
Contents
What is trimethylaminuria
Trimethylaminuria also known as “fish odor syndrome”, is a rare metabolic disorder in which the body is unable to break down trimethylamine, a nitrogen-containing compound that has a pungent fishy odor 1. Trimethylamine has been described as smelling like rotting fish, rotting eggs, garbage, or urine. As trimethylamine compound builds up in the body, it causes affected people to give off a strong odor in their sweat, urine, and breath. The intensity of the odor may vary over time. The odor can interfere with many aspects of daily life, affecting a person’s relationships, social life, and career. Some people with trimethylaminuria experience depression and social isolation as a result of this condition.
Trimethylamine is normally formed by bacteria in the gut from reduction of compounds such as trimethylamine-N-oxide (TMAO) found in salt water fish and choline (found in foods such as soya, liver, kidneys, wheat germ, legumes, brewer’s yeast, and egg yolk) are digested 1. Trimethylamine is a chemical found in fish, sharks and rays, molluscs, and crustaceans, and is the main odorant that is characteristic of degrading seafood.
Trimethylamine is normally transported to the liver to be metabolized into non-odorous Trimethylamine-N-oxide (TMAO) by the flavin-containing monooxygenase 3 (FMO3) metabolic enzyme produced in the liver. Both trimethylamine-N-oxide (TMAO) and trimethylamine are normally excreted in the urine. However, when there is a deficiency of the FMO3 enzyme due to an autosomal recessive condition resulting in an FMO3 mutation, trimethylamine is then not oxidized, and thus remains in an odorous state. Over time, trimethylamine compound accumulate in the body and causing the excretion of a strong and offensive odor to leave the body through every single pore, breath, urine, and reproductive fluids. This leads to the characteristic odor of trimethylaminuria. Trimethylaminuria seems to be more common in women. Researchers think this may be due to higher hormone levels aggravating symptoms of the disorder 2.
Primary trimethylaminuria (TMAU1) sufferers have an inherited enzyme deficiency where trimethylamine is not efficiently converted to the non-odorous TMAO (trimethylamine-N-oxide) in the liver.
Secondary trimethylaminura (TMAU2) is an acquired form of trimethylaminuria usually involving an overproduction of trimethylamine by gut flora in a patient with normal FMO3 function (i.e. does not have an FMO3 deficiency). Experts believe that secondary trimethylaminura from overproduction of trimethylamine by bacterial overgrowth in the gut can be experienced for many years, but if the correct antibiotic therapy is applied, it can be cured by eradication of the bacteria responsible, though the bacterial overgrowth tends to return. Secondary trimethylaminura or TMAU2, has been recognized for many years, particularly in the US, although much of the trimethylaminuria interest has been in the inherited metabolic disorder FMO3 deficiency, TMAU1.
Diagnosis of trimethylaminuria requires the measurement of trimethylamine and trimethylamine-N-oxide (TMAO) in urine, which should be collected after a high substrate meal in milder or intermittent cases, most simply, a marine-fish meal. The symptoms of trimethylaminuria can be improved by changes in the diet to avoid precursors, in particular trimethylamine-N-oxide (TMAO) which is found in high concentrations in marine fish.
There is currently no cure for trimethylaminuria. However, it is possible for people with this condition to live normal, healthy lives. Both primary trimethylaminuria (TMAU1) and secondary trimethylaminura (TMAU2) can be controlled with periodic antibiotic therapy as well as dietary choline restriction of eggs, liver, beans, carnitine (meat), and TMAO rich foods (seafood). Choline and TMAO (trimethylamine-N-oxide) are broken down by gut bacteria into trimethylamine, which creates a favorable environment for the bacteria to grow in. The odor effects of trimethylamine may also be reduced by activated charcoal or copper chlorophyllin tablets to adsorb the trimethylamine in the gut, which is later eliminated in the feces. The alkaline pH of trimethylamine in the sweat can be neutralized with the use of pH skin cleansers and creams.
The following are some ways a person with trimethylaminuria can lower symptoms of odor:
- Avoiding foods containing trimethylamine and its precursors (choline, lecithin and trimethylamine N-oxide).
- Trimethylamine is present in high levels in milk obtained from wheat-fed cows
- Choline is present in high amounts in:
- Eggs
- Liver
- Kidney
- Peas
- Beans
- Peanuts
- Soy products
- Brassicas (brussel sprouts, broccoli, cabbage, and cauliflower)
- Lecithin and lecithin-containing fish oil supplements
- Trimethylamine N-oxide (TMAO) is present in seafood (fish, cephalopods, crustaceans). Freshwater fish have lower levels of trimethylamine N-oxide and it’s OK to eat.
- Taking low doses of antibiotics to reduce the amount of bacteria in the gut. This suppresses the production of trimethylamine.
- Taking laxatives can decrease intestinal transit time and reduce the amount of trimethylamine produced in the gut.
- Taking supplements to decrease the concentration of free trimethylamine in the urine.
- Activated charcoal taken at a dose of 750mg twice daily for ten days. Copper chlorophyllin taken at a dose of 60mg three times a day after meals for three weeks.
- Using soaps with a moderate pH, between 5.5 and 6.5. Trimethylamine is a strong base (pH 9.8), thus soaps with pH closer to that of normal skin help retain the secreted trimethylamine in a less volatile form that can be removed by washing.
- Taking riboflavin (vitamin B2) supplements to enhance any residual FMO3 enzyme activity. Recommended intake is 30-40mg taken 3-5 times per day with food.
- Avoiding factors that promote sweating, such as exercise, stress, and emotional upsets.
It is important that a person who has trimethylamuinuria follow the treatment advice of their health care provider. They should not attempt to self-administer these treatment approaches. Medications and supplements can have unintended interactions, and dietary restrictions can result in nutritional deficits. Choline is essential for nerve and brain development in fetuses and infants, therefore, pregnant and breast-feeding women should consult with their health care provider before restricting their dietary choline.
See a doctor if you notice a strong, unpleasant smell that doesn’t go away. They can check for more common causes, such as body odour, gum disease, a urinary tract infection or bacterial vaginosis.
Tell your doctor if you think it might be trimethylaminuria. It’s an uncommon condition and they may not have heard of it.
They may refer you to a specialist for tests to check for trimethylaminuria.
One of the most traumatic aspects of living with trimethylaminuria is that the affected person has no recourse in the medical system to find treatment, and a cure is nowhere in sight due to lack of research in this field for lack of funding. It is difficult to understand why the medical community is not well versed in this condition as a result of there being little dispersion of the limited research information sporadically published in professional journals. Thus, a sufferer very rarely receives the necessary appropriate medical attention and treatment to control his or her odor.
Can trimethylaminuria be cured by replacing the enzyme FMO3?
Unfortunately at this time, enzyme replacement therapy with the enzyme FMO3, which when absent, is believed to cause the condition, is not an option in the management of trimethylaminuria.
Trimethylaminuria causes
Mutations in the FMO3 (Flavin-containing monooxygenase 3) gene cause trimethylaminuria 3. The FMO3 gene provides instructions for making Flavin-containing monooxygenase 3 (FMO3) enzyme that breaks down nitrogen-containing compounds from the diet, including trimethylamine. This compound is produced by bacteria in the intestine during the digestion of proteins from eggs, liver, legumes (such as soybeans and peas), certain kinds of fish, and other foods. Normally, the Flavin-containing monooxygenase 3 (FMO3) enzyme converts strong-smelling trimethylamine into another molecule trimethylamine-N-oxide (TMAO) that has no odor. If the Flavin-containing monooxygenase 3 (FMO3) enzyme is missing or its activity is reduced because of a mutation in the FMO3 gene, trimethylamine is not processed properly and can build up in the body. As excess trimethylamine is released in a person’s sweat, urine, and breath, it causes the odor characteristic of trimethylaminuria. Researchers believe that stress and diet also play a role in triggering symptoms.
Although FMO3 gene mutations account for most cases of trimethylaminuria, the condition can also be caused by other factors. The strong body odor may result from an excess of certain proteins in the diet or from an abnormal increase in bacteria that produce trimethylamine in the digestive system. A few cases of the disorder have been identified in adults with liver or kidney disease. Temporary symptoms of this condition have been reported in a small number of premature infants and in some healthy women at the start of menstruation.
Secondary trimethylaminuria occurs as the result of large oral doses of dietary precursors of the offending chemical, L-carnitine, choline or lecithin. Symptoms develop when the ability of the liver enzyme (flavin-containing monooxygenase 3) to break down (metabolize) trimethylamine is inhibited. L-carnitine is used in the treatment of carnitine-deficiency syndromes and is sometimes used by athletes who believe it enhances physical strength. Choline is used in the treatment of Huntington disease and Alzheimer disease. Choline and lecithin are present in certain food supplements and ‘health’ foods. When secondary trimethylaminuria develops as a result of large oral doses of L-carnitine, choline or lecithin, the symptoms disappear as the dosage is lowered.
- Choline is present in the diet both as choline and as a component of lecithin and originates from peas and beans, organ meats and egg yolks. The total daily free choline intake is about 9 mg, and the 2.1 g of choline which provoked an increased methylamine excretion were well in excess of this 4. It is absorbed throughout the small intestine 5 and excess choline transiting to the large bowel is metabolised to methylamines by colonic bacteria 4. The absorbed fraction is either used directly, for example as a component in cell membranes, or is metabolised to glycine betaine in the mammalian liver 6. Choline is not converted directly to methylamines in human tissues; intraperitoneal injection of choline into rats does not provoke an increase in methylamine excretion 7.
- Lecithin or phosphotidylcholine is 13% choline by weight. However, even the large doses of 11.65 g lecithin used in a study did not provoke an increase in urinary total methylamine excretion, so although it is still not clear if lecithin is hydrolysed in the jejunum, it would not appear to be a factor anyway. Colonic bacteria could generate methylamines from lecithin, but it must require very large oral intake 8.
- Carnitine is produced both naturally as the L isomer in human tissues and absorbed actively and passively from the diet 9. It is excreted unchanged in the urine. Dietary L-carnitine in meat does not cause excretion of total methylamines 4. It is used medically as a supplement in some fatty acid oxidation and organic acid disorders and can also be converted to TMA by colonic bacteria 8, such that large doses are anecdotally known to provoke a fishy odor in some susceptible individuals 10.
Figure 1. Dietary sources of trimethylamine (TMA) and normal trimethylamine-N-oxide (TMAO) metabolism
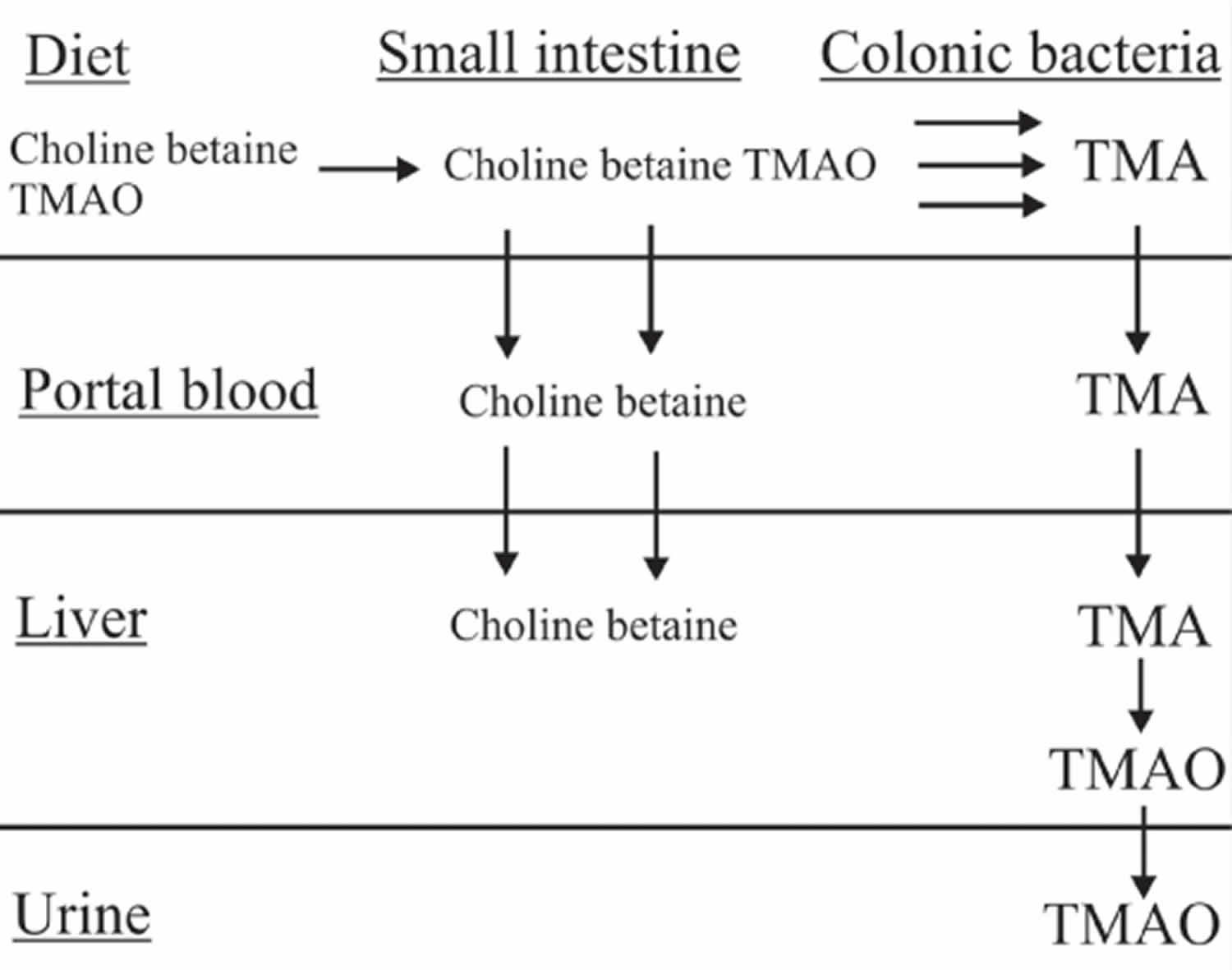 Footnote: In the human, dietary trimethylamine-N-oxide (TMAO) ingested in marine fish is reduced to trimethylamine (TMA) by the colonic microflora and absorbed by passive diffusion across the cell membranes. It enters the enterohepatic circulation and is removed by the liver. In normal liver cells, TMA is oxygenated back to the odourless TMAO by the microsomal flavin-containing monooxygenase FMO3. TMAO so formed is very water soluble and in the normal subject is excreted mainly in the urine. [Source 11]
Footnote: In the human, dietary trimethylamine-N-oxide (TMAO) ingested in marine fish is reduced to trimethylamine (TMA) by the colonic microflora and absorbed by passive diffusion across the cell membranes. It enters the enterohepatic circulation and is removed by the liver. In normal liver cells, TMA is oxygenated back to the odourless TMAO by the microsomal flavin-containing monooxygenase FMO3. TMAO so formed is very water soluble and in the normal subject is excreted mainly in the urine. [Source 11]Inheritance Pattern
Most cases of trimethylaminuria appear to be inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. Most often, the parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but typically do not show signs and symptoms of the condition. Carriers of an FMO3 mutation, however, may have mild symptoms of trimethylaminuria or experience temporary episodes of strong body odor.
Many people with trimethylaminuria inherit a faulty version of a gene called FMO3 from both their parents. This means they have 2 copies of the faulty gene.
The parents themselves might only have 1 copy of the faulty gene. This is known as being a “carrier”. They usually won’t have symptoms, although some may have mild or temporary ones.
If you have trimethylaminuria, any children you have will be carriers of the faulty gene so are unlikely to have problems. There’s only a risk they could be born with the condition if your partner is a carrier.
Genetic counseling may help you understand the risks of passing trimethylaminuria on to any children you have.
- To find a medical professional who specializes in genetics, you can ask your doctor for a referral or you can search for one yourself. Online directories are provided by the American College of Medical Genetics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) and the National Society of Genetic Counselors (https://www.findageneticcounselor.com/).
Figure 2. Trimethylaminuria autosomal recessive inheritance pattern
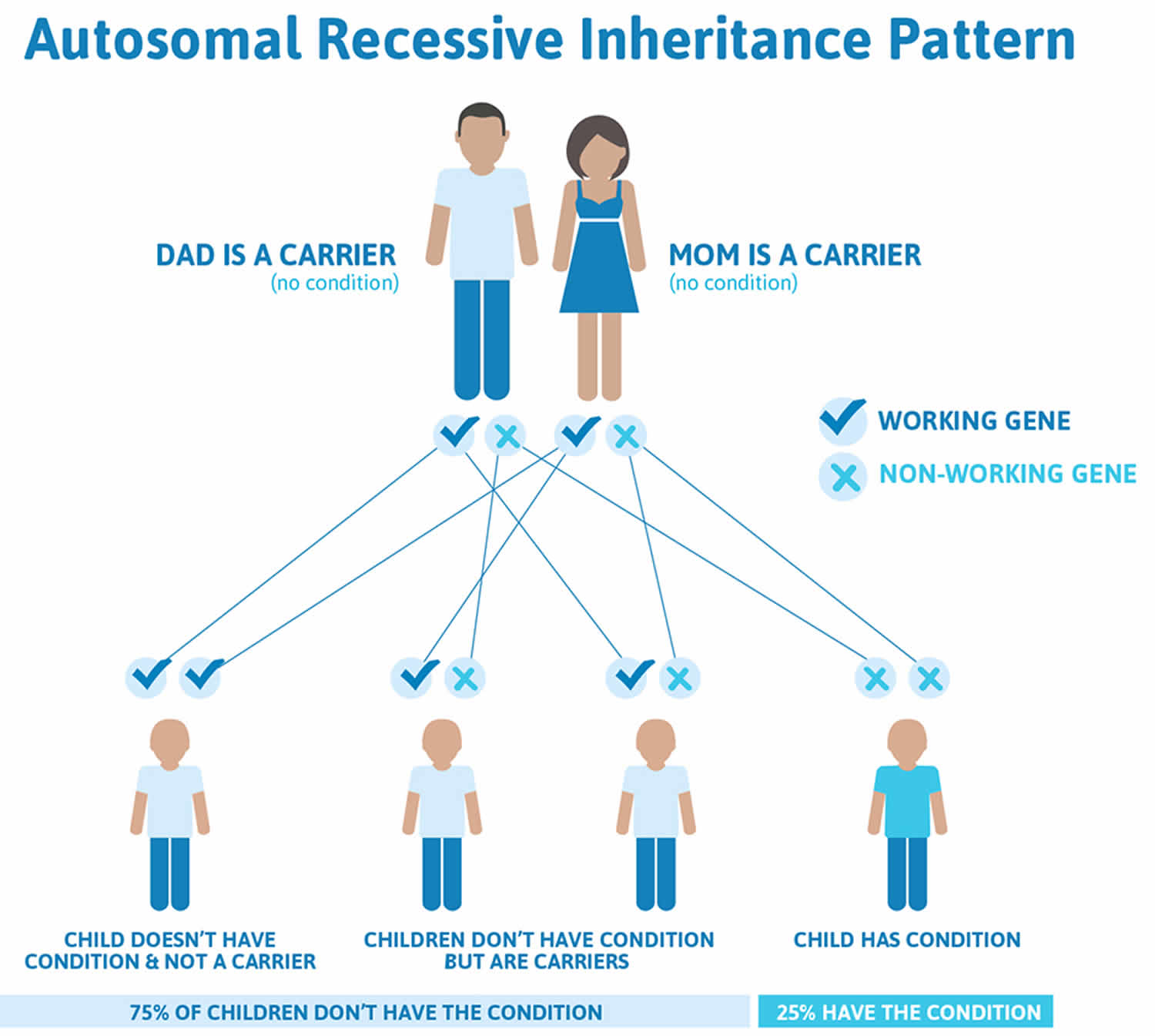
Footnote: Recessive genetic disorders occur when an individual inherits the same abnormal gene for the same trait from each parent. If an individual receives one normal gene and one gene for the disease, the person will be a carrier for the disease, but usually will not show symptoms. The risk for two carrier parents to both pass the altered gene and, therefore, have an affected child is 25% with each pregnancy. The risk of having a child who is a carrier like the parents is 50% with each pregnancy. The chance for a child to receive normal genes from both parents and be genetically normal for that particular trait is 25%. The risk is the same for males and females.
Trimethylaminuria symptoms
The fish-odor smell is the obvious symptom; otherwise affected individuals appear normal and healthy.
Trimethylaminuria symptoms can be present from birth, but they may not start until later in life, often around puberty.
The only symptom is an unpleasant smell, typically of rotting fish – although it can be described as smelling like other things – that can affect the:
- breath
- sweat
- pee
- vaginal fluids
The smell may be constant or may come and go. Things that can make it worse include:
- sweating
- stress
- certain foods – such as fish, eggs and beans
- periods
According to some experts, 40% of persons affected with trimethylaminuria report they have remained housebound for at least a week at a time out of fear of offending others. Two-thirds have contemplated suicide, and a third have attempted it. The vast majority suffer from depression or some other mental disorder, and sometimes substance abuse, since most develop a very lonely lifestyle without relief in sight at a very young age, and are struck by the overwhelming distress that comes with being socially ostracized. Erroneously, due to total lack of social awareness and understanding of trimethylaminuria, most in society perceive it as an offensive lack of personal hygiene on the part of the affected individual. As a result, people normally tend to respond to the sufferer in an overtly or covertly hostile manner.
Trimethylaminuria diagnosis and testing
The presence of the rotten-fish odor is indicative, especially in severe cases. However, diagnosis based on smell is unreliable because the odor is often episodic and not everyone can detect the smell of trimethylamine. In addition, on the basis of smell, trimethylaminuria can be difficult to distinguish from other conditions that give rise to an unpleasant body odor.
A urine test is used to diagnose trimethylaminuria. The person’s urine is tested to look for higher levels of trimethylamine. Testing can be done by giving choline by mouth followed by urine collection a certain number of times over a 24 hour period. Urine testing should be performed on two separate occasions when the individual is on a non-restricted diet. The test measures the ratio of trimethylamine to trimethylamine N-oxide present in the urine.
A carrier of this condition can be identified by the “TMA challenge” or a “TMA load” test. This involves giving an individual a 600 dmg pill of trimethylamine (TMA). Carriers of trimethylaminuria excrete 20-30 percent of total trimethylamine as the free unmetabolized amine and the rest as trimethylamine N-oxide. Non-carriers excrete less than 13% of the dose as trimethylamine. Gene testing called gene sequencing can be used to look for mutations in the FMO3 gene. Gene testing is currently available only through research laboratories.
Trimethylaminuria treatment
There’s currently no cure for primary trimethylaminuria, but there are things that can help with the smell.
Your doctor may recommend:
- Short courses of antibiotics – this can help reduce the amount of trimethylamine produced in your gut. Some severe cases may require the administration of a gut-sterilizing antibiotic such as metronidazole. This treatment reduces the number of intestinal bacteria that break down choline and trimethylamine N-oxide into trimethylamine.
- Metronidazole: particularly effective against anaerobic bacteria and protozoa
- Neomycin: appears to be the most effective in preventing formation of trimethylamine from choline
- Taking certain supplements – such as charcoal, copper chlorophyllin or riboflavin (vitamin B2).
- In the case of FMO3 gene mutations that do not completely abolish FMO3 activity, supplements of riboflavin (vitamin B2) might help maximize residual enzyme activity. Recommended riboflavin (vitamin B2) intake is 30-40mg taken 3-5 times per day with food.
- Dietary supplements such as activated charcoal and copper chlorophyllin can bind trimethylamine in the gut and hence reduce the amount available for absorption.
- Activated Charcoal: 750mg twice daily for 10 days
- Copper Chlorophyllin: 60mg three times/day after meals for 3 weeks
- Laxatives, such as Lactulose: decrease intestinal transit time, may reduce the amount of trimethylamine produced in the gut. Neutralizes the pH of trimethylamine odorous chemicals. Lactulose is a prescription item in the USA.
Things you can do that may also be helpful to:
- Avoid strenuous exercise – try gentle exercises that don’t make you sweat as much
- Try to find ways to relax – stress can make your symptoms worse
- Wash your skin with slightly acidic soap or shampoo – look for products with a pH of 5.5 to 6.5 (Normal skin pH 5.5-6.5). The use of slightly acidic soaps and body lotions can convert trimethylamine on the skin into a less volatile form that can be removed by washing.
- Use anti-perspirant
- Wash your clothes frequently
Trimethylaminuria diet
Foods to avoid
It can help to avoid certain foods that make the smell worse, such as:
- Cows’ milk
- Seafood, salt water fish, shellfish, cephalopods, and crustaceans
- Eggs
- Offal
- Legumes
- Brassicas
- Soya products
- Beans
- Peanuts
- Liver and kidney
- Food supplements and health foods that contain high doses of the trimethylamine precursors choline, L-carnithine and lecithin
Notes: Fresh water fish have a lower content of trimethylamine N-oxide (TMAO) AND THUS ARE NOT A PROBLEM.
It’s not a good idea to make any big changes to your diet on your own, particularly if you’re pregnant or planning a pregnancy, or are breastfeeding.
Your specialist can refer you to a dietitian for advice. They’ll help you make sure your diet still contains all the nutrients you need.
Choline foods
Many foods contain choline 12. The main dietary sources of choline in the United States consist primarily of animal-based products that are particularly rich in choline—meat, poultry, fish, dairy products, and eggs 12. Cruciferous vegetables and certain beans are also rich in choline, and other dietary sources of choline include nuts, seeds, and whole grains.
About half the dietary choline consumed in the United States is in the form of phosphatidylcholine 13. Many foods also contain lecithin, a substance rich in phosphatidylcholine that is prepared during commercial purification of phospholipids; lecithin is a common food additive used as an emulsifying agent in processed foods, such as gravies, salad dressings, and margarine 14. Choline is also present in breast milk and is added to most commercial infant formulas 14. Precise estimates of the percentage absorption of the different forms of dietary choline in humans are not available 14.
The U.S. Department of Agriculture’s (USDA’s) National Nutrient Database for Standard Reference 15 lists the nutrient content of many foods and provides a comprehensive list of foods containing choline arranged by choline content (https://ndb.nal.usda.gov/ndb/nutrients/report/nutrientsfrm?max=25&offset=0&totCount=0&nutrient1=421&nutrient2=&nutrient3=&subset=0&fg=&sort=c&measureby=m) and by food name (https://ndb.nal.usda.gov/ndb/nutrients/report/nutrientsfrm?max=25&offset=0&totCount=0&nutrient1=421&nutrient2=&nutrient3=&subset=0&fg=&sort=f&measureby=m).
Several food sources of choline are listed in Table 1.
Table 1. Foods high in choline
| Food | Milligrams (mg) per serving | Percent DV* |
|---|---|---|
| Beef liver, pan fried, 3 ounces | 356 | 65 |
| Egg, hard boiled, 1 large egg | 147 | 27 |
| Beef top round, separable lean only, braised, 3 ounces | 117 | 21 |
| Soybeans, roasted, ½ cup | 107 | 19 |
| Chicken breast, roasted, 3 ounces | 72 | 13 |
| Beef, ground, 93% lean meat, broiled, 3 ounces | 72 | 13 |
| Fish, cod, Atlantic, cooked, dry heat, 3 ounces | 71 | 13 |
| Mushrooms, shiitake, cooked, ½ cup pieces | 58 | 11 |
| Potatoes, red, baked, flesh and skin, 1 large potato | 57 | 10 |
| Wheat germ, toasted, 1 ounce | 51 | 9 |
| Beans, kidney, canned, ½ cup | 45 | 8 |
| Quinoa, cooked, 1 cup | 43 | 8 |
| Milk, 1% fat, 1 cup | 43 | 8 |
| Yogurt, vanilla, nonfat, 1 cup | 38 | 7 |
| Brussels sprouts, boiled, ½ cup | 32 | 6 |
| Broccoli, chopped, boiled, drained, ½ cup | 31 | 6 |
| Cottage cheese, nonfat, 1 cup | 26 | 5 |
| Fish, tuna, white, canned in water, drained in solids, 3 ounces | 25 | 5 |
| Peanuts, dry roasted, ¼ cup | 24 | 4 |
| Cauliflower, 1” pieces, boiled, drained, ½ cup | 24 | 4 |
| Peas, green, boiled, ½ cup | 24 | 4 |
| Sunflower seeds, oil roasted, ¼ cup | 19 | 3 |
| Rice, brown, long-grain, cooked, 1 cup | 19 | 3 |
| Bread, pita, whole wheat, 1 large (6½ inch diameter) | 17 | 3 |
| Cabbage, boiled, ½ cup | 15 | 3 |
| Tangerine (mandarin orange), sections, ½ cup | 10 | 2 |
| Beans, snap, raw, ½ cup | 8 | 1 |
| Kiwifruit, raw, ½ cup sliced | 7 | 1 |
| Carrots, raw, chopped, ½ cup | 6 | 1 |
| Apples, raw, with skin, quartered or chopped, ½ cup | 2 | 0 |
*DV = Daily Value. DVs were developed by the U.S. Food and Drug Administration (FDA) to help consumers compare the nutrient contents of products within the context of a total diet. The DV for choline is 550 mg for adults and children age 4 and older 16. However, the FDA does not require food labels to list choline content unless a food has been fortified with this nutrient. Foods providing 20% or more of the DV are considered to be high sources of a nutrient.
- Trimethylaminuria. https://rarediseases.info.nih.gov/diseases/6447/trimethylaminuria[↩][↩]
- Learning About Trimethylaminuria. https://www.genome.gov/11508983/[↩]
- Trimethylaminuria. https://ghr.nlm.nih.gov/condition/trimethylaminuria[↩]
- Zhang AQ, Mitchell SC, Smith RL. Dietary precursors of trimethylamine in man: a pilot study. Food Chem Toxicol. 1999;37:515–20.[↩][↩][↩]
- De La Huerga J, Popper H. Factors influencing choline absorption in the intestinal tract. J Clin Invest. 1952;31:598–603[↩]
- Lever M, Slow S. The clinical significance of betaine, an osmolyte with a key role in methyl group metabolism. Clin Biochem. 2010;43:732–44[↩]
- Norris ER, Benoit GJ. Studies on trimethylamine oxide. III. Trimethylamine oxide excretion by the rat. J Biol Chem. 1945;158:443–8.[↩]
- Zeisel SH, Gettner S, Youssef M. Formation of aliphatic amine precursors of N-nitrosodimethylamine after oral administration of choline and choline analogues in the rat. Food Chem Toxicol. 1989;27:31–4.[↩][↩]
- Bremer J. Carnitine—metabolism and functions. Physiol Rev. 1983;63:1420–80.[↩]
- Chalmers RA, Bain MD, Michelakakis H, Zschocke J, Iles RA. Diagnosis and management of trimethylaminuria (FMO3 deficiency) in children. J Inherit Metab Dis. 2006;29:162–72.[↩]
- Mackay RJ, McEntyre CJ, Henderson C, Lever M, George PM. Trimethylaminuria: Causes and Diagnosis of a Socially Distressing Condition. The Clinical Biochemist Reviews. 2011;32(1):33-43. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3052392/[↩]
- Zeisel SH. Choline. In: Ross AC, Caballero B, Cousins RJ, Tucker KL, Ziegler TR, eds. Modern Nutrition in Health and Disease. 11th ed. Baltimore, MD: Lippincott Williams & Wilkins; 2014:416-26.[↩][↩]
- Leermakers ET, Moreira EM, Kiefte-de Jong JC, Darweesh SK, Visser T, Voortman T, et al. Effects of choline on health across the life course: a systematic review. Nutr Rev 2015;73:500-22. https://www.ncbi.nlm.nih.gov/pubmed/26108618[↩]
- Zeisel SH. Choline. In: Coates PM, Betz JM, Blackman MR, et al., eds. Encyclopedia of Dietary Supplements. 2nd ed. London and New York: Informa Healthcare; 2010:136-43.[↩][↩][↩]
- U.S. Department of Agriculture, Agricultural Research Service. USDA National Nutrient Database for Standard Reference. https://ndb.nal.usda.gov/ndb/[↩]
- U.S. Food and Drug Administration. https://www.federalregister.gov/documents/2016/05/27/2016-11867/food-labeling-revision-of-the-nutrition-and-supplement-facts-labels[↩]

{kind=link}