Contents
What is chondrosarcoma
Chondrosarcoma is a bone cancer that starts in cartilage cells. To date, the cell type of chondrosarcoma origin is not clearly defined. However, it seems that mesenchymal stem and progenitor cells in the bone marrow facing a pro-proliferative as well as predominantly chondrogenic differentiation milieu, as is implicated in early stage osteoarthritis at that age, are the source of chondrosarcoma genesis 1. Chondrosarcoma is the second most common primary bone cancer accounting for about 20-25% of primary bone cancer 2. However, chondrosarcoma is rare in terms of cancer overall accounting for 0.2% of all cancers. In Europe, the incidence of chondrosarcoma is 1 to 1000,000 3.Chondrosarcoma is rare in people younger than 20. After age 20, the risk of getting a chondrosarcoma goes up until about age 75. Peak incidence of chondrosarcoma is in the 4th to 7th decades. However, the clear cell chondrosarcoma and mesenchymal chondrosarcoma variants can occur in younger patients with sex incidence being a 2:1 male predominance 4. Geographically, this chondrosarcoma is found worldwide.
Cartilage is a key tissue in the development and maintenance of most bones in the body. During fetal development, the long bones of the body develop from cartilage models. In adults, cartilage forms the articular surface of many joints and the rings of the trachea. There are three different types of cartilage classified on the basis of the extracellular matrix: hyaline, elastic and fibrocartilage.
Chondrosarcomas can start anywhere there’s cartilage. Most develop in bones like the pelvis, legs, or arms . Sometimes chondrosarcoma starts in the trachea, larynx, or chest wall. Other sites are the scapula (shoulder blade), ribs, or skull.
Benign (not cancer) tumors are more common in the cartilage than malignant ones. These are called enchondromas. Another type of benign cartilage tumor is a bony projection capped by cartilage called an osteochondroma. These benign tumors rarely turn into cancer. People who have many of these tumors have a slightly higher chance of developing cancer, but this isn’t common.
Chondrosarcomas are classified by grade, which measures how fast they grow. The grade is assigned by the pathologist (a doctor specially trained to examine and diagnose tissue samples with a microscope). The lower the grade, the slower the cancer grows. When a cancer is slow growing, the chance that it will spread is lower, so the outlook is better. Most chondrosarcomas are either low grade (grade I) or intermediate grade (grade II). High-grade (grade III) chondrosarcomas, which are the most likely to spread, are less common.
Chondrosarcoma grading allows the division of chondrosarcoma into 3 (sometimes 4) grades.
Grade 1 – low grade
- low cellularity
- mostly chondroid matrix
- little if any myxoid
- difficult to distinguish from enchondroma (see enchondroma vs. low grade chondrosarcoma for imaging distinguishing features)
- Examples:
- conventional chondrosarcoma
- juxtacortical chondrosarcoma
- Examples:
Grade 2 – intermediate grade
- increased cellularity
- little chondroid matrix
- necrosis and more common prominent myxoid
- Examples:
- conventional chondrosarcoma
- juxtacortical chondrosarcoma
- myxoid chondrosarcoma
- Examples:
Grade 3 – high grade
- highly cellular
- nuclear pleomorphism
- absent chondroid matrix
- stroma present is myxoid
- Examples:
- conventional chondrosarcoma
- juxtacortical chondrosarcoma
- mesenchymal chondrosarcoma
- dedifferentiated chondrosarcoma
- Examples:
Some chondrosarcomas have distinctive features which can be seen with a microscope. These sub-types of chondrosarcoma often have a different prognosis (outlook):
- Dedifferentiated chondrosarcomas start out as typical chondrosarcomas but then some parts of the tumor change into cells like those of a high-grade sarcoma (such as high grade forms of malignant fibrous histiocytoma, osteosarcoma, or fibrosarcoma). This type of chondrosarcoma tends to develop in older patients and grows faster than usual chondrosarcomas.
- Clear cell chondrosarcomas are rare and grow slowly. They seldom spread to other parts of the body unless they have already come back several times in the original location.
- Mesenchymal chondrosarcomas can grow rapidly, but are sensitive to treatment with radiation and chemotherapy.
Chondrosarcoma spreads by direct extension into adjacent tissues with characteristic scalloping on x-rays due to the nodular growth patterns of the lesions. When metastases occur, they tend to be to the lungs and other bones. Other sites can include regional lymph nodes (more common in mesenchymal chondrosarcoma), skin and soft tissues.
Chondrosarcoma is largely considered to be resistant to conventional chemotherapy and radiotherapy 5. As such, surgical resection has been the cornerstone of treatment for over 50 years 5. However, in recent years several novel therapeutic approaches have been evaluated in experimental studies 6.
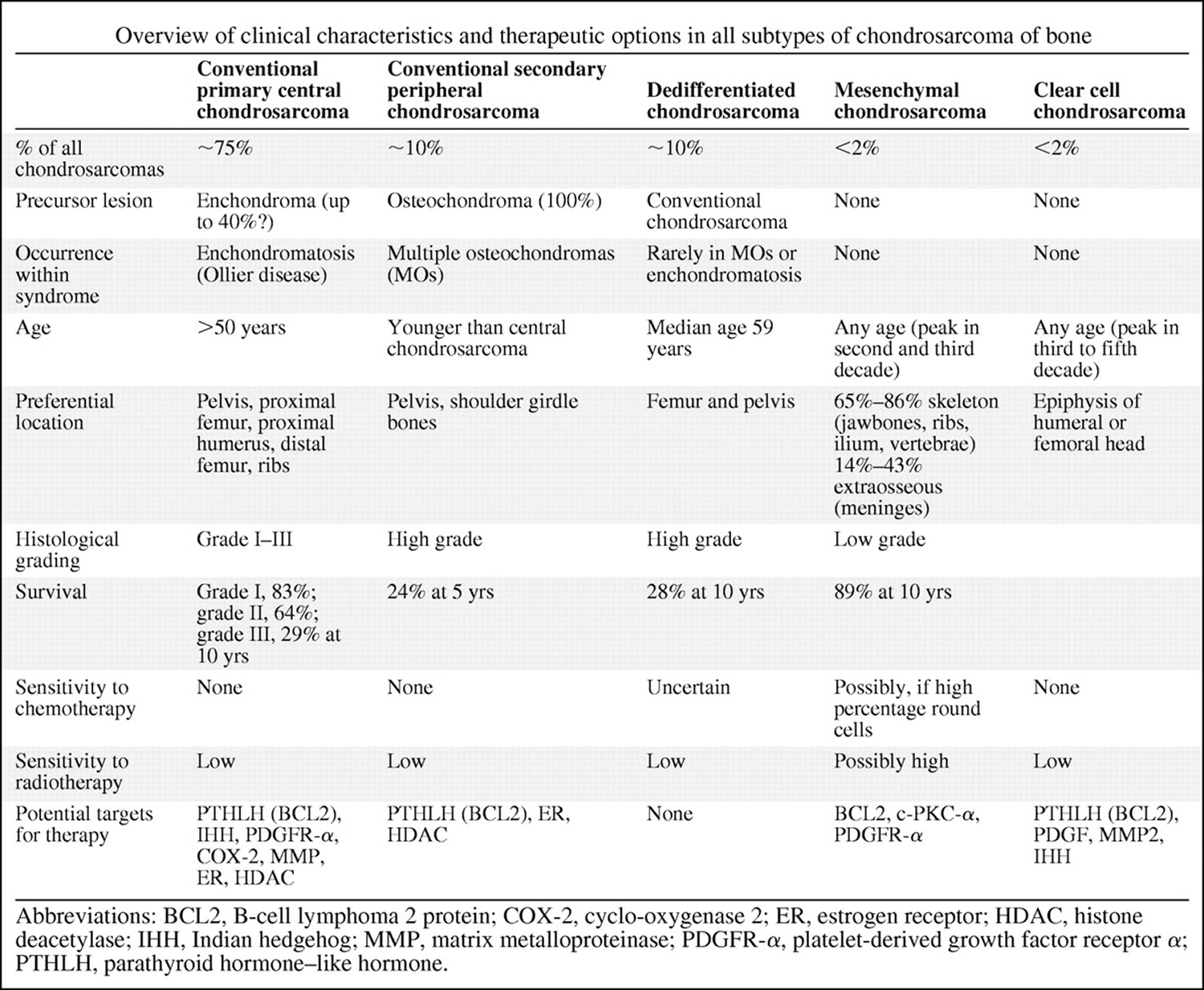
Figure 1. Overview of chondrosarcoma sub-types and their therapeutic options
Conventional Chondrosarcoma
Conventional chondrosarcoma of bone constitutes approximately 80-90% of all chondrosarcomas 8. Conventional chondrosarcomas can be categorized according to their location in bone into primary central and secondary peripheral chondrosarcomas. The vast majority (>85%) are primary central chondrosarcomas, designated as such based on their location centrally within the medullar cavity. A minority (up to 15%) of conventional chondrosarcomas develop from the surface of bone, most of them as a result of malignant transformation within the cartilage cap of a pre-existing osteochondroma. These are therefore called secondary peripheral chondrosarcomas 9. A minority (<1%) occur at the surface of bone, possibly of periosteal origin, and are designated periosteal chondrosarcoma. Conventional chondrosarcomas start to grow intramedullary (central) and often affect the pelvis, femur and humerus, but also ribs and ilium 10. Their histology is similar to that of conventional chondrosarcoma. Juxtacortical chondrosarcoma should be distinguished from secondary peripheral chondrosarcoma because juxtacortical chondrosarcomas have a better prognosis, which is excellent after adequate local surgery alone 11.
Central and peripheral chondrosarcomas are histologically similar, and for both, three different grades are discerned, which is at present the best predictor of clinical behavior 12. Grade I chondrosarcomas are lowly cellular, with an abundant hyaline cartilage matrix, and rarely metastasize 13. In contrast, grade III chondrosarcomas are highly cellular, with a mucomyxoid matrix and mitoses, with metastases developing in 70% of patients 12. Up to 13% of recurrent chondrosarcomas exhibit a higher grade of malignancy than the original neoplasm 13, suggesting chondrosarcomas may biologically progress. Moreover, recurrence of low-grade chondrosarcoma bears the risk for tumor progression toward a higher grade or even dedifferentiation, with a severe adverse prognosis.
Dedifferentiated chondrosarcoma
Dedifferentiated chondrosarcoma is a high-grade noncartilaginous sarcoma next to a (usually low-grade) malignant cartilage-forming tumor, with a remarkably sharp junction between the two components 14. The highly aggressive dedifferentiated chondrosarcomas, which make up about 10% of all chondrosarcomas 15 most often arise in the long bones, namely the femur and humerus or the pelvis 16. Dedifferentiated chondrosarcomas consist of chondroid and non-chondroid parts often resembling fibroblastic or osteoblastic tissue indicating two types of mesenchymal differentiation in one tumor 17. Notably, the non-cartilaginous part of these tumors seems to determine local growth and recurrence 18. The tumor bears a dismal prognosis 19. The rare genetic reports on dedifferentiated chondrosarcoma demonstrate that both components share identical genetic aberrations 20, with additional genetic changes in the anaplastic component 21, indicating a common precursor cell with early diversion of the two components. No targets for therapy have been reported for dedifferentiated chondrosarcoma.
Mesenchymal chondrosarcoma
Mesenchymal chondrosarcoma is a highly malignant lesion that can occur in bone and soft tissue of relatively young patients and is characterized by varying amounts of differentiated cartilage admixed with undifferentiated small round cells 22. A chromosomal abnormality der(13;21)(q10;q10) was found in two cases 23. Although 61% of the tumors demonstrate p53 overexpression, no mutations were found 24. In three cases, expression of the antiapoptotic BCL2, protein kinase C (PKC)-α, and platelet-derived growth factor receptor (PDGFR)-α pathways was found, suggesting targets for treatment, including interferon-α and ciprofloxacin inhibiting PKC-α activity or imatinib inhibiting PDGFR-α signaling 25.
Clear cell chondrosarcoma
Clear cell chondrosarcoma is a low-grade malignant tumor characterized by tumor cells with clear, empty cytoplasms. Clear cell chondrosarcomas often involve the epiphysis of the proximal femur and humerus and extend to the articular cartilage of the acetabulofemoral joint and glenohumeral joint 4. Metastases are rare, but may occur up to 24 years after initial diagnosis; thus, long term follow-up is mandatory 26. A cytogenetic study on four cases suggested that extra copies of chromosome 20 and loss or rearrangements of 9p may be recurrent 27. Expression of PTHLH, PDGF 28, IHH, Runt related transcription factor 2 29, and matrix metalloproteinase 2 30 was demonstrated, suggesting potential targets for future therapy.
Myxoid chondrosarcoma
Myxoid chondrosarcomas are rare, intermediate grade subtype of chondrosarcoma (see chondrosarcoma grading above). Myxoid chondrosarcoma is found in both bone and soft tissues (extraskeletal myxoid chondrosarcoma). Myxoid chondrosarcoma typically affects patients in their 30s to 60s with a male predilection 31. They may account for as many as 12% of chondrosarcomas of bone 32. It is generally debated whether true myxoid chondrosarcoma of bone exists. Although myxoid chondrosarcoma of bone has similar light microscopic features as extraskeletal myxoid “chondrosarcoma”, extraskeletal myxoid chondrosarcoma is unrelated to skeletal high-grade (myxoid) chondrosarcoma of bone 33. The original classification of extraskeletal myxoid chondrosarcoma as chondrosarcoma was based on positivity for S100, resemblance to chondroblasts on electron microscopy, and focal cartilage formation. Well-formed hyaline cartilage is, however, found in a minority of extraskeletal myxoid chondrosarcomas 34. In fact, expression of collagen II and aggrecan, two markers of cartilaginous differentiation, was absent in 86% of extraskeletal myxoid chondrosarcomas, while S100 expression was very focal or absent 34. Therefore, the term “chondrosarcoma” in extraskeletal myxoid chondrosarcoma is a misnomer and confusing. The 2002 World Health Organization classification classified this entity in the “tumors of uncertain differentiation” category. The translocation (9;22), which is specific for extraskeletal myxoid chondrosarcoma, is generally absent in myxoid chondrosarcoma of bone and its ultrastructure is different 35. The few reported cases of bone with proven t(9;22) have a large soft tissue component, which makes the distinction from extraskeletal myxoid chondrosarcoma with bone destruction extremely difficult 36. Thus, they represent two different entities, and myxoid chondrosarcoma of bone should be regarded as a myxoid variant of intermediate- or high-grade conventional chondrosarcoma.
Extraskeletal chondrosarcoma
Extraskeletal chondrosarcomas make up only 2% of soft-tissue sarcomas and only 1% of all chondrosarcomas. They tend to be of higher grade than run-of-the-mill conventional intramedullary chondrosarcomas, with the majority being of the myxoid (most common) or mesenchymal varieties 37.
Extra skeletal myxoid chondrosarcomas typically occur in the extremities, with the thigh being most common. They occur at all ages but typically around the age of 50 37.
Extra skeletal mesenchymal chondrosarcomas on the other hand tend to occur in young adults. They are seen also in the thigh, but also have a predilection for the head and neck, occurring in the meninges, the orbit and even in the brain.
Extra skeletal chondrosarcomas have been reported in most other parts of the body, although with less frequency, including:
- meninges of the brain and spinal cord (most common)
- lower limb (particularly thigh)
- soft tissues of the head and neck
- orbit
- larynx
- sinonasal cavity 38
- solid organs
- pancreas 39
Chondrosarcoma prognosis
There have been few long-term studies on the outcome of chondrosarcoma and the findings regarding prognostic factors are controversial. Chondrosarcoma tumor grade and localization were found to be statistically significant independent predictors of chondrosarcoma-related deaths. The majority of conventional chondrosarcomas tend to be of low grade and are thus associated with an improved prognosis. Low grade chondrosarcomas rarely metastasize. In the 2013 World Health Organization (WHO) classification system, grade I chondrosarcomas have been renamed as atypical cartilaginous tumors describing their clinical behavior as an intermediate type of tumor 40. In contrast, high grade chondrosarcomas, which make up 5–10% of all conventional chondrosarcomas, are very aggressive and frequently metastasize to the lung 41. Ten year survival rates are 83% for grade I, 64% for grade II and 30–40% for grade III 42. Yet, also about 20% of low grade tumors locally recur 42. Tumor size also plays an important role in chondrosarcoma prognosis with smaller lesions behaving less aggressively than larger chondrosarcoma tumors (irrespective of grade). Therefore, early chondrosarcoma diagnosis and chondrosarcoma treatment are associated with an improved prognosis. Unfortunately, chondrosarcomas often present as gross, unresectable masses.
In a recent review 43 involving 115 patients with primary central chondrosarcoma of bone who presented with localized disease and who had a minimum follow-up of 5 years after diagnosis. 68 tumors were localized in the extremities and 47 in the axial skeleton or pelvis. 59 patients had a high-grade (II and III) and 56 a low-grade (I) tumor. 94 patients underwent surgical resection with adequate (wide or radical) margins, while 21 patients had inadequate (marginal or intralesional) margins. At the time of the study (between 1982 and 2004), 73 patients were alive with no evidence of disease. 32 patients had died from disease, 6 patients from treatment-related complications and 4 patients from other causes. Overall survival rates of the entire group at 5 and 10 years were 72% and 69%, respectively 43. Event-free survival at 5 and 10 years amounted to 57% and 53%, respectively 43. Both overall and event-free survival were statistically significantly better for patients with chondrosarcomas of the extremities (E group) than for those with tumors of the axial skeleton and pelvic girdle (AP group) (Figure 2).
Figure 2. Chondrosarcoma survival rate according to the tumor location
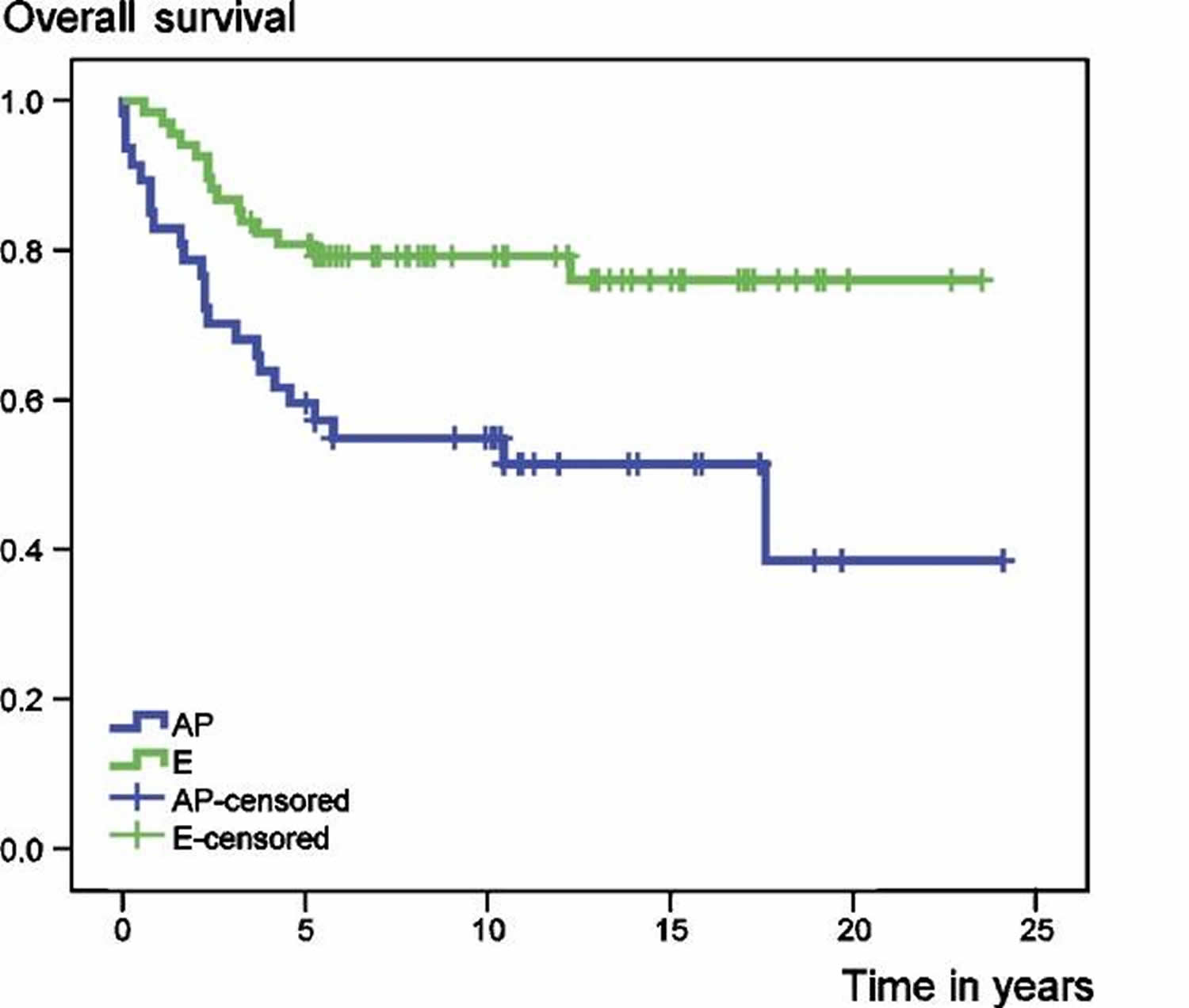
Figure 3. Chondrosarcoma survival rate according to surgical margin
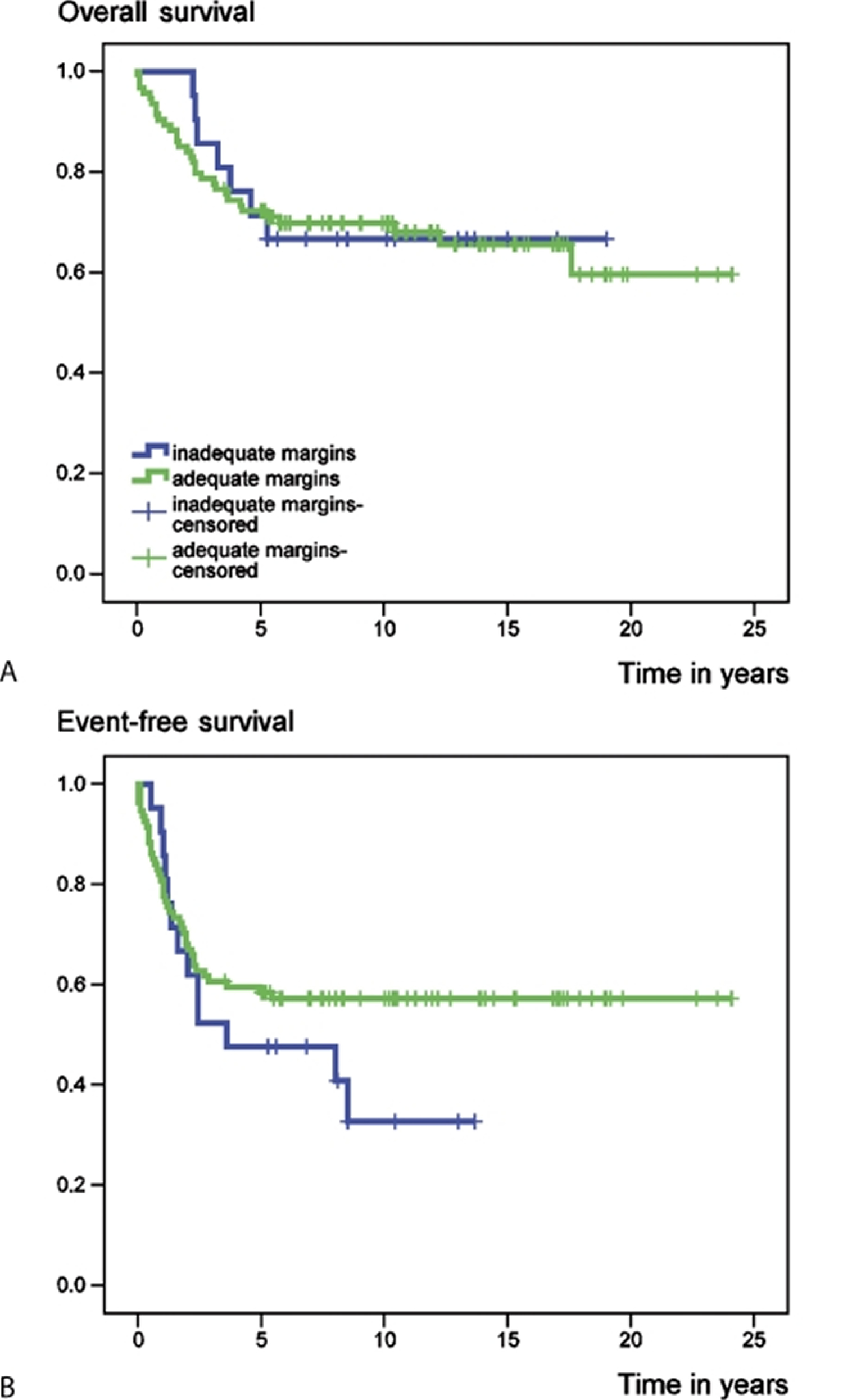
Figure 4. Chondrosarcoma overall survival rate and survival rate according to the American Joint Committee on Cancer (AJCC) staging system
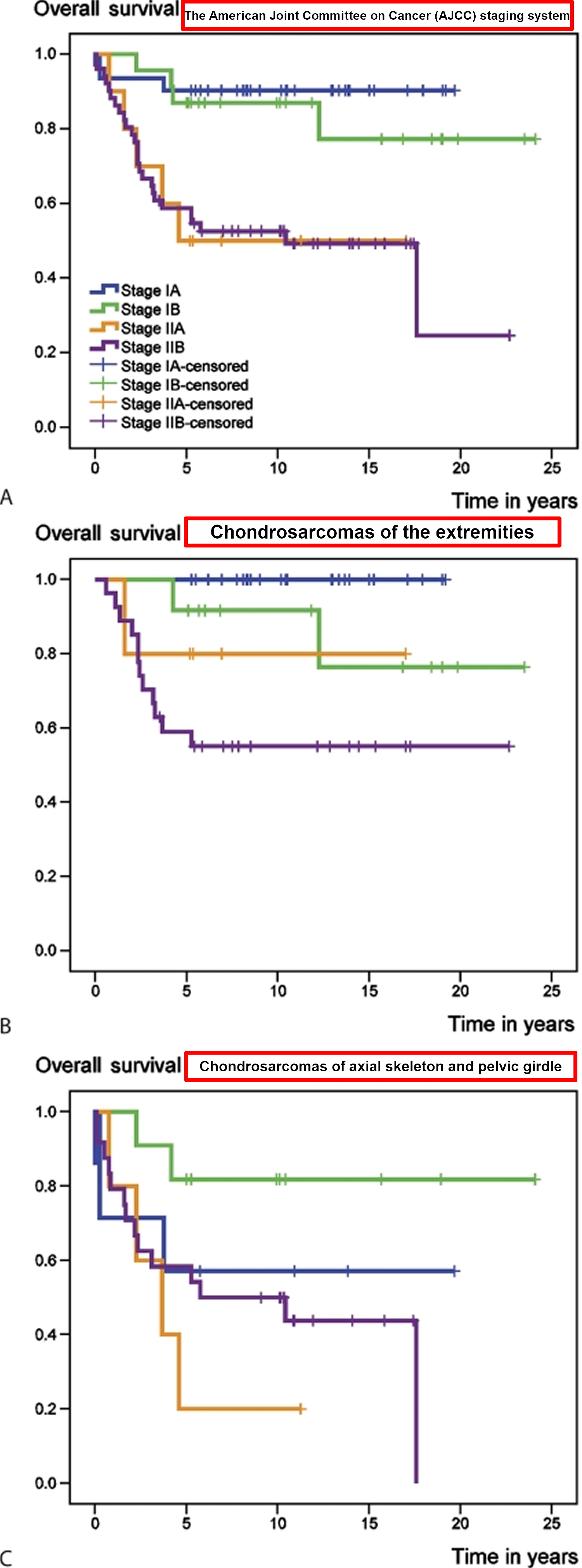
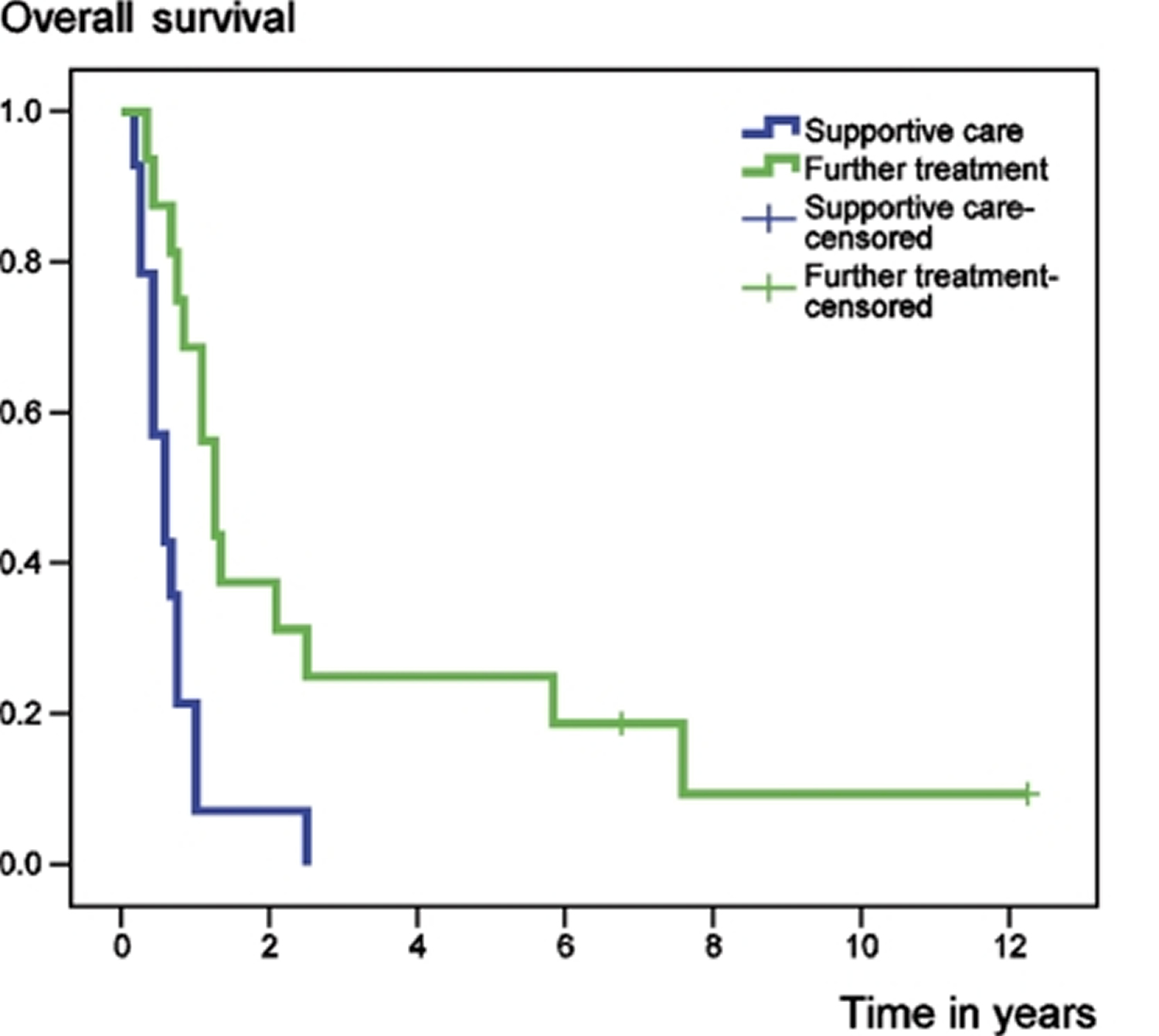
Figure 5. Chondrosarcoma survival rate following the development of metastatic disease, according to further treatment
Chondrosarcoma causes
The exact cause of chondrosarcoma is unknown. However there are some rare conditions that have a small risk of developing chondrosarcoma. Some varieties of chondrosarcoma arise from the transformation of benign cartilage lesions into cancers.
Risk Factors for chondrosarcoma
Increasing age, being of the male sex and the presence of a pre-existing enchondroma (benign tumors of hyaline cartilage occurring within the medullary cavity) – although only 10% of chondrosarcomas arise in these pre-existing lesions. Ollier disease and Mafucci syndrome are conditions marked by an increased number of benign cartilage lesions (enchondromas) in the body. Enchondroma sometimes transform into chondrosarcoma.
Chondrosarcoma symptoms
Pain is the most common symptom of chondrosarcoma. This depends on where the cancer is located. There may be some swelling over the site, or it may be sore to touch. Sometimes because the bone is weakened from the cancer, it can break easily. This is sometimes how people find out about their cancer.
If the tumor is pressing on the spinal cord, you might experience weakness, numbness or incontinence.
Chondrosarcoma diagnosis
Because most chondrosarcoma tumors grow so slowly, they may not be diagnosed for years. In some cases, tumors are discovered during imaging tests for unrelated problems. A biopsy may be needed to confirm the diagnosis.
Imaging tests
An X-ray may identify a suspicious area of bone for further examination. Other imaging tests, such as magnetic resonance imaging (MRI) and computerized tomography (CT), can offer additional information about the tumor.
Biopsy
Doctors can confirm a diagnosis of chondrosarcoma by removing a sample of suspicious tissue with a needle or a scalpel and testing it in a lab. A biopsy must be performed in a certain way so that it doesn’t make it more difficult to remove the cancer during a later operation.
Chondrosarcoma grading and staging
Chondrosarcoma Grading:
Once the cancer is diagnosed, it will be graded. This means that cells will be looked at under a microscope and the rate in which they grow will be determined.
Cells that look mostly normal (and are slow-growing) are given a lower grade than cells that look abnormal (and are fast-growing).
Chondrosarcomas are graded from 1-3. Grade 1 is the slowest growing, and grade 3 is the fastest growing. Grade 3 sarcomas can spread to other parts of the body.
Chondrosarcoma Staging:
Chondrosarcomas are staged according to their size and whether they have spread from their primary location. Staging is used to help doctors organize treatment:
- Stage 1A: Low-grade and is contained within the bone.
- Stage 1B: Low-grade cancer found outside the bone. No metastasis.
- Stage 2A: High-grade and is contained within the hard coating of the bone. No metastasis
- Stage 2B: High-grade cancer extending outside the bone and into the soft tissue spaces. No metastasis
- Stage 3: The cancer can be low-grade or high-grade and it is found either within the bone or outside it. The cancer has metastasized and spread to other parts of the body.
Chondrosarcoma treatment
Chondrosarcoma treatments depend on the grading and staging of the tumor. Surgery is the primary treatment for chondrosarcoma, because most chondrosarcomas are resistant to chemotherapy, so treatment of chondrosarcomas revolves around wide surgical excision where clinically possible. Chemotherapy and radiotherapy are less effective treatments for chondrosarcoma.
An orthopedic surgeon will perform the surgery. They will work out if chemotherapy or radiotherapy is required in consultation with an oncologist (a doctor who specializes in treating cancer with chemotherapy) and/or radiologist (a doctor who specializes in treating cancer with radiation).
Surgery for chondrosarcoma
The aim of surgery is to remove the tumor. Because of the type of tumor (being in the bone) it can often be difficult to remove.
Sometimes some of the bone will be removed, and either a prosthesis (a metal replacement bone), or a bone graft (bone taken from another part of the body) will be inserted. This is known as limb-sparing surgery.
Unfortunately because of the location of the chondrosarcoma tumor, sometimes limb-sparing surgery doesn’t work and a limb might have to be amputated. It only ever happens if it is completely unavoidable. This is because the chondrosarcoma cancer has spread from the bone and into the nearby blood vessels.
Chemotherapy for chondrosarcoma
Chondrosarcoma is typically a very slow-growing cancer and chemotherapy targets cells that grow very quickly, so this treatment is generally not very effective. Chemotherapy is advocated in the treatment of the histological variants – mesenchymal chondrosarcomas and dedifferentiated chondrosarcomas because they grow swiftly so chemotherapy may be helpful in these cases, but its role in the treatment of conventional chondrosarcoma remains to be defined.
Radiotherapy for chondrosarcoma
Radiotherapy is used in chondrosarcoma patients where the primary chondrosarcoma tumor is not resectable. For example if your tumor is in a location such as the base of the skull, which makes it difficult to remove all the cancer, your doctor might suggest using radiation therapy before or after the operation.
Improvement in chondrosarcoma symptoms is an important measurement. Specific monitoring may be by serial imaging to detect any recurrence or metastatic spread. The chondrosarcoma symptoms that may require attention are somatic pain from bone metastases, visceral pain from lung metastases and neurogenic pain if nerve tissue is compressed. Coughing and breathlessness from lung involvement may require specific treatment.
Coping and support
A cancer diagnosis can change your life forever. Each person finds his or her own way of coping with the emotional and physical changes cancer brings. But when you’re first diagnosed with cancer, sometimes it’s difficult to know what to do next.
Here are some ideas to help you cope:
- Learn enough about cancer to make decisions about your care. Ask your doctor about your cancer, including your treatment options and, if you like, your prognosis. As you learn more about cancer, you may become more confident in making treatment decisions.
- Keep friends and family close. Keeping your close relationships strong will help you deal with your cancer. Friends and family can provide the practical support you’ll need, such as helping take care of your house if you’re in the hospital. And they can serve as emotional support when you feel overwhelmed by cancer.
- Find someone to talk with. Find a good listener who is willing to listen to you talk about your hopes and fears. This may be a friend or family member. The concern and understanding of a counselor, medical social worker, clergy member or cancer support group also may be helpful.
- Boehme KA, Schleicher SB, Traub F, Rolauffs B. Chondrosarcoma: A Rare Misfortune in Aging Human Cartilage? The Role of Stem and Progenitor Cells in Proliferation, Malignant Degeneration and Therapeutic Resistance. International Journal of Molecular Sciences. 2018;19(1):311. doi:10.3390/ijms19010311. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5796255/[↩]
- Bertoni F, Bacchini P, Hogendoorn PCW. Chondrosarcoma. In: Fletcher CDM, Unni KK, Mertens F, editors. World Health Organization Classification of Tumours. Pathology and Genetics of Tumours of Soft Tissue and Bone. IARC Press; Lyon: 2002.[↩]
- Bone sarcomas: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Hogendoorn PC, ESMO/EUROBONET Working Group., Athanasou N, Bielack S, De Alava E, Dei Tos AP, Ferrari S, Gelderblom H, Grimer R, Hall KS, Hassan B, Hogendoorn PC, Jurgens H, Paulussen M, Rozeman L, Taminiau AH, Whelan J, Vanel D. Ann Oncol. 2010 May; 21 Suppl 5():v204-13. https://academic.oup.com/annonc/article/21/suppl_5/v204/194341[↩]
- Corradi D., Bacchini P., Campanini N., Bertoni F. Aggressive clear cell chondrosarcomas: Do distinctive characteristics exist? A report of 4 cases. Arch. Pathol. Lab. Med. 2006;130:1673–1679. https://www.ncbi.nlm.nih.gov/pubmed/17076530[↩][↩]
- Gelderblom H, Hogendoorn PC, Dijkstra SD, van Rijswijk CS, Krol AD, Taminiau AH, Bovée JV. The clinical approach towards chondrosarcoma. Oncologist. 2008;13:320–9. http://theoncologist.alphamedpress.org/content/13/3/320.long[↩][↩]
- Schrage YM, Machado I, Meijer D, Briaire-de Bruijn I, van den Akker BE, Taminiau AH, Kalinski T, Llombart-Bosch A, Bovée JV. COX-2 expression in chondrosarcoma: a role for celecoxib treatment? Eur J Cancer. 2010;46:616–24. https://www.ncbi.nlm.nih.gov/pubmed/20004565[↩]
- The Clinical Approach Towards Chondrosarcoma. The Oncologist March 2008 vol. 13 no. 3 320-329. doi: 10.1634/theoncologist.2007-0237 http://theoncologist.alphamedpress.org/content/13/3/320.long[↩]
- Novel therapeutic approaches in chondrosarcoma. Polychronidou G, Karavasilis V, Pollack SM, Huang PH, Lee A, Jones RL. Future Oncol. 2017 Mar; 13(7):637-648. https://www.futuremedicine.com/doi/10.2217/fon-2016-0226[↩]
- Bertoni F, Bacchini P, Hogendoorn PCW. Chondrosarcoma. In: Fletcher CDM, Unni KK, Mertens F, editors. World Health Organisation Classification of Tumours. Pathology and Genetics of Tumours of Soft Tissue and Bone. Lyon, France: IARC Press; 2002. p. 247-251.[↩]
- Risk factors for survival and local control in chondrosarcoma of bone. Fiorenza F, Abudu A, Grimer RJ, Carter SR, Tillman RM, Ayoub K, Mangham DC, Davies AM. J Bone Joint Surg Br. 2002 Jan; 84(1):93-9. https://www.ncbi.nlm.nih.gov/pubmed/11837841/[↩]
- Vanel D, De Paolis M, Monti C, et al. Radiological features of 24 periosteal chondrosarcomas. Skeletal Radiol 2001;30:208-212[↩]
- Evans HL, Ayala AG, Romsdahl MM. Prognostic factors in chondrosarcoma of bone: A clinicopathologic analysis with emphasis on histologic grading. Cancer 1977;40:818-831.[↩][↩]
- Bjornsson J, McLeod RA, Unni KK, et al. Primary chondrosarcoma of long bones and limb girdles. Cancer 1998;83:2105-2119.[↩][↩]
- Milchgrub S, Hogendoorn PCW. Dedifferentiated chondrosarcoma. In: Fletcher CDM, Unni KK, Mertens F, editors. World Health Organization Classification of Tumours. Pathology and Genetics. Tumours of Soft Tissue and Bone. Lyon, France: IARC Press; 2002. p. 252-254.[↩]
- Qasem S.A., DeYoung B.R. Cartilage-forming tumors. Semin. Diagn. Pathol. 2014;31:10–20. doi: 10.1053/j.semdp.2014.01.006. https://www.ncbi.nlm.nih.gov/pubmed/24680178[↩]
- Grimer R.J., Gosheger G., Taminiau A., Biau D., Matejovsky Z., Kollender Y., San-Julian M., Gherlinzoni F., Ferrari C. Dedifferentiated chondrosarcoma: Prognostic factors and outcome from a european group. Eur. J. Cancer. 2007;43:2060–2065. doi: 10.1016/j.ejca.2007.06.016. https://www.ncbi.nlm.nih.gov/pubmed/17720491[↩]
- Dornauer K., Soder S., Inwards C.Y., Bovee J.V., Aigner T. Matrix biochemistry and cell biology of dedifferentiated chondrosarcomas. Pathol. Int. 2010;60:365–372. doi: 10.1111/j.1440-1827.2010.02530.x. https://www.ncbi.nlm.nih.gov/pubmed/20518886[↩]
- Frassica F.J., Unni K.K., Beabout J.W., Sim F.H. Dedifferentiated chondrosarcoma. A report of the clinicopathological features and treatment of seventy-eight cases. J. Bone Jt. Surg. Am. 1986;68:1197–1205. doi: 10.2106/00004623-198668080-00008. https://www.ncbi.nlm.nih.gov/pubmed/3021775[↩]
- Grimer RJ, Gosheger G, Taminiau A, et al. Dedifferentiated chondrosarcoma: Prognostic factors and outcome from a European group. Eur J Cancer 2007;43:2060-2065.[↩]
- Bovée JVMG, Cleton-Jansen AM, Rosenberg C, et al. Molecular genetic characterization of both components of a dedifferentiated chondrosarcoma, with implications for its histogenesis. J Pathol 1999;189:454-462.[↩]
- Ropke M, Boltze C, Neumann HW, et al. Genetic and epigenetic alterations in tumor progression in a dedifferentiated chondrosarcoma. Pathol Res Pract 2003;199:437-444.[↩]
- Nakashima Y, Park YK, Sugano O. Mesenchymal chondrosarcoma. In: Fletcher CDM, Unni KK, Mertens F, editors. World Health Organization Classification of Tumours. Pathology and Genetics. Tumours of Soft Tissue and Bone. Lyon, France: IARC Press; 2002. p. 255-256.[↩]
- Naumann S, Krallman PA, Unni KK, et al. Translocation der(13;21)(q10;q10) in skeletal and extraskeletal mesenchymal chondrosarcoma. Mod Pathol 2002;15:572-576.[↩]
- Park YK, Park HR, Chi SG, et al. Overexpression of p53 and rare genetic mutation in mesenchymal chondrosarcoma. Oncol Rep 2000;7:1041-1047.[↩]
- Brown RE, Boyle JL. Mesenchymal chondrosarcoma: Molecular characterization by a proteomic approach, with morphogenic and therapeutic implications. Ann Clin Lab Sci 2003;33:131-141.[↩]
- Donati D, Yin JQ, Colangeli M, et al. Clear cell chondrosarcoma of bone: Long time follow-up of 18 cases. Arch Orthop Trauma Surg 2008;128:137-142.[↩]
- Nishio J, Reith JD, Ogose A, et al. Cytogenetic findings in clear cell chondrosarcoma. Cancer Genet Cytogenet 2005;162:74-77.[↩]
- Masui F, Ushigome S, Fujii K. Clear cell chondrosarcoma: A pathological and immunohistochemical study. Histopathology 1999;34:447-452.[↩]
- Park HR, Park YK. Differential expression of runx2 and Indian hedgehog in cartilaginous tumors. Pathol Oncol Res 2007;13:32-37.[↩]
- Corradi D, Bacchini P, Campanini N, et al. Aggressive clear cell chondrosarcomas: Do distinctive characteristics exist? A report of 4 cases. Arch Pathol Lab Med 2006;130:1673-1679.[↩]
- Pearls and Pitfalls in Musculoskeletal Imaging: Variants and Other Difficult Diagnoses. Cambridge University Press. ISBN:0521196329[↩]
- Murphey MD, Walker EA, Wilson AJ et-al. From the archives of the AFIP: imaging of primary chondrosarcoma: radiologic-pathologic correlation. Radiographics. 23 (5): 1245-78. doi:10.1148/rg.235035134 https://www.ncbi.nlm.nih.gov/pubmed/12975513[↩]
- Goh YW, Spagnolo DV, Platten M, et al. Extraskeletal myxoid chondrosarcoma: A light microscopic, immunohistochemical, ultrastructural and immuno-ultrastructural study indicating neuroendocrine differentiation. Histopathology 2001;39:514-524.[↩]
- Aigner T, Oliveira AM, Nascimento AG. Extraskeletal myxoid chondrosarcomas do not show a chondrocytic phenotype. Mod Pathol 2004;17:214-221.[↩][↩]
- Antonescu CR, Argani P, Erlandson RA, et al. Skeletal and extraskeletal myxoid chondrosarcoma: A comparative clinicopathologic, ultrastructural, and molecular study. Cancer 1998;83:1504-1521[↩]
- Kilpatrick SE, Inwards CY, Fletcher CD, et al. Myxoid chondrosarcoma (chordoid sarcoma) of bone: A report of two cases and review of the literature. Cancer 1997;79:1903-1910.[↩]
- Murphey MD, Walker EA, Wilson AJ et-al. From the archives of the AFIP: imaging of primary chondrosarcoma: radiologic-pathologic correlation. Radiographics. 2003;23 (5): 1245-78. doi:10.1148/rg.235035134 https://www.ncbi.nlm.nih.gov/pubmed/12975513[↩][↩]
- Kim Y, Im S, Lim G et-al. Korean Journal of Radiology. 2007;8 (5): . doi:10.3348/kjr.2007.8.5.452 https://synapse.koreamed.org/DOIx.php?id=10.3348/kjr.2007.8.5.452[↩]
- Oh B, Han Y, Lee B et-al. Korean Journal of Radiology. 2007;8 (6): . doi:10.3348/kjr.2007.8.6.541 https://synapse.koreamed.org/DOIx.php?id=10.3348/kjr.2007.8.6.541[↩]
- Fletcher C.D.M., Bridge J.A., Hogendoorn P.C.W., Mertens F. Who Classification of Tumours of Soft Tissue and Bone. 4th ed. Volume 5 IARC WHO Classification of Tumours; Lyon, France: 2013. WHO 2013.[↩]
- Polychronidou G., Karavasilis V., Pollack S.M., Huang P.H., Lee A., Jones R.L. Novel therapeutic approaches in chondrosarcoma. Future Oncol. 2017;13:637–648. doi: 10.2217/fon-2016-0226. https://www.ncbi.nlm.nih.gov/pubmed/28133974[↩]
- Fiorenza F., Abudu A., Grimer R.J., Carter S.R., Tillman R.M., Ayoub K., Mangham D.C., Davies A.M. Risk factors for survival and local control in chondrosarcoma of bone. J. Bone Jt. Surg. Br. 2002;84:93–99. doi: 10.1302/0301-620X.84B1.11942. https://www.ncbi.nlm.nih.gov/pubmed/11837841[↩][↩]
- Andreou D, Ruppin S, Fehlberg S, Pink D, Werner M, Tunn P-U. Survival and prognostic factors in chondrosarcoma: Results in 115 patients with long-term follow-up. Acta Orthopaedica. 2011;82(6):749-755. doi:10.3109/17453674.2011.636668. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3247897/[↩][↩][↩][↩][↩][↩][↩]





{kind=link}