Contents
What is bullous pemphigoid
Bullous pemphigoid is an autoimmune, chronic supepidermal blistering skin disease. Autoimmune disorders are generated when your body’s natural defenses (e.g., the immune system with its antibodies) against “foreign” or invading organisms, attack healthy tissue for unknown reasons. In bullous pemphigoid, patients have autoantibodies that are against the basement membrane of the epidermis. This means that the blisters (bullae) are actually subepidermal, so they are less fragile than those of pemphigus vulgaris. If you see a patient with bullous pemphigoid, you’ll see lots of intact, tense blisters (bullae), rather than a bunch of ruptured bullae covered with scabs. The immunofluorescence pattern is correspondingly different – you’ll see just a line at the base of the epidermis (rather than the lace-like outlining of epidermal cells you see in pemphigus vulgaris). Generalized blistering occurs in and under the upper layers of the skin and usually subsides spontaneously within several months or years.
Patients with bullous pemphigoid are generally elderly presenting with crops of tense pruritic (itchy) blisters and the clinical presentation varies a lot (but usually it doesn’t start in the mouth, like pemphigus vulgaris). Bullous pemphigoid is a less serious disease than pemphigus, usually, since the bullae (blisters) often don’t rupture so there’s less chance of infection and scarring. Mucosal involvement may occur and a number of clinical subtypes exist. Bullous pemphigoid affects males and females in equal numbers.
The first symptom of bullous pemphigoid is usually redness of the skin surrounding a lesion, scar, and/or the navel (umbilicus). Within weeks, thin walled blisters with clear fluid centers (bullae) appear on the undersurfaces of the arms and legs (flexor surfaces), in the armpits (axillae), on the abdomen, and/or around the groin. These blisters are create red, itchy patches. Unlike pemphigus, bullous pemphigoid blisters usually do not affect the mucous membrane lining the mouth; if they do they heal rapidly.
The blisters are usually hard and tight, and contain clear or blood-tinged fluid; they do not rupture easily. If the blisters do rupture, pain may occur but healing is usually rapid.
Bullous pemphigoid usually itches and in its early phase, itching and hive-like patches may be the only symptoms.
After a few months, the symptoms of bullous pemphigoid often disappear spontaneously, but they may recur for no apparent reason.
Figure 1. Skin structure
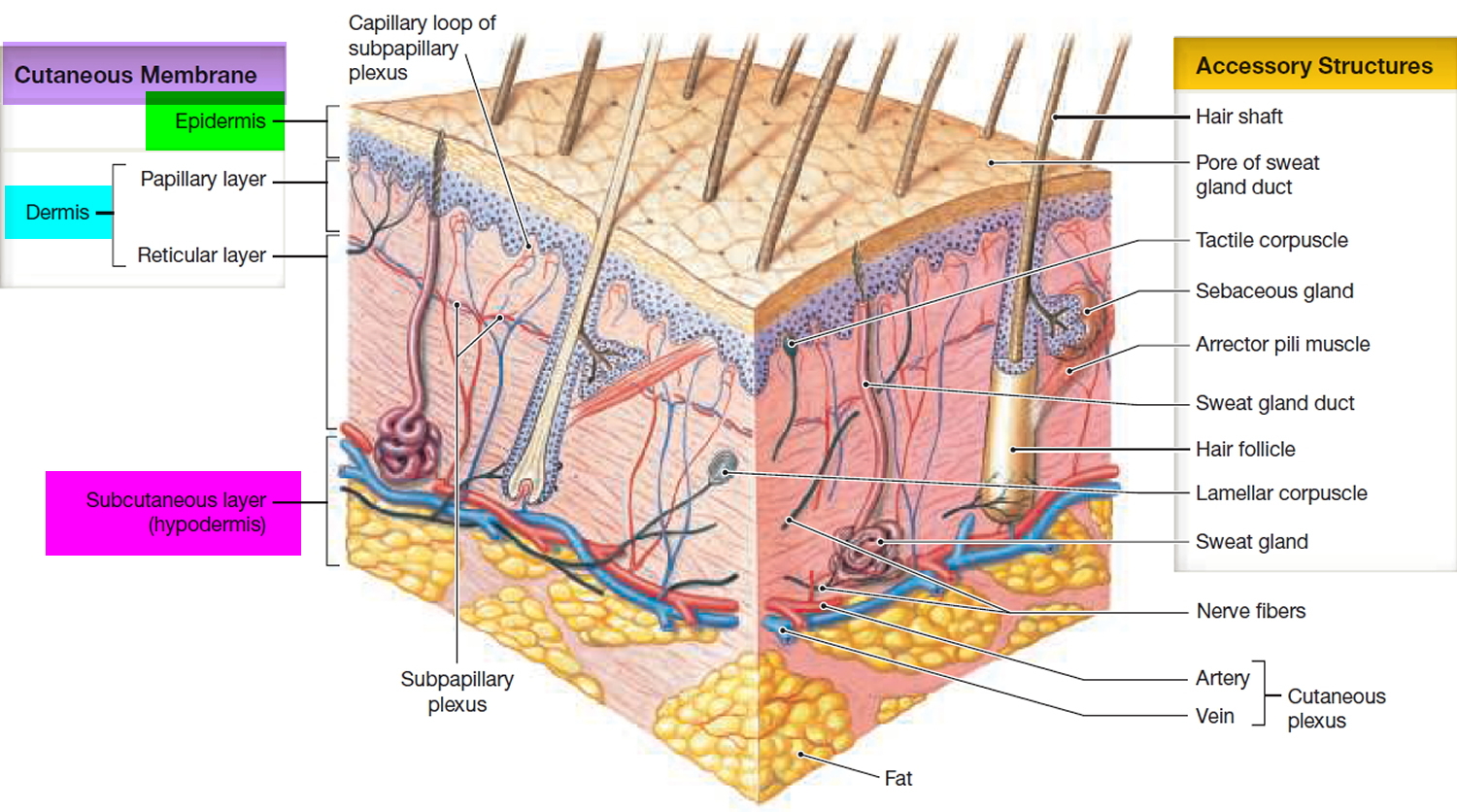
Figure 2. Bullous pemphigoid
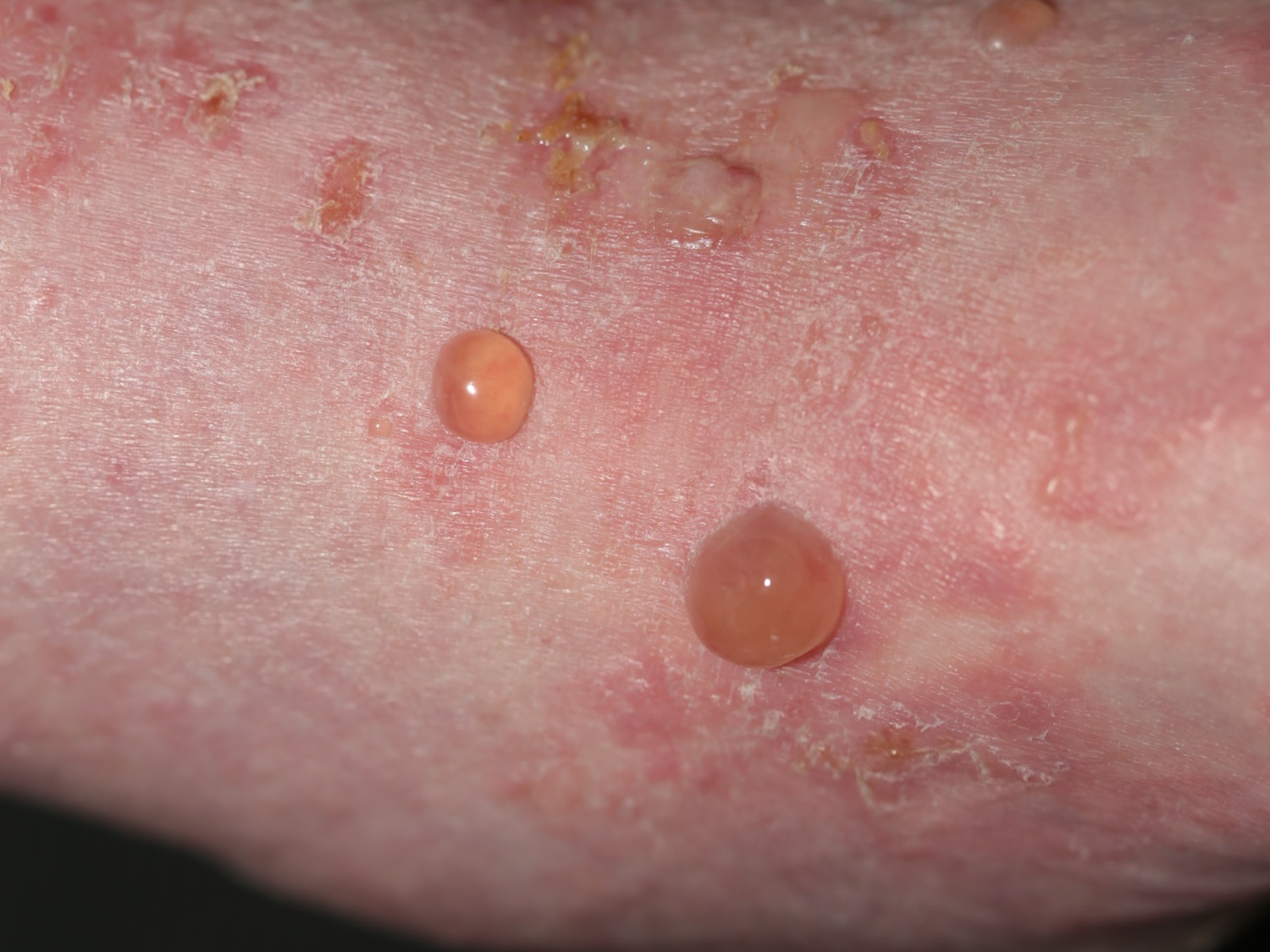
Figure 3. Bullous pemphigoid (diffuse)
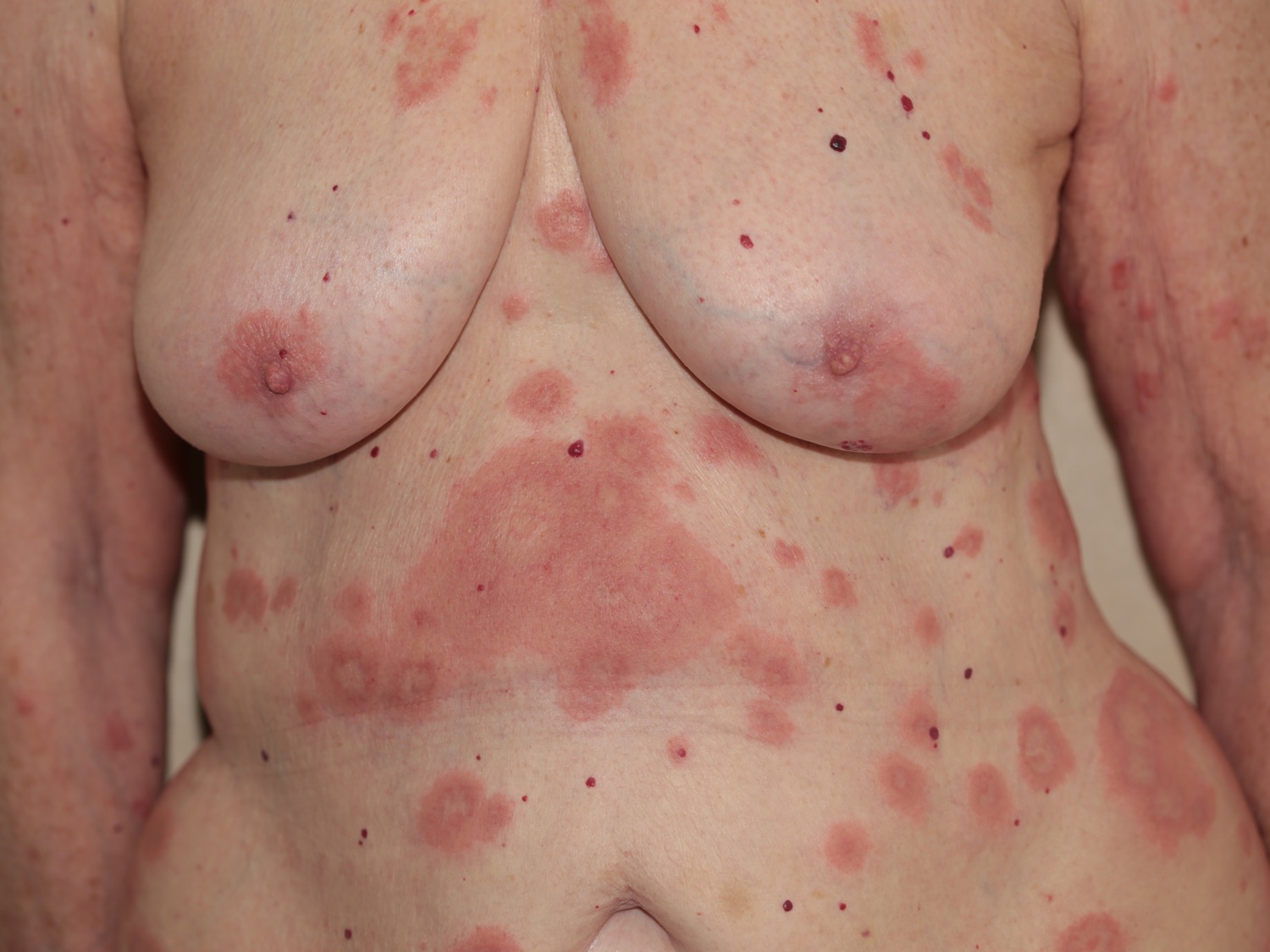
Figure 4. Bullous pemphigoid (localized)
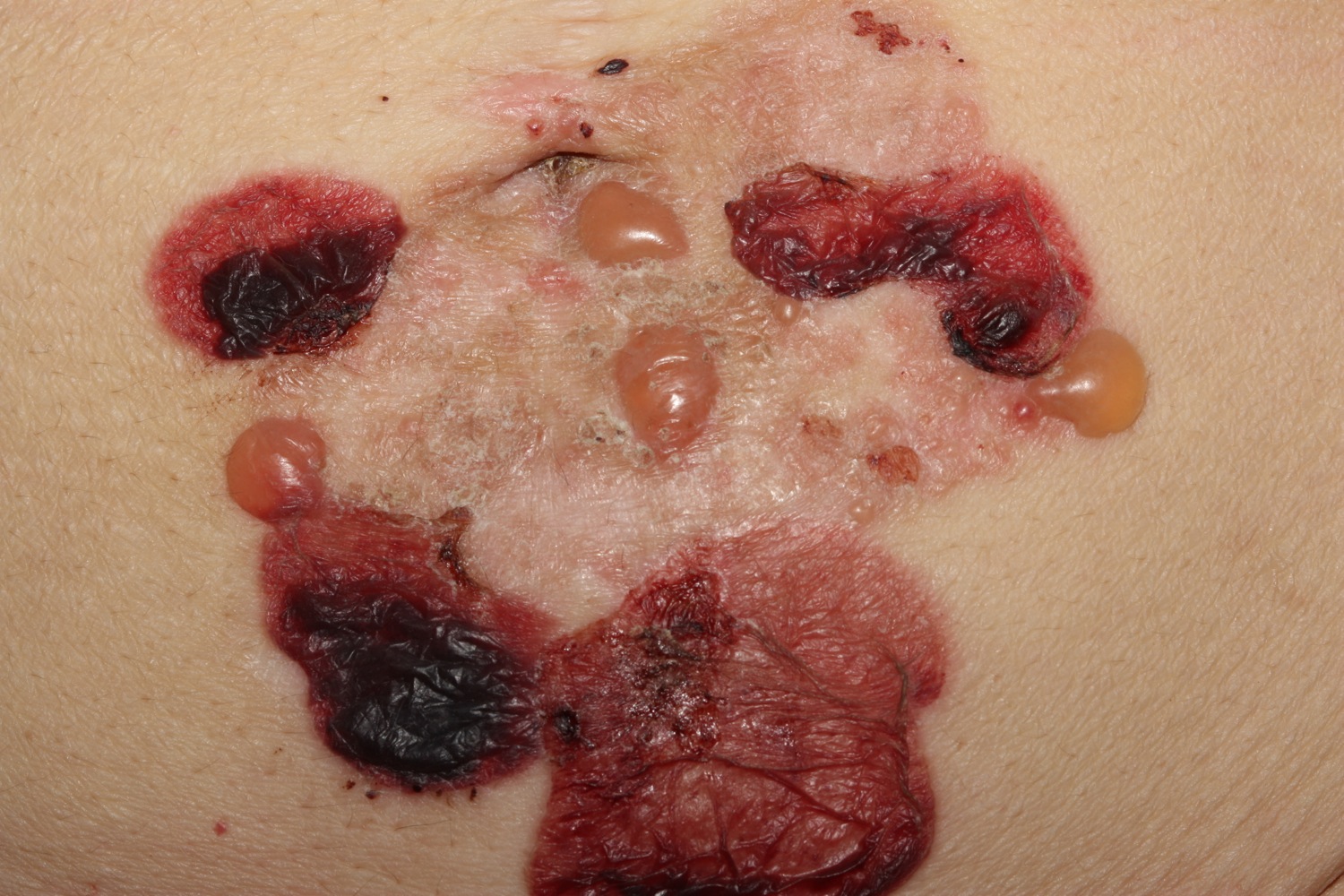
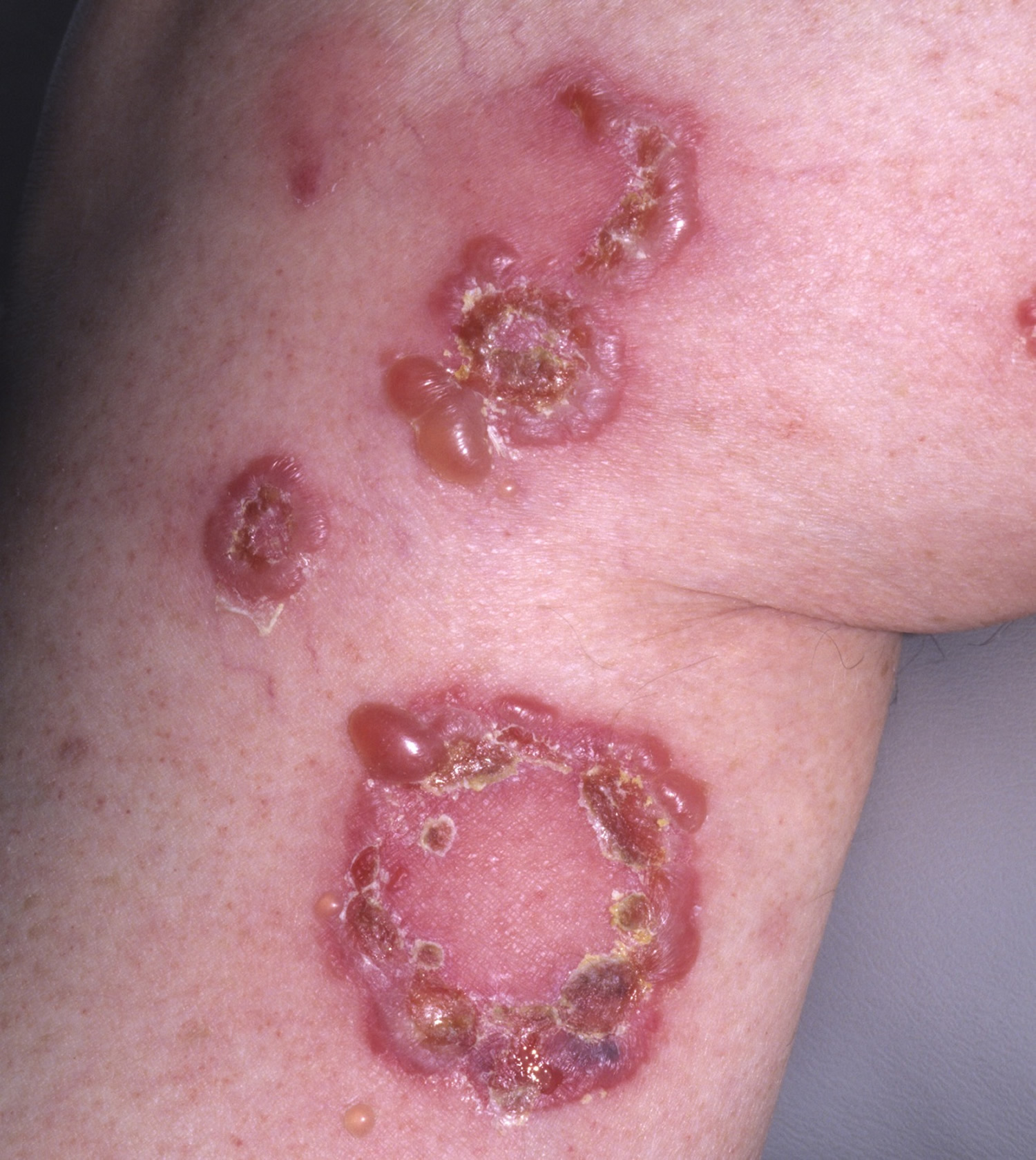
Figure 5. Bullous pemphigoid (rings may form)
Is bullous pemphigoid contagious?
No. bullous pemphigoid is an autoimmune skin condition where your body’s immune system produces antibodies to the fibers that connect the outer layer of skin (epidermis) and the next layer of skin (dermis). These antibodies trigger inflammation that produces the blisters and itching of bullous pemphigoid.
Who gets bullous pemphigoid?
Bullous pemphigoid often presents in people over 80 years of age, and mostly affects people over 50. It can occur in younger adults, but bullous pemphigoid in infants and children is rare.
- Bullous pemphigoid occurs equally in males and females.
- There are HLA associations indicating genetic predisposition to the disease.
- It is more prevalent in elderly patients with neurological disease, particularly stroke, dementia and Parkinson disease.
- The risk of developing bullous pemphigoid is greater in people with the skin disease, psoriasis, and it can be precipitated by treatment of psoriasis with phototherapy.
- There may be an association with internal malignancy in some patients.
- A drug, an injury, or skin infection can trigger the onset of disease.
Bullous pemphigoid causes
Bullous pemphigoid is the most common autoimmune dermatosis where your body produces antibodies (IgG +/- IgE immunoglobulins) and activated T lymphocytes (white blood cells) that attack components of the basement membrane of the epidermis, particularly the bullous pemphigoid antigens BP180 (also called Type XVII collagen) and less frequently BP230 (a plakin). These bullous pemphigoid antigen proteins are within the NC16A domain of collagen XVII. They are associated with the hemidesmosomes, structures that ensure the epidermal keratinocyte cells stick to the dermis to make a waterproof seal.
The binding of the autoantibodies to the proteins and/or release of cytokines from the T cells lead to complement activation, recruitment of neutrophils (acute inflammatory cells) and the release of proteolytic enzymes. These destroy the hemidesmosomes and cause the formation of subepidermal blisters.
The association of neurological disease with the skin disease is thought to relate to the presence of collagen XVII in the central nervous system and in skin hemidesmosomes.
Drug-Induced Bullous Pemphigoid
Both topical and oral iodine have been reported to cause bullous pemphigoid. Rarely, bullous pemphigoid may be drug-induced (e.g. everolimus, sirolimus furosemide, penicillamine, captopril, etc.). Dipeptidyl peptidase-4 inhibitors used to treat diabetes have been reported to trigger bullous pemphigoid, specifically linagliptin, sitagliptin and vildagliptin. Immune checkpoint inhibitors may cause bullous pemphigoid, e.g., nivolumab.
Bullous pemphigoid symptoms
Bullous pemphigoid causes severe itch and (usually) large, tense bullae (fluid-filled blisters), which rupture forming crusted erosions.
Other variable features include:
- Nonspecific rash for several weeks before blisters appear
- Eczematous areas resembling nummular dermatitis
- Urticaria-like red skin
- Annular (ring-shaped) lesions
- Smaller blisters (vesicles)
- Prurigo nodules (pemphigoid nodularis)
- Clear or cloudy, yellowish or blood stained blister fluid
- Postinflammatory pigmentation
- Milia in healed areas
Bullous pemphigoid may be localized to one area, or widespread on the trunk and proximal limbs.
- Frequently it affects the skin around skin folds.
- Blisters inside the mouth and in genital sites are uncommon.
Some patients have a diagnosis of bullous pemphigoid made despite not having any bullae (non-bullous pemphigoid). This can affect any body site.
Oral involvement is less common than in pemphigus vulgaris. Rarely, bullous pemphigoid may preferentially affect the sun-exposed areas or be induced by UVB. In patients who scratch too much, prurigo nodularis lesions may develop, called pemphigoid nodularis. The clinical presentation of bullous pemphigoid in children is similar to that in adults except there is more frequently involvement acrally and of the mucous membranes. A very rare form of erythrodermic bullous pemphigoid has been reported 1. bullous pemphigoid is the most common bullous disease to coexist with psoriasis 2.
Neurologic disorders, e.g. Parkinson’s disease, dementia, stroke, multiple sclerosis, are much higher in patients with bullous pemphigoid than general population. It is thought that neurodegenerative processes uncover bullous pemphigoid Ag1-n, an alternatively spliced form of bullous pemphigoid Ag1-e that stabilizes the cytoskeleton of sensory neurons, generating autoantibodies that may subsequently lead to bullous pemphigoid by cross-reacting with bullous pemphigoid Ag1-e. A study reported an associated occurrence of bullous pemphigoid in 21% cases of neurological disorder (8% stroke, 7% dementia, 3% Parkinsonism, 2% epilepsy and 1% multiple sclerosis) of more than a year’s duration 3.
Bullous pemphigoid prognosis
Overall mortality is increased with the diagnosis of bullous pemphigoid. Older age > 70 years, low Karnofsky performance scale and neurologic disease confer a higher mortality. Death is attributed to infection in the majority of cases.
Infection is seen in about half of patients and is associated with demential and poor general function 4.
Bullous pemphigoid complications
Bullous pemphigoid can be a serious disease, particularly when widespread or resistant to treatment. Morbidity and mortality result from:
- Bacterial staphylococcal and/or streptococcal skin infection and sepsis
- Complications of treatment
- Underlying and associated diseases
Risk of death is 2-6 times higher with bullous pemphigoid compared with the general population. Infection is the cause of death in up to 60% of cases. Pneumonia is 3 times higher than the general population. Older age, poor general condition, dementia and the use of high-dose corticosteroids are risk factors 5.
Bullous pemphigoid diagnosis
When typical bullae are present, the diagnosis is suspected clinically. In most cases, the diagnosis will be confirmed by a skin biopsy of an early blister. The diagnosis can also be made from non-blistered, inflamed skin.
Pathological examination of bullous pemphigoid shows a split under the epidermis. A dermal neutrophilic infiltrate is usual but not always present. Eosinophils may be prominent.
Direct immunofluorescence staining of a skin biopsy taken adjacent to a blister highlights antibodies along the basement membrane that lies between the epidermis and dermis.
Blood tests include an indirect immunofluorescence test for both circulating BP230 IgG as well as BP180 IgG antibodies.
Other tests will relate to planning and monitoring treatment.
Bullous pemphigoid treatment
If the bullous pemphigoid is very widespread, hospital admission may be arranged to dress blisters and erosions.
Most patients with bullous pemphigoid are treated with steroid tablets, usually prednisone. The dose is adjusted until the blisters have stopped appearing, which usually takes several weeks. The dose of prednisone is then slowly reduced over many months or years. As systemic steroids have many undesirable side effects, other medications are added to ensure the lowest possible dose (aiming for 5 to 10 mg prednisone daily).
Treatment is usually needed for several years. In many cases, the pemphigoid eventually completely clears up and the treatment can be stopped. If it recurs, it can be started again.
Medical treatment involves:
- Ultrapotent topical steroids to treat limited disease (eg clobetasol proprionate)
- Moderate potency topical steroids and emollients to relieve itch and dryness
- Systemic steroids
- Steroid-sparing medications
- Antibiotics for secondary bacterial infection
- Pain relief
- First Steps
- Oral prednisone alone at 40-60 mg/ day in a single morning dose. A dramatic clinical response will be seen in 70-80% of patients after 2-3 weeks.
- Clobetasol ointment or another superpotent topical steroid is applied to the affected areas twice daily.
- Subsequent Steps
- Over 2-8 weeks, taper the prednisone dose to 30 mg/day by reduction of the daily dose by 10 mg every 2-3 weeks. Then, over the next 2-3 months, reduce prednisone to 10-20 mg daily. After several months, shift the patient to alternate steroid therapy, reaching 10 mg every other day. When 10 mg every other day is reached, taper dose by 1 mg every other day every 1-4 weeks until the lowest amount of prednisone controlling the appearance of new lesions is reached.
These other medications may include:
- Tetracycline antibiotics, eg, doxycycline
- Nicotinamide
- Dapsone
- Methotrexate
- Azathioprine
- Mycophenolate
- Intravenous immunoglobulin
- Rituximab
People on systemic steroids may also receive additional medicines to alleviate their potential side effects (such as gastritis, hypertension, diabetes and osteoporosis).
Alternative Steps
Alternative agents may be used in patients who do not rapidly respond to systemic steroids or in patients in whom the use of systemic steroids is contraindicated.
- If the patient has complications or side effects from the prednisone, has persistent severe disease, or if sufficient clinical response is not achieved after 3 weeks, begin an immunosuppressive agent in combination with prednisone. In each case, continue the immunosuppressive agent for at least 2 months before abandoning it as ineffective. Once the steroid-sparing agent has begun to work, the dose of systemic steroids is gradually tapered following the protocol above. The choice of immunosuppressive agent is determined by the patient’s coexistent medical conditions, the risk of complications, and the cost. There is no evidence that one agent is superior to another. Steroid-sparing immunosuppressives include:
- Mycophenolate mofetil 500 mg – 1.5 mg twice daily (1.0-3.0 g daily)
- Azathioprine 100-150 mg/day
- Methotrexate 5mg to 25 mg once weekly as a single oral dose
- Cyclophosphamide 50-150 mg/day
- Adjunctive measures that may be added to prednisone or prednisone plus immunosuppressive agents.
- Tetracycline 500 mg three or four times daily plus niacinamide 500 mg three times daily. This may be adequate alone to control mild disease or may provide additional antiinflammatory effect without causing additional immunosuppression.
- Plasmapheresis to remove circulating pemphigoid autoantibody may be of value, but should be combined with cyclophosphamide 50-150 mg to inhibit antibody production.
- Intravenous immunoglobin infusions (IVIG) may be used in refractory cases. The dose is 2g/kg given over 3-5 days. This is very costly and is less frequently required than in pemphigus vulgaris.
Assessment and monitoring
As systemic treatment may be required for bullous pemphigoid for long periods, the extent and severity of the disease should be recorded carefully at baseline and at follow-up appointments. The following aspects may be considered.
- Body sites affected (skin and mucous membranes)
- Type of lesion: transient and non-transient
- Numbers of lesions: blisters, urticarial weals, eczematous plaques,
- Severity of itch
- Observation point: initial phase, active treatment, reducing treatment, maintenance phase on minimal treatment, or complete remission off treatment
- Current treatment
The Bullous Pemphigoid Disease Area Index has separate scores for skin and mucous membrane activity.
Blood pressure, body weight, DEXA bone scan, and blood tests are required to monitor therapy, as medications used for bullous pemphigoid may have serious side effects in some patients.
Pitfalls of treatments
- Patients with bullous pemphigoid are often elderly and frail. They tolerate high-dose systemic steroid therapy poorly. Do not be reluctant to add low doses of a steroid-sparing agent to allow for reduction in the systemic steroid dose. Complications usually occur when systemic steroid dose is not reduced judiciously.
- Potential complications of systemic steroids and immunosuppressants must be carefully sought in all patients so treated.
- Other bullous diseases such as epidermolysis bullosa acquisita, erythema multiforme, and bullous drug eruptions may clinically resemble pemphigoid and may explain atypical therapeutic responses.
- Immunosuppression may result in reactivation of herpes simplex or varicella zoster virus infection. These bullous infectious diseases may be generalized and simulate a flare of the pemphigoid.
Bullous pemphigoid treatment diet
There is no diet specifically for bullous pemphigoid. However, a regular exercise combined with a healthy diet that’s rich in vitamin D, calcium and phosphates will help minimize the side effects of long term corticosteroids use.





{kind=link}