Contents
Variegate porphyria
Variegate porphyria (VP) also known as South African genetic porphyria is a rare genetic disorder that involve defects in heme or ‘haem’ biosynthetic pathway caused by mutations (changes) in the PPOX gene which results in deficiency of the enzyme protoporphyrinogen oxidase (PPOX), the seventh enzyme in the heme biosynthetic pathway that produces porphyrins and heme. Variegate porphyria (VP) is a type of acute hepatic porphyria that is characterized by both a cutaneous porphyria with chronic skin blistering lesions (bullae) resembling porphyria cutanea tarda (PCT) and an acute porphyria with severe episodic neurovisceral symptoms resembling acute intermittent porphyria (AIP) 1, 2, 3, 4, 5, 6, 7, 8. Hepatic (liver) porphyrias are porphyrias in which the enzyme deficiency occurs in the liver. Hepatic porphyrias include acute intermittent porphyria (AIP), variegate porphyria (VP), aminolevulinic acid dehydratase deficiency porphyria (ALAD), hereditary coproporphyria (HCP), and porphyria cutanea tarda (PCT) 5. Protoporphyrinogen and coproporphyrinogen accumulate in the liver in variegate porphyria because protoporphyrinogen oxidase (PPOX) enzyme is deficient, and become oxidized to protoporphyrin and coproporphyrin, which are transported in the blood plasma and cause the skin to be sensitive to sunlight. Variegate porphyria is characterized by elevated urinary porphobilinogen (PBG) and elevated plasma and fecal porphyrins. The neurological symptoms are associated with accumulation of porphyrin precursors, namely, delta-aminolevulinic acid (ALA) and porphobilinogen (PBG).
The majority of individuals who inherit the PPOX gene for variegate porphyria remain asymptomatic throughout life, especially when known triggering factors are avoided 9, 10, 11. Triggering factors include a variety of drugs, hormones (especially progesterone), reduced intake of calories and carbohydrate, alcohol, and stress induced by infection or other illness. Only a minority of people with the protoporphyrinogen oxidase (PPOX) enzyme defect develop clinical signs and symptoms.
Variegate porphyria (VP) specific symptoms can vary greatly from one person to another, but typically begin in adulthood (occur after puberty or later); cases reported in infancy or early childhood are rare 12. Some affected individuals present with skin symptoms, some with neurological symptoms and some with both. Blistering skin lesions (bullae)) and skin fragility of sun-exposed skin are the most common skin (cutaneous) symptoms, which are chronic in many people with variegate porphyria. Common neurological symptoms include abdominal pain, nausea, vomiting, constipation, extremity pain and weakness, anxiety, restlessness and convulsions. Many different PPOX gene mutations have been identified in different families with variegate porphyria. The PPOX genetic mutation in a family is inherited as an autosomal dominant trait meaning that a mutation is present in only one of the pair of PPOX (protoporphyrinogen oxidase) genes, but many individuals who inherit a PPOX gene mutation do not develop any symptoms (asymptomatic).
In people with variegate porphyria (VP), acute attacks almost always start with severe abdominal pain but sometimes pain in the chest, back, or thighs, and are often accompanied by nausea, vomiting, and constipation 13. Heart rate and blood pressure are commonly increased 13. These symptoms and signs are all due to the effects of the disease on your nervous system. Confusion, convulsions, and muscular weakness, due to impairment of the nerves controlling your muscles (peripheral neuropathy), may lead to paralysis. An acute attack usually lasts for days or weeks. Recovery from severe paralysis is generally slow 13.
Although, in most cases, the symptoms of variegate porphyria (VP) occur after puberty or later, very rare cases have been described where symptoms developed during infancy or childhood. Most such cases are homozygous cases who have inherited the same PPOX gene mutation from both parents. Homozygous cases may have impaired mental development and photosensitivity, but acute attacks are not prominent. Mental retardation is prevalent in the neonatal form of variegate porphyria 4.
Variegate porphyria is especially common in South Africa in individuals of Dutch ancestry due to a founder effect, where it has been estimated that 1 to 3 cases per 1,000 individuals of the white population are affected and are heterozygous for the PPOX genetic variant p.Arg59Trp 14, 15. A founder effect is when a small isolated population of settlers (original settlers from the Netherlands) expands over several generations leading to a high prevalence of a genetic trait. Most individuals with variegate porphyria in South Africa carry the same PPOX gene mutation and are descendents of a Dutch settler from the late 1600s.
Variegate porphyria is much less prevalent in other countries. Some reports suggest that variegate porphyria affects more women than men. The incidence is estimated to occur in 3.2 in 1,000,000 individuals in the general population in European populations 16.
Rare cases of homozygous variegate porphyria (HVP) also called “biallelic variegate porphyria” which include biallelic PPOX gene mutations in homozygous and compound heterozygous form have been described with disease onset in infancy 17, 18. Usually the affected children became symptomatic in the first days or months of life. Heterozygous and homozygous variegate porphyria cases do not only differ concerning the age at onset, but also regarding the clinical presentation. Most of the homozygous variegate porphyria (HVP) cases lead to a variable neurodevelopmental disorder while acute neurovisceral attacks have not been reported 3.
A diagnosis of variegate porphyria is suspected based upon your symptoms and examination of your skin. None of the symptoms are specific, so the diagnosis must be confirmed by biochemical testing. In the evaluation of neurological symptoms, the other acute porphyrias need to be considered. For initial screening, a spot urine sample should be obtained for measurement of porphobilinogen (PBG), delta aminolevulinic acid (ALA) and total porphyrins. If none of these is elevated, acute porphyrias can be excluded as a cause of recent or concurrent symptoms. Urine porphobilinogen (PBG) measurement is most important and specific for acute porphyrias. However, porphobilinogen (PBG) and aminolevulinic acid (ALA) may be less elevated and return to normal more quickly after an attack of variegate porphyria or hereditary coproporphyria (HCP) than in acute intermittent porphyria (AIP). Therefore, measurement of total urine porphyrins is important, keeping in mind that an elevation of urine porphyrins can occur in many other medical conditions.
When blistering skin signs and symptoms are present, porphyria cutanea tarda (PCT), hereditary coproporphyria (HCP) and even congenital erythropoietic porphyria (CEP) are possibilities to differentiate. Measurement of plasma and fecal porphyrins and determining the wavelength of the fluorescence peak of plasma porphyrins is useful in differentiating these conditions.
Molecular genetic testing to identify a PPOX gene mutation is recommended for all biochemically confirmed cases of variegate porphyria (VP). Molecular testing is sometimes useful when symptoms have been absent for months or years and biochemical abnormalities are no longer present. Knowing the PPOX gene mutation in a family enables other family members to be tested reliably for the same gene mutation.
There is no recognized treatment effective for variegate porphyria (VP), and avoidance of sunlight and precipitating factors are the current forms of management. Acute attacks of variegate porphyria often require hospitalization for managing symptoms like severe pain, nausea, and vomiting. Strong pain medications and drugs to control nausea are commonly used. Doctors also monitor for muscle weakness and breathing issues. Identifying and stopping any triggers, like unsafe medications, is important for recovery.
The first steps of variegate porphyria (VP) treatment are:
- Stopping medications that may trigger the attack
- Staying hydrated with IV fluids that contain a high amount of carbohydrates (like dextrose)
- Managing pain with narcotic painkillers (opiates)
- Treating nausea and vomiting with medications like phenothiazines (such as chlorpromazine or prochlorperazine)
If these steps don’t help, an intravenous (IV) heme infusion like Panhematin, which is available in the U.S. as lyophilized hematin, is recommended for 3-14 days. Heme infusions are important because they prevent porphyria attacks from worsening and causing permanent nerve damage. In the U.S., Panhematin is the FDA approved treatment, but a similar medication, heme arginate (Normosang), is used in Europe and South Africa.
“Heme infusion” refers to the oxidized form of iron protoporphyrin IX (PPIX), but is also the generic term for heme preparations used as intravenous (IV) therapies for acute porphyrias, such as lyophilized hematin (Panhematin) and heme arginate (Normosang). When these hemin preparations are infused intravenously, the heme is bound to circulating albumin as heme albumin. The heme albumin is taken up by liver cells and decreases the synthesis of liver ALAS1 (aminolevulinic acid synthase 1), the rate-controlling enzyme for heme synthesis, which limits the formation of porphyrin in the liver and marrow. Hemin represses the heme pathway in the liver and lowers aminolevulinic acid, porphobilinogen and porphyrins, and is associated with more rapid recovery from an attack. The duration of this heme treatment depends on the condition and how well the affected person is responding. If the symptoms worsen, immediate treatment with a heme infusion is necessary to prevent nerve damage.
While receiving heme treatment, doctors may monitor certain chemicals in the urine such as aminolevulinic acid (ALA), porphobilinogen (PBG) and porphyrins to see if heme treatment is working.
It’s crucial that the doctor follows the right dosage guidelines, as giving too much heme in a single dose can cause temporary kidney problems, though normal doses are generally safe.
If the attack is mild, IV glucose may help, as it works similarly to heme but is less potent. Glucose given intravenously has a similar effect, but because it is less potent, glucose is only a temporary solution until heme can be administered.
If seizures occur during an attack, controlling them can be complicated because many anti-seizure medications are processed by the liver and can make the attack worse. The following treatments are generally safe for seizures during porphyria 7:
- Magnesium sulfate or diazepam are usually the first choices for controlling seizures.
- Lorazepam is often used for status epilepticus (a severe, prolonged seizure).
Doctors will also check for any imbalances in the body’s electrolytes, which can cause or worsen seizures.
For long-term seizure control, medications like gabapentin may be preferred because they are not metabolized by the liver.
In 2019, the FDA approved Givosiran (Givlaari), a small interfering RNA (siRNA), a medication designed to prevent porphyria attacks. Monthly subcutaneous injections of givosiran can be effective for prevention of frequently recurring attacks 19. Givosiran (Givlaari) works by reducing levels of a liver enzyme that triggers these attacks and has been shown to reduce the frequency of attacks by 74% in clinical trials.
Hospitalization is often necessary for managing severe symptoms of an acute attack, such as 7:
- Severe pain, which may require strong pain medications
- Nausea and vomiting, treated with medications like Zofran (ondansetron) or chlorpromazine
- Muscle weakness and breathing problems, which require careful monitoring
After an acute attack, it’s important to avoid anything that might trigger future episodes. Common triggers include certain medications, stress, or fasting. The American Porphyria Foundation provides a list of drugs that are safe or unsafe for individuals with porphyria. It’s a good idea for patients to wear a Medic Alert bracelet or carry a wallet card that lists the condition and any medications you need to avoid.
For those who have frequent attacks, there are preventive treatments 7:
- Gonadotropin-releasing hormone (GnRH) analogues can help prevent attacks that follow a monthly cycle in women around menstruation.
- Some people may also receive prophylactic heme infusions (once a week or twice a week) to prevent attacks.
People with variegate porphyria who have chronic skin symptoms should avoid exposure to sunlight to help prevent flare-ups. Wearing protective clothing like hats and long-sleeved shirts and using opaque sunscreen can reduce skin damage. Traditional sunscreens may not be effective, so it’s important to choose products specifically designed for porphyria patients. Iron supplementation is likely to cause a skin flare-up and should preferably be administered in the vein.
Since porphyria is hereditary, genetic counseling can help the patient and their families to understand the condition and how it may affect future generations.
Figure 1. Variegate porphyria (VP)
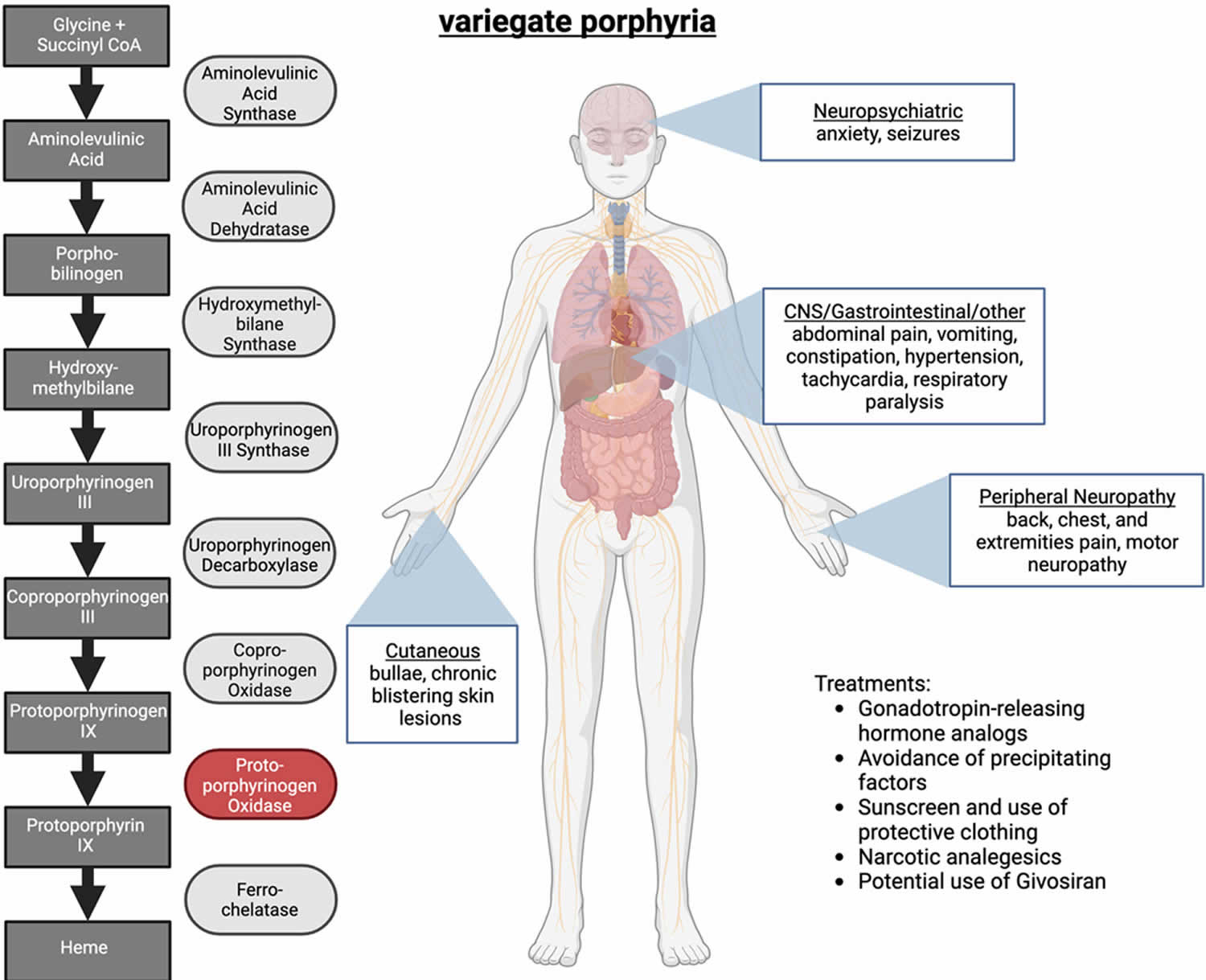
Footnotes: Variegate porphyria (VP) is caused by decreased activity of protoporphyrinogen oxidase (PPOX), which causes accumulation of protoporphyrinogen and coproporphyrinogen. Variegate porphyria (VP) is both an acute neurovisceral and skin porphyria, with acute episodes resembling acute intermittent porphyria (AIP) and skin signs and symptoms resembling porphyria cutanea tarda (PCT). There is no recognized treatment effective for variegate porphyria (VP), and avoidance of sunlight and precipitating factors are the current forms of management.
[Source 2 ]Figure 2. Variegate porphyria (VP) signs and symptoms
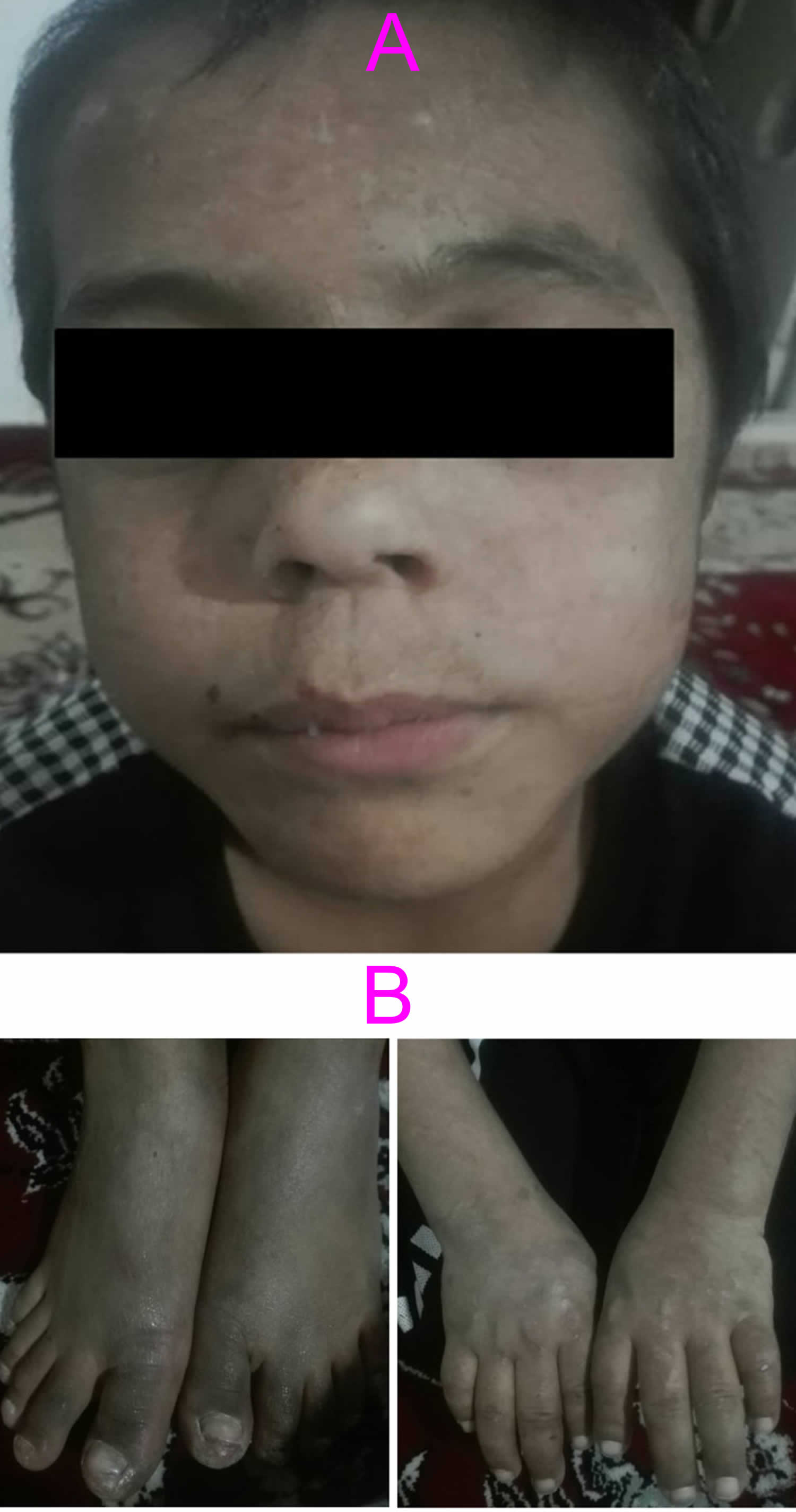
Footnotes: A 7-year-old boy was admitted to our hospital with three episodes of generalized tonic seizure in the last 6 months. He didn’t have fever in each episode. His standard laboratory test results were normal without electrolytes imbalance. On physical examination, he was shorter than average. He had coarse and hairy facial features (A). Cutaneous symptoms, including erosive lesions scars, hyperpigmentation, fragility and blistering of sun-exposed skin, and thickened skin on hands and feet were observed (B). He was the second child of the family from parents with consanguineous marriage.
[Source 8 ]Figure 3. Porphyrin molecular structure
Footnote: Molecular structure of porphyrin (M represent metal ions, such as Mg, Cu, Fe, Zn, etc.).
[Source 20 ]Figure 4. Hemoglobin molecular structure
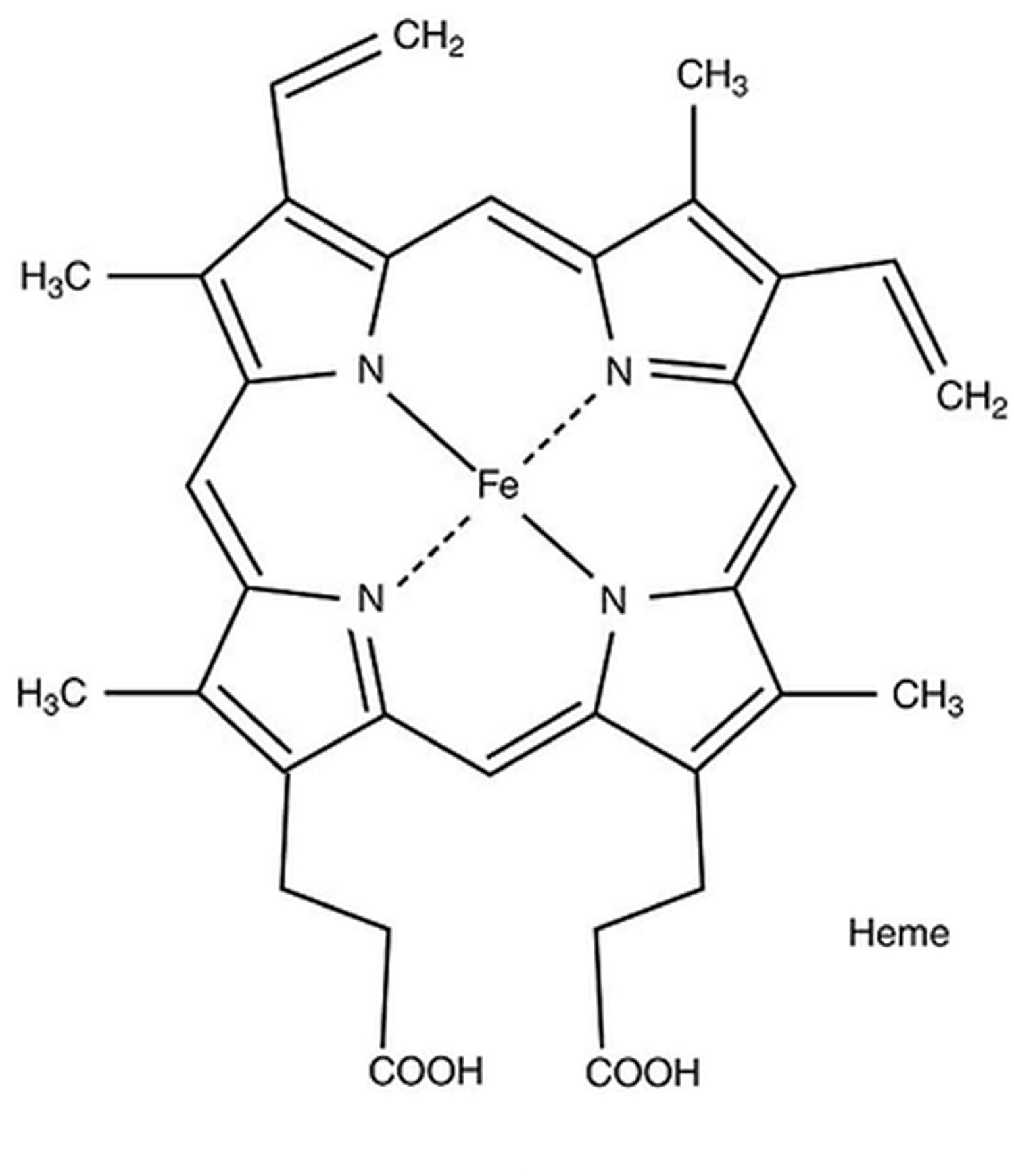
Figure 5. Heme biosynthesis pathway
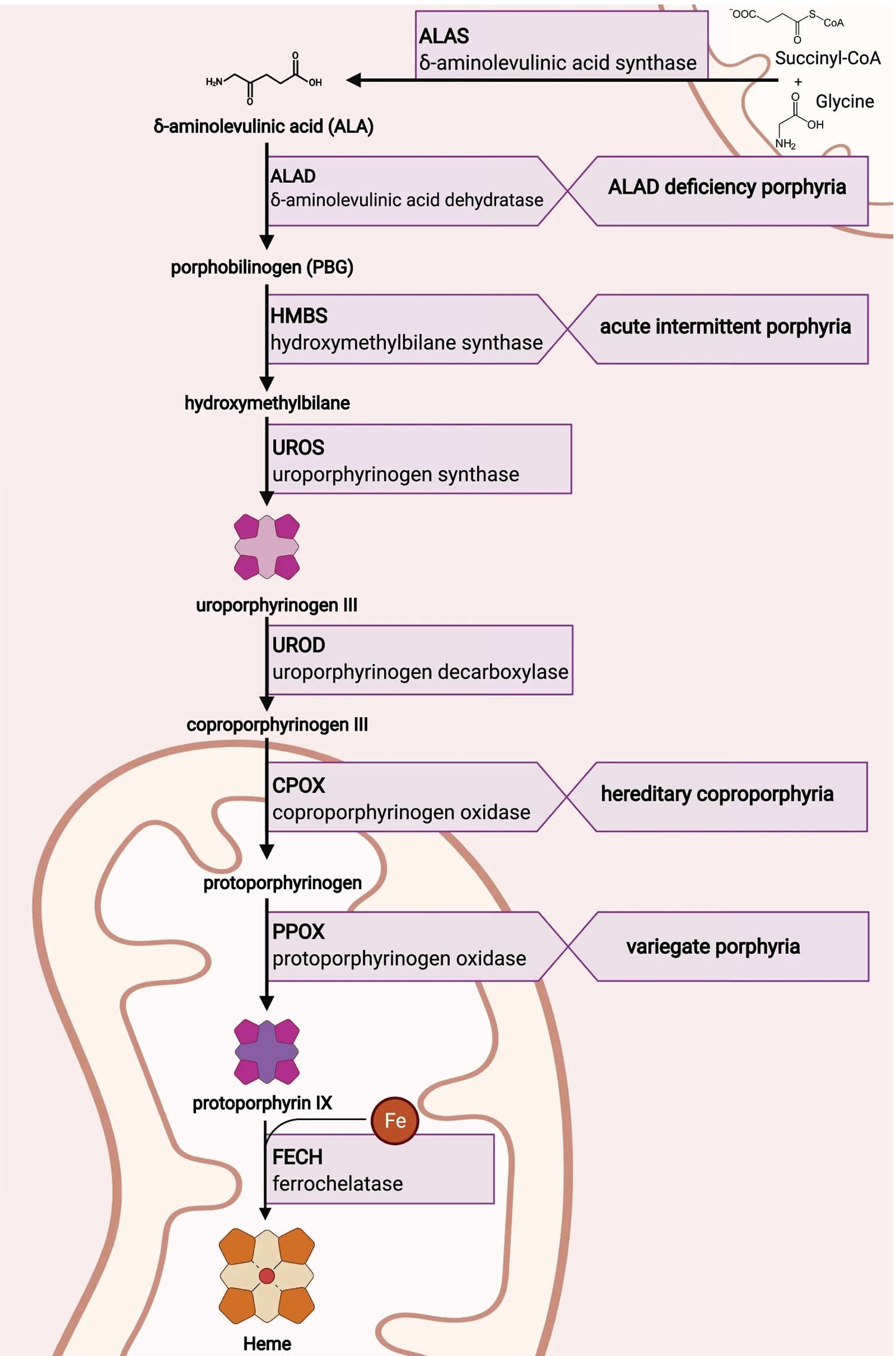
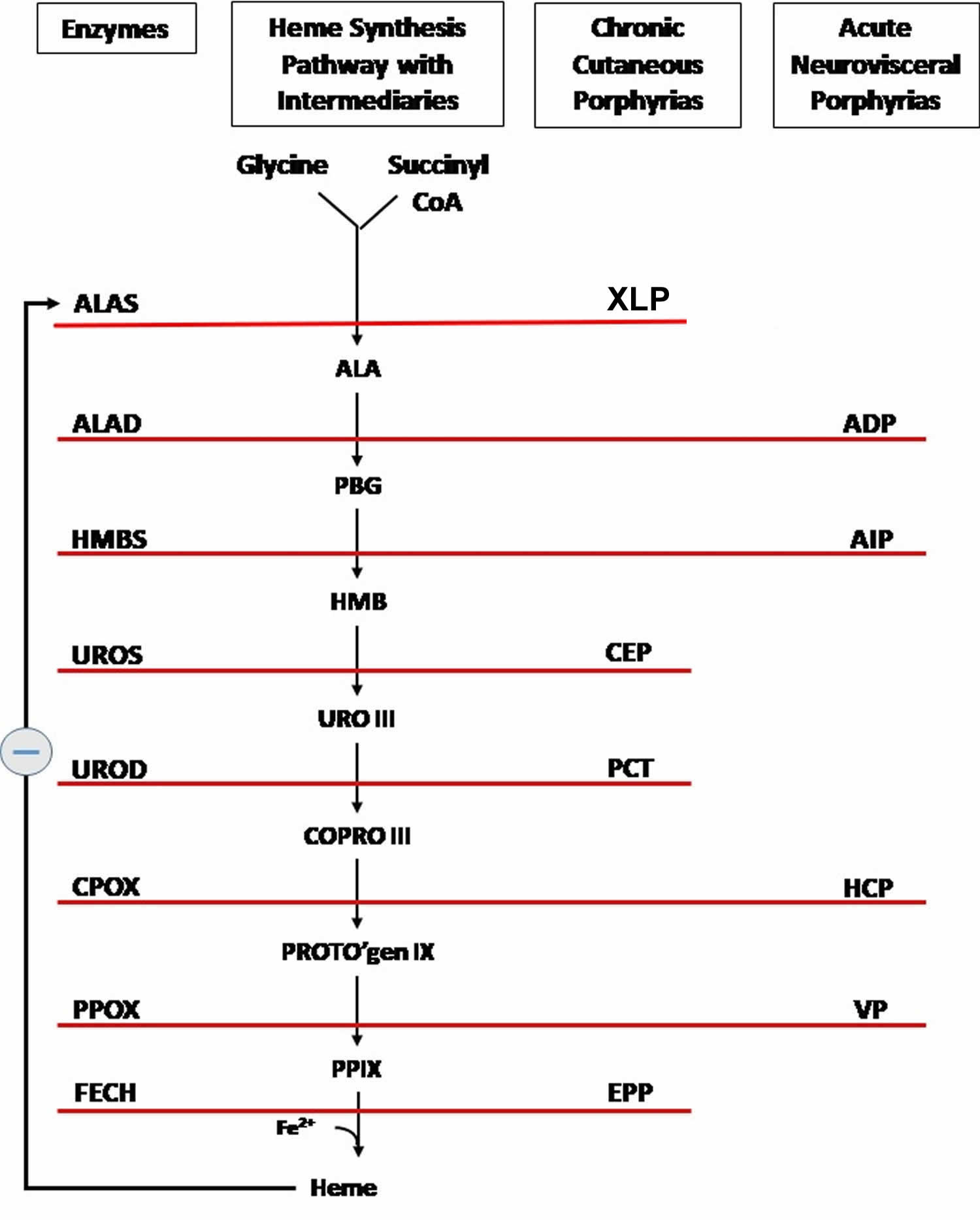
Footnotes: The heme biosynthetic pathway requires 8 enzymatic steps. Heme synthesis pathway showing the enzymes involved in the heme synthesis pathway and the associated porphyrias with the disruption of each specific enzyme. Gain-of-function variants in ALAS2 result in X-linked protoporphyria (XLP), and loss-of-functions variants in FECH result in erythropoietic protoporphyria (EPP). In both X-linked protoporphyria (XLP) and erythropoietic protoporphyria (EPP), metal-free protoporphyrin IX (PPIX) accumulates in erythroblasts, erythrocytes, the plasma, and the biliary system. Metal-free protoporphyrin IX (PPIX) is photosensitive, particularly to visible light in the blue range, and the light-mediated activation of metal-free protoporphyrin IX (PPIX) produces free radicals that damage the surrounding tissues.
Enzymes, encoded by genes, catalyze each of the steps. Gene mutations cause deficient enzyme production. Disruptions are indicated by red lines connecting enzymes with the resultant porphyrias. ALAS (ALAS2) = aminolevulinate synthase (aminolevulinate synthase 2); ALAD = delta-aminolevulinic acid dehydratase; PBGD = porphobilinogen dehydratase; HMBS = hydroxymethylbilane synthase; UROS = uroporphyrinogen-III synthase; UROD = uroporphyrinogen III decarboxylase; CPOX = coproporphyrinogen-III oxidase; PPOX = protoporphyrinogen oxidase; FECH = ferrochelatase.
Porphyrias resulting from disruption of enzyme production. XLP (X-linked protoporphyria); ADP (aminolevulinic acid dehydratase porphyria); AIP (acute intermittent porphyria); CEP (congenital erythropoietic porphyria); PCT (porphyria cutanea tarda); HCP (hereditary coproporphyria); VP (variegate porphyria); EPP (erythropoietic protoporphyria).
Abbreviations: ALA = aminolevulinic acid; PBG = porphobilinogen; HMB = hydroxymethylbilane; URO III = uroporphyrinogen III; COPRO III = coproporphyrinogen III; PROTO’gen IX protoporphyrinogen IX; PPIX = protoporphyrin IX; Fe2+ = iron.
[Source 22 ]Figure 6. Variegate porphyria pathophysiology
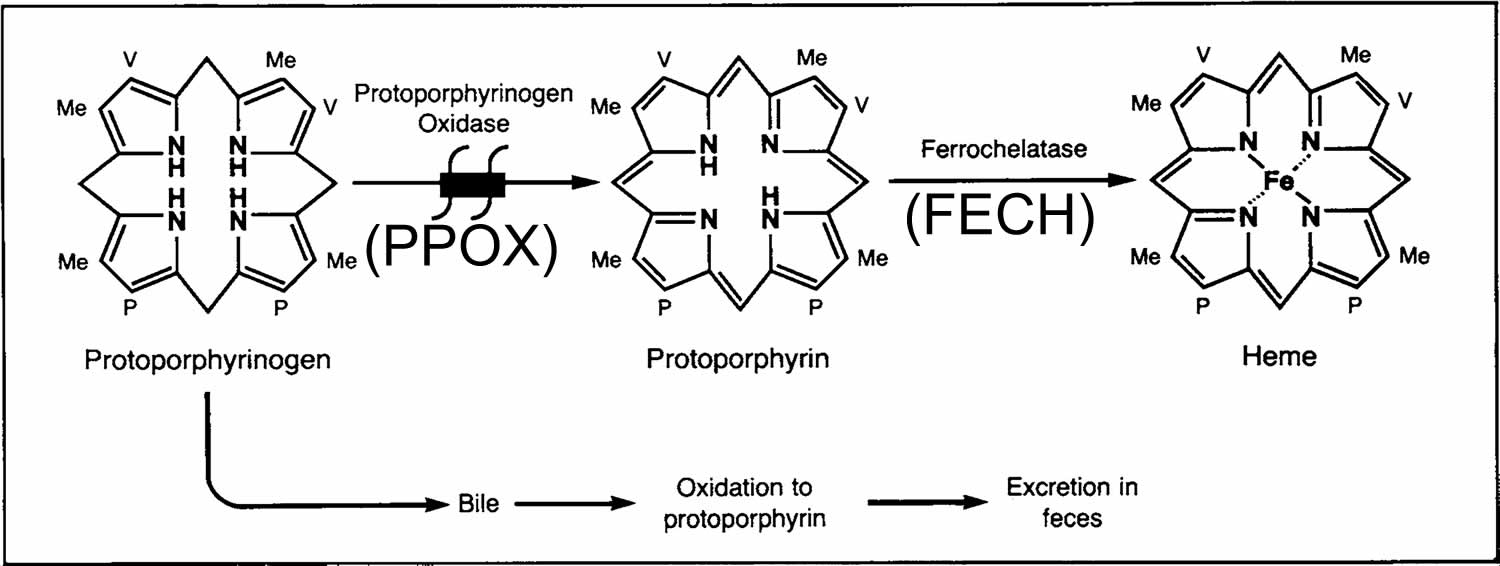
Footnotes: The primary enzyme abnormality in variegate porphyria (VP) is a deficiency of protoporphyrinogen oxidase (PPOX) enzyme in the mitochondria, which causes excessive accumulation and excretion of protoporphyrinogen. The subsequent nonenzymatic oxidation of protoporphyrinogen to protoporphyrin leads to increased levels of protoporphyrin in bile and the feces.
[Source 6 ]Variegate porphyria cause
Variegate porphyria (VP) is caused by mutations of the protoporphyrinogen oxidase (PPOX) gene. The PPOX gene provides instructions for making an enzyme known as protoporphyrinogen oxidase (PPOX). The protoporphyrinogen oxidase (PPOX) enzyme is the seventh enzyme in the heme biosynthetic pathway that produces porphyrins and heme. Heme (haem) is an iron (Fe2+) containing porphyrin (iron protoporphyrin) and is a part of many heme-containing proteins (hemoproteins) in the body. Hemoproteins interact with oxygen, and some are involved in electron transport and energy metabolism. The best-known hemoprotein is hemoglobin (the protein that carries oxygen in the blood), which is made in the bone marrow, makes red blood cells red, and transports oxygen from the lungs to other tissues. However, your bone marrow and hemoglobin are not affected in variegate porphyria (VP). In variegate porphyria the heme pathway in the liver, which makes heme for other important hemoproteins, is affected. The production of heme is a multi-step process that requires 8 different enzymes. Protoporphyrinogen oxidase (PPOX) enzyme is responsible for the seventh step in this process, in which two hydrogen atoms are removed from protoporphyrinogen IX (the product of the sixth step) to form protoporphyrin IX. In the final step, another enzyme called ferrochelatase (FECH) modifies protoporphyrin IX by inserting an iron (Fe2+) atom to produce heme.
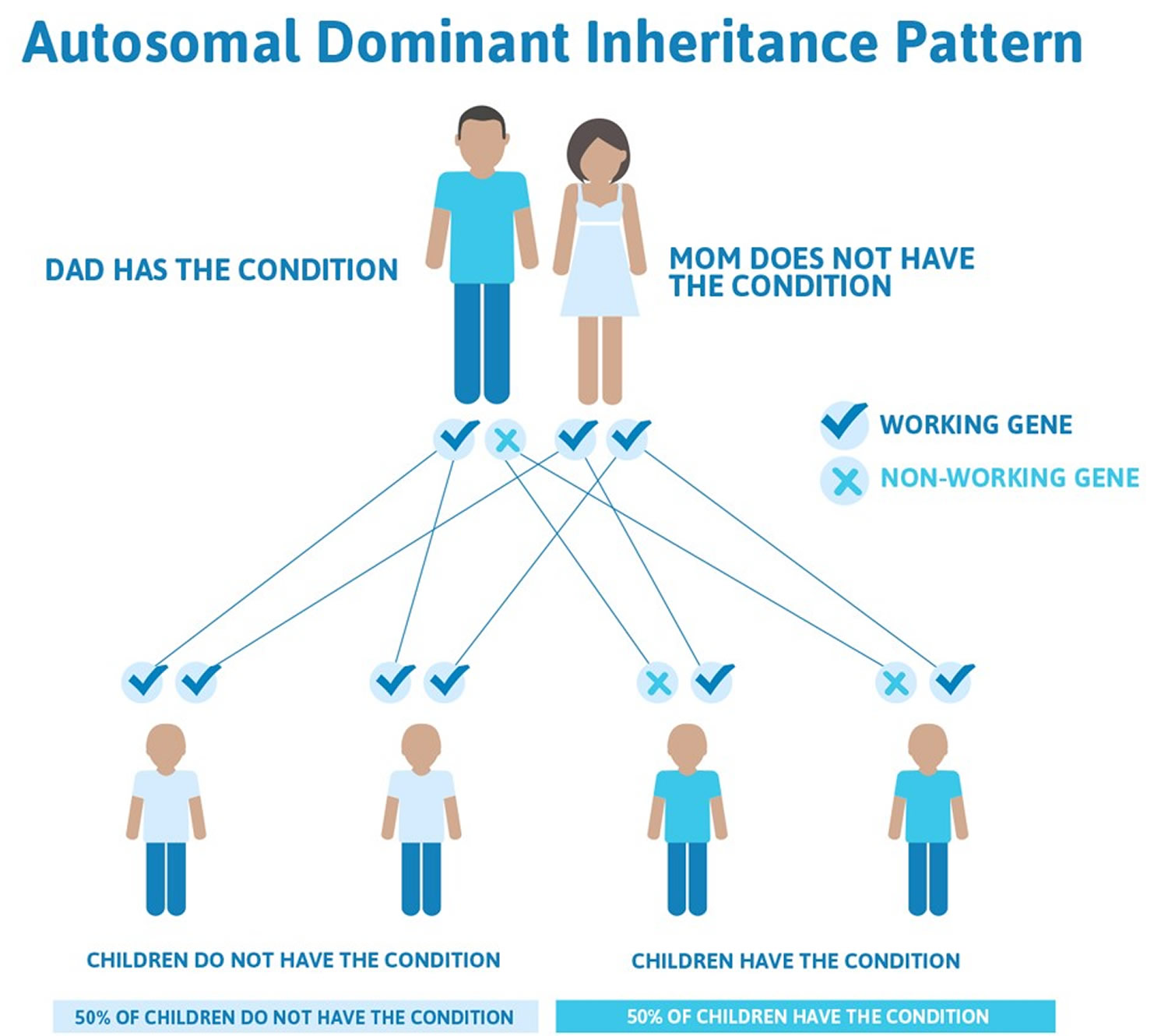
The PPOX gene mutation is inherited as an autosomal dominant trait within a family, which means that a single PPOX gene mutation is inherited from one parent and, in the presence of other triggering factors, is sufficient to cause variegate porphyria (VP). The abnormal PPOX gene can be inherited from either parent, or on rare occasions can be the result of a new mutation (de novo mutation) in the affected individual for the first time and is not inherited from either parent. In autosomal dominant inheritance pattern the risk of passing the abnormal gene from affected parent to offspring is 50 percent for each pregnancy regardless of the sex of the resulting child.
The PPOX (protoporphyrinogen oxidase) gene is located on the long arm (q) of chromosome 1 (1q22). Chromosomes, which are present in the nucleus of human cells, carry the genetic information for each individual. Human body cells normally have 46 chromosomes. Pairs of human chromosomes are numbered from 1 through 22 and the sex chromosomes are designated X and Y. Males have one X and one Y chromosome and females have two X chromosomes. Each chromosome has a short arm designated “p” and a long arm designated “q”. Chromosomes are further sub-divided into many bands that are numbered. For example, “chromosome 1q22” refers to band 22 on the long arm of chromosome 1. The numbered bands specify the location of the thousands of genes that are present on each chromosome.
The PPOX (protoporphyrinogen oxidase) gene contains instructions for creating protoporphyrinogen oxidase (PPOX) enzyme, one of the eight enzymes necessary for the production of heme. Mutations of the PPOX gene result in deficient levels of protoporphyrinogen oxidase (PPOX) enzyme, which, in turn, disrupts the biochemical process to create heme in the liver. This disruption causes porphyrins and porphyrin precursors to accumulate in the liver and these are then transported to other parts of the body to affect the nervous system and skin.
Most patients with autosomal dominantly inherited variegate porphyria (VP) are genetically heterozygous for a pathogenic variant in the PPOX gene 1. As heterozygous variegate porphyria shows an incomplete penetrance of approximately 40%, many carriers may be asymptomatic during their whole life 23. If symptomatic, heterozygous variegate porphyria usually presents in adulthood and rarely before puberty 24, 1, with a combination of severe skin disease and acute symptoms with attacks of severe abdominal pain, neurologic symptoms including peripheral nerve palsies, seizures and nausea as well as nonspecific psychiatric abnormalities.
A variety of different triggers are known to lead to attacks in individuals with variegate porphyria. Many of these triggers act by increasing heme synthesis in the liver, which makes the protoporphyrinogen oxidase (PPOX) enzyme deficiency more significant and increases the accumulation of porphyrins and porphyrin precursors. As noted above, triggers include a variety of drugs, hormones (especially progesterone), reduced intake of calories and carbohydrate, alcohol, and stress induced by infection or other illness.
The American Porphyria Foundation offers a drug database with safety information about the interaction of specific drugs in patients with porphyria (https://porphyriafoundation.org/for-healthcare-professionals/ahp-drug-safety-database/) or https://porphyriafoundation.org/drugdatabase/drug-safety-database-search/.
The EPNET/NAPOS Database should also be consulted. The Norwegian Porphyria Centre (NAPOS), with the European Porphyria Network (EPNET), has created a list of medications that clinicians must avoid using in porphyria patients (https://drugsporphyria.net/). These drugs include ketamine, thiopental, chloramphenicol, erythromycin, nitrofurantoin, rifampicin, trimethoprim/sulfamethoxazole, spironolactone, methyldopa, valproic acid, carbamazepine, phenytoin, phenobarbital, primidone, and risperidone 25. For information on prescribing medication in the context of certain conditions (e.g., HIV, epilepsy, malaria), see https://porphyria.uct.ac.za/porphyria-professionals/prescribing-porphyria-treatment-specific-disorders-poprhyria/therapy-epilepsy.
Variegate porphyria inheritance pattern
Variegate porphyria (VP) is inherited in an autosomal dominant pattern manner with reduced penetrance 1. All individuals inherit two copies of each gene. In autosomal dominant conditions, having a mutation in just one copy of the PPOX gene causes the person to have variegate porphyria (VP). The mutation can be inherited from either parent. Some people are born with variegate porphyria due to a new genetic mutation (de novo mutation) and do not have a history of this condition in their family. There is nothing either parent can do, before or during pregnancy, to cause a child to have this.
Each child of an individual with an autosomal dominant condition has a 50% or 1 in 2 chance of inheriting the mutation and the condition. Offspring who inherit the mutation may develop variegate porphyria, although they could be more or less severely affected than their parent. Sometimes a person may have a PPOX gene mutation for variegate porphyria and show no signs or symptoms of it.
Often autosomal dominant conditions can be seen in multiple generations within the family. If one looks back through their family history they notice their mother, grandfather, aunt/uncle, etc., all had the same condition. In cases where the autosomal dominant condition does run in the family, the chance for an affected person to have a child with the same condition is 50% regardless of whether it is a boy or a girl. These possible outcomes occur randomly. The chance remains the same in every pregnancy and is the same for boys and girls.
- When one parent has the abnormal gene, they will pass on either their normal gene or their abnormal gene to their child. Each of their children therefore has a 50% (1 in 2) chance of inheriting the changed gene and being affected by the condition.
- There is also a 50% (1 in 2) chance that a child will inherit the normal copy of the gene. If this happens the child will not be affected by the disorder and cannot pass it on to any of his or her children.
There are cases of autosomal dominant gene changes, or mutations, where no one in the family has it before and it appears to be a new thing in the family. This is called a de novo mutation. For the individual with the condition, the chance of their children inheriting it will be 50%. However, other family members are generally not likely to be at increased risk.
Rare cases of homozygous variegate porphyria (HVP) also called “biallelic variegate porphyria” which include biallelic PPOX gene mutations in homozygous and compound heterozygous form have been described with disease onset in infancy 17, 18. Usually the affected children became symptomatic in the first days or months of life. Heterozygous and homozygous variegate porphyria cases do not only differ concerning the age at onset, but also regarding the clinical presentation. Most of the homozygous variegate porphyria (HVP) cases lead to a variable neurodevelopmental disorder while acute neurovisceral attacks have not been reported 3.
Genetic counseling is recommended for affected individuals and their families.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://abgc.learningbuilder.com/Search/Public/MemberRole/Verification) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (https://clinics.acmg.net/) has a searchable database of medical genetics clinic services in the United States.
Figure 7. Variegate porphyria autosomal dominant inheritance pattern
Variegate porphyria signs and symptoms
The symptoms and severity of variegate porphyria can vary greatly from one person to another. Symptoms are rarely apparent before puberty. Affected individuals often develop skin (cutaneous) or neurovisceral (neurological) abnormalities or both. It is important to note that affected individuals may not have all of the symptoms discussed below. Affected individuals should talk to their doctor about their specific case, associated symptoms and overall prognosis.
Many individuals with variegate porphyria may not develop any notable symptoms (asymptomatic) for all or most of their lives 13. Other individuals can develop a variety of symptoms. Skin (cutaneous) symptoms are chronic and commonly last for months or years. Neurological symptoms usually occur as acute attacks lasting days or weeks and occasionally become chronic. Acute attacks can be severe and may occur in the absence of skin abnormalities.
Symptoms during an acute attack may include intense abdominal discomfort or pain, which is usually steady but may be cramping and is often accompanied by nausea, vomiting, constipation (occasionally diarrhea) and trouble urinating 9. These symptoms are due to effects on the nerves of your bowel and bladder. The central nervous system (brain and spinal cord) is often affected with insomnia, restlessness, anxiety, agitation, confusion, hallucinations and seizures. The level of sodium in your blood may decrease (hyponatremia) and cause convulsions. The peripheral nerves are affected, leading to severe pain in the extremities, back or chest and, especially with more prolonged attacks, paralysis of muscles (motor neuropathy). This may progress to involve all extremities (quadriparesis) and the muscles that control breathing (respiratory paralysis). These acute neurovisceral symptoms may be severe and are potentially deadly 1.
Your urine may be reddish due to increased porphyrins, and dark due to porphobilin, which is a brownish degradation product of porphobilinogen (PBG) 13.
Increases in heart rate (tachycardia) and blood pressure are very common on examination during attacks. Fever is usually absent or slight, because the nerve damage (neuropathy) is not inflammatory. Reflexes may be increased initially and decreased or absent if motor neuropathy (nerves that control muscle movement are damaged) advances.
A variety of triggers are known to set off an acute attack. These include a variety of drugs, steroid hormones, alcohol, decreased intake of calories or carbohydrates, and metabolic or possibly psychological stress. Women may have attacks during the second half of the menstrual cycle when progesterone levels are highest. In some cases, no trigger can be identified.
Chronic skin abnormalities result from photosensitivity, a condition in which the skin is abnormally sensitive to sunlight, causing blistering skin lesions (bullae). Symptoms include abnormally fragile skin, blisters (bullae), milia, which are tiny, white bumps or cysts, and excessive hair growth (hypertrichosis). Blisters are slow to heal and can scar leaving patches of skin that are abnormally dark (hyperpigmentation) or light (hypopigmentation). Skin symptoms may be less common in individuals who live in nontropical climates. Some individuals with Variegate Porphyria only develop skin abnormalities, others only develop neurological symptoms, and some have both.
Individuals with variegate porphyria are at an increased risk for developing a form of liver cancer known as hepatocellular carcinoma (HCC). There is also risk of developing chronic kidney disease.
Skin signs and symptoms
Cutaneous symptoms (skin-related) are due to photosensitivity, where the skin is overly sensitive to sunlight (photosensitivity) and they include:
- Fragile skin that blisters easily
- Blisters (bullae) that heal slowly and may leave scars
- Milia (tiny white bumps or cysts)
- Excessive hair growth (hypertrichosis), especially on exposed areas
- Scarring may result in darkened (hyperpigmented) or lightened (hypopigmented) skin patches.
Cutaneous symptoms (skin-related) can last months or years.
Skin symptoms may be less common in individuals living in non-tropical climates.
Chronic blistering skin lesions on sun exposed skin (photosensitivity), typically on the backs of the hands, is the most common manifestation of variegate porphyria (VP) 1. The lesions result from sun exposure that activates porphyrins and makes the skin fragile and prone to blister formation. Lesions are located on sun-exposed areas, especially the back of your hands and less frequently your face, neck, ears, and lower extremities. Because sun-induced damage is not acute, the role of sunlight is often not recognized. Skin signs and symptoms may improve in winter and be less prevalent in northern regions and in dark-skinned individuals 1.
Skin signs and symptoms and other manifestations of variegate porphyria (VP) appear typically in adulthood and rarely before puberty 1.
The subepidermal vesicles, bullae, and erosions crust over and heal slowly. When blisters rupture they may become infected and painful 1.
Other chronic skin findings include milia (tiny, white bumps or cysts), scarring, thickening, and areas of decreased and increased skin pigmentation. Facial hyperpigmentation and excessive hair growth (hypertrichosis) may occur.
The skin signs and symptoms are identical to those seen in porphyria cutanea tarda (PCT) and hereditary coproporphyria (HCP), and less severe than those seen in congenital erythropoietic porphyria (CEP) and hepatoerythropoietic porphyria (HEP). They contrast with the acute non-blistering photocutaneous manifestations of erythropoietic protoporphyria (EPP).
Of note, the great majority of individuals who are heterozygous for a PPOX pathogenic gene variant are asymptomatic and are unlikely to be recognized unless they are screened for variegate porphyria (VP) based on a family history of variegate porphyria (VP) 1.
In South Africa the frequency of acute attacks has decreased in recent decades 1. This may be due to less common use of harmful drugs such as barbiturates and sulfonamide antibiotics in clinical practice and perhaps better case recognition and better dissemination of information on how to avoid future attacks. Variegate porphyria (VP) now more commonly presents in South Africa with skin rather than acute neurovisceral signs and symptoms 14, 26, 27.
Acute neurological attacks
Acute neurological attacks can be severe and occur without skin symptoms. Common symptoms during an attack include 7:
- Abdominal symptoms:
- Severe abdominal pain (constant or cramping) lasting hours or days
- Nausea and vomiting
- Constipation, sometimes diarrhea
- Trouble urinating, due to nerve effects on the bladder
- Neurological problems:
- Insomnia, restlessness, agitation, confusion
- Hallucinations and, in severe cases, convulsions
- Low blood sodium levels can also contribute to convulsions
- Severe pain in the limbs, back, or chest
- Muscle paralysis, especially if the attack is prolonged
- In severe cases, muscle weakness may affect the arms, legs and even the muscles that control breathing
- Other signs and symptoms during an attack:
- Reddish urine due to increased porphyrins
- Dark urine due to porphobilin, a byproduct of the condition
- Increased heart rate and blood pressure.
- Reflexes may start out strong (hyperreflexia) but decrease or disappear as motor neuropathy progresses
Several factors can trigger acute attacks, including:
- Certain medications
- Steroid hormones
- Alcohol
- Caloric or carbohydrate restriction
- Stress (both metabolic and possibly psychological)
- Hormonal changes in females, particularly during the second half of the menstrual cycle (when progesterone levels are highest)
Acute neurological attack symptoms can occur at any age after puberty as acute attacks, but may become chronic. Symptoms are more common in women than men, and occur less often in the elderly 1. The frequency and severity of attacks vary considerably and are determined, in part, by exacerbating factors such as certain drugs, hormones, and nutritional deficits 26. The proportion of persons heterozygous for a PPOX genetic mutation who experience acute attacks decreased from about 30%-40% in the 1980s to 5% to 10% in 2005 27.
The neurovisceral symptoms are identical to those in the other acute porphyrias.
Acute signs and symptoms vary. The most common symptoms are abdominal pain; nausea and vomiting; constipation; pain in the back, chest, and extremities; anxiety; seizures; and a predominantly motor peripheral neuropathy resulting in muscle weakness that may progress to quadriparesis and respiratory paralysis 26, 27, 14, 28. Psychiatric disturbances and autonomic neuropathy can also be observed 1. Not all symptoms are present in a single episode and symptoms can vary from episode to episode; however, recurrent attacks are often similar 1. Acute attacks may be severe and are potentially fatal 1, but on average are less frequent and less severe than those observed in acute intermittent porphyria (AIP) 27. An acute attack can be fatal in the presence of severe signs and symptoms including neuropathy, seizures, and respiratory compromise. If managed properly, the outcome of an acute attack is generally good 1. Even severe motor neuropathy is reversible with recovery over a variable period of months and sometimes over several years 1.
Motor neuropathy usually manifests initially as proximal upper-extremity muscle weakness and can be difficult to detect 1. Hyperreflexia may be seen initially, followed by hyporeflexia as the motor neuropathy progresses 1. The motor neuropathy may be accompanied by sensory loss. Note: Motor neuropathy due to acute porphyrias is accompanied by little or no elevation of cerebrospinal fluid protein, which helps to differentiate it from the Guillain-Barré syndrome (GBS) 26.
Because abdominal pain is neuropathic rather than inflammatory, abdominal findings are minimal compared to the severity of the pain. Ileus and bladder distension may be present.
Factors that predispose to acute attacks that are often identified include exposure to a harmful drug, alcohol, reduced dietary intake, or stress from an infection or other illness. Most harmful drugs are known to be inducers of hepatic delta-aminolevulinic acid synthase (ALAS) and hepatic cytochrome P450 enzymes.
- The American Porphyria Foundation offers a drug database with safety information about the interaction of specific drugs in patients with porphyria (https://porphyriafoundation.org/for-healthcare-professionals/ahp-drug-safety-database/) or https://porphyriafoundation.org/drugdatabase/drug-safety-database-search/.
- The EPNET/NAPOS Database should also be consulted. The Norwegian Porphyria Centre (NAPOS), with the European Porphyria Network (EPNET), has created a list of medications that clinicians must avoid using in porphyria patients (https://drugsporphyria.net/). These drugs include ketamine, thiopental, chloramphenicol, erythromycin, nitrofurantoin, rifampicin, trimethoprim/sulfamethoxazole, spironolactone, methyldopa, valproic acid, carbamazepine, phenytoin, phenobarbital, primidone, and risperidone 25. For information on prescribing medication in the context of certain conditions (e.g., HIV, epilepsy, malaria), see https://porphyria.uct.ac.za/porphyria-professionals/prescribing-porphyria-treatment-specific-disorders-poprhyria/therapy-epilepsy.
Pregnancy is usually well tolerated but can precipitate acute attacks in some women.
Physical findings such as rapid heart rate (tachycardia), high blood pressure (hypertension), restlessness, and agitation result from autonomic neuropathy and increased circulating catecholamines 1.
Chronic pain may be a manifestation of variegate porphyria (VP) and other acute porphyrias 1. Depression may be more difficult to link to the disease. Chronic pain and depression may become important management issues 1.
Chronic liver abnormalities, particularly mild elevation of serum transaminases (AST and ALT), are common. Risks for development of hepatocellular carcinoma (HCC) and chronic kidney disease are increased in variegate porphyria (VP). Hepatocellular carcinoma (HCC) may develop, especially after age 50 years in persons with persistent elevations in porphobilinogen and porphyrins.
Variegate porphyria diagnosis
Variegate porphyria (VP) should be suspected in individuals with the following clinical findings and initial laboratory findings.
Clinical findings
- Skin (cutaneous) signs and symptoms include chronic blistering skin lesions (bullae) on sun-exposed skin (photosensitivity), most commonly on the backs of your hands. Chronic skin features include blisters (bullae), milia (tiny, white bumps or cysts), scarring, thickening, and areas of decreased and increased skin pigmentation. Facial hyperpigmentation (increased skin pigmentation) and excessive hair growth (hypertrichosis) may occur. The skin lesions are identical to those of porphyria cutanea tarda (PCT) and other blistering cutaneous porphyrias 14.
- Neurovisceral (neurological) symptoms most commonly include the following 1:
- Abdominal pain. The abdominal pain is typically severe, steady rather than cramping, and diffuse rather than localized. Because the abdominal pain is neuropathic (related to your nerve) rather than inflammatory, abdominal findings are minimal compared to the severity of the pain. Ileus (a temporary condition where the intestines stop moving food and waste through the digestive tract, leading to a buildup of fluids and gas) and bladder distension may be present. Acute hepatic porphyrias should be suspected whenever abdominal pain remains unexplained after an initial workup for common causes.
- Constipation
- Pain in the back, chest, and extremities
- Anxiety
- Seizures
- Muscle weakness due to a primarily motor neuropathy that usually begins in the proximal upper extremities and may progress to quadriparesis (weakness in all four limbs or arms and legs) and the muscles that control breathing (respiratory paralysis). This is accompanied by pain and sometimes sensory loss. Hyperreflexia may be seen initially, followed by hyporeflexia as motor neuropathy progresses.
- Hyponatremia (a condition where the sodium level in your blood is too low), which increases the risk for seizures. It may be a manifestation of hypothalamic involvement and the syndrome of inappropriate antidiuretic hormone secretion (SIADH) 26.
Biochemical laboratory findings
As variegate porphyria (VP) may present with blistering skin lesions on sun-exposed skin, neurovisceral symptoms or both, initial first-line testing aims to detect all porphyrias that can cause either skin or neurovisceral manifestations.
- Blistering cutaneous porphyrias including variegate porphyria (VP). When variegate porphyria (VP) or any other blistering cutaneous porphyria is suspected, the recommended initial test is measurement of plasma or urine porphyrins. If elevated, further testing is needed to determine the type of porphyria or whether the porphyrin elevation – particularly in urine – represents nonspecific porphyrinuria rather than porphyria.
- Acute porphyrias including variegate porphyria (VP). Measurement of urinary porphobilinogen (PBG) and total porphyrins. Urine delta-aminolevulinic acid (ALA) is often measured at the same time as porphobilinogen (PBG) but this is not necessary for initial screening.
- Note:
- (1) If an acute porphyria is confirmed by substantial elevation of urinary porphobilinogen (PBG), treatment can be started, if appropriate, for symptoms of an acute attack while further biochemical testing is being performed to determine the type of acute porphyria.
- (2) If urinary porphobilinogen (PBG) is normal, total porphyrins and delta-aminolevulinic acid (ALA) should be measured in the same urine sample, because total porphyrins often remain elevated longer than porphobilinogen (PBG). In ALA dehydratase-deficiency porphyria (ADP or aminolevulinic acid dehydratase porphyria), the rarest type of porphyria, aminolevulinic acid (ALA) and total porphyrins (but not porphobilinogen [PBG]) are markedly elevated 26.
- Note:
- Substantial elevation in erythrocyte porphyrins is not consistent with variegate porphyria (VP), and points to an erythropoietic porphyria as a cause of blistering skin signs and symptoms and elevation of urine and plasma porphyrins. Alternatively, substantial erythrocyte protoporphyrin in an individual with variegate porphyria (VP) may suggest a concurrent condition that elevates zinc protoporphyrin, such as iron deficiency, lead poisoning, or another erythrocyte disorder.
Establishing the Diagnosis
When initial biochemical laboratory findings support an acute porphyria (i.e., elevated urine porphobilinogen [PBG] or porphyrins) or a blistering cutaneous porphyria (i.e., elevated plasma or urine porphyrins), further diagnostic biochemical testing is required to differentiate variegate porphyria (VP) from other acute and cutaneous porphyrias and from other conditions (e.g., liver disease) that cause nonspecific porphyrinuria 1:
- PPOX (protoporphyrinogen oxidase) enzyme oxidizes protoporphyrinogen to protoporphyrin and PPOX (protoporphyrinogen oxidase) deficiency leads to accumulation of protoporphyrinogen in the liver, which subsequently is autoxidized to protoporphyrin. The PPOX enzyme assay is not needed for diagnostic purposes and is not widely available 1.
- Urine porphobilinogen (PBG) elevation should be detected by a quantitative method such as that described by Mauzerall & Granick 29 which also measures delta-aminolevulinic acid (ALA) or mass spectrometry. Results of qualitative methods such as the Watson-Schwartz and Hoesch tests, which are considered obsolete, should be confirmed on the same sample by a quantitative method. Aminolevulinic acid (ALA) is less elevated than porphobilinogen (PBG). Note: Aminolevulinic acid (ALA) is elevated in ALA dehydratase-deficiency porphyria (ADP or aminolevulinic acid dehydratase porphyria), in which porphobilinogen (PBG) is normal or only slightly increased.
- Active variegate porphyria (VP) is suggested by a quantitative urinary porphobilinogen (PBG) that is substantially elevated.
- Plasma fluorescence scanning can establish or exclude variegate porphyria (VP) when urine porphobilinogen (PBG) is elevated since a fluorescence peak at ~626 nm is not found in any other type of porphyria.
- For screening, it is also useful to measure total porphyrins in the same urine sample, since levels of porphobilinogen (PBG) can be less elevated in variegate porphyria (VP) and hereditary coproporphyria (HCP) than in acute intermittent porphyria (AIP) and decrease to normal more rapidly. Note: Unlike a substantial increase in urine porphobilinogen (PBG), a substantial increase in urinary porphyrins does not indicate porphyria, as urinary porphyrins are increased in many other medical conditions, especially when the hepatobiliary system or bone marrow is affected.
- Porphobilinogen (PBG) and total porphyrins may not be elevated in persons whose symptoms have resolved. If an acute porphyria is suspected to have caused past symptoms, full biochemical testing to include urinary aminolevulinic acid (ALA), porphobilinogen (PBG), and porphyrins, fecal porphyrins, and plasma porphyrins may be indicated.
- Fecal porphyrin analysis can differentiate variegate porphyria (VP), acute intermittent porphyria (AIP), and hereditary coproporphyria (HCP), the only diseases that substantially elevate urine porphobilinogen (PBG).
- Fecal porphyrins are markedly elevated in hereditary coproporphyria (HCP) and variegate porphyria (VP), whereas in acute intermittent porphyria (AIP) there is little or no elevation. The pattern of fecal porphyrins differentiates hereditary coproporphyria (HCP) and variegate porphyria (VP), with marked predominance of coproporphyrin III in hereditary coproporphyria (HCP), and roughly equal elevations of coproporphyrin III and protoporphyrin in variegate porphyria (VP).
- Fecal porphyrin analysis and plasma fluorescence scanning can also reliably distinguish variegate porphyria (VP) from porphyria cutanea tarda (PCT) and other porphyrias that cause blistering skin lesions.
- A fluorescence scan of diluted plasma at neutral pH provides a fluorescence peak at wavelength ~626 nm in variegate porphyria (VP) that is highly sensitive and specific for this porphyria 30. This is the most sensitive biochemical method for establishing variegate porphyria (VP) in the absence of symptoms. Fecal porphyrin analysis is somewhat less sensitive than plasma fluorescence scanning.
Molecular genetic testing to identify a PPOX (protoporphyrinogen oxidase) genetic mutation is recommended for all biochemically confirmed cases of variegate porphyria (VP). Molecular genetic testing is sometimes useful when symptoms have been absent for months or years and biochemical abnormalities are no longer present. Knowing the PPOX (protoporphyrinogen oxidase) mutation is a family enables other family members to be tested reliably for the same mutation.
Variegate porphyria (VP) is especially common in South Africa, where the founder variant p.Arg59Trp 31 accounts for about 95% of cases 15.
Multiexon deletions of PPOX gene have been reported 32; however, no data on detection rate of gene-targeted deletion/duplication analysis are available.
Variegate porphyria differential diagnosis
Acute attacks of variegate porphyria (VP) are identical to those that occur in three other porphyrias. Therefore, these other porphyrias are often classified as acute porphyrias based on their symptoms.
Acute intermittent porphyria (AIP) is the most common of the acute porphyrias in countries other than South Africa and is due to a deficiency of the enzyme porphobilinogen deaminase (PBGD), also known as hydroxymethylbilane synthase (HMBS) and formerly as uroporphyrinogen I-synthase. The factors that trigger acute attacks are the same as in variegate porphyria (VP). Cutaneous symptoms do not occur in acute intermittent porphyria (AIP).
Hereditary coproporphyria (HCP) also shares the same clinical features of variegate porphyria (VP) and is due to a deficiency or coproporphyrinogen oxidase (CPOX) activity. Blistering skin lesions may occur, but much less commonly than in variegate porphyria.
The fourth and least common acute porphyria is delta-aminolevulinic acid dehydratase porphyria (ALAD porphyria or ADP), which results from the deficiency of the second enzyme (delta-aminolevulinic acid dehydratase or ALAD) in the pathway to make heme. Aminolevulinic acid dehydratase porphyria (ALAD porphyria or ADP), in contrast to the other three acute porphyrias, is autosomal recessive. Only six cases are documented in the literature.
The skin lesions caused by variegate porphyria (VP) are identical to those in porphyria cutanea tarda (PCT), which is the most common of all the porphyrias. Porphyria cutanea tarda (PCT) does not cause neurological symptoms. Porphyria cutanea tarda (PCT) is caused by a deficiency of the uroporphyrinogen decarboxylase (UROD), the fifth enzyme in the heme pathway, in the liver. The severe deficiency of uroporphyrinogen decarboxylase (UROD) enzyme in the liver is acquired and requires the presence of iron, but some patients have an inherited UROD gene variant that acts as a predisposing susceptibility factor. Other susceptibility factors include alcohol, smoking, estrogens, hepatitis C, HIV and hemochromatosis (HFE) gene variants. This porphyria responds to treatment by repeated phlebotomies to remove excess iron, or to a low-dose regimen of hydroxychloroquine or chloroquine (antimalarial drug).
Congenital erythropoietic porphyria (CEP) is a very rare inherited disorder resulting from the deficient function of the enzyme uroporphyrinogen III synthase (UROS), the fourth enzyme in the heme biosynthetic pathway. Congenital erythropoietic porphyria (CEP) is an autosomal recessive genetic disorder. The skin lesions in congenital erythropoietic porphyria (CEP) are similar to those found in porphyria cutanea tarda (PCT) and variegate porphyria (VP), but are usually much more severe, reflecting much higher porphyrin levels.
Erythropoietic protoporphyria (EPP) is the third most common porphyria and the most common in children. Erythropoietic protoporphyria (EPP) is usually due to a deficiency of ferrochelatase (FECH), the last enzyme in the heme pathway. Some cases are due to gene variants that increase production of delta-aminolevulinic acid synthase (ALAS), the first enzyme in the pathway specifically in the marrow. Erythropoietic protoporphyria (EPP) causes an acute nonblistering photosensitivity that is quite different from the skin manifestations of the other cutaneous porphyrias.
Variegate porphyria treatment
There is no recognized treatment effective for variegate porphyria (VP), and avoidance of sunlight and identifying and stopping precipitating factors are the current forms of management. Doctors also monitor for muscle weakness and breathing issues. Acute attacks of variegate porphyria are treated like the acute attacks seen in other forms of porphyria. When experiencing an acute porphyria attack, it’s important to start with conservative (non-invasive) treatment before moving to more aggressive options like heme infusion (adding heme or glucose directly into a vein).
The first steps of variegate porphyria (VP) treatment are:
- Stopping medications that may trigger the attack
- Staying hydrated with IV fluids that contain a high amount of carbohydrates (like dextrose)
- Managing pain with narcotic painkillers (opiates)
- Treating nausea and vomiting with medications like phenothiazines (such as chlorpromazine or prochlorperazine)
If these steps don’t help, an intravenous (IV) heme infusion like Panhematin, which is available in the U.S. as lyophilized hematin, is recommended for 3-14 days. Heme infusions are important because they prevent porphyria attacks from worsening and causing permanent nerve damage. In the U.S., Panhematin is the FDA approved treatment, but a similar medication, heme arginate (Normosang), is used in Europe and South Africa.
“Heme infusion” refers to the oxidized form of iron protoporphyrin IX (PPIX), but is also the generic term for heme preparations used as intravenous (IV) therapies for acute porphyrias, such as lyophilized hematin (Panhematin) and heme arginate (Normosang). When these hemin preparations are infused intravenously, the heme is bound to circulating albumin as heme albumin. The heme albumin is taken up by liver cells and decreases the synthesis of liver ALAS1 (aminolevulinic acid synthase 1), the rate-controlling enzyme for heme synthesis, which limits the formation of porphyrin in the liver and marrow. Hemin represses the heme pathway in the liver and lowers aminolevulinic acid, porphobilinogen and porphyrins, and is associated with more rapid recovery from an attack. The duration of this heme treatment depends on the condition and how well the affected person is responding. If the symptoms worsen, immediate treatment with a heme infusion is necessary to prevent nerve damage.
While receiving heme treatment, doctors may monitor certain chemicals in the urine such as aminolevulinic acid (ALA), porphobilinogen (PBG) and porphyrins to see if heme treatment is working.
It’s crucial that the doctor follows the right dosage guidelines, as giving too much heme in a single dose can cause temporary kidney problems, though normal doses are generally safe.
If the attack is mild, IV glucose may help, as it works similarly to heme but is less potent. Glucose given intravenously has a similar effect, but because it is less potent, glucose is only a temporary solution until heme can be administered.
If seizures occur during an attack, controlling them can be complicated because many anti-seizure medications are processed by the liver and can make the attack worse. The following treatments are generally safe for seizures during porphyria 7:
- Magnesium sulfate or diazepam are usually the first choices for controlling seizures.
- Lorazepam is often used for status epilepticus (a severe, prolonged seizure).
Doctors will also check for any imbalances in the body’s electrolytes, which can cause or worsen seizures.
For long-term seizure control, medications like gabapentin may be preferred because they are not metabolized by the liver.
In 2019, the FDA approved Givosiran (Givlaari), a small interfering RNA (siRNA), a medication designed to prevent porphyria attacks. Monthly subcutaneous injections of givosiran can be effective for prevention of frequently recurring attacks 19. Givosiran (Givlaari) works by reducing levels of a liver enzyme that triggers these attacks and has been shown to reduce the frequency of attacks by 74% in clinical trials.
Hospitalization is often necessary for managing severe symptoms of an acute attack, such as 7:
- Severe pain, which may require strong pain medications
- Nausea and vomiting, treated with medications like Zofran (ondansetron) or chlorpromazine
- Muscle weakness and breathing problems, which require careful monitoring
After an acute attack, it’s important to avoid anything that might trigger future episodes. Common triggers include certain medications, stress, or fasting. The American Porphyria Foundation provides a list of drugs that are safe or unsafe for individuals with porphyria. It’s a good idea for patients to wear a Medic Alert bracelet or carry a wallet card that lists the condition and any medications you need to avoid.
For those who have frequent attacks, there are preventive treatments 7:
- Gonadotropin-releasing hormone (GnRH) analogues can help prevent attacks that follow a monthly cycle in women around menstruation.
- Some people may also receive prophylactic heme infusions (once a week or twice a week) to prevent attacks.
People with variegate porphyria who have chronic skin symptoms should avoid exposure to sunlight to help prevent flare-ups. Wearing protective clothing like hats and long-sleeved shirts and using opaque sunscreen can reduce skin damage. Traditional sunscreens may not be effective, so it’s important to choose products specifically designed for porphyria patients. Iron supplementation is likely to cause a skin flare-up and should preferably be administered in the vein.
Since porphyria is hereditary, genetic counseling can help the patient and their families to understand the condition and how it may affect future generations.
Evaluations Following Initial Diagnosis
To establish the extent of disease and to plan the management of an individual diagnosed with variegate porphyria (VP), the following clinical and laboratory evaluations (if not performed as part of the evaluation that led to the diagnosis) are recommended 1:
- Degree of elevations on plasma and urine porphyrins and urine porphobilinogen (PBG), if not determined at the time of diagnosis
- Clinical evaluation of any current acute neurovisceral manifestations to determine the need for hospital admission and treatment with hemin.
- Nervous system. Assessment of the extent of neurologic involvement causing paresis, pain or sensory changes
- Psychiatric evaluation if depression or other psychiatric features are present
- Liver. Liver function tests to indicate chronic liver involvement and liver imaging in patients older than age 50 years
- Kidneys. Kidney function tests to assess for presence and progression of kidney damage
- Skin. Assessment of blistering cutaneous lesions to assess their relationship to variegate porphyria (VP)
- Contributions of medications, diet, and concurrent conditions to severity of variegate porphyria (VP)
- Consultation with a clinical geneticist and/or genetic counselor
Neurological Symptoms Treatment
Most acute neurovisceral attacks require hospital admission; patients with mild attacks not requiring narcotic analgesics and without hyponatremia, seizures, or muscle weakness are sometimes treated as outpatients. A rapid, thorough, and multidisciplinary evaluation is optimized by in-patient management.
As with other acute porphyrias, evaluation should include identification of exacerbating drugs and other precipitating factors. Harmful medications include barbiturates, sulfonamide antibiotics, griseofulvin, rifampin, most anticonvulsants including phenytoin and carbamazepine, alcohol, ergot alkaloids, metoclopramide, and progestins. Harmful medications should be discontinued 33.
Seizures, motor neuropathy, and hyponatremia suggest severe disease and should be managed in the ICU with adequate supportive treatment. Evidence of reversible cerebral vasospasm may be found by MRI 34.
Narcotic analgesics are usually required for pain and ondansetron or a related drug for nausea and vomiting. A phenothiazine is also effective for nausea and for psychiatric symptoms (e.g., agitation, hallucinations) 35, 26.
Mild attacks not requiring narcotics and without hyponatremia, seizures, or motor neuropathy can be treated with glucose loading, but most attacks should be treated with intravenous hemin 26, 33.
Note: “Hemin” refers to the oxidized form of iron protoporphyrin IX (PPIX), but is also the generic term for heme preparations used as intravenous (IV) therapies for acute porphyrias, such as lyophilized hematin (heme hydroxide) and heme arginate. When these hemin preparations are infused intravenously, the heme is bound to circulating albumin as heme albumin. The heme albumin is taken up by liver cells and decreases the synthesis of liver ALAS1 (aminolevulinic acid synthase 1), the rate-controlling enzyme for heme synthesis, which limits the formation of porphyrin in the liver and marrow.
Givosiran (Givlaari), a small interfering RNA (siRNA) therapeutic, was recently approved by the FDA for treatment of acute porphyrias, including variegate porphyria (VP). Monthly subcutaneous injections of givosiran can be effective for prevention of frequently recurring attacks 19.
Individuals who are prone to attacks should consume a normal balanced diet. Despite on-line discussion, there is no evidence that pushing carbohydrate prevents attacks, and it has the side effect of weight gain, which is undesirable for most people. Fasting, fad diets (for example, high protein) and gastric reduction surgery should be avoided. If weight loss is desired, it is advisable to consult a physician and a dietitian about an individualized diet with modest caloric restriction (ca. 10%), which will produce gradual weight loss without increasing the risk of an attack of porphyria. Exercise is safe in porphyria, and recommended.
Patients with acute attack should be carefully monitored for muscle weakness and respiratory impairment that may require ventilatory support.
Hyponatremia should be corrected slowly and seizures treated with medications that do not exacerbate porphyria.
Liver transplantation, which has been effective in persons with acute intermittent porphyria (AIP) with severe repeated acute attacks that respond poorly to medical therapy, is also a consideration in variegate porphyria (VP) 36.
Progression of kidney disease may be prevented to some degree by controlling high blood pressure.
Skin Treatment
Porphyrin levels may decrease and photosensitivity improve if exacerbating factors can be identified and removed; otherwise, there is no effective treatment that lowers porphyrin levels. Treatment with hemin may lower porphyrins in the short term only.
Patients should wear protective clothing and avoid exposure to sunlight.
Analgesics may be needed for painful lesions and antibiotics for superimposed infection. Topical steroids are of little or no benefit.
Specific measures effective in the treatment of porphyria cutanea tarda (i.e., phlebotomy and low-dose hydroxychloroquine or chloroquine) are not effective in the management of variegate porphyria (VP) 1.
Surveillance
Hepatocellular carcinoma (HCC) may develop especially after age 50 years in patients with acute porphyrias and persistent elevations in porphobilinogen or porphyrins; liver imaging at six-month intervals beginning at age 50 years may detect early lesions 37, 38.
Agents and Circumstances to Avoid
Precipitating factors that should be avoided include: barbiturates, sulfonamide antibiotics, griseofulvin, rifampin, most anticonvulsants including phenytoin and carbamazepine, alcohol, ergot alkaloids, metoclopramide, and progestins.
Updated lists are maintained at the websites of the American Porphyria Foundation and the European Porphyria Network.
- The American Porphyria Foundation offers a drug database with safety information about the interaction of specific drugs in patients with porphyria (https://porphyriafoundation.org/for-healthcare-professionals/ahp-drug-safety-database/) or https://porphyriafoundation.org/drugdatabase/drug-safety-database-search/.
- The EPNET/NAPOS Database should also be consulted. The Norwegian Porphyria Centre (NAPOS), with the European Porphyria Network (EPNET), has created a list of medications that clinicians must avoid using in porphyria patients (https://drugsporphyria.net/). These drugs include ketamine, thiopental, chloramphenicol, erythromycin, nitrofurantoin, rifampicin, trimethoprim/sulfamethoxazole, spironolactone, methyldopa, valproic acid, carbamazepine, phenytoin, phenobarbital, primidone, and risperidone 25. For information on prescribing medication in the context of certain conditions (e.g., HIV, epilepsy, malaria), see https://porphyria.uct.ac.za/porphyria-professionals/prescribing-porphyria-treatment-specific-disorders-poprhyria/therapy-epilepsy.
Although birth control pills should generally be avoided, low-dose hormonal preparations may be tolerated.
Fasting and very low calorie diets should be avoided. Weight loss surgery (bariatric surgery) should be avoided in patients who have had frequent exacerbations of variegate porphyria (VP) and other acute porphyrias. Patients who wish to lose weight should do so gradually with moderate, long-term reductions in calorie intake under guidance of a dietician.
Pregnancy Management
Pregnancy is usually well tolerated in women with variegate porphyria (VP); however, some women with variegate porphyria (VP) may experience exacerbations during pregnancy.
Badminton & Deybach 39 published an anecdotal report of successful treatment of several pregnant women experiencing attacks of variegate porphyria (VP) or other acute porphyrias during pregnancy with hemin (in the form of heme arginate) without adverse fetal effect. They emphasize that interruption of pregnancy is almost never indicated for management of acute porphyria.
Experience with heme hydroxide (hematin) is also limited but suggests no adverse effects during pregnancy 40. Hemin is delivered to tissues as heme albumin when administered as either heme arginate or heme hydroxide, and these preparations are expected to have similar safety profiles.
A fetus heterozygous for a PPOX genetic mutation has a good prognosis, because current postnatal management involves genetic counseling of the family regarding appropriate use of drugs and avoidance of known precipitating factors.
Variegate porphyria prognosis
Variegate porphyria prognosis is usually good if the disease is recognized and treated promptly, before nerve damage develops. Although symptoms usually resolve after an attack, recovery of neuromuscular function (in a severe case) may require several months. Mental symptoms may occur during attacks but are not chronic. Premenstrual attacks often resolve quickly with the onset of menses.
Can attacks be prevented?
Yes, attacks are less likely to occur in the future if exacerbating factors are corrected or avoided particularly with regard to drugs and diet. Genetic variegate porphyria carriers should become informed on medications to avoid and should be prepared to point their healthcare providers to on-line drug lists that are regularly updated.
A Medic Alert bracelet is useful for a situation in which the patient is incapacitated. Very frequent premenstrual attacks can be prevented by a gonadotropin-releasing hormone (GnRH) analogue (Lupron, Zoladex, others) administered with expert guidance 41, 42. In selected cases, frequent noncyclical attacks can be prevented by once- or twice-weekly infusions of hemin; however, published experience is lacking 43.
Givosiran (Givlaari), a small interfering RNA (siRNA) therapeutic, was recently approved by the FDA for treatment of acute porphyrias, including variegate porphyria (VP). Monthly subcutaneous injections of givosiran can be effective for prevention of frequently recurring attacks 19.
Individuals who are prone to attacks should consume a normal balanced diet. Despite on-line discussion, there is no evidence that pushing carbohydrate prevents attacks, and it has the side effect of weight gain, which is undesirable for most people. Fasting, fad diets (for example, high protein) and gastric reduction surgery should be avoided. If weight loss is desired, it is advisable to consult a physician and a dietitian about an individualized diet with modest caloric restriction (ca. 10%), which will produce gradual weight loss without increasing the risk of an attack of porphyria. Exercise is safe in porphyria, and recommended.
Prevention of the skin manifestations of variegate porphyria (VP) requires protection from sunlight. Avoidance of exacerbating factors may also be beneficial.
- Singal AK, Anderson KE. Variegate Porphyria. 2013 Feb 14 [Updated 2019 Dec 12]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK121283[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Balogun O, Nejak-Bowen K. Understanding Hepatic Porphyrias: Symptoms, Treatments, and Unmet Needs. Semin Liver Dis. 2024 May;44(2):209-225. doi: 10.1055/s-0044-1787076[↩][↩]
- Kaiser N, Magg J, Nägele T, Wolf N, Krägeloh-Mann I. Neurodevelopmental retardation and neurological symptoms in homozygous variegate porphyria: two new cases and a literature review. Orphanet J Rare Dis. 2025 Mar 20;20(1):139. doi: 10.1186/s13023-025-03606-6[↩][↩][↩]
- Gounden V, Jialal I. Acute Porphyria. [Updated 2023 Jul 17]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK537352[↩][↩]
- Kothadia JP, LaFreniere K, Shah JM. Acute Hepatic Porphyria. [Updated 2023 May 1]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK537178[↩][↩]
- Logan GM, Weimer MK, Ellefson M, Pierach CA, Bloomer JR. Bile porphyrin analysis in the evaluation of variegate porphyria. N Engl J Med. 1991 May 16;324(20):1408-11. doi: 10.1056/NEJM199105163242005[↩][↩]
- Variegate Porphyria. https://rarediseases.org/rare-diseases/variegate-porphyria/[↩][↩][↩][↩][↩][↩][↩][↩]
- Vafaee-Shahi M, Ghasemi S, Riahi A, Sadr Z. A boy with blistering of sun-exposed skin and finger shortening: the first case of Variegate Porphyria with a novel mutation in protoporphyrinogen oxidase (PPOX) gene in Iran: a case report and literature review. Ital J Pediatr. 2022 Feb 14;48(1):27. doi: 10.1186/s13052-022-01215-8[↩][↩]
- Eales L, Day RS, Blekkenhorst GH. The clinical and biochemical features of variegate porphyria: an analysis of 300 cases studied at Groote Schuur Hospital, Cape Town. Int J Biochem. 1980;12(5-6):837-53. doi: 10.1016/0020-711x(80)90173-1[↩][↩]
- von und zu Fraunberg M, Timonen K, Mustajoki P, Kauppinen R. Clinical and biochemical characteristics and genotype-phenotype correlation in Finnish variegate porphyria patients. Eur J Hum Genet. 2002 Oct;10(10):649-57. doi: 10.1038/sj.ejhg.5200860[↩]
- Long C, Smyth SJ, Woolf J, Murphy GM, Finlay AY, Newcombe RG, Elder GH. Detection of latent variegate porphyria by fluorescence emission spectroscopy of plasma. Br J Dermatol. 1993 Jul;129(1):9-13. doi: 10.1111/j.1365-2133.1993.tb03303.x[↩]
- Pinder VA, Holden ST, Deshpande C, Siddiqui A, Mellerio JE, Wraige E, Powell AM. Homozygous variegate porphyria presenting with developmental and language delay in childhood. Clin Exp Dermatol. 2013 Oct;38(7):737-40. doi: 10.1111/ced.12071[↩]
- Variegate Porphyria (VP). https://porphyriafoundation.org/for-patients/types-of-porphyria/vp/[↩][↩][↩][↩][↩]
- Meissner P, Hift RJ, Corrigall A. Variegate porphyria. In: Kadish KM, Smith K, Guilard R, eds. Porphyrin Handbook, Part II. Vol 14. San Diego, CA: Academic Press; 2003:93-120.[↩][↩][↩][↩]
- Meissner PN, Dailey TA, Hift RJ, Ziman M, Corrigall AV, Roberts AG, Meissner DM, Kirsch RE, Dailey HA. A R59W mutation in human protoporphyrinogen oxidase results in decreased enzyme activity and is prevalent in South Africans with variegate porphyria. Nat Genet. 1996 May;13(1):95-7. doi: 10.1038/ng0596-95[↩][↩]
- Elder G, Harper P, Badminton M, Sandberg S, Deybach JC. The incidence of inherited porphyrias in Europe. J Inherit Metab Dis. 2013 Sep;36(5):849-57. doi: 10.1007/s10545-012-9544-4[↩]
- Puy H, Gouya L, Deybach JC. Porphyrias. Lancet. 2010 Mar 13;375(9718):924-37. doi: 10.1016/S0140-6736(09)61925-5[↩][↩]
- Yasuda M, Chen B, Desnick RJ. Recent advances on porphyria genetics: Inheritance, penetrance & molecular heterogeneity, including new modifying/causative genes. Mol Genet Metab. 2019 Nov;128(3):320-331. doi: 10.1016/j.ymgme.2018.11.012[↩][↩]
- Sardh E, Harper P, Balwani M, Stein P, Rees D, Bissell DM, Desnick R, Parker C, Phillips J, Bonkovsky HL, Vassiliou D, Penz C, Chan-Daniels A, He Q, Querbes W, Fitzgerald K, Kim JB, Garg P, Vaishnaw A, Simon AR, Anderson KE. Phase 1 Trial of an RNA Interference Therapy for Acute Intermittent Porphyria. N Engl J Med. 2019 Feb 7;380(6):549-558. doi: 10.1056/NEJMoa1807838[↩][↩][↩][↩]
- Lin, Jou & Shi, Donglu. (2021). Photothermal and photovoltaic properties of transparent thin films of porphyrin compounds for energy applications. Applied Physics Reviews. 8. 011302. https://doi.org/10.1063/5.0036961[↩]
- Panawala, Lakna. (2017). What is the Function of Hemoglobin in the Human Body. https://www.researchgate.net/publication/313841668_What_is_the_Function_of_Hemoglobin_in_the_Human_Body[↩]
- Edel Y, Mamet R. Porphyria: What Is It and Who Should Be Evaluated? Rambam Maimonides Med J. 2018 Apr 19;9(2):e0013. doi: 10.5041/RMMJ.10333[↩]
- Hift RJ, Peters TJ, Meissner PN. A review of the clinical presentation, natural history and inheritance of variegate porphyria: its implausibility as the source of the ‘Royal Malady’. J Clin Pathol. 2012 Mar;65(3):200-5. doi: 10.1136/jclinpath-2011-200276[↩]
- Elder GH. Hepatic porphyrias in children. J Inherit Metab Dis. 1997 Jun;20(2):237-46. doi: 10.1023/a:1005313024076[↩]
- Roveri G, Nascimbeni F, Rocchi E, Ventura P. Drugs and acute porphyrias: reasons for a hazardous relationship. Postgrad Med. 2014 Nov;126(7):108-20. doi: 10.3810/pgm.2014.11.2839[↩][↩][↩]
- Anderson KE, Bloomer JR, Bonkovsky HL, Kushner JP, Pierach CA, Pimstone NR, Desnick RJ. Recommendations for the diagnosis and treatment of the acute porphyrias. Ann Intern Med. 2005 Mar 15;142(6):439-50. doi: 10.7326/0003-4819-142-6-200503150-00010. Erratum in: Ann Intern Med. 2005 Aug 16;143(4):316.[↩][↩][↩][↩][↩][↩][↩][↩]
- Hift RJ, Meissner PN. An analysis of 112 acute porphyric attacks in Cape Town, South Africa: Evidence that acute intermittent porphyria and variegate porphyria differ in susceptibility and severity. Medicine (Baltimore). 2005 Jan;84(1):48-60. doi: 10.1097/01.md.0000152454.56435.f3[↩][↩][↩][↩]
- Kauppinen R, Mustajoki P. Prognosis of acute porphyria: occurrence of acute attacks, precipitating factors, and associated diseases. Medicine (Baltimore). 1992 Jan;71(1):1-13.[↩]
- MAUZERALL D, GRANICK S. The occurrence and determination of delta-amino-levulinic acid and porphobilinogen in urine. J Biol Chem. 1956 Mar;219(1):435-46. https://www.jbc.org/article/S0021-9258(18)65809-0/pdf[↩]
- Poh-Fitzpatrick MB. A Plasma Porphyrin Fluorescence Marker for Variegate Porphyria. Arch Dermatol. 1980;116(5):543–547. doi:10.1001/archderm.1980.01640290053010[↩]
- Dean G. The Porphyrias: A Study of Inheritance and Environment. 2 ed. London, UK: Pitman Medical; 1971.[↩]
- Barbaro M, Kotajärvi M, Harper P, Floderus Y. Partial protoporphyrinogen oxidase (PPOX) gene deletions, due to different Alu-mediated mechanisms, identified by MLPA analysis in patients with variegate porphyria. Orphanet J Rare Dis. 2013 Jan 16;8:13. doi: 10.1186/1750-1172-8-13[↩]
- Balwani M, Wang B, Anderson KE, Bloomer JR, Bissell DM, Bonkovsky HL, Phillips JD, Desnick RJ; Porphyrias Consortium of the Rare Diseases Clinical Research Network. Acute hepatic porphyrias: Recommendations for evaluation and long-term management. Hepatology. 2017 Oct;66(4):1314-1322. doi: 10.1002/hep.29313[↩][↩]
- Webb AJ, Ingale H, Irani SR, Hu MT. Acute variegate porphyria presenting with reversible cerebral vasoconstriction. Clin Neurol Neurosurg. 2016 Jul;146:102-4. doi: 10.1016/j.clineuro.2016.04.018[↩]
- Harper P, Wahlin S. Treatment options in acute porphyria, porphyria cutanea tarda, and erythropoietic protoporphyria. Curr Treat Options Gastroenterol. 2007 Dec;10(6):444-55. doi: 10.1007/s11938-007-0044-9[↩]
- Dowman JK, Gunson BK, Mirza DF, Bramhall SR, Badminton MN, Newsome PN; UK Liver Selection and Allocation Working Party. Liver transplantation for acute intermittent porphyria is complicated by a high rate of hepatic artery thrombosis. Liver Transpl. 2012 Feb;18(2):195-200. doi: 10.1002/lt.22345[↩]
- Andant C, Puy H, Bogard C, Faivre J, Soulé JC, Nordmann Y, Deybach JC. Hepatocellular carcinoma in patients with acute hepatic porphyria: frequency of occurrence and related factors. J Hepatol. 2000 Jun;32(6):933-9. doi: 10.1016/s0168-8278(00)80097-5[↩]
- Schneider-Yin X, van Tuyll van Serooskerken AM, Went P, Tyblewski W, Poblete-Gutiérrez P, Minder EI, Frank J. Hepatocellular carcinoma in variegate porphyria: a serious complication. Acta Derm Venereol. 2010 Sep;90(5):512-5. doi: 10.2340/00015555-0870[↩]
- Badminton MN, Deybach JC. Treatment of an acute attack of porphyria during pregnancy. Eur J Neurol. 2006 Jun;13(6):668-9. doi: 10.1111/j.1468-1331.2006.01238.x[↩]
- Isenschmid M, König C, Fässli C, Haenel A, Hänggi W, Schneider H. Akute intermittierende Porphyrie in der Schwangerschaft: Therapie mit Glukose oder Hämatin? [Acute intermittent porphyria in pregnancy: glucose or hematin therapy?]. Schweiz Med Wochenschr. 1992 Nov 14;122(46):1741-5. German.[↩]
- Schulenburg-Brand D, Gardiner T, Guppy S, Rees DC, Stein P, Barth J, Felicity Stewart M, Badminton M. An Audit of the Use of Gonadorelin Analogues to Prevent Recurrent Acute Symptoms in Patients with Acute Porphyria in the United Kingdom. JIMD Rep. 2017;36:99-107. doi: 10.1007/8904_2017_2[↩]
- Anderson KE, Spitz IM, Bardin CW, Kappas A. A Gonadotropin Releasing Hormone Analogue Prevents Cyclical Attacks of Porphyria. Arch Intern Med. 1990;150(7):1469–1474. doi:10.1001/archinte.1990.00390190115018[↩]
- Marsden JT, Guppy S, Stein P, Cox TM, Badminton M, Gardiner T, Barth JH, Stewart MF, Rees DC. Audit of the Use of Regular Haem Arginate Infusions in Patients with Acute Porphyria to Prevent Recurrent Symptoms. JIMD Rep. 2015;22:57-65. doi: 10.1007/8904_2015_411[↩]





{kind=link}