Contents
What is pemphigus
Pemphigus is a group of life-threatening blistering disorders characterized by “acantholysis” that results in blisters in mucous membranes and skin 1. Blisters tend to form when the skin is rubbed (Nikolsky sign). Acantholysis is when skin cells are no longer being held together. Pemphigus patients develop mucosal erosions and/or blisters, erosions, or small bumps that fill with pus or fluid.
The four major types of the pemphigus include:
- Pemphigus vulgaris: Mucosal and/or skin involvement with blisters on the upper skin layers. This happens when autoantibodies attack desmoglein 3 or both desmoglein 1 and desmoglein 3 (the “glue” holding the cells together)
- Pemphigus foliaceus : Skin involvement only with blisters caused when autoantibodies attack desmoglein 1
- IgA pemphigus : Grouped blisters with crusts in the epidermis when autoantibodies attack desmocollin 1
- Paraneoplastic pemphigus : Extensive oral and skin blistering associated with underlying malignancies when autoantibodies weaken cell adhesion and inflammatory cells infiltrate the skin, damaging the structure of adjacent cells
The different forms of pemphigus are distinguished by their clinical features, associated autoantigens, and laboratory findings. Benign familial pemphigus, or Hailey-Hailey, is a genetic condition caused by cell mutation. It is not autoimmune like the other forms mentioned.
Typically, pemphigus vulgaris and nonendemic pemphigus foliaceous typically occur in adults between 40-60 years old 2. Pemphigus is pretty rare in children (except for endemic pemphigus foliaceous, which affects children and young adults in endemic areas) 3. Neonatal pemphigus is a rare form of pemphigus that happens when an affected mother’s autoantibodies are transferred to the fetus.
Epidemiological information on IgA pemphigus is limited. It may occur at any age and might be more common in females 4.
Paraneoplastic pemphigus is very rare and more common in middle-aged adults, but some children have been diagnosed with it.
What causes pemphigus
Why autoantibodies attack cell adhesion is still intensively debated. Several reasons have been proposed, including events that cause cell separation and damage to adhesive molecule function 5, 6, 7. One theory suggests acantholysis results from autoantibody caused disruption of cellular signals leading to a cell’s structural collapse and shrinkage 8.
Autoantibodies that attack a variety of cell surface antigens have been identified in patients with pemphigus.
Desmogleins are the antigens most studied in pemphigus vulgaris and pemphigus foliaceous. Desmogleins are components of desmosomes, key components for cell-to-cell adhesion.
Like other autoimmune diseases, what causes the pemphigus diseases is not really understood. Researches believe genetic and environmental factors may influence the diseases 9.
Some suggest that ultraviolet radiation could lead to pemphigus foliaceous and pemphigus vulgaris activity 10. Pemphigus has even developed following burns or electrical injury 11. Others have suggested viral infections, certain food compounds, ionizing radiation, and pesticides may trigger or worsen the disease 12.
Pemphigus vulgaris
Pemphigus vulgaris is a rare autoimmune disease that is characterized by blisters and erosions on the skin and mucous membranes. “Vulgaris” means “common” so this type of pemphigus is the most common form of pemphigus. Almost all pemphigus vulgaris patients will have some mucosal involvement. The mouth is the most common location of mucosal lesions, and often is the first area the disease manifests 13. Other mucous membranes areas are also often affected (e.g., eyes, nose, esophagus, vulva, vagina, cervix, and anus) 14. In women with cervical involvement, pemphigus vulgaris may be mistaken for cervical dysplasia during Papanicolaou (Pap) smears 6.
Because mucosal blisters erode quickly, erosions are often the only clinical findings. The inner mouth (cheeks, lips, and floor of the mouth) are the most common areas for oral lesions 7.
Many patients also have skin involvement. These soft blisters occur on normal or reddened, irritated skin. The blisters pop easily, resulting in painful sores that bleed. There usually isn’t a lot of itching. While any area of the skin may be affected, the palms and soles are usually not. The Nikolsky sign (blistering via mechanical pressure at the edge of a blister or on normal skin) often can be concluded 8.
Pemphigus vulgaris is the most common subtype of pemphigus, accounting for 70% of all pemphigus cases worldwide although it is extremely rare in America occuring between 0.1 and 2.7 per 100,000 people per year. Pemphigus vulgaris is categorized as an ultra-orphan disease (meaning it is very rare), affecting approximately 10,000–30,000 people in the United States. Its frequency is influenced by geographic location and ethnicity. Studies have found certain populations (e.g., people of Jewish ancestry, particularly Ashkenazi Jews, and inhabitants of India, Southeast Europe, and the Middle East) are at a greater risk for pemphigus vulgaris.
People with pemphigus vulgaris, together with members of their family, have an increased risk of developing other autoimmune diseases. The most common of these is autoimmune thyroid disease.
Pemphigus vulgaris prognosis (outlook)
Pemphigus is a serious condition which had a 90% mortality rate before the advent of steroids. Currently the mortality rate is 5-10%. Death is mainly due to infections that may result from the immunosuppression caused by the medications given to control the disease. It is important, therefore, to tailor the regimen to minimise side effects.
Currently there is no cure for pemphigus but it can be managed successfully. The aim of treatment is to put the disease into remission so that it has minimal impact on the person’s quality of life.
Figure 1. Pemphigus vulgaris
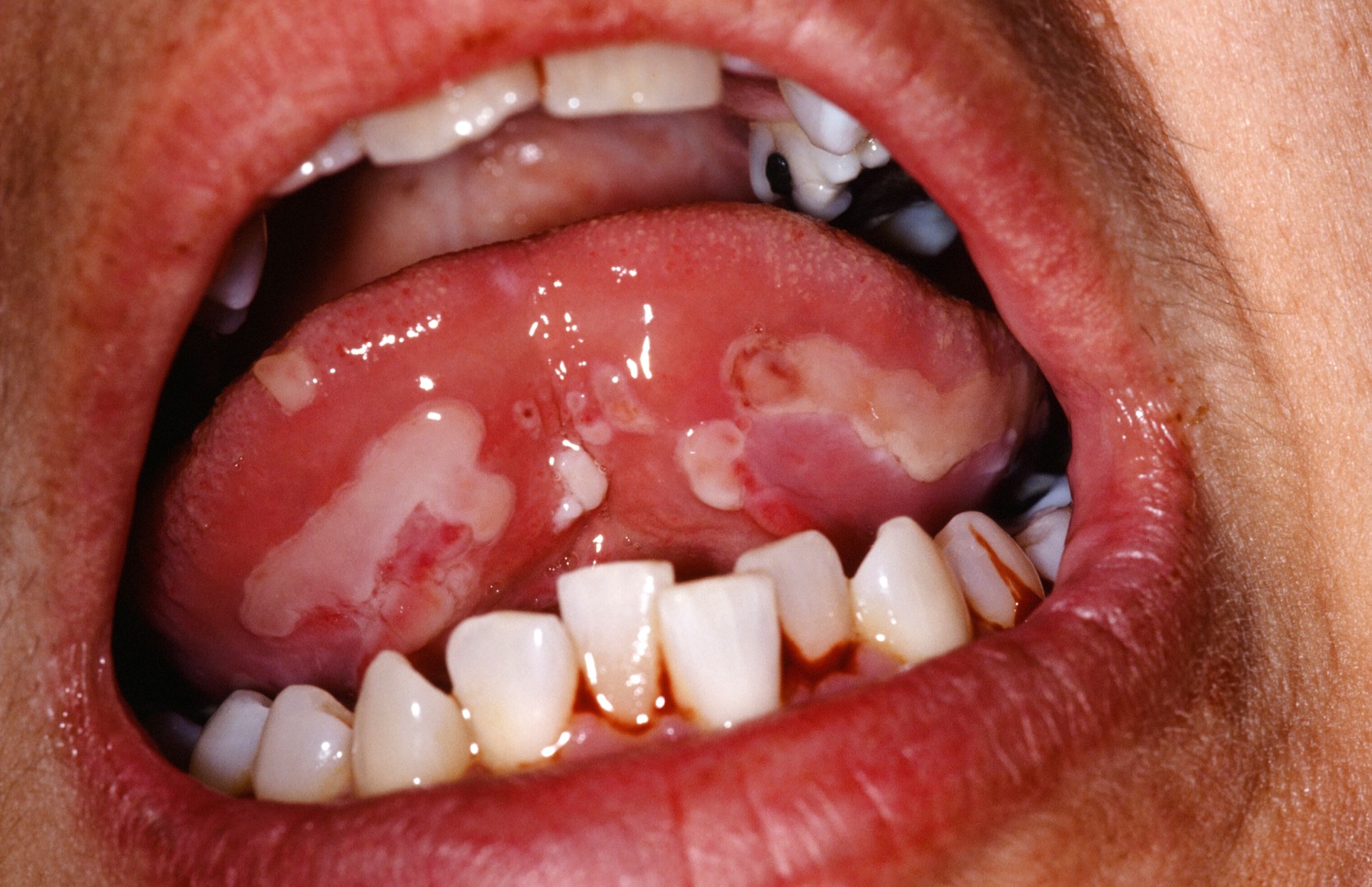
Figure 2. Pemphigus vulgaris
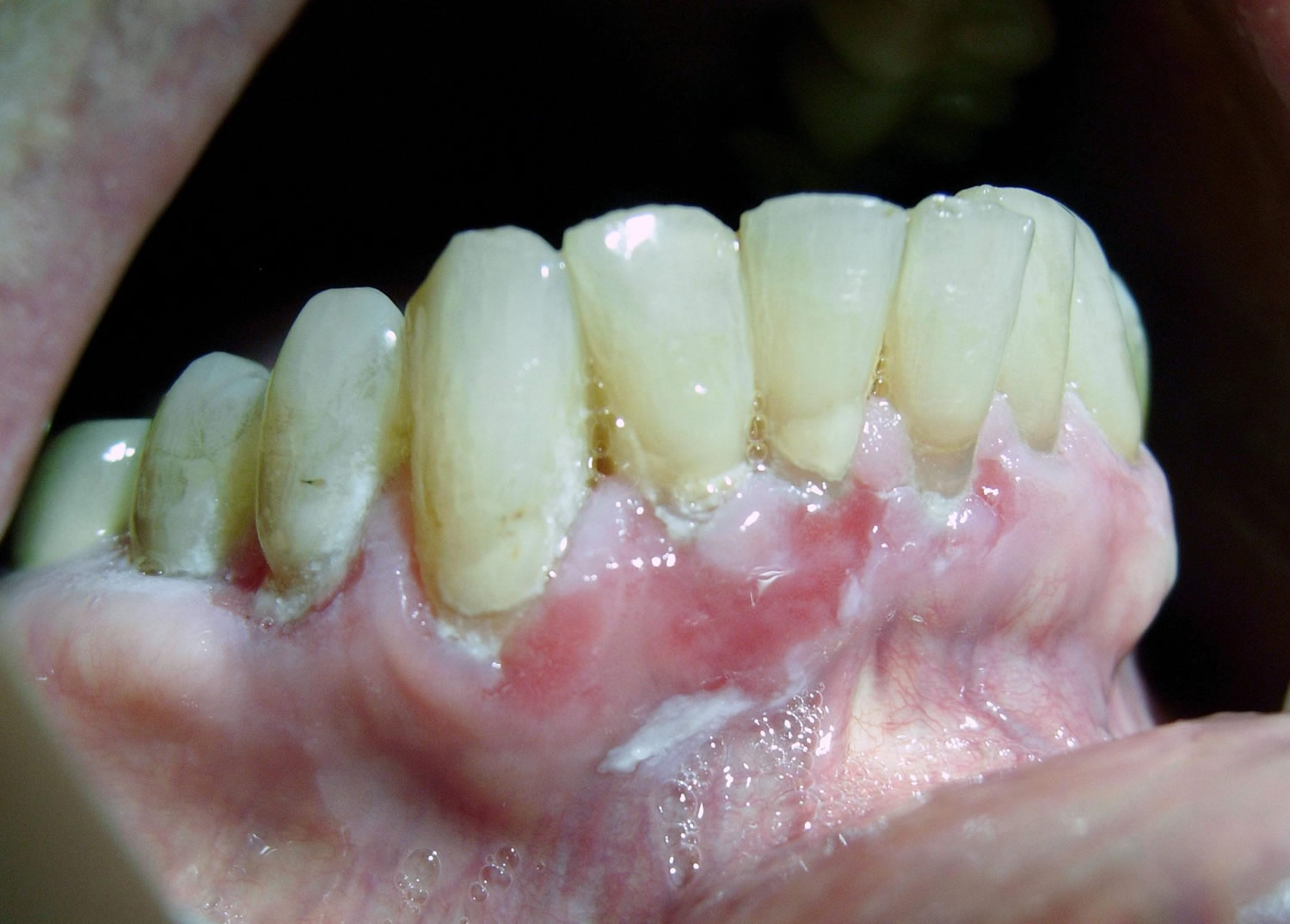
Figure 3. Pemphigus vulgaris on the skin, flaccid bullae and erosions later develop and may become extensive.
What causes pemphigus vulgaris?
Pemphigus vulgaris is an autoimmune blistering disease, which basically means the body’s immune system produce proteins (autoantibodies) that damage the adhesion points between the skin cells. These adhesion points act like press studs holding the top layer of the skin cells (epidermis) together. If they are damaged, the skin cells break apart and are unable to form a proper barrier.
The cause of the immune attack on the skin is unknown. Genetic factors are thought to play a role.
The building block cells of the epidermis are called keratinocytes. These cells are cemented together at special sticky spots called desmosomes. In pemphigus vulgaris immunoglobulin type G (IgG) autoantibodies bind to a protein called desmoglein 3, which is found in desmosomes in the keratinocytes near the bottom of the epidermis. The result is the keratinocytes separate from each other, and are replaced by fluid, the blister.
Thiol containing drugs, vaccines and radiotherapy may trigger the development of the blisters of pemphigus vulgaris.
Who gets pemphigus vulgaris?
Pemphigus vulgaris affects people of all races, age and sex. It appears most commonly between the ages of 50-60 years, but childhood cases can occur and is more common in Ashkenazi Jews, those of Mediterranean origin and Indians presumably for genetic reasons.
What are the signs and symptoms of pemphigus vulgaris?
Pemphigus vulgaris often first presents with mouth ulcers or tenderness, redness and bleeding of the gums, which make it difficult to eat or drink. This phase may progress unrecognized for many months. It may not be until blistering or erosions on the skin develop, that the diagnosis may be considered.
Mucosal areas such as inside of mouth are most commonly affected but other sites include the conjunctiva, esophagus, labia, vagina, cervix, penis, urethra and anus. If the condition has spread to the larynx, it may cause hoarseness when talking.
Blistering is superficial and often appears as erosions. Skin lesions appear as thin walled flaccid blisters filled with clear fluid that easily rupture causing painful erosions.
In rare cases, pemphigus vulgaris can present with skin-only lesions.
Oral erosions are often an initial sign of pemphigus vulgaris.
Common features of oral mucosal pemphigus include:
- 50-70% of patients get oral lesions
- blistering superficial and often appears as erosions
- widespread involvement in the mouth
- painful and slow to heal
- may spread to the larynx causing hoarseness when talking
- may make it difficult to eat or drink
Skin lesions appear as thin walled flaccid blisters filled with clear fluid that easily rupture causing painful erosions. Erosions in the skin folds may develop into vegetative lesions which are granular and crusty looking (known as pemphigus vegetans).
Pemphigus vulgaris diagnosis
Skin biopsies are usually required to confirm the diagnosis. Two kinds of biopsies will be needed – one biopsy involving the edge of a blister for routine histology in formalin and a second perilesional (normal skin) biopsy for direct immunofluorescence on frozen sections in Michel’s transport media or normal saline if the lab is on site.
A blood test may show evidence of circulating autoantibodies.
Pemphigus vulgaris treatment
The pemphigus group of diseases are rare and treatment regimens have developed over years based on smaller case studies and physician experience rather than large controlled studies.
Some people affected with pemphigus vulgaris may require admission to hospital to stabilize their condition initially.
Rest, reducing stress levels and attention to oral hygiene are important. The involvement of a dentist and hygienist is often needed. Following a soft diet is important to avoid trauma to the fragile skin of the mouth. Appropriate wound healing dressings are needed and antibiotics are given to prevent infection.
Oral prednisolone at doses around 1mg/kg/day is generally used as first line therapy to gain control of disease activity. Intermittent doses of intravenous methylprednisolone are used in some cases.
High doses of prednisolone for prolonged periods can cause side effects which need to be anticipated and managed.
Steroid sparing agents are generally introduced early in the course of the disease. These are additional medications used to reduce the dose of prednisone needed to control the disease. No single medication has been shown to be routinely superior. The choice of agent will depend on the severity of the condition and the person’s other medical problems, medications and blood test results.
Options include:
- Anti-inflammatory agents (such as minocycline/doxycycline and dapsone)
- Immunosuppressive agents(such as azathioprine, mycophenolate mofetil, cyclosporine, methotrexate and cyclophosphamide both oral and intravenous)
- Immunomodulators (such as human IVIG and plasmapheresis)
- Biologic response modifiers (such as intravenous rituximab)
Pemphigus foliaceus
Pemphigus foliaceus is a rare superficial autoimmune blistering disease which is characterized by superficial blisters, erosions and crusts on the skin and/or mucous membranes of which there are two main types: 1) Classic, 2) Endemic.
Pemphigus foliaceus is less common than pemphigus vulgaris in America but in some parts of the world it is “endemic” and very common. Pemphigus foliaceus usually first manifests in adulthood (average 50 to 60 years) but childhood cases can occur. In some places (e.g., North Africa, Turkey, and South America), pemphigus foliaceous is more common than pemphigus vulgaris 9. Some studies indicate women in Tunisia are four times more likely to have pemphigus foliaceous than men 15. However, one study identified a 19:1 ratio of males to females in an one location in Columbia 16.
People with pemphigus foliaceus, together with members of their family, have an increased risk of other autoimmune diseases. The most common of these is autoimmune thyroid disease.
The condition often first presents with an eruption on the face or scalp which looks red and scaly. This may easily be mistaken for more common problems such as eczema or fungal infections or seborrheic dermatitis, but does not respond to treatments for these conditions. It may spread to the body and become painful. The rash may last for many months unrecognized because the blisters are very difficult to see. It may not be until the affected person sees a dermatologist that the diagnosis may be suspected.
Unlike other forms of pemphigus, the mouth and other mucosal sites are not affected in pemphigus foliaceus.
Facial involvement is often the initial sign of pemphigus foliaceus.
What causes pemphigus foliaceus?
Pemphigus foliaceus occurs when cells of the body’s immune system produce proteins (autoantibodies) – primarily to the type 1 isoform of desmoglein, an important component of desmosomes (specialized domains of the plasma membrane that play a fundamental role in intercellular adhesion) – that damage the adhesion points between skin cells. These adhesion points act like press studs holding one of the top layers of the skin cells (epidermis) together. If they are damaged, the skin cells break apart and the top layer peels off. As the top layer is very thin, the blisters may not be noticed and the skin may appear to be peeling only.
The cause of the immune attack on the skin is not known. Genetic and environmental factors are thought to play a role: some medications containing sulfur groups are thought to be involved; in countries where the condition is endemic in young people, certain parasitic infections and insects are thought to be involved.
Triggers which may initiate pemphigus foliaceus include sunburn, drugs containing thiol, vaccines and radiotherapy.
- Drug-induced Pemphigus foliaceus may occur. Causative medications include fosinopril, an angiotensin-converting enzyme inhibitor (ACE inhibitor), nifedipine and penicillamine.
- Epitope spread is the idea that an autoimmune blistering disease creates skin injury, exposing other antigens to the body’s immune system, with can create new auto antigens. Thus for example, patients with pemphigus foliaceus, over time may develop pemphigus vulgarism as well.
Endemic pemphigus foliaceus: An endemic type also known as fogo selvageum occurs primarily in Brazil and mainly affects children and young adults. Exposure to hematophagous black flies possibly is related to the cause of the disease. Direct immunofluorescence shows intercellular IgG and/or C3. A new focus of endemic pemphigus foliaceus has been identified in El Bagre, Colombia 17. It has features similar to Senear-Usher syndrome.
What does pemphigus foliaceus look like?
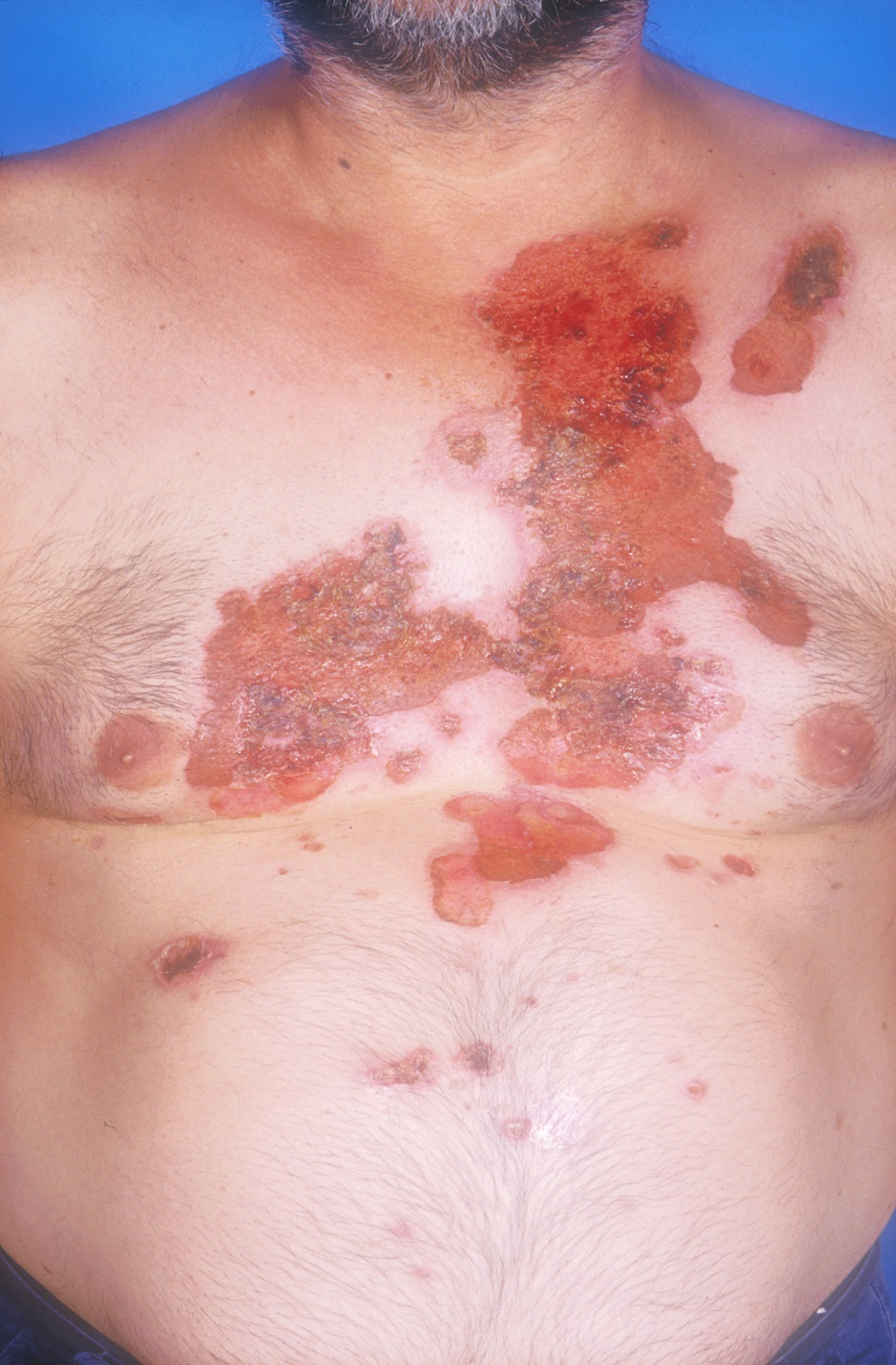
Erosions, bulla and crusted plaques affect the face, chest and elsewhere. Some patients may have their disease exacerbated by UV exposure. Annular plaques with vesicles at the periphery occur in the pemphigus herpetiformis variant.
Some cases without bulla are initially misdiagnosed as eczema, or intertrigo (when occurring in the folds).
Rarely, the patient may show prominent pustular skin lesions 18.
Pemphigus foliaceus outlook (prognosis)
Pemphigus is a serious condition, which had a 90% mortality rate before the advent of steroids. Currently the mortality rate is 5-10%, mainly due to infections that may result from the immunosuppression required to control the disease. It is important, therefore, to tailor the regimen to minimize side effects.
Currently there is no cure for pemphigus foliaceus but it can be managed successfully. The aim of treatment is to put the disease into remission so that it has minimal impact on the person’s quality of life.
Pemphigus foliaceus diagnosis
Skin biopsies are usually required to confirm the diagnosis. Two biopsies will be needed – one biopsy involving the edge of a blister for routine histology in formalin and a second perilesional (normal skin) biopsy which looks for evidence of the abnormal proteins in the skin.
Blood tests may allow identification of the abnormal proteins in the blood.
Pemphigus foliaceus treatment
The pemphigus group of diseases are rare and treatment regimens have developed over years based on smaller case studies and physician experience rather than large controlled studies.
Some people affected with pemphigus foliaceus may initially require admission to hospital to stabilize their condition.
Rest, reducing stress levels and reviewing medications are important. Any potential triggering drugs should be ceased. Appropriate wound healing dressings are needed and antibiotics are given to prevent infection.
Prednisone 20-60 mg/day is usually tried initially. A typical dosing schedule might be 1 gram is generally used as first line therapy to gain control of disease activity on days 1 and 15. To avoid secondary infection, the authors 19 recommended avoiding prolonged high-dose steroids and as well as avoiding a third immunosuppressive agent.
Steroid sparing agents are generally introduced early in the course of the disease to reduce the overall side effects of long term, high dose steroids. No single medication has been shown to be routinely superior. The choice of agent will depend on the severity of the condition and the person’s other medical problems, medications and blood test results.
Options include:
- Anti-inflammatory agents such as:
- Dapsone – in milder cases of pemphigus foliaceus and in children, dapsone alone without prednisolone may be sufficient
- Minocycline/doxycycline
- Immunosuppressive agents (such as azathioprine, mycophenolate mofetil, cyclosporine, methotrexate, cyclophosphamide – oral and intravenous)
- Immunomodulators (such as human intravenous immunoglobulin (IVIG) and plasmapheresis)
- Biologic response modifiers (such as intravenous rituximab)
Rituximab is an excellent second line agent 19. Add infection prophylaxis (co-trimoxazole and valaciclovir) when more immunosuppression than prednisone and rituximab is needed.
Azathioprine may be added to prednisone. Hydroxychloroquine can be an effective steroid-sparing agent especially in patients whose disease is exacerbated by sunlight. Prednisone and methotrexate have been used in combination.
Plasma exchange successfully controlled one case of pemphigus foliaceus over a 10 year period 20. Intravenous immunoglobulin (IVIG) was very effective in one study of 11 patients 21.
Figure 4. Pemphigus foliaceus on the skin, very superficial erosions and desquamation/scaling develop and may become extensive

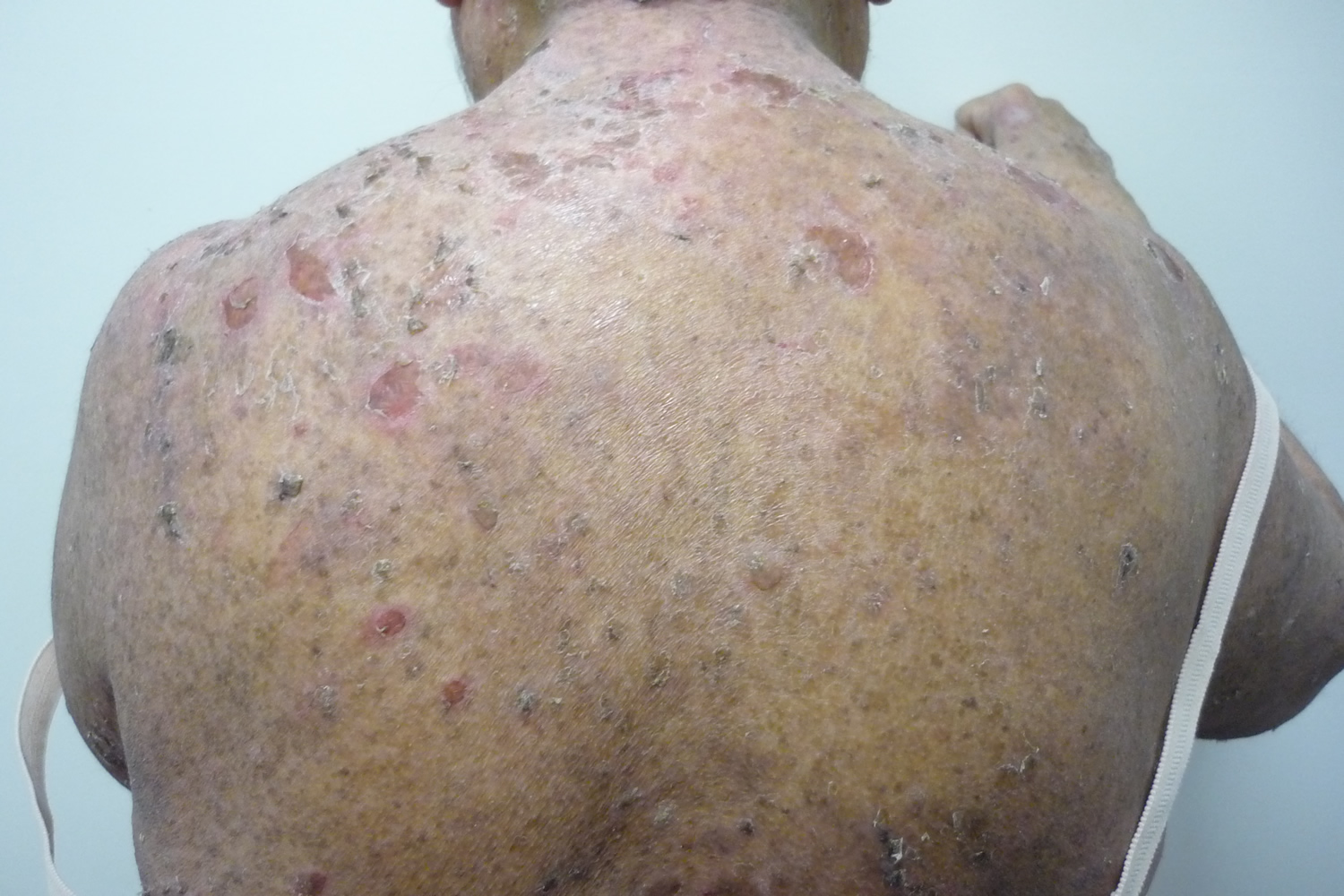
Figure 5. Pemphigus foliaceus (face)
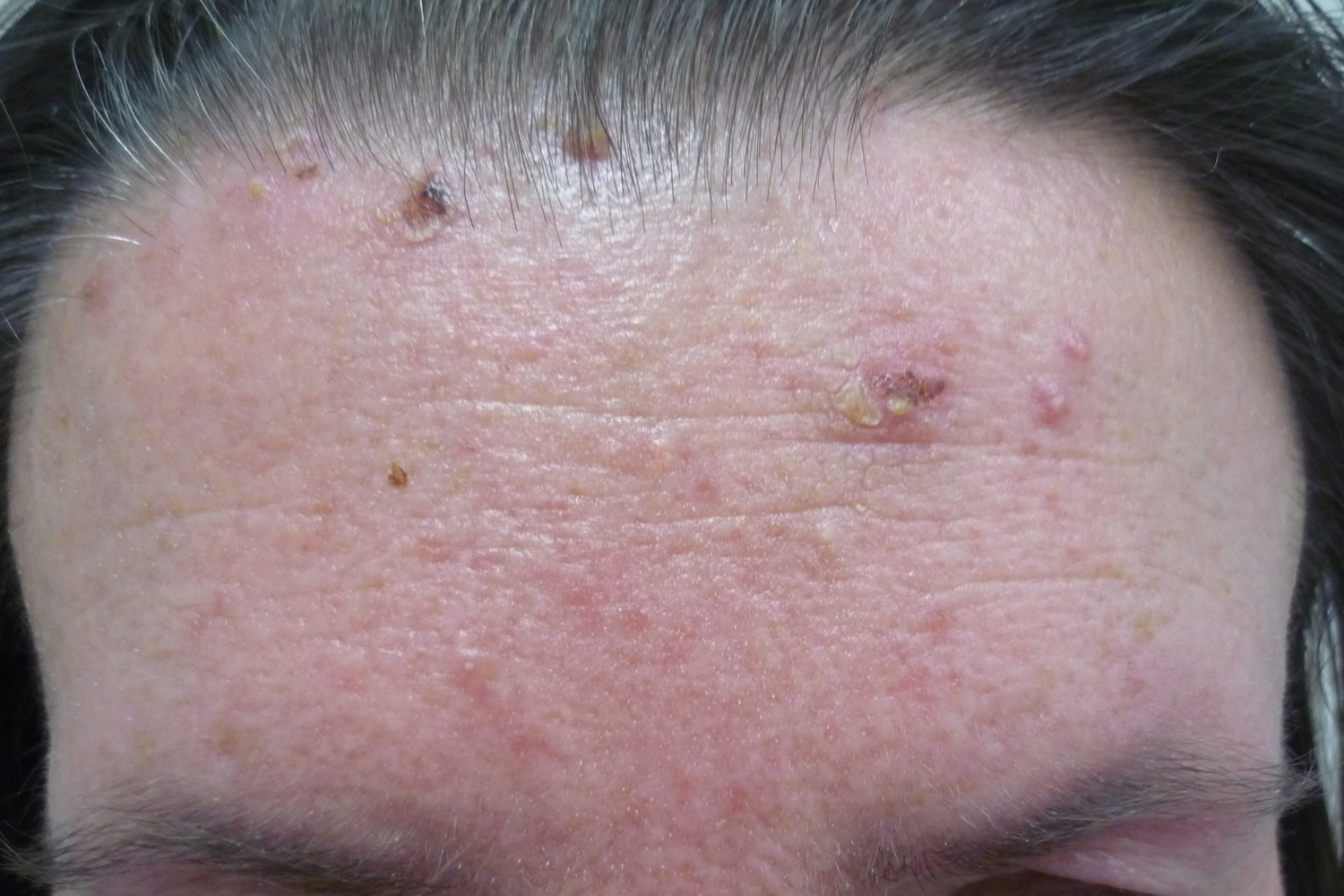
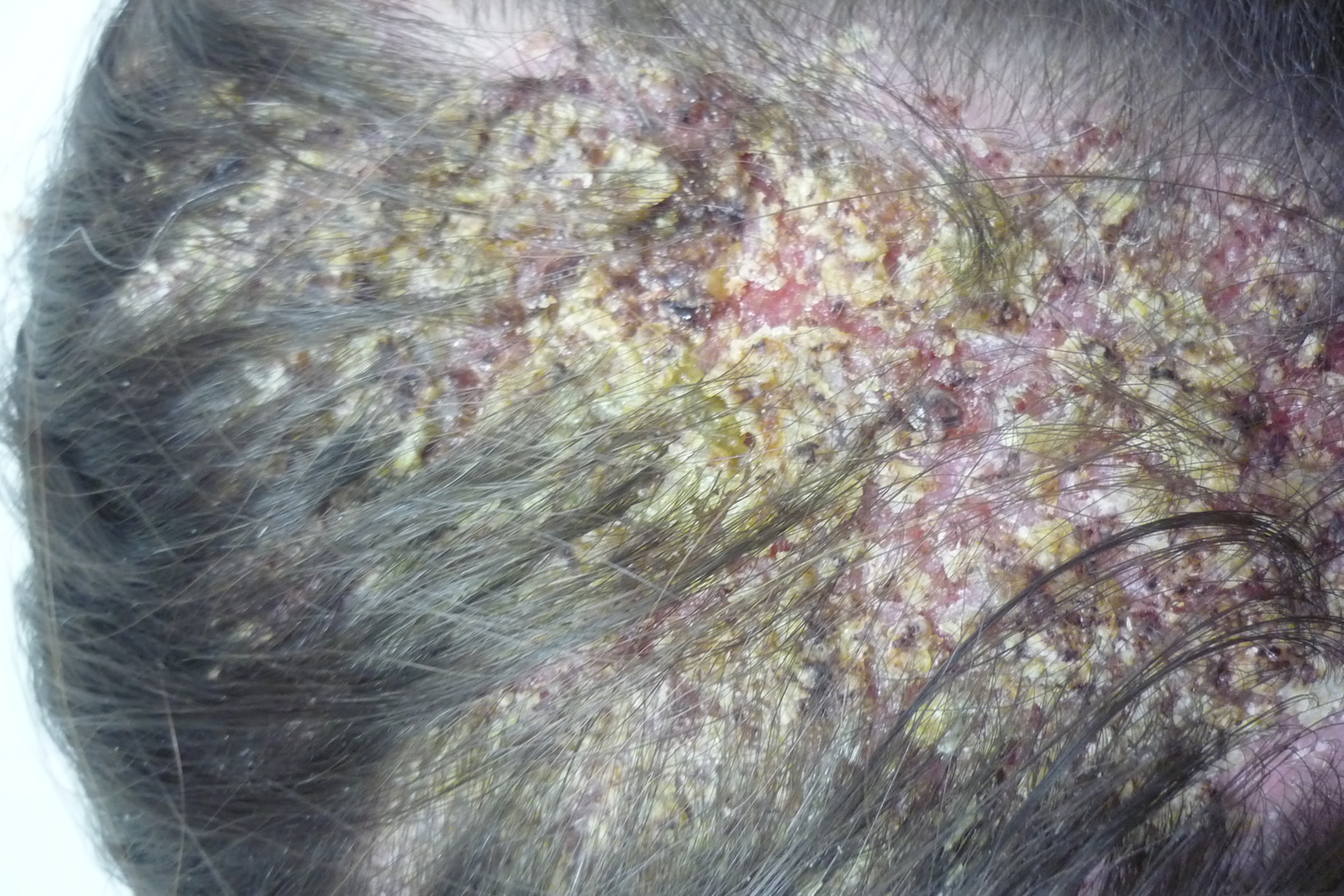
Figure 6. Pemphigus foliaceus (scalp)
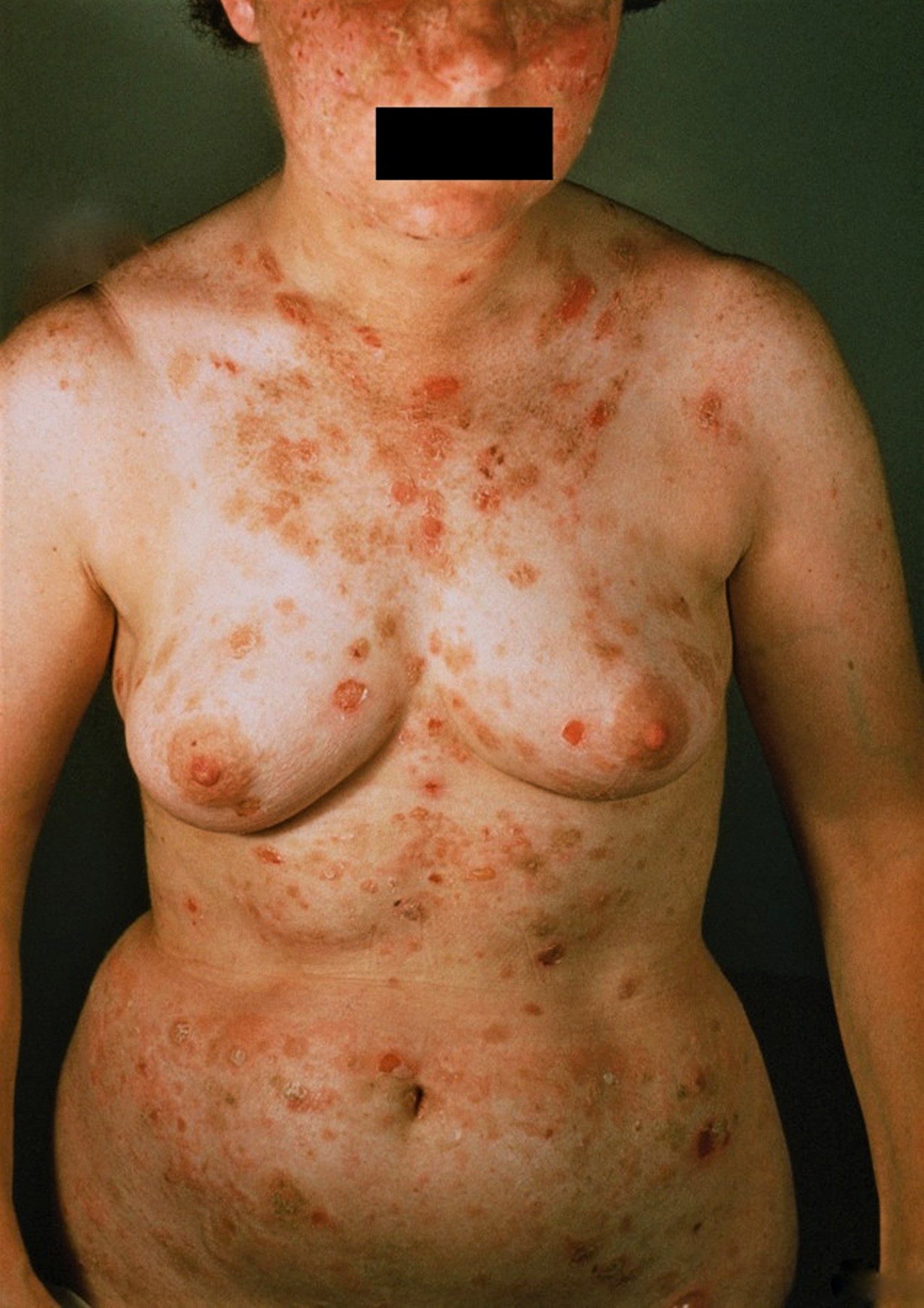
Pemphigus erythematosus
Pemphigus erythematosus is the combination of pemphigus foliaceus and lupus erythematosis. It is characterized by crusted erosions in sun-exposed areas. Mucosal involvement occurs but is uncommon. It is also known as Senear-Usher syndrome.
What does pemphigus erythematosus look like?
Red, scaly, erosive bullous or hyperkeratotic lesions occur on the nose and cheeks–an appearance and distribution similar to lupus erythematosus. Other photoexposed sites such as the upper back and V of the chest may be involved as well. Sunlight may aggravate the condition. Direct immunofluorescence shows IgG and/or C3 intercellularly as well as along the basement membrane zone. Indirect immunofluorescence is usually positive. A low-positive antinuclear antibodies (ANA) may be present in approximately 30% of patients.
A 70-year-old man developed a relapse of pemphigus erythematosus after atorvastatin intake 22.
Pemphigus erythematosus treatment
Photoprotection is key. Using sunscreen containing a physical block is best. See sunscreen for more information. A potent topical steroid may be tried. Otherwise, a systemic corticosteroid is used. Dapsone may be added if needed. Dexamethasone-cyclophosphamide pulse therapy has put patients in remission. Some have suggested the use of rituximab.
Figure 7. Pemphigus erythematosus
Paraneoplastic pemphigus
Paraneoplastic pemphigus is an autoimmune multi-organ syndrome associated with neoplastic disease 23. Typically, patients suffer from severe and critical mucosal involvement with extensive, stubborn inflammation of the mucous linings. The skin indicators vary and include blisters, erosions, and lichen-type lesions that may resemble other autoimmune blistering diseases, erythema multiforme, graft versus host disease, or lichen planus.
What causes paraneoplastic pemphigus?
The cancer is thought to trigger paraneoplastic pemphigus but the mechanism is not known. The cancer releases antibodies to particular proteins found in many tissue surfaces which result in the inflammation and blistering. There is a genetic susceptibility in some individuals.
Cancer is associated with all cases of paraneoplastic pemphigus. In two thirds of these, the cancer is present at the time of diagnosis and the remainder will be identified after diagnosis.
The most common associated cancers include:
- Hematological cancers such as:
- Non Hodgkins lymphoma (38.6%),
- Castleman tumor (18.4%),
- Chronic lymphocytic leukaemia (18.4%),
- Waldenstrom macroglobulinaemia (1.2%),
- Hodgkin’s lymphoma (0.6%)
- Mmonoclonal gammopathy (0.6%);
- Other types of cancer (8.6%) and sarcoma (7%), malignant and benign thymoma (5.5%), and rarely others.
Paraneoplastic pemphigus prognosis (outlook)
Paraneoplastic pemphigus has a very high mortality rate (75-90%) due to septicaemia, multi-organ failure, respiratory failure (including bronchiolitis obliterans and obstructive lung disease), gastrointestinal bleeding, the cancer itself or complications of the cancer therapies at the same time as immunosuppression. Typically those affected with paraneoplastic pemphigus succumb from disease 1 month to 2 years post diagnosis.
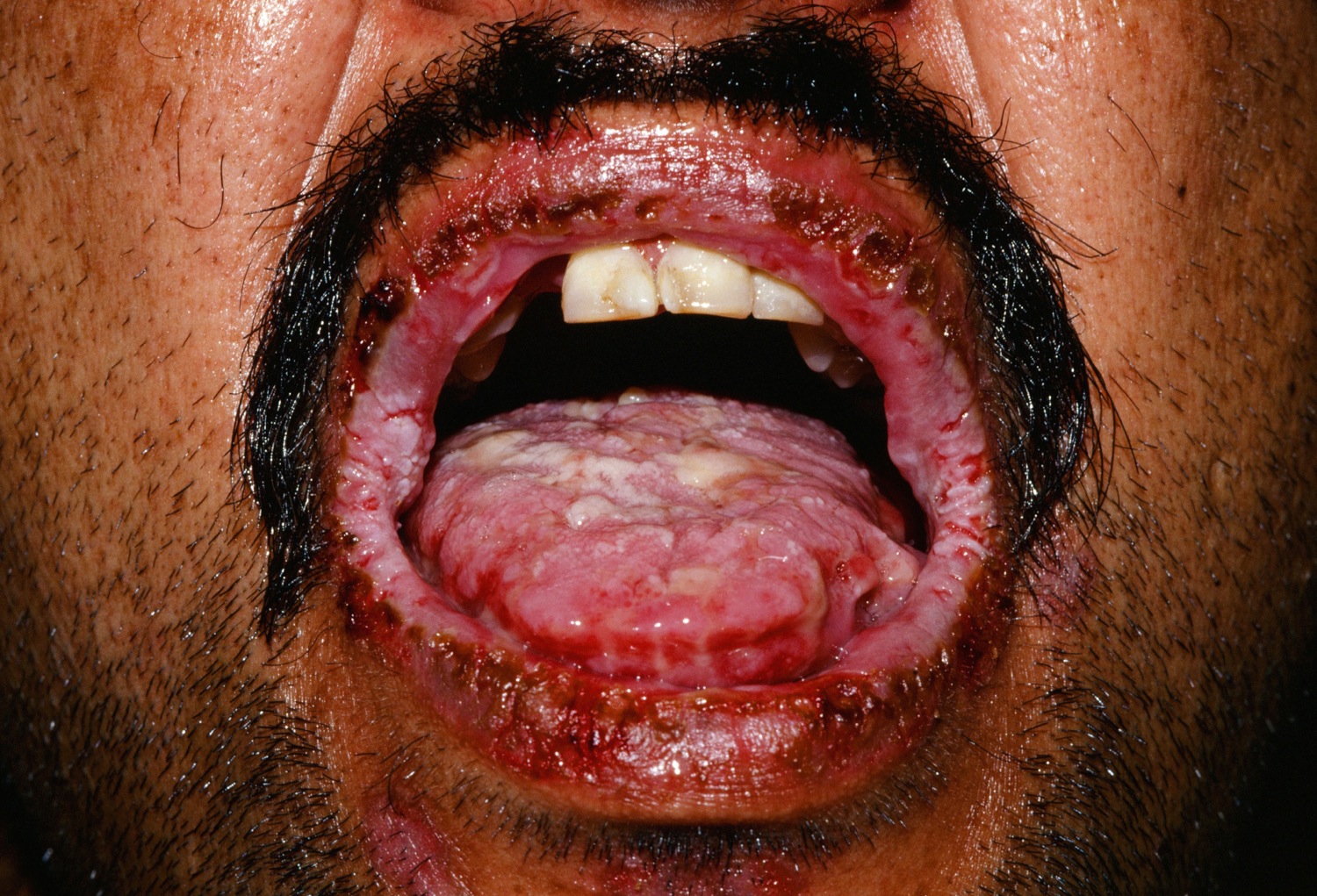
What does paraneoplastic pemphigus look like?
Intractable and painful sores on the lips and in the mouth (stomatitis) as well as other mucosal linings (e.g. eyes, nose, esophagus, genitals) are the predominant features of paraneoplastic pemphigus. The associated discomfort often results in severe exhaustion and a lack of appetite.
A skin rash develops later with many different features, possibly including blisters. It typically involves the face and hands and then spreads to the rest of the body. The rash often looks like other conditions such as pemphigus, pemphigoid, erythema multiforme, lichen planus or graft versus host disease.
Other organ systems may be affected.
Secondary infection and septicaemia are common complications.
Figure 8. Paraneoplastic pemphigus
Paraneoplastic pemphigus diagnosis
Skin biopsies are usually required to confirm the diagnosis. Routine samples are taken. These may show that the cells in the surface skin are not adhering to each other and the underlying deeper skin, in a normal manner. Tests from adjacent but not blistered skin may show immune proteins and another blood complement deposited between cells and along the basement membrane zone.
Blood tests may show evidence of special pemphigus autoantibodies (immune proteins active against one’s own tissues).
Screening for the most common associated cancers should be performed including imaging such as MRI of chest, abdomen and pelvis. Pulmonary function tests should be performed to exclude respiratory complications and regular swabs taken to exclude super infection such as herpes simplex.
Paraneoplastic pemphigus treatment
Paraneoplastic pemphigus is generally difficult to treat and requires a multi-disciplinary approach.
General measures include good oral hygiene, pain control, chlorhexidine mouthwash, lubricants for eyes and nose. Pureed and soft foods may be required as affected individuals are often unable to eat and swallow normally.
Intensive supportive nursing care is required including regular observation monitoring, barrier nursing (strict procedures to avoid transmitting infection from carers and visitors), non-stick dressings and intravenous fluids.
Specific treatment is aimed at suppressing the blistering disorder using immunosuppressive agents that may include oral steroids, intravenous immunoglobulin, azathioprine, cyclosporine, mycophenolate mofetil, cyclophosphamide, plasmapheresis and rituximab.
In addition, the underlying cancer needs management with combinations of surgery and chemotherapy.
If the underlying cancer is not apparent at the time of diagnosis, regular review is required.
Many medical, nursing and paramedical experts are required to look after those affected.
Severe infections and respiratory problems are frequent and other organ systems may also be involved further complicating the management of the condition.
Pemphigus vegetans
Pemphigus vegetans is a rare variant of pemphigus vulgaris in which vegetative lesions occur, typically in the intertriginous areas (between skin folds in armpits and groin).
Occasionally drugs may be associated, e.g. enalopril 24 and captopril 25. Involvement of the esophagus has been reported 26.
Pemphigoid vegetans which appears to be a variant of bullous pemphigoid is clinically similar. Pemphigus vegetans clinically have flacid bulla and erosions which progress to vegetating, malodorous plaques in the intertriginous areas (e.g. groin, axilla) is characteristic of pemphigus vegetans whose histology and immunoflourescence show features of pemphigus vulgaris. Occasionally other areas may be involved, e.g. scalp 27. Sulci and gyri on the dorsa of the tongue (called cerebriform tongue) have been described.
Pemphigus vegetans treatment
Prednisone and azathioprine are typically used. For example, one may use prednisone 40-60 mg/day and azathiprine 2-3 mg/kg/day with a maximum of 250 mg/day. Oral antibiotics are often given to treat any secondary infection. Cultures may direct therapy. Minocycline 100 mg/day and nicotinamide 500 three times daily cleared a case of pemphigus vegetans in one week 28. Tetracycline 500 four times daily and niacinamide 500 three times daily has been used. For the vegetative or acanthotic changes, low dose acitretin may be used 29. Spontaneous resolution may occur.
Figure 9. Pemphigoid vegetans
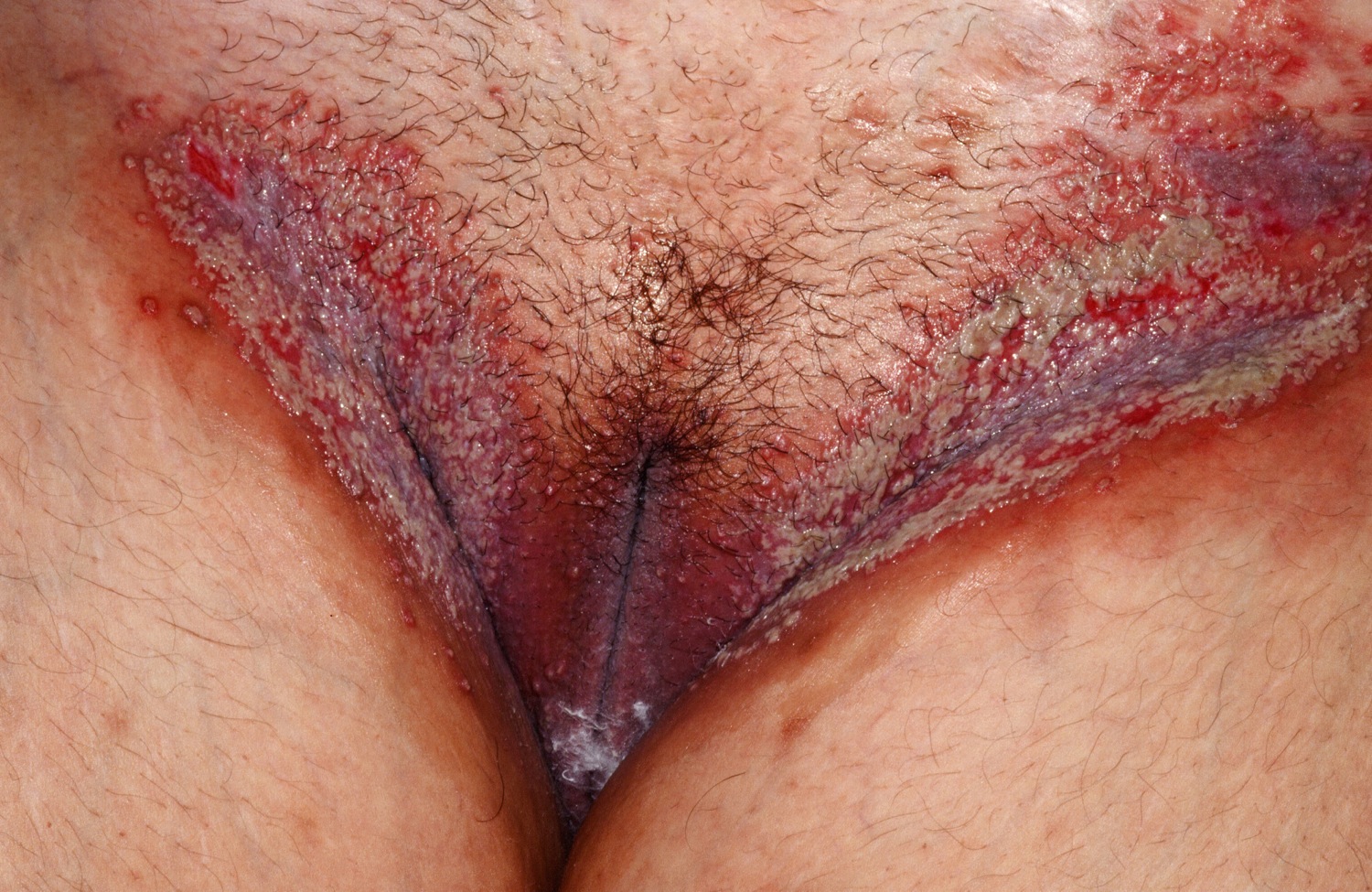
Figure 10. Pemphigoid vegetans
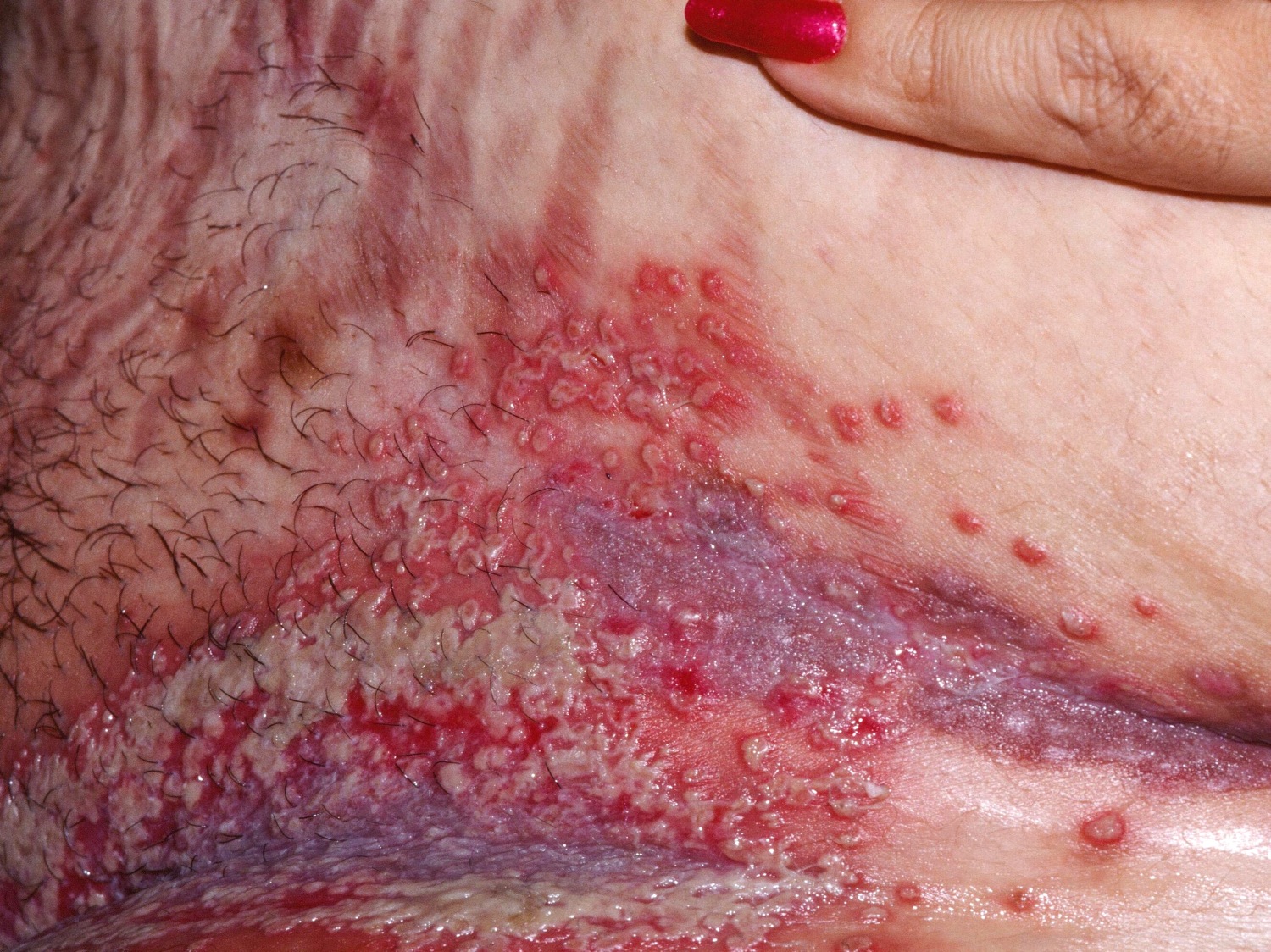
Pemphigus vulgaris vs Bullous pemphigoid
Pemphigus is characterized by autoantibodies against the connections between epidermal cells. In bullous pemphigoid, patients have autoantibodies – but they are against the basement membrane of the epidermis, not against epidermal cell junctions. This means that the bullae are actually subepidermal, so they are less fragile than those of pemphigus vulgaris (if you see a patient with bullous pemphigoid, you’ll see lots of intact, tense bullae, rather than a bunch of ruptured bullae covered with scabs). The immunofluorescence pattern is correspondingly different – you’ll see just a line at the base of the epidermis (rather than the lace-like outlining of epidermal cells you see in pemphigus vulgaris).
Patients with bullous pemphigoid are generally elderly, and the clinical presentation varies a lot (but usually it doesn’t start in the mouth, like pemphigus vulgaris). It’s a less serious disease, usually, since the bullae often don’t rupture (so there’s less chance of infection and scarring).
Bullous pemphigoid is a subepidermal autoimmune blistering disease. “Bullous” means blistering and “pemphigoid” comes from the Greek word pemphix and means bubbles. Bullous pemphigoid is the commonest type of autoimmune blistering disease, with an incidence of 12.1 to 66 new cases per million per year.
Approximately half to one third of affected individuals with bullous pemphigoid also have neurological disease such as Parkinson’s disease, dementia, stroke, multiple sclerosis and cerebrovascular disease. It is thought that neurodegenerative processes uncover BPAg1-n, an alternatively spliced form of BPAg1-e that stabilizes the cytoskeleton of sensory neurons, generating autoantibodies that may subsequently lead to bullous pemphigoid by cross-reacting with BPAg1-e. A study reported an associated occurrence of bullous pemphigoid in 21% cases of neurological disorder (8% stroke, 7% dementia, 3% Parkinsonism, 2% epilepsy and 1% multiple sclerosis) of more than a year’s duration 30.
Other possible associations include malignancy, psoriasis, primary diabetes mellitus, as well as neuroleptic and diuretic drug use.
What does bullous pemphigoid look like?
Bullous pemphigoid can occur at any age but it usually affects older individuals with onset in the late 70s. There have been rare cases reported in infants and children.
Bullous pemphigoid can look initially like dermatitis or urticaria (hives) with red itchy bumps and patches. Unlike hives, these patches do not move around or disappear. The condition may stay like this for months until blisters eventually start to form. The blisters are typically large, tense, fluid filled and very itchy. They occur most frequently on the skin of the body folds and trunk. There is typically no mucosal membrane involvement.
Figure 11. Bullous pemphigoid
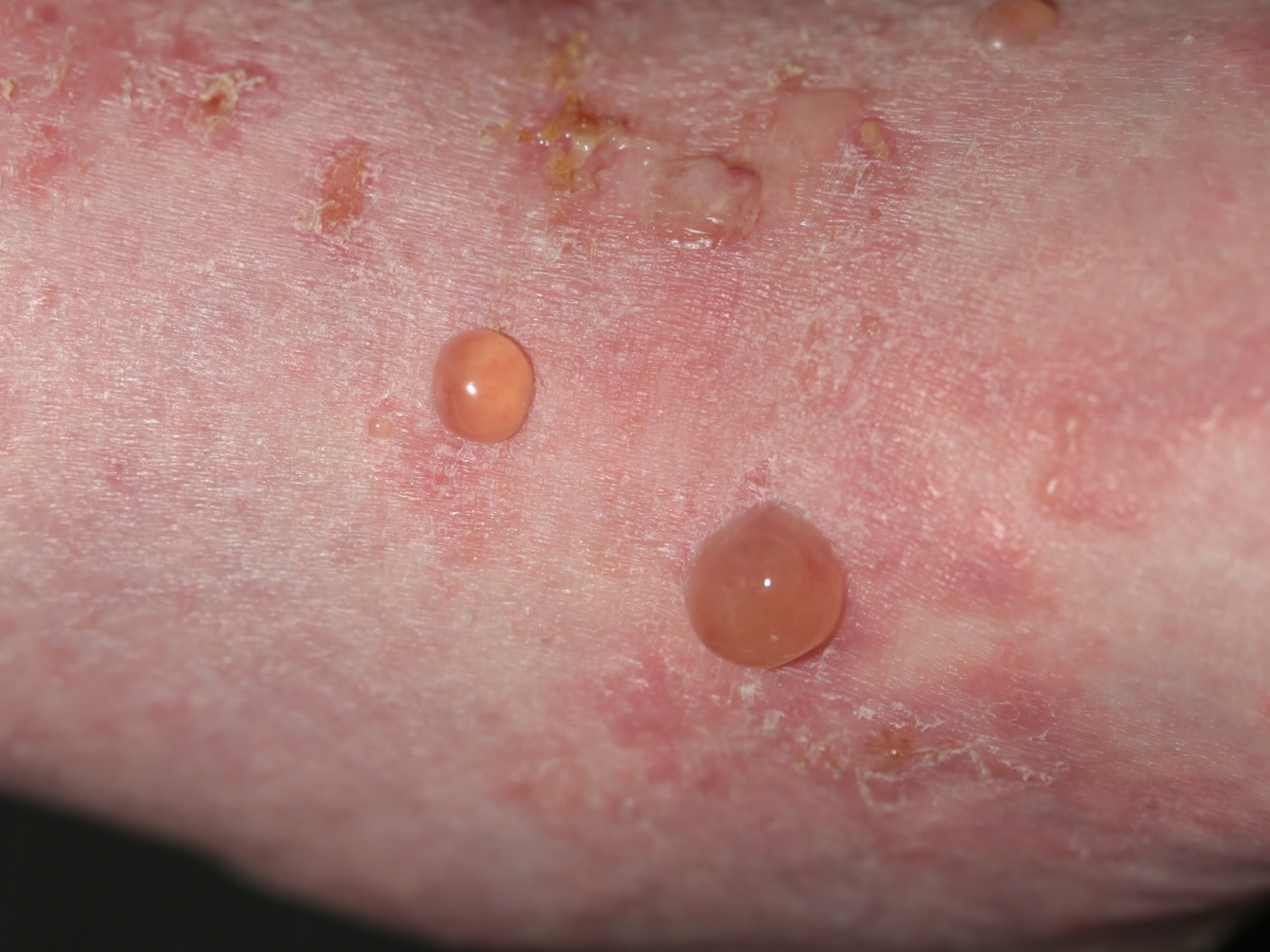
Figure 12. Bullous pemphigoid
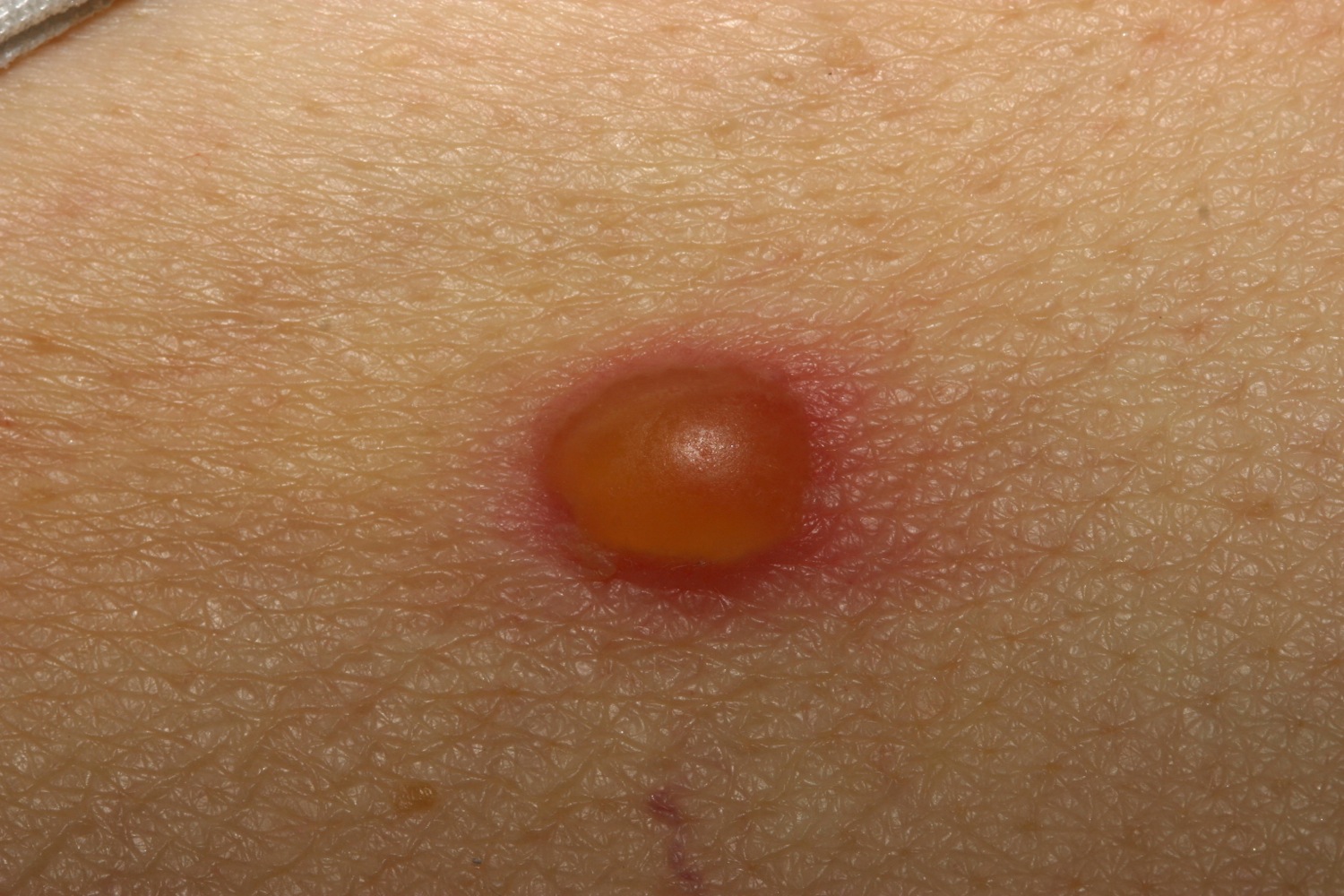
Figure 13. Bullous pemphigoid
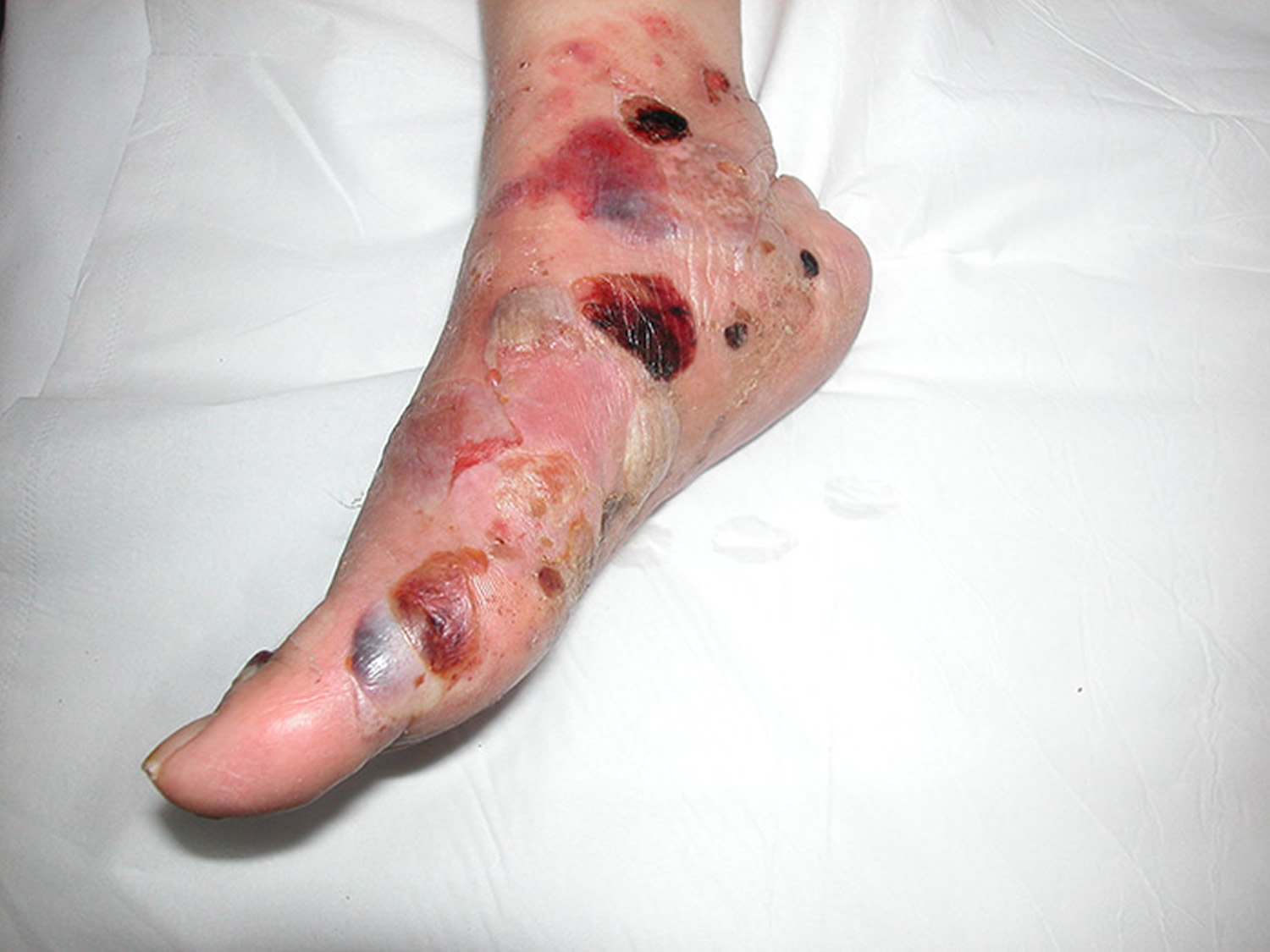
Bullous pemphigoid causes
There are two main layers of the skin, the epidermis (the very top layer) and the dermis (the deep layer). The area between these layers is known as the basement membrane zone. Bullous pemphigoid occurs when cells of the body’s immune system produce proteins (autoantibodies) which attack and damage specific proteins of the basement membrane zone (known as BP 180 and BP 230), which are critical in attaching the top layer of skin cells (the epidermis) to the underlying layer (the dermis). If the basement membrane proteins are damaged, the top and bottom layers of the skin separate, causing blister formation.
The cause of the immune attack on the skin is unknown. It may occur several years after damage to the nervous system or muscle system although how these events are related is unknown.
Both topical and oral iodine have been reported to cause bullous pemphigoid. Rarely, bullous pemphigoid may be drug-induced (e.g. everolimus, sirolimus furosemide, penicillamine, captopril, etc.). Dipeptidyl peptidase-4 inhibitors used to treat diabetes have been reported to trigger bullous pemphigoid, specifically linagliptin, sitagliptin and vildagliptin. Immune checkpoint inhibitors may cause bullous pemphigoid, e.g., nivolumab.
Bullous pemphigoid outlook (prognosis)
Unfortunately, bullous pemphigoid has a one-year mortality rate of 20-40% which is approximately two to three times higher than the aged-matched normal population. Factors associated with a worse prognosis include: advanced age; more extensive disease; higher prednisolone dosage use; and the presence of other medical conditions.
Infection is the cause of mortality in up to 60% of cases. Pneumonia is 3 times higher than the general population. Older age, poor general condition, dementia and the use of high-dose corticosteroids are risk factors 31.
How is bullous pemphigoid diagnosed?
As it often takes some time for the blisters to emerge, diagnosis may be delayed.
The following tests are usually required:
- Skin biopsies – biopsy of the edge of a blister for routine pathology; biopsy of skin close to a blister can be sent for direct immunofluorescence which looks for deposition of the autoantibodies in the skin.
- Blood tests – these test for autoantibodies in the blood.
How is bullous pemphigoid treated?
There is currently no cure for bullous pemphigoid. Treatments are personalised to the individual and directed towards reducing blister formation, relieving the pruritus (itchiness), promoting wound healing and minimising the side effects of treatment.
Individuals with mild disease
- Potent topical corticosteroid
Individuals with more aggressive disease
Oral medications are often required. These can include:
- First-line treatment
- Oral prednisolone
- Second-line treatments
- Topical immunomodulators (e.g. tacrolimus)
- Tetracyclines
- Dapsone
- Azathioprine
- Mycophenolate
- Methotrexate
- Intravenous immunoglobulin
- Rituximab
Pemphigus vs Pemphigoid
Pemphigus and pemphigoid are NOT contagious. Not by blood. Not by fluids. Not by anything. Pemphigus and pemphigoid are not genetic.
Pemphigus and pemphigoid are rare, autoimmune diseases that affect a very small percentage of the population. There are many different autoimmune diseases, and they can each affect the body in different ways. Pemphigus vulgaris, the most common of the pemphigus diseases, affects the skin and mucous membranes.
Blistering may be accompanied by severe pain, itching, burning, and stinging. If extensive, blistering can lead to life-threatening fluid loss, infection, and disfigurement. PV can also cause significant damage to the skin, including nail loss and pigmentary alteration, making timeliness of intervention and treatment essential to prevention of disability.
Pemphigus and pemphigoid are chronic illnesses that, with rare exception, do not improve without active treatment. Treatment approaches include a control phase and then a maintenance phase, with the possibility of complete remission or disease relapse (flare).
Pemphigus and pemphigoid patients are normal, everyday people and can live normal, everyday lives. It takes time, treatment, and perseverance, but it is possible.
- Mihai S, Sitaru C. Immunopathology and molecular diagnosis of autoimmune bullous diseases. J Cell Mol Med 2007; 11:462.[↩]
- Joly P, Litrowski N. Pemphigus group (vulgaris, vegetans, foliaceus, herpetiformis, brasiliensis). Clin Dermatol 2011; 29:432.[↩]
- Diaz LA, Sampaio SA, Rivitti EA, et al. Endemic pemphigus foliaceus (Fogo Selvagem): II. Current and historic epidemiologic studies. J Invest Dermatol 1989; 92:4.[↩]
- Tsuruta D, Ishii N, Hamada T, et al. IgA pemphigus. Clin Dermatol 2011; 29:437.[↩]
- Sitaru C, Zillikens D. Mechanisms of blister induction by autoantibodies. Exp Dermatol 2005; 14:861.[↩]
- Waschke J. The desmosome and pemphigus. Histochem Cell Biol 2008; 130:21.[↩][↩]
- Getsios S, Waschke J, Borradori L, et al. From cell signaling to novel therapeutic concepts: international pemphigus meeting on advances in pemphigus research and therapy. J Invest Dermatol 2010; 130:1764.[↩][↩]
- Grando SA. Pemphigus autoimmunity: hypotheses and realities. Autoimmunity 2012; 45:7.[↩][↩]
- Meyer N, Misery L. Geoepidemiologic considerations of auto-immune pemphigus. Autoimmun Rev 2010; 9:A379.[↩][↩]
- Reis VM, Toledo RP, Lopez A, et al. UVB-induced acantholysis in endemic Pemphigus foliaceus (Fogo selvagem) and Pemphigus vulgaris. J Am Acad Dermatol 2000; 42:571.[↩]
- Tan SR, McDermott MR, Castillo CJ, Sauder DN. Pemphigus vulgaris induced by electrical injury. Cutis 2006; 77:161.[↩]
- Tur E, Brenner S. Contributing exogenous factors in pemphigus. Int J Dermatol 1997; 36:888.[↩]
- Futamura S, Martins C, Rivitti EA, et al. Ultrastructural studies of acantholysis induced in vivo by passive transfer of IgG from endemic pemphigus foliaceus (Fogo Selvagem). J Invest Dermatol 1989; 93:480.[↩]
- Amagai M, Hashimoto T, Shimizu N, Nishikawa T. Absorption of pathogenic autoantibodies by the extracellular domain of pemphigus vulgaris antigen (Dsg3) produced by baculovirus. J Clin Invest 1994; 94:59.[↩]
- Brenner S, Wohl Y. A survey of sex differences in 249 pemphigus patients and possible explanations. Skinmed 2007; 6:163.[↩]
- Bastuji-Garin S, Souissi R, Blum L, et al. Comparative epidemiology of pemphigus in Tunisia and France: unusual incidence of pemphigus foliaceus in young Tunisian women. J Invest Dermatol 1995; 104:302.[↩]
- JAAD 2003;49;599 and 609[↩]
- BJD 2001;145;132[↩]
- BJD 2015;172;1420[↩][↩]
- ADV 1998;78;295[↩]
- JAAD 2002;46;42[↩]
- Drug Des Devel Ther 2014;8:1463-5[↩]
- Roscoe JT, Diaz L, Sampaio SA, et al. Brazilian pemphigus foliaceus autoantibodies are pathogenic to BALB/c mice by passive transfer. J Invest Dermatol 1985; 85:538.[↩]
- IJD 1994;33;168[↩]
- JAAD 1992;27;281[↩]
- IJD 1998;25;195[↩]
- JEADV 2016;30;368[↩]
- BJD 1995;132;668[↩]
- CED 1998;23;178[↩]
- J Invest Dermatol. 2011;131:631–6[↩]
- JAAD 2015;72;834[↩]





{kind=link}