{kind=link}
Contents
- The pituitary gland
- Pituitary Gland Disorders
- Pituitary gland tumor
- Clinical Presentation of pituitary tumor
- Stage Information for Pituitary Tumors
- Treatment of Pituitary Tumor Option Overview
- Standard Treatment Options for Prolactin-Producing Pituitary Tumors
- Standard Treatment Options for Adrenocorticotropic Hormone (ACTH)-Producing Pituitary Tumors
- Standard Treatment Options for Growth Hormone (GH)-Producing Pituitary Tumors
- Standard Treatment Options for Thyrotropin-Producing Tumors
- Standard Treatment Options for Nonfunctioning Pituitary Tumors
- Standard Treatment Options for Pituitary Carcinomas
- Standard Treatment Options for Recurrent Pituitary Tumors
The pituitary gland
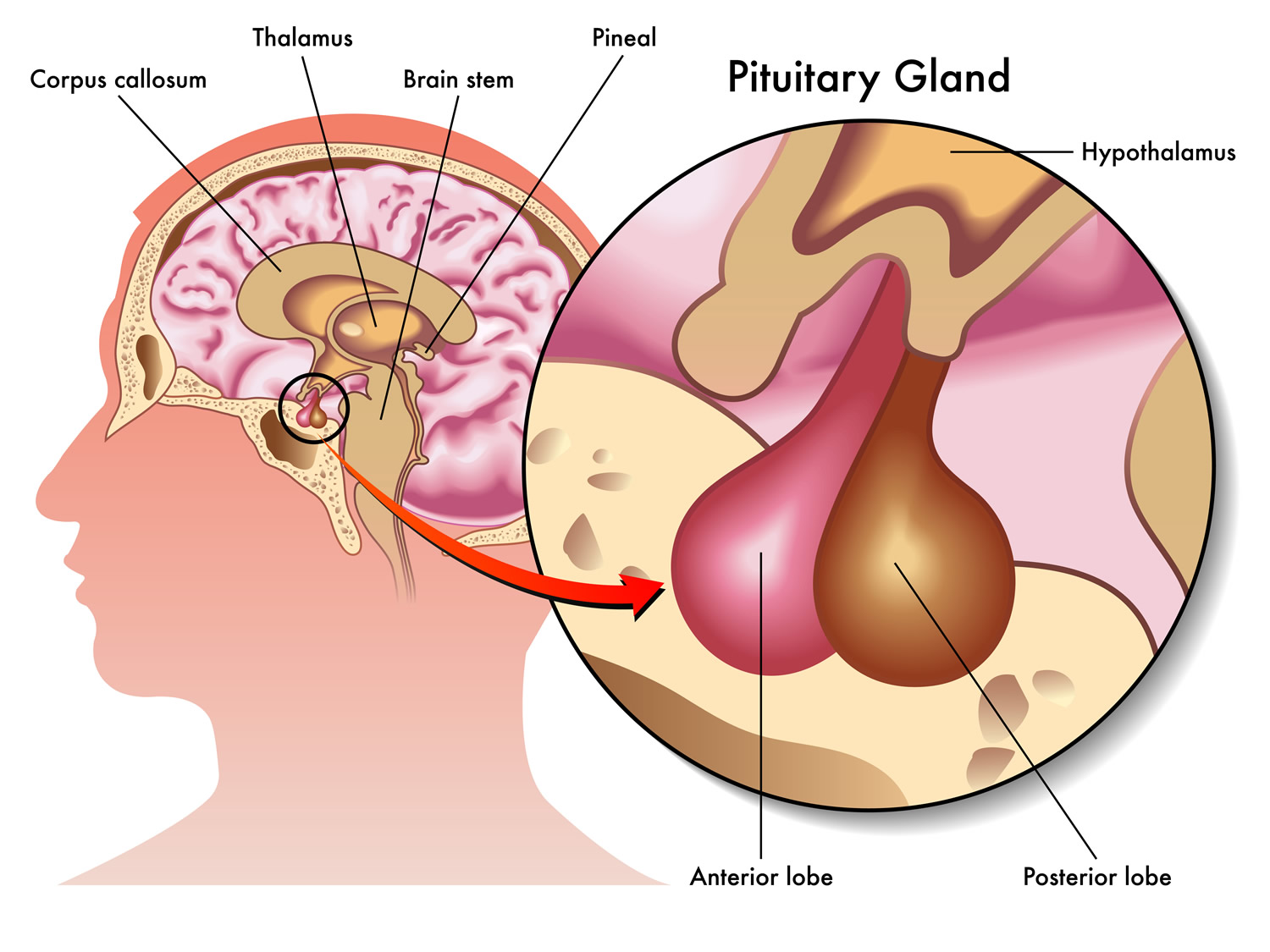
The pituitary gland (hypophysis) is located at the base of the brain, where a pituitary stalk (infundibulum) attaches it to the hypothalamus. The gland is about 1 centimeter in diameter and weighs about 0.5 to 1 g and is divided into anterior pituitary or anterior lobe, and a posterior pituitary, or posterior lobe. The pituitary stalk (infundibulum) contains both blood vessels and nerves. The pituitary gland controls a system of hormones in the body that regulate growth, metabolism, the stress response, and functions of the sex organs via the thyroid gland, adrenal gland, ovaries, and testes.
Figure 1. The pituitary gland location
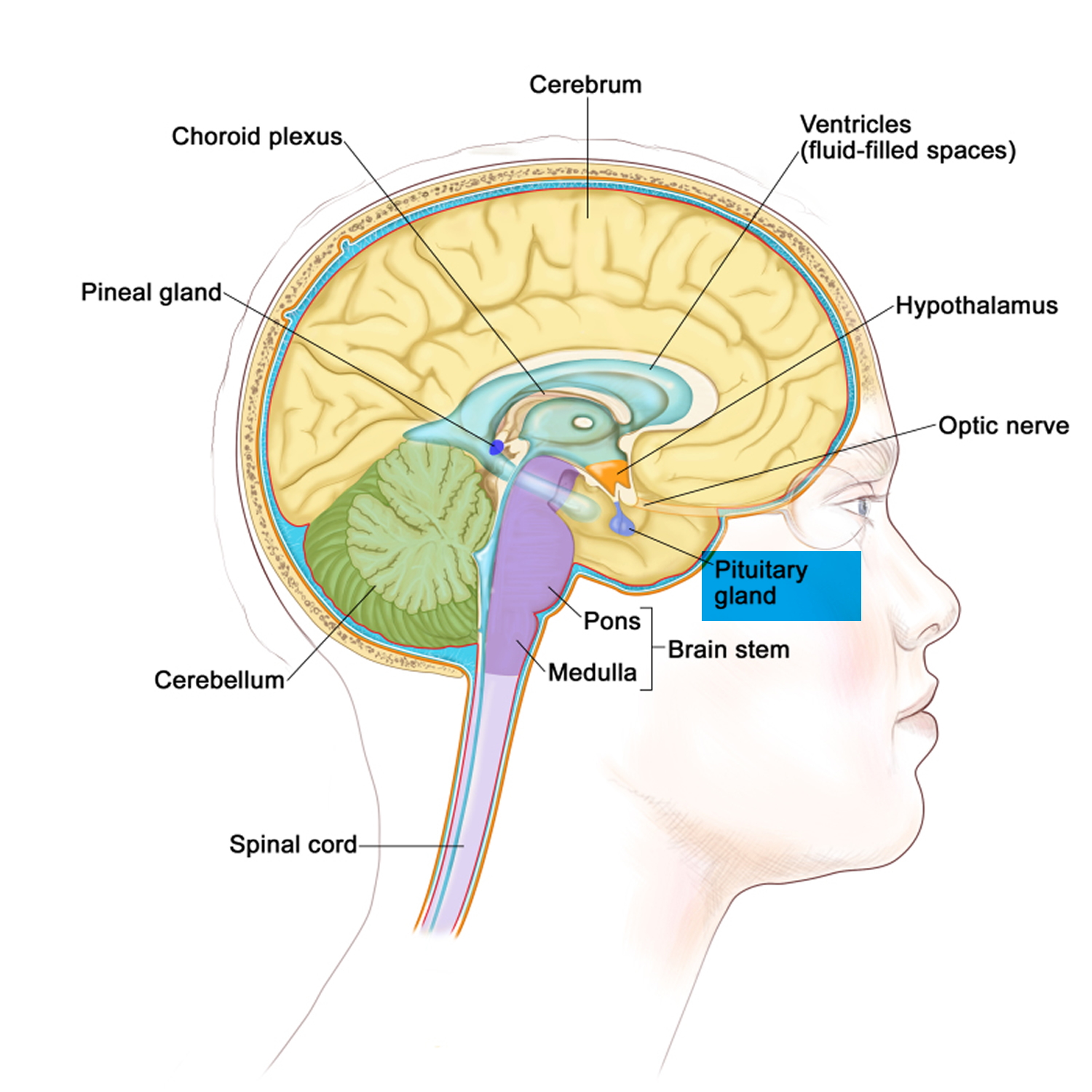
Figure 2. Pituitary gland
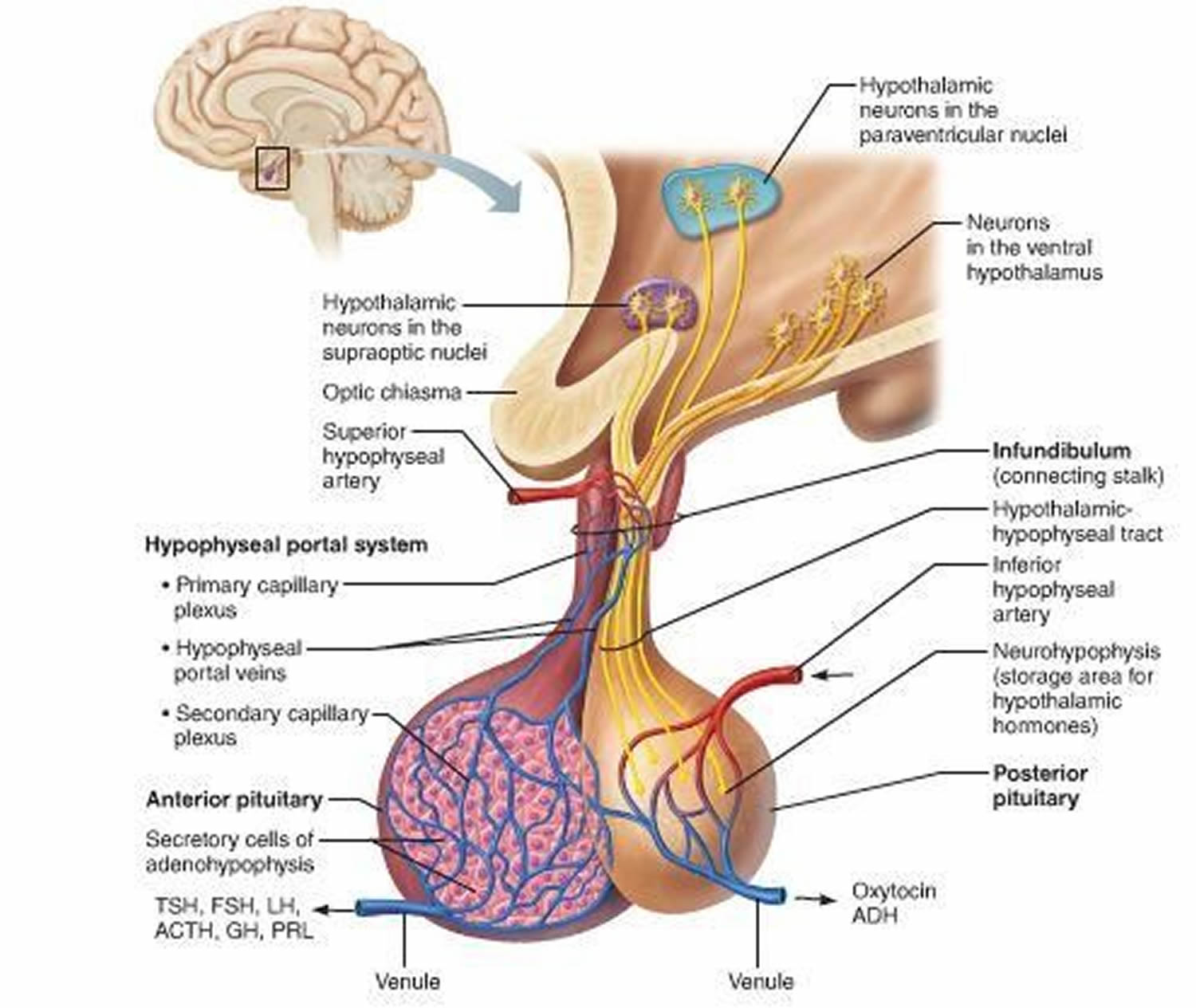
Figure 3. The hypothalamus and pituitary gland (anterior and posterior) endocrine pathways and target organs
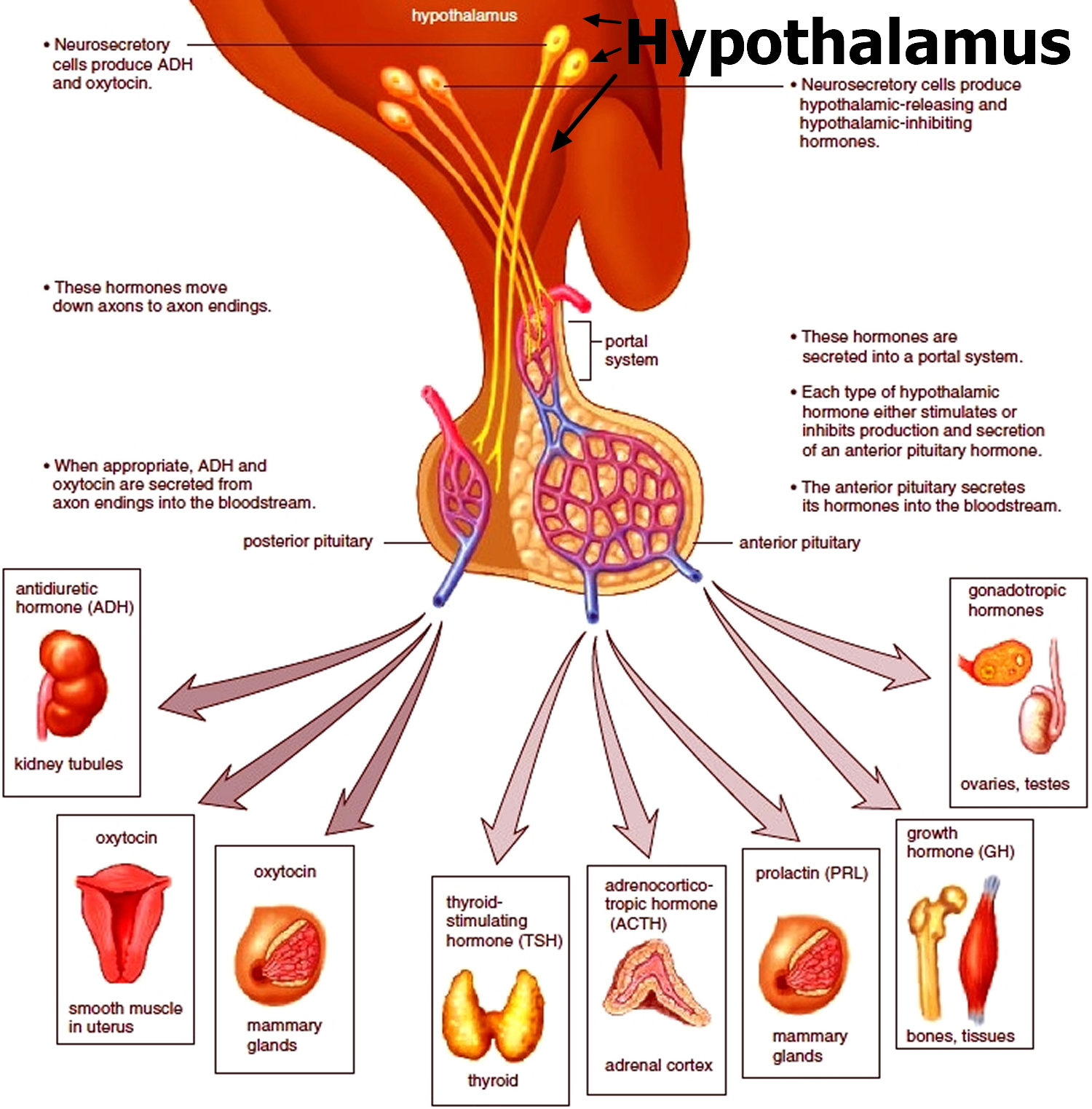
Anterior Pituitary Function and Hormones
The anterior pituitary is enclosed in a capsule of dense connective tissue. It consists largely of epithelial tissue organized in blocks around many thin-walled blood vessels. The cells of the anterior pituitary lobe (which constitutes 80% of the pituitary by weight) synthesize and release several hormones necessary for normal growth and development and also stimulate the activity of several target glands.
Anterior pituitary hormones are regulated by hypothalamic releasing and inhibitory hormones and by negative feedback of the target glandular hormones at the pituitary and hypothalamic levels (Table 1). Among pituitary hormones, only the secretion of prolactin is increased in the absence of hypothalamic influence, because it is mainly under tonic suppression by dopamine, the main inhibitory factor. Antidiuretic hormone (ADH, vasopressin) is produced by the supraoptic and paraventricular nuclei of the hypothalamus and travels in the axons through the pituitary stalk to the posterior pituitary gland.
Table 1. Relationship Among Hypothalamic, Pituitary, and Feedback Hormones and Target Glands
| Target Gland | Hypothalamic Regulatory Hormone | Pituitary Hormone | Feedback Hormone |
|---|---|---|---|
| Thyroid gland | TRH (thyrotropin-releasing hormone) | TSH (thyroid-stimulating hormone) | T4 (thyroxine), T3 (triiodothyronine) |
| Gonad | LHRH (luteinizing hormone-releasing hormone) | LH (luteinizing hormone) | E2 (estradiol), T (testosterone) |
| Gonad | LHRH (luteinizing hormone-releasing hormone) | FSH (follicle-stimulating hormone) | Inhibin, E2 (estradiol), T (testosterone) |
| Many organs | GHRH (growth hormone-releasing hormone), SMS (somatostatin) | GH (growth hormone) | IGF-1 (insulin-like growth factor 1) |
| Breast | PIF (prolactin release inhibitory factor) | Prolactin | ? |
| Adrenal | CRH (corticotropin-releasing hormone), ADH (antidiuretic hormone or vasopressin) | ACTH (adrenocorticotropic hormone) | Cortisol |
Adrenocorticotropic hormone (ACTH)
Adrenocorticotropic hormone (ACTH) is also known as corticotropin. Corticotropin-releasing hormone (CRH) is the primary stimulator of adrenocorticotropic hormone (ACTH) release, but vasopressin plays a role during stress. Adrenocorticotropic hormone (ACTH) induces the adrenal cortex to release cortisol and several weak androgens, such as dehydroepiandrosterone (DHEA). Circulating cortisoland other corticosteroids (including exogenous corticosteroids) inhibit the release of CRH and ACTH. The CRH-ACTH-cortisol axis is a central component of the response to stress. Without adrenocorticotropic hormone (ACTH), the adrenal cortex atrophies and cortisol release virtually ceases.
Thyroid-stimulating hormone (TSH)
Thyroid-stimulating hormone (TSH) regulates the structure and function of the thyroid gland and stimulates synthesis and release of thyroid hormones. Thyroid-stimulating hormone (TSH) synthesis and release are stimulated by the hypothalamic hormone thyrotropin-releasing hormone (TRH) and suppressed (by negative feedback) by circulating thyroid hormones.
Luteinizing hormone (LH) and follicle-stimulating hormone (FSH)
Luteinizing hormone (LH) and follicle-stimulating hormone (FSH) control the production of the sex hormones. Synthesis and release of luteinizing hormone (LH) and follicle-stimulating hormone (FSH) are stimulated mainly by gonadotropin-releasing hormone (GnRH) and suppressed by estrogen and testosterone. One factor controlling GnRH release is kisspeptin, a hypothalamic peptide that is triggered by increased leptin levels at puberty. Two gonadal hormones, activin and inhibin, affect only FSH; activin is stimulative, and inhibin is inhibitory.
In women, luteinizing hormone (LH) and follicle-stimulating hormone (FSH) stimulate ovarian follicular development and ovulation. In men, FSH acts on Sertoli cells and is essential for spermatogenesis; LH acts on Leydig cells of the testes to stimulate testosterone biosynthesis.
Growth hormone (GH)
Growth hormone (GH) stimulates somatic growth and regulates metabolism. Growth hormone–releasing hormone (GHRH) is the major stimulator and somatostatin is the major inhibitor of the synthesis and release of growth hormone. Growth hormone (GH) controls synthesis of insulin-like growth factor 1 (IGF-1, also called somatomedin-C), which largely controls growth. Although IGF-1 is produced by many tissues, the liver is the major source. A variant of IGF-1 occurs in muscle, where it plays a role in enhancing muscle strength. It is less under control of growth hormone than is the liver variant.
The metabolic effects of growth hormone are biphasic. Growth hormone initially exerts insulin-like effects, increasing glucose uptake in muscle and fat, stimulating amino acid uptake and protein synthesis in liver and muscle, and inhibiting lipolysis in adipose tissue. Several hours later, more profound anti–insulin-like metabolic effects occur. They include inhibition of glucose uptake and use, causing blood glucose and lipolysis to increase, which increases plasma free fatty acids. Growth hormone levels increase during fasting, maintaining blood glucose levels and mobilizing fat as an alternative metabolic fuel. Production of growth hormone decreases with aging. Ghrelin, a hormone produced in the fundus of the stomach, promotes growth hormone release from the pituitary, increases food intake, and improves memory.
Insufficient secretion of growth hormone (GH) during childhood limits growth, causing pituitary dwarfism. Body parts are normally proportioned, and mental development is normal—the individual is just very small. Typically, hormone therapy can stimulate some growth. Oversecretion of growth hormone during childhood causes gigantism, in which height may exceed 8 feet. This rare condition is usually a result of a pituitary gland tumor, which may also cause oversecretion of other pituitary hormones. As a result, a person with gigantism often has several metabolic disturbances. Acromegaly is the overproduction of growth hormone in adulthood. The many symptoms attesting to the wide effects of this hormone include enlargement of the heart, the bones, the thyroid gland, facial features, the hands, the feet, and the head. Early symptoms include headache, joint pain, fatigue, and depression.
Prolactin
Prolactin is produced in cells called lactotrophs that constitute about 30% of the cells of the anterior pituitary. The pituitary doubles in size during pregnancy, largely because of hyperplasia and hypertrophy of lactotrophs. In humans, the major function of prolactin is stimulating milk production. Also, prolactin release occurs during sexual activity and stress. Prolactin may be a sensitive indicator of pituitary dysfunction; prolactin is the hormone most frequently produced in excess by pituitary tumors, and it may be one of the hormones to become deficient from infiltrative disease or tumor compression of the pituitary.
Other hormones
Several other hormones are produced by the anterior pituitary. These include pro-opiomelanocortin (POMC, which gives rise to ACTH), alpha- and beta-melanocyte-stimulating hormone (MSH), beta-lipotropin (β-LPH), the enkephalins, and the endorphins. Pro-opiomelanocortin (POMC) and melanocyte-stimulating hormone (MSH) can cause hyperpigmentation of the skin and are only significant clinically in disorders in which ACTH levels are markedly elevated (eg, Addison disease, Nelson syndrome). The function of β-LPH is unknown. Enkephalins and endorphins are endogenous opioids that bind to and activate opioid receptors throughout the brain.
Posterior Pituitary Function and Hormones
The posterior pituitary consists mostly of axons and neuroglia, unlike the anterior pituitary, which is composed primarily of glandular epithelial cells. Neuroglia support the axons, which originate from neurons in the hypothalamus. The secretions of these neurons function not as neurotransmitters, but as hormones.
The posterior pituitary releases vasopressin (also called arginine vasopressin or antidiuretic hormone [ADH]) and oxytocin. These hormones are transported down axons through the pituitary stalk to the posterior pituitary lobe, and are stored in vesicles (secretory granules) near the ends of the axons. Impulses from the hypothalamus release the hormones into the blood and have half-lives of about 10 min. Thus, though synthesized in the hypothalamus, antidiuretic hormone (ADH) and oxytocin are considered posterior pituitary hormones because they enter the bloodstream from the posterior pituitary gland.
Antidiuretic Hormone (ADH)
A diuretic is a chemical that increases urine production, whereas an antidiuretic decreases urine formation. Antidiuretic hormone (ADH) produces an antidiuretic effect by reducing the volume of water the kidneys excrete. In this way, antidiuretic hormone helps regulate the water concentration of body fluids. Antidiuretic hormone (ADH) acts primarily to promote water conservation by the kidneys by increasing the permeability of the distal tubular epithelium to water. At high concentrations, antidiuretic hormone (ADH) also causes vasoconstriction. Like aldosterone, antidiuretic hormone plays an important role in maintaining fluid homeostasis and vascular and cellular hydration. The main stimulus for antidiuretic hormone release is increased osmotic pressure of water in the body, which is sensed by osmoreceptors in the hypothalamus. The other major stimulus is volume depletion, which is sensed by baroreceptors in the left atrium, pulmonary veins, carotid sinus, and aortic arch, and then transmitted to the brain through the vagus and glossopharyngeal nerves. Other stimulants for antidiuretic hormone release include pain, stress, emesis, hypoxia, exercise, hypoglycemia, cholinergic agonists, beta-blockers, angiotensin, and prostaglandins. Inhibitors of antidiuretic hormone release include alcohol, alpha-blockers, and glucocorticoids.
A lack of antidiuretic hormone causes central diabetes insipidus. An inability of the kidneys to respond normally to antidiuretic hormone causes nephrogenic diabetes insipidus. Removal of the pituitary gland usually does not result in permanent diabetes insipidus because some of the remaining hypothalamic neurons produce small amounts of vasopressin.
Copeptin is coproduced with antidiuretic hormone in the posterior pituitary. Measuring it may be useful in distinguishing the cause of hyponatremia.
Oxytocin
Oxytocin has 2 major targets:
- Myoepithelial cells of the breast, which surround the alveoli of the mammary gland
- Smooth muscle cells of the uterus
Suckling stimulates the production of oxytocin, which causes the myoepithelial cells to contract. This contraction causes milk to move from the alveoli to large sinuses for ejection (ie, the milk letdown reflex of nursing mothers). Oxytocin stimulates contraction of uterine smooth muscle cells, and uterine sensitivity to oxytocin increases throughout pregnancy. However, plasma levels do not increase sharply during parturition, and the role of oxytocin in the initiation of labor is unclear.
In males, oxytocin may play a role in the sexual response, including erection of the penis and movement of sperm. In addition, oxytocin is an antidiuretic, but it is much weaker than antidiuretic hormone (ADH). Men have extremely low levels of oxytocin.
Pituitary Gland Disorders
Patients with hypothalamic-pituitary lesions generally present with some combination of 2:
- Symptoms and signs of a mass lesion: headaches, altered appetite, thirst, visual field defects—particularly bitemporal hemianopia or the hemifield slide phenomenon (images drifting apart)
- Imaging evidence of a mass lesion as an incidental finding
- Hypersecretion or hyposecretion of one or more pituitary hormones
The most common cause of hypopituitary or hyperpituitary secretion is a pituitary or hypothalamic tumor. A pituitary tumor tends to produce an enlarged sella (sella turcica). Alternatively, an enlarged sella may represent empty sella syndrome.
Empty sella syndrome
In this disorder, the sella appears empty because it is filled with cebrospinal fluid (CSF), which flattens the pituitary gland against the wall of the sella. The syndrome may be:
- Congenital
- Primary
- Secondary to injury (eg, ischemia after childbirth, surgery, head trauma, radiation therapy)
The typical patient is female (> 80%), obese (about 75%), and hypertensive (30%) and may have idiopathic intracranial hypertension (10%) or spinal fluid rhinorrhea (10%).
Pituitary function in patients with empty sella syndrome is frequently normal. However, hypopituitarism may occur, as may headaches and visual field defects. Occasionally, patients have small coexisting pituitary tumors that secrete growth hormone (GH), prolactin, or ACTH.
Diagnosis can be confirmed by CT or MRI.
No specific therapy is needed for an empty sella alone.
Anterior pituitary lobe lesions
Hypersecretion of anterior lobe hormones (hyperpituitarism) is almost always selective, although occasionally a tumor hypersecretes both growth hormone and prolactin. The anterior pituitary hormones most commonly secreted in excess are GH (as in acromegaly, gigantism), prolactin (as in galactorrhea), and ACTH (resulting in Cushing disease).
Hyposecretion of anterior lobe hormones (hypopituitarism) may be generalized, usually due to a pituitary tumor, or is idiopathic or may involve the selective loss of one or a few pituitary hormones.
Selective Pituitary Hormone Deficiencies
Selective deficiencies of pituitary hormones may represent an early stage in the development of more generalized hypopituitarism 3. Patients must be observed for signs of other pituitary hormone deficiencies, and sellar imaging should be done at intervals to check for signs of a pituitary tumor.
Isolated growth hormone (GH) deficiency is responsible for many cases of pituitary short stature. Although one autosomal dominant form of complete growth hormone deficiency is associated with a deletion of the GH structural gene, such gene defects probably account for a minority of cases. Treatment of growth hormone deficiency in adults < 50 yr deficient in growth hormone are now sometimes treated with growth hormone doses of 0.002 to 0.012 mg/kg sc once/day. Benefits of treatment include improved energy and quality of life, increased body muscle mass, and decreased body fat mass. Suggestions that growth hormone replacement can prevent an acceleration of atherosclerosis induced by growth hormone deficiency are unproved 4.
Isolated gonadotropin deficiency occurs in both sexes and must be distinguished from primary hypogonadism; men have low serum testosterone levels and infertility, and women have amenorrhea, low serum estrogenlevels, and infertility. A eunuchoid habitus is generally present. However, patients with primary hypogonadism have elevated levels of luteinizing hormone (LH) and follicle-stimulating hormone (FSH), whereas those with gonadotropin deficiency, either secondary (pituitary) or tertiary (hypothalamic), have low-normal, low, or unmeasurable levels of LH and FSH. Although most cases of hypogonadotropic hypogonadism involve deficiencies of both LH and FSH, in rare cases the secretion of only one is impaired. Isolated gonadotropin deficiency must also be distinguished from hypogonadotropic amenorrhea secondary to exercise, diet, or mental stress. Although the history may be helpful, differential diagnosis may be impossible.
In Kallmann syndrome, the specific lack of gonadotropin-releasing hormone (GnRH) is associated with midline facial defects, including anosmia and cleft lip or palate, and with color blindness. Embryologic studies have shown that GnRH neurons originally develop in the epithelium of the olfactory placode and migrate into the septal-preoptic region of the hypothalamus early in development. In at least some cases, gene defects, localized to the X chromosome in the X-linked form of the disorder and termed the KALIG-1(Kallmann syndrome interval gene 1) gene, have been found in the adhesion proteins facilitating this neuronal migration. Administration of GnRH is not indicated.
Isolated ACTH deficiency is rare. Weakness, hypoglycemia, weight loss, and decreased axillary and pubic hair suggest the diagnosis. Blood and urinary steroid levels are low and rise to normal after ACTH replacement. Clinical and laboratory evidence of other hormonal deficiencies is absent. Treatment is with cortisol replacement, as for Addison disease; mineralocorticoid replacement is not required.
Isolated thyroid-stimulating hormone deficiencyis likely when clinical features of hypothyroidism exist, serum thyroid-stimulating hormone (TSH) levels are low or not elevated, and no other pituitary hormone deficiencies exist. Serum TSH levels, as measured by immunoassay, are not always lower than normal, suggesting that the TSH secreted is biologically inactive. Administration of recombinant human TSH increases thyroid hormone levels.
Isolated prolactin deficiency has been noted rarely in women who fail to lactate after delivery. Basal prolactin levels are low and do not increase in response to provocative stimuli, such as thyroid-releasing hormone. Administration of prolactin is not indicated.
Generalized Hypopituitarism
Generalized hypopituitarism refers to endocrine deficiency syndromes due to partial or complete loss of anterior lobe pituitary function 5. Various clinical features occur depending on the specific hormones that are deficient. Diagnosis involves imaging tests and measurement of pituitary hormone levels basally and after various provocative stimuli. Treatment depends on cause but generally includes removal of any tumor and administration of replacement hormones.
Hypopituitarism is divided into:
- Primary hypopituitarism: Caused by disorders that affect the pituitary gland
- Secondary hypopituitarism: Caused by disorders of the hypothalamus.
The different causes of primary and secondary hypopituitarism are listed in the table below (see Table 2. Causes of Hypopituitarism)
Table 2. Causes of Hypopituitarism
| Cause | Examples |
|---|---|
| Causes primarily affecting the pituitary gland (primary hypopituitarism) | |
| Pituitary tumors | Adenoma Craniopharyngioma |
| Infarction or ischemic necrosis | Hemorrhagic infarction (pituitary apoplexy) Shock, especially postpartum (Sheehan syndrome), or in diabetes mellitus or sickle cell anemia Vascular thrombosis or aneurysm, especially of the internal carotid artery |
| Inflammatory processes | Meningitis (tubercular, other bacterial, fungal, malarial) Pituitary abscess Sarcoidosis |
| Infiltrative disorders | Hemochromatosis Langerhans cell histiocytosis |
| Idiopathic isolated or multiple pituitary hormone deficiencies | — |
| Iatrogenic | Drugs (eg hypophysitis due to antimelanoma monoclonal antibodies) Irradiation Surgical extirpation |
| Autoimmune dysfunction | Lymphocytic hypophysitis |
| Causes primarily affecting the hypothalamus (secondary hypopituitarism) | |
| Hypothalamic tumors | Craniopharyngioma Ependymoma Meningioma Metastatic tumor Pinealoma |
| Inflammatory processes | Sarcoidosis |
| Neurohormone deficiencies of the hypothalamus | Isolated Multiple |
| Iatrogenic | Surgical transection of the pituitary stalk |
| Trauma | Basal skull fracture |
Generalized Hypopituitarism Signs and Symptoms
Symptoms and signs relate to the underlying disorder and to the specific pituitary hormones that are deficient or absent. Onset is usually insidious and may not be recognized by the patient; occasionally, onset is sudden or dramatic.
Most commonly, growth hormone (GH) is lost first, then gonadotropins, and finally thyroid-stimulating hormone (TSH) and adrenocorticotropic hormone (ACTH). Vasopressin (ADH) deficiency is rare in primary pituitary disorders but is common with lesions of the pituitary stalk and hypothalamus. Function of all target glands decreases when all hormones are deficient (panhypopituitarism).
Lack of luteinizing hormone (LH) and follicle-stimulating hormone (FSH) in children leads to delayed puberty. Premenopausal women develop amenorrhea, reduced libido, regression of secondary sexual characteristics, and infertility. Men develop erectile dysfunction, testicular atrophy, reduced libido, regression of secondary sexual characteristics, and decreased spermatogenesis with consequent infertility.
Growth hormone (GH) deficiency may contribute to decreased energy but is usually asymptomatic and clinically undetectable in adults. Suggestions that growth hormone (GH) deficiency accelerates atherosclerosis are unproved. Growth hormone (GH) deficiency is the most common pituitary hormone deficiency in children and can be isolated or accompanied by deficiency of other pituitary hormones. GH deficiency typically results in abnormally slow growth and short stature with normal proportions. Diagnosis involves measurement of pituitary hormone levels and CT or MRI to detect structural pituitary anomalies or brain tumors. Treatment usually involves specific hormone replacement and removal of any causative tumor.
Thyroid-stimulating hormone (TSH) deficiency leads to hypothyroidism, with such symptoms as facial puffiness, hoarse voice, bradycardia, and cold intolerance.
Adrenocorticotropic hormone (ACTH) deficiency results in hypoadrenalism with attendant fatigue, hypotension, and intolerance to stress and infection. ACTH deficiency does not result in the hyperpigmentation characteristic of primary adrenal failure.
Hypothalamic lesions, which can result in hypopituitarism, can also disturb the centers that control appetite, causing a syndrome resembling anorexia nervosa, or sometimes hyperphagia with massive obesity.
Sheehan syndrome, which affects postpartum women, is pituitary necrosis due to hypovolemia and shock occurring in the immediate peripartum period. Lactation does not start after childbirth, and the patient may complain of fatigue and loss of pubic and axillary hair.
Pituitary apoplexy is a symptom complex caused by hemorrhagic infarction of either a normal pituitary gland or, more commonly, a pituitary tumor. Acute symptoms include severe headache, stiff neck, fever, visual field defects, and oculomotor palsies. The resulting edema may compress the hypothalamus, resulting in somnolence or coma. Varying degrees of hypopituitarism may develop suddenly, and the patient may present with vascular collapse because of deficient ACTH and cortisol. The CSF often contains blood, and MRI documents hemorrhage.
Generalized Hypopituitarism Diagnosis
- MRI or CT
- Free thyroxine (T4), TSH, prolactin, LH, FSH, and testosterone (in men) or estradiol (in women) levels
- Cortisol levels plus provocative testing of pituitary-adrenal axis
- Sometimes other provocative testing
Clinical features are often nonspecific, and the diagnosis must be established with certainty before committing the patient to a lifetime of hormone replacement therapy. Pituitary dysfunction must be distinguished from anorexia nervosa, chronic liver disease, myotonia dystrophica, polyglandular autoimmune disease and disorders of the other endocrine glands. The clinical picture may be particularly confusing when the function of more than one gland decreases at the same time. Evidence of structural pituitary abnormalities and of hormonal deficiencies should be sought with imaging and laboratory tests.
- Imaging tests
Patients should undergo high-resolution CT or MRI, with contrast media as required to rule out structural abnormalities, such as pituitary adenomas. PET is a research tool used in a few specialized centers and therefore is rarely done. When no modern neuroradiologic facilities are available, a simple cone-down lateral x-ray of the sella turcica can identify pituitary macroadenomas with a diameter > 10 mm. Cerebral angiography is indicated only when other imaging tests suggest perisellar vascular anomalies or aneurysms.
- Laboratory testing
Initial evaluation should include testing for TSH and ACTH deficiencies, because both conditions are potentially life threatening. Testing for deficiencies of other hormones is also discussed elsewhere.
Free T4 and TSH levels should be determined. Levels of both are usually low in generalized hypopituitarism; a pattern of normal TSH level with low free T4 may also occur. In contrast, elevated TSH levels with low free T4 indicate a primary abnormality of the thyroid gland.
Synthetic thyrotropin-releasing hormone (TRH), 200 to 500 mcg IV given over 15 to 30 sec, may help identify patients with hypothalamic as opposed to pituitary dysfunction, although this test is not often done. Serum TSH levels are generally measured at 0, 20, and 60 min after injection. If pituitary function is intact, TSH should rise by > 5 mU/L, peaking by 30 min after injection. A delayed rise in serum TSH levels may occur in patients with hypothalamic disease. However, some patients with primary pituitary disease also show a delayed rise.
Serum cortisol levels alone are not reliable indicators of ACTH-adrenal axis function, although a very low morning serum cortisol level (< 3.5 μg/dL between 7:30 and 9:00 am) almost certainly indicates cortisol deficiency. One of several provocative tests should be done. The short ACTH stimulation test is a safer and less labor-intensive test for cortisol deficiency than the insulin tolerance test. In the short ACTH stimulation test, synthetic ACTH 250 mcg IV or IM (standard-dose test) or 1 mcg IV (low-dose test) is given, and the blood cortisol level is measured immediately before and 30 and 60 min after administration of synthetic ACTH. Cortisol should rise significantly; a peak of < 20 µg/dL is abnormal. However, the short ACTH stimulation test is abnormal in secondary cortisol deficiency only when the test done at least 2 to 4 wk after onset of the deficiency; before this time, the adrenal glands have not atrophied and remain responsive to exogenous ACTH.
The insulin tolerance test is considered the most accurate way of evaluating ACTH (as well as GH and prolactin) reserve, but because of its demands, it is probably best reserved for patients who fail the short ACTH stimulation test (if confirmation is needed) or when a test must be done within 2 to 4 wk of a possible pituitary injury. Regular insulin at a dosage of 0.1 units/kg body weight IV is given over 15 to 30 sec, and venous blood samples are obtained to determine GH, cortisol, and glucose levels at baseline (before insulin administration) and 20, 30, 45, 60, and 90 min later. If glucose drops to < 40 mg/dL (< 2.22 mmol/L) or symptoms of hypoglycemia develop, cortisol should increase by > 7 μg/dL or to >20 μg/dL. (Caution: This test is hazardous in patients with severe documented panhypopituitarism or diabetes mellitus and in the elderly and is contraindicated in patients with coronary artery disease or epilepsy. A health care practitioner should be present during the test.) Usually, only transient perspiration, tachycardia, and nervousness occur. If the patient complains of palpitations, loses consciousness, or has a seizure, the test should be stopped promptly by giving 50 mL of 50% glucose solution IV.
Neither the short ACTH stimulation test nor the insulintolerance test alone will differentiate between primary (Addison disease) and secondary (hypopituitary) adrenal insufficiency. Tests to make this distinction and to evaluate the hypothalamic-pituitary-adrenal axis are described under Addison disease.
The corticotropin-releasing hormone (CRH) test is done to distinguish between primary, secondary (pituitary), and tertiary (hypothalamic) causes of adrenal insufficiency. CRH 1 mcg/kg IV is given by rapid injection. Serum ACTH and cortisol levels are measured 15 min before, then at baseline, and 15, 30, 60, 90, and 120 min after the injection. Adverse effects include temporary flushing, a metallic taste in the mouth, and slight and transient hypotension.
Prolactin levels are routinely measured. These levels are often elevated up to 5 times normal values when a large pituitary tumor is present, even if it does not produce prolactin. The tumor compresses the pituitary stalk, preventing dopamine, which inhibits pituitary prolactin production and release, from reaching the pituitary. Patients with such hyperprolactinemia often have hypogonadotropism and secondary hypogonadism.
Measurement of basal levels of LH and FSH is most helpful in evaluating hypopituitarism in postmenopausal women not taking exogenous estrogens in whom circulating gonadotropin concentrations are normally high (> 30 mIU/mL). Although gonadotropin levels tend to be low in other patients with panhypopituitarism, overlap exists with the normal range. Levels of both hormones should increase in response to synthetic gonadotropin-releasing hormone (GnRH) at a dose of 100 mcg IV, with LH peaking about 30 min and FSH peaking 40 min after GnRH administration. However, normal, diminished, or absent responses to GnRH may occur in hypothalamic-pituitary dysfunction. Normal increases in LH and FSH in response to GnRH vary. Administration of exogenous GnRH is not helpful in distinguishing primary hypothalamic disorders from primary pituitary disorders.
Screening for GH deficiency in adults is not recommended unless GH treatment is contemplated (eg, for unexplained reduced energy and quality of life in patients with hypopituitarism in which other hormones have been fully replaced). GH deficiency is suspected if ≥ 2 other pituitary hormones are deficient. Because GH levels vary by time of day and other factors and are difficult to interpret, levels of insulin-like growth factor 1 (IGF-1), which reflect GH, are used; low levels suggest GH deficiency, but normal levels do not rule it out. A provocative test of GH release (see Growth Hormone Deficiency in Children : Diagnosis) may be necessary.
Although the usefulness of provocative testing of pituitary function using releasing hormones remains to be established, if such testing is elected, it is most efficient to evaluate multiple hormones simultaneously. Growth hormone–releasing hormone (1 mcg/kg), CRH (1 mcg/kg), TRH (200 mcg), and GnRH (100 mcg) are given together IV over 15 to 30 sec. Glucose, cortisol, GH, TSH, prolactin, LH, FSH, and ACTH are measured at frequent intervals for the ensuing 180 min. The normal responses are the same as those delineated earlier for individual testing.
Generalized Hypopituitarism Treatment
- Hormone replacement
- Treatment of cause (eg, tumor)
Treatment is replacement of the hormones of the hypofunctioning target glands, as discussed in the pertinent chapters in this section and elsewhere in The Manual. Adults ≤ 50 yr deficient in GH are now sometimes treated with GH doses of 0.002 to 0.012 mg/kg sc once/day. Benefits of treatment include improved energy and quality of life, increased body muscle mass, and decreased body fat mass. Suggestions that GH replacement can prevent an acceleration of atherosclerosis induced by GH deficiency are unproved.
In pituitary apoplexy, immediate surgery is warranted if visual field disturbances or oculomotor palsies develop suddenly or if somnolence progresses to coma because of hypothalamic compression. Although management with high-dose corticosteroids and general support may suffice in a few cases, transsphenoidal decompression of the tumor should generally be undertaken promptly.
Surgery and radiation therapy may be followed by the loss of other pituitary hormone functions. Irradiated patients may lose endocrine function slowly over years. Therefore, posttreatment hormonal status should be evaluated frequently, preferably at 3 and 6 mo and yearly thereafter for at least 10 yr and preferably up to 15 yr after radiation therapy. Such evaluation should include at least assessment of thyroid and adrenal function. Patients may also develop visual difficulties related to fibrosis of the optic chiasm. Sellar imaging and visual field assessment should be done at least every 2 yr initially for about 10 yr, particularly if residual tumor tissue is present.
Cushing Syndrome
Cushing syndrome is a constellation of clinical abnormalities caused by chronic high blood levels of cortisol or related corticosteroids. Cushing disease is Cushing syndrome that results from excess pituitary production of adrenocorticotropic hormone (ACTH), generally secondary to a pituitary adenoma 6. Typical symptoms and signs include moon face and truncal obesity, easy bruising, and thin arms and legs. Diagnosis is by history of receiving corticosteroids or by finding elevated serum cortisol. Treatment depends on the cause.
Gigantism and Acromegaly
Gigantism and acromegaly are syndromes of excessive secretion of growth hormone (hypersomatotropism) that are nearly always due to a pituitary adenoma 7. Before closure of the epiphyses, the result is gigantism. Later, the result is acromegaly, which causes distinctive facial and other features. Diagnosis is clinical and by skull and hand x-rays and measurement of growth hormone levels. Treatment involves removal or destruction of the responsible adenoma.
Galactorrhea
Galactorrhea is lactation in men or in women who are not breastfeeding. It is generally due to a prolactin-secreting pituitary adenoma 8. Diagnosis is by measurement of prolactin levels and imaging tests. Treatment involves tumor inhibition with dopamine agonist drugs and sometimes removal or destruction of the adenoma.
Posterior pituitary lobe lesions
The 2 posterior lobe hormones are:
- Oxytocin
- Vasopressin (ADH)
In women, oxytocin causes myoepithelial cells of the breast and myometrial cells of the uterus to contract. Oxytocin is present in men but has no proven function.
Deficiency of antidiuretic hormone (ADH) results in central diabetes insipidus. Excess vasopressin secretion results in the syndrome of inappropriate ADH secretion (SIADH).
Central diabetes insipidus
Diabetes insipidus results from a deficiency of antidiuretic hormone (ADH) due to a hypothalamic-pituitary disorder (central diabetes insipidus) or from resistance of the kidneys to vasopressin (nephrogenic diabetes insipidus) 9. Polyuria and polydipsia develop. Diagnosis is by water deprivation test showing failure to maximally concentrate urine; antidiuretic hormone (ADH) levels and response to exogenous antidiuretic hormone (ADH) help distinguish central diabetes insipidus from nephrogenic diabetes insipidus. Treatment is with intranasal desmopressin or lypressin. Nonhormonal treatment includes use of diuretics (mainly thiazides) and vasopressin-releasing drugs, such as chlorpropamide.
Syndrome of inappropriate ADH secretion (SIADH)
Syndrome of inappropriate antidiuretic hormone secretion also called SIADH or syndrome of inappropriate antidiuresis (SIAD), is a condition in which your body makes too much antidiuretic hormone (ADH) also called vasopressin (arginine vasopressin or AVP) from the pituitary gland or nonpituitary sources such as lung cancer despite normal or increased plasma volume 10, 11, 12, 13. Antidiuretic hormone (ADH) is naturally produced in an area of your brain called the hypothalamus (anterior hypothalamus). Antidiuretic hormone (ADH) is then stored and released by the posterior pituitary gland at the base of your brain. Antidiuretic hormone (ADH) helps your kidneys control the amount of water your body loses through the urine. When antidiuretic hormone (ADH) is released by the posterior pituitary gland at the base of your brain more water is reabsorbed from the distal and collecting tubule in your kidneys; it increases water reabsorption and leads to concentrated urine and more water in the blood circulation. SIADH (syndrome of inappropriate antidiuretic hormone secretion) causes your body to retain too much water, resulting in low blood sodium also called hyponatremia (ie, serum sodium [Na+] less than 135 mmol/L) with serum hypo-osmolality (a condition where the levels of electrolytes, proteins, and nutrients in your blood are lower than normal, serum osmolality < 280 mOsm/kg) and high urine osmolality (a condition where the levels of electrolytes, proteins, and nutrients in your urine are higher than normal) which are the hallmark of SIADH 10.
Sodium (Na+) is an electrolyte (mineral). Sodium (Na+) is very important for maintaining your blood pressure. Sodium is also needed for nerves, muscles, and other body tissues to work properly. A normal blood sodium level is between 136 and 145 millimoles per liter (mmol/L). When the amount of sodium in fluids outside cells drops below normal such as in hyponatremia (serum sodium [Na+] less than 135 mmol/L), water moves into the cells. This causes the cells to swell with too much water. Brain cells are especially sensitive to swelling, and this causes many of the symptoms of low blood sodium (hyponatremia).
Hyponatremia signs and symptoms may include:
- Nausea and vomiting
- Headache
- Problems with balance that may result in falls
- Mental changes, such as confusion, memory problems, strange behavior
- Loss of energy, drowsiness and fatigue
- Restlessness and irritability
- Muscle weakness, spasms or cramps
- Seizures or coma in severe cases.
With SIADH not enough water is excreted in your urine resulting in very concentrated urine and there is too much water in your blood. This dilutes many substances in the blood such as sodium (hyponatremia). A low blood sodium level (hyponatremia) is the most common cause of symptoms of too much ADH. It is also the most common clue that a person may have SIADH.
When your body’s sodium level drops too much (hyponatremia), it can be a life-threatening emergency. Contact your doctor right away if you have symptoms of SIADH.
The first published cases of SIADH were 2 patients with lung cancer with hyponatremia, yet ongoing renal sodium loss, described by Schwartz et al in 1957 13. They developed the classic Schwartz and Bartter criteria for the diagnosis of SIADH, which has not changed. SIADH is characterized by impaired water excretion leading to hyponatremia with fluid overload (hypervolemia) or normal volume of fluids in the body (euvolemia) 14, 15, 16.
The incidence of SIADH increases with age but, recently, a higher incidence of SIADH has been reported in children 11. Children and older adults are more hyponatremic, particularly when hospitalized for lung and central nervous system (brain and spinal cord) infections like pneumonia or meningitis. SIADH is also more common in hospitalized, post-operative patients due to the administration of hypotonic fluids, drugs, and the body’s response to stress 11.
SIADH treatment depends on the cause of the problem. For example, surgery is done to remove a tumor producing ADH. Or, if a medicine is the cause, its dosage may be changed or another medicine may be tried.
In all cases, the first step is to limit fluid intake. This helps prevent excess fluid from building up in the body. Your doctor will tell you what your total daily fluid intake should be. The restriction is not just for water, but for almost all fluids (coffee, tea, juice, soda, etc.).
If you have severe symptoms, it is a medical emergency. This is usually treated with salt solution (3% saline) given through an IV into the veins (intravenous) in the hospital.
Medicines may be needed to block the effects of ADH on the kidneys so that excess water is excreted by the kidneys. These medicines may be given as pills or as injections given into the veins.
Figure 4. SIADH causes
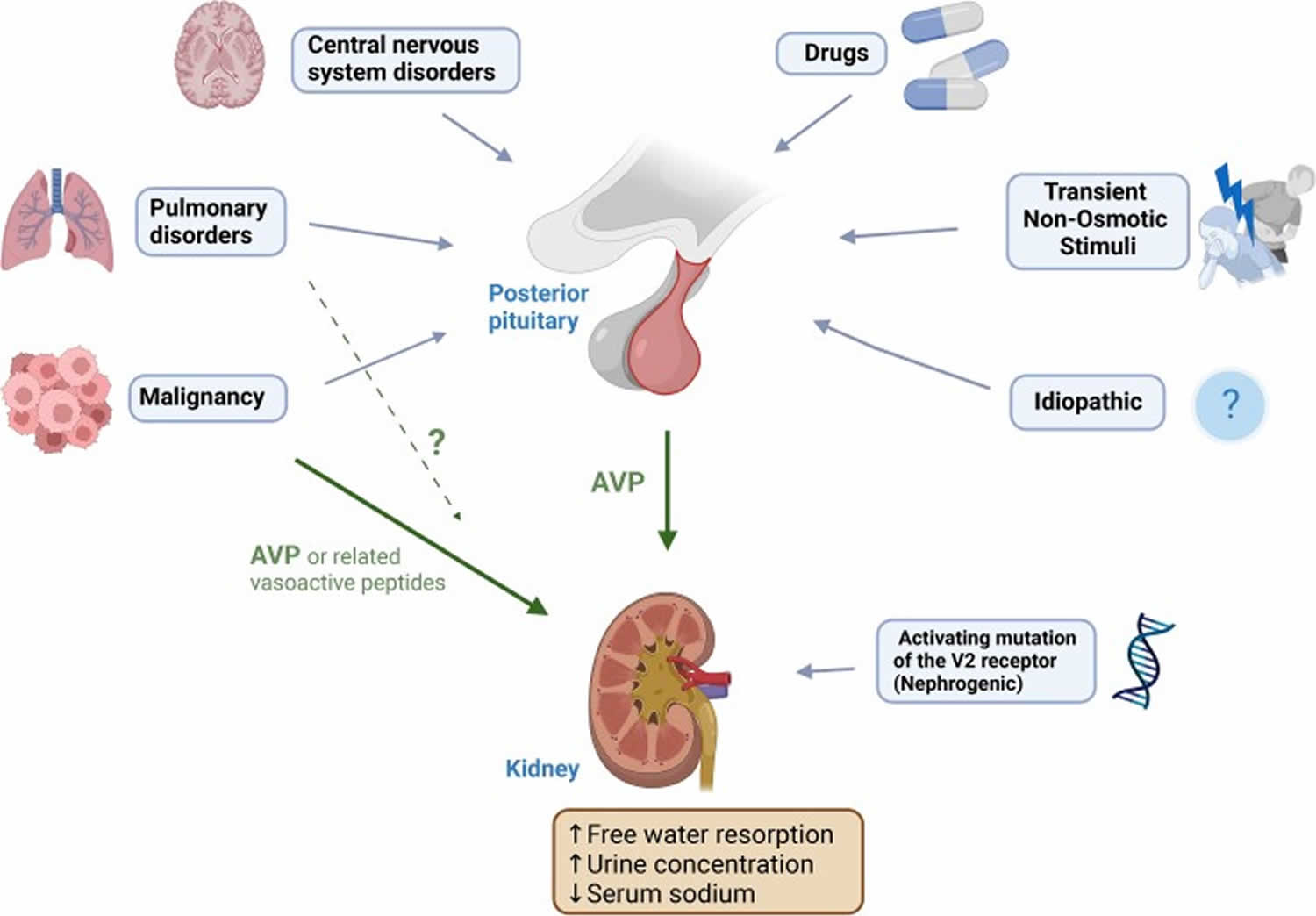
Footnotes: Causes of SIADH. Cancer (solid organ particularly lung and nasopharyngeal, lymphoma). Lung disorders (infection, asthma, cystic fibrosis, respiratory failure). Central nervous system disorders (infection, hemorrhage, thrombosis, trauma, tumour, hydrocephalus, autoimmune (multiple sclerosis, Guillain-Barré syndrome), multiple system atrophy, delirium tremens). Transient stimuli (nausea, pain, stress, prolonged endurance exercise, general anesthesia, pituitary surgery). Drugs. Idiopathic (“reset osmostat,” cause not yet apparent). Hereditary (nephrogenic SIAD).
Abbreviation: AVP = arginine vasopressin.
[Source 10 ]Table 3. Disorders associated with Syndrome of Inappropriate ADH Secretion (SIADH)
| Disorder | Examples |
|---|---|
| Cancer | BrainDuodenum Lung (especially, small cell carcinoma) Lymphoma Pancreas |
| Neurologic disorders | Acute intermittent porphyriaAcute psychosis Brain abscess Encephalitis Guillain-Barré syndrome Head trauma Meningitis Stroke Subdural or subarachnoid hemorrhage |
| Pulmonary disorders and treatments | AspergillosisLung abscess Pneumonia Positive-pressure breathing TB |
| Miscellaneous | Protein-energy undernutrition |
Syndrome of inappropriate ADH secretion (SIADH) causes
SIADH is most often caused by either inappropriate hypersecretion of ADH from its normal hypothalamic source or by ectopic production. Most commonly, SIADH occurs secondary to another disease process elsewhere in your body. Common causes of SIADH (syndrome of inappropriate antidiuretic hormone secretion) include:
- Medicines. A number of drugs associated with SIADH act by enhancing the release or effect of ADH. The most common drugs include carbamazepine, oxcarbazepine, chlorpropamide, cyclophosphamide, and selective serotonin reuptake inhibitors (SSRI). Carbamazepine and oxcarbazepine act in part by increasing the sensitivity to ADH. Chlorpropamide increases the number of V2 receptors in collecting tubules. As high-dose intravenous cyclophosphamide is given with a fluid load to prevent hemorrhagic cystitis, SIADH in such patients is a particular problem, leading to potentially fatal hyponatremia. SSRIs cause SIADH by an unknown mechanism, but people above 65 years of age are more at risk. “Ecstasy” (methylenedioxymethamphetamine), a drug of abuse, is particularly associated with the direct release of ADH. (It also stimulates thirst, which further worsens hyponatremia.) Less commonly, non-steroidal anti-inflammatory drugs (NSAIDs), opiates, interferons, methotrexate, vincristine, vinblastine, ciprofloxacin, haloperidol, and high dose imatinib have been linked with SIADH.
- Surgery under general anesthesia. Surgical procedures are often associated with hypersecretion of ADH, a response that is probably mediated by pain afferents 18.
- Any central nervous system (CNS) abnormality can enhance ADH-release from the pituitary gland, leading to SIADH. Central nervous system disorders include stroke, hemorrhage, infection, trauma, mental illness, and psychosis.
- Brain surgery in the region of the hypothalamus
- Tuberculosis, cancer, chronic infections, and lung disease, such as pneumonia
- Pulmonary diseases, particularly pneumonia (viral, bacterial, tuberculous), can lead to SIADH by unknown mechanisms. A similar response has infrequently been seen in patients with asthma, atelectasis, acute respiratory failure, and pneumothorax.
- Substance use disorder
Rare causes of SIADH (syndrome of inappropriate antidiuretic hormone secretion) include:
- Rare diseases of the hypothalamus or pituitary
- Leukemia and ectopic production of the ADH, like carcinoma of the lung, cancer of the small intestine, thymus, lymphoma, carcinoma of the pancreas and brain cancer. Small cell lung cancer (SCLC) is the most common tumor leading to ectopic ADH production. Less commonly, extrapulmonary small cell carcinomas, head and neck cancers, and olfactory neuroblastomas also cause ectopic ADH release.
- Mental disorders
- Hormone deficiency: Both hypopituitarism and hypothyroidism may be accompanied by hyponatremia and a SIADH picture that can be corrected by hormone replacement.
- Hormone administration: SIADH can be induced by exogenous hormone administration, as with vasopressin (to control gastrointestinal bleeding), desmopressin (dDAVP, to treat von Willebrand disease, hemophilia, or platelet dysfunction), and oxytocin (to induce labor). All three act by increasing the activity of the vasopressin-2 (V2; antidiuretic) receptors.
- Human Immunodeficiency Virus (HIV) infection: A common laboratory manifestation seen in HIV infection, either with the acquired immune deficiency syndrome (AIDS) or early symptomatic HIV infection, is hyponatremia. It can be due to SIADH, or it can be due to volume depletion, secondary to adrenal insufficiency or gastrointestinal losses. Pneumonia, due to Pneumocystis carinii or other organisms and central nervous system infections by opportunistic pathogens, is also responsible for SIADH 19.
- Hereditary SIADH, also known as nephrogenic SIADH, has been ascribed to the gain of function mutation in vasopressin 2 (V2) receptors in the kidneys 20. A gain of function mutation in the AVPR2 gene that encodes the V2 receptor (located on the X chromosome) is responsible for hereditary SIADH. The AVPR2 gene mutation leads to constant activation of the V2 receptors in the renal collecting duct, leading to excessive water absorption and hyponatremia resembling chronic SIADH, which in turn is resistant to vasopressin receptor antagonists. Overt cases present in infancy and can be associated with severe cerebral edema. There is high phenotypic variability, however, with some cases minimally symptomatic and diagnosed only after challenge with a water load 21. Genetic testing in patients with SIADH is infrequent, hence the proportion of nephrogenic SIADH is unknown, but it is estimated to affect fewer than 1 per million population. A low serum copeptin concentration (eg, < 2.0 pmol/L, with low serum sodium and osmolality) may provide a clue to this diagnosis and prompt genetic testing 22.
Central nervous system disorders
Central nervous system (brain and spinal cord) disorders causing SIADH include:
- Acute psychosis
- Acute intermittent porphyria
- Brain abscess
- Cavernous sinus thrombosis
- Cerebellar and cerebral atrophy
- Cerebrovascular accident
- Central nervous system lupus
- Delirium tremens
- Encephalitis (viral or bacterial)
- Epilepsy
- Guillain-Barré syndrome
- Head trauma
- Herpes zoster (chest wall)
- Hydrocephalus
- Hypoxic ischemic encephalopathy
- Meningitis (viral, bacterial, tuberculous, and fungal)
- Midfacial hypoplasia
- Multiple sclerosis
- Perinatal hypoxia
- Rocky Mountain spotted fever
- Schizophrenia
- Shy-Drager syndrome
- Subarachnoid hemorrhage
- Subdural hematoma
- Ventriculoatrial shunt obstruction
- Wernicke encephalopathy
Medications
Medications are a common cause of SIADH 23, 24. The list of drugs that can induce SIADH is long. However, a study of 146 cases of drug‐associated SIADH found that the following five drug classes were implicated in 82.3% of patients 25:
- Antidepressants (eg, citalopram, escitalopram, venlafaxine, amitriptyline)
- Anticonvulsants (eg, carbamazepine, phenytoin, valproate)
- Antipsychotic agents (eg, risperidone, haloperidol, quetiapine)
- Cytotoxic agents (eg, vincristine, cyclophosphamide, cisplatin, ifosfamide)
- Pain medications (eg, duloxetine, pregabalin, tramadol)
Many chemotherapeutic drugs cause nausea, which is a powerful stimulus of ADH release or vasopressin secretion. SIADH is also a leading cause of hyponatremia in children following chemotherapy or stem cell transplantation.
Drugs that stimulate ADH release (AVP release) are as follows:
- Acetylcholine
- Chemotherapy agents – Adenine arabinoside, cyclophosphamide, ifosfamide, vincristine, vinblastine
- Barbiturates
- Bromocriptine
- Carbachol
- Chlorpropamide
- Clofibrate
- Cyclopropane
- Dibenzazepines (eg, carbamazepine, oxcarbazepine)
- Halothane
- Haloperidol
- Histamine
- Isoproterenol
- Lorcainide
- Opiates (eg, morphine)
- Nicotine (inhaled tobacco smoke)
- Nitrous oxide
- Phenothiazines (eg, thioridazine)
- Thiopental
- Monoamine oxidase inhibitors (eg, tranylcypromine)
- Tricyclic antidepressants (eg, amitriptyline, desipramine)
Drugs that potentiate the effects of ADH action (primarily facilitate peripheral action of ADH) are as follows:
- Clofibrate
- Griseofulvin
- Hypoglycemic agents – Metformin, phenformin, tolbutamide
- Oxytocin (large doses)
- Prostaglandin synthetase inhibitors (inhibit renal PGE 2 synthesis) – Indomethacin, aspirin, nonsteroidal anti-inflammatory drugs
- Theophylline
- Triiodothyronine
- Vasopressin analogs (eg, AVP, DDAVP)
Drugs with an uncertain mechanism are as follows:
- Amiodarone 26
- Antineoplastic agents – Cisplatin, melphalan, methotrexate, imatinib
- Ciprofloxacin
- Clomipramine
- 3,4-methylenedioxymethamphetamine (MDMA; ecstasy)
- Phenoxybenzamine
- Antiepilepsy drugs – Sodium valproate, lamotrigine, levetiracetam, gabapentin
- Selective serotonin reuptake inhibitors (SSRIs; eg, sertraline, fluoxetine, paroxetine)
- Thiothixene
Cancer
Neoplastic disorders causing SIADH include:
- Pulmonary – Lung carcinoma and mesothelioma
- Gastrointestinal – Carcinomas of the duodenum, pancreas, and colon
- Genitourinary – Adrenocortical carcinoma; carcinomas of cervix, ureter/bladder, and prostate; and ovarian tumors
- Other – Brain tumors, carcinoid tumors, Ewing sarcoma, leukemia, lymphoma, nasopharyngeal carcinoma, neuroblastoma (olfactory), and thymoma
Lung disorders
Pulmonary disorders causing SIADH include:
- Acute bronchitis/bronchiolitis
- Acute respiratory failure
- Aspergillosis (cavitary lesions)
- Asthma
- Atelectasis
- Bacterial pneumonia
- Chronic obstructive lung disease
- Cystic fibrosis
- Emphysema
- Empyema
- Pneumonia (viral including COVID-19, bacterial [mycoplasmal], fungal)
- Pneumothorax
- Positive pressure ventilation
- Pulmonary abscess
- Pulmonary fibrosis
- Sarcoidosis
- Tuberculosis
- Viral pneumonia
Miscellaneous causes
Miscellaneous causes causing SIADH include:
- Giant cell arteritis
- HIV infection – Hyponatremia has been reported in as many as 40% of adult patients with HIV infection. Patients with acquired immunodeficiency syndrome (AIDS) can have many potential causes for increased ADH secretion, including volume depletion and infection of the lungs and the central nervous system 19. Although one third of the hyponatremic patients with AIDS are clinically hypovolemic, the remaining hyponatremic patients fulfill most of the criteria for SIADH.
Pituitary gland tumor
A pituitary tumor is an abnormal growth of cells within the pituitary gland 27. Most pituitary tumors are benign, which means they are non-cancerous, grow slowly and do not spread to other parts of the body; however they can make the pituitary gland produce either too many or too few hormones, which can cause problems in the body 27.
Pituitary tumors represent from 10% to 25% of all intracranial neoplasms 28. Depending on the study cited, pituitary tumors can be classified into three groups according to their biological behavior 29, 30:
- Benign adenoma.
- Invasive adenoma.
- Carcinoma.
Adenomas comprise the largest portion of pituitary neoplasms with an overall estimated prevalence of approximately 17%. Only a minority of adenomas are symptomatic 31. In addition, pituitary adenomas may be distinguished anatomically as intrapituitary, intrasellar, diffuse, and invasive 32. Invasive adenomas, which account for approximately 35% of all pituitary neoplasms, may invade the dura mater, cranial bone, or sphenoid sinus 33. Carcinomas account for 0.1% to 0.2% of all pituitary tumors 34, 35.
Clinical Presentation of pituitary tumor
The most characteristic-presenting features of pituitary adenomas include inappropriate pituitary hormone secretion and visual field deficits 36.
Rare signs and symptoms of pituitary disease include 36:
- Cranial nerve palsies.
- Temporal lobe epilepsy.
- Hydrocephalus.
- Cerebrospinal fluid rhinorrhea.
The signs and symptoms commonly associated with pituitary tumors derived from each specific cell type (i.e., prolactinomas, corticotroph adenomas, somatotroph adenomas, thyrotroph adenomas, and nonfunctioning adenomas) are as follows:
Prolactin-producing pituitary tumors
Prolactin producing pituitary tumors, also known as prolactinomas and lactotroph adenomas, secrete prolactin and are typically an intrasellar tumor. In women, these adenomas are often small (<10 mm). In either sex, however, they can become large enough to enlarge the sella turcica. These adenomas represent the most common hormone-producing pituitary tumors and account for 25% to 41% of tumor specimens 31.
Signs and symptoms of prolactin-producing pituitary tumors, also known as prolactinomas and lactotroph adenomas, may include 36:
- Headache.
- Visual field deficits.
- Oligomenorrhea or amenorrhea.
- Reduced fertility.
- Loss of libido.
- Erectile dysfunction.
- Galactorrhea in the estrogen-primed female breast.
Adrenocorticotrophic hormone-producing pituitary tumors
Adrenocorticotrophic producing pituitary tumors, also know as corticotroph adenomas, is secretion of adrenocorticotropic hormone (ACTH), which results in Cushing syndrome. These tumors are initially confined to the sella turcica, but they may enlarge and become invasive after bilateral adrenalectomy (i.e., Nelson syndrome). These adenomas represent the second or third most common hormone-producing pituitary tumors, depending on the series; in one series, these tumors accounted for 10% of all tumor specimens 32, 31.
Signs and symptoms of adrenocorticotrophic hormone (ACTH)-producing pituitary tumors, also known as corticotroph adenomas, may include 36:
- Headache.
- Visual field deficits.
- Proximal myopathy.
- Centripetal fat distribution.
- Neuropsychiatric symptoms.
- Striae.
- Ability to easily bruise.
- Skin thinning.
- Hirsutism.
- Osteopenia.
Growth hormone-producing pituitary tumors
Growth hormone producing pituitary tumors, also known as somatotroph adenomas, produce growth hormone, resulting in gigantism in younger patients and acromegaly in others. Suprasellar extension is not uncommon. These adenomas represent the second or third most common hormone-producing pituitary tumors, depending on the series; in one series these adenomas accounted for 13% of tumor specimens 32, 31.
Signs and symptoms of growth hormone (GH)-producing pituitary tumors, also known as somatotroph adenomas, may include 36:
- Headache.
- Visual field deficits.
- Growth of hands and feet.
- Coarsening of facial features.
- Carpal tunnel syndrome.
- Snoring and obstructive sleep apnea.
- Jaw growth and prognathism.
- Osteoarthritis and arthralgia.
- Excessive sweating.
- Dysmorphophobia.
Thyrotropin-producing pituitary tumors
Thyrotropin producing pituitary tumors, also known as thyrotroph adenomas, secrete thyroid-stimulating hormone (TSH), also known as thyrotropin, which results in hyperthyroidism without thyroid-stimulating hormone suppression. Many are large and invasive, may be plurihormonal, and secrete both growth hormone (GH) and/or prolactin 37. These tumors are rare and account for no more than 2% of tumor specimens 37.
Signs and symptoms of thyrotropin (thyroid-stimulating hormone [TSH])-producing tumors, also known as thyrotroph adenomas, may include 38:
- Palpitations.
- Tremor.
- Weight loss.
- Insomnia.
- Hyperdefecation.
- Sweating.
Plurihormonal Adenomas
Plurihormonal tumors produce more than one hormone. Morphologically, they can be either monomorphous or plurimorphous. Monomorphous plurihormonal adenomas consist of one cell population that produces two or more hormones. The adenoma cells often differ from nontumorous adenohypophysial cells, and their cellular derivation may remain obscure despite extensive morphological studies. Plurimorphous plurihormonal adenomas consist of two or more distinct cell types, and each produces one hormone 32. Thyrotroph adenomas are often plurihormonal 37.
Nonfunctioning (endocrine-inactive) adenomas
These tumors arise from the adenohypophysis (anterior pituitary) and cause symptoms when they extend beyond the sella, which results in pressure on the surrounding structures rather than secretion of a hormonally active substance 39. Endocrine-inactive adenomas show positive immunostaining for one or more pituitary hormones 32; however, they are not associated with clinical and biochemical evidence of hormone excess. Gonadotrophic hormones, as detected by antisera to beta-FSH and beta-LH, are present in many clinically nonfunctioning adenomas. Some of these adenomas are recognized by electron microscopy to have gonadotrophic differentiation, but some have characteristics of less well-differentiated cells and resemble the null cells that were initially thought to be undifferentiated precursors of adenohypophysial cells 29. Endocrine-inactive pituitary adenomas comprise approximately 30% to 35% of the pituitary tumors in most series and are the most common type of macroadenoma 40.
Signs and symptoms of nonfunctioning adenomas (most commonly gonadotroph adenomas) may include 41:
- Headache.
- Visual field deficits.
- Pituitary insufficiency, which is due to compression of the pituitary stalk or destruction of normal pituitary tissue by the tumor, and predominantly manifests as secondary hypogonadism.
- Rarely, ovarian overstimulation, testicular enlargement, or increased testosterone levels.
In addition to cell-type specific presentations, pituitary apoplexy (i.e., pituitary adenoma apoplexy) represents another important clinical presentation of pituitary adenomas. Pituitary apoplexy can result from an acute hemorrhagic or ischemic infarction of the pituitary in patients harboring often unrecognized secreting or nonfunctioning pituitary adenomas. In a series analyzing 40 cases of pituitary apoplexy, the presenting signs and symptoms included headache (63%), vomiting (50%), visual field defects (61%), ocular paresis (40%), mental deterioration (13%), hyponatremia (13%), and syncope (5%); in only four cases pituitary tumor was diagnosed prior to presentation 42.
The development of pituitary adenomas may also occur as a component of three familial cancer syndromes 36:
- Multiple endocrine neoplasia 1 (MEN 1).
- Carney complex (e.g., cardiac myxomas, spotty skin pigmentation, and tumors of the adrenal gland and anterior pituitary).
- Isolated familial acromegaly.
A number of other lesions should be considered in the differential diagnosis of sellar masses. Although rare, lymphocytic (i.e., autoimmune) hypophysitis should be considered in the differential diagnosis of any nonsecreting pituitary mass, especially when occurring during pregnancy or postpartum 43. In addition, the clinician should consider craniopharyngioma and Rathke cleft cyst in the differential diagnosis of pituitary tumors. Sellar masses may also result from tumors that are metastatic to the pituitary. This typically occurs as a part of a generalized metastatic spread and is usually associated with five or more additional metastatic sites, especially osseous; breast and lung cancer are the most common primary neoplasms metastasizing to the pituitary 44.
Oncocytic Tumors
Oncocytic tumors of the pituitary, also known as pituitary oncocytomas, are characterized by an abundance of mitochondria, which may fill up to 50% of the cytoplasmic area, which is normally around 8%, and obscure other organelles. These tumors are usually unassociated with clinical and biochemical evidence of hormone excess; in some cases, they may be accompanied by various degrees of hypopituitarism and/or mild hyperprolactinemia. Oncocytic change may occur in several other pituitary tumor types 32.
Carcinomas
Pituitary carcinomas are usually endocrinologically functional, and ACTH-producing and prolactin-producing tumors are the most frequent 45, 35. The histological and cytological characteristics of pituitary carcinomas vary from bland and monotonous to frankly malignant 46. Carcinomas show a variable degree of nuclear atypia and cellular pleomorphism, but they also show significantly higher mitotic rates and cell proliferation indices than adenomas 45. Carcinomas account for 0.1% to 0.2% of all pituitary tumors 35, 34.
Metastatic Tumors
Breast and lung cancer are the most common primary neoplasms metastasizing to the pituitary. Although tumors that are metastatic to the pituitary have been reported to be as high as 28% in autopsy series, the majority of metastatic tumors are clinically silent 44.
Other Tumors
Other tumors that arise in the pituitary include craniopharyngiomas, meningiomas, and germ cell tumors; the rare granular cell tumors, pituicytomas, and gangliogliomas; and the even rarer gangliocytomas, lymphomas, astrocytomas, and ependymomas 45.
Stage Information for Pituitary Tumors
As with other tumors of the central nervous system (brain), no tumor, nodes, metastases-based American Joint Committee on Cancer classification and staging system for pituitary tumors exists 47. Pituitary tumors are classified according to size and divided into microadenomas (i.e., the greatest diameter is <10 mm) and macroadenomas (i.e., the greatest diameter is ≥10 mm) 31. Most pituitary adenomas are microadenomas.
The most widely used radioanatomical classification was based primarily on a neuroradiological examination including skull x-rays, pneumoencephalography, polytomography, and carotid angiography 48. Subsequently validated by the application of more accurate magnetic resonance imaging (MRI) and computed tomography, this radioanatomical classification places adenomas into 1 of 4 grades (I–IV) and has been augmented by additional studies including immunohistochemistry and electron microscopy 29.
Currently, MRI is considered the imaging modality of choice for the diagnosis of pituitary disorders because of its multiplanar capability and good soft tissue contrast enhancement 31. Because no unequivocal histopathologic features of pituitary carcinoma exist, the diagnosis of malignancy is reserved for pituitary neoplasms that have metastasized to remote areas of the brain or to outside of the brain 33, 49, 50.
The radiographical classification for pituitary adenomas is as follows 48, 40:
- 0: Normal pituitary appearance.
- I: Enclosed within the sella turcica, microadenoma, smaller than 10 mm.
- II: Enclosed within the sella turcica, macroadenoma, 10 mm or larger.
- III: Invasive, locally, into the sella.
- IV: Invasive, diffusely, into the sella.
The grading schema for suprasellar extensions is as follows 48, 40:
- A: 0 to 10 mm suprasellar extension occupying the suprasellar cistern.
- B: 10 mm to 20 mm extension and elevation of the third ventricle.
- C: 20 mm to 30 mm extension occupying the anterior of the third ventricle.
- D: A larger than 30 mm extension, beyond the foramen of Monro, or Grade C with lateral extensions.
Treatment of Pituitary Tumor Option Overview
The goals of treatment of pituitary adenomas include normalization of hormonal secretion (i.e., normalization of hypersecretion and improvement in hypofunction) and resolution or cessation of the progression of neurological defects.
Standard treatments for patients with pituitary tumors include 39:
- Surgery.
- Radiation therapy.
- Medical therapy.
- A combination of surgery, radiation therapy, and medical therapy.
The treatment of choice must be individualized and is dictated by the type of tumor, the nature of the excessive hormonal expression, and whether or not the tumor extends into the brain around the pituitary 29, 30.
The transsphenoidal microsurgical approach to a pituitary lesion is the most widely employed surgical approach to pituitary lesions and represents a major development in the safe surgical treatment of both hormonally active and nonfunctioning tumors 40, 51, 52. This approach is often successful in debulking tumors, even those that have a significant suprasellar extension.
A contraindication to this approach includes tumors with a significant suprasellar extension with an hourglass-shaped narrowing between the intrasellar and suprasellar component because blind attempts to reach the suprasellar tumor may lead to cerebral damage. In addition, an infection in the sphenoid sinus is potentially a contraindication to the transsphenoidal approach. In such cases, craniotomies via a pterional or subfrontal approach may be performed. Rapid deterioration of vision is an immediate indication for surgery to relieve pressure produced by an expanding tumor mass, except in the case of macroprolactinomas (where intensive observation with a patient on dopaminergic agonists may be an acceptable alternative). Progressive deterioration of visual fields is often the primary neurological criterion on which surgical management decisions are based 36.
Conventional radiation therapy is an effective adjunct to the treatment of pituitary tumors 40. The advantages of radiation therapy are that it is noninvasive and suitable for surgically high-risk patients. The clinical and biochemical response, however, is slow and may require from 2 years to 10 years for complete and sustained remission. In addition, radiation therapy carries a substantial risk of hypopituitarism (i.e., approximately 30% at 10 years).
Hormone-secreting tumors may be treated with surgery or radiation therapy. Surgical therapy is the treatment of choice for growth hormone-(GH) producing, adrenocorticotropic hormone-(ACTH) producing, and endocrine-inactive adenomas. GH-secreting tumors can be treated with somatostatin analogues, dopamine analogues, and the newer GH-receptor antagonists, such as pegvisomant 36. Ketoconazole, an inhibitor of steroidogenesis, is considered the first drug of choice as adjunctive medical therapy for ACTH-producing tumors 40. Somatostatin analogues are the drugs of choice for treatment of thyroid-stimulating, hormone-producing adenomas; however, the efficacy of treatment may wane with time 36.
The natural history of growth hormone-secreting and ACTH-secreting pituitary tumors is usually one of slowly progressive enlargement 40. Microprolactinomas, however, often remain unchanged, or decrease in size over time, and have been observed to undergo complete, spontaneous resolution on occasion 36.
Treatments under clinical evaluation for patients with pituitary tumors include:
- Stereotactic radiation surgery 53.
Standard Treatment Options for Prolactin-Producing Pituitary Tumors
Standard treatment options for prolactin-producing pituitary tumors include the following:
- Dopamine agonists, such as cabergoline and bromocriptine 54, 55.
- Surgery (second-line) 40, 36.
- Radiation therapy (occasionally) 40, 36.
When the pituitary tumor secretes prolactin, treatment will depend on tumor size and the symptoms that result from excessive hormone production. Patients with prolactin-secreting tumors are treated with surgery and radiation therapy 40.
Most microprolactinomas and macroprolactinomas respond well to medical therapy with ergot-derived dopamine agonists, including bromocriptine and cabergoline 36. For many patients, cabergoline has a more satisfactory side effect profile than bromocriptine. Cabergoline therapy may be successful in treating patients whose prolactinomas are resistant to bromocriptine or who cannot tolerate bromocriptine, and this treatment has a success rate of more than 90% in patients with newly diagnosed prolactinomas 56. In a prospective study, cabergoline was safely withdrawn in patients with normalized prolactin levels and no evidence of tumor, which may effect a cure rate of approximately 70% 57. On the basis of its safety record in pregnancy, however, bromocriptine is the treatment of choice when restoration of fertility is the patient’s goal 58.
Microprolactinomas change little in size with treatment, but macroprolactinomas can be expected to shrink, sometimes quite dramatically. Microprolactinomas may decrease in size over time and have been observed to undergo complete, spontaneous resolution on occasion 31. Surgery is typically reserved for those patients who cannot tolerate dopamine agonists, who suffer pituitary apoplexy during treatment, or whose macroprolactinomas are not responsive to medical therapy 36. Occasionally, these tumors may ultimately require radiation therapy 59.
Standard Treatment Options for Adrenocorticotropic Hormone (ACTH)-Producing Pituitary Tumors
Standard treatment options for ACTH-producing pituitary tumors include the following:
- Surgery (usually a transsphenoidal approach) 40, 60.
- Surgery plus radiation therapy 40, 36.
- Radiation therapy 40, 36.
- Steroidogenesis inhibitors, including mitotane, metyrapone, ketoconazole, and aminoglutethimide 40, 36.
For patients with corticotroph adenomas, transsphenoidal microsurgery is the treatment of choice 40, 36. Remission rates reported in most series are approximately 70% to 90% 40. In a series of 216 patients, who were operated on using a transsphenoidal approach, 75% experienced long-term remission, 21% experienced persistence of Cushing disease, and 9% had recurrence after the initial correction of the hypercortisolism 60. The average time interval for reoperation was 3.8 years. Seventy-nine percent of the tumors were microadenomas, and 18% were macroadenomas; 86% of the cases with microadenoma had long-term remission, whereas, only 46% of those with macroadenoma had remission. In cases in which hypercortisolemia persists, early repeat exploration and/or radiation therapy or laparoscopic bilateral adrenalectomy may be required 36.
Radiation therapy has been used in patients who are deemed to be poor surgical candidates and has also been used as adjunctive therapy in patients with residual or recurrent active tumor 61.
Drug therapy is considered to be an adjunct to transsphenoidal microsurgery in cases in which there is a residual tumor and in cases in which one is awaiting the effects of the radiation therapy 40. Steroidogenesis inhibitors, including mitotane, metyrapone, ketoconazole, and aminoglutethimide are used. Ketoconazole is the best tolerated of these agents and is effective as monotherapy in about 70% of patients 62.
If untreated, patients frequently succumb to cardiovascular disease or infection.
Treatment Options Under Clinical Evaluation for ACTH-Producing Pituitary Tumors
Standard Treatment Options for Growth Hormone (GH)-Producing Pituitary Tumors
Standard treatment options for GH-producing pituitary tumors include the following:
- Surgery (usually a transsphenoidal approach).
- Dopamine analogues, such as bromocriptine.
- Somatostatin analogues, such as octreotide.
- The GH-receptor antagonist, pegvisomant 65, 66.
- Surgery and postoperative radiation therapy.
Treatment for patients with acromegaly includes surgical, radiation, and medical therapies 36. Treatment will depend on the size and extent of the tumor and the need for rapid cessation of hormone function that results in serious clinical sequelae (i.e., hypertension and cardiomyopathy).
Microadenomectomy or macroadenoma decompression is approached transsphenoidally in most patients. Increasingly, endoscopic surgery is used to allow the entire surgical field to be viewed and to allow tumor tissue that would otherwise be inaccessible with rigid instruments to be safely resected. Complete return of grwoth hormone concentrations to normal, however, is not often achieved. Increasingly, adjunctive radiation therapy is reserved for tumors that extend beyond the safe operative area and appear to pose an ongoing threat.
Drug treatment, whether used as an adjuvant or primary therapy in appropriately selected patients, which is advocated by some 67, includes the use of somatostatin analogues, such as octreotide; dopamine analogues, such as bromocriptine; and, the GH-receptor antagonist, pegvisomant. As the first of a new class of GH-receptor antagonists, pegvisomant works by inhibiting functional dimerization of growth hormone receptors and thereby inhibits GH action. Preliminary results indicate that it may be the most effective medical treatment for acromegaly reported to date 65, 66.
In acromegalic patients, impaired glucose tolerance, hypertension, and hyperlipidemia should be vigorously treated concurrently with definitive therapy. A multidisciplinary clinical approach may be required for the treatment of arthritis, carpal tunnel syndrome, obstructive sleep apnea, and prognathism 68. Mortality is related primarily to cardiovascular and respiratory diseases 68.
Standard Treatment Options for Thyrotropin-Producing Tumors
Standard treatment options for thyrotropin-producing tumors include the following:
- Surgery (usually a transsphenoidal approach), with or without adjuvant radiation therapy 69, 36.
- Somatostatin analogues, such as octreotide 70, 37.
Transsphenoidal surgery is the treatment of choice for patients with thyrotropic adenomas 69. Adjuvant radiation therapy may be employed when surgery is known to be noncurative even if the patient is still euthyroid because relapse is inevitable, and the full effect of radiation therapy requires months or years.
Medical therapy may be required for patients who still have hyperthyroid symptoms despite surgery and external radiation. Somatostatin analogues are the drugs of choice for treatment; however, the efficacy of treatment may wane with time 69, 36, 70, 37.
Standard Treatment Options for Nonfunctioning Pituitary Tumors
Standard treatment options for nonfunctioning pituitary tumors include the following:
- Surgery (preferably with a transsphenoidal approach) followed by close observation with radiation therapy reserved for recurrence 41, 40.
- Radiation therapy 71, 41, 40.
- Surgery and postoperative radiation therapy 41, 40.
The selection of treatment for patients with nonfunctioning (endocrine-inactive) tumors will depend on tumor size, the progressive course of the disease, and anatomical structures affected by the tumor extension. The majority of patients present with suprasellar extension and visual field deficits. In addition, many have hormone deficits prior to treatment. The initial treatment of patients with gonadotroph adenomas is usually by transsphenoidal surgery, particularly if the adenoma presents with neurological symptoms, because the effect of radiation therapy occurs too slowly, and no reliable medical therapy exists 72.
Surgical management is typically considered the first choice of treatment for patients with endocrine inactive pituitary adenomas because of its effectiveness in ameliorating symptoms of chiasmal compression and headache 41. Radical removal of the tumor, however, is difficult to obtain because of the frequent invasiveness into the cavernous sinus. Seventy percent to 80% of patients experience normalization or improvement of visual field defects, and almost 100% of patients with headache as a presenting symptom experience relief. Regrowth of the tumor after radiologically confirmed gross total removal appears to be uncommon. In a series of 32 patients, only 2 (6.2%) with gross total tumor removal and no postoperative radiation therapy showed radiological recurrence of the tumor at a mean follow-up of 5.5 years 73.
Radiation therapy has been administered routinely in the postoperative period and after clear radiological evidence of residual or recurrent tumor has been demonstrated 41, 40, 71. Drug therapy appears to be of limited value 41, 40, 71.
Standard Treatment Options for Pituitary Carcinomas
Standard treatment options for pituitary carcinomas include the following:
- Surgery.
- Dopamine agonists, such as bromocriptine, pergolide, quinagolide, and cabergoline, for prolactin (PRL)-producing carcinomas.
- Somatostatin analogues, such as octreotide, for growth hormone (GH)-producing and thyroid-stimulating hormone (TSH)-producing carcinomas.
- Adjuvant radiation therapy, which does not appear to change the disease’s outcome.
- Chemotherapy, which is of little benefit.
Some reports indicate that as many as 88% of pituitary carcinomas are endocrinologically active, and adrenocorticotrophin hormone-secreting tumors are the most common 35. Treatments for patients with pituitary carcinomas are palliative, with the mean survival time ranging from 2 years to 2.4 years, though several case reports of long-term survivors have been published 34, 74, 30, 75.
Treatment options for patients with pituitary carcinomas include resection and dopamine agonists for prolactin-producing tumors; somatostatin analogues for GH-producing and TSH-producing tumors; radiation therapy, and chemotherapy 35. These treatments are palliative with the mean survival time ranging from 2 years to 2.4 years, though several case reports of long-term survivors have been published 34, 74, 30, 75.
Standard Treatment Options for Recurrent Pituitary Tumors
Standard treatment options for recurrent pituitary tumors include the following:
- Radiation therapy for postsurgical recurrence, which offers a high likelihood of local control 71, 76.
- Reirradiation, which provides long-term local control and control of visual symptoms 77.
The question and selection of further treatment for patients who relapse is dependent on many factors, including the specific type of pituitary tumor, prior treatment, visual and hormonal complications, and individual patient considerations.
Treatment Options Under Clinical Evaluation for Recurrent Pituitary Tumors
- Merck Sharp & Dohme Corp. Merck Manual. Overview of the Endocrine System. https://www.merckmanuals.com/professional/endocrine-and-metabolic-disorders/principles-of-endocrinology/overview-of-the-endocrine-system#v27411775[↩]
- Merck Sharp & Dohme Corp. Merck Manual. Pituitary Lesions. https://www.merckmanuals.com/professional/endocrine-and-metabolic-disorders/pituitary-disorders/pituitary-lesions[↩]
- Merck Sharp & Dohme Corp. Merck Manual. Selective Pituitary Hormone Deficiencies. https://www.merckmanuals.com/professional/endocrine-and-metabolic-disorders/pituitary-disorders/selective-pituitary-hormone-deficiencies[↩]
- Merck Sharp & Dohme Corp. Merck Manual. Generalized Hypopituitarism. https://www.merckmanuals.com/professional/endocrine-and-metabolic-disorders/pituitary-disorders/generalized-hypopituitarism[↩]
- Merck Sharp & Dohme Corp. Merck Manual. Generalized hypopituitarism. https://www.merckmanuals.com/professional/endocrine-and-metabolic-disorders/pituitary-disorders/generalized-hypopituitarism[↩][↩]
- Merck Sharp & Dohme Corp. Merck Manual. Cushing Syndrome. https://www.merckmanuals.com/professional/endocrine-and-metabolic-disorders/adrenal-disorders/cushing-syndrome[↩]
- Merck Sharp & Dohme Corp. Merck Manual. Gigantism and Acromegaly. https://www.merckmanuals.com/professional/endocrine-and-metabolic-disorders/pituitary-disorders/gigantism-and-acromegaly[↩]
- Merck Sharp & Dohme Corp. Merck Manual. Galactorrhea. https://www.merckmanuals.com/professional/endocrine-and-metabolic-disorders/pituitary-disorders/galactorrhea[↩]
- Merck Sharp & Dohme Corp. Merck Manual. Central Diabetes Insipidus. https://www.merckmanuals.com/professional/endocrine-and-metabolic-disorders/pituitary-disorders/central-diabetes-insipidus[↩]
- Warren AM, Grossmann M, Christ-Crain M, Russell N. Syndrome of Inappropriate Antidiuresis: From Pathophysiology to Management. Endocr Rev. 2023 Sep 15;44(5):819-861. doi: 10.1210/endrev/bnad010[↩][↩][↩]
- Yasir M, Mechanic OJ. Syndrome of Inappropriate Antidiuretic Hormone Secretion. [Updated 2023 Mar 6]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK507777[↩][↩][↩]
- Syndrome of Inappropriate Antidiuretic Hormone Secretion (SIADH). https://emedicine.medscape.com/article/246650-overview[↩]
- SCHWARTZ WB, BENNETT W, CURELOP S, BARTTER FC. A syndrome of renal sodium loss and hyponatremia probably resulting from inappropriate secretion of antidiuretic hormone. Am J Med. 1957 Oct;23(4):529-42. doi: 10.1016/0002-9343(57)90224-3[↩][↩]
- Lockett J, Berkman KE, Dimeski G, Russell AW, Inder WJ. Urea treatment in fluid restriction-refractory hyponatraemia. Clin Endocrinol (Oxf). 2019 Apr;90(4):630-636. doi: 10.1111/cen.13930[↩]
- Monden MAH, van der Vorst LP, Martens HJM, van der Wolk A. Het syndroom van inadequate secretie van het antidiuretisch hormoon (SIADH) met letale afloop tijdens gebruik van paliperidon en lamotrigine [The syndrome of inappropriate diuretic hormone secretion (SIADH) ending lethal during the use of paliperidon and lamotrigine]. Tijdschr Psychiatr. 2018;60(12):848-851. Dutch.[↩]
- Baba Y, Harada H, Shimada S, Sasaki Y, Murai S, Abe M, Fujiwara S, Arai N, Kawaguchi Y, Kabasawa N, Tsukamoto H, Uto Y, Ariizumi H, Yanagisawa K, Hattori N, Saito B, Nakamaki T. [Syndrome of inappropriate secretion of antidiuretic hormone in multiple myeloma patients treated with bortezomib, lenalidomide, and dexamethasone combination therapy]. Rinsho Ketsueki. 2018;59(11):2423-2427. Japanese. doi: 10.11406/rinketsu.59.2423[↩]
- Merck Sharp & Dohme Corp. Merck Manual. Hyponatremia. https://www.merckmanuals.com/professional/endocrine-and-metabolic-disorders/electrolyte-disorders/hyponatremia#v1149316[↩]
- Steele A, Gowrishankar M, Abrahamson S, Mazer CD, Feldman RD, Halperin ML. Postoperative hyponatremia despite near-isotonic saline infusion: a phenomenon of desalination. Ann Intern Med. 1997 Jan 1;126(1):20-5. doi: 10.7326/0003-4819-126-1-199701010-00003[↩]
- Vitting KE, Gardenswartz MH, Zabetakis PM, et al. Frequency of Hyponatremia and Nonosmolar Vasopressin Release in the Acquired Immunodeficiency Syndrome. JAMA. 1990;263(7):973–978. doi:10.1001/jama.1990.03440070061033[↩][↩]
- Feldman BJ, Rosenthal SM, Vargas GA, Fenwick RG, Huang EA, Matsuda-Abedini M, Lustig RH, Mathias RS, Portale AA, Miller WL, Gitelman SE. Nephrogenic syndrome of inappropriate antidiuresis. N Engl J Med. 2005 May 5;352(18):1884-90. doi: 10.1056/NEJMoa042743[↩]
- Decaux G, Vandergheynst F, Bouko Y, Parma J, Vassart G, Vilain C. Nephrogenic syndrome of inappropriate antidiuresis in adults: high phenotypic variability in men and women from a large pedigree. J Am Soc Nephrol. 2007 Feb;18(2):606-12. doi: 10.1681/ASN.2006090987[↩]
- Fenske WK, Christ-Crain M, Hörning A, Simet J, Szinnai G, Fassnacht M, Rutishauser J, Bichet DG, Störk S, Allolio B. A copeptin-based classification of the osmoregulatory defects in the syndrome of inappropriate antidiuresis. J Am Soc Nephrol. 2014 Oct;25(10):2376-83. doi: 10.1681/ASN.2013080895[↩]
- Ellison DH, Berl T. Clinical practice. The syndrome of inappropriate antidiuresis. N Engl J Med. 2007 May 17;356(20):2064-72. doi: 10.1056/NEJMcp066837[↩]
- Verbalis JG, Goldsmith SR, Greenberg A, Korzelius C, Schrier RW, Sterns RH, Thompson CJ. Diagnosis, evaluation, and treatment of hyponatremia: expert panel recommendations. Am J Med. 2013 Oct;126(10 Suppl 1):S1-42. doi: 10.1016/j.amjmed.2013.07.006[↩]
- Shepshelovich D, Schechter A, Calvarysky B, Diker-Cohen T, Rozen-Zvi B, Gafter-Gvili A. Medication-induced SIADH: distribution and characterization according to medication class. Br J Clin Pharmacol. 2017 Aug;83(8):1801-1807. doi: 10.1111/bcp.13256[↩]
- Marcelino GP, Collantes CMC, Oommen JK, Wang S, Baldassari H, Muralidharan R, Hanna A. Amiodarone-Induced Syndrome of Inappropriate Antidiuretic Hormone: A Case Report and Review of the Literature. P T. 2019 Jul;44(7):416-423 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6590929[↩]
- The National Institute of Neurological Disorders and Stroke. Pituitary tumor. https://www.ninds.nih.gov/Disorders/All-Disorders/Pituitary-Tumors-Information-Page[↩][↩]
- National Cancer Institute. Pituitary Tumors Treatment. https://www.cancer.gov/types/pituitary/hp/pituitary-treatment-pdq[↩]
- Asa SL, Ezzat S: The cytogenesis and pathogenesis of pituitary adenomas. Endocr Rev 19 (6): 798-827, 1998. https://www.ncbi.nlm.nih.gov/pubmed/9861546[↩][↩][↩][↩]
- Landman RE, Horwith M, Peterson RE, et al.: Long-term survival with ACTH-secreting carcinoma of the pituitary: a case report and review of the literature. J Clin Endocrinol Metab 87 (7): 3084-9, 2002. https://www.ncbi.nlm.nih.gov/pubmed/12107205[↩][↩][↩][↩]
- Ezzat S, Asa SL, Couldwell WT, et al.: The prevalence of pituitary adenomas: a systematic review. Cancer 101 (3): 613-9, 2004. https://www.ncbi.nlm.nih.gov/pubmed/15274075[↩][↩][↩][↩][↩][↩][↩]
- Kovacs K, Horvath E, Vidal S: Classification of pituitary adenomas. J Neurooncol 54 (2): 121-7, 2001. https://www.ncbi.nlm.nih.gov/pubmed/11761429[↩][↩][↩][↩][↩][↩]
- Scheithauer BW, Kovacs KT, Laws ER Jr, et al.: Pathology of invasive pituitary tumors with special reference to functional classification. J Neurosurg 65 (6): 733-44, 1986. https://www.ncbi.nlm.nih.gov/pubmed/3095506[↩][↩]
- Pernicone PJ, Scheithauer BW, Sebo TJ, et al.: Pituitary carcinoma: a clinicopathologic study of 15 cases. Cancer 79 (4): 804-12, 1997. https://www.ncbi.nlm.nih.gov/pubmed/9024719[↩][↩][↩][↩]
- Ragel BT, Couldwell WT: Pituitary carcinoma: a review of the literature. Neurosurg Focus 16 (4): E7, 2004. https://www.ncbi.nlm.nih.gov/pubmed/15191336[↩][↩][↩][↩][↩]
- Levy A: Pituitary disease: presentation, diagnosis, and management. J Neurol Neurosurg Psychiatry 75 (Suppl 3): iii47-52, 2004. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1765669/pdf/v075piii47.pdf[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Teramoto A, Sanno N, Tahara S, et al.: Pathological study of thyrotropin-secreting pituitary adenoma: plurihormonality and medical treatment. Acta Neuropathol (Berl) 108 (2): 147-53, 2004. https://www.ncbi.nlm.nih.gov/pubmed/15185102[↩][↩][↩][↩][↩]
- Vance ML: Treatment of patients with a pituitary adenoma: one clinician’s experience. Neurosurg Focus 16 (4): E1, 2004. https://www.ncbi.nlm.nih.gov/pubmed/15191330[↩]
- National Cancer Institute. Pituitary Tumors Treatment–Health Professional Version. https://www.cancer.gov/types/pituitary/hp/pituitary-treatment-pdq[↩][↩]
- Yeh PJ, Chen JW: Pituitary tumors: surgical and medical management. Surg Oncol 6 (2): 67-92, 1997. https://www.ncbi.nlm.nih.gov/pubmed/9436654[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Losa M, Mortini P, Barzaghi R, et al.: Endocrine inactive and gonadotroph adenomas: diagnosis and management. J Neurooncol 54 (2): 167-77, 2001. https://www.ncbi.nlm.nih.gov/pubmed/11761433[↩][↩][↩][↩][↩][↩][↩]
- Lubina A, Olchovsky D, Berezin M, et al.: Management of pituitary apoplexy: clinical experience with 40 patients. Acta Neurochir (Wien) 147 (2): 151-7; discussion 157, 2005. https://www.ncbi.nlm.nih.gov/pubmed/15570437[↩]
- Caturegli P, Newschaffer C, Olivi A, et al.: Autoimmune hypophysitis. Endocr Rev 26 (5): 599-614, 2005. https://www.ncbi.nlm.nih.gov/pubmed/15634713[↩]
- Komninos J, Vlassopoulou V, Protopapa D, et al.: Tumors metastatic to the pituitary gland: case report and literature review. J Clin Endocrinol Metab 89 (2): 574-80, 2004. https://www.ncbi.nlm.nih.gov/pubmed/14764764[↩][↩]
- Ironside JW: Best Practice No 172: pituitary gland pathology. J Clin Pathol 56 (8): 561-8, 2003. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1770019/[↩][↩][↩]
- Pernicone PJ, Scheithauer BW: Invasive pituitary adenoma and pituitary carcinoma. In: Thapar K, Kovacs K, Scheithauer BW, et al., eds.: Diagnosis and Management of Pituitary Tumors. Totowa, NJ: Humana Press, 2001, pp 369-86.[↩]
- Brain and spinal cord. In: Edge SB, Byrd DR, Compton CC, et al., eds.: AJCC Cancer Staging Manual. 7th ed. New York, NY: Springer, 2010, pp 593-7.[↩]
- Hardy J: Transsphenoidal surgery of hypersecreting pituitary tumors. In: Kohler PO, Ross GT, eds.: Diagnosis and treatment of pituitary tumors: proceedings of a conference sponsored jointly by the National Institute of Child Health and Human Development and the National Cancer Institute, January 15-17, 1973, Bethesda, Md. Amsterdam, The Netherlands: Excerpta medica, 1973, pp 179-98.[↩][↩][↩]
- Della Casa S, Corsello SM, Satta MA, et al.: Intracranial and spinal dissemination of an ACTH secreting pituitary neoplasia. Case report and review of the literature. Ann Endocrinol (Paris) 58 (6): 503-9, 1997. https://www.ncbi.nlm.nih.gov/pubmed/9686010[↩]
- Kemink SA, Wesseling P, Pieters GF, et al.: Progression of a Nelson’s adenoma to pituitary carcinoma; a case report and review of the literature. J Endocrinol Invest 22 (1): 70-5, 1999. https://www.ncbi.nlm.nih.gov/pubmed/10090141[↩]
- Hardy J: Transsphenoidal microsurgery of the normal and pathological pituitary. Clin Neurosurg 16: 185-217, 1969. https://www.ncbi.nlm.nih.gov/pubmed/5811707[↩]
- Hardy J: Transsphenoidal hypophysectomy. J Neurosurg 34 (4): 582-94, 1971. https://www.ncbi.nlm.nih.gov/pubmed/5554367[↩]
- Laws ER, Sheehan JP, Sheehan JM, et al.: Stereotactic radiosurgery for pituitary adenomas: a review of the literature. J Neurooncol 69 (1-3): 257-72, 2004 Aug-Sep. https://www.ncbi.nlm.nih.gov/pubmed/15527095[↩][↩]
- Cannavò S, Curtò L, Squadrito S, et al.: Cabergoline: a first-choice treatment in patients with previously untreated prolactin-secreting pituitary adenoma. J Endocrinol Invest 22 (5): 354-9, 1999. https://www.ncbi.nlm.nih.gov/pubmed/10401709[↩]
- Colao A, Di Sarno A, Landi ML, et al.: Macroprolactinoma shrinkage during cabergoline treatment is greater in naive patients than in patients pretreated with other dopamine agonists: a prospective study in 110 patients. J Clin Endocrinol Metab 85 (6): 2247-52, 2000. https://www.ncbi.nlm.nih.gov/pubmed/10852458[↩]
- Colao A, Di Sarno A, Landi ML, et al.: Long-term and low-dose treatment with cabergoline induces macroprolactinoma shrinkage. J Clin Endocrinol Metab 82 (11): 3574-9, 1997. https://www.ncbi.nlm.nih.gov/pubmed/9360509[↩]
- Colao A, Di Sarno A, Cappabianca P, et al.: Withdrawal of long-term cabergoline therapy for tumoral and nontumoral hyperprolactinemia. N Engl J Med 349 (21): 2023-33, 2003. https://www.ncbi.nlm.nih.gov/pubmed/14627787[↩]
- Schlechte JA: Clinical practice. Prolactinoma. N Engl J Med 349 (21): 2035-41, 2003. https://www.ncbi.nlm.nih.gov/pubmed/14627789[↩]
- Nomikos P, Buchfelder M, Fahlbusch R: Current management of prolactinomas. J Neurooncol 54 (2): 139-50, 2001. https://www.ncbi.nlm.nih.gov/pubmed/11761431[↩]
- Mampalam TJ, Tyrrell JB, Wilson CB: Transsphenoidal microsurgery for Cushing disease. A report of 216 cases. Ann Intern Med 109 (6): 487-93, 1988. https://www.ncbi.nlm.nih.gov/pubmed/2843068[↩][↩]
- Mahmoud-Ahmed AS, Suh JH: Radiation therapy for Cushing’s disease: a review. Pituitary 5 (3): 175-80, 2002. https://www.ncbi.nlm.nih.gov/pubmed/12812309[↩][↩]
- Nieman LK: Medical therapy of Cushing’s disease. Pituitary 5 (2): 77-82, 2002. https://www.ncbi.nlm.nih.gov/pubmed/12675504[↩]
- Devin JK, Allen GS, Cmelak AJ, et al.: The efficacy of linear accelerator radiosurgery in the management of patients with Cushing’s disease. Stereotact Funct Neurosurg 82 (5-6): 254-62, 2004. https://www.ncbi.nlm.nih.gov/pubmed/15665560[↩]
- Wong GK, Leung CH, Chiu KW, et al.: LINAC radiosurgery in recurrent Cushing’s disease after transsphenoidal surgery: a series of 5 cases. Minim Invasive Neurosurg 46 (6): 327-30, 2003. https://www.ncbi.nlm.nih.gov/pubmed/14968397[↩]
- Stewart PM: Pegvisomant: an advance in clinical efficacy in acromegaly. Eur J Endocrinol 148 (Suppl 2): S27-32, 2003. https://www.ncbi.nlm.nih.gov/pubmed/12670298[↩][↩]
- Muller AF, Kopchick JJ, Flyvbjerg A, et al.: Clinical review 166: Growth hormone receptor antagonists. J Clin Endocrinol Metab 89 (4): 1503-11, 2004. https://www.ncbi.nlm.nih.gov/pubmed/15070905[↩][↩]
- Kleinberg DL: Primary therapy for acromegaly with somatostatin analogs and a discussion of novel peptide analogs. Rev Endocr Metab Disord 6 (1): 29-37, 2005. https://www.ncbi.nlm.nih.gov/pubmed/15711912[↩]
- Colao A, Ferone D, Marzullo P, et al.: Systemic complications of acromegaly: epidemiology, pathogenesis, and management. Endocr Rev 25 (1): 102-52, 2004. https://www.ncbi.nlm.nih.gov/pubmed/14769829[↩][↩]
- Brucker-Davis F, Oldfield EH, Skarulis MC, et al.: Thyrotropin-secreting pituitary tumors: diagnostic criteria, thyroid hormone sensitivity, and treatment outcome in 25 patients followed at the National Institutes of Health. J Clin Endocrinol Metab 84 (2): 476-86, 1999. https://www.ncbi.nlm.nih.gov/pubmed/10022404[↩][↩][↩]
- Caron P, Arlot S, Bauters C, et al.: Efficacy of the long-acting octreotide formulation (octreotide-LAR) in patients with thyrotropin-secreting pituitary adenomas. J Clin Endocrinol Metab 86 (6): 2849-53, 2001. https://www.ncbi.nlm.nih.gov/pubmed/11397898[↩][↩]
- Tsang RW, Brierley JD, Panzarella T, et al.: Radiation therapy for pituitary adenoma: treatment outcome and prognostic factors. Int J Radiat Oncol Biol Phys 30 (3): 557-65, 1994. https://www.ncbi.nlm.nih.gov/pubmed/7928486[↩][↩][↩][↩]
- Snyder PJ: Extensive personal experience: gonadotroph adenomas. J Clin Endocrinol Metab 80 (4): 1059-61, 1995. https://www.ncbi.nlm.nih.gov/pubmed/7714066[↩]
- Lillehei KO, Kirschman DL, Kleinschmidt-DeMasters BK, et al.: Reassessment of the role of radiation therapy in the treatment of endocrine-inactive pituitary macroadenomas. Neurosurgery 43 (3): 432-8; discussion 438-9, 1998. https://www.ncbi.nlm.nih.gov/pubmed/9733298[↩]
- Sironi M, Cenacchi G, Cozzi L, et al.: Progression on metastatic neuroendocrine carcinoma from a recurrent prolactinoma: a case report. J Clin Pathol 55 (2): 148-51, 2002. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1769584/[↩][↩]
- Vaquero J, Herrero J, Cincu R: Late development of frontal prolactinoma after resection of pituitary tumor. J Neurooncol 64 (3): 255-8, 2003. https://www.ncbi.nlm.nih.gov/pubmed/14558601[↩][↩]
- Kovalic JJ, Grigsby PW, Fineberg BB: Recurrent pituitary adenomas after surgical resection: the role of radiation therapy. Radiology 177 (1): 273-5, 1990. https://www.ncbi.nlm.nih.gov/pubmed/2399329[↩]
- Schoenthaler R, Albright NW, Wara WM, et al.: Re-irradiation of pituitary adenoma. Int J Radiat Oncol Biol Phys 24 (2): 307-14, 1992. https://www.ncbi.nlm.nih.gov/pubmed/1526869[↩]
- Sheehan JP, Kondziolka D, Flickinger J, et al.: Radiosurgery for residual or recurrent nonfunctioning pituitary adenoma. J Neurosurg 97 (5 Suppl): 408-14, 2002. https://www.ncbi.nlm.nih.gov/pubmed/12507066[↩]
- Picozzi P, Losa M, Mortini P, et al.: Radiosurgery and the prevention of regrowth of incompletely removed nonfunctioning pituitary adenomas. J Neurosurg 102 (Suppl): 71-4, 2005. https://www.ncbi.nlm.nih.gov/pubmed/15662784[↩]