Contents
What is xeroderma pigmentosum
Xeroderma pigmentosum has also been called DeSanctis-Cacchione syndrome, is a very rare inherited skin disorder where a person is extremely sensitivity to ultraviolet (UV) rays from sunlight, has premature skin ageing and is prone to developing skin cancers. In affected individuals, exposure to sunlight often causes dry skin (xeroderma) and changes in skin coloring (pigmentation). This combination of features gives the condition its name, xeroderma pigmentosum. Xeroderma pigmentosum mostly affects the eyes and areas of skin exposed to the sun. Some affected individuals also have problems involving the nervous system.
Xeroderma pigmentosum is a rare disorder; it is estimated to affect about 1 in 1 million people in the United States and Europe 1. Xeroderma pigmentosum is more common in Japan, North Africa, and the Middle East 2.
- In Japan prevalence is estimated at 1:22,000 3.
- In North Africa (Tunisia, Algeria, Morocco, Libya, and Egypt 4 and the Middle East (Turkey, Israel, and Syria) 5 prevalence is increased, especially in communities in which consanguinity is common.
Xeroderma pigmentosum is caused by cellular hypersensitivity to ultraviolet (UV) radiation, as a result of a defect in the DNA repair system caused by mutations in genes that are involved in repairing damaged DNA 6. Inherited mutations in at least nine genes have been identified 6. Xeroderma pigmentosum is inherited in an autosomal recessive manner 6. People with Xeroderma pigmentosum need total protection from sunlight. This includes protective clothing, sunscreen, and dark sunglasses when out in the sun. To prevent skin cancer, medications like retinoid creams may be prescribed. Skin cancers that do develop should be treated using standard practices 7.
The signs of xeroderma pigmentosum usually appear in infancy or early childhood. Xeroderma pigmentosum symptoms typically develop by the time a child is 2 years old 2. Many affected children develop a severe sunburn after spending just a few minutes in the sun. The sunburn causes redness and blistering that can last for weeks. Other affected children do not get sunburned with minimal sun exposure, but instead tan normally. By age 2, almost all children with xeroderma pigmentosum develop freckling of the skin in sun-exposed areas (such as the face, arms, and lips); this type of freckling rarely occurs in young children without the disorder.
People with xeroderma pigmentosum have a greatly increased risk of developing skin cancer. Without sun protection, about half of children with this condition develop their first skin cancer by age 10. Most people with xeroderma pigmentosum develop multiple skin cancers during their lifetime. These cancers occur most often on the face, lips, and eyelids. Cancer can also develop on the scalp, in the eyes, and on the tip of the tongue. Studies suggest that people with xeroderma pigmentosum may also have an increased risk of other types of cancer, including brain tumors. Additionally, affected individuals who smoke cigarettes have a significantly increased risk of lung cancer.
The eyes of people with xeroderma pigmentosum may be painfully sensitive to UV rays from the sun. If the eyes are not protected from the sun, they may become bloodshot and irritated, and the clear front covering of the eyes (the cornea) may become cloudy. In some people, the eyelashes fall out and the eyelids may be thin and turn abnormally inward or outward. In addition to an increased risk of eye cancer, xeroderma pigmentosum is associated with noncancerous growths on the eye. Many of these eye abnormalities can impair vision.
About 30 percent of people with xeroderma pigmentosum develop progressive neurological abnormalities in addition to problems involving the skin and eyes. These abnormalities can include hearing loss, poor coordination, difficulty walking, movement problems, loss of intellectual function, difficulty swallowing and talking, and seizures. When these neurological problems occur, they tend to worsen with time.
Xeroderma pigmentosum is characterized by 6:
- Sun sensitivity (severe sunburn with blistering, persistent erythema on minimal sun exposure in ~60% of affected individuals), with marked freckle-like pigmentation of the face before age two years in most affected individuals;
- Sunlight-induced ocular involvement (photophobia, keratitis, atrophy of the skin of the lids);
- Greatly increased risk of sunlight-induced cutaneous neoplasms (basal cell carcinoma, squamous cell carcinoma, melanoma).
People with xeroderma pigmentosum need total protection from sunlight. Even the light coming through windows or from fluorescent bulbs can be dangerous. However, UV protection does not prevent the neurodegeneration 8.
The main goal of treatment is protection from UV from the sun and unshielded fluorescent lamps (sun avoidance).
- Outdoor activities should be restricted to night-time; stay indoors during the day.
- Wear protective clothing (long sleeves and pants, shirts with collars, tightly woven fabrics, wide-brimmed hat).
- Shield eyes with UV–absorbing wraparound sunglasses.
- Apply broad-spectrum sunscreens with SPF of 50 or greater to all exposed areas.
Patients should undergo frequent skin examinations by someone who has been taught to recognise signs of skin cancer. Any suspicious spots or growths should be immediately reported to the doctor.
- Examination by a doctor (preferably a dermatologist) at least every 3 to 6 months.
- Actinic keratoses may be treated by cryotherapy or 5-fluorouracil cream. Any suspicious growths should undergo biopsy. Skin cancers are usually excised.
- Frequent eye examinations by an ophthalmologist.
- Yearly testing (through to age 20) for potential neurological problems.
Some patients with xeroderma pigmentosum patients may be prescribed isotretinoin. This is a vitamin A derivative that may prevent the formation of new cancers by altering keratinocyte differentiation.
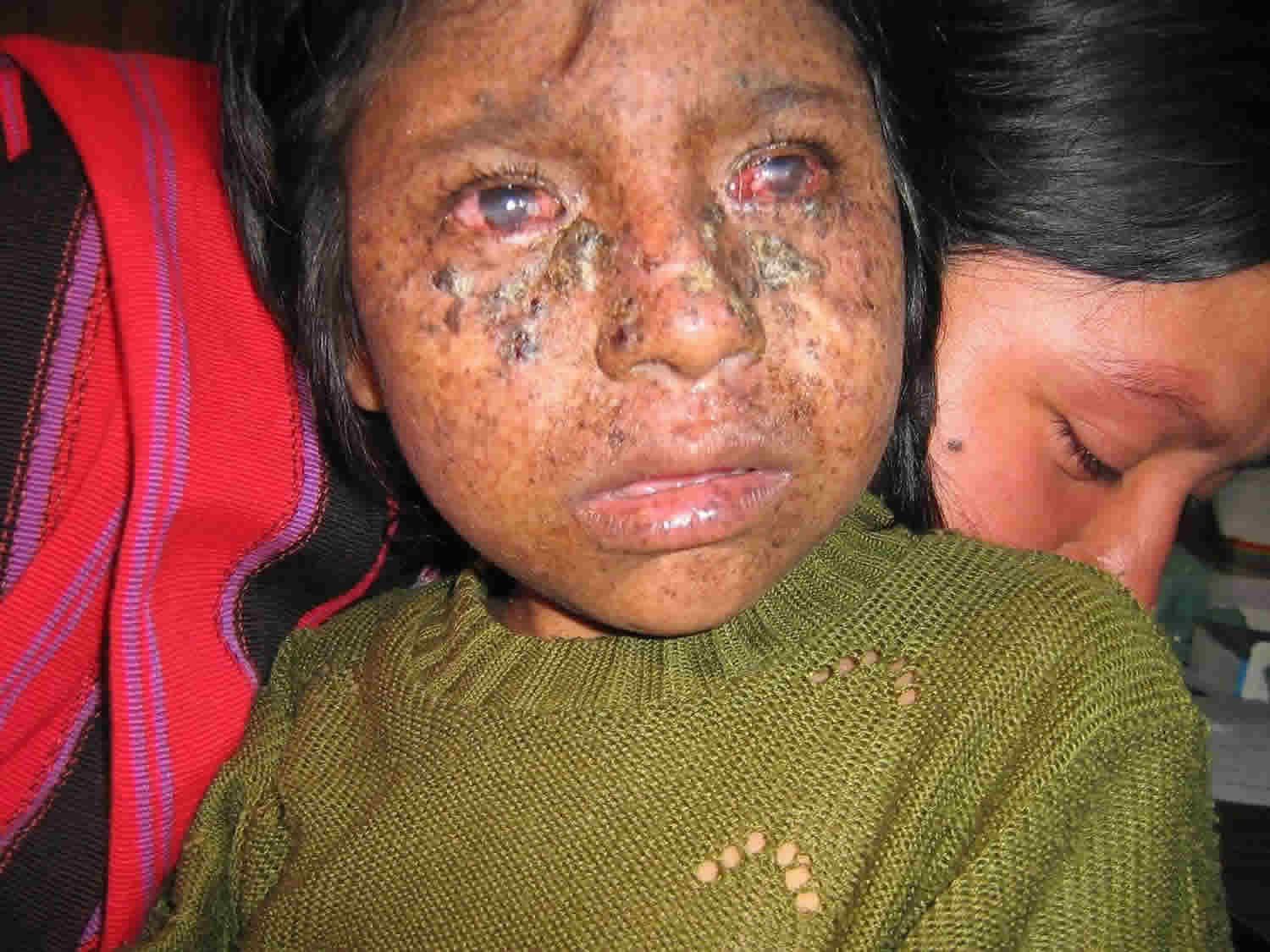
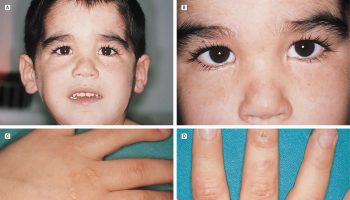
Figure 1. Xeroderma pigmentosum
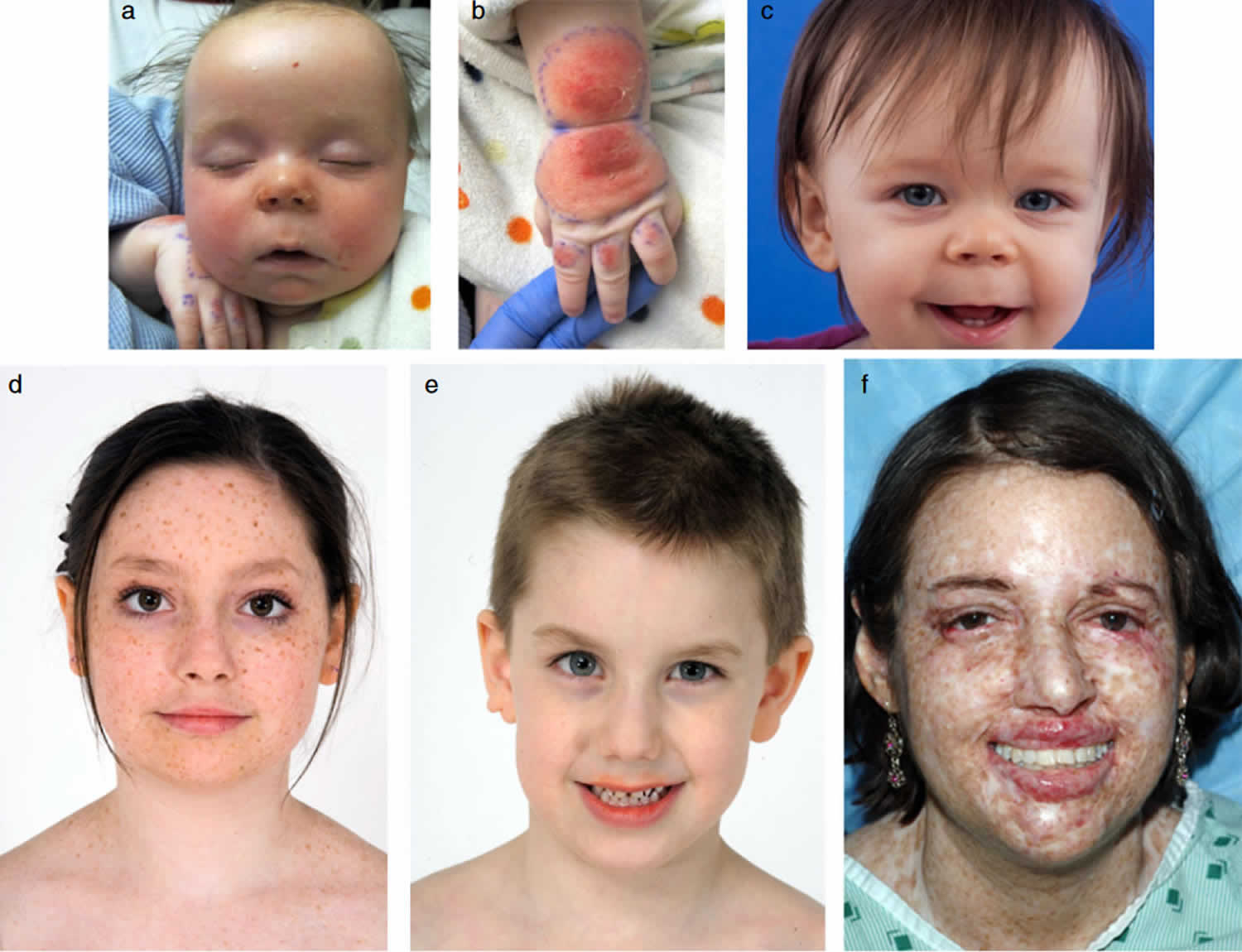
Footnote: Sunburning in XP-D patient and lack of burning in XP-C patients. (a–c) XP-D patient (XP499BE) who sustained severe blistering sunburn following intermittent evening sun exposure. (a) Five months of age: day 8 after sun exposure – swelling and burns of face resolving with delayed erythema on dorsum of right arm and hand. (b) Day 8 after sun exposure – delayed swelling, erythema, blistering, and peeling on dorsum of left hand and wrist. (c) Age 11 months: excellent sun protection and normal skin exam. (d and e) XP-C siblings with no history of burning on minimal sun exposure. (d) Older sister age 12 (XP198BE) – diagnosed at age 1 year secondary to freckling on face, hands, and arms. She had excellent UV protection since diagnosis and had 1 BCC in her scalp; (e) Patient XP338BE, 7-year-old brother of patient XP198BE. He had excellent UV protection since diagnosis at birth and no clinical evidence of XP. (f) XP-C patient (XP24BE) age 32 years, did not have a history of burning on minimal sun exposure. She had a relatively late age of diagnosis (age 8 years) and poor UV protection. She has multiple lentigines, chelitis, telangiectasias, and > 200 skin cancers. She died at age 35 years of a glioblastoma of her brain 9.
[Source 10 ]Xeroderma pigmentosum facts
- At present there is no known cure for xeroderma pigmentosum.
- Xeroderma pigmentosum is an autosomal recessive condition. This means a person with xeroderma pigmentosum would need to inherit one copy of the xeroderma pigmentosum gene from each parent. Parents often have no idea they carry the xeroderma pigmentosum gene, however once it is known that they do, they are advised that there is a 1 in 4 risk in every further pregnancy of having a child with xeroderma pigmentosum. Xeroderma pigmentosum can affect both sexes and all racial groups. Genetic counselling and advice is available to people with xeroderma pigmentosum and parents of children with xeroderma pigmentosum. Eight of the genes make up the nucleotide excision repair pathway (NER) that identities and repairs UV induced DNA damage. The ninth gene acts to bypass unrepaired damage.
- The diagnosis of xeroderma pigmentosum is made on the basis of clinical findings and family history and/or by the identification of biallelic pathogenic variants in DDB2, ERCC1, ERCC2, ERCC3, ERCC4, ERCC5, POLH, XPA, or XPC 6.
- People with xeroderma pigmentosum have a 10,000-fold increased risk for developing skin cancer including basal cell carcinoma, squamous cell carcinoma and melanoma. They also have a 2000-fold increased risk for cancer of the eye and surrounding ocular tissues. These symptoms appear early in life, typically before age 10 years.
- Xeroderma pigmentosum is managed by preventative techniques (i.e., avoiding the sun, using sunscreen, wearing protective clothing) and regular screening for changes in the skin, vision, and neurologic status. Many symptoms can be treated with medication and/or surgery, but some cancers and neurologic problems can be life threatening.
- It is recommended someone diagnosed with xeroderma pigmentosum should have:
- Skin checks by a dermatologist who looks for early signs of any skin cancers. This can reduce the risk of them spreading and causing further harm. If there are any concerning skin lesions these can be biopsied and surgically removed. In some cases topical treatments (creams to skin) may be prescribed which can help treat some early signs of skin cancers.
- Eye checks by an ophthalmologist who looks for eye damage associated with ultraviolet radiation. Sometimes eye lubricants are prescribed to treat dryness. Various types of glasses may be suggested to reduce ultraviolet radiation damage and in some cases, surgery may be recommended if there are any suspicious areas, which could indicate eye cancer, although this is rare.
- The minority of people with xeroderma pigmentosum who show signs of balance problems or learning difficulties or a loss of skills they previously had can benefit from seeing a neurologist (a doctor skilled in problems of the nerves and brain). They would examine the patient and take a detailed history and may also suggest a MRI (brain scan), hearing test and/or nerve conduction study. These will help them understand the extent of the problem so they can give advice. This advice may include things such as organising help at school to maximise a child’s learning potential, hearing aids if required, better footwear to help walking.
- It can sometimes be hard to understand and adapt to live with xeroderma pigmentosum; it can help to have access to a psychologist who patients can talk to in confidence about their feelings. They may be able to give suggestions as to what may be helpful to that person.
- If a patient is protecting themselves from ultraviolet radiation then Vitamin D levels in their blood can be low, due to lack of sunlight which is needed to help the body produce this vitamin. Vitamin D levels can be checked with a simple blood test and supplements prescribed if required. Low vitamin D could lead to bone pain and long term bone problems.
- Self care:
- Protection from ultraviolet radiation is very important to help reduce any further damage to your skin. ultraviolet radiation is present at the same time as daylight, from the moment the sun comes over the horizon to when it falls at night. ultraviolet radiation is part of the electromagnetic spectrum but unlike sunlight, it is invisible. Although there is less ultraviolet radiation present when it is cloudy or rainy, it is still there and therefore a person would still need to protect themselves in the following ways all year round, whatever the weather.
- Think about when to go outside. ultraviolet radiation levels are highest when the sun is directly overhead. ultraviolet radiation is not present outside at night and therefore no extra precautions would be needed. Understandably people have to be able to go out during the day but avoiding the highest ultraviolet radiation levels would be advised. In addition there are a number of other protective steps recommended.
- Clothing to cover the whole body including long sleeves and long trousers to protect the arms and legs and gloves to protect the hands. Tight weave materials let through less ultraviolet radiation and thick clothing or multiple layers can block ultraviolet radiation completely. Some manufacturers produce clothing to protect from ultraviolet radiation and this is marked with a 50+ UDF (a measure of how much ultraviolet radiation a material lets through) label, these can help, but can be expensive. Some people opt to wear two thinner layers of clothing instead.
- Hat with a wide brim that covers the forehead, ears and neck for example a legionnaire’s style hat.
- Sunglasses to protect the eyes with a high ultraviolet radiation protection rating to protect against UVA and UVB wavelengths. If a person prefers clear non-tinted glasses these can now be bought with UVA and UVB coating. The style of glasses that wrap around the face or have side covers to limit ultraviolet radiation reaching the eye are suggested.
- Ultraviolet radiation protective face visor is very good for full protection of the face.
- Sunscreen should be broad spectrum 50+ sun protective factor (SPF) which protects against UVB as well as UVA. Sunscreens come in a variety of forms, sprays, creams, lotions, gels, roll-on along with different smells and colours/tints. It is important to choose one that a person will be happy to use all the time. Some are available on prescription for people with xeroderma pigmentosum. Sunscreen should be applied 20 minutes before going out, and should be reapplied 2-3 hourly. Reapply the sunscreen if the cream is removed, e.g. after hand washing. Care should be taken in application to ensure no areas are missed and the correct amount is used. Sunscreen is tested for its SPF rating by testing with 2 mg/cm2 so if using a lotion, this equates to six heaped teaspoons of lotion or 35 ml to cover an adult body. This can vary if using different formulations so check the manufacturer’s recommendations. Lips should not be forgotten, SPF 50+ protection lipblock should also be used.
- Skin checks. If you notice anything different on your skin or a freckle or mole looks like it is changing in some way then make an appointment to see your doctor or local dermatologist to get it checked out. The sooner something is noticed and treated the better. Your dermatologist/family doctor or specialist nurse can teach you what to look for and how to do a skin check.
- Eye concerns. If you notice any changes with your eyes or notice any lumps, inform your medical team as soon as possible to get them checked.
- Environment. ultraviolet radiation, in particular UVA, can travel through some window glass, although not all. It all depends on how the glass was manufactured which can be difficult to know if it is already in a window. ultraviolet radiation protective film is a transparent film that can be applied to a window to reduce UVA coming through. It is available from companies that supply window films and can be up to 99.8% effective at blocking UVA. Some patients may be eligible for a disability facilities grant to help fund this (https://www.gov.uk/disabled-facilities-grants) or the xeroderma pigmentosum charities may be able to help. Cars may also need this protection. Remember an open window will let in ultraviolet radiation. Drawing curtains and blinds is an effective way of reducing ultraviolet radiation but makes a room dark.
- Light bulbs. In particular, halogen, fluorescent and compact fluorescent bulbs are known to produce some UVA. This risk is relatively low but can be a problem if a bulb is close to a person with xeroderma pigmentosum. Bulbs can be covered with protective ultraviolet radiation sleeves or changed to ones that produce less ultraviolet radiation such as LEDs.
- UV meter. As ultraviolet radiation is invisible it can be hard to know when you are at risk. A UV meter, can help with a more accurate assessment of the amount of ultraviolet radiation in the environment.
Is xeroderma pigmentosum hereditary?
Yes it is. Xeroderma pigmentosum is an autosomal recessive condition. This means a person with xeroderma pigmentosum would need to inherit one copy of the xeroderma pigmentosum gene from each parent. The parents are known as carriers as they only carry one copy of the xeroderma pigmentosum gene and therefore do not themselves have xeroderma pigmentosum. Parents often have no idea they carry the xeroderma pigmentosum gene, however once it is known that they do, they are advised that there is a 1 in 4 risk in every further pregnancy of having a child with xeroderma pigmentosum. Xeroderma pigmentosum can affect both sexes and all racial groups. Genetic counselling and advice is available to people with xeroderma pigmentosum and parents of children with xeroderma pigmentosum.
It there a cure for xeroderma pigmentosum?
There is no cure for xeroderma pigmentosum, but there are ways to prevent and treat some of the problems associated with it. Some of the strategies employed in the management of xeroderma pigmentosum include 6:
- Protection from ultraviolet light
- Frequent skin and eye examinations
- Prompt removal of cancerous tissue
- Neurological examination
- Psychosocial care
Small, premalignant skin lesions such as actinic keratoses can be frozen with liquid nitrogen. Larger areas of sun-damaged skin can be treated with topical 5-fluorouracil or imiquimod. In rare cases, therapeutic dermatome shaving or dermabrasion is used to remove damaged superficial epidermal layers. Skin cancers can be treated using standard treatment protocols, including electrodesiccation and curettage (scrapes away the lesion and uses electricity to kill any remaining cells), surgical excision, or chemosurgery. High dose oral isotretinoin or acitretin can be used to prevent new cancers. Cancers of the eyelids, conjunctiva, and cornea are usually treated surgically. Corneal transplantation may be necessary for those with severe keratitis and corneal opacity 6.
Xeroderma pigmentosum causes
Xeroderma pigmentosum is caused by mutations in genes that are involved in repairing damaged DNA. DNA can be damaged by UV rays from the sun and by toxic chemicals such as those found in cigarette smoke. Normal cells are usually able to fix DNA damage before it causes problems. However, in people with xeroderma pigmentosum, DNA damage is not repaired normally. As more abnormalities form in DNA, cells malfunction and eventually become cancerous or die.
Many of the genes related to xeroderma pigmentosum are part of a DNA-repair process known as nucleotide excision repair (NER). The proteins produced from these genes play a variety of roles in this process. They recognize DNA damage, unwind regions of DNA where the damage has occurred, snip out (excise) the abnormal sections, and replace the damaged areas with the correct DNA. Inherited abnormalities in the NER-related genes prevent cells from carrying out one or more of these steps. The POLH gene also plays a role in protecting cells from UV-induced DNA damage, although it is not involved in NER; mutations in this gene cause the variant type of xeroderma pigmentosum.
The major features of xeroderma pigmentosum result from a buildup of unrepaired DNA damage. When UV rays damage genes that control cell growth and division, cells can either die or grow too fast and in an uncontrolled way. Unregulated cell growth can lead to the development of cancerous tumors. Neurological abnormalities are also thought to result from an accumulation of DNA damage, although the brain is not exposed to UV rays. Researchers suspect that other factors damage DNA in nerve cells. It is unclear why some people with xeroderma pigmentosum develop neurological abnormalities and others do not.
Inherited mutations in at least eight genes have been found to cause xeroderma pigmentosum. More than half of all cases in the United States result from mutations in the XPC, ERCC2, or POLH genes. Mutations in the other genes generally account for a smaller percentage of cases.
Xeroderma pigmentosum genetics
There are 9 different genes that may be altered in patients with xeroderma pigmentosum and include: the DDB2 (XP-E) gene, located on the short arm of chromosome 11 (11p11.2), the ERCC1 gene, located on the long arm of chromosome 19 (19q13.32), the ERCC2 (XP-D) gene, located on the long arm of chromosome 19 (19q13.32), the ERCC3 gene (XP-G), located on the long arm of chromosome 2 (2q14.3), the ERCC4 gene (XP-F), located on the short arm of chromosome 16 (16p13.12), the ERCC5 (XP-B) gene, located on the long arm of chromosome 13 (13q33.1), the POLH gene (XP-V or variant), located on the short arm of chromosome 6 (6p21.1), the XPA gene, located on the long arm of chromosome 9 (9q22.33), and the XPC gene, located on the short arm of chromosome 3 (3p25.1). The proteins resulting from normal expression of these genes are involved in DNA repair and serve to recognize damaged DNA, remove the damage and fill in the resulting gap. The types are distinguished by their genetic cause. All of the types increase skin cancer risk, although some are more likely than others to be associated with neurological abnormalities.
Table 1. Xeroderma pigmentosum Associated Genes and Their Complementation Groups
| Gene | Complementation Group |
|---|---|
| DDB2 | E |
| ERCC1 | See footnote 1 |
| ERCC2 | D |
| ERCC3 | B |
| ERCC4 | F |
| ERCC5 | G |
| POLH | Variant |
| XPA | A |
| XPC | C |
Footnote: Previously called group H, a designation that was subsequently withdrawn
[Source 11 ]Related Disorders
There are several genetically related disorders caused by mutations in genes in the nucleotide excision (NER) pathway. People with these diseases demonstrate very different symptoms despite having mutations in some of the same genes as xeroderma pigmentosum patients. These conditions include Cockayne syndrome (CS), cerebro-oculo-facio-skeletal (COFS) syndrome, trichothiodystrophy (TTD), and UV-sensitive syndrome.
Cockayne syndrome is a rare form of dwarfism characterized by short stature, UV sensitivity, and prematurely aged appearance (progeria). Although prenatal growth is normal, developmental abnormalities usually appear within two years of life; height, weight, and head circumferences tend to fall below the 5th percentile, and death usually occurs within the first two decades.
Cerebro-oculo-facio-skeletal syndrome (COFS) is a genetic neuro-degenerative disorder of the brain and spinal cord that begins before birth. The disorder is characterized by growth failure at birth, little or no neurological development, structural abnormalities of the eye, and fixed bending of the spine and joints. Abnormalities of the skull, face, limbs, and other parts of the body may also occur. COFS syndrome is inherited as an autosomal recessive genetic trait and is now considered to be part of the spectrum of disorders within Cockayne syndrome. For more information on this disorder, choose “cerebro oculo facio skeletal syndrome” as your search term in the Rare Disease Database.
Trichothiodystrophy (TTD) is a hereditary disorder characterized by short brittle hair that demonstrates alternating dark and light ‘tiger tail’ banding when examined through a polarized microscope. Symptoms may include photosensitivity, intellectual impairment, short stature, and microcephaly. Unlike, xeroderma pigmentosum patients, TTD patients do not have an increased cancer risk. Individuals with TTD have a 20x increased risk of death before the age of 10 (typically due to infections). Patients with TTD tend to have complications during their gestations and exhibit neonatal abnormalities. For more information on this disorder, choose “trichothiodystrophy” as your search term in the Rare Disease Database.
UV-sensitive syndrome is a form of photosensitivity that does not involve pigmentary abnormalities or nervous system deficits. People with UV sensitive syndrome develop sun burns after very minimal sun exposure but do not have increased cancer risk.
In addition, there are patients who demonstrate combinations of xeroderma pigmentosum with other NER disorders, most notably, xeroderma pigmentosum with Cockayne syndrome (XP/CS) and xeroderma pigmentosum with trichothiodystrophy (XP/TTD). There have been a few patients reported with cerebraloculofacioskeletal syndrome and trichothiodystrophy (COFS/TTD) and Cockayne syndrome and trichothiodystrophy (CS/TTD). Individuals with these ‘overlap’ syndromes show a mixture of the symptoms that are normally present in both of the disorders. For example, those with XP/CS show facial freckling (typical of xeroderma pigmentosum), as well as short stature, sunken eyes and wasting (typical of CS). XP/CS differs from xeroderma pigmentosum alone in that there is dysmyelination (defective structure/function of the myelin sheath) along with the neuronal degeneration typically seen in people with xeroderma pigmentosum and neurologic disease.
Table 2. Phenotype Correlations by Gene in Xeroderma pigmentosum and Related Disorders
| Gene | Cutaneous Neoplasia | Phenotype |
|---|---|---|
| DDB2 | + 1 | Xeroderma pigmentosum with no neurologic abnormalities |
| ERCC1 | – | Cerebrooculofacioskeletal syndrome 2 |
| ERCC2 3 | + | Xeroderma pigmentosum with neurologic abnormalities ranging from none to severe |
| + | Xeroderma pigmentosum-Cockayne syndrome complex | |
| + | Trichothiodystrophy with xeroderma pigmentosum | |
| – | Trichothiodystrophy (without xeroderma pigmentosum) | |
| – | Cerebrooculofacioskeletal syndrome | |
| ERCC3 4 | + | Xeroderma pigmentosum-Cockayne syndrome complex |
| – | Trichothiodystrophy (without xeroderma pigmentosum) | |
| + | Xeroderma pigmentosum with mild neurologic abnormalities | |
| ERCC4 | + | Xeroderma pigmentosum with no neurologic abnormalities or severe late-onset neurologic abnormalities 5; Fanconi anemia (FA) 6; 1 individual with features of xeroderma pigmentosum , Cockayne syndrome, and Fanconi anemia, and 2 individuals with Cockayne syndrome 7 |
| ERCC5 | + | Xeroderma pigmentosum with no neurologic abnormalities or severe neurologic abnormalities |
| + | Xeroderma pigmentosum-Cockayne syndrome complex | |
| POLH | + | Xeroderma pigmentosum with no neurologic abnormalities 8 |
| XPA | + | Xeroderma pigmentosum with neurologic abnormalities ranging from mild to severe |
| XPC | + | Xeroderma pigmentosum with no neurologic abnormalities 9, 10 |
Footnotes:
- 1. Adults with large numbers of skin cancers have been reported 12.
- 2. Only one person with cerebrooculofacioskeletal syndrome has been reported to have biallelic pathogenic variants in ERCC1 13. One individual with a homozygous ERCC1 pathogenic variant had severe Cockayne syndrome type II and died at age 2.5 years 14.
- 3. Individuals with xeroderma pigmentosum caused by mutation of ERCC2 (XP-D) have xeroderma pigmentosum, xeroderma pigmentosum with neurologic abnormalities, the xeroderma pigmentosum-Cockayne syndrome complex, trichothiodystrophy without xeroderma pigmentosum, or trichothiodystrophy with xeroderma pigmentosum 12.
- 4. Fassihi et al 12.
- 5. Most individuals are from Japan. Several have been seen in UK 12.
- 6. Bogliolo et al 15
- 7. Kashiyama et al 14
- 8. Individuals with xeroderma pigmentosum variant are clinically identical to other individuals with xeroderma pigmentosum with cutaneous symptoms without neurologic abnormalities 12.
- 9. “Xeroderma pigmentosum neurologic abnormalities” refers to progressive loss of motor, sensory, and cognitive function thought to result from neuronal loss.
- 10. Most individuals with xeroderma pigmentosum caused by mutation of XPC (XP-C) have xeroderma pigmentosum without xeroderma pigmentosum neurologic abnormalities 12.
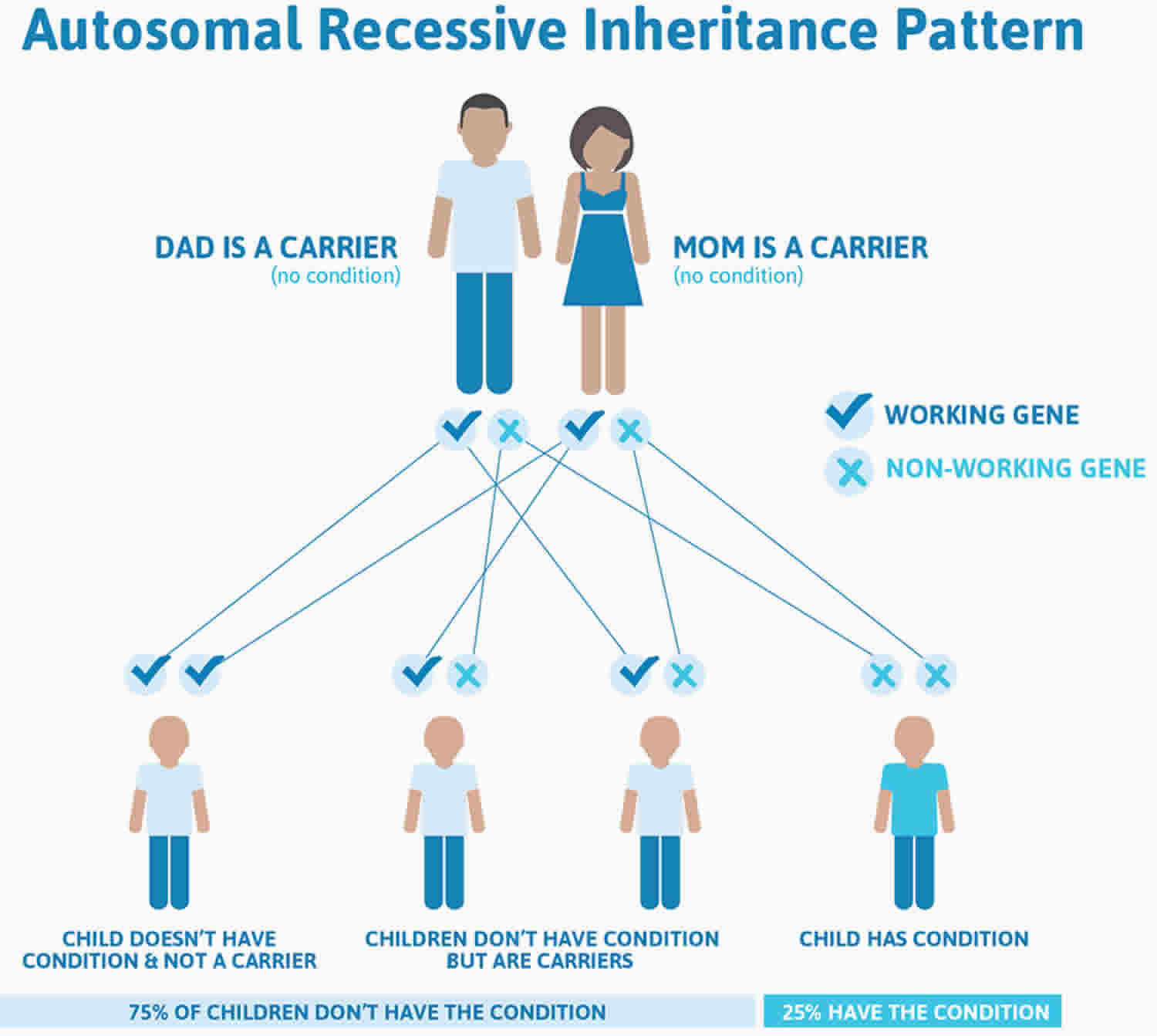
Xeroderma pigmentosum inheritance pattern
Xeroderma pigmentosum is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
Autosomal recessive means two copies of the abnormal gene, one from each parent (one abnormal gene from mum and one abnormal gene from dad), is needed to cause the disorder or disease. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of xeroderma pigmentosum. Autosomal recessive disorders are typically not seen in every generation of an affected family.
Autosomal recessive conditions are individually pretty rare, so the chance that you and your partner are carriers for the same recessive genetic condition are likely low. Even if both partners are a carrier for the same condition, there is only a 25% chance that they will both pass down the non-working copy of the gene to the baby, thus causing a genetic condition. This chance is the same with each pregnancy, no matter how many children they have with or without the condition.
- If both partners are carriers of the same abnormal gene, they may pass on either their normal gene or their abnormal gene to their child. This occurs randomly.
- Each child of parents who both carry the same abnormal gene therefore has a 25% (1 in 4) chance of inheriting a abnormal gene from both parents and being affected by the condition.
- This also means that there is a 75% ( 3 in 4) chance that a child will not be affected by the condition. This chance remains the same in every pregnancy and is the same for boys or girls.
- There is also a 50% (2 in 4) chance that the child will inherit just one copy of the abnormal gene from a parent. If this happens, then they will be healthy carriers like their parents.
- Lastly, there is a 25% (1 in 4) chance that the child will inherit both normal copies of the gene. In this case the child will not have the condition, and will not be a carrier.
These possible outcomes occur randomly. The chance remains the same in every pregnancy and is the same for boys and girls.
Figure 2 illustrates xeroderma pigmentosum autosomal recessive inheritance. The example below shows what happens when both dad and mum is a carrier of the abnormal gene, there is only a 25% chance that they will both pass down the abnormal gene to the baby, thus causing a genetic condition.
Figure 2. Xeroderma pigmentosum autosomal recessive inheritance pattern
Genetics counseling
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Xeroderma pigmentosum prevention
Experts recommend genetic counseling for people with a family history of xeroderma pigmentosum who wish to have children.
Xeroderma pigmentosum symptoms
Xeroderma pigmentosum symptoms usually appear by the time a child is 2 years old.
Skin symptoms include:
- Sunburn that does not heal after just a little bit of sun exposure
- Blistering after just a little bit of sun exposure
- Spider-like blood vessels under the skin
- Patches of discolored skin that get worse, resembling severe aging
- Crusting of the skin
- Scaling of the skin
- Oozing raw skin surface
- Discomfort when being in bright light (photophobia)
- Skin cancer at a very young age (including melanoma, basal cell carcinoma, squamous cell carcinoma)
Eye symptoms include:
- Dry eye
- Clouding of the cornea
- Ulcers of the cornea
- Swelling or inflammation of the eyelids
- Cancer of eyelids, cornea or sclera
Nervous system (neurologic) symptoms, which develop in some children, include:
- Intellectual disability
- Delayed growth
- Loss of hearing
- Muscle weakness of the legs and arms
Xeroderma pigmentosum usually progresses through 3 stages. The first stage occurs around 6 months after birth (skin appears normal at birth).
- Areas exposed to the sun such as the face show reddening of the skin with scaling and freckling. Irregular dark spots may also begin to appear.
- These skin changes progress to the neck and lower legs, and in severe cases, to the trunk.
- Over the winter months, these changes may diminish.
Continued sun exposure will lead to the second stage, which is characterised by:
- Poikiloderma
- Skin atrophy
- Telangiectasia
- Mottled hyperpigmentation and hypopigmentation.
The third stage is the development of actinic keratoses and skin cancers. These may occur as early as age 4–5 years and a mean of 8 years. They are more prevalent in sun-exposed areas such as the face. They include:
- Basal cell carcinoma
- Squamous cell carcinoma
- Melanoma.
Eye problems occur in nearly 80% of xeroderma pigmentosum patients.
- Eyes become painfully sensitive to the sun (photophobia).
- Eyes easily irritated, bloodshot and clouded. Conjunctivitis may occur.
- Non-cancerous and cancerous growths on the eyes may occur.
Neurological problems occur in about 20% of xeroderma pigmentosum patients.
- These can be mild or severe and include spasticity, poor coordination, developmental delay, deafness, and short stature.
- May develop in late childhood or adolescence. Once they do occur, they tend to worsen over time.
Xeroderma pigmentosum life expectancy
Many patients with xeroderma pigmentosum die at an early age from skin cancers. Over one half of people with this condition die of skin cancer early in adulthood. However, if a person is diagnosed early, does not have severe neurological symptoms or has a mild variant, and takes all the precautionary measures to avoid exposure to UV light, they may survive beyond middle age.
Patients with xeroderma pigmentosum and their families will face many challenges in daily living. Constant educating and reminding of the need to protect oneself from sunlight is paramount to the management of xeroderma pigmentosum.
Due to good UV protection and improved medical care, xeroderma pigmentosum patients are living longer and increasingly more public lives. Planning for college and careers is something that xeroderma pigmentosum patients and families now need to consider. xeroderma pigmentosum patients can work at jobs that do not involve prolonged day-time outdoor activities. Adult xeroderma pigmentosum patients enrolled in the National Institute of Health natural history study 16 hold positions such as physician assistant, computer scientist, and flooring business owner. These patients are able to be active in the community while maintaining an adequate level of UV protection 17.
Xeroderma pigmentosum diagnosis
Usually, xeroderma pigmentosum is detected in early infancy, around 1–2 years. A child presenting with severe sunburn after their first exposure to sun may be a clue to the diagnosis of xeroderma pigmentosum. Xeroderma pigmentosum can usually be conclusively diagnosed by measuring the DNA repair factor from skin or blood samples.
If xeroderma pigmentosum is suspected, a small skin biopsy can be taken from a non-sun exposed site, often the buttock. After a person has given their consent, the biopsy site would be made numb by injecting some local anaesthetic into the skin and removing a small piece of skin. This would be sent to the diagnostic laboratory to be tested. Results can take up to 4 months as it is a very complicated laboratory test.
The xeroderma pigmentosum group can then be diagnosed with a blood test which would be sent to a genetic laboratory.
The diagnosis of xeroderma pigmentosum is made on the basis of clinical findings and family history and/or by the identification of biallelic pathogenic variants in DDB2, ERCC1, ERCC2, ERCC3, ERCC4, ERCC5, POLH, XPA, or XPC 6.
Tests that may be done include:
- Skin biopsy in which skin cells are studied in the laboratory
- DNA testing for the problem gene
The following tests can help diagnose the condition in a baby before the birth:
- Amniocentesis
- Chorionic villous sampling
- Culture of amniotic cells
Family members and known carriers should undergo genetic counselling and avoid consanguineous marriage.
A fetus at risk of xeroderma pigmentosum can undergo prenatal diagnosis by amniocentesis or chorionic villi sampling and chromosomal breakage studies.
To establish the extent of disease and needs in an individual diagnosed with xeroderma pigmentosum, the following evaluations are recommended 18.
Skin
- Perform baseline examination of the skin (including all sun-exposed as well as sun-shielded areas) for evidence of sunlight-induced damage including pigmentary changes, precancerous lesions, and skin cancers.
- For examination of the scalp, use a hair dryer (on a cool setting) to blow the hair aside.
- Examination of the lip and adjacent tip of the tongue for signs of sun damage, including actinic cheilitis (a type of actinic keratosis or leukoplakia occurring on the lips) and prominent telangiectasia, which may precede the development of cancer in these areas 19.
- Baseline clinical color photographs of the entire skin surface with close-ups (including a ruler) of individual lesions to facilitate follow-up and detection of early skin cancers.
Eyes
- Examine the lids and anterior UV-exposed portions of the globe for evidence of sun-induced damage including ectropion, entropion, inflammatory masses (pterygia, pinguecula), clouding of the cornea, and cancer of the lids, conjunctiva, or cornea. Eversion of the lids may be necessary to detect cancers of the mucosal surface.
- Use the Schirmer test to detect dry eyes. This test involves measurement of the extent of absorption of tears into filter paper placed under eyelids for a few minutes 20.
Neurologic
- Deep tendon reflex testing
- Measurement of the occipital frontal circumference (OFC), to determine if microcephaly is present
- MRI of the brain and nerve conduction velocities, if other neurologic problems are detected
Auditory
- Baseline audiometry evaluation to screen for the sensorineural hearing loss, which may be present as a component of the xeroderma pigmentosum-associated neurologic abnormalities 21
Genetic
- Consultation with a clinical geneticist and/or genetic counselor
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Xeroderma pigmentosum treatment
There is no cure for xeroderma pigmentosum, but there are ways to prevent and treat some of the problems associated with it.
People with xeroderma pigmentosum need total protection from sunlight. Even the light coming through windows or from fluorescent bulbs can be dangerous.
Some of the strategies employed in the management of xeroderma pigmentosum include 6:
- Protection from ultraviolet light
- Frequent skin and eye examinations
- Prompt removal of cancerous tissue
- Neurological examination
- Psychosocial care
Small, premalignant skin lesions such as actinic keratoses can be frozen with liquid nitrogen. Larger areas of sun-damaged skin can be treated with topical 5-fluorouracil or imiquimod. In rare cases, therapeutic dermatome shaving or dermabrasion is used to remove damaged superficial epidermal layers. Skin cancers can be treated using standard treatment protocols, including electrodesiccation and curettage (scrapes away the lesion and uses electricity to kill any remaining cells), surgical excision, or chemosurgery. High dose oral isotretinoin or acitretin can be used to prevent new cancers. Cancers of the eyelids, conjunctiva, and cornea are usually treated surgically. Corneal transplantation may be necessary for those with severe keratitis and corneal opacity 6.
Skin
Premalignant lesions (e.g., small actinic keratosis) may be treated by freezing with liquid nitrogen.
Larger areas of sun-damaged skin can be treated with field treatments such as topical 5-fluorouracil or imiquimod preparations. Rarely, therapeutic dermatome shaving or dermabrasion has been used to remove the more damaged superficial epidermal layers. This procedure permits repopulation by relatively UV-shielded cells from the follicles and glands.
Cutaneous neoplasms are treated in the same manner as in individuals who do not have xeroderma pigmentosum. This involves electrodesiccation/curettage or surgical excision. Skin cancers which are recurrent or in locations at high risk for recurrence are best treated with Mohs micrographic surgery. Because multiple surgical procedures are often necessary, removal of undamaged skin should be minimized. Severe cases have been treated by excision of large portions of the facial surface and grafting with sun-protected skin.
While individuals with xeroderma pigmentosum are not abnormally sensitive to therapeutic x-rays, and individuals with xeroderma pigmentosum have responded normally to full-dose therapeutic x-radiation for treatment of inoperable neoplasms 22, cultured cells from a few individuals with xeroderma pigmentosum were found to be hypersensitive to x-radiation 23. When x-radiation therapy is indicated, an initial small dose is advisable to test for clinical hypersensitivity.
Oral isotretinoin or acitretin can be effective in preventing new neoplasms in individuals with multiple skin cancers 24. Because of its toxicity (hepatic, hyperlipidemic, and teratogenic effects; calcification of ligaments and tendons; premature closure of the epiphyses), oral isotretinoin or acitretin should be reserved for individuals with xeroderma pigmentosum who are actively developing large numbers of new tumors. Some individuals may respond to lower doses of isotretinoin or acitretin with less toxicity.
A few case reports have described regression of skin cancers with use of imiquimod cream in individuals with xeroderma pigmentosum 25; however, no controlled studies have been reported.
Eyes
Methylcellulose eye drops or soft contact lenses have been used to keep the cornea moist and to protect against mechanical trauma in individuals with deformed eyelids.
Corneal transplantation has restored vision in individuals with severe keratitis with corneal opacity. However, the immunosuppression necessary to prevent rejection of the transplant may increase the risk for skin cancer.
Neoplasms of the lids, conjunctiva, and cornea are usually treated surgically.
Hearing
Hearing aids can be of great help for individuals who have sensorineural hearing loss with learning difficulties in school 21.
Neurologic Abnormalities
Neurologic abnormalities are associated with increased high frequency sensory-neural hearing loss. The hearing loss is progressive (gets worse over time) and can be treated with hearing aids. Cognitive delays can be seen in childhood and special education classes, physical and occupational therapies along with UV safe accommodations at school are very helpful for xeroderma pigmentosum children. As they get older, people with xeroderma pigmentosum neurologic disease also experience increasing ataxia, dysphagia (difficulty swallowing) and dysarthria (difficulty speaking) as the condition progresses. They may require wheel chairs, feeding tubes and long term nursing care.
Long-Term Monitoring
Skin. A physician should examine the skin of an affected individual at frequent intervals (every ~3-12 months, depending on the severity of skin disease).
Affected individuals or their parents should be educated to look for abnormal pigmented lesions or the appearance of basal cell or squamous cell carcinoma. Individuals should be examined frequently by a family member who has been instructed in recognition of cutaneous neoplasms.
Eyes should be examined regularly for signs of UV exposure and damage.
Neurologic. Routine neurologic examination is indicated because of progressive neurologic abnormalities that are present in a minority of individuals with xeroderma pigmentosum and may not be detected in young children.
Hearing. Periodic audiograms. Serial audiograms at regular intervals may also be useful for assessing the presence or absence of progressive neurologic degeneration, especially in those with a history of acute burning on minimal sun exposure 21.
Prevention of secondary complications
Vitamin D is produced in the skin by a reaction involving exposure to UV radiation. Active adults with xeroderma pigmentosum and skin cancers received sufficient vitamin D in their diet in the past to result in normal serum concentrations of the active form (1,25 dihydroxy vitamin D) 26. However, children protected from sunlight very early in life have had low serum concentration of 25 hydroxy vitamin D; one child became susceptible to bone fractures 27. Dietary supplementation with oral vitamin D is recommended for persons with low serum concentration of serum vitamin D 28.
Agents or Circumstances to Avoid
UV exposure from sunlight and artificial sources of UV radiation should be avoided.
Artificial sources of UV
Certain light sources (e.g., mercury arc, halogen, and other lamps) can be unrecognized sources of UV. Although such light sources are often shielded, in open areas such as gymnasiums they can be a source of UV if the shield has been breached. UV meters are readily available to enable monitoring of areas to identify unexpected UV sources.
Cigarette smoke
Because cells from individuals with xeroderma pigmentosum are also hypersensitive to environmental mutagens, such as benzo[a]pyrene found in cigarette smoke, prudence dictates that individuals with xeroderma pigmentosum should be protected against these agents. One individual with xeroderma pigmentosum who smoked cigarettes for more than ten years died of bronchogenic carcinoma of the lungs at age 35 years 29. The authors recently cared for another individual with xeroderma pigmentosum who smoked and developed lung cancer in the fifth decade of life.
Pregnancy Management
The systemic retinoids isotretinoin and acitretin are used as skin cancer chemopreventive agents in individuals who are actively developing large numbers of skin cancers, and thus may be used by some women with xeroderma pigmentosum 24. Systemic retinoids are known to be teratogenic to a developing fetus and pose a high risk for birth defects. Therefore, women who are using systemic retinoids should be appropriately counseled about pregnancy risks and the need for effective contraception; regular monitoring with pregnancy tests is indicated. Systemic retinoids should be administered only by physicians who are knowledgeable regarding their risks and benefits.
To access isotretinoin in the US, women and their prescribing providers must be enrolled in the iPLEDGE program to minimize the potential for fetal exposure. Pregnancy avoidance is initiated before therapy, continues during therapy, and extends post-treatment until the drug is cleared from the body. While both drugs may be effective in preventing skin cancers, acitretin may take longer to be eliminated from the body, requiring an extended period (3 years) of post-therapy pregnancy avoidance to minimize teratogenic risk.
Therapies Under Investigation
The bacterial DNA repair enzyme T4 endonuclease V in a topical liposome-containing preparation has been reported to reduce the frequency of new actinic keratoses and basal cell carcinomas in individuals with xeroderma pigmentosum in one research study 30. As of 2016, this treatment is not approved by the US Food and Drug Administration.
Oral vismodegib (Erivedge®), an inhibitor of the hedgehog pathway, has been approved by the FDA for treatment of metastatic basal cell carcinoma or locally advanced basal cell carcinoma that has recurred following surgery. This drug has also been approved for use in individuals with basal cell carcinoma who are not candidates for surgery or for radiation therapy (see FDA package insert). This treatment may be appropriate for some individuals with xeroderma pigmentosum, however, no studies on the efficacy of this drug in those with xeroderma pigmentosum have been published. Oral vismodegib is also a teratogen, leading to embryo-fetal death, midline defects, missing digits, and other birth defects in an exposed embryo or fetus; effective contraception during and after vismodegib treatment is advised in both women and men.
- Kleijer WJ, Laugel V, Berneburg M, Nardo T, Fawcett H, Gratchev A, Jaspers NG, Sarasin A, Stefanini M, Lehmann AR. Incidence of DNA repair deficiency disorders in western Europe: Xeroderma pigmentosum, Cockayne syndrome and trichothiodystrophy. DNA Repair (Amst). 2008;7:744–50.[↩]
- Xeroderma pigmentosum. https://ghr.nlm.nih.gov/condition/xeroderma-pigmentosum[↩][↩]
- Hirai Y, Kodama Y, Moriwaki S, Noda A, Cullings HM, Macphee DG, Kodama K, Mabuchi K, Kraemer KH, Land CE, Nakamura N. Heterozygous individuals bearing a founder mutation in the XPA DNA repair gene comprise nearly 1% of the Japanese population. Mutat Res. 2006;601:171–8.[↩]
- Messaoud O, Ben Rekaya M, Kefi R, Chebel S, Boughammoura-Bouatay A, Bel Hadj Ali H, Gouider-Khouja N, Zili J, Frih-Ayed M, Mokhtar I, Abdelhak S, Zghal M. Identification of a primarily neurological phenotypic expression of xeroderma pigmentosum complementation group A in a Tunisian family. Br J Dermatol. 2010;162:883–6.[↩]
- Jerbi M, Ben Rekaya M, Naouali C, Jones M, Messaoud O, Tounsi H, Nagara M, Chargui M, Kefi R, Boussen H, Mokni M, Mrad R, Boubaker MS, Abdelhak S, Khaled A, Zghal M, Yacoub-Youssef H. Clinical, genealogical and molecular investigation of the xeroderma pigmentosum type C complementation group in Tunisia. Br J Dermatol. 2016;174:439–43.[↩]
- Kraemer KH, DiGiovanna JJ. Xeroderma Pigmentosum. 2003 Jun 20 [Updated 2016 Sep 29]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2019. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1397[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Xeroderma pigmentosum. https://medlineplus.gov/ency/article/001467.htm[↩]
- Bradford PT, Goldstein AM, Tamura D et al. Cancer and neurologic degeneration in xeroderma pigmentosum: long term follow-up characterises the role of DNA repair. J Med Genet 2011; 48: 168–176.[↩]
- Lai J-P, Liu Y-C, Alimchandani M et al. The influence of DNA repair on neurologic degeneration, cachexia, skin cancer and internal neoplasms: autopsy report of four xeroderma pigmentosum patients (XP-A, XP-C and XP-D). Acta Neuropathol Commun 2013; 1: 4.[↩]
- Living with xeroderma pigmentosum: comprehensive photoprotection for highly photosensitive patients. Deborah Tamura, John J. DiGiovanna, Sikandar G. Khan & Kenneth H. Kraemer. Photodermatol Photoimmunol Photomed 2014; 30: 146–152 https://onlinelibrary.wiley.com/doi/pdf/10.1111/phpp.12108[↩]
- DiGiovanna JJ, Kraemer KH. Shining a light on xeroderma pigmentosum. J Invest Dermatol. 2012;132:785–96.[↩]
- Fassihi H, Sethi M, Fawcett H, Wing J, Chandler N, Mohammed S, Craythorne E, Morley AM, Lim R, Turner S, Henshaw T, Garrood I, Giunti P, Hedderly T, Abiona A, Naik H, Harrop G, McGibbon D, Jaspers NG, Botta E, Nardo T, Stefanini M, Young AR, Sarkany RP, Lehmann AR. Deep phenotyping of 89 xeroderma pigmentosum patients reveals unexpected heterogeneity dependent on the precise molecular defect. Proc Natl Acad Sci U S A. 2016;113:E1236–45.[↩][↩][↩][↩][↩][↩]
- Jaspers NG, Raams A, Silengo MC, Wijgers N, Niedernhofer LJ, Robinson AR, Giglia-Mari G, Hoogstraten D, Kleijer WJ, Hoeijmakers JH, Vermeulen W. First reported patient with human ERCC1 deficiency has cerebro-oculo-facio-skeletal syndrome with a mild defect in nucleotide excision repair and severe developmental failure. Am J Hum Genet. 2007;80:457–66.[↩]
- Kashiyama K, Nakazawa Y, Pilz DT, Guo C, Shimada M, Sasaki K, Fawcett H, Wing JF, Lewin SO, Carr L, Li TS, Yoshiura K, Utani A, Hirano A, Yamashita S, Greenblatt D, Nardo T, Stefanini M, McGibbon D, Sarkany R, Fassihi H, Takahashi Y, Nagayama Y, Mitsutake N, Lehmann AR, Ogi T. Malfunction of nuclease ERCC1-XPF results in diverse clinical manifestations and causes Cockayne syndrome, xeroderma pigmentosum, and Fanconi anemia. Am J Hum Genet. 2013;92:807–19.[↩][↩]
- Bogliolo M, Schuster B, Stoepker C, Derkunt B, Su Y, Raams A, Trujillo JP, Minguillón J, Ramírez MJ, Pujol R, Casado JA, Baños R, Rio P, Knies K, Zúñiga S, Benítez J, Bueren JA, Jaspers NG, Schärer OD, de Winter JP, Schindler D, Surrallés J. Mutations in ERCC4, encoding the DNA-repair endonuclease XPF, cause Fanconi anemia. Am J Hum Genet. 2013;92:800–6.[↩]
- Totonchy MB, Tamura D, Pantell MS et al. Auditory analysis of xeroderma pigmentosum 1971–2012: hearing function, sun sensitivity and DNA repair predict neurological degeneration. Brain 2013; 136: 194–208.[↩]
- Milota M, Jones DL, Cleaver J, Jamall IS. Xeroderma pigmentosum family support group: helping families and promoting clinical initiatives. DNA Repair (Amst) 2011; 10: 792–797.[↩]
- Tamura D, Kraemer KH, DiGiovanna JJ. Xeroderma pigmentosum. In: Lebwohl MG, Heymann WR, Berth-Jones J, Coulson I, eds. Treatment of Skin Disease: Comprehensive Therapeutic Strategies. 4 ed. London, UK: Elsevier; 2014.[↩]
- Butt FM, Moshi JR, Owibingire S, Chindia ML. Xeroderma pigmentosum: a review and case series. J Craniomaxillofac Surg. 2010;38:534–7.[↩]
- Brooks BP, Thompson AH, Bishop RJ, Clayton JA, Chan CC, Tsilou ET, Zein WM, Tamura D, Khan SG, Ueda T, Boyle J, Oh KS, Imoto K, Inui H, Moriwaki S, Emmert S, Iliff NT, Bradford P, Digiovanna JJ, Kraemer KH. Ocular manifestations of xeroderma pigmentosum: long-term follow-up highlights the role of DNA repair in protection from sun damage. Ophthalmology. 2013;120:1324–36.[↩]
- Totonchy MB, Tamura D, Pantell MS, Zalewski C, Bradford PT, Merchant SN, Nadol J, Khan SG, Schiffmann R, Pierson TM, Wiggs E, Griffith AJ, Digiovanna JJ, Kraemer KH, Brewer CC. Auditory analysis of xeroderma pigmentosum 1971-2012: hearing function, sun sensitivity and DNA repair predict neurological degeneration. Brain. 2013;136:194–208.[↩][↩][↩]
- DiGiovanna JJ, Patronas N, Katz D, Abangan D, Kraemer KH. Xeroderma pigmentosum: spinal cord astrocytoma with 9-year survival after radiation and isotretinoin therapy. J Cutan Med Surg. 1998;2:153–8.[↩]
- Arlett CF, Plowman PN, Rogers PB, Parris CN, Abbaszadeh F, Green MH, McMillan TJ, Bush C, Foray N, Lehmann AR. Clinical and cellular ionizing radiation sensitivity in a patient with xeroderma pigmentosum. Br J Radiol. 2006;79:510–7[↩]
- Kraemer KH, DiGiovanna JJ, Moshell AN, Tarone RE, Peck GL. Prevention of skin cancer in xeroderma pigmentosum with the use of oral isotretinoin. N Engl J Med. 1988;318:1633–7.[↩][↩]
- Giannotti B, Vanzi L, Difonzo EM, Pimpinelli N. The treatment of basal cell carcinomas in a patient with xeroderma pigmentosum with a combination of imiquimod 5% cream and oral acitretin. Clin Exp Dermatol. 2003;28 Suppl 1:33–5.[↩]
- Sollitto RB, Kraemer KH, DiGiovanna JJ. Normal vitamin D levels can be maintained despite rigorous photoprotection: Six years’ experience with xeroderma pigmentosum. J Am Acad Dermatol. 1997;37:942–7.[↩]
- Ali JT, Mukasa Y, Coulson IH. Xeroderma pigmentosum: early diagnostic features and an adverse consequence of photoprotection. Clin Exp Dermatol. 2009;34:442–3.[↩]
- Reichrath J. Sunlight, skin cancer and vitamin D: What are the conclusions of recent findings that protection against solar ultraviolet (UV) radiation causes 25-hydroxyvitamin D deficiency in solid organ-transplant recipients, xeroderma pigmentosum, and other risk groups? J Steroid Biochem Mol Biol. 2007;103:664–7.[↩]
- Kraemer KH, Lee MM, Andrews AD, Lambert WC. The role of sunlight and DNA repair in melanoma and nonmelanoma skin cancer. The xeroderma pigmentosum paradigm. Arch Dermatol. 1994;130:1018–21.[↩]
- Yarosh D, Klein J, O’Connor A, Hawk J, Rafal E, Wolf P. Effect of topically applied T4 endonuclease V in liposomes on skin cancer in xeroderma pigmentosum: a randomised study. Xeroderma Pigmentosum Study Group. Lancet. 2001;357:926–9.[↩]

{kind=link}