
Contents
What is polymyositis
Polymositis also known as idiopathic inflammatory myopathy, is a type of inflammatory myopathy, which refers to a group of muscle diseases characterized by chronic muscle inflammation and degenerative changes in the muscles, leading to symmetric weakness and some degree of muscle wasting (atrophy). Polymyositis is part of a larger group of diseases called myositis. The muscles affected by polymyositis are the skeletal muscles (those involved with making movements) principally affecting those muscles closest to and within the trunk of the body such as the hip, shoulders, arms, pharynx and neck on both sides of the body. Although polymyositis may appear at any time from infancy through the age of 80 years, most cases are seen in adults between the ages of 31 and 60 years, especially those aged 45 to 60 years. Juvenile polymyositis is very rare (much less common than juvenile dermatomyositis) and the symptoms usually appear between the ages of 5 to 15 years. Females are affected twice as often as males and polymyositis is more common in African Americans than in Caucasians.
Polymyositis is a systemic disease, which means it affects the whole body. Muscle weakness and tenderness can be signs of polymyositis. Muscle weakness usually happens over days, weeks or months. Progressive muscle weakness starts in the proximal muscles (muscles closest to the trunk of the body) which eventually leads to difficulties climbing stairs, rising from a seated position, lifting objects, or reaching overhead. Some affected people have muscle pain, arthritis, breathing problems, difficulty swallowing and speakingand heart arrhythmias. In some cases of polymyositis, distal muscles (muscles further away from the trunk of the body, such as those in the forearms and around the ankles and wrists) may be affected as the disease progresses. Polymyositis may be associated with collagen-vascular or autoimmune diseases, such as lupus. Polymyositis may also be associated with infectious disorders, such as HIV-AIDS.
Polymyositis is rare. Incidence is estimated to be somewhere between 1-8 cases per million people.
The exact cause of polymyositis is unknown (idiopathic). Polymositis shares many characteristics with autoimmune disorders, which occur when your body’s immune system mistakenly attacks healthy body tissues for unknown reasons, leading to inflammation or swelling. In some cases, polymyositis may be associated with viral infections, connective tissue disorders, or an increased risk for malignancies (cancer). Diagnosis is based on a clinical examination that may include laboratory tests, imaging studies, electromyography, and a muscle biopsy. Although there is no cure for polymyositis, treatment with corticosteroids or immunosuppressants can improve muscle strength and function. Physical therapy is usually recommended to prevent muscle atrophy and to regain muscle strength and range of motion. Occupational therapists can prepare an assessment of daily activities to help address issues such as bathing and eating.
Polymyositis vs Dermatomyositis
Dermatomyositis is a rare acquired skeletal (voluntary) muscle disease (myositis) that is accompanied by a rash. It is one of a group of muscle diseases called inflammatory myopathies. Polymyositis is myositis without the skin rash. Muscle inflammation and weakness occur in both conditions while patients with dermatomyositis also have a rash.
Dermatomyositis may affect people of any race, age or sex, although it is twice as common in women than in men. The onset of the disease is most common in those aged 50–70 years.
Dermatomyositis muscle abnormalities may begin with aches and weakness of the muscles of the trunk, upper arms, hips, and thighs (proximal muscles). Muscles may be stiff, sore, tender and, eventually, show signs of degeneration (atrophy). Affected individuals may experience difficulty in performing certain functions, such as raising their arms and/or climbing stairs or develop speech and swallowing difficulties. Skin abnormalities associated with dermatomyositis often include a distinctive reddish-purple rash (heliotrope rash) on the upper eyelid or across the cheeks and bridge of the nose in a “butterfly” distribution and on the forehead and scalp. Other characteristic rashes include scaling and redness of the knuckles, elbows, knees, and/or other extensor regions (Gottron papules and sign); an abnormal accumulation of fluid (edema) in body tissues surrounding the eyes; and/or other features. The symptoms of childhood (juvenile) dermatomyositis are similar to those associated with the adult form of the disorder. However, onset is usually more sudden. In addition, abnormal accumulations of calcium deposits (calcifications) in muscle and skin tissues as well as involvement of the digestive (gastrointestinal [GI]) tract are more common in juvenile dermatomyositis.
Figure 1. Dermatomyositis – photodistributed rash
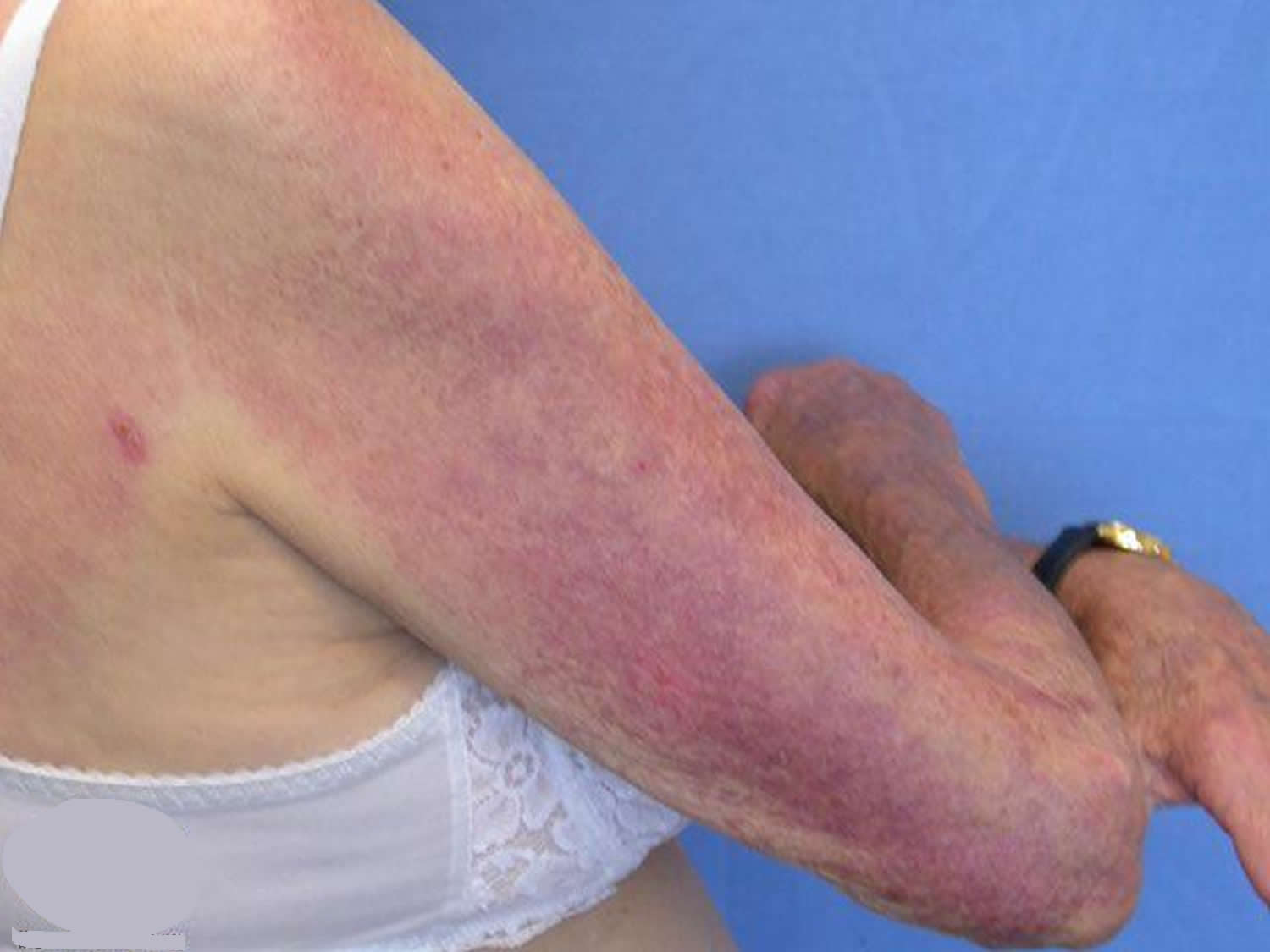
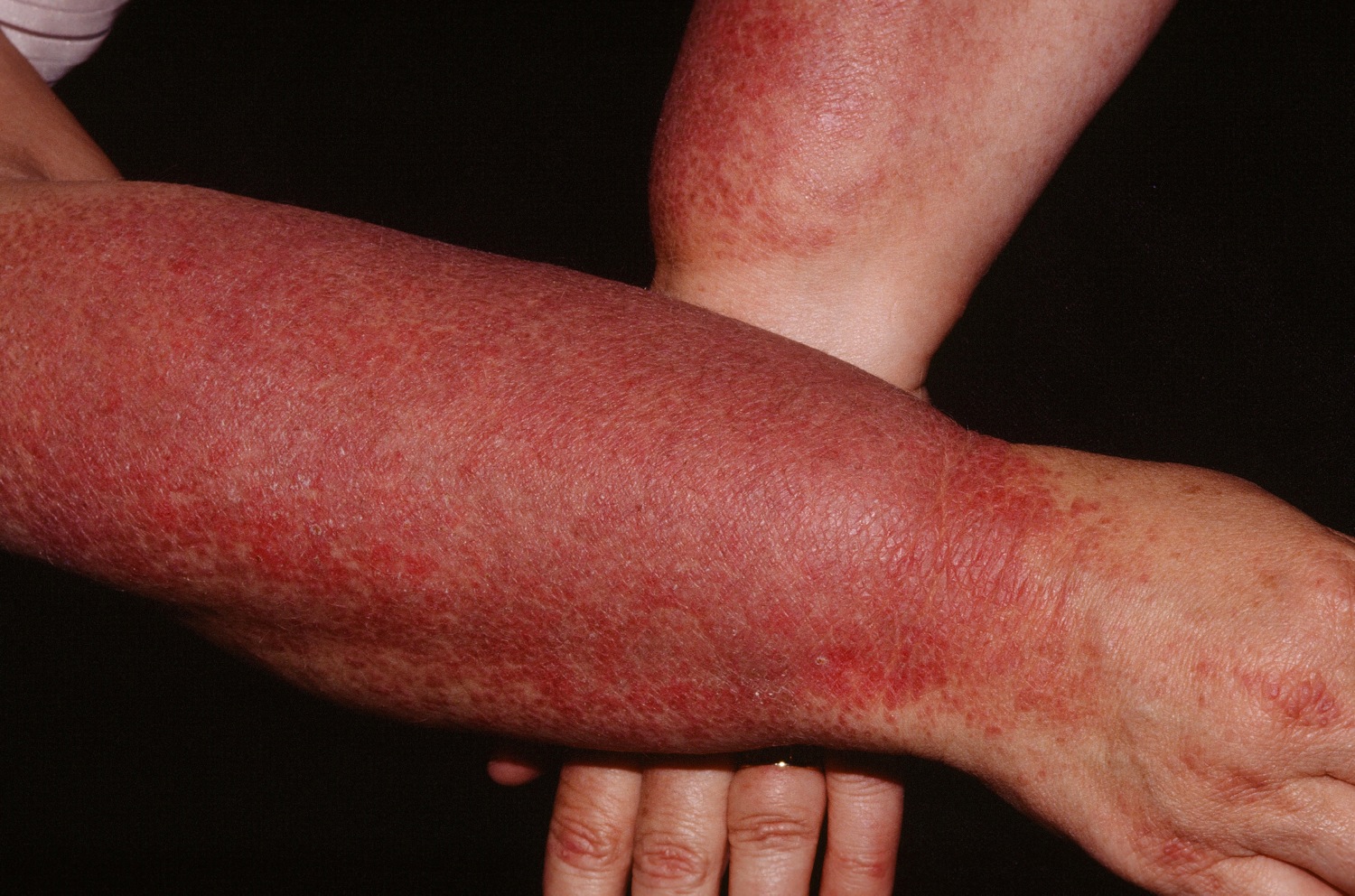
Figure 2. Dermatomyositis – periorbital swelling
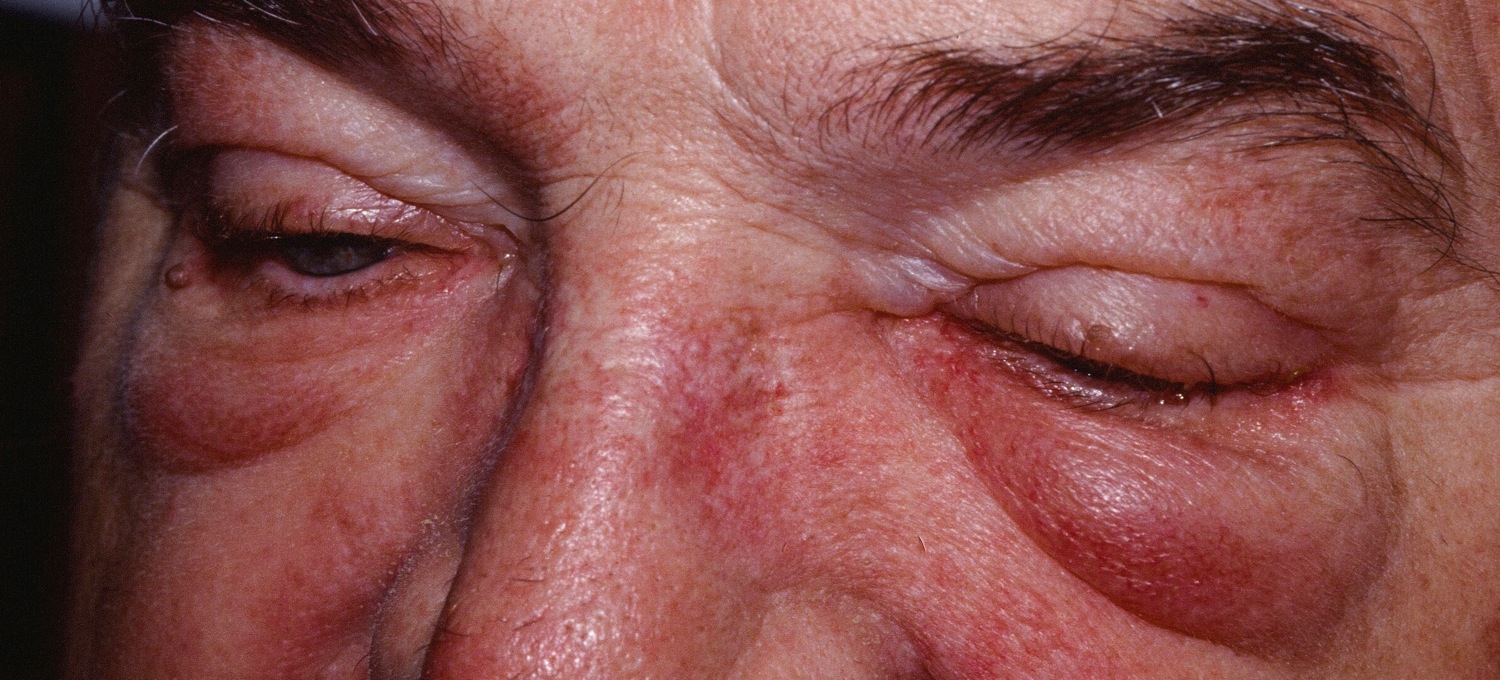
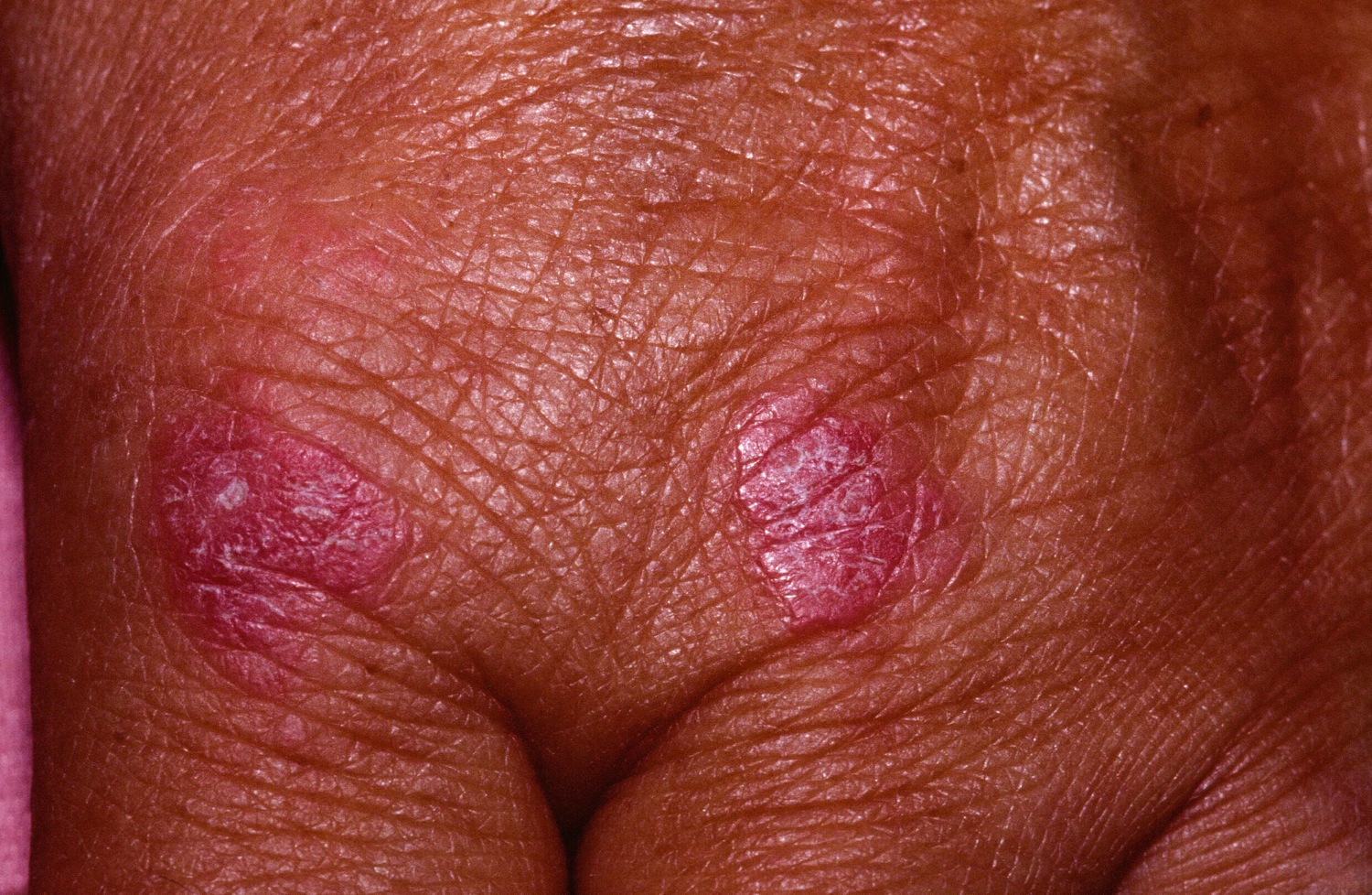
Figure 3. Dermatomyositis – violaceous plaques over the knuckles
Dermatomyositis signs and symptoms
In many patients the first sign of dermatomyositis is the presence of a symptomless, itchy or burning rash. The rash often, but not always, develops before the muscle weakness.
- Reddish or bluish-purple patches mostly affect sun exposed areas.
- A violaceous rash may also affect cheeks, nose, shoulders, upper chest and elbows.
- Purple eyelids, which are described as heliotrope, as they resemble the heliotrope flower, Heliotropium peruvianum, which has small purple petals.
- Purple papules or plaques are found on bony prominences, especially the knuckles (Gottron papules).
- Ragged cuticles and prominent blood vessels on nail folds are best seen by capillaroscopy or dermoscopy.
- A scaly scalp and thinned out hair may occur.
- Less commonly, there is poikiloderma, in which the skin is atrophic (pale, thin skin), red (dilated blood vessels) and brown (post-inflammatory pigmentation).
Muscle weakness may arise at the same time as the rash, or it may occur weeks, months or years later. Proximal muscles are affected, that is, those closest to the trunk (upper arms, thighs). The first indication of myositis is when the following everyday movements become difficult.
- Climbing stairs or walking
- Rising from a sitting or crouching position
- Lifting objects
- Raising arms above the shoulders, e.g. combing hair
- Difficulty swallowing (dysphagia)
Occasionally the affected muscles ache and become tender to touch.
Calcinosis affects some people, especially children and adolescents
- It presents as hard yellow or white lumps under the skin.
- These usually appear on fingers or over joints.
- Sometimes these nodules may poke through the skin and ulcerate.
- The ulcers may become infected.
Some patients have swollen joints and Raynaud phenomenon (this term refers to fingers that go very white and stiff in cold conditions, then purple as they warm again).
Polymyositis prognosis
The prognosis for polymyositis varies. Most people respond fairly well to therapy, but some have a more severe disease that does not respond adequately to therapies and are left with significant disability. In rare cases individuals with severe and progressive muscle weakness will develop respiratory failure or pneumonia. Difficulty swallowing may cause weight loss and malnutrition.
Polymyositis rarely causes death, but is associated with lung, and heart disease, as well as an increased risk of certain cancers, such as bladder cancer and lymphoma.
Polymyositis life expectancy
Onset of polymyositis is gradual over several months. Initially, patients may experience fevers, weight loss and a general feeling of unwellness. This is followed by wasting of the shoulder and hip muscles, leading to weakness in these areas. Severity of polymyositis is variable, from a mild condition to severe disability. In progressive disease, widespread weakness and wasting may develop, with heart and lung involvement.
Response to treatment varies based on the complications. Proper diagnosis and treatment raise the chance of living life fully despite this illness. The 5-year mortality rate can be as high as 1 in 5 patients. Many people, especially children, have a period when no symptoms are present and recover. For most other people, immunosuppressant drugs can control the disease.
Polymyositis complications
Possible complications of polymyositis include:
- Difficulty swallowing. If the muscles in your esophagus are affected, you may have problems swallowing (dysphagia), which in turn may cause weight loss and malnutrition.
- Aspiration pneumonia. Difficulty swallowing may also cause you to breathe food or liquids, including saliva, into your lungs (aspiration), which can lead to pneumonia.
- Breathing problems. If your chest muscles are affected by the disease, you may experience breathing problems, such as shortness of breath or, in severe cases, respiratory failure.
Associated conditions
Although these are not complications, polymyositis is often associated with other conditions that may cause further complications of their own, or in combination with polymyositis symptoms. Associated conditions include:
- Raynaud’s phenomenon. This is a condition in which your fingers, toes, cheeks, nose and ears initially turn pale when exposed to cold temperatures.
- Other connective tissue diseases. Other conditions, such as lupus, rheumatoid arthritis, scleroderma and Sjogren’s syndrome, can occur in combination with polymyositis.
- Cardiovascular disease. Polymyositis may cause the muscular walls of your heart to become inflamed (myocarditis). In a small number of people who have polymyositis, congestive heart failure and heart arrhythmias may develop.
- Lung disease. A condition called interstitial lung disease may occur with polymyositis. Interstitial lung disease refers to a group of disorders that cause scarring (fibrosis) of lung tissue, making lungs stiff and inelastic. Signs and symptoms include a dry cough and shortness of breath.
- Cancer. People who have polymyositis have an elevated risk of cancer.
Polymyositis causes
The exact cause of polymyositis is unknown. Like other autoimmune diseases, the body’s natural immune defense mechanisms attack its own tissue in polymyositis. This can occur through a T-cell (T lymphocyte) mediated destructive process directed against unidentified muscle antigens, but the finding of myositis-specific autoantibodies in many polymyositis patients also supports disorders associated with B cells (B lymphocytes).
Exposure to certain drugs has been shown to increase the likelihood of developing myopathy. A common drug known to induce a necrotizing form of myositis are the statin agents and a specific autoantibody (anti-HMGCR) has been found to be associated with statin-induced myopathy.
The injection of collagen for cosmetic use has been implicated in the onset of a polymyositis-like syndrome. Injectable bovine collagen has been used to reduce wrinkles and facial scars, usually in women. The onset of polymyositis in such patients is felt to result from an autoimmune reaction to the collagen.
Polymyositis symptoms
The symptoms of polymyositis may start gradually or suddenly and often wax and wane for no apparent reason.
The major symptom of polymyositis is muscle weakness, most often in the hip and shoulder areas, eventually making it difficult for patients to lift their arms, get out of a chair or to climb steps. Other muscles which may be affected are the neck and throat muscles, which may result in difficulty swallowing and cause changes in the voice due to muscle weakness. Rarely, chest muscles are affected.
The muscle weakness may appear suddenly and progress over weeks to months. Additionally, the degree of muscle weakness may fluctuate over time as well. The muscles of the hands, feet and face are rarely involved in polymyositis, but generalized muscle pain may occur. Atrophy (loss of bulk) of muscle and contractures of the muscle around larger joints may develop late in the chronic stage.
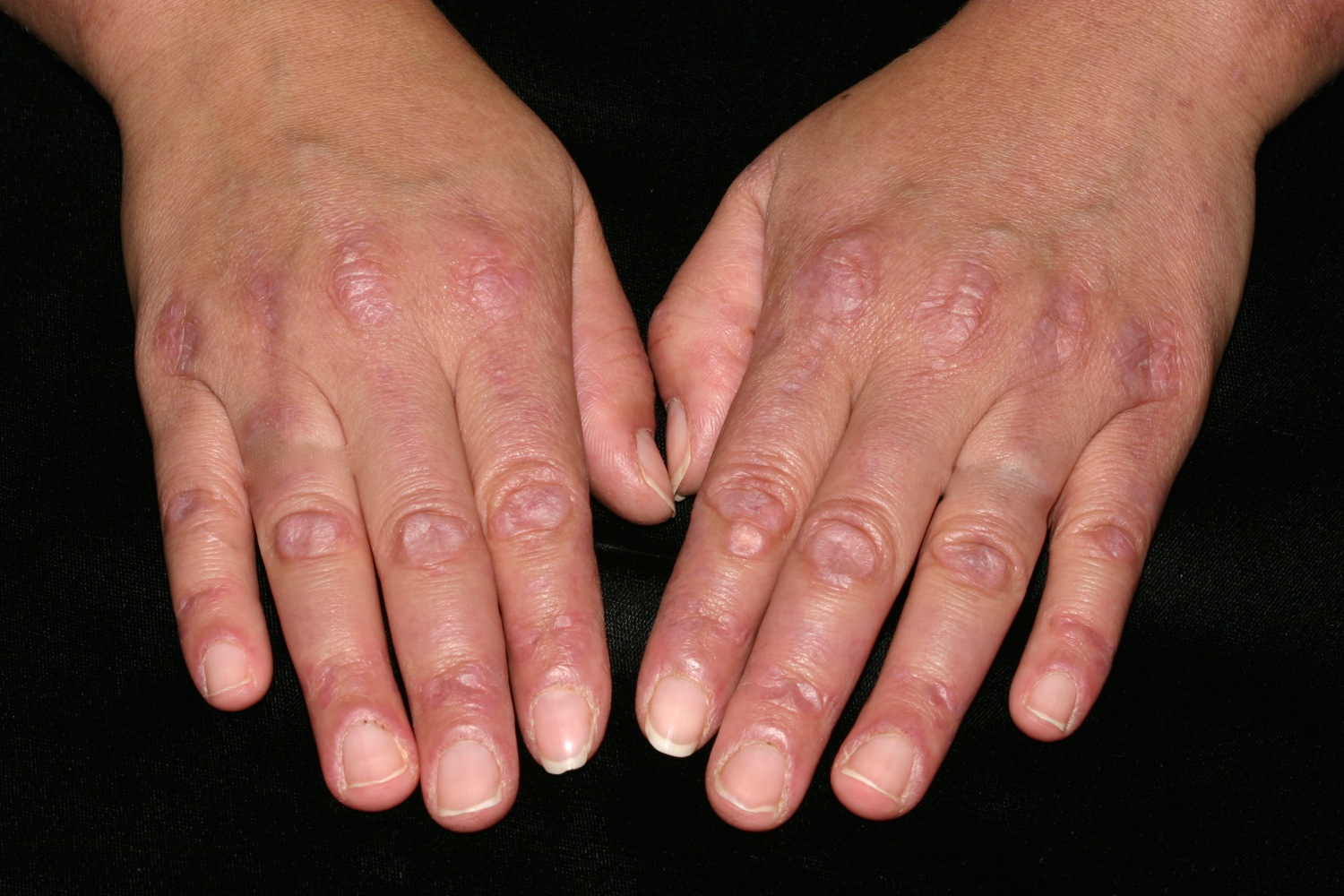
Other symptoms of polymyositis may include fever, weight loss, fatigue, joint pain and Raynaud phenomenon which is a sensitivity to the cold that is most often felt in the fingers. Raynaud phenomenon is caused by constriction (narrowing) of the blood vessels in the fingers upon exposure to the cold. Cracking of the fingers, termed “mechanic’s hands” may also develop. Mechanic’s hands is a term used to describe skin changes of the hands seen in a variety of connective tissue diseases with an inflammatory myopathy.
Figure 4. Polymyositis – mechanic’s hands
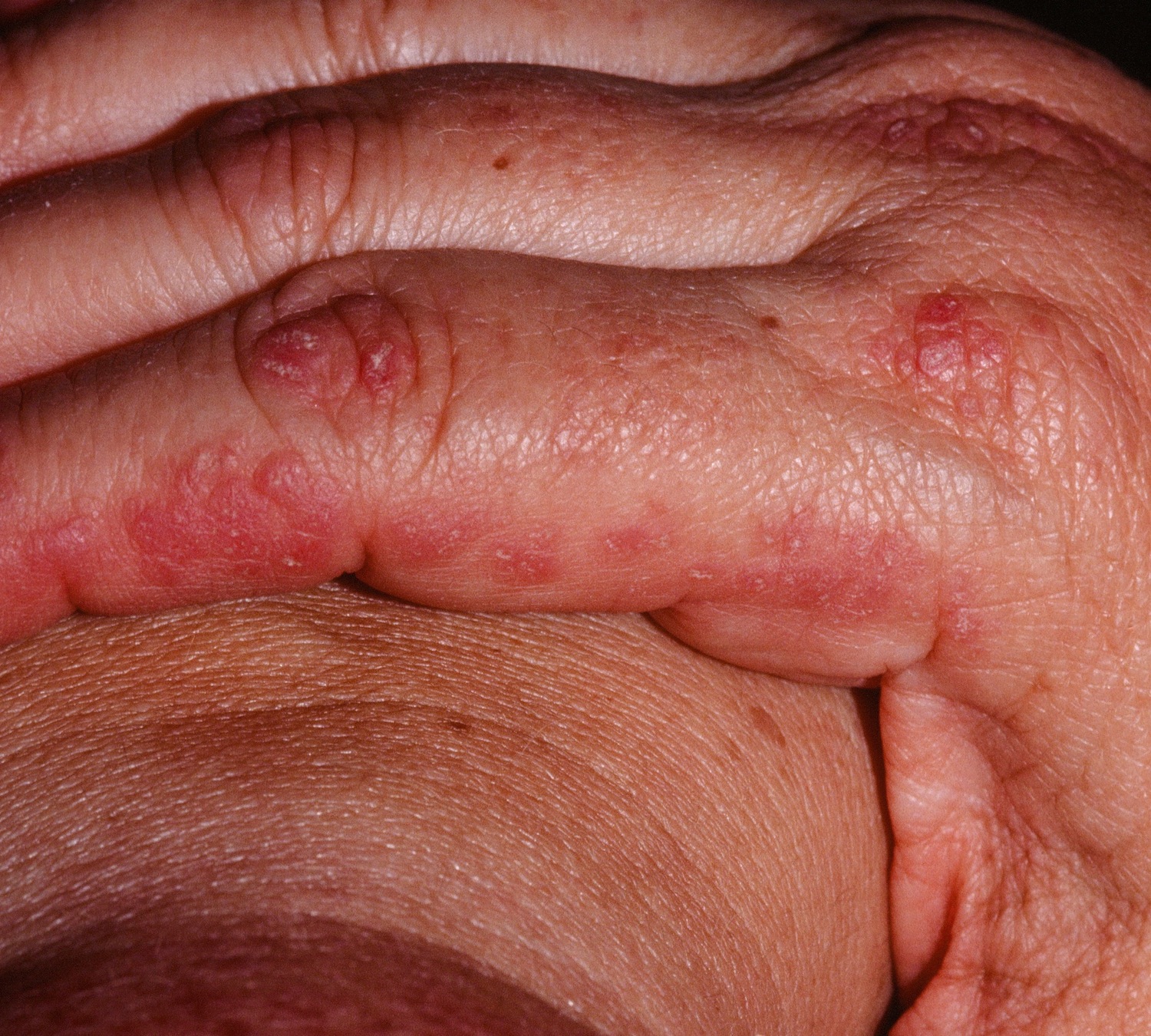
The joint pain seen in polymyositis (polyarthralgia) may be accompanied by swelling and mimic the findings seen in patients with rheumatoid arthritis but generally these rheumatic complaints are mild and respond well to glucocorticoids (i.e. prednisone). Gastrointestinal involvement can lead to difficulty swallowing as the muscles involved in swallowing can become inflamed and weak, and the esophagus can be affected leading to heartburn but lower GI (gastrointestinal) involvement is uncommon. Involvement of the heart, detected chiefly by irregularities in the electrocardiogram (ECG), has been reported but inflammation and weakening of the heart muscle (myocarditis) is rare.
Interstitial lung disease is a serious complication seen in polymyositis and leads to cough with shortness of breath as inflammation occurs in the lung tissue. This may progress to scarring (fibrosis) of the lung tissue making the lungs stiff and inelastic. In some instances lung disease may precede myositis and dominate the clinical picture.
Symptoms may worsen during pregnancy in women whose disease is active and active polymyositis can increase the risk of premature birth or stillbirth.
Myositis can occur in overlap with other autoimmune or rheumatic diseases, including Sjogren’s syndrome or scleroderma (systemic sclerosis). In addition, malignancy may complicate the course and presentation of polymyositis but is more common in dermatomyositis. This is more common in males and females over the age of 50.
Abdominal symptoms, more common in children with juvenile dermatomyositis, may be associated with the passage of dark stools or the vomiting of blood from gastrointestinal ulcerations that may progress to perforation and require surgical intervention. Calcinosis (the deposition of calcium deposits in the skin and soft tissue or muscle) is also more frequent in juvenile dermatomyositis.
Polymyositis diagnosis
The diagnosis of polymyositis is often delayed due to the lack of physical findings before the onset of muscle disease. Both family history and medication history are important in excluding other causes of myopathy. Additionally, various tests may be performed to establish a diagnosis.
Tests may include:
- Electromyography done by a specialist in neuromuscular diseases which detects characteristic electrical patterns in muscle tissue and are abnormal in almost all patients with polymyositis;
- Muscle biopsy which reveals inflammation in the muscle tissue;
- Magnetic resonance imaging (MRI) of the affected muscle(s) which demonstrates inflammation within the muscle tissue.
- Blood tests can be performed to detect elevated levels of muscle enzymes, predominantly creatine kinase (CK) and aldolase among others, which are indicative of muscle damage. Autoantibodies have been identified in many polymyositis patients consistent with an autoimmune cause as discussed earlier.
Polymyositis treatment
There is no cure for polymyositis, but the symptoms can be treated with medication, physical and occupational therapy, exercise, heat therapy (including microwave and ultrasound), orthotics and assistive devices, and rest. Polymyositis generally responds well to treatment in most patients, although, residual weakness may occur in approximately 30% of patients.
Corticosteroids
Corticosteroids (i.e. prednisone or prednisolone) are widely used as the initial form of treatment in polymyositis. They control the inflammation, lessen pain, and increase muscle strength through their ability to suppress youre body’s immune system. The specific dose is individualized; however, the starting dose of prednisone is generally high at about 1mg/kg of body weight (often equating to about 60mg/day). They are then tapered depending on the response of the patient and associated side effects. Measurement of the serum creatine kinase (CK) is used to gauge the effectiveness of therapy and reduction of the muscle enzymes to normal values is noted in a majority of patients with this disorder within 4 to 8 weeks after treatment is started. This is followed by an improvement in muscle strength. As the steroids are reduced other medications that also suppress the immune system may be added so that the corticosteroids can be tapered. This is necessary to reduce the side effects (i.e. osteoporosis, weight gain, change in mental status, increased blood sugar, cataracts, stomach upset, etc.) that the steroids cause. In many cases of adult polymyositis prolonged maintenance therapy with prednisone or other corticosteroids may be necessary indefinitely. A major concern with these agents is osteoporosis, which may cause significant morbidity. In this instance, additional calcium and vitamin D supplementation may be beneficial.
How prednisone works
Prednisone (prednisolone) works by blocking the effects of certain chemicals that cause inflammation inside your body. It doesn’t cure polymyositis, but it can help relieve the symptoms.
When used to treat polymyositis, prednisone is taken as a tablet. Most people will be prescribed several tablets to take once a day.
You’ll be prescribed a high dose of prednisone to start with, and the dose will be gradually reduced every one to two months.
Side effects of prednisolone
About 1 in 20 people who take prednisolone will experience changes in their mental state when they take the medication.
You may feel depressed and suicidal, anxious or confused. Some people also experience hallucinations, which is seeing or hearing things that aren’t there.
Contact your doctor as soon as possible if you experience changes to your mental state.
Other side effects of prednisolone include:
- increased appetite, which often leads to weight gain
- increased blood pressure
- mood changes, such as becoming aggressive or irritable with people
- weakening of the bones (osteoporosis)
- stomach ulcers
- increased risk of infection, particularly with the varicella-zoster virus, the virus that causes chickenpox and shingles
You should seek immediate medical advice if you think you’ve been exposed to the varicella-zoster virus or if a member of your household develops chickenpox or shingles.
The risk of these side effects should improve as your dose of prednisolone is decreased.
Monitoring side effects
Long-term use of corticosteroids can result in a number of serious side effects. Your doctor will monitor you closely for problems. He or she may adjust your dosage and prescribe treatments to manage these reactions to corticosteroid treatment. Possible side effects include:
- Weight gain
- Osteoporosis — the loss of bone density and weakening of bones
- High blood pressure (hypertension)
- Diabetes
- Thinning of the skin and bruising
- Cataracts — a clouding of the lenses of your eyes
It is recommended that clinicians assess bone mineral density (BMD) before initiating corticosteroid treatment, which is likely to last longer than 3 months. The following steps should be taken at the commencement of steroid therapy:
- Start adequate supplementation of calcium (1200 mg/day) and vitamin D3 (cholecalciferol, 800 IU/day)
- Assess lumber and hip spine bone mineral density
- If bone mineral density T-score is –1.5 or less consider an oral bisphosphonate such as alendronate (70 mg/week) or risedronate sodium (35 mg/week).
Immunosuppressive drugs
Immunosuppressive drugs such as methotrexate, azathioprine, mycophenolate mofetil, tacrolimus, cyclosporine cyclophosphamide, and others have been beneficial to patients who fail to respond to steroids alone and can be used as second-line therapies or in combination with prednisone. Cyclophosphamide and tacrolimus are used more frequently in patients with interstitial lung disease.
Acthar is a synthetic adrenocorticotropic hormone (ACTH) that is FDA-approved for the treatment of polymyositis and dermatomyositis but the exact mechanism of action is unknown and this agent acts through melanocortin receptors. Currently, there is limited data on its effectiveness.
Intravenous immunoglobulin (IVIG)
Intravenous immunoglobulin (IVIG) is a purified blood product that contains healthy antibodies from thousands of blood donors, that enhances the body’s immune system response. Intravenous immunoglobulin (IVIG) is usually used in patients who do not respond to other therapies. Biologic agents provide a more targeted therapy and are often monoclonal antibodies or proteins targeting various immune system mediators. Anti-tumor necrosis factor (anti-TNF) agents such as etanercept, adalimumab, and infliximab have potential for use in polymyositis as they suppress tumor-necrosis factor proteins that are associated with inflammation but data supporting their effectiveness are limited. Rituximab is another biologic monoclonal antibody that targets B cells which play a role in inflammation. This agent has been studied in treating polymyositis and may provide some benefit to these patients; however, more studies are required.
Alternative medicine
Various vitamins and supplements, although not approved specifically for use in polymyositis, have been useful to patients. Examples include: coenzyme Q10 (CoQ10), creatine, fish oil, and others.
Physical therapy
Physical therapy and a regular exercise routine are often recommended even when the myositis is active as the beneficial effects of exercise are being increasingly reported. Patients also benefit from heat therapy, passive range-of-motion exercises, and splints to avoid contractures. Speech therapy may be useful if the swallowing muscles become weakened. Additionally, a registered dietician can be recommended if chewing and swallowing become problematic.
Speech therapy
If your swallowing muscles are weakened by polymyositis, speech therapy can help you learn how to compensate for those changes.
Dietetic assessment
Later in the course of polymyositis, chewing and swallowing can become more difficult. A registered dietitian can teach you how to prepare easy-to-eat, nutritious foods.
Coping and support
Living with a chronic autoimmune disease can make you wonder at times whether you’re up to the challenge. To help you cope, try supplementing your medical care with the following suggestions:
- Know your illness. Read all you can about polymyositis and other muscle and autoimmune disorders. Talk to other people who have a similar condition. Don’t be afraid to ask your doctor any questions that you may have concerning your illness, diagnosis or treatment plan.
- Be a part of your medical team. Consider yourself, your doctor and any other medical experts involved as a united front in the fight against your disease.
- Following the treatment plan you agreed to is vital. Keep your doctor updated on any new signs or symptoms you may experience.
- Know and assert your limits. Learn to say no effectively and ask for help when you need it.
- Rest when you’re tired. Don’t wait until you’re exhausted. This will only set you back further as your body tries to recuperate. Learning to pace yourself can help you maintain a consistent level of energy, accomplish just as much and feel better emotionally.
- Acknowledge your emotions. Denial, anger and frustration are normal feelings when you must deal with an illness. Things don’t seem normal or fair and likely seem out of your control. Feelings of fear and isolation are common, so stay close to your family and friends. Try to maintain your daily routine as best you can and don’t neglect doing those things you enjoy. Many people find support groups to be a helpful resource.





{kind=link}