Contents
What is medulloblastoma
Medulloblastoma is an aggressive cancerous (malignant) brain tumor that only develops in the posterior fossa of the brain – classically in the midline of the cerebellum. The cerebellum is involved in muscle coordination, balance and movement. Medulloblastoma is an important tumor because medulloblastoma occurs in childhood and medulloblastoma can be very aggressive. Medulloblastoma is made up of small cells that are believed to have a neuro-ectodermal origin. Medulloblastoma is thought to arise from neural stem cell precursors in the granular cell layer of the cerebellum.
The brain is contained within the cranial vault, and is divided into several sections by folds of dura mater (one of the three membranes encasing the brain). The posterior fossa of the skull contains the cerebellum and brainstem.
The medulloblastoma is the second most common brain tumor in children. But the most common malignant (high grade) childhood brain tumor 1. Approximately 350 to 1,000 new cases are diagnosed in children and adults each year in the United States. Medulloblastoma accounts for 16-20% of all pediatric brain tumors, and 40-50% of all cerebellar tumors in childhood are medulloblastoma. In contrast, only 1% of brain tumors in adults are medulloblastomas. Medulloblastoma occurs bimodally, with 50% occur in the fist decade of life between 3 and 4 years and 8 and 9 years of age. The incidence of medulloblastoma peaked in those age 9 years and younger. Approximately 10 to 15% of medulloblastomas are diagnosed in infancy. Medulloblastoma accounts for less than 1% of central nervous system (CNS) tumors in adults, with highest incidence in adults 20 to 34 years of age. Medulloblastomas are slightly more common in males than in females. In 1 to 2% of patients, medulloblastoma is associated with Gorlin syndrome, a nevoid basal carcinoma syndrome. Medulloblastoma also occurs in up to 40% of patients with Turcot syndrome.
Incidence of medulloblastoma decreased with age 2:
- Incidence was 0.53 per 100,000 population in children age-groups 0–4,
- 0.56 per 100,000 population in children age-groups 5–9,
- 0.33 per 100,000 population in children age-groups 10–14, and
- 0.16 per 100,000 population in adolescents age 15–19 years.
Based on different types of gene mutations, there are at least four subtypes of medulloblastoma. Though medulloblastoma is not inherited, syndromes such as Gorlin’s syndrome or Turcot’s syndrome might increase the risk of medulloblastoma.
The term Primitive Neuro Ectodermal Tumor or PNET is a relatively new term that is used to describe a group of tumors. In these Primitive Neuro Ectodermal Tumor (PNET), the type of cell seen is very similar. Their locations are quite different.
Tumors that fall under the heading Primitive Neuro Ectodermal Tumor (PNET) include:
- medulloblastoma (the most common),
- pineoblastoma,
- ependymoblastoma,
- retinoblastoma,
- neuroblastoma and
- esthesioneuroblastoma.
Other than the medulloblastoma, these are all rare tumors. These tumors generally occur in children.
Medulloblastomas initially grow into the cerebellum – the past of the brain that controls balance and posture. After this medulloblastoma may spread via the cerebrospinal fluid (CSF) into the spinal cord and other areas around the brain. As medulloblastoma grows it may block the flow of cerebrospinal fluid (CSF), leading to the development of hydrocephalus (increased head size caused by increased fluid in the brain) in young infants and children, or raised intracranial pressure in older children and adults. Medulloblastoma rarely spreads to other areas of the body.
The most common presenting symptoms of medulloblastoma cancer are headache, vomiting, and ataxia. Ataxia describes a lack of muscle control or coordination of voluntary movements, such as walking or picking up objects. Additional features that may be observed include lethargy, motor or cranial nerve impairment, gaze palsy, visual impairment due to hydrocephalia, vertigo/hearing loss, behavioral changes/irritability, and extracranial pain (e.g. back pain in those with spinal metastases). Around 30% of pediatric medulloblastoma cases present with metastases at diagnosis. Most metastases occur within the central nervous system by seeding via the cerebrospinal fluid (cranial or spinal), while spread to extracranial organs (e.g. bone marrow, liver, lungs) is very rare at diagnosis.
Children need to be seen at a center that has a team of pediatric specialists with expertise and experience in pediatric brain tumors, with access to the latest technology and treatments for children. Standard treatment includes surgery, chemotherapy, and, depending on the age of the patient, radiation therapy 3.
Figure 1. Human brain
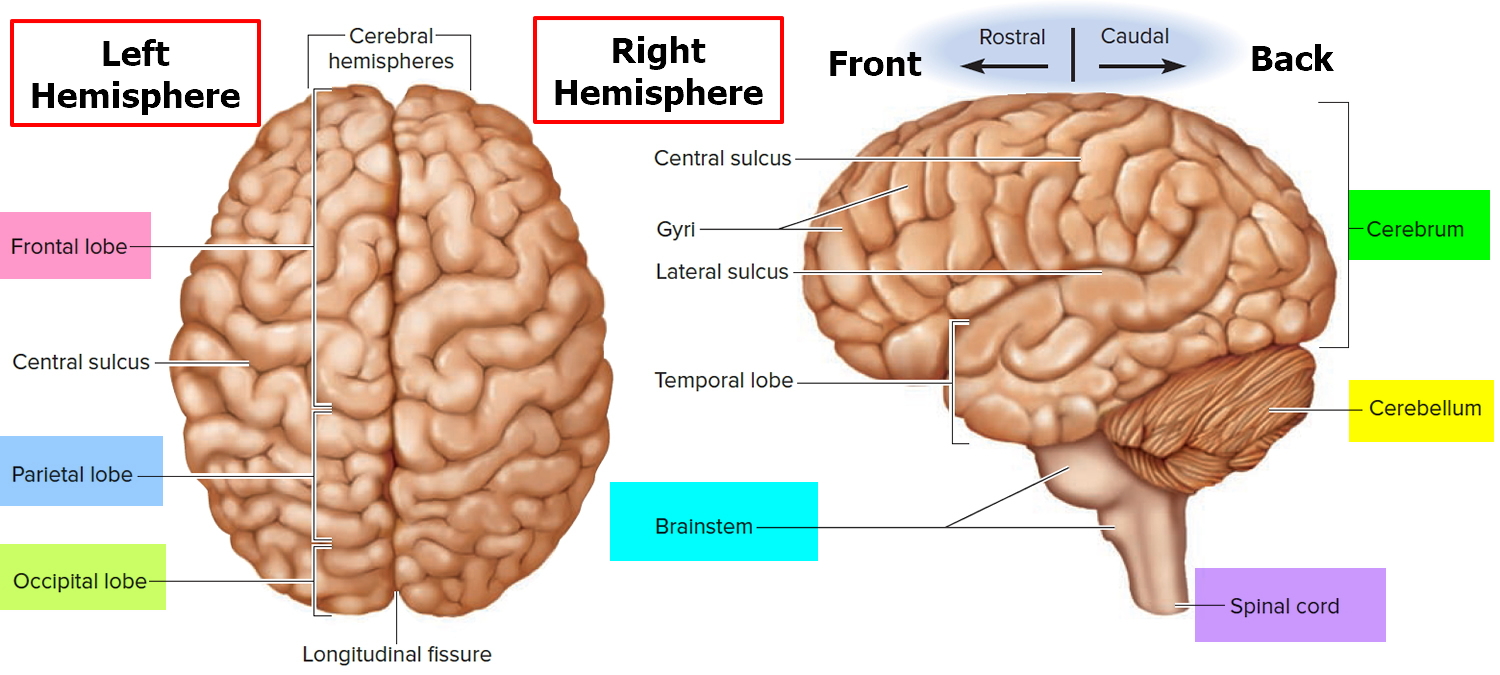
Figure 2. Medial aspect of the human brain
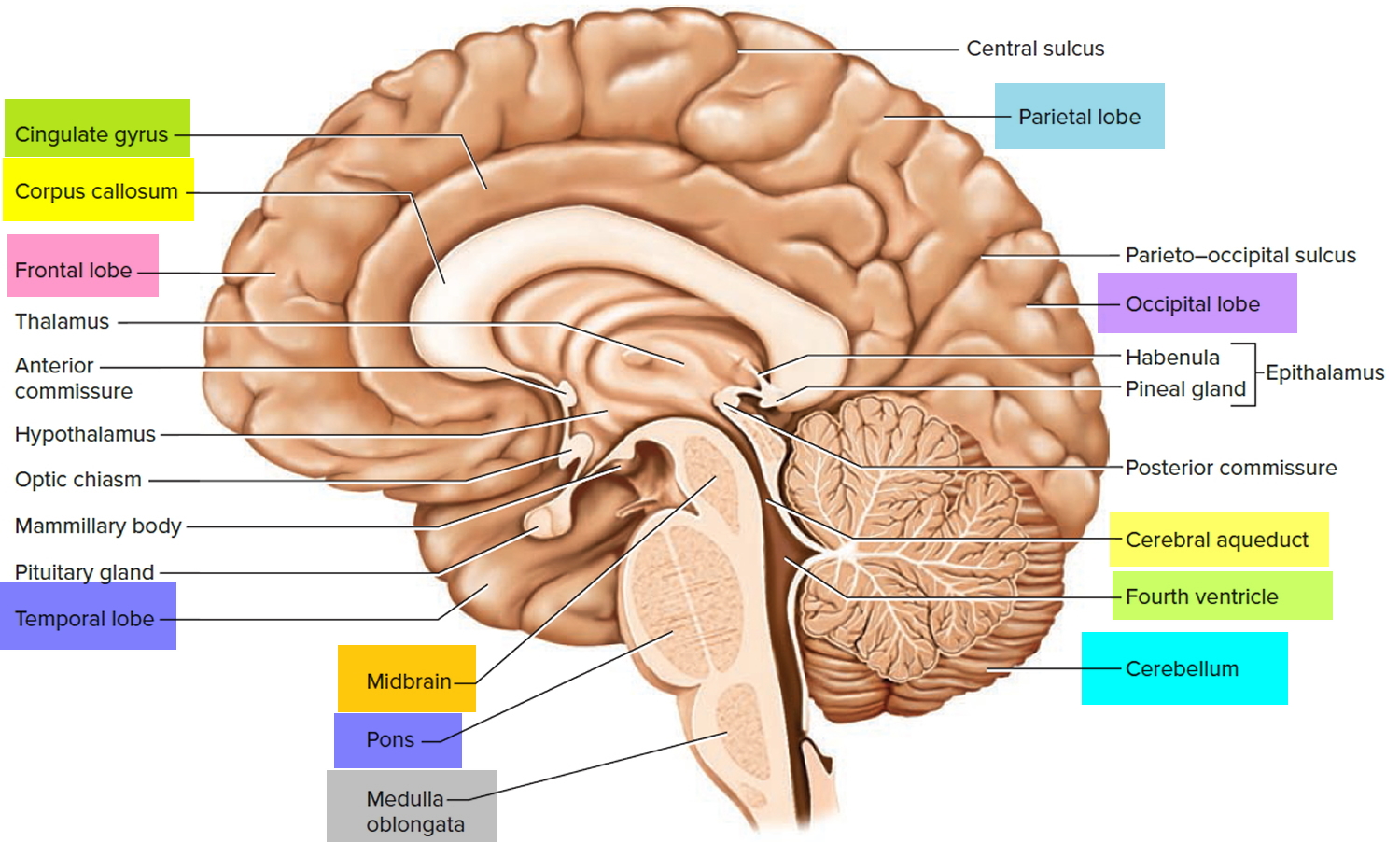
Figure 3. Cerebellum of brain
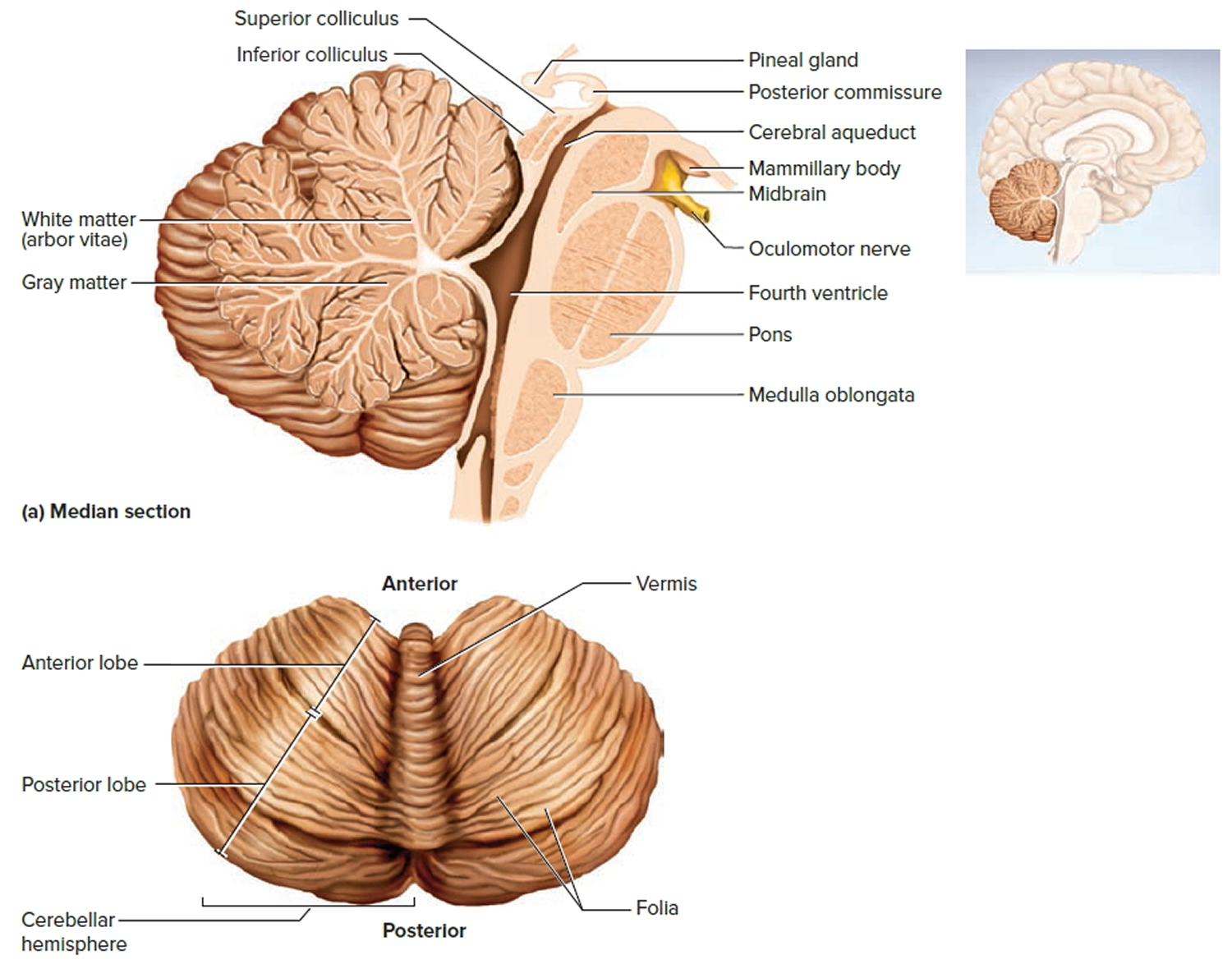
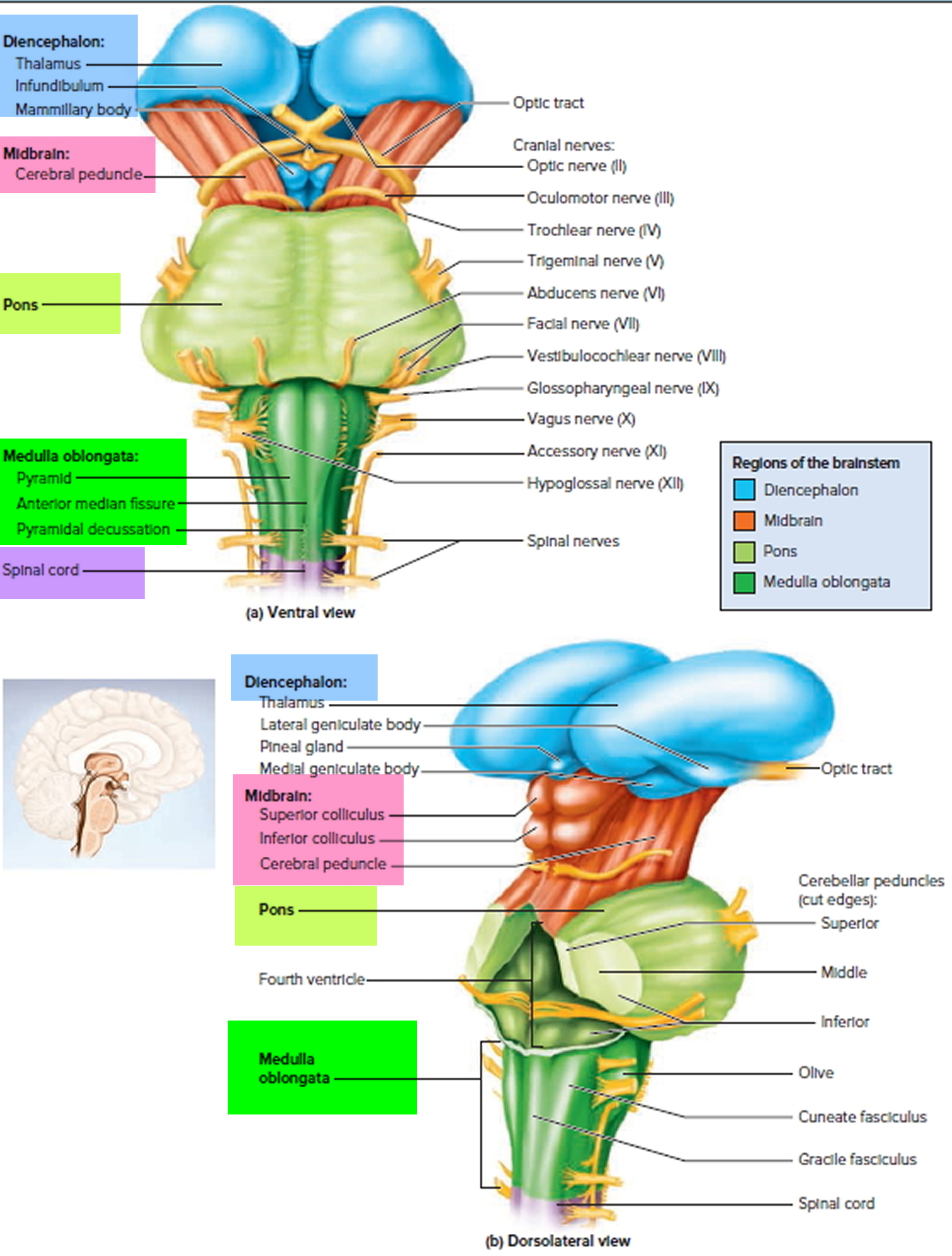
Figure 4. The Brainstem
Medulloblastomas classification
Successfully treating your child’s medulloblastoma depends largely on whether the tumor can be completely removed through surgery and whether the tumor has spread to other parts of the brain or spinal cord. Typically, post-surgery medulloblastomas are divided into three risk assessment groups:
- Infants (children under age 3)
- Standard risk (no evidence of disease with a complete removal of the tumor)
- High risk (evidence of incomplete removal or tumor spread elsewhere in the nervous system)
Medulloblastoma subtypes
Clinical researchers involved in international collaborations that have revealed that medulloblastomas are comprised of at least four different subtypes.
Medulloblastoma subtypes include 4:
- Wingless (WNT) Medulloblastoma
- Sonic Hedgehog (SHH) Medulloblastoma
- Group 3 (also called group C) Medulloblastoma
- Group 4 (also called group D) Medulloblastoma
These groups are associated with specific age-groups, with SHH being most common in infants and adults, and all other groups being more common in childhood. Several review articles have elaborated on the details of these subgroups and their implications for diagnosis and treatment 5.
Figure 5. Medulloblastoma
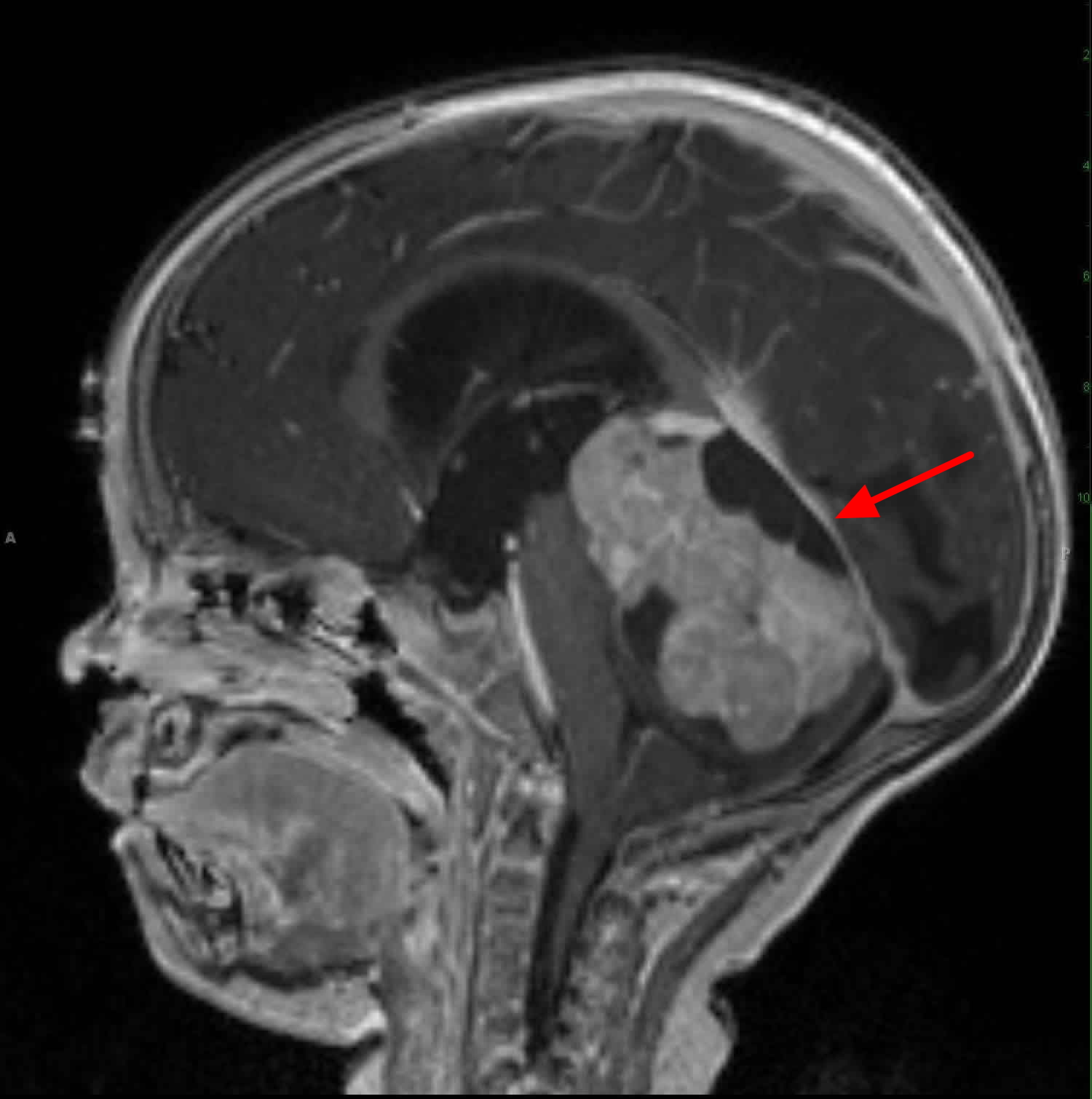
Figure 6. Medulloblastoma subtypes
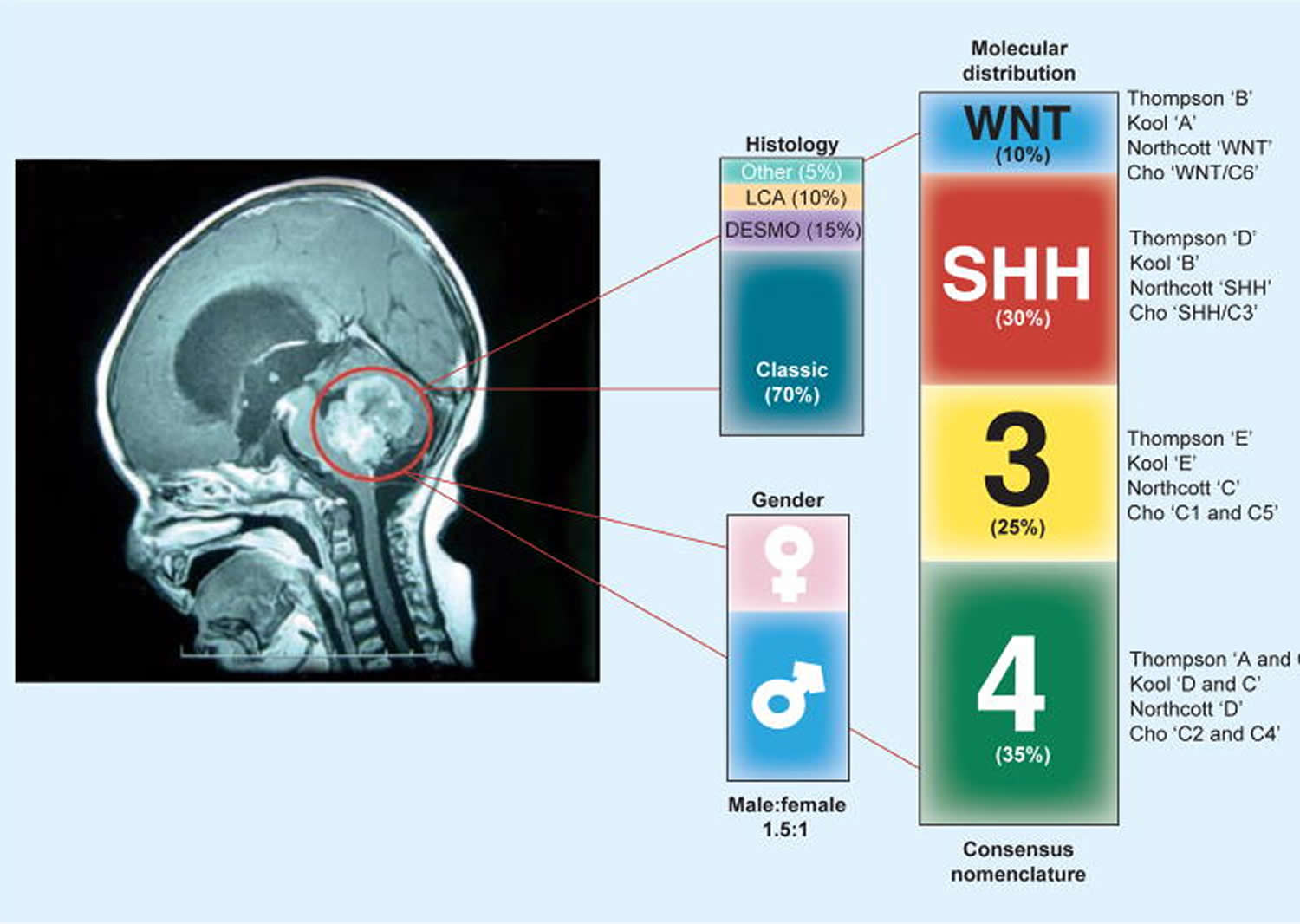
Each of these subtypes has a different biological driver, and some subtypes require more intensive treatments than others. These findings are guiding clinical trials exploring new treatments for children with medulloblastoma.
After your doctors complete all necessary tests, they meet to review and discuss what they have learned about your child’s condition. Then your doctors will meet with you and your family to discuss the results and outline the best treatment options.
Medulloblastoma prognosis
The prognosis (chance of recovery) and treatment options depend on:
- the tumor’s molecular subtype (the specific genetic mutations within the tumor)
- the age of your child at the time of diagnosis
- the location of the tumor
- the amount of tumor remaining after surgery
- whether the cancer has spread to other parts of the central nervous system (brain and spinal cord), or to other parts of the body, such as the bones
The outcome for children with medulloblastoma has improved dramatically over the past several decades.
The outcomes in infants remain poor and many studies are underway to evaluate new treatment strategies in infants. Some include aggressive chemotherapy, including high-dose chemotherapy and stem cell transplant, and localized radiation therapy to minimize the harmful effects of radiation therapy on the developing brain.
Medulloblastoma survival rate
Provided the medulloblastoma is not widespread at diagnosis, it is often responsive to treatment, otherwise it is quite an aggressive tumor with a poor outcome. More than 73% of children survive longer than 5 years after diagnosis with 10-year survival was 64.7% for medulloblastoma 2.
Survival rates are often used by doctors as a standard way of discussing a person’s prognosis (outlook). The 5-year survival rate refers to the percentage of children who live at least 5 years after their cancer is diagnosed. Of course, many children live much longer than 5 years (and many are cured).
To get 5-year survival rates, doctors have to look at children who were treated at least 5 years ago. Improvements in treatment since then might result in a better outlook for children now being diagnosed with brain tumors.
The numbers below come from the Central Brain Tumor Registry of the United States (CBTRUS) and are based on children aged 19 or younger who were treated between 2010 and 2014. There are some important points to note about these numbers 2:
- In some cases, the numbers include a wide range of different types of tumors that can have different outlooks. For example, the survival rate for PNETs below includes medulloblastomas, pineoblastomas, and PNETs in other parts of the brain. Medulloblastomas tend to have a better outlook than the other PNETs. Therefore the actual survival rate for medulloblastomas would be expected to be higher than the number below, while the number for other PNETs would likely be lower.
- Medulloblastoma 5 year survival rate is about 73% (between 70.6-75.2 percent)
- Medulloblastoma 10 year survival rate is about 64.7% (between 61.8-67.4 percent)
Note that many other factors can also affect a child’s outlook, such as the location and extent of the tumor and how well it responds to treatment. Even taking these other factors into account, survival rates are at best rough estimates. Your child’s doctor knows your child’s situation and is your best source of information on this topic.
Medulloblastoma causes
The exact underlying cause of medulloblastoma is unknown. Most cases occur randomly for no apparent reason (sporadically). In some cases, medulloblastoma is associated with certain inherited diseases, including:
- Li-Fraumeni syndrome
- Nevoid basal cell carcinoma syndrome (Gorlin syndrome)
- Turcot syndrome
It’s important to understand that these and other brain tumors most often occur with no known cause. There’s nothing that you could have done or avoided doing that would have prevented the tumor from developing.
Many cases of medulloblastoma are associated with chromosomal abnormalities. These abnormalities are not inherited (i.e., are not passed on from one generation to the next), but occur at some unknown point during a child’s development, even during the development of a fetus or embryo. Although medulloblastomas are associated with chromosomal changes, they are not inherited.
In individuals with cancer, malignancies may develop due to abnormal changes in the structure and orientation of certain cells. As mentioned above, the specific cause or causes of such changes are unknown. However, research suggests that abnormalities of DNA (deoxyribonucleic acid), which is the carrier of the body’s genetic code, are the underlying basis of cellular malignant transformation. Depending upon the form of cancer present and several other factors, these abnormal genetic changes may occur spontaneously for unknown reasons (sporadically).
Evidence suggests that, in approximately one-third to one-half of individuals with a medulloblastoma, tumor cells may have a specific chromosomal abnormality, known as isochromosome 17q, with associated loss or inactivation of certain genetic information. Chromosomes, which are present in the nucleus of human cells, carry the genetic characteristics of each individual. Pairs of human chromosomes are numbered from 1 through 22, with an unequal 23rd pair of X and Y chromosomes for males and two X chromosomes for females. Each chromosome has a short arm designated as “p” a long arm identified by the letter “q” and a narrowed region at which the two arms are joined (centromere).
An isochromosome is an abnormal chromosome with identical arms on each side of the centromere. More specifically, in certain cases of medulloblastoma, there is duplication of the long arm and deletion of the short arm of chromosome 17. Some researchers suggest that such structural abnormalities of chromosome 17 may lead to inactivation of a gene on the chromosome that normally acts as a tumor suppressor, potentially leading to malignant transformation of certain cells. However, the implications of such findings remain unclear.
Additional chromosomal abnormalities have been identified in individuals with medulloblastoma including abnormalities on chromosome 1, 7, 8, 9, 10q, 11, and 16. How these various abnormalities play a role in the development of medulloblastoma is unknown. Further research is needed to determine the complex underlying mechanisms responsible for the development of a medulloblastoma.
In individuals with cancer, including medulloblastoma, malignancies may develop due to abnormal changes in the structure and orientation of certain cells known as oncogenes or tumor suppressor genes. Oncogenes control cell growth; tumor suppressor genes control cell division and ensure that cells die at the proper time. Oncogenes that are associated with medulloblastoma include ERBB2, MYCC, and OTX2. Many medulloblastomas are characterized by alterations in specific molecular signaling pathways that result in uncontrolled cell growth. Pathways implicated in medulloblastoma include the WNT pathway, the SHH pathway and the myc pathway.
In extremely rare cases, medulloblastomas occur in individuals who have certain inherited disorders including Gorlin syndrome (nevoid basal cell carcinoma), Turcot syndrome, Li Fraumeni syndrome, Rubinsten-Taybi syndrome, Nijmegen breakage syndrome, neurofibromatosis and ataxia-telangiectasia. Individuals with these disorders have an increased risk of developing a medulloblastoma.
Researchers theorize that medulloblastoma originates from immature cells that are somehow prevented from maturing (i.e., differentiating) into more specialized cells, which have “intended”, specific functions within the tissue in question. Such immature or incompletely differentiated cells may grow and divide at an unusually rapid, uncontrolled rate that cannot be contained by the body’s natural immune defenses. Eventually, such proliferation of abnormal cells may result in formation of a mass known as a tumor (neoplasm).
Several different subtypes of medulloblastoma have been identified including anaplastic (large cell) medulloblastoma; classic medulloblastoma; desmoplastic nodular medulloblastoma; medulloblastoma with extensive nodularity (MBEN); medullomyoblastoma; and melanotic medulloblastoma. The various subtypes of medulloblastoma appear different on a cellular level, but as yet to not influence treatment options. However, in the future such distinctions may be used to develop novel, targeted therapies based on a particular subtype and other factors.
Extensive transcriptional profiling of human medulloblastomas has recently yielded a second and more precise classification system that stratifies medulloblastomas according to their mRNA expression profiles. Four subgroups with distinct mRNA signatures have been identified, and are presently categorized as WNT, Sonic hedgehog (SHH), Group 3 and Group 4.
Medulloblastomas in the WNT subgroup feature genetic alterations that affect members of the Wnt signaling pathway, which are linked to the processes of embryogenesis and oncogenesis. Mutations of the ß-catenin gene and monosomy 6 are among the more common genetic events that define this subgroup, and the incidence rate among males and females is approximately equal. WNT subgroup medulloblastomas tend to affect older children and are rare in adults. Among the different subgroups, WNT tumors have the best prognosis and clinical outcomes. The SHH subgroup is characterized by up-regulation of members of the SHH signaling family. Common genetic events exclusive to this subgroup are mutations in the genes for PTCH, the receptor of SHH, and SUFU, a negative regulator of SHH signaling pathway. SHH tumors are the most common subgroup of medulloblastoma found in infants and adults, and they carry an intermediate prognosis. Like the WNT subgroup, the incidence of SHH tumors is equal for males and females. Group 3 tumors are characterized by over-amplification of MYC and genes related to phototransduction and glutamate signaling. These tumors are also known for their high frequency of metastasis and have the worst prognosis of any medulloblastoma subtype. Group 3 tumors are extremely rare in adults, and are more prevalent in males than females. The last subgroup, currently known as Group 4, is characterized by up-regulation of genes related to neuronal or glutameminergic signaling. Although these tumors are common across all age groups, comparatively little is known about them. Like Group 3, Group 4 tumors are more prevalent in males and have a high tendency to metastasize. Their prognosis is considered intermediate.
Medulloblastoma is sometimes classified as a primitive neuroectodermal tumor or PNET. PNETs are a group of tumors that arise from primitive nerve cells in the brain. A medulloblastoma is sometimes referred to as a primitive neuroectodermal tumor of the posterior fossa.
Risk factors for medulloblastoma
As for most brain tumors the cause of medulloblastoma is unknown. However, like all other brain tumors, there are a few known risk factors:
- Ionizing radiation is known to be a possible cause;
- Genetic studies show a familial risk
Cerebellar medulloblastoma is a feature of basal cell nevus syndrome, von Hippel-Lindau syndrome and familial adenomatous polyposis. In a formal risk analysis for brain tumors in familial adenomatous polyposis, Hamilton et al. 6 found that the relative risk of cerebellar medulloblastoma in patients with familial adenomatous polyposis was 92 times that for the general population.
Medulloblastoma symptoms
The specific symptoms associated with a medulloblastoma will vary from one person to another based upon the exact location and size of a medulloblastoma and whether the tumor has spread to other areas. Affected individuals may not have all of the symptoms discussed below. Affected individuals should talk to their physician and medical team about their specific case, associated symptoms and overall prognosis.
Medulloblastomas typically involve the fluid-filled fourth cavity (ventricle) of the brain. The brain has four cavities called ventricles that are filled with cerebrospinal fluid (CSF) and joined by channels, through which CSF circulates. Because the tumor often fills the fourth ventricle, CSF circulation is obstructed, resulting in hydrocephalus. Hydrocephalus is a condition in which the accumulation of excess CSF in the brain causes a variety of symptoms, including repeated, often severe vomiting, lethargy and headaches that frequently occur in the morning and improve as the day goes on. Additional symptoms may include irritability, increased head size, and paralysis (paresis) of the muscles that help control eye movements (extraocular muscles).
Many infants and children with a medulloblastoma develop papilledema, a condition in which the optic nerve swells because of increased intracranial pressure. The optic nerve is the nerve that transmits impulses from the retina to the brain. Papilledema can cause reduced clarity of vision. Because many the symptoms associated with a medulloblastoma are nonspecific and often subtle, papilledema may the first sign that brings affected infants and children to the attention of a neurologist.
Children with medulloblastoma often have evidence of cerebellar dysfunction. Symptoms may include poor coordination, difficulty walking, and clumsiness (ataxia). Affected children may fall frequently and develop an unsteady, clumsy manner of walking (unsteady gait). They may tend to stand with their feet widely separated, stagger or sway when walking and easily lose their balance.
As a tumor grows or spreads, additional symptoms can develop. Such symptoms may include double vision (diplopia), rapid, jerky movements of the eyes (nystagmus), facial weakness, ringing in the ears (tinnitus), hearing loss and a stiff neck. Some children with double vision may tilt their heads in an effort align the two images.
Signs and symptoms of medulloblastoma may include:
- Nighttime or morning headache (generally upon awakening in the morning)
- Poor coordination and unsteady walk (ataxia)
- Nausea and vomiting
- Dizziness
- Double vision (diplopia)
- Head bobbing or neck tilt
- Nystagmus (an abnormal, side-to-side movement of the eyes)
- Lethargy (tiredness) or confusion
- Hydrocephalus due to obstruction of normal cerebrospinal fluid circulation
- Changes in personality or behavior
- Seizures
Rarely, medulloblastoma can spread into the central nervous system or the spinal canal, and your child may experience:
- loss of strength in the lower extremities
- back pain
- bowel and bladder control issues
- difficulty walking
These symptoms may be related to the tumor itself or be due to the buildup of pressure within the brain.
Medulloblastoma diagnosis
Your doctor may start with a neurological exam of your child.
Tests and procedures used to diagnose medulloblastoma include:
- Neurological exam. During this procedure, vision, hearing, balance, coordination and reflexes are tested. This helps determine which part of the brain might be affected by the tumor.
- Imaging tests. Imaging tests can help determine the location and size of the brain tumor. These tests are also very important to identify pressure or blockage of the CSF pathways. A computerized tomography (CT) scan or magnetic resonance imaging (MRI) may be done right away. These tests are often used to diagnose brain tumors. Advanced techniques, such as perfusion MRI and magnetic resonance spectroscopy, also may be used.
- Tissue sample testing (biopsy). A biopsy is usually not done, but it may be recommended if the imaging tests are not typical of medulloblastoma. The sample of suspicious tissue is analyzed in a lab to determine the types of cells.
- Removal of cerebrospinal fluid for testing (lumbar puncture). Also called a spinal tap, this procedure involves inserting a needle between two bones in the lower spine to draw out cerebrospinal fluid from around the spinal cord. The fluid is tested to look for tumor cells or other abnormalities. This test is only done after managing the pressure in the brain or removing the tumor.
A medulloblastoma may appear similar to other kinds of brain tumors. After surgery, a pathologist can examine the tumor cells under a microscope and make a definitive diagnosis. Once the diagnosis of medulloblastoma is clear, your doctors can determine the most appropriate treatment for your child.
To see if the medulloblastoma has spread, the doctor may recommend a lumbar puncture to test your child’s cerebrospinal fluid for cancer cells.
Medulloblastoma treatment
The treatment for medulloblastoma depends on a number of factors including the general health of the patient and the size and position of the tumor. Treatment for medulloblastoma usually includes surgery followed by radiation or chemotherapy, or both.
Your child’s physician will determine a specific course of medulloblastoma treatment based on several factors, including:
- your child’s age, overall health and medical history
- type, location, and size of the tumor
- extent of the disease
- your child’s tolerance for specific medications, procedures or therapies
- how your child’s doctors expects the medulloblastoma cancer to behave
Medulloblastomas occur much more commonly in children than adults, and treatment is not without risk. There may be some long-term effects of treatment including growth and hormonal changes, behavioral changes and possible learning problems, and these need to be discussed with the treating doctor.
Treatment for medulloblastoma focuses on removing as much of the tumor as safely possible and relieving pressure in the child’s skull (intracranial pressure) due to swelling or hydrocephalus. In addition to surgical removal of the tumor, the doctor may sometimes recommend a shunt to help drain cerebrospinal fluid buildup and steroid treatments to reduce tumor swelling.
Surgery is followed by radiation and chemotherapy. These therapies address cancer cells that might have been unreachable by surgery and those that have spread from the tumor to other parts of the brain or spinal cord. Medulloblastoma spread and recurrence is common; radiation and chemotherapy can reduce the risks.
This three-part approach – surgery, radiation and chemotherapy – can offer survival in up to 75 percent of patients. It is important to understand that each of the three treatments, especially radiation treatment of the brain, may cause complications that could affect your child’s development.
It is essential to discuss each stage of your child’s therapy thoroughly with your doctors so you can make informed choices for your child and understand potential benefits and risks.
Enrolling your child in a clinical trial may offer additional options for treatment. Your doctor can refer you to studies if they are appropriate.
- Surgery to relieve fluid buildup in the brain. A medulloblastoma may grow to block the flow of cerebrospinal fluid, which can cause a buildup of fluid that puts pressure on the brain (hydrocephalus). Surgery to create a pathway for the fluid to flow out of the brain (external ventricular drain or ventriculoperitoneal shunt) may be recommended. Sometimes this procedure can be combined with surgery to remove the tumor.
- Endoscopic third ventriculostomy (ETV) or ventriculo-peritoneal shunt (VP shunt): In an endoscopic third ventriculostomy, surgeons create a small hole that allows fluid to flow around the blockage and into the spinal column. About 90 percent of children with symptoms of hydrocephalus will undergo this procedure. In some cases, children may have an alternative procedure in which a tube is installed to drain excess fluid into the abdomen (VP shunt).
- Surgery to remove the medulloblastoma. A pediatric or adult brain surgeon (neurosurgeon) removes the tumor, taking care not to harm nearby tissue. But sometimes it’s not possible to remove the tumor entirely because medulloblastoma forms near critical structures deep within the brain. All patients with medulloblastoma should receive additional treatments after surgery to target any remaining cells.
- Radiation therapy. You might have radiotherapy to the brain and sometimes the whole of the spinal cord. About 1 in 5 people (20%) with meduloblastoma have spread to the spinal cord when they are diagnosed. In other people, there is a risk that it will spread. So you have radiotherapy to reduce this risk or treat spread that is already there. A pediatric or adult radiation oncologist administers radiation therapy to the brain and spinal cord using high-energy beams, such as X-rays or protons, to kill cancer cells. Standard radiation therapy can be used, but proton beam therapy — available at a limited number of major health care centers in the United States — delivers higher targeted doses of radiation to brain tumors, minimizing radiation exposure to nearby healthy tissue.
- Chemotherapy. Chemotherapy uses drugs to kill tumor cells. Typically, children and adults with medulloblastoma receive these drugs as an injection into the vein (intravenous chemotherapy). Chemotherapy may be recommended after surgery or radiation therapy, or in certain cases, at the same time as radiation therapy. In some cases, high dose chemotherapy followed by stem cell rescue (a stem cell transplant using the patient’s own stem cells) may be used.
- Clinical trials. Clinical trials enroll eligible participants to study the effectiveness of new treatments or to study new ways of using existing treatments, such as different combinations or timing of radiation therapy and chemotherapy. These studies provide a chance to try the latest treatment options, though the risk of side effects may not be known. Talk with your doctor for advice.
Posterior fossa syndrome
About 25 out of 100 (25%) people have particular symptoms after medulloblastoma surgery. The symptoms are called posterior fossa syndrome and they can be very mild or severe. Symptoms include difficulty talking, swallowing or walking. This syndrome is thought to be more unusual in children.
“Posterior fossa mutism” is a condition that may occur after surgery. Within 24 hours, the child develops an inability to speak, has problems with balance and has difficulty with swallowing. The condition may range from mild to severe. The cause of this condition is not entirely known but it is unique to this area of the brain.
Posterior fossa syndrome might develop from one day to a week after medulloblastoma surgery. The symptoms usually improve slowly over a few weeks or months. But they may not go away completely in some people. Research is trying to find out what causes posterior fossa syndrome.
Complications of treatment
Other complications include post-operative infection, paralysis, nerve palsies, cognitive dysfunction and growth retardation.
Treatment for children under 3
The specialist will usually avoid using radiotherapy to the whole brain and spine if your child is younger than 3. Their young age makes them more likely to develop long term side effects.
The specialist might recommend chemotherapy instead. This aims to keep your child’s tumor under control until radiotherapy is likely to cause less damage.
In general your child is likely to have high dose chemotherapy with a number of drugs. They might also have radiotherapy just to the area containing the tumor. This way, radiotherapy to the whole brain and spinal cord can be delayed until your child is older. Or it might be avoided altogether.
Your child has chemotherapy using a number of drugs given into their vein. They also have chemotherapy into the fluid around the spinal cord (intrathecal chemotherapy).
Follow up
You will have regular check ups once you finish your treatment. Your doctor will examine you and ask about your general health.
This is your chance to ask questions and to tell your doctor if anything is worrying you.
How often you have check ups depends on your individual situation.
Coping and support
Finding out you or your child has a brain tumor may feel overwhelmed, as though things are out of your control, just when you need to make crucial decisions. Coping with the shock, fear and sadness that come with a cancer diagnosis can take time. With time, each person finds a way of coping and coming to terms with the diagnosis.
You are likely to have a range of emotions that change very quickly. You might feel upset, frightened and confused. One day you might feel positive and able to cope but the next day feel the exact opposite. This is natural.
Counseling can help you to cope with the difficulties you’ll face. It can help to reduce your stress and improve your quality of life.
You are more able to cope and make decisions if you have information about your brain tumor and its treatment. Information also helps you to know what to expect.
Taking in information can be difficult at first. Make a list of questions before you see your doctor. Take someone with you to remind you what you want to ask and help remember the answers.
Ask your doctors and nurses to explain things again if you need them to.
Remember, you don’t have to sort everything out at once. It might take some time to deal with each issue. Ask for help if you need it.
Treatment causes side effects. These can be mild or more severe. Tell your doctor or nurse if you have any or if they get worse. They can treat them and help you find ways of coping.
What questions should I ask if my child has medulloblastoma?
You and your family are key players in your child’s medical care. It’s important that you share your observations and ideas with your child’s health care provider and that you understand your doctor’s recommendations. If your child has been diagnosed with medulloblastoma, you probably have a lot on your mind. So it’s often helpful to write questions down. Some of the questions you may want to ask include:
- What does a diagnosis of medulloblastoma mean for my child?
- How will you manage my child’s symptoms?
- What are my child’s treatment options?
- How many other children with medulloblastoma does your team treat each year?
- What are the possible short and long-term complications of treatment?
- What is the long-term outlook for my child?
- How likely is it that the tumor will come back?
- What services are available to help my child and my family cope?
Until you find what brings you the most comfort, consider trying to:
- Find out enough about the cancer to make decisions about your care. Ask your doctor for the specifics about your cancer, such as its type and stage. And ask for recommended sources of information where you can learn more about your treatment options. The National Cancer Institute 7 and the American Cancer Society 8 are good places to start.
- Stay connected to friends and family. Your friends and family can provide a crucial support network for you during your cancer treatment. As you begin telling people about your cancer diagnosis, you’ll likely get offers for help. Think ahead about things you may like help with, whether it’s having someone to talk to if you’re feeling low or getting help preparing meals.
- Find someone to talk to. You might have a close friend or family member who’s a good listener. Or talk to a counselor, medical social worker, or pastoral or religious counselor.
Consider joining a support group for people with cancer. You may find strength and encouragement in being with people who are facing the same challenges you are. Ask your doctor, nurse or social worker about groups in your area. Or try online message boards, such as those available through the American Cancer Society 8.
Talking to other people
Talking to your friends and relatives about your brain tumor can help and support you. But some people are scared of the emotions this could bring up and won’t want to talk. They might worry that you won’t be able to cope with your situation.
It can strain relationships if your family or friends don’t want to talk. But talking can help increase trust and support between you.
Help your family and friends by letting them know if you would like to talk about what’s happening and how you feel.
You might find it easier to talk to someone outside your own friends and family. For example your specialist nurse, or other people in a similar situation to you. You could join a support group, or contact one of the brain tumor charities.
- Central Brain Tumor Registry of the United States. Statistical Report: Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2004–2007 http://www.cbtrus.org/2011-NPCR-SEER/WEB-0407-Report-3-3-2011.pdf[↩]
- CBTRUS Statistical Report: Primary brain and other central nervous system tumors diagnosed in the United States in 2010–2014. Neuro-Oncology, Volume 19, Issue suppl_5, 6 November 2017, Pages v1–v88. https://academic.oup.com/neuro-oncology/article/19/suppl_5/v1/4596648[↩][↩][↩]
- Crawford, J. R., MacDonald, T. J., Packer, R. J. Medulloblastoma in childhood: new biological advances. Lancet Neurol. 6: 1073-1085, 2007. http://www.thelancet.com/journals/laneur/article/PIIS1474-4422(07)70289-2/fulltext[↩]
- Kool M, Korshunov A, Remke M, et al. Molecular subgroups of medulloblastoma: an international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta Neuropathologica. 2012;123(4):473-484. doi:10.1007/s00401-012-0958-8. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3306778/[↩]
- Northcott PA, Dubuc AM, Pfister S, Taylor MD. Molecular subgroups of medulloblastoma. Expert review of neurotherapeutics. 2012;12(7):871-884. doi:10.1586/ern.12.66. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4334443/[↩]
- Hamilton, S. R., Liu, B., Parsons, R. E., Papadopoulos, N., Jen, J., Powell, S. M., Krush, A. J., Berk, T., Cohen, Z., Tetu, B., Burger, P. C., Wood, P. A., Taqi, F., Booker, S. V., Petersen, G. M., Offerhaus, G. J. A., Tersmette, A. C., Giardiello, F. M., Vogelstein, B., Kinzler, K. W. The molecular basis of Turcot’s syndrome. New Eng. J. Med. 332: 839-847, 1995 http://www.nejm.org/doi/full/10.1056/NEJM199503303321302[↩]
- National Cancer Institute. https://www.cancer.gov/[↩]
- American Cancer Society. https://www.cancer.org/[↩][↩]





{kind=link}