Collins-Franceschetti syndrome, is a rare genetic disorder that affects the development of bones and other tissues of the head and face. Treacher Collins syndrome may affect the size and shape of the ears, eyelids, cheek bones, and upper and lower jaws 1. The signs and symptoms of Treacher Collins syndrome vary greatly, ranging from almost unnoticeable to severe. Most affected individuals have underdeveloped facial bones, particularly the cheek bones, and a very small jaw and chin (micrognathia). Some people with Treacher Collins syndrome are also born with an opening in the roof of the mouth called a cleft palate. In severe cases, underdevelopment of the facial bones may restrict an affected infant’s airway, causing potentially life-threatening respiratory problems.
Craniofacial abnormalities tend to involve underdevelopment of the zygomatic complex, cheekbones, jaws, palate and oral cavity (mouth) which can lead to breathing (respiratory) and feeding difficulties. In addition, affected individuals may also have malformations of the eyes (ocular) including a downward slant of the opening between the upper and lower eyelids (palpebral fissures), sparse eyelashes, and a notch in the lower eyelids called an eyelid coloboma. Some affected individuals have additional eye abnormalities that can lead to vision loss. Treacher Collins syndrome is also characterized by absent, small, or unusually formed ears. Hearing loss occurs in about half of all affected individuals; hearing loss is caused by defects of the three small bones in the middle ear, which transmit sound, or by underdevelopment of the ear canal.
People with Treacher Collins syndrome usually have normal intelligence. Brain and behavioral anomalies such as microcephaly and psychomotor delay have also been occasionally reported as part of Treacher Collins syndrome. The specific symptoms and physical characteristics associated with Treacher Collins syndrome can vary greatly from one individual to another. Some individuals may be so mildly affected as to go undiagnosed, while others may develop serious, life-threatening complications.
Treacher Collins syndrome is named after Edward Treacher Collins, a London ophthalmologist who first described the disorder in the medical literature in 1900 2.
Treacher Collins syndrome affects an estimated 1 in 10,000 to 50,000 people. Some mildly affected individuals may go undiagnosed, making it difficult to determine the disorder’s true frequency in the general population.
Treacher Collins syndrome is caused by a mutation in the TCOF1, POLR1C or POLR1D genes. In the case of TCOF1 the mode of inheritance is autosomal dominant, while for POLR1C it is autosomal recessive. In contrast, both autosomal dominant and recessive mutations in POLR1D have been reported in association with Treacher Collins syndrome.
There is no cure for Treacher Collins syndrome. Treatment is directed toward the specific symptoms that are apparent in each individual. Treatment may require the coordinated efforts of a team of specialists. Pediatricians, pediatric ear, nose and throat specialists (pediatric otolaryngologists), pediatric dentist, pediatric nurse, plastic surgeon, speech pathologists, audiologists, ophthalmologists, psychologists, geneticists, and other healthcare professionals may need to systematically and comprehensively plan an affect child’s treatment.
Figure 1. Treacher Collins syndrome
Treacher Collins syndrome facts
Treacher Collins syndrome is inherited
You must have the Treacher Collins syndrome to pass it on. For those that have Treacher Collins syndrome there is a 50-50 chance of having children affected by it. The severity of a parent’s condition is not an indication of the possible severity of the child. This means that a severely affected parent may have a mildly affected child and vice-versa.
While most cases of Treacher Collins syndrome are inherited in an autosomal dominant manner, about 60% of Treacher Collins syndrome cases are not inherited from a parent, but are due to a new mutation that occurred for the first time in the affected person 1. These children in turn will have a 50-50 chance of passing it on.
Every case of Treacher Collins syndrome is unique and needs to be assessed individually.
There is significant variability among people with Treacher Collins syndrome (Treacher Collins syndrome) – both within families, and between families. Some people with Treacher Collins syndrome are so mildly affected that they are never diagnosed. Others can have severe facial involvement and life-threatening complications 4. This phenomenon is called variable expressivity.
Additionally, not all people with genetic mutations associated with Treacher Collins syndrome have features of the condition. This phenomenon is called reduced penetrance. Most people with a Treacher Collins syndrome mutation are affected to some degree (i.e. penetrance is high), but cases of non-penetrance (i.e. no signs or symptoms) have been reported 4.
Many features of Treacher Collins syndrome can be improved by surgery and other supportive treatments. A well-planned treatment can produce excellent results for complete restoration of the form and function of the patient. When confirmed with Treacher Collins syndrome, it is important to pay particular attention to the psychological needs too. This would in turn help to build self-esteem in a child, thereby enabling him to lead a normal life. The psychological affects of Treacher Collins Syndrome are significant due to facial malformations. It is important to have correct consultation on everything involved.
Does this mean that Treacher Collins syndrome can happen again in my family?
If both parents are normal, the chances of a second child being born with Treacher Collins syndrome are extremely small. However, if one parent is affected, the chances that any pregnancy with result in a child with Treacher Collins syndrome is 1 out of 2 (50% risk). For this reason, it is very important that both parents of an affected child be thoroughly examined before any recurrence risks are quoted to them.
If my child, who has Treacher Collins syndrome, marries and has children, will all the children have it, too?
No. The risk is 50% for each pregnancy.
What are the risks that my other children will transmit Treacher Collins syndrome to their own children?
If your other children are not affected (showing no signs of the syndrome), there is no increased risk to their children. If another family member shows any feature of the syndrome, the occurrence risk for each pregnancy is 50%.
Is there a treatment for Treacher Collins syndrome?
There is no cure for Treacher Collins syndrome. Treatment is directed toward the specific symptoms that are apparent in each individual. Treatment is tailored to the specific needs of each affected person. Ideally, treatment is managed by a multidisciplinary team of craniofacial specialists.
Newborns may need special positioning or tracheostomy to manage the airway. Hearing loss may be treated with bone conduction amplification, speech therapy, and/or educational intervention 4.
In many cases, craniofacial reconstruction is needed. Surgery may be performed to repair cleft palate, to reconstruct the jaw, or to repair other bones in the skull. The specific surgical procedures used and the age when surgery is performed depends on the severity of the abnormalities, overall health and personal preference 2.
There are some possible treatments that are being investigated. Researchers are looking for ways to inhibit a protein called p53, which helps the body to kill off unwanted cells 2. In people with Treacher Collins syndrome, p53 is abnormally activated, leading to the loss of specific cells and ultimately causing features of Treacher Collins syndrome. It has been proposed that inhibiting the production of p53 (or blocking its activation) may help to treat affected people. However, more research is needed to determine if this type of treatment is effective and safe 2.
Researchers are also studying the use of stems cells found in fat tissue to be used alongside surgery in people with Treacher Collins syndrome and other craniofacial disorders. Early studies have shown that surgical outcomes may be improved using these stem cells to help stimulate the regrowth of affected areas. However, this therapy is still experimental and controversial 4.
Will my child be intellectually disabled?
There is no evidence that intellectual disability is a feature of Treacher Collins syndrome. Hearing loss, however, is present in most affected individuals to some degree. Early diagnosis and treatment of the hearing loss can prevent associated developmental and educational handicaps.
Will my child be deaf?
The term “deaf” applies only to very severe hearing losses in which the nerves for hearing, in the ear or the brain, do not function properly. The hearing loss in Treacher Collins syndrome is due to abnormalities in the structures of the outer and middle ear which conduct sound to the nerve endings in the inner ear. Thus, the hearing loss in Treacher Collins syndrome is usually termed “conductive” and in the majority of children it is not of sufficient severity to be termed “deafness.” However, any degree of hearing loss may affect the development of speech and language ability to succeed in school.
What kinds of problems can I expect?
First, Treacher Collins syndrome, like nearly all birth defects, varies in severity from patient to patient. In fact, some cases are so mild that they are never recognized unless they are seen by specialists experienced in making such a diagnosis. In other children, the physical abnormalities of the face and ears are much more obvious and functional problems may develop.
Second, both the oral cavity (mouth) and the air passage (nose and throat) tend to be small in persons who have this syndrome. This may produce problems for the affected infant with breathing and feeding. You should be on the alert for such problems. If your infant has difficulty breathing or feeding, or has weight loss or poor weight gain, discuss your observations and concerns with your child’s primary care provider or craniofacial center. Some children who have severe breathing difficulties require an operation to improve breathing and/or feeding.
Third, cleft palate is a condition frequently associated with this syndrome. Cleft palate itself sometimes can cause feeding problems and increase the risk of middle ear problems. Your child’s primary care provider or cleft palate or craniofacial center can assist you with the management of feeding problems.
The next concern after breathing and feeding is hearing. The hearing loss in Treacher Collins syndrome is usually bilateral (meaning that both ears are affected) and, while it is not severe enough to be termed “deafness,” it is severe enough to affect the ability to hear the human voice. Hearing levels can and should be measured. Depending upon the results of the testing, your child may be fitted with a hearing aid to restore his or her access to the world of sound. An early childhood program of speech and language therapy may also be recommended.
The fact that a hearing loss is present does not mean that your child will be dependent upon sign language. The great majority of children with this syndrome do learn to talk. However, there are several features of the syndrome, besides the hearing loss, which can affect speech and language development. Particularly in the severely affected child, the size and position of various structures inside the mouth (e.g. the relationship of the upper and lower teeth) may affect the ability to learn certain speech sounds.
You can facilitate your child’s speech and language development by (1) seeking early evaluation by a specialist in hearing (an audiologist) and a specialist in communication development (a speech/language pathologist), and (2) follow their advice with regard to the need for a hearing aid and for early therapy programs. The specialists most prepared to evaluate and manage your child are those who are members of a multidisciplinary craniofacial team.
The facial deformity and need for treatment of Teacher Collins syndrome may create problems in family and social relationships, in school adjustment, and so on. The craniofacial center may have a psychologist or social worker, or the center can refer you to someone for evaluation and counseling if needed. Remember that children with Treacher Collins syndrome, like all other children, are individuals. They vary in social adjustment, academic achievement, and their ability to cope with adults. The professionals of craniofacial centers try to maximize each child’s potential by offering early diagnosis and treatment when indicated.
Treacher Collins syndrome life expectancy
Children with Treacher Collins syndrome typically grow to become functioning adults with normal intelligence and life expectancy 5.
How other people act around a child with Treacher Collins syndrome can have a big impact on the child’s life and self-esteem. If your child has Treacher Collins syndrome, offering your love and support will help ensure your child’s emotional well-being. If feelings of self-consciousness keep your child from enjoying social events or other activities, find a counselor or psychologist who can help your child work through these difficult emotions.
Treacher Collins syndrome symptoms
Symptoms of Treacher Collins syndrome can be mild or severe. The same Treacher Collins syndrome mutation can affect one family member much more than another, a difference called penetrance. Treacher Collins syndrome symptoms can be so mild that a parent may have the mutation and not notice the symptoms (low penetrance) until the mutation is passed to a child who has more obvious symptoms (higher penetrance).
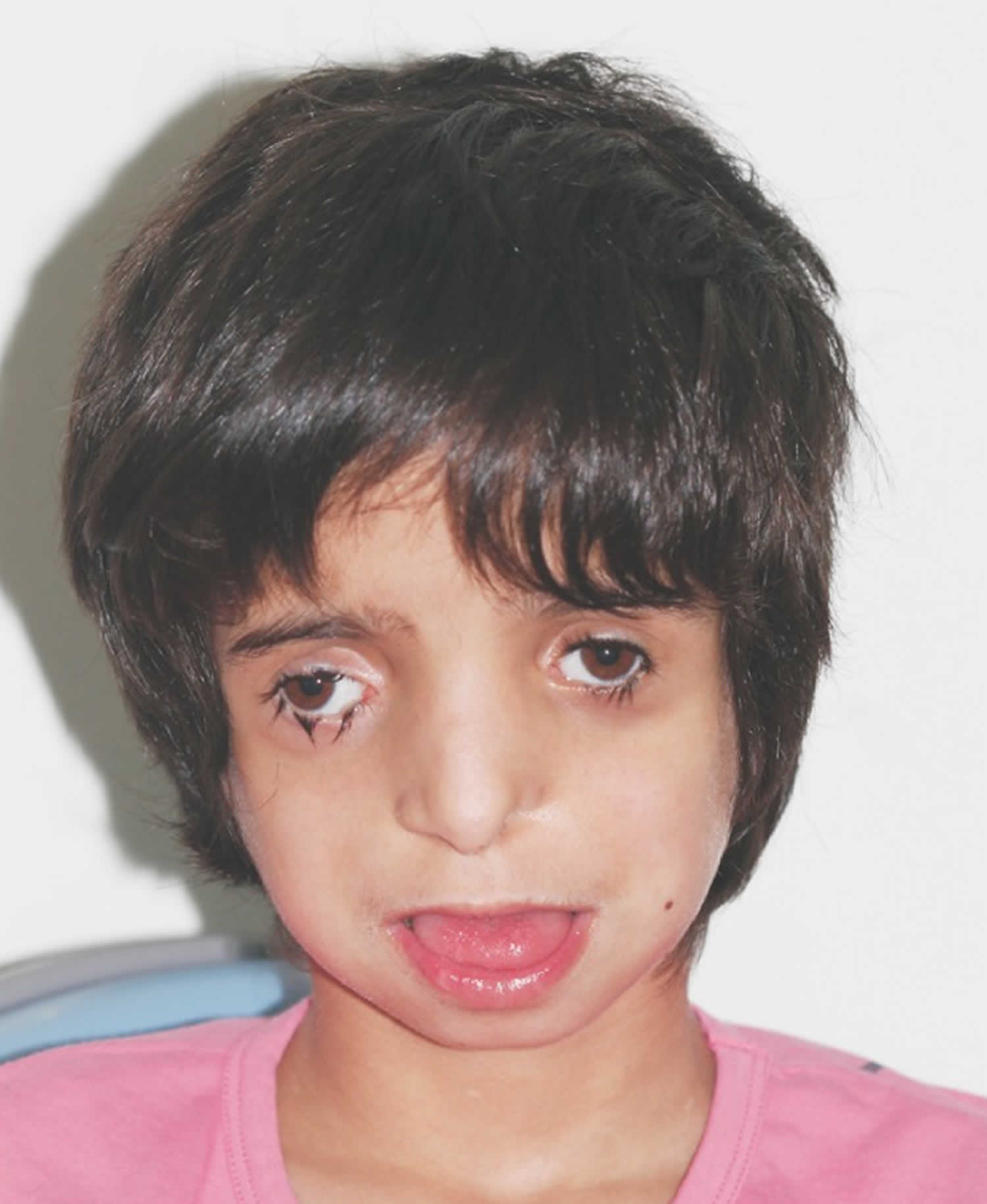
Treacher Collins syndrome causes changes that are usually symmetrical, meaning both sides of the body look the same. Features of Treacher Collins syndrome include:
- downward slant of the outer corners of the eyes
- drooping upper eyelids
- notches in the lower eyelids with few, if any, lower lid eyelashes
- small cheekbones
- fewer teeth than usual; they may be crooked and have patchy coloring
- small mandible (lower jaw) causing an overbite (the chin and lower teeth sit back from the upper teeth)
- open or split roof of the mouth (cleft palate) and upper lip (cleft lip)
- small, unusually shaped ears
- small or missing ear openings
- hearing loss because sound is poorly transferred by the tiny bones in the middle ear
The major characteristic features of Treacher Collins syndrome encompass certain bones of the face, ears and soft tissues around the eyes. Affected individuals present with distinctive facial features and potentially develop hearing and vision problems. The abnormalities of Treacher Collins syndrome are typically symmetric (almost identical on both sides of the face) and are present at birth (congenital). Speech and language development can be compromised by hearing loss, cleft palate or jaw and airway problems. Intelligence is usually unaffected but brain and behavioral anomalies such as microcephaly and cognitive delay have been reported infrequently as part of the condition.
Someone with Treacher Collins Syndrome may have malformed cheekbones, chin, nose, jaw and temples. Eyelids are often drooping, seeming not to support the eyes, and there may be a small nick in the lower lid. The ears may be malformed or completely absent. Hairline and palate may also be unusual.
Depressed Cheekbones
The cheekbones can be underdeveloped or absent. This means that the cheek muscles join onto the lower jaw muscles and so cause sloping eyes. This can be corrected by surgery using implants and bone grafts.
Malformed or Absent Ears
This will vary greatly from case to case. Some may just have small ears, others may have no ears at all. The outer ear may be folded or squashed. The middle ear can also be malformed or missing. Any hearing loss is almost always conductive and can vary from partial to severe.
Corrective surgery will depend on the individual and on which parts of the ear are missing. Artificial ears can be attached using Titanium, a metal that uniquely bonds with bone. Hearing may be improved with hearing aids. Most often a bone conduction hearing aid is used, attached to a headband, or a Bone Anchored Hearing Aid attached directly to the mastoid bone.
Receded chin
The Treacher Collins syndrome baby is often born with a much receded lower jaw. This can improve as the child grows.
Hair growth
This again is quite common at birth. The hairline continues across the cheek forming a definite sideburn towards the mouth. This may only be temporary and varies from child to child.
Other possible problems
Breathing problems can occur. In small children and babies breathing can be very noisy. When colds or infections occur the usual problems are sometimes more severe. This is because the roof of the mouth may be high and the nasal passages very small. Snoring is a common problem and may be loud. Eye infections are common in children.
Dental problems may occur because of the smallness of the mouth. A receded jaw could cause overcrowded teeth or an incorrect bite. All this can be improved by orthodontic treatment. Cleft palate or choanal atresia conditions may be present in severe cases.
Infants with Treacher Collins syndrome exhibit underdeveloped (hypoplastic) or absent cheekbones (malars), causing this area of the face to appear flat or sunken. The bone of the lower jaw (mandible) is incompletely developed (mandibular hypoplasia), causing the chin and the lower jaw to appear abnormally small (micrognathia). Certain bony structures (e.g., coronoid and condyloid processes) that anchor portions of the lower jaw bone to muscle can be unusually flat or absent. Affected infants may also exhibit underdevelopment of the throat (pharyngeal hypoplasia). Pharyngeal hypoplasia with underdevelopment of the lower jaw (mandibular hypoplasia) and/or abnormal smallness of the jaw (micrognathia) may contribute to feeding problems and/or breathing difficulties (respiratory insufficiency) during early infancy. Children may experience obstructive sleep apnea which is characterized by repeated short interruptions of normal breathing and air movement during sleep. In some severely-affected individuals, life-threatening respiratory difficulties may develop.
Additional abnormalities that may contribute to respiratory or feeding difficulties include narrowing or obstruction of the nasal airways (choanal stenosis or atresia). Some children may be described as having features of “Pierre Robin Sequence” which include severe micrognathia, a tongue that is displaced farther back in the mouth than normal (glossoptosis) with or without incomplete closure of the roof of the mouth (cleft palate). In addition, malformations of the mouth and the jaw may result in dental abnormalities, such as teeth that are underdeveloped (hypoplastic) and/or misaligned (malocclusion). Additional dental abnormalities have also been reported including missing teeth (tooth agenesis), clouding or discoloration of the enamel of teeth (enamel opacity), and improper (ectopic) eruption of certain upper teeth (maxillary molars)Individuals with Treacher Collins syndrome may develop hearing loss due to the failure of sound waves to be conducted through the middle ear (conductive hearing loss). Conductive hearing loss usually results from abnormalities affecting structures within the middle ear and individuals with Treacher Collins syndrome may also have malformed or absent ossicles, the three small bones through which sound waves are transmitted in the middle ear (i.e., incus, malleus, and stapes). In addition, the external ear structures are often absent, small or malformed (microtia), with narrowing (stenosis) or blockage (atresia) of the external ear canals. The outer ears may be crumpled or rotated. In contrast, the inner ear is usually unaffected, although malformation of the bony spiral organ in the inner ear (cochlea) and the structures within the inner ear that play a role in balance (vestibular apparatus) have been reported. Additional symptoms may include the presence of small growths of skin or pits just in front of the external ear (preauricular tags) and an abnormal passage that is closed on one end (blind fistula) that normally drains the ears to the nose.
Many infants with Treacher Collins syndrome have abnormalities of the tissue surrounding the eyes. These eye differences can give affected individuals a saddened facial appearance. The most common ocular symptom is a downward slant to the opening between the upper and lower eyelids (palpebral fissures). Additional symptoms include a lower eyelid notch or cleft of missing lid tissue (lid coloboma), partial absence of eyelashes on the lower eyelid, crossed eyes (strabismus) and narrowed tear ducts (dacrostenosis). Occasionally malformations of the globe are seen and can include notch or cleft of missing tissue of the iris or abnormally small eyes (microphthalmia). Vision loss may occur in some patients. The degree of visual impairment varies depending upon the severity and combination of ocular abnormalities. Lower eyelid abnormalities can cause the eyes to dry out, which increases the risk of chronic irritation and eye infections.
Some individuals with Treacher Collins syndrome exhibit additional physical abnormalities such as an widely spaced eyes, notching of the upper eyelid, nasal deformity, an abnormally wide mouth (macrostomia), a highly-arched roof of the mouth (palate), unusual growth of the scalp hair toward the cheeks, and/or congenital heart defect.
Approximately 5% of individuals with Treacher Collins syndrome display development deficits or neurological problems such as psychomotor delay. However, intelligence is generally unaffected with normal language development. Nonetheless, issues with speech development can occur because of hearing loss, cleft palate or difficulties producing sounds because of structural distortion.
Treacher Collins syndrome causes
Almost all children with Treacher Collins syndrome have a mutation of one of three genes that control bone growth in and around the face. The Treacher Collins syndrome mutation causes a change in a baby’s growth very early in pregnancy. For a few people with Treacher Collins syndrome, the gene causing the problem is not known.
Mutations in the TCOF1, POLR1C, or POLR1D gene can cause Treacher Collins syndrome. TCOF1 gene mutations are the most common cause of the disorder, accounting for 81 to 93 percent of all cases. POLR1C and POLR1D gene mutations cause an additional 2 percent of cases. In individuals without an identified mutation in one of these genes, the genetic cause of the condition is unknown.
The proteins produced from the TCOF1, POLR1C, and POLR1D genes all appear to play important roles in the early development of bones and other tissues of the face. These proteins are involved in the production of a molecule called ribosomal RNA (rRNA), a chemical cousin of DNA. Ribosomal RNA helps assemble protein building blocks (amino acids) into new proteins, which is essential for the normal functioning and survival of cells. Mutations in the TCOF1, POLR1C, or POLR1D gene reduce the production of rRNA. Researchers speculate that a decrease in the amount of rRNA may trigger the self-destruction (apoptosis) of certain cells involved in the development of facial bones and tissues. The abnormal cell death could lead to the specific problems with facial development found in Treacher Collins syndrome. However, it is unclear why the effects of a reduction in rRNA are limited to facial development.
Treacher Collins syndrome inheritance pattern
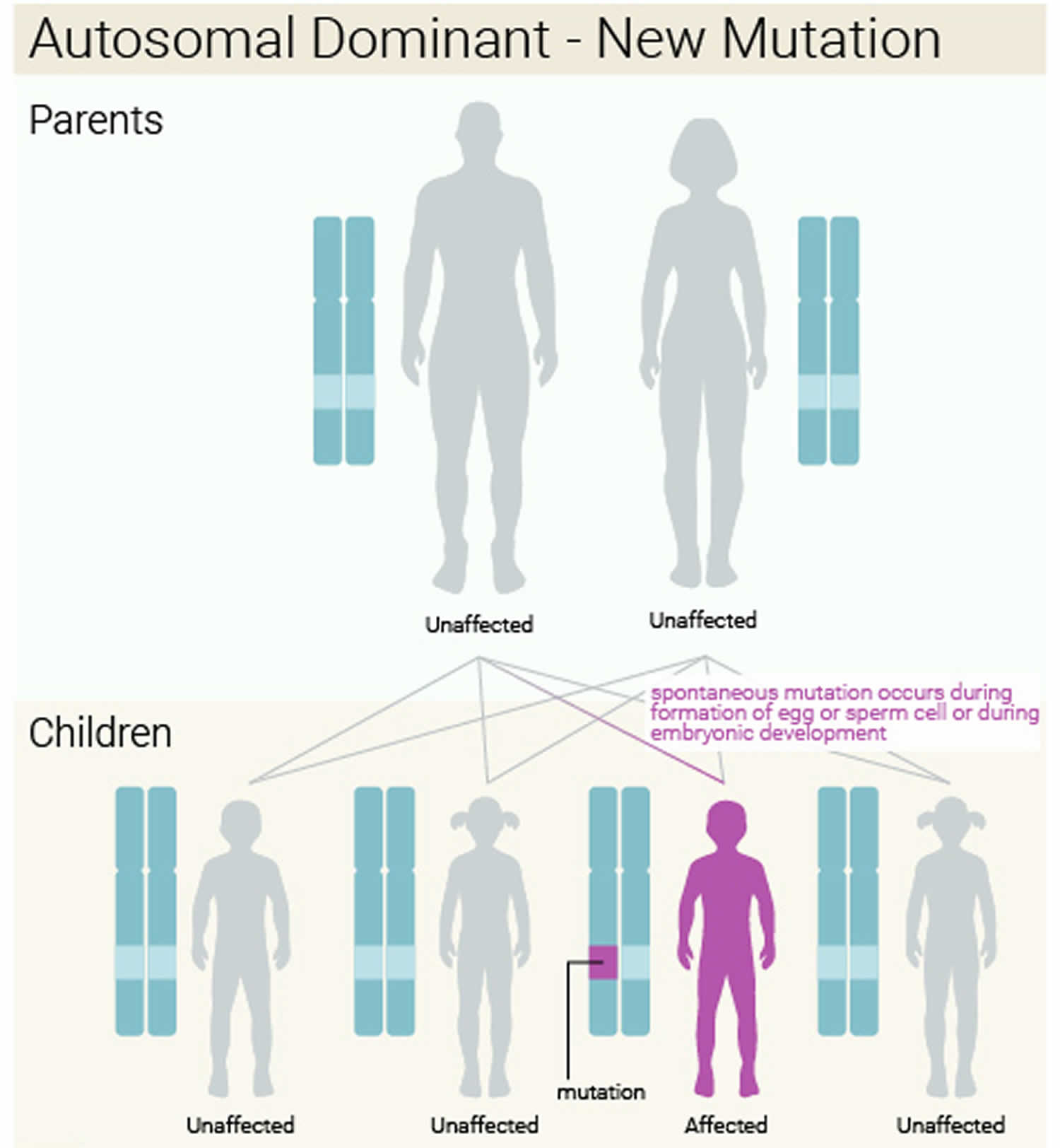
When Treacher Collins syndrome results from mutations in the TCOF1 or POLR1D gene, it is considered an autosomal dominant condition, which means one copy of the altered gene in each cell is sufficient to cause the disorder. About 60 percent of these cases result from new mutations in the gene and occur in people with no history of the disorder in their family. In the remaining autosomal dominant cases, a person with Treacher Collins syndrome inherits the altered gene from an affected parent.
When Treacher Collins syndrome is caused by mutations in the POLR1C gene, the condition has an autosomal recessive pattern of inheritance. Autosomal recessive inheritance means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
Figure 2. Treacher Collins syndrome autosomal dominant inheritance pattern
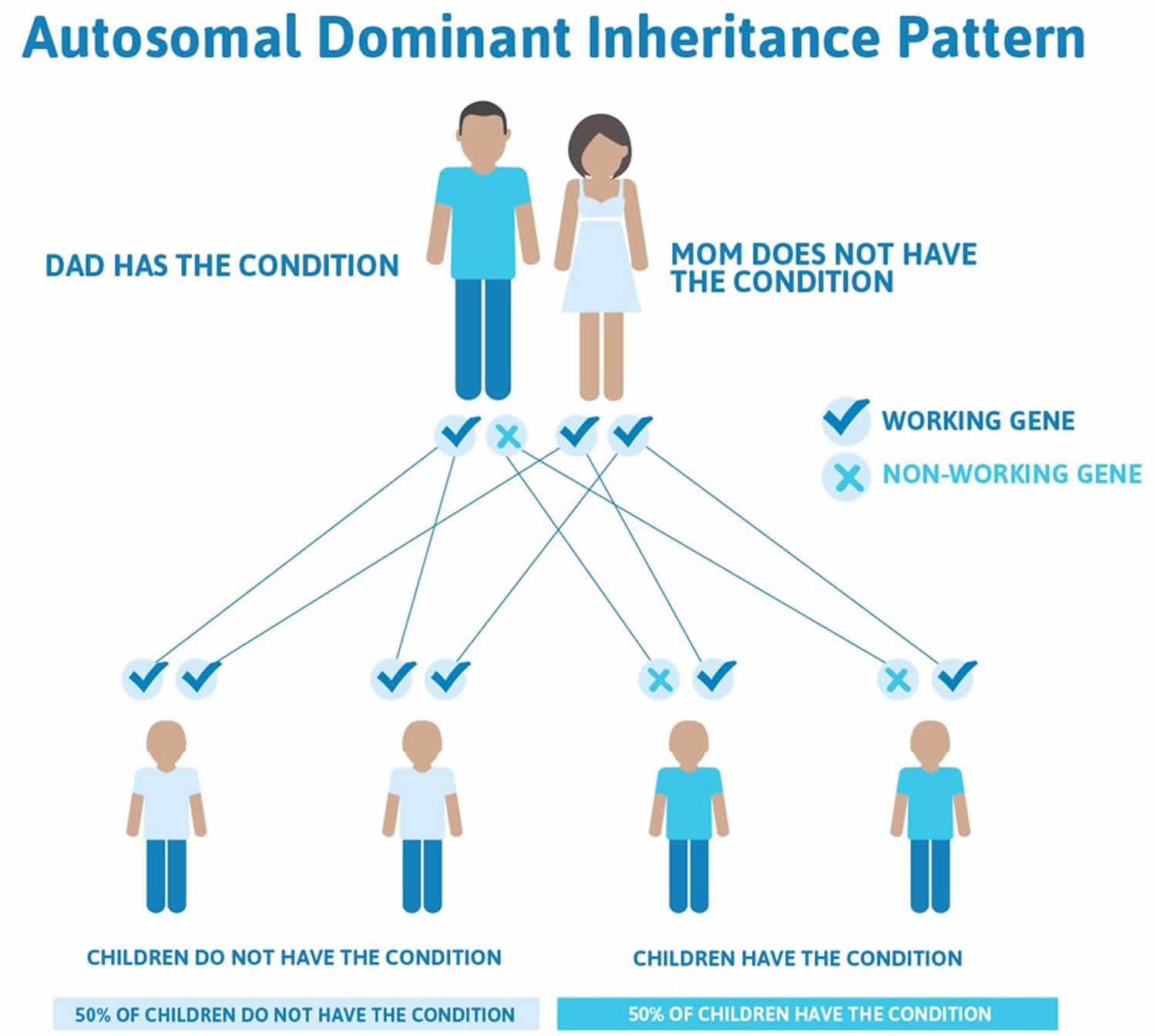
Figure 3. Treacher Collins syndrome autosomal dominant with a new (de novo) mutation
Treacher Collins syndrome diagnosis
Making a diagnosis for a genetic or rare disease can often be challenging. Healthcare professionals typically look at a person’s medical history, symptoms, physical exam, and laboratory test results in order to make a diagnosis.
The child most often will show normal intelligence. An exam of the infant may reveal a variety of problems, including:
- Abnormal eye shape
- Flat cheekbones
- Cleft palate or lip
- Small jaw
- Low-set ears
- Abnormally formed ears
- Abnormal ear canal
- Hearing loss
- Defects in the eye (coloboma that extends into the lower lid)
- Decreased eyelashes on the lower eyelid
Genetic tests can help identify gene changes linked to this condition.
Clinical testing and work-up
Specialized X-ray studies will confirm the presence and/or extent of certain observed craniofacial abnormalities. For example, such imaging tests show the abnormal smallness of the jaw (micrognathia) due to underdevelopment of the lower jaw bone (mandibular hypoplasia), the presence and/or extent of hypoplasia affecting certain parts of the skull, and/or the presence of additional malformations of the ear that cannot be seen during clinical evaluation.
In addition, in those affected individuals who exhibit few signs, a thorough clinical examination and X-ray imaging of the craniofacial area can demonstrate the subtle presence of certain characteristic features (e.g., hypoplasia of zygomatic arches) associated with Treacher Collins syndrome. Because Treacher Collins syndrome shares several physical features that may occur in other craniofacial syndromes, many researchers recommend that diagnostic confirmation be made through molecular genetic testing and/or, in some cases, a careful, detailed family history.
Molecular genetic testing to confirm a diagnosis is available through commercial and academic research laboratories to detect mutations in the TCOF1, POLR1C and POLR1D genes. Approximately 90-95% of individuals have an identifiable mutation of the TCOF1 gene. Furthermore, genetic confirmation of a TCOF1, POLR1C, or POLR1D mutation can be detected before birth (prenatally) by amniocentesis and chorionic villus sampling if a mutation has been identified in an affected family member. In certain cases, fetal ultrasonography, which uses reflected sound waves to create an image of the developing fetus, can reveal characteristic findings suggestive of Treacher Collins syndrome. Relatives, especially parents and siblings, of an individual diagnosed with Treacher Collins syndrome should be carefully examined because mild cases often go unrecognized and undiagnosed.
Treacher Collins syndrome treatment
Treatment depends on the symptoms, and requires a team of medical specialists, including pediatricians, plastic surgeons, ear nose and throat specialists (ENTs), dentists ophthalmologists, and audiologists.
Treatment begins at birth
Newborns with Treacher Collins syndrome may have trouble breathing because their airways are narrow. Certain positions, like lying on the stomach, can help make breathing easier. For severe breathing problems, a child might need a tube inserted into the windpipe (called a tracheostomy). Some babies with Treacher Collins syndrome have problems with feeding, especially when it interferes with breathing. So they might need a feeding tube into the stomach through the nose.
Unless a child with Treacher Collins syndrome has breathing or feeding problems, most facial reconstruction surgery is done over a number of years when the child is older. Surgery of the face and jaw can improve appearance, and have a positive effect on a child’s self-esteem and social functioning.
Hearing should be checked at birth and routinely as the child grows. A full assessment should be done early during life, even before one year of age and then yearly, in order to ensure proper speech development. Because the inner ear still works well in most children with Treacher Collins syndrome, hearing aids that transmit sound through the bone instead of the middle ear can work well. Speech-language therapy is often needed.
Kids with Treacher Collins syndrome need regular eye exams to check for problems with vision, eye movements, and cornea exposure (because they can’t close their eyelids completely). An instrument (ophthalmoscope) is used to visualize the interior of the eye to detect any possibility of visual impairment. This examination is important to ensure appropriate preventive steps and/or prompt treatment for those who exhibit abnormalities of the eyes in association with Treacher Collins syndrome (e.g., colobomas, strabismus, microphthalmia). Affected individuals should also be monitored for jaw and dental abnormalities.
Early intervention is important to ensure that affected children reach their potential. Special services that may be beneficial may include speech therapy, special social support, and other medical, social, and/or vocational services. Genetic counseling will be of benefit for affected individuals and their families.
Surgery
In some patients, surgical reconstruction of craniofacial malformations may be necessary. Surgery may be performed to repair cleft palate, to reconstruct the jaw, or to repair other bones in the skull (e.g., malar bones, zygomatic complex). The specific surgical procedures used and the age when surgery is performed depends upon the severity of the malformations, overall health and personal preference.
For example, different abnormalities may be treated at different ages. Cleft palate is often corrected around 1-2 years of age. Zygomatic and orbital reconstruction usually occurs around 5-7 years of age. External and inner ear reconstruction usually occurs around 6 years of age. Jawbone lengthening or reconstruction can range from newborn to teenager years depending upon the extent and severity of the condition.
Obstructive airways can be a serious problem not always obvious to parents or clinicians. A sleep or nap study may be used to help determine the severity of the obstruction and may influence the treatment plan. In severe affected individuals, a tube may be surgically inserted into the windpipe (trachea) to maintain an effective airway, a procedure called a tracheostomy. A procedure known as mandibular distraction, which is used to increase the length of the jawbone, may be necessary. A tube may be surgically implanted into the stomach to assure that affected infants experiencing feeding difficulties receive a sufficient amount of calories (gastrostomy).
Multiple surgeries may be required to treat the various craniofacial abnormalities that are potentially associated with Treacher Collins syndrome. Despite the number of surgeries, results vary from one person to another and the end result is rarely fully corrective.
In some individuals, an operation may be performed to help correct middle ear malformations and associated conductive hearing loss. However, specialized hearing aids such as bone-anchored hearing aids (BAHA) may suffice rather than surgery in most patients. Bone-anchored hearing aids transmit sound directly through bone into the inner ear, bypassing the external ear canal and the middle ear (both of which are often affected in individuals with Treacher Collins syndrome. Reconstructive surgery may be performed to help correct outer ear malformations for functional and cosmetic reasons. Generally, reconstruction of the external ear should be performed first.
Additional therapies
In individuals with Treacher Collins syndrome who exhibit eye abnormalities and associated visual impairment, corrective glasses, contact lenses, surgery, and/or other supportive techniques may be used to help improve vision in some cases. Artificial teeth (dentures), dental implants, braces, dental surgery, and/or other corrective procedures may be used to correct dental abnormalities.
Anesthesia considerations
Structural airway problems associated with Treacher Collins syndrome can make it difficult for anesthesiologists to manage and maintain an airway during surgery. Proper evaluation including a comprehensive preoperative assessment and complete clinical history should be performed to best plan an anesthetic strategy.
- Treacher Collins syndrome. https://ghr.nlm.nih.gov/condition/treacher-collins-syndrome[↩][↩]
- Treacher Collins Syndrome. https://rarediseases.org/rare-diseases/treacher-collins-syndrome/[↩][↩][↩][↩]
- Damlar İ, Altan A, Turgay B, Kiliç S. Management of obstructive sleep apnea in a Treacher Collins syndrome patient using distraction osteogenesis of the mandible. Journal of the Korean Association of Oral and Maxillofacial Surgeons. 2016;42(6):388-392. doi:10.5125/jkaoms.2016.42.6.388. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5206247/[↩]
- Katsanis SH, Jabs EW. Treacher Collins Syndrome. 2004 Jul 20 [Updated 2018 Sep 27]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2018. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1532[↩][↩][↩][↩]
- Treacher Collins Syndrome. https://kidshealth.org/en/Parents/tcs.html[↩]

{kind=link}